

Abstract
The aggregation of the amyloid beta (Aβ) peptide is associated with Alzheimer’s disease (AD) pathogenesis. Cell membrane composition, especially monosialotetrahexosylganglioside (GM1), is known to promote the formation of Aβ fibrils, yet little is known about the roles of GM1 in the early steps of Aβ oligomer formation. Here, by using GM1-contained liposomes as a mimic of the neuronal cell membrane, we demonstrate that GM1 is a critical trigger of Aβ oligomerization and aggregation. We find that GM1 not only promotes the formation of Aβ fibrils but also facilitates the maintenance of Aβ42 oligomers on liposome membranes. We structurally characterize the Aβ42 oligomers formed on the membrane and find that GM1 captures Aβ by binding to its arginine-5 residue. To interrogate the mechanism of Aβ42 oligomer toxicity, we design a new liposome-based Ca2+-encapsulation assay and provide new evidence for the Aβ42 ion channel hypothesis. Finally, we determine the toxicity of Aβ42 oligomers formed on membranes. Overall, by uncovering the roles of GM1 in mediating early Aβ oligomer formation and maintenance, our work provides a novel direction for pharmaceutical research for AD.
Keywords: amyloid beta, GM1, amyloid beta–membrane interaction, Aβ aggregation, Aβ oligomer toxicity, ion channel
Graphical Abstract
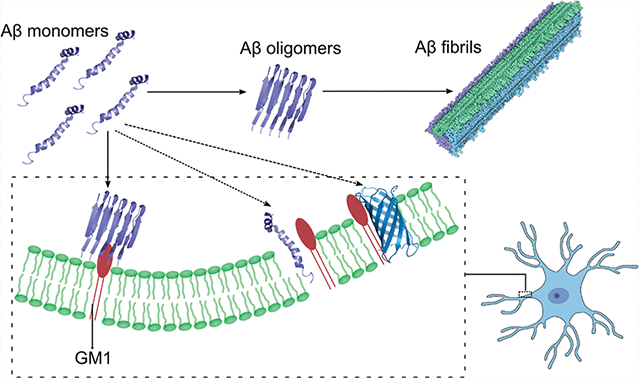
INTRODUCTION
Alzheimer’s disease (AD) is the most common neuro-degenerative disorder, responsible for 60–70% of dementia cases.1 The economic burden associated with caring for nearly 50 million people worldwide with AD is estimated in the hundreds of billions of dollars annually.2 Except for a few cases,3 most AD cases are characterized by the accumulation of extracellular plaques, predominantly composed of amyloid beta (Aβ), and cytoplasmic neurofibrillary tangles, mostly composed of the tau protein. Despite multiple proposed hypotheses,4–14 the molecular etiology of the disease remains a mystery. One of the oldest and most central hypotheses, the amyloid cascade hypothesis, posits that the accumulation of Aβ proteins in the brain is the primary cause of AD pathogenesis, leading to tau pathology, neuroinflammation, synapse loss, and ultimately neuron death.15–19 The amyloid cascade hypothesis was subsequently revised to the Aβ oligomer cascade hypothesis,20–24 stating that small Aβ oligomers25,26 rather than fibrils27,28 are the main toxic species. The oligomer hypothesis has been gaining significant momentum since numerous studies have shown that Aβ oligomers are toxic to primary neurons, inhibit hippocampal long-term potentiation, and cause memory impairment in rat and mouse models.29,30 Mounting evidence, stemming from other neurodegenerative diseases, also supports the oligomer hypothesis. For example, in amyotrophic lateral sclerosis, soluble superoxide dismutase (SOD1) oligomers31–33 of disease-associated proteins, rather than insoluble aggregates, are shown to be responsible for cytotoxicity. Insoluble SOD1 aggregates have been found to be protective against neuronal toxicity, 34 potentially due to competition with soluble oligomers.34,35
The mechanism of Aβ oligomer toxicity is unknown, but several studies23,36–39 show that Aβ oligomers may disrupt the plasma membrane to upset ionic (especially calcium) homeostasis. A number of mechanisms by which Aβ oligomers induce cell membrane disruption have been proposed, including membrane thinning,40 excessive membrane tabulation,41,42 membrane extraction through amyloid–lipid co-aggregation,43,44 and formation of ion channels to disrupt Ca2+ homeostasis.45,46 Apart from Aβ oligomer toxicity, the lipidchaperone hypothesis has also been proposed.47 Since cellular membranes are highly heterogeneous, containing many constituents, deciphering which cellular membrane components are catalyzing Aβ cytotoxic action is challenging to untangle. In the past decade, several findings have implied that GM1 (monosialotetrahexosylganglioside), a glycosphingolipid in cell membranes,48 may play an important role in Aβ oligomer toxicity pathways. These findings include the following: (1) AD patients have much higher amounts of GM1 than cognitively normal patients in their cerebrospinal fluid,49 (2) GM1 clusters48,50–52 affect the conformational transition of Aβ40, and (3) GM1-bound Aβ has been found in early pathological changes of the AD brain.53 Spurred by these findings, several natural questions that arise are (1) whether GM1 promotes the formation of Aβ42 oligomers, which are the most toxic Aβ species, (2) how does GM1 bind Aβ, and (3) whether GM1 levels are correlated with Aβ oligomer toxicity.
In this work, we employ in vitro, in silico, and cellular studies to interrogate the interaction between GM1 and Aβ42 oligomers and to uncover the mechanisms driving the assembly and toxicity of Aβ42 oligomers (Figure S1). We utilize GM1-contained liposomes to mimic GM1 clusters and find that GM1 not only promotes the formation of Aβ42 oligomers but also prevents specific oligomer species from further aggregating into fibrils, which are not as toxic as Aβ42 oligomers.54,55 By performing molecular dynamics (MD) simulations, we observe the binding between Aβ42 and GM1 and identify the most critical residue, R5, involved in the binding. We perform mutagenesis and verify the role of R5. We utilize a fluorescence assay to interrogate the mechanism of how Aβ42 disrupts the membrane and uncover the role of various membrane constituents in Aβ42 aggregation. These results provide new evidence supporting the permeabilization of lipid membranes through the formation of Aβ ion channels or membrane thinning. We purify Aβ42 oligomers formed on liposomes and find that the most likely molecular weight range is 45–100 kDa and that the membrane-associated Aβ42 oligomers are rich in β-sheets. Finally, we perform cellular studies to demonstrate the neuronal toxicity of membrane-associated Aβ42 oligomers. Overall, our results provide new insights into the mechanisms of Aβ42 aggregation by revealing the role of GM1 in mediating early Aβ42 oligomer formation and maintenance. Uncovering the mechanisms of Aβ42 aggregation and determining critical cellular players responsible for downstream neurotoxicity are essential for the development of novel pharmaceutical strategies to treat AD.
RESULTS
GM1 Liposomes Promote Early Aβ42 Oligomer Formation and Maintenance.
We first investigate whether GM1 promotes the formation of Aβ42 oligomers. To answer this question, we use GM1-containing liposomes to interrogate the interaction between GM1 and Aβ42 oligomers. Liposomes are artificial vesicles that form phospholipid bilamellar membranes, and we can control the composition of liposome membranes to mimic the GM1 clusters in neuron membranes. We incubate Aβ42 peptides in the presence and absence of liposomes consisting of GM1, sphingomyelin, and cholesterol (GM1 liposomes) for 0.5, 4, 24, and 72 h and then perform 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and silver staining (Figure 1a). We use cholesterol and sphingomyelin in liposomes because (1) they are indispensable components of GM1 clusters,56 and (2) cholesterol has been reported to catalyze Aβ42 aggregation in the presence of lipid membranes.57 When we start the incubation, some Aβ42 oligomers that are smaller than tetramers form promptly (Figure S2). In the absence of GM1 liposomes, more Aβ42 oligomer species with different molecular weights are formed within 0.5 h, but the amount of Aβ42 oligomer species then reduces with the increase in time. After 72 h, there are effectively no Aβ oligomers. We speculate that the oligomers have formed larger assemblies, such as amyloid fibrils. In the presence of GM1 liposomes, we still observe abundant Aβ oligomer species formed within 0.5 h, and the amount of oligomer species reduces even more rapidly than when Aβ is incubated without GM1 liposomes. However, after 72 h, we still observe a few clear Aβ oligomer bands (30, 35, 40, 45, and 60 kDa), indicating that some oligomers are maintained in their oligomeric state, thus suggesting some distinct oligomer species from those formed without GM1. Since the cellular membrane includes not only GM1 but also sphingomyelin, we prepare liposomes that are sphingomyelin-rich and do not contain GM1. We observe no massive Aβ oligomer formation in 24 h when incubating Aβ with the sphingomyelin-rich liposome (Figure S3). Thus, GM1 can promote the formation of fibrils by catalyzing oligomers to form fibrils, but GM1 can also facilitate the maintenance of certain oligomer species.
Figure 1.
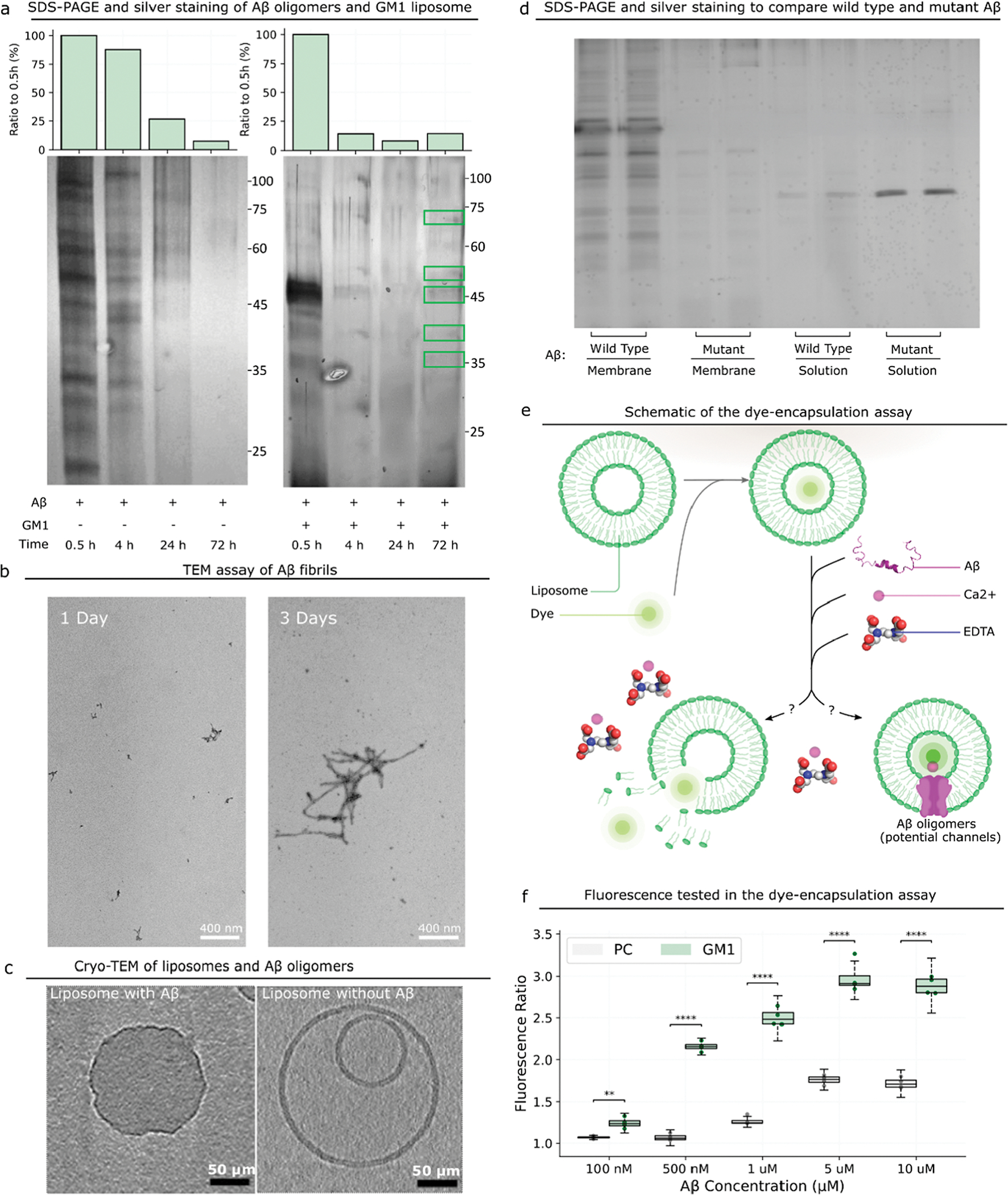
Interaction between Aβ and the GM1 liposome. (a) SDS-PAGE and silver staining results of Aβ oligomers formed in the absence and presence of GM1 liposomes at different time points. On the top is the quantitative analysis of Aβ oligomers at different time points. The lipid bilayer is visible in the untreated sample, and only a monolayer is visible after treatment. (b) The formation of Aβ fibrils is identified by TEM. (c) An approximately 10 nm-thick digital slices through tomograms of liposomes incubated with or without Aβ. Scale bars represent 50 μm. (d) Comparison of the amount of the membrane-bound Aβ wild type to that of R5G through SDS-PAGE and silver staining. (e) Schematic of the experimental design to prove the formation of Aβ ion channels with dye-encapsulated GM1 liposomes. (f) Ratio of the fluorescence intensity of dye-encapsulated liposomes incubated with Aβ to that without Aβ. P-value: NS (0.05 < p ≤ 1), * (0.01 < p ≤ 0.05), ** (0.001 < p ≤ 0.01), *** (0.0001 < p ≤ 0.001), and **** (p ≤ 0.0001).
To interrogate if the “missing” oligomers form fibrils, we determine the formation of fibrils (Figure 1b) through transmission electron microscopy (TEM). We incubate 80 μM Aβ42 for 1 and 3 days and find that scattered small fibrils (<200 nm) are formed after 1 day. After 3 days, large fibrils (≫400 nm) are formed (Figure 1b), indicating that 80 μM Aβ42 monomers aggregate into fibrils as the time increases. We also use thioflavin T (ThT)-binding assay to detect Aβ fibrils and find that ThT fluorescence intensities increase with time (Figure S4). Overall, previous studies have reported that Aβ binding to GM1 is a seeding step to form larger aggregates,53 and our work further shows that (1) Aβ42 oligomers form rapidly in 0.5 h but then partially disappear because they begin to assemble into higher-molecular weight aggregates and fibrils, and (2) GM1 maintains some Aβ42 oligomer species to avert further formation of aggregates and fibrils, potentially because these species have direct interaction with GM1. These oligomers are likely off the pathway to form fibrils and structurally distinct from oligomers formed on-pathway to fibrils.
Not only does GM1 affect Aβ42 oligomer formation but Aβ42 oligomers may also impact GM1 membranes; thus, we use cryogenic TEM (cryo-TEM) to analyze the morphology of liposomes incubated with or without Aβ42. In the absence of Aβ42, GM1 liposomes form bilamellar vesicles with smooth surfaces (Figure 1c), while in the presence of Aβ, GM1 liposomes become unilamellar vesicles, and the membranes of these liposomes are disordered and deformed. Thus, the presence of Aβ42 oligomers significantly affects the surface morphology of GM1 liposome membranes by deforming the membranes. These findings are consistent with previous reports.58,59 We posit that for neurons, Aβ42 may interact with GM1 clusters in cell membranes23,36–38 in a similar fashion in the brain, thereby resulting in a disrupted membrane and, ultimately, in neurotoxicity and synaptic loss.60
R5 of Aβ42 Plays an Important Role in the Interaction of Aβ and GM1 Membranes.
We perform 100 ns MD simulations of GM1 membranes and five Aβ42 monomers (Figure 2a). We calculate the minimum distance between atoms of each residue of Aβ and all atoms of GM1 on the membrane. We find that the fifth residue (arginine, R5) of Aβ maintains a distance of 1–2 Å from GM1 (Figure 2c,d), suggesting that R5 stably binds GM1. We also calculate the distance between Aβ and cholesterol (Figure S5), and we find that the average distance between Aβ and SM is larger than 10 Å, indicating no significant physical interactions between Aβ and GM1. Thus, we posit that R5 plays a critical role in the interaction between Aβ and GM1, specifically, the guanidine group in R5 interacting with the O11 and O12 atoms in GM1 (Figure 2b). We further computationally substitute R5 with glycine, and upon performing MD simulations, we find that the average distance between G5 and GM1 increases to >10 Å (Figure 2e) and is within binding distance (<2 Å) for as little as 2% of the time (Figure 2f), indicating that the mutation of R5G disrupts the tight binding interaction between the fifth residue and GM1. This result strongly suggests the critical role of R5 in GM1–Aβ interaction.
Figure 2.
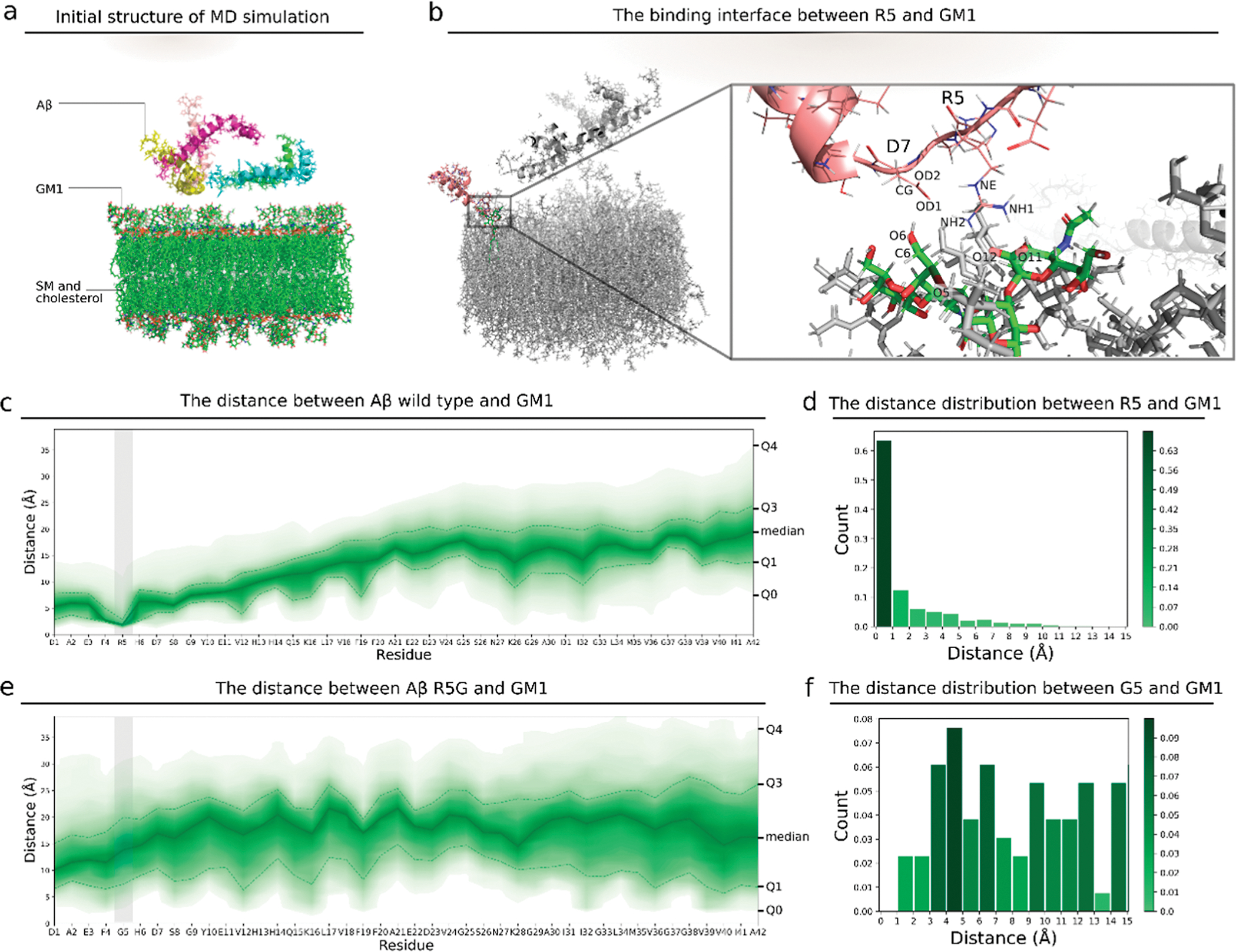
Interaction between the wild type, mutant Aβ, and GM1 liposome. (a) Initial structure for the MD simulation of the GM1 membrane and 5 Aβ monomers. (b) 3D view of the atoms at the interacting interface between R5 and GM1. (c) Distances between atoms of each residue in wild type Aβ and atoms of GM1 in the membrane. The upper area edge, the upper dashed line, the solid line, the lower dashed line, and the lower area edge are the 100, 75, 50, 25, and 0 percentiles, respectively, for the distance between each residue and GM1. The color corresponds to the percentile. The gray vertical bar indicates the region of the fifth residue. (d) Histogram of the minimum distances between R5 in Aβ and GM1 in the membrane of all steps in the MD simulation. (e) Distances between the Aβ mutant R5G and GM1. (f) Histogram of the minimum distances between G5 and GM1. G5 is the fifth residue in the Aβ mutant R5G.
To confirm the role of R5 in GM1–Aβ interaction experimentally, we mutate the fifth residue from arginine to glycine (R5G) and compare the amount of the membrane-bound Aβ wild type to that of R5G. We obtain membrane-bound Aβ by removing Aβ in solution through a 1000 kDa-molecular weight cut off (MWCO) centrifugation tube, and we compare the amount through SDS-PAGE and silver staining. We found remarkably more the wild type than R5G bound to the membrane, indicating a lower binding affinity between R5G and the GM1 liposome membrane. Thus, the mutation of R5G compromises the binding affinity between Aβ42 and GM1 liposomes.
GM1-Catalyzed Channel Formation by Aβ42 Oligomers.
Various mechanisms40–44 by which Aβ oligomers disrupt cell membranes have been proposed. Our work strongly supports the ion channel hypothesis,61 where Aβ oligomers damage neurons by forming ion channels,45,46 and the membrane thinning hypothesis,40 where Aβ oligomers increase the membrane permeability by thinning the membrane. Although the two hypotheses provide a biophysical mechanism for explaining the Aβ oligomer toxicity,62 it needs further validation. We utilize a fluorescence assay to interrogate (1) if the Aβ42 oligomers maintained by GM1 form ion channels or cause membrane thinning to disrupt the plasma membrane and (2) if various membrane constituents (PC, SM, and GM1) affect the disruption process. We encapsulate Ca2+ inside liposomes and dialyze away excess Ca2+ from outside of the liposomes (Figure S6). In support of the ion channel hypothesis or the membrane thinning hypothesis, we expect that membrane thinning or the formation of Aβ ion channels would lead to the efflux of Ca2+, thus resulting in an increase in the Ca2+ concentration outside the liposomes, so we utilize a calcium-sensitive dye (Fluo-4) to detect the Ca2+ concentration change outside of the liposomes (Figure S7a). We calculate the ratio of the fluorescence intensity of the Ca2+-encapsulated GM1 liposome after adding Aβ42 to that without adding Aβ42. We find that the ratio is >1 (Figure S7b), suggesting that the concentration of Ca2+ outside of the liposome increases because the liposome membranes are disrupted so that Ca2+ can efflux. When increasing the concentration of Aβ42, the ratio of fluorescence increases, indicative of the increased toxicity of Aβ42 with higher concentrations.
Although we observe the Ca2+ efflux, Ca2+ may actually exit liposomes through two potential mechanisms: (i) Aβ42 forms channels on liposome membranes through which calcium can pass, or (ii) Aβ42 disrupts the integrity of the liposome membranes. To validate the formation of Aβ42 ion channels or membrane thinning, we encapsulate Fluo-4, instead of Ca2+, inside liposomes (Figure 1e). We add Ca2+ to the solution and incubate them for 1 h to make sure that Ca2+ has time to enter the liposomes. Then, we add an excess of ethylenediaminetetraacetic acid (EDTA) to chelate Ca2+ outside the liposomes. EDTA has a higher binding affinity to Ca2+ than Fluo-4.63 If Aβ disrupts the liposomes, the encapsulated Fluo-4 dye will diffuse into the solution outside of the liposomes, where it will be inert because all extracellular Ca2+ has been chelated by EDTA. Conversely, if ion channels are formed or the membrane is thinned, Ca2+ will enter the liposomes through the channels to interact with Fluo-4 and emit fluorescent signals. Importantly, we assume that EDTA is too large to enter the liposomes through the ion channels or thinned membranes. We still calculate the ratio of the fluorescence intensity of the liposome after adding Aβ42 to that without adding Aβ42. Again, the ratio is >1 (Figure 1f), suggesting that Aβ42 forms ion channels or thins the membrane so that Ca2+ can influx. When increasing the concentration of Aβ42, the fluorescence intensity ratio also increases. We further replace GM1 liposomes with PC liposomes (l-α-phosphatidylcholine, sphingomyelin, and cholesterol), which increases the fluorescence intensity ratio much slower with the increase in the Aβ42 concentration compared to the GM1 liposome, suggesting that GM1, rather than sphingomyelin or cholesterol, is the key component of the liposome membrane which affects the formation of Aβ channels. Similarly, upon using a Fura-2 assay and 6-carboxyfluorescein dye leakage assay,64 Aβ40 has been shown to form ion-selective pores on membranes followed by fibrils growing that leads to nonspecific fragmentation of the lipid membrane.
GM1 Membrane Promotes Conformational Change of Oligomeric Aβ42.
To determine the potential size of Aβ42 oligomer species that can form channels, we utilize photoinduced cross-linking of unmodified proteins (PICUP, Figure S8)65 to cross-link Aβ42 oligomers. To determine the most probable number of monomers in Aβ42 oligomers that can form channels, we divide the Aβ42 oligomers formed with GM1 liposomes (Figure 1a) into four groups according to the following molecular weight: <25, 25–45, 45–100, and >100 kDa (Figure 3a). We extract these four different Aβ42 samples from gel, incubate them at the same concentration with Ca2+-encapsulated liposomes, and measure the fluorescence intensity as described previously. We find that the 45–100 kDa sample has the highest fluorescence intensity, indicating that the most likely molecular weight range of Aβ42 oligomers that can form channels is 45–100 kDa (Figure 3a).
Figure 3.
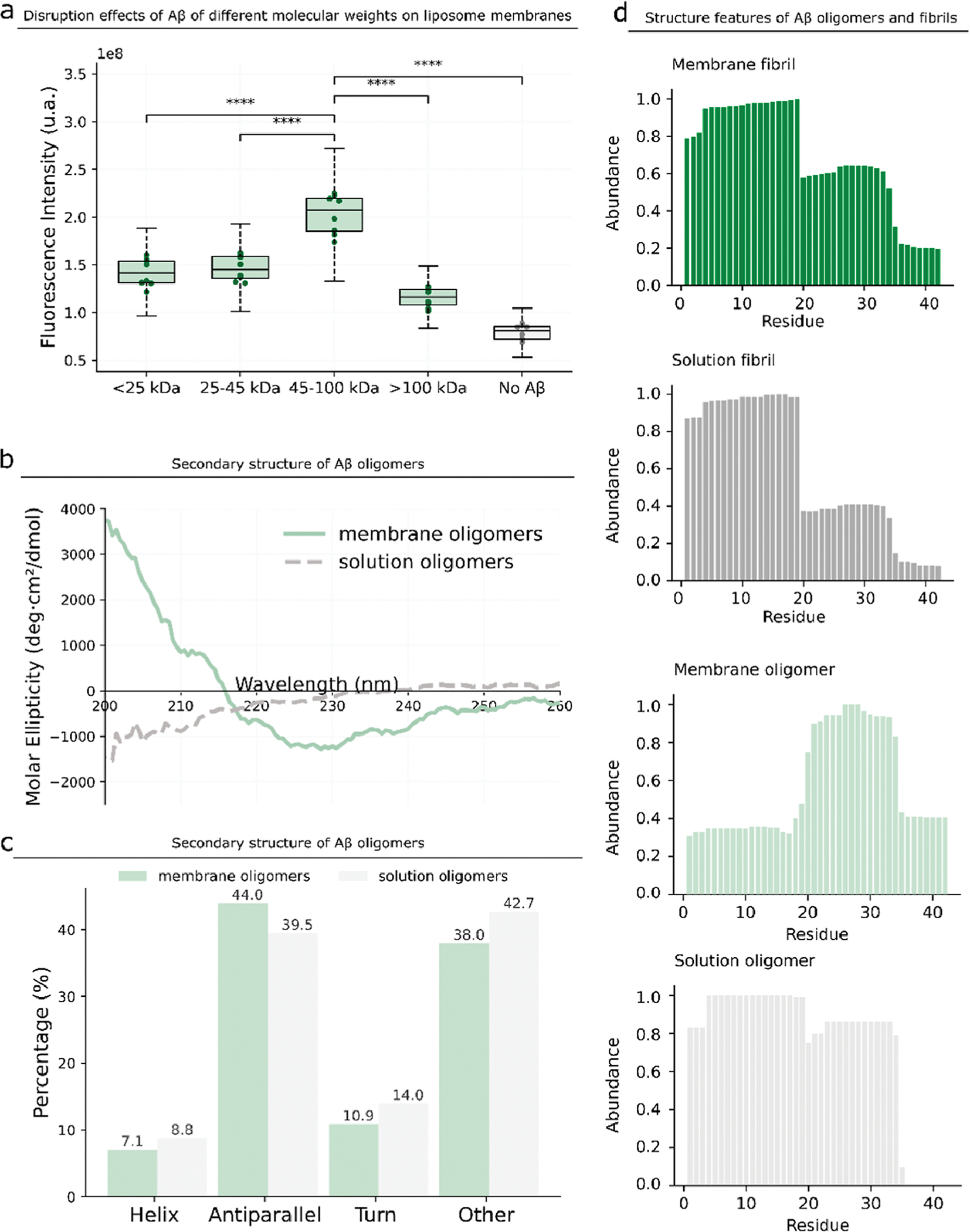
Structure characterization of membrane Aβ oligomers. (a) We divide the membrane Aβ oligomers into four samples according to the molecular weights and purify them from gels. We incubate the four samples at the same concentration with the Ca2+-encapsulated liposome and determine the fluorescence intensity. P-value: NS (0.05 < p ≤ 1), * (0.01 < p ≤ 0.05), ** (0.001 < p ≤ 0.01), *** (0.0001 < p ≤ 0.001), and **** (p ≤ 0.0001). (b,c) Secondary structure of the Aβ oligomers formed in the presence and absence of GM1 liposomes. (d) Residue abundance of Aβ fibrils formed in the presence and absence of GM1 liposomes and the residue abundance of Aβ oligomers formed on the membrane and in the absence of GM1 liposomes.
We further determine the secondary structure of Aβ42 oligomers by circular dichroism (CD). We prepare two oligomer samples, one obtained from the liposome membrane (membrane Aβ42 oligomers) and the other from the buffer solution (solution Aβ oligomers) (Figure 3b,c). Solution Aβ42 oligomers have 39.5% antiparallel β-sheets, while membrane Aβ42 oligomers have a higher percentage (44%) of antiparallel β-sheet structures. Consistent with previous reports,49,66,67 our results show that GM1 membranes can promote a conformational change of oligomeric Aβ42 to form β-sheet-rich structures.
We compare the structural features of Aβ42 oligomers and fibrils by digesting all purified Aβ42 oligomers and fibrils with pepsin and measuring the abundance of fragments by mass spectrometry (MS)32 (Figure 3d). Theoretically, higher-abundance peptides are likely to appear on the surface of the protein because surface residues have a higher chance to be digested, while lower-abundant peptides are buried inside the structure.32 In both membrane Aβ42 fibrils and solution Aβ42 fibrils, the abundances of Aβ4–19 peptides are high, suggesting that they are on the surface; the abundances of Aβ36–42 peptides are low, suggesting that they are internal. Regardless of the presence or absence of GM1 liposomes, the abundances of peptides of the two fibrils are similar, indicating that although GM1 liposomes promote the formation of fibrils, they do not affect the structure of fibrils. However, the peptide abundances of the solution Aβ42 oligomers and membrane Aβ oligomers are drastically different. In membrane Aβ42 oligomers, Aβ20–34 peptides feature high abundances, while Aβ1–17 peptides feature low abundances. In solution Aβ42 oligomers, Aβ4–19 peptides have the highest abundances, and Aβ35–42 peptides have the lower abundances. Thus, the structures of membrane Aβ42 oligomers, promoted by GM1 liposomes, are distinct from the structures of solution Aβ42 oligomers. Finally, based on the abundances of peptides, the structures of Aβ42 oligomers and fibrils are also drastically different. Overall, the presence of GM1 liposomes changes Aβ42 oligomer structures but does not change Aβ42 fibril structures.
Toxicity of Membrane Aβ42 Oligomers In Vitro.
We interrogate the toxicity of these GM1-bound Aβ42 oligomers by cellular studies (Figure 4a). We incubate Aβ42 with GM1 liposomes, crosslink them, and remove solution Aβ oligomers. We then add the retained membrane Aβ42 oligomers to differentiated PC12 cells. Tong and coworkers68 have shown that the viability of PC12 cells is not reduced when incubating with 1 μM or even 5 μM Aβ42, slightly reduced with 10 μM Aβ42, and significantly reduced with 20 μM Aβ42. We incubate Aβ42 with the cells for 3 days and then measure the toxicity by calcein AM and ethidium homodimer-1 live/dead viability assay. We find that membrane Aβ42 oligomers at a concentration of 5 μM exhibit toxicity (increased dead/live cell ratio) in PC12 cells (Figure 4b,c), while at the same concentration, solution Aβ42 oligomers do not exhibit toxicity. We further find that the addition of membrane Aβ42 oligomers induces increased levels of cleaved caspase-3, a reliable marker for cells that are dying, compared to the live control (Figure S9). The toxicity of membrane Aβ oligomers is consistent with our fluorescence results: The increase in the Aβ oligomer concentration leads to the increase in Ca2+ influx/efflux caused by membrane disruption, suggesting that the toxicity of membrane Aβ42 oligomers may be related to the dysregulation of calcium homeostasis. The GM1 content in the membrane of differentiated PC 12 cell lines has increased remarkably after nerve growth factor stimulation,69 so we also performed a cell viability assay in post-natal day 1 (P1) CD1 mouse cortical neurons, where the ganglioside profile is dominated by GM3 and GD3 rather than GM1.70 We find that membrane Aβ42 oligomers do not result in reduced viability of P1 (Figure S10), supporting that GM1 is critically necessary for the toxicity of Aβ42 oligomers.
Figure 4.
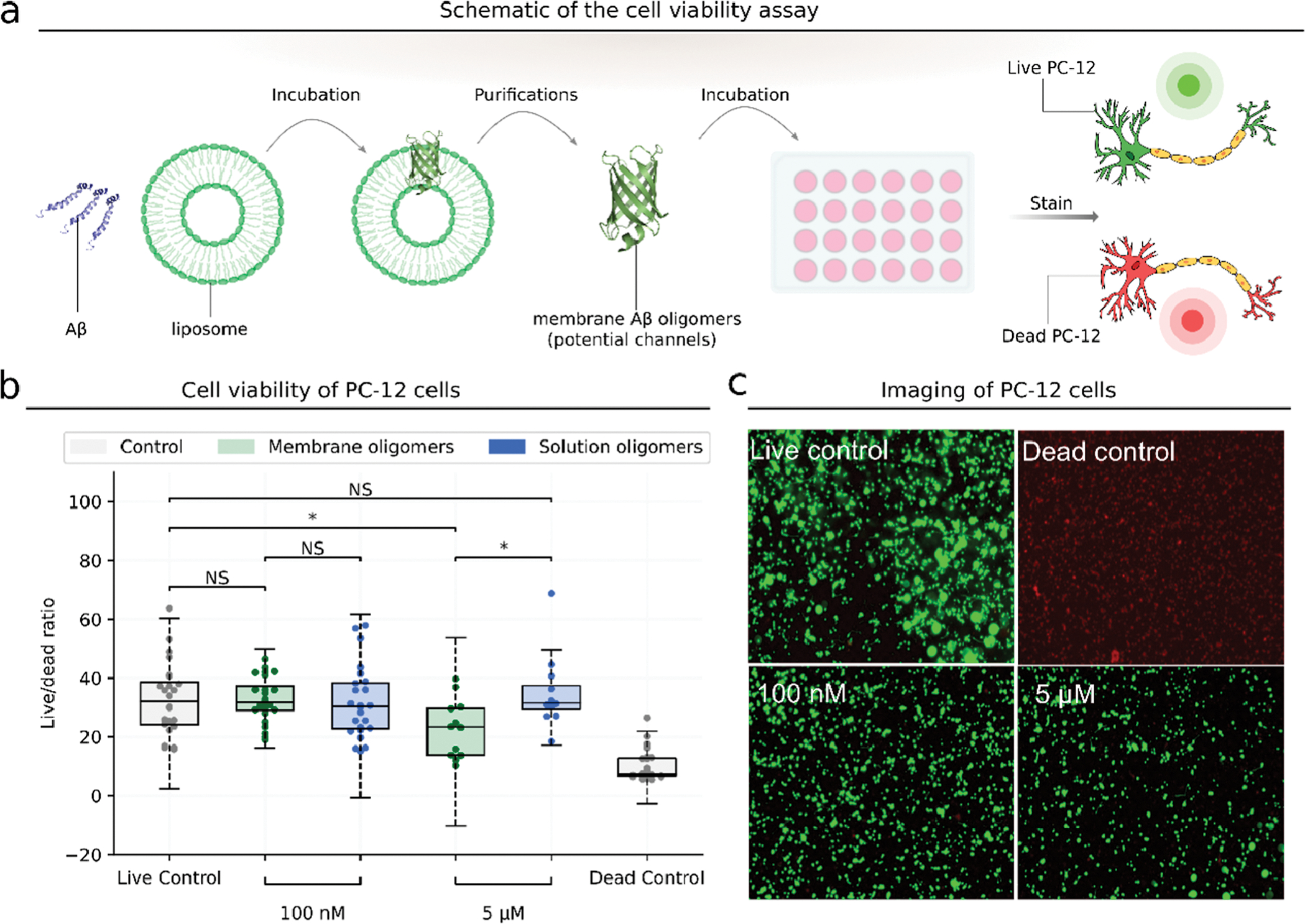
Aβ oligomers formed on liposome membranes are cytotoxic. (a) Schematic of cell viability experiment. (b) Cell viability of PC-12 cells incubated with membrane or solution Aβ oligomers of different concentrations. The live control has no added Aβ. The dead control is treated with 70% methanol. Cell viability is assessed by calcein AM/ethidium homodimer-1 and calculated using the ratio of dead cells to live cells. P-value: NS (0.05 < p ≤ 1), * (0.01 < p ≤ 0.05), ** (0.001 < p ≤ 0.01), *** (0.0001 < p ≤ 0.001), and **** (p ≤ 0.0001). (c) PC-12 cells incubated with membrane Aβ oligomers of different concentrations. Live cells are stained by calcein AM in green, and dead cells are stained by ethidium homodimer-1 in red.
DISCUSSION
The concentration of Aβ42 is important to the formation of oligomers.71,72 Most in vitro aggregation studies and Aβ42 ion channel studies have been conducted with Aβ peptide concentrations far beyond physiological concentrations. For example, previous studies have demonstrated the disruptive effects of Aβ42 on calcium homeostasis of PC12 cells with Aβ42 concentrations as high as 20 μM.68 Here, we demonstrate that PC12 cells can be disrupted by membrane Aβ42 oligomers at 5 μM, and phospholipid bilayer membranes can be disrupted by Aβ42 at nanomolar concentrations with GM1 serving as the catalyst. Cells have some mechanisms to protect themselves from Aβ-induced neurotoxicity; for example, neuroglobin has been shown to protect PC12 cells against β-amyloid-induced cell injury,73 so the needed concentration of Aβ42 to disrupt an artificial phospholipid bilayer is much lower than the needed concentration of Aβ42 to disrupt cell membranes.
GM1 is distributed as clusters on neuronal membranes in the mouse brain,67 and the binding of Aβ42 to GM1 clusters may increase the local concentration of Aβ42 and cause a conformational change that promotes aggregation.48,50,51 The presence of GM1 clusters aiding the maintenance of Aβ42 oligomers may be due to the high binding affinity between GM1 clusters and Aβ42. Previous reports and our results both suggest that GM1 can bind to Aβ4274 and promote the formation of Aβ42 fibrils,53 and we further find that some Aβ oligomer species are stable and not inclined to further aggregate into fibrils in the presence of GM1. Our liposome encapsulation experiments demonstrate that these stable oligomers may form channel-like structures, which provides new evidence for the hypothesis that Aβ forms channel-like oligomeric structures that disrupt cellular calcium homeostasis.38 The formation of channel-like structures may be contributed by the conformational change of Aβ42 to β-sheet-rich states, which has been demonstrated in both previous studies67,75 and our results. Thus, the mechanism of the disrupted calcium homeostasis may be that GM1 catalyzes the formation of Aβ42 ion channels or membrane thinning, thereby decreasing the calcium gradient.76 Furthermore, although some studies38,46,77 have utilized channel conductance measurements on planar lipid bilamellar membranes to suggest that Aβ forms ion channels or thins the membranes, the size and structure information of the channel oligomers was unclear. In our work, we utilize MS and CD to measure the possible size and secondary structure of Aβ42 oligomers, and we also find that the binding of Aβ42 to the GM1 membrane deforms the GM1 membrane. Finally, the ion channel hypothesis and the membrane thinning hypothesis are two of several hypotheses of the mechanism of how Aβ disrupts Ca2+ homeostasis. Both hypotheses propose that Aβ oligomers permeabilize, instead of completely disassembling, lipid membranes to disrupt Ca2+ homeostasis, but the key difference between these hypotheses is how Aβ oligomers permeabilize the lipid membrane. Despite a long-standing debate between these two hypotheses,45 there is no direct evidence that can unequivocally support one of them and rule out the other one. Our experiments also cannot distinguish the ion channel hypothesis and the membrane thinning hypothesis, but our primary finding excludes hypotheses that suggest complete disintegration of lipid membranes, such as the excessive membrane tabulation hypothesis and the membrane extraction hypothesis.
Overall, we integrate computational, biochemical, and cellular studies to uncover the molecular etiology of early Aβ42 aggregation modulated by GM1 in AD pathology and identify processes responsible for neuronal toxicity. Such a comprehensive approach is necessary to decouple multiple processes accompanying AD pathophysiology.
METHODS
Preparation of Liposomes.
We dissolve GM1 (Sigma-Aldrich), l-α-phosphatidylcholine (PC, from egg yolk, Sigma-Aldrich), sphingomyelin (VWR International), and cholesterol (Fisher Scientific) in a chloroform/methanol (1:1, v/v) mixture at a total concentration of 1 mM. To mimic the GM1 cluster on the cell membrane, the molar ratio of GM1, sphingomyelin, and cholesterol is 1:2:2 for GM1 liposomes. For PC liposomes, the molar ratio of PC, sphingomyelin, and cholesterol is 1:2:2. For sphingomyelin-rich liposomes, the molar ratio of sphingomyelin and cholesterol is 1:1. To prepare liposomes, we first dry the organic solvents under a gentle stream of nitrogen for 2 h and then under vacuum overnight. Then, we rehydrate the resulting lipid film with N-(2-hydroxyethyl)-piperazine-N′-ethanesulfonic acid (HEPES) buffer (10 mM HEPES and 150 mM NaCl, pH 7.4) and incubate it for 1 h in a 45 °C water bath, vortexing every 15 min. In order to prepare Ca2+-encapsulated liposomes, we replace the rehydrated lipid HEPES buffer with 10 mM CaCl2 (Fisher Scientific), 10 mM HEPES, and 140 mM NaCl, pH 7.4.78,79 To prepare dye-encapsulated liposomes, we replace the rehydrated lipid buffer with 0.03 mM Fluo-4 (Life Technologies) and HEPES buffer. The dye-encapsulated liposomes are always protected from light. We sonicate the rehydrated suspension for 15 min and then freeze-thaw for five cycles in the liquid nitrogen and 60 °C water bath. To obtain uniform liposomes, we extrude the resulting suspension 16 times with an Avanti extruder. To encapsulate calcium and dye, we use 800 nm-pore size polycarbonate filters to filter the liposomes. For the Ca2+-encapsulated and dye-encapsulated liposomes, we use a 10000 Da-MWCO dialysis cassette with HEPES buffer changed three times a day to remove Ca2+ and dye in the buffer outside the liposomes.
Fluorescence Intensity Determination.
We dissolve Aβ42 (Fisher Scientific) in dimethyl sulfoxide at a concentration of 1 mM and sonicate for 10 min. For the Ca2+-encapsulated liposomes, we add different concentrations (100 nM, 500 nM, 1 μM, 10 μM, 20 μM, and 30 μM) of Aβ42 and incubate with liposomes for 3 days at room temperature. We add 50 μM Fluo-4 into the incubated Aβ42 and liposome solution and determine the fluorescence intensity using a SpectraMax i3 plate reader. We treat the control sample in the same way, except without adding Aβ42. We analyze the fluorescence intensity ratio of samples incubated with different concentrations of Aβ42 and the control.
We protect the dye-encapsulated liposomes from light throughout the experiment to prevent photo-bleaching. We add different concentrations of Aβ42 (500 nM, 1 μM, 5 μM, and 10 μM) into the liposome solutions and incubate for 3 days at room temperature. We add 14 mM CaCl2 into the incubated Aβ42 and liposomes and incubate for 1 h to ensure that calcium ions have enough time to enter the liposomes through ion channels or thinned membranes. Then, we add 16.5 mM EDTA (excess) to bind all the Ca2+ ions in the solution. In this way, only Ca2+ that enters the liposomes can bind with Fluo-4. Similarly, we treat the control sample in the same way, except without adding Aβ42. Finally, we determine the fluorescence intensity using a SpectraMax i3 plate reader. After incubation with GM1 liposomes, Aβ42 oligomers were divided into >100, 45–100, 25–45, and <25 kDa samples according to a trident pre-stained protein ladder (GTX50875), and then, the same concentration- and volume-resulted sample was incubated with Ca2+-encapsulated GM1 liposomes overnight. After this, the fluorescence intensity was determined, as previously mentioned.
Transmission Electron Microscopy.
We incubate 80 μM Aβ42 in HEPES buffer for 1 day and 3 days at 37 °C and characterize the formation of fibrils in negative-stain TEM images using a JEF 1400 and a NANOSPORT43 camera at the TEM facility of Penn State COM.
Cross-Linking.
To obtain stable Aβ oligomers, particularly the Aβ oligomers formed on membranes, we utilize the PICUP method80 to cross-link. We add ammonium persulfate (1 mM, 10 μL, Dot Scientific) and Tris (2,2-bipyridyl)dichlororuthenium(II) hexahydrate [Ru(II)Bpy32+ 0.05 mM, 10 μL, Sigma-Aldrich] in 10 mM HEPES (pH 7.4) to the incubated Aβ oligomer solution (180 μL). We irradiate the resulting solution with a 500 W lamp for 5 s and quench the cross-linking process with 47.6 mM dithiothreitol (1 M, 10 μL, Dot Scientific).
Analysis of the Size of Aβ Oligomers by SDS-PAGE and Silver Staining.
We incubate Aβ in the presence or absence of GM1 liposomes and analyze samples at 0.5, 4, 24, and 72 h. We cross-link all samples using the PICUP method. For the sample in the presence of GM1 liposomes, we add 40 μM N-octylglucoside (Dot Scientific) into the samples and then incubate at room temperature for 1 h to dissolve the lipids of the liposomes. To compare wild-type and mutant Aβ with respect to the binding affinity to GM1 liposomes, we incubate GM1 liposomes with 10 μM wild-type and mutant Aβ, respectively. We obtain membrane-bound Aβ by removing Aβ in solution through 1000 kDa-MWCO centrifugation tubes, spun at 1300 rpm. We add Laemmli SDS sample buffer dye (Fisher Scientific) to the resulting solution and heat at 95 °C for 10 min. We use 10% SDS-PADE and silver staining (Fisher Scientific) to analyze the formed Aβ oligomers.
Purification of Aβ42 Oligomers on the Liposomes.
We incubate Aβ42 monomers with GM1 liposomes at room temperature for 3 days and cross-link Aβ before further purification. We use 1000 kDa-MWCO centrifugation tubes, spun at 1300 rpm, to remove Aβ oligomers that were not inserted into liposomes. We treat the sample with N-octylglucoside, Laemmli SDS sample buffer dye, a heater, SDS-PAGE, and silver staining as described above. After gel electrophoresis, we cut out the gel portion according to the reference results of silver staining. We crush the cut gel portion containing the Aβ oligomers of interest and dissolve them in HEPES buffer overnight in a shaker at 4 °C. We centrifuge the resulting samples at 10,000g for 10 min and save the supernatants for further concentration by centrifugation. To measure the size of the Aβ channels, we cut the gel according to various molecular weight ranges (>100, 45–100, 25–45, and <25 kDa) and purify Aβ oligomers in the gel as described above. We determine the concentration of the purified Aβ sample using Nanodrop and a bicinchoninic acid (BCA) protein assay kit.
Secondary Structure.
We incubate Aβ42 monomers (10 μM) for 3 days at room temperature in the presence or absence of GM1 liposomes and then cross-link samples using PICUP. For the sample of Aβ42 incubated in the presence of GM1 liposomes, we remove Aβ oligomers in the solution and add N-octylglucoside to dissolve the lipids as described previously. We remove the fibrils in the resulting solution using 1000 kDa-MWCO centrifugation tubes spun at 3900 rpm and wash with HEPES buffer. We remove the monomers, dimers, and lipids using 10 kDa-MWCO centrifugation tubes, spun at 3900 rpm, and wash with HEPES buffer with 40 μM N-octylglucoside and then wash again with HEPES buffer. For the control sample of Aβ42 monomers (10 μM) incubated in HEPES without liposomes, we remove the fibrils, monomers, and dimers in the same way except adding N-octylglucoside to dissolve the lipids and washing with 40 μM N-octylglucoside HEPES buffer. We determine the concentration of Aβ oligomers using the BCA protein assay kit. We determine the secondary structure characteristics of Aβ42 oligomers using a Jasco J-1500 CD spectrophotometer. We place samples in 0.1 mm-pathlength (200 μL) cuvettes and record the spectral range at 185–240 nm with 0.5 nm data pitch and a scanning speed of 50 nm/min.
PC-12 Cell Culture.
We obtained PC-12 cells from the American Type Culture Collection (ATCC). For the PC-12 cells, we use Dulbecco’s modified Eagle’s medium (DMEM) (VWR International) supplemented with 5% heat-inactivated horse serum (Life Technologies), 5% fetal bovine serum (Fisher Scientific), and 1% penicillin/streptomycin (Fisher Scientific). We culture cells at 37 °C with 5% CO2. For the neuronal differentiation, we seed PC-12 cells in 96-well plates at 2000 cells/well. Prior to seeding, we treat the 96-well plate using a UVO cleaner (Jelight Company, model 18) for 30 min, coat with 10× diluted collagen, and incubate at 4 °C for 2 h before use. For the differentiated PC-12 cells, we use DMEM supplemented with 1% heat-inactivated horse serum, 1% penicillin/streptomycin, and 100 ng/mL nerve growth factor (Sigma-Aldrich) and culture for 1 week at 37 °C with 5% CO2. Before further analysis, we confirm differentiation by visual observation using a phase contrast microscope.
Cell Viability Assay.
We determine cell viability using calcein AM and ethidium homodimer-1 dye. After differentiation, we add purified Aβ oligomer samples to the differentiation cells and incubate for 3 days. For the dead cell controls, we add 70% methanol to cells 2 h before plate read. We remove the differentiation medium and wash the cells twice with phosphate buffered saline (PBS). For each well, we add 4 μM ethidium homodimer-1 and 2 μM calcein AM in PBS to label live and dead cells. Cells then incubate for 30 min at room temperature. We determine the fluorescence intensity using SpectraMax i3. For calcein AM, the excitation wavelength is 495 nm, and the emission wavelength is 525 nm. For ethidium homodimer-1, the excitation wavelength is 495 nm, and the emission wavelength is 645 nm. We take the fluorescent images on a Keyence BZ-X800 fluorescence microscope.
Mass Spectrometry.
After cross-linking and purification, we measure the molecular weight of the Aβ oligomers with MS using the Sciex 5600 TripleTOF (AB SCIEX). We digest purified Aβ oligomers and fibrils with pepsin. We add 1 N HCl to the Aβ samples to a final concentration of 0.04 N and suspend pepsin in 100 mM acetate buffer (pH 3.5). We add the pepsin to Aβ solution at a ratio of 1:20 (enzyme/protein W/W) and incubate them for 1 h at 37 °C. We add 1 M Tris buffer (pH 8) to a final concentration of 150 mM to stop the reaction. Then, the samples are run on a Bruker timsTOF fleX using a short PASEF protocol, on a PepSep 75, 3 μm, 15 cm UPLC column heated at 45 °C using nanoElute liquid chromatography-MS. The data are analyzed using BYOS software (4.0.1) from Protein Metrics. The identified Aβ peptide sequences are determined using BYOS software (4.0.1) from Protein Metrics with the UniProt Proteomes-Homo sapiens database (UP000005640, version March 7, 2021). ProteinPilot software is used to reanalyze the peak area intensity of the peptides identified using BYOS software of the Byonics group file (Table S1). The cleavage residues are set to tyrosine, phenylalanine, tryptophan, and leucine. Mass tolerance for precursor ions is set to 30 ppm, and mass tolerance for fragment ions is set to 30 ppm. Fixed modifications are carbamidomethyl. Variable modifications: oxidation +15.994915 at M, deamidated +0.984016 at N, Gln- > pyro-Glu −17.026549 at N-terminal Q, Gln- > pyro-Glu −18.010565 at N-terminal E, and acetyl +42.010565 at K and the protein N-terminal. Two missed cleavages are allowed. The abundance of each residue is calculated by summing up the intensities of all peptides that contain this residue. Finally, the intensity of each residue is divided by the sum of intensities of all residues to be normalized. The MS proteomics data have been deposited into the ProteomeXchange consortium via the PRIDE partner repository with the data set identifier PXD029117.
Cryo-Electron Tomography.
To increase the ratio of Aβ/lipid to 1:10, we dilute GM1 liposomes 10 times and incubate the resulting GM1 liposomes with or without 10 μM Aβ for 3 days. Then, we concentrate the samples using a 100 kDa-MWCO centrifuge tube, centrifuging at 1300 rpm to reduce sample volume from 1 mL to 50 μL to increase the concentration of liposomes. We mix the samples with 10 nm gold fiducials before pipetting 4 μL onto a freshly glow-discharged Quantifoil R2/2 holey carbon grid. We hand grids blotted from behind and plunge grids into liquid ethane using a Mark IV Vitrobot (Thermo Fisher Scientific) for vitrification. We then transfer samples under liquid nitrogen into a Titan Krios G3i cryo transmission electron microscope (Thermo Fisher Scientific) operating at 300 kV for the acquisition of tilt series. We target holes containing liposomes contained within vitreous ice data collection and collect tilt series in 2° increments from −60 to +60° at −6.0 μm defocus. Tilt series images are acquired with a Bioquantum energy filter (Gatan) operating at the zero-loss peak using a K3 direct electron detector (Gatan) in the single electron counting mode. We use a nominal magnification of ×33,000 which corresponds to a pixel size of 3.3 Å /pixel. We set exposure time to achieve a total electron dose of ~120 e−/Å2 for a complete tilt series. We collect 6 to 10 tilt series from each grid. We reconstruct tomograms using the IMOD81 software suite.
Computational Modeling.
We first model the GM1 membrane structure through CHARMM-GUI82 with a ratio of SM, cholesterol, and GM1 as 4:4:2. We then model the intact structure of Aβ using SWISS-MODEL83 with PDB ID 1IYT as the template. The missing residues in the N-terminal region are completed using SWISS-MODEL. Next, we perform MD simulation of the GM1 membrane–Aβ system using GROMACS.84 The CHARMM36m85 force field for the protein and lipid parameters is used. Hydrogens for heavy atoms were added using the pdb2gmx module in the GROMACS simulation package. The system is subsequently energy-minimized for 2000 steps using the conjugate gradient algorithm and another 2000 steps using the steepest descent algorithm. The structure is solvated using explicit water in a cubic periodic box with water molecules extending 10 Å outside the protein on all sides. Water molecules are described using a simple point charge water model. The system is solvated using TIP3 water molecules, then energy-minimized again, and heated up gradually to reach a temperature of 310 K using a V-rescale thermostat with a coupling constant of 0.1 ps. The solvent density is adjusted under isobaric and isothermal conditions at 1 bar and 310 K, respectively. A Parrinello–Rahman barostat with isotropic pressure coupling and a coupling constant of 0.1 ps was used to set the pressure at 1 bar. The system is equilibrated for 10 ns in the NPT ensemble with a simulation time step of 2 fs. Finally, the production run is carried out for 100 ns for the primary system. The long-range electrostatic interactions are treated using a particle-mesh Ewald sum with a cutoff of 1.0 nm. The van der Waals interactions are terminated beyond the cut-off value of 1.0 nm. The LINCS algorithm is used to constrain all bonds involving hydrogen atoms. All simulations were performed using the GROMACS simulation program. The analyses of the trajectories are performed using GROMACS and MDAnalysis.86
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge support from the National Institutes of Health 1R35 GM134864, the Huck Institutes of the Life Sciences, and the Passan Foundation. The project described was also supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant UL1 TR002014. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We also acknowledge the help from Brianna L. Hnath, Sophie E. Dokholyan, and Martin V. Dokholyan with the experiments.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.2c00221.
Additional methods about western blot, primary neuron culture, and the measurement of Ca2+ influx in primary neurons, Aβ oligomers and fibril formation in the presence of various liposomes, computational analyses of the distances between Aβ and SM, validation of the encapsulation of Ca2+ in liposomes, disruption effect of Aβ oligomers on the liposome membrane, schema of the PICUP reaction mechanism, and cell assays about the cell viability and Ca2+ influx in primary neurons (PDF) Additional data of the peptide peak area intensity identified with the ProteinPilot software (XLSX)
Contributor Information
Dong Yan Zhang, Department of Pharmacology, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States.
Jian Wang, Department of Pharmacology, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States.
Rebecca M. Fleeman, Department of Pharmacology, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States; Department of Neurosurgery, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States; Center for Neural Engineering, Pennsylvania State University, State College, Pennsylvania 16801, United States
Madison K. Kuhn, Department of Pharmacology, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States; Department of Neurosurgery, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States; Center for Neural Engineering and Department of Biomedical Engineering, Pennsylvania State University, State College, Pennsylvania 16801, United States
Matthew T. Swulius, Department of Biochemistry & Molecular Biology, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States
Elizabeth A. Proctor, Department of Pharmacology, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States; Department of Neurosurgery, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States; Center for Neural Engineering, Department of Biomedical Engineering, and Department of Engineering Science & Mechanics, Pennsylvania State University, State College, Pennsylvania 16801, United States
Nikolay V. Dokholyan, Department of Pharmacology, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States; Department of Biomedical Engineering and Department of Chemistry, Pennsylvania State University, State College, Pennsylvania 16801, United States; Department of Biochemistry & Molecular Biology, Penn State College of Medicine, Hershey, Pennsylvania 17033-0850, United States
Data Availability:
DOI: 10.17632/38tt768dkd.2.
REFERENCES
- (1).Burns A; Iliffe S Alzheimer’s Disease. BMJ 2009, 338, b158. [DOI] [PubMed] [Google Scholar]
- (2).Vos T; Allen C; Arora M; Barber RM; Bhutta ZA; Brown A; Carter A; Casey DC; Charlson FJ; Chen AZ Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 310 Diseases and Injuries, 1990–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sengupta U; Nilson AN; Kayed R The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Francis PT; Palmer AM; Snape M; Wilcock GK The Cholinergic Hypothesis of Alzheimer’s Disease: A Review of Progress. J. Neurol., Neurosurg. Psychiatry 1999, 66, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hardy J; Allsop D Amyloid Deposition as the Central Event in the Aetiology of Alzheimer’s Disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [DOI] [PubMed] [Google Scholar]
- (6).Mudher A; Lovestone S Alzheimer’s Disease–Do Tauists and Baptists Finally Shake Hands? Trends Neurosci. 2002, 25, 22–26. [DOI] [PubMed] [Google Scholar]
- (7).Deane R; Zlokovic B Role of the Blood-Brain Barrier in the Pathogenesis of Alzheimer’s Disease. Curr. Alzheimer Res. 2007, 4, 191–197. [DOI] [PubMed] [Google Scholar]
- (8).Miklossy J Alzheimer’s Disease—a Neurospirochetosis. Analysis of the Evidence Following Koch’s and Hill’s Criteria. J. Neuroinflammation 2011, 8, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pisa D; Alonso R; Rábano A; Rodal I; Carrasco L Different Brain Regions Are Infected with Fungi in Alzheimer’s Disease. Sci. Rep. 2015, 5, 15015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Alves GS; Oertel Knöchel V; Knöchel C; Carvalho AF; Pantel J; Engelhardt E; Laks J Integrating Retrogenesis Theory to Alzheimer’s Disease Pathology: Insight from DTI-TBSS Investigation of the White Matter Microstructural Integrity. BioMed Res. Int. 2015, 2015, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bartzokis G Alzheimer’s Disease as Homeostatic Responses to Age-Related Myelin Breakdown. Neurobiol. Aging 2011, 32, 1341–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cataldo JK; Prochaska JJ; Glantz SA Cigarette Smoking Is a Risk Factor for Alzheimer’s Disease: An Analysis Controlling for Tobacco Industry Affiliation. J. Alzheimer’s Dis. 2010, 19, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Su B; Wang X; Nunomura A; Moreira P; Lee H.-g.; Perry G; Smith M; Zhu X Oxidative Stress Signaling in Alzheimer’s Disease. Curr. Alzheimer Res. 2008, 5, 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kandimalla R; Vallamkondu J; Corgiat EB; Gill KD Understanding Aspects of Aluminum Exposure in Alzheimer’s Disease Development. Brain Pathol. 2016, 26, 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tanzi RE; Bertram L Twenty Years of the Alzheimer’s Disease Amyloid Hypothesis: A Genetic Perspective. Cell 2005, 120, 545–555. [DOI] [PubMed] [Google Scholar]
- (16).Iversen LL; Mortishire-Smith RJ; Pollack SJ; Shearman MS The Toxicity in Vitro of Beta-Amyloid Protein. Biochem. J. 1995, 311Pt1, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jakob-Roetne R; Jacobsen H; Jakob-Roetne R; Jacobsen H Alzheimer’s Disease: From Pathology to Therapeutic Approaches. Angew. Chem., Int. Ed. 2009, 48, 3030–3059. [DOI] [PubMed] [Google Scholar]
- (18).Hardy J Has the Amyloid Cascade Hypothesis for Alzheimer’s Disease Been Proved? Curr. Alzheimer Res. 2006, 3, 71–73. [DOI] [PubMed] [Google Scholar]
- (19).Hardy JA; Higgins GA Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [DOI] [PubMed] [Google Scholar]
- (20).Bernstein SL; Dupuis NF; Lazo ND; Wyttenbach T; Condron MM; Bitan G; Teplow DB; Shea J-E; Ruotolo BT; Robinson CV; Bowers MT Amyloid-β Protein Oligomerization and the Importance of Tetramers and Dodecamers in the Aetiology of Alzheimer’s Disease. Nat. Chem. 2009, 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Butterfield SM; Lashuel HA Amyloidogenic Protein-Membrane Interactions: Mechanistic Insight from Model Systems. Angew. Chem., Int. Ed. Engl. 2010, 49, 5628–5654. [DOI] [PubMed] [Google Scholar]
- (22).Glabe CG Structural Classification of Toxic Amyloid Oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Quist A; Doudevski I; Lin H; Azimova R; Ng D; Frangione B; Kagan B; Ghiso J; Lal R Amyloid Ion Channels: A Common Structural Link for Protein-Misfolding Disease. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Matsumura S; Shinoda K; Yamada M; Yokojima S; Inoue M; Ohnishi T; Shimada T; Kikuchi K; Masui D; Hashimoto S; Sato M; Ito A; Akioka M; Takagi S; Nakamura Y; Nemoto K; Hasegawa Y; Takamoto H; Inoue H; Nakamura S; Nabeshima Y.-i.; Teplow DB; Kinjo M; Hoshi M Two Distinct Amyloid β-Protein (Aβ) Assembly Pathways Leading to Oligomers and Fibrils Identified by Combined Fluorescence Correlation Spectroscopy, Morphology, and Toxicity Analyses. J. Biol. Chem. 2011, 286, 11555–11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).DeToma AS; Salamekh S; Ramamoorthy A; Lim MH Misfolded Proteins in Alzheimer’s Disease and Type II Diabetes. Chem. Soc. Rev. 2012, 41, 608–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Haass C; Selkoe DJ Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid Beta-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [DOI] [PubMed] [Google Scholar]
- (27).Klein W; Krafft GA; Finch CE Targeting Small Abeta Oligomers: The Solution to an Alzheimer’s Disease Conundrum? Trends Neurosci. 2001, 24, 219–224. [DOI] [PubMed] [Google Scholar]
- (28).Lambert MP; Viola KL; Chromy BA; Chang L; Morgan TE; Yu J; Venton DL; Krafft GA; Finch CE; Klein WL Vaccination with Soluble Abeta Oligomers Generates Toxicity-Neutralizing Antibodies. J. Neurochem. 2001, 79, 595–605. [DOI] [PubMed] [Google Scholar]
- (29).Karran E; Mercken M; Strooper BD The Amyloid Cascade Hypothesis for Alzheimer’s Disease: An Appraisal for the Development of Therapeutics. Nat. Rev. Drug Discovery 2011, 10, 698. [DOI] [PubMed] [Google Scholar]
- (30).Martinez Hernandez A; Urbanke H; Gillman AL; Lee J; Ryazanov S; Agbemenyah HY; Benito E; Jain G; Kaurani L; Grigorian G; Leonov A; Rezaei-Ghaleh N; Wilken P; Arce FT; Wagner J; Fuhrman M; Caruana M; Camilleri A; Vassallo N; Zweckstetter M; Benz R; Giese A; Schneider A; Korte M; Lal R; Griesinger C; Eichele G; Fischer A The Diphenylpyrazole Compound Anle138b Blocks Abeta Channels and Rescues Disease Phenotypes in a Mouse Model for Amyloid Pathology. EMBO Mol. Med. 2018, 10, 32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Redler RL; Fee L; Fay JM; Caplow M; Dokholyan NV Non-Native Soluble Oligomers of Cu/Zn Superoxide Dismutase (SOD1) Contain a Conformational Epitope Linked to Cytotoxicity in Amyotrophic Lateral Sclerosis (ALS). Biochemistry 2014, 53, 2423–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Proctor EA; Fee L; Tao Y; Redler RL; Fay JM; Zhang Y; Lv Z; Mercer IP; Deshmukh M; Lyubchenko YL; Dokholyan NV Nonnative SOD1 Trimer Is Toxic to Motor Neurons in a Model of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Choi ES; Dokholyan NV SOD1 Oligomers in Amyotrophic Lateral Sclerosis. Curr. Opin. Struct. Biol. 2021, 66, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhu C; Beck MV; Griffith JD; Deshmukh M; Dokholyan NV Large SOD1 Aggregates, Unlike Trimeric SOD1, Do Not Impact Cell Viability in a Model of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bieschke J; Herbst M; Wiglenda T; Friedrich RP; Boeddrich A; Schiele F; Kleckers D; Lopez del Amo JM; Grüning BA; Wang Q; Schmidt MR; Lurz R; Anwyl R; Schnoegl S; Fändrich M; Frank RF; Reif B; Günther S; Walsh DM; Wanker EE Small-Molecule Conversion of Toxic Oligomers to Nontoxic β-Sheet-Rich Amyloid Fibrils. Nat. Chem. Biol. 2011, 8, 93–101. [DOI] [PubMed] [Google Scholar]
- (36).Bhowmik D; Mote KR; MacLaughlin CM; Biswas N; Chandra B; Basu JK; Walker GC; Madhu PK; Maiti S Cell-Membrane-Mimicking Lipid-Coated Nanoparticles Confer Raman Enhancement to Membrane Proteins and Reveal Membrane-Attached Amyloid-Beta Conformation. ACS Nano 2015, 9, 9070–9077. [DOI] [PubMed] [Google Scholar]
- (37).Jang H; Zheng J; Nussinov R Models of β-Amyloid Ion Channels in the Membrane Suggest That Channel Formation in the Bilayer Is a Dynamic Process. Biophys. J. 2007, 93, 1938–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Jang H; Arce FT; Ramachandran S; Capone R; Azimova R; Kagan BL; Nussinov R; Lal R Truncated β-Amyloid Peptide Channels Provide an Alternative Mechanism for Alzheimer’s Disease and Down Syndrome. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 6538–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Anekonda TS; Quinn JF; Harris C; Frahler K; Wadsworth TL; Woltjer RL L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol. Dis. 2011, 41, 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Dante S; Hauss T; Brandt A; Dencher NA; Hauß T; Brandt A; Dencher NA Membrane Fusogenic Activity of the Alzheimer’s Peptide Aβ(1–42) Demonstrated by Small-Angle Neutron Scattering. J. Mol. Biol. 2008, 376, 393–404. [DOI] [PubMed] [Google Scholar]
- (41).Pandey AP; Haque F; Rochet J-C; Hovis JS α-Synuclein-Induced Tubule Formation in Lipid Bilayers. J. Phys. Chem. B 2011, 115, 5886–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Varkey J; Isas JM; Mizuno N; Jensen MB; Bhatia VK; Jao CC; Petrlova J; Voss JC; Stamou DG; Steven AC; Langen R Membrane Curvature Induction and Tubulation Are Common Features of Synucleins and Apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Reynolds NP; Soragni A; Rabe M; Verdes D; Liverani E; Handschin S; Riek R; Seeger S Mechanism of Membrane Interaction and Disruption by α-Synuclein. J. Am. Chem. Soc. 2011, 133, 19366–19375. [DOI] [PubMed] [Google Scholar]
- (44).Hellstrand E; Nowacka A; Topgaard D; Linse S; Sparr E Membrane Lipid Co-Aggregation with α-Synuclein Fibrils. PLoS One 2013, 8, No. e77235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Shirwany NA; Payette D; Xie J; Guo Q The Amyloid Beta Ion Channel Hypothesis of Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2007, 3, 597. [PMC free article] [PubMed] [Google Scholar]
- (46).Arispe N; Pollard HB; Rojas E Giant Multilevel Cation Channels Formed by Alzheimer Disease Amyloid Beta-Protein [A Beta P-(1–40)] in Bilayer Membranes. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 10573–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sciacca MF; Lolicato F; Tempra C; Scollo F; Sahoo BR; Watson MD; García-Viñuales S; Milardi D; Raudino A; Lee JC; Ramamoorthy A; La Rosa C Lipid-Chaperone Hypothesis: A Common Molecular Mechanism of Membrane Disruption by Intrinsically Disordered Proteins. ACS Chem. Neurosci. 2020, 11, 4336–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yagi-Utsumi M; Matsuo K; Yanagisawa K; Gekko K; Kato K Spectroscopic Characterization of Intermolecular Interaction of Amyloid β Promoted on GM1 Micelles. J. Alzheimer’s Dis. 2011, 2011, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Okada T; Wakabayashi M; Ikeda K; Matsuzaki K Formation of Toxic Fibrils of Alzheimer’s Amyloid β-Protein-(1–40) by Monosialoganglioside GM1, a Neuronal Membrane Component. J. Mol. Biol. 2007, 371, 481–489. [DOI] [PubMed] [Google Scholar]
- (50).Ikeda K; Yamaguchi T; Fukunaga S; Hoshino M; Matsuzaki K Mechanism of Amyloid β-Protein Aggregation Mediated by GM1 Ganglioside Clusters. Biochemistry 2011, 50, 6433–6440. [DOI] [PubMed] [Google Scholar]
- (51).Fernández-Pérez EJ; Sepúlveda FJ; Peoples R; Aguayo LG Role of Membrane GM1 on Early Neuronal Membrane Actions of Aβ during Onset of Alzheimer’s Disease. Biochim. Biophys. Acta, Mol. Basis Dis. 2017, 1863, 3105–3116. [DOI] [PubMed] [Google Scholar]
- (52).Tachi Y; Okamoto Y; Okumura H Conformational Change of Amyloid-β 40 in Association with Binding to GM1-Glycan Cluster. Sci. Rep. 2019, 9, 6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Yanagisawa K; Odaka A; Suzuki N; Ihara Y GM1 Ganglioside–Bound Amyloid β–Protein (Aβ): A Possible Form of Preamyloid in Alzheimer’s Disease. Nat. Med. 1995, 1, 1062. [DOI] [PubMed] [Google Scholar]
- (54).Verma M; Vats A; Taneja V Toxic Species in Amyloid Disorders: Oligomers or Mature Fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Goure WF; Krafft GA; Jerecic J; Hefti F Targeting the Proper Amyloid-Beta Neuronal Toxins: A Path Forward for Alzheimer’s Disease Immunotherapeutics. Alzheimer’s Res. Ther. 2014, 6, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Mori K; Mahmood MI; Neya S; Matsuzaki K; Hoshino T Formation of GM1 Ganglioside Clusters on the Lipid Membrane Containing Sphingomyeline and Cholesterol. J. Phys. Chem. B 2012, 116, 5111–5121. [DOI] [PubMed] [Google Scholar]
- (57).Habchi J; Chia S; Galvagnion C; Michaels TCT; Bellaiche MMJ; Ruggeri FS; Sanguanini M; Idini I; Kumita JR; Sparr E; Linse S; Dobson CM; Knowles TPJ; Vendruscolo M Cholesterol Catalyses Aβ42 Aggregation through a Heterogeneous Nucleation Pathway in the Presence of Lipid Membranes. Nat. Chem. 2018, 10, 673–683. [DOI] [PubMed] [Google Scholar]
- (58).Pannuzzo M; Milardi D; Raudino A; Karttunen M; La Rosa C Analytical Model and Multiscale Simulations of Aβ Peptide Aggregation in Lipid Membranes: Towards a Unifying Description of Conformational Transitions, Oligomerization and Membrane Damage. Phys. Chem. Chem. Phys. 2013, 15, 8940–8951. [DOI] [PubMed] [Google Scholar]
- (59).Pannuzzo M; Raudino A; Milardi D; La Rosa C; Karttunen M α-Helical Structures Drive Early Stages of Self-Assembly of Amyloidogenic Amyloid Polypeptide Aggregate Formation in Membranes. Sci. Rep. 2013, 3, 2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Du Y; Du Y; Zhang Y; Huang Z; Fu M; Li J; Pang Y; Lei P; Wang YT; Song W; He G; Dong Z MKP-1 Reduces Aβ Generation and Alleviates Cognitive Impairments in Alzheimer’s Disease Models. Signal Transduction Targeted Ther. 2019, 4, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kagan BL; Hirakura Y; Azimov R; Azimova R; Lin M-C The Channel Hypothesis of Alzheimer’s Disease: Current Status. Peptides 2002, 23, 1311–1315. [DOI] [PubMed] [Google Scholar]
- (62).Hane F; Leonenko Z Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation. Biomolecules 2014, 4, 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Park JG; Palmer AE Measuring the in Situ Kd of a Genetically Encoded Ca2+ Sensor. Cold Spring Harb. Protoc. 2015, 2015, pdb.prot076554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Sciacca MFM; Kotler SA; Brender JR; Chen J; Lee D.-k.; Ramamoorthy A Two-Step Mechanism of Membrane Disruption by Aβ through Membrane Fragmentation and Pore Formation. Biophys. J. 2012, 103, 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Fancy DA; Kodadek T Chemistry for the Analysis of Protein–Protein Interactions: Rapid and Efficient Cross-Linking Triggered by Long Wavelength Light. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 6020–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Devarajan S; Sharmila JS Molecular Dynamics Study of GM1 Ganglioside Complex with Amyloid β Peptide (Aβ42) in Lipid Membrane. J. Mol. Liq. 2014, 195, 59–64. [Google Scholar]
- (67).Matsuzaki K How Do Membranes Initiate Alzheimer’s Disease? Formation of Toxic Amyloid Fibrils by the Amyloid β-Protein on Ganglioside Clusters. Acc. Chem. Res. 2014, 47, 2397–2404. [DOI] [PubMed] [Google Scholar]
- (68).Tong Y; Bai L; Gong R; Chuan J; Duan X; Zhu Y Shikonin Protects PC12 Cells against β-Amyloid Peptide-Induced Cell Injury through Antioxidant and Antiapoptotic Activities. Sci. Rep. 2018, 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Shibahara M; Zhao X; Wakamatsu Y; Nomura N; Nakahara T; Jin C; Nagaso H; Murata T; Yokoyama KK Mannosylerythritol Lipid Increases Levels of Galactoceramide in and Neurite Outgrowth from PC12 Pheochromocytoma Cells. Cytotechnology 2000, 33, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Sipione S; Monyror J; Galleguillos D; Steinberg N; Kadam V Gangliosides in the Brain: Physiology, Pathophysiology and Therapeutic Applications. Front. Neurosci. 2020, 14, 572965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Hu X; Crick SL; Bu G; Frieden C; Pappu RV; Lee J-M Amyloid Seeds Formed by Cellular Uptake, Concentration, and Aggregation of the Amyloid-Beta Peptide. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 20324–20329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Thal DR; Walter J; Saido TC; Fändrich M Neuropathology and Biochemistry of Aβ and Its Aggregates in Alzheimer’s Disease. Acta Neuropathol. 2015, 129, 167–182. [DOI] [PubMed] [Google Scholar]
- (73).Li RC; Pouranfar F; Lee SK; Morris MW; Wang Y; Gozal D Neuroglobin Protects PC12 Cells against β-Amyloid-Induced Cell Injury. Neurobiol. Aging 2008, 29, 1815–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Kakio A; Nishimoto S.-i.; Yanagisawa K; Kozutsumi Y; Matsuzaki K Interactions of Amyloid β-Protein with Various Gangliosides in Raft-like Membranes: Importance of GM1 Ganglioside-Bound Form as an Endogenous Seed for Alzheimer Amyloid. Biochemistry 2002, 41, 7385–7390. [DOI] [PubMed] [Google Scholar]
- (75).Chen G.-f.; Xu T.-h.; Yan Y; Zhou Y.-r.; Jiang Y; Melcher K; Xu HE Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Ledeen RW; Wu G The Multi-Tasked Life of GM1 Ganglioside, a True Factotum of Nature. Trends Biochem. Sci. 2015, 40, 407–418. [DOI] [PubMed] [Google Scholar]
- (77).Arispe N; Rojas E; Pollard HB Alzheimer Disease Amyloid Beta Protein Forms Calcium Channels in Bilayer Membranes: Blockade by Tromethamine and Aluminum. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Sanghera N; Correia BEFS; Correia JRS; Ludwig C; Agarwal S; Nakamura HK; Kuwata K; Samain E; Gill AC; Bonev BB; Pinheiro TJT Deciphering the Molecular Details for the Binding of the Prion Protein to Main Ganglioside GM1 of Neuronal Membranes. Chem. Biol. 2011, 18, 1422–1431. [DOI] [PubMed] [Google Scholar]
- (79).Colletier J-P; Chaize B; Winterhalter M; Fournier D Protein Encapsulation in Liposomes: Efficiency Depends on Interactions between Protein and Phospholipid Bilayer. BMC Biotechnol. 2002, 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Bitan G; Teplow DB Rapid Photochemical Cross-Linking–a New Tool for Studies of Metastable, Amyloidogenic Protein Assemblies. Acc. Chem. Res. 2004, 37, 357–364. [DOI] [PubMed] [Google Scholar]
- (81).Kremer JR; Mastronarde DN; McIntosh JR Computer Visualization of Three-Dimensional Image Data Using IMOD. J. Struct. Biol. 1996, 116, 71–76. [DOI] [PubMed] [Google Scholar]
- (82).Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- (83).Schwede T; Kopp J; Guex N; Peitsch MC SWISSMODEL: An Automated Protein Homology-Modeling Server. Nucleic Acids Res. 2003, 31, 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Van Der Spoel D; Lindahl E; Hess B; Groenhof G; Mark AE; Berendsen HJC GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- (85).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; De Groot BL; Grubmüller H; MacKerell AD CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Michaud-Agrawal N; Denning EJ; Woolf TB; Beckstein O MDAnalysis A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DOI: 10.17632/38tt768dkd.2.