

Summary
Long-term potentiation (LTP), which underlies learning and memory, can be induced by high-frequency electrical stimulation (HFS or HFES) and is thought to occur at the synapses of efferent projection. Here, the contralateral connectivity in mice auditory cortex was investigated to reveal the fundamental corticocortical connection properties. After HFES, plasticity was not observed at the terminal synapses at the recording site. The optogenetic HFS at the recording site of the interhemispheric cortical projections could not induce LTP, but HFES at the recording site could induce the interhemispheric cortical LTP. Our subsequent results uncovered that it is the cholecystokinin (CCK) released from the entorhino-neocortical pathway induced by HEFS that modulates the neuroplasticity of the afferent projections, including interhemispheric auditory cortical afferents. Our study illustrates a heterosynaptic mechanism as the basis for cortical plasticity. This regulation might contribute new spots for the understanding and treatment of neurological disorders.
Subject areas: Cellular neuroscience, Physiology, Sensory neuroscience
Graphical abstract
Highlights
-
•
Optogenetic HFS of interhemispheric projection could not induce homosynaptic LTP
-
•
Interhemispheric LTP requires heterosynaptic activation of entorhinal projections
-
•
Entorhinal afferents could release CCK which modulates interhemispheric plasticity
Cellular neuroscience; Physiology; Sensory neuroscience
Introduction
Brain connections determine the brain’s functional organization, hence complex cognition and associative behavior require an integration of cortical areas orchestrated by densely yet precisely connected networks.1 The cortical network connectivity can be shaped by experience to adapt to both the complexity and changes in the environment. The developing brain exhibits a high possibility of change and cortical plasticity progressively declines across the life span.2,3,4 The investigation of plasticity mechanisms may shed light on the neurobiological mechanisms underlying normative aging and neurodegenerative processes.
Interhemispheric commissural circuits are essential in high-level associative complexity through the bilateral integration of information.5 The callosal projection is the largest connection system of the mammalian brain that can directly activate contralateral pyramidal neurons, as well as inhibitory interneurons.6,7,8 These callosal inputs can drive excitation or inhibition in cortical circuits to achieve cortical integration or segregation of lateralized functions.9 In the auditory cortex (AC), callosal communication between the two cerebral hemispheres is necessary for sound localization processes underlying spatial hearing.7,10,11 The formation of callosal synapses is supported by presynaptic and postsynaptic neuronal co-activities based on the Hebbian rule in post-natal periods.12,13 Nevertheless, how an adult brain forms interhemispheric cortical plasticity needs to be elucidated. Herein, we focus on the induction of interhemispheric cortical long-term potentiation (LTP) in the auditory cortex of adult mice using in vivo methodology attempting to answer the proposed question.
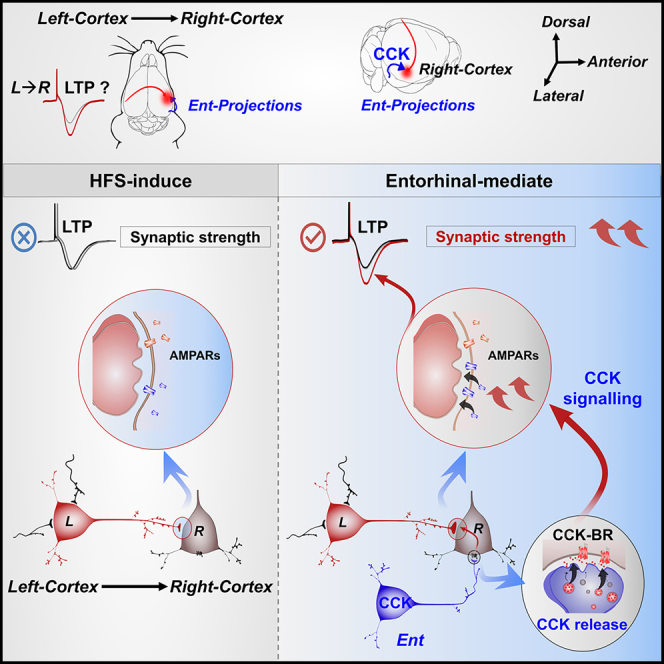
LTP, a form of synaptic change, is considered to be the basis of memory traces in different brain regions.14,15,16,17,18,19 Impairment of LTP-like cortical plasticity is a key indicator in some neurological diseases such as Alzheimer’s diseases.20,21 LTP can be induced by high-frequency stimulation (HFS), which activates both presynaptic fibers and the postsynaptic neurons and thus produces homosynaptic Hebbian LTP.14,17,18,19,22,23,24 Although a few studies reported that the interhemispheric LTP could be inducted in slices, the requirements to trigger this LTP at the system-level (in vivo) are yet to be discovered.25,26 In the current study, we found that HFS could not induce the homosynaptic LTP of interhemispheric cortical pathway. Conversely, the interhemispheric cortical LTP could be induced in a heterosynaptic manner with the assistance of other projections. The studied heterosynaptic plasticity, in some ways, assists in preventing runaway dynamics of basic Hebbian plasticity by introducing another force to drive the interhemispheric cortical pathway cooperatively.27 According to previous studies, the entorhinal cortex enables neocortical plasticity via its CCK-positive cortical projection neurons, and this plasticity can be suppressed by the infusion of a CCK-B receptor (CCK-BR) antagonist.28,29 Moreover, CCK plays an essential role in inducing LTP and enabling associative memory formation.29 Therefore, we hypothesize that the entorhino-neocortical projections are necessary to mediate the heterosynaptic LTP of interhemispheric cortical pathway through CCK release.
This work might depict a heterosynaptic mechanism underlying the formation of interhemispheric cortical plasticity that could foster the diversity of research on complicated cortical plasticity.
Results
Interhemispheric cortical LTP is not homosynaptic but heterosynaptic LTP
The presynaptic and postsynaptic neuronal activity enables callosal cortical plasticity in the developing brain,12,13 on which homosynaptic plasticity is based, according to the Hebbian rule. HFS is the most adopted methodology to induce a homosynaptic Hebbian-type LTP,14,17,18,19,22,23,24 whose plasticity is thought to occur at the synapses that terminate from the stimulation site projecting onto neurons at the recording site.30,31 To investigate the requirements for cortical plasticity in a mature brain, we first utilize HFS to induce interhemispheric LTP in the auditory cortex of adult mice. Contralateral cortical projection illustrates a “mirror-like” symmetrical but attenuated connectivity.32 Both hemispheric auditory cortices served as the stimulation site L and the recording site R, respectively. We combined optogenetics and electrical stimulation through in-vivo electrophysiology recordings.
To confirm the topological homosynaptic cortical projections, we injected the virus (AAV-hSyn-Chronos-GFP) into the auditory cortex (site L) of wild-type C57 mice (Figure 1A left panel). Projections labeled by the virus injected in site L were found in the mirrored location of the contralateral auditory cortex (site R, Figure 1A right panel). The commissural fibers were only found via corpus callosum, meaning most of the interhemispheric projection neurons in the auditory cortex are callosal projection neurons (Figure 1A right panel). The majority, accounting for 93.5 ± 3.8% of the GFP+ neurons, were CamKll+ neurons (Figures S1A and S1B). This result confirmed that the neurons of one hemisphere directly projected to the contralateral auditory cortex, and these projections are excitatory.
Figure 1.

Interhemispheric cortical LTP could not be homosynaptic but heterosynaptic LTP
(A) Left panel: AAV-hSyn-Chronos-GFP was injected into the auditory cortex at site L of C57 mice. Right panel: Images of virus-expressing neurons in injection site L and site R. Confocal images presenting the afferent fibers were detected in site R in the enlarged image (scale bars: 500 and 100 μm).
(B) Left panel: The diagram of the experiment design. A laser of 473-nm wavelength was used to activate the afferent terminals with opsins expression at site R, which was projected from the contralateral auditory cortex. Glass pipette electrodes at site R were used to record the field EPSPs evoked by electrical stimulation ES-L and laser stimulations LS-R. Right panel: Experimental protocol. ES was applied to site L (ES-L). Laser was applied to site R (LS-R). HFES-L was applied to site L.
(C) Left: Representative fEPSP traces evoked by ES-L and LS-R (473 nm, right) before (dotted) and after (solid) HFES-L. Right: Normalized fEPSP slopes in response to ES-L (red) and LS-R (473 nm, blue) before and after HFES-L (ES-L: ∗∗, p < 0.001, two-way RM ANOVA; LS-R: ns, p = 0.809, two-way RM ANOVA; ES-L vs. LS-R after HFES-L:∗∗, two-way RM ANOVA, p < 0.001).
(D) A schematic Diagram. Upper: Stronger outputs from the stimulation (left) site due to the enhanced neural activity in response to ES after the HFS. Lower: Strengthened synaptic connectivity within the recording site after the HFS.
(E) AAV-hSyn-Chronos-GFP was injected into the auditory cortex at site L of C57 mice. 473 nm laser was used to activate the afferent terminals with opsins expression at site R, which was projected from the contralateral auditory cortex. Glass pipette electrodes at site R were used to record the field EPSPs evoked by ES-L and LS-R.
(F) Experimental protocol. ES was applied to site L. Laser was applied to site R. HFLS was applied to site R. Lower: The fEPSPs evoked by HFLS-R.
(G) fEPSPs in response to HF 473 nm laser pulse train at site R. Left: Representative fEPSP traces evoked by ES-L and LS-R (473 nm) before (dotted) and after (solid) HFLS-R. Right: Normalized fEPSP slopes in response to ES-L (red) and LS-R (473 nm, blue) before and after HFLS-R (ES-L: ns, p = 0.970, two-way RM ANOVA; LS-R: ns, p = 0.906, two-way RM ANOVA).
(H) Upper panel: AAV-hSyn-ChrimsonR-tdTomato was injected into the auditory cortex at site L of C57 mice. Lower panel: Images of virus-expressing neurons in injection site L and site R. Confocal images presenting the afferent fibers were detected in site R in the enlarged image (Scale bars: 500 and 100 μm).
(I) Upper panel: The diagram of the experiment design. A laser of 620-nm wavelength was used to activate the afferent terminals with opsins expression at site R, which was projected from the contralateral auditory cortex. Glass pipette electrodes at site R were used to record the field EPSPs evoked by electrical stimulation ES-L and laser stimulation LS-R. Lower panel: Experimental protocol. ES was applied to site L. Laser was applied to site R. HFES-R was applied to site R.
(J) Left: Representative fEPSP traces evoked by ES-L and LS-R (620 nm) before (dotted) and after (solid) HFES-R. Right: Normalized fEPSP slopes in response to ES-L (black) and LS-R (620 nm, red) before and after HFES-R (Response to ES-L, before vs. after HFES-R: ∗∗, p < 0.001, two-way RM ANOVA; response to LS-R, before vs. after HFES-R, ∗∗, p < 0.001, two-way RM ANOVA).
(K) HFLS-R could neither potentiate the neural response to ES-L nor to LS-R; HFES-L could potentiate the response to ES-L but not to LS-R; HFES-R could potentiate the response to both ES-L and LS-R.
(L) Besides the cortical neurons, HFS could also activate the afferent fibers from other brain regions and cortical fibers locally.
During the electrophysiology experiment (Figure 1B left panel), we first inserted low-impedance electrodes into the layer V of the left auditory cortex where the virus had been injected (site L) for ES (ES-L), as well as glass electrodes into the symmetrical site of the right hemisphere (site R) for neural responses recording. Then we attached an optic fiber on the cortex’s surface near site R for laser stimulation (LS-R), which could specifically activate the projection terminals of the pathway L→R, to examine the possible contribution of the plasticity in the recording site R to the HFES-L-induced LTP of pathway L→R. Since we inserted the ES electrode into the same location where the virus was injected, LS-R activated many fiber terminals that ES-L also activated. We applied ES-L and LS-R alternatively and detected neural responses to ES-L or LS-R at site R by the slope of the field excitatory postsynaptic potentials (fEPSPs). ES-L could evoke extracellular neuronal spikes (Figure S1C). High-frequency electrical stimulation at site L (HFES-L, Figure S1D) was delivered to induce the LTP of the pathway L→R after baseline probe testing (Figure 1B right panel). The fEPSPs in response to ES-L were potentiated, but fEPSPs to LS-R remained unchanged after HFES-L (two-way RM ANOVA, significant interaction F[1,138] = 116.3, p < 0.001; responses to ES-L: 99.0 ± 1.4% vs. 151.0 ± 3.1%, before vs. after HFES-L, pairwise comparison, p < 0.001; LS-R activation: 98.4 ± 1.9% vs. 99.3 ± 3.0%, before vs. after HFES-L, pairwise comparison, p = 0.809, n = 14 from 9 mice, Figure 1C). The difference between the fEPSPs in response to ES-L and LS-R was significant after HFES-L (151.0 ± 3.1% vs. 99.3 ± 3.0%, responses to ES-L vs. LS-R activation, pairwise comparison, p < 0.001, Figure 1C). These results indicated that HFES-L-induced neuroplasticity might not occur within the synapses at site R but in the stimulation site L (Figure 1D). We reasoned that HFES-L at the neocortex enabled neuroplasticity around the stimulation site L; the enhanced local cortical network at site L resulted in stronger outputs generated by ES after HFS than before HFS (Figure 1D upper panel). Therefore, the LTP observed in site R is not an interhemispheric cortical LTP (Figure 1D lower panel).
These results differ from our current understanding of neuroplasticity occurring in synapses at the recording site of projections from the stimulation site in the HFS-induced LTP. Previous studies have demonstrated that the inactivation of a targeted neuron prevents neuroplasticity from happening.33,34,35 In the next experiment, we silenced site R while applying HFS to site L to ensure our results were reliable. If the neuroplasticity occurred at site L as we expected, the potentiation of the fEPSPs could still be recorded.
We infused lidocaine (1.0%, 0.5 μL) into site R after baseline probe ES testing (Figure S1E). Lidocaine temporarily suppressed the neuronal activity at site R (Figure S1F, time at 0 min). In the control group without HFES-L, the fEPSP slope dropped initially and gradually recovered after lidocaine infusion. After the recovery of the neural activity, the fEPSP slope remained the same as before (Figure S1F, blue dots; 99.8 ± 0.7% vs. 100.9 ± 2.0%, before vs. after, pairwise comparison, p = 0.594, n = 19 from 5 mice). In the experimental group, HFES-L was applied after the lidocaine infusion. The neural responses to ES-L were successfully potentiated even when the neurons at recording site R were temporarily silenced during HFES-L (Figure S1F, red dots; two-way RM ANOVA, significant interaction F [1,158] = 208.9, p < 0.001; 99.3 ± 0.9% vs. 147.9 ± 2.4% before vs. after lidocaine pairwise comparison, p < 0.001, n = 13 from 6 mice). The result of the control group excluded the possibility of LTP caused by any rebound effects after neural suppression by lidocaine. This cortical LTP occurred even though the neural activities at site R were silenced. Compared to Figure 1C ES group, the mean difference of LTP induced by HFES-L with or without neural activities at site R is 5.1 ± 4.4%, which is nonsignificant (two-way RM ANOVA, pairwise comparison, p = 0.241), implying that neural plasticity could only occur within site L.
Previously, we adopted a traditional method to induce interhemispheric cortical LTP, the HFES-L had proved herein to induce a local LTP but not the interhemispheric cortical LTP. The plasticity did not occur at site R, where synaptic pathway L→R connections are located, but at the HFS site L. Hence, how could the real LTP of the interhemispheric cortical pathway L→R be formed?
First, we tried to conduct high-frequency activation of the interhemispheric cortical projections directly. The same virus-injected mice were employed as those in the experiment in Figures 1A–1C. The experimental protocol is shown in Figures 1E and 1F. Neural responses to ES-L and LS-R were recorded at site R. We applied high-frequency laser stimulation at site R (HFLS-R) to directly activate the commissural terminals of pathway L→R. The fEPSPs response to HFLS-R showed that the activation of the projection could follow HFLS-R at 80 Hz (Figure 1F). Still, the fEPSPs evoked by either ES-L or LS-R were not potentiated after HFLS-R (ES-L: 98.7 ± 0.9% vs. 98.6 ± 2.0%, before vs. after HFLS-R, pairwise comparison, p = 0.970; LS-R: 98.6 ± 2.6% vs. 99.0 ± 2.5%, before vs. after HFLS-R, pairwise comparison, p = 0.906, n = 13 from 8 mice, Figure 1G). It further proved that the high-frequency activation of terminals of commissural projection neurons failed to strengthen the synaptic connectivity of the pathway L→R. Interhemispheric cortical LTP could not be formed in a homosynaptic manner.
In the next experiment, we tried to apply the high-frequency electrical stimulation at site R (HFES-R) to induce LTP of the interhemispheric cortical pathway L→R. HFES-R could induce high-frequency activation of all the afferent projections to site R non-specifically. Since HFS could enable neuroplasticity around the HFS site (Figure 1C), we aimed to verify if the cortical HFS could enable the plasticity of the afferents from the contralateral hemisphere.
We injected the virus (AAV-hSyn-ChrimsonR-tdTomato) into the auditory cortex (site L) of C57 mice (Figure 1H). Projections labeled by the virus from site L were found in the contralateral auditory cortex at site R. We delivered HFES-R to the recording site R after baseline probe testing (Figure 1I). The fEPSPs to both ES-L and LS-R were potentiated after HFES-R this time (response to ES-L: increased by 34.2 ± 3.8%, pairwise comparison p < 0.001; response to LS-R: increased by 36.6 ± 3.8%, pairwise comparison p < 0.001, Figure 1J), which suggested neural plasticity occurred at recording site R (two-way RM ANOVA, significant interaction F[1,168] = 38.1, p < 0.001; response to ES-L: 99.6 ± 0.5% vs. 133.9 ± 3.7% before vs. after HFES-R, n = 17 from 12 mice; response to LS-R: 100.8 ± 0.5% vs. 137.4 ± 3.7%, n = 17 from 12 mice, Figure 1J). The mean difference of LTP induced by HFES-R to ES or laser at site R is 3.5 ± 5.2%, which is nonsignificant (two-way RM ANOVA, pairwise comparison, p = 0.496). The real interhemispheric cortical LTP of pathway L→R was successfully induced by HFES-R.
Figure 1G showed that high-frequency laser stimulation of the interhemispheric afferent terminals (at site R) could not induce the interhemispheric cortical LTP of pathway L→R. However, the high-frequency electrical stimulation at the recording site R could induce this interhemispheric cortical LTP (Figures 1J and 1K). Besides the specific projection from site L, HFES-R could activate other afferent projections at site R (Figure 1L), which might help modulate this LTP. These results imply that the interhemispheric cortical LTP of pathway L→R could not be homosynaptic but heterosynaptic LTP.
Interhemispheric cortical LTP could be generated in a heterosynaptic manner through the modulation of entorhino-neocortical projections
At first, we surprisingly found that HFES-L could not induce the homosynaptic LTP of the interhemispheric cortical pathway but only triggered the local neural plasticity around the stimulation site L. Further, we confirmed that the interhemispheric cortical LTP could only be induced in a heterosynaptic manner. Next, we aimed to find which pathway was responsible for modulating this synaptic plasticity. Previous studies showed that the entorhinal cortex enables neocortical plasticity via its CCK-positive cortical projection neurons and this plasticity can be suppressed by the infusion of a CCK-B receptor (CCK-BR) antagonist.28,29 Therefore, we hypothesize that the entorhino-neocortical CCK projections are necessary to modulate the heterosynaptic LTP of the interhemispheric cortical pathway.
In the following experiment, we examined whether the high-frequency stimulation of entorhino-neocortical CCK projections induced hetero-synaptic LTP in the commissural terminals of the pathway L→R. We injected two viruses, one into the entorhinal cortex, ipsilateral to site R (AAV-EF1ɑ-FLEX-Chronos-eYFP) and the other into site L (AAV-hSyn-ChrimsonR-tdTomato) in CCK-ires-Cre mice (Figure 2A). The virus in the entorhinal cortex selectively infected the CCK neurons, and the virus in the auditory cortex transfected neurons non-specifically (Figures 2B and 2C). Virus-labeled afferents from both the auditory cortex and the entorhinal cortex overlapped in site R, where optical fiber was placed (Figure 2C). In the entorhinal cortex, 90.1 ± 3.1% of eYFP+ neurons were CamKll+, 5.0 ± 1.4% were GAD67+, and 92.7 ± 0.93% of eYFP expressing neurons were CCK+ (Figures S2A and S2C: total 1,474 neurons, 1,326 eYFP+ neurons, n = 12 slices, N = 3 mice; Figures S2B and S2C: 1,665 eYFP+ neurons, n = 12 slices, N = 3 mice, correspondingly), implying most entorhino-neocortical projections at site R were from excitatory CCK neurons.
Figure 2.

Interhemispheric cortical LTP could be generated in a heterosynaptic manner through the modulation of entorhino-neocortical afferents
(A) AAV-hSyn-ChrimsonR-tdTomato was injected into the auditory cortex at site L; AAV-EF1α-FLEX-chronos-eYFP or AAV-EF1α-DIO-eYFP was injected into the entorhinal cortex on the contralateral side of CCK-Cre mice.
(B) Locations of tdTomato expressing somas in site L and projecting fibers in site R are revealed (scale bar: 500 μm).
(C) Enlarged images show the AAV-hSyn-Chrimson-tdTomato injection site (left panel) and both afferents from the auditory cortex (red) and entorhinal cortex (green, middle panel, scale bars: 100 μm). Right: Confocal image shows the virus injection position in the entorhinal cortex (Ent, scale bar: 500 μm).
(D) Diagram of the experiment design; 473 nm and 620 nm lasers were used to activate the afferent terminals with opsins expression at site R, which was projected from the entorhinal and contralateral auditory cortex, respectively. Glass pipette electrodes at site R were used to record the field EPSPs evoked by ES-L and 620 nm laser stimulations (LS-R).
(E) Experimental protocol. ES was applied to site L. Laser of 620 nm was applied to site R. High-frequency laser of 473 nm (5 pulses-50 Hz × 10 burst-0.1Hz, HFLS) were applied to site R, followed by low-frequency ES-L (5 pulse-1Hz × 10 burst-0.1Hz, ES).
(F) Upper: fEPSPs evoked by HF 473 nm laser pulse train at site R of CCK-ires-Cre mice from the experimental group. Lower: fEPSPs in response to HF 473 nm laser pulse train at site R of CCK-ires-Cre mice from the control group.
(G) Experimental group. Upper: Representative fEPSP traces evoked by ES-L and LS-R (620 nm, right) before (dotted) and after (solid) HFLS-ES pairing. Lower: Normalized fEPSP slopes in response to ES-L (black) and LS-R (620 nm, red light) before and after HFLS-ES pair (ES-L: ∗∗ Two-way RM ANOVA, p < 0.001; LS-R: ∗∗ Two-way RM ANOVA, p < 0.001; ES-L vs. LS-R after HFLS-ES pair: ns, two-way RM ANOVA, p = 0.132).
(H) Control group. Upper: Representative fEPSP traces evoked by ES-L and LS-R (620 nm) before (dotted) and after (solid) HFLS-ES pair. Lower: Normalized fEPSP slopes in response to ES-L (black) and LS-R (620 nm, red) before and after HFLS-ES pair (ES-L: ns, two-way RM ANOVA, p = 0.923; LS-R: ns, two-way RM ANOVA, p = 0.816).
We delivered ES to site L (ES-L) and monitored the neural activity at site R. The optic fiber at site R activated the afferents of L→R with red light or the afferents of entorhino-neocortex with blue light (Figure 2D). We applied ES-L and red light at site R (LS-R) alternatively for the baseline fEPSPs, before delivering high-frequency laser stimulation of blue light to site R (HFLS, five pulses at 50 Hz for each burst, ten bursts at 0.1 Hz). Five pulses of ES-L were presented after each blue light HFLS burst at 1 Hz to activate the pathway L→R (HFLS-ES, Figure 2E, pairing protocol). The fEPSPs evoked by blue light HFLS showed that entorhino-neocortical afferent terminals reliably followed the high-frequency laser stimulation (Figure 2F upper panel). After the pairing of blue-light HFLS and ES-L (Figure 2E), the fEPSPs response to ES-L and red light LS-R were both potentiated in a similar level (fEPSP traces and time course, Figure 2G; two-way RM ANOVA, significant interaction F[1,198] = 2.04, p = 0.155; responses to ES-L: pairwise comparison, 99.8 ± 0.9% vs. 130.2 ± 2.9%, before vs. after paring, p < 0.001, n = 20 from 6 mice; response to red light LS-R: pairwise comparison, 99.5 ± 2.6% vs. 139.0 ± 5.1%, before vs. after paring, p < 0.001, n = 20 from 6 mice; pairwise comparison between fEPSPs to ES-L and red light LS-R at 60 min after the pairing, p = 0.132; Figure 2G), indicating that LS-R and ES-L may activate similar projections at site R. Different from the experiments in Figure 1C, the neural plasticity occurred at the recording site R but not site L. The interhemispheric cortical LTP was successfully induced under the modulation of entorhino-neocortical CCK projections.
For the control group, we injected the AAV-EF1ɑ-DIO-eYFP virus into the entorhinal cortex, so that the entorhino-neocortical CCK projections would not be activated by the blue laser (Figure 2F lower panel). The other conditions remained unchanged. The same HFLS-ES pairing induced no LTP of pathway L→R via either ES-L or red light LS-R (two-way RM ANOVA, significant interaction F[1,168] = 0.055, p = 0.816; fEPSP to ES-L: pairwise comparison, 99.6 ± 0.5% vs. 99.2 ± 1.0%, before vs. after pairing, p = 0.923; fEPSP to red light LS-R: pairwise comparison, 100.0 ± 3.6% vs. 100.9 ± 3.8%, before vs. after pairing, p = 0.816; n = 17 from 6 mice; Figure 2H). We also specified the expression of the AAV-EF1ɑ-DIO-eYFP virus in the entorhinal cortex (Figures S2D–S2K) and found the same results as the experimental group.
The interhemispheric cortical LTP of pathway L→R could not be induced by high-frequency activation of its commissural terminals, which means that the LTP is not homosynaptic LTP. In contrast, this LTP could be induced in the commissural terminals when their low-frequency activation was paired with high-frequency laser stimulation of entorhino-neocortical CCK projections. These results indicate that the interhemispheric cortical LTP could be generated in a heterosynaptic manner through the modulation of entorhino-neocortical afferents.
High-frequency activation of the entorhino-neocortical projections could release CCK that enables the interhemispheric cortical plasticity
In the final experiment, we aimed to explore how entorhino-neocortical CCK projections could modulate the LTP of the interhemispheric cortical pathway. Immunohistochemistry showed that 37.9 ± 2.8% of eYFP+ and excitatory postsynaptic density protein 95 (PSD95) labeled interhemispheric cortical terminals were CCK positive (Figures S3A and S3C). We inferred that very limited CCK, if any, was released during the current stimulation protocol of commissural projection terminals of the interhemispheric pathway. However, 90.3 ± 0.6% of PSD95 and eYFP co-labeled entorhino-neocortical terminals were CCK positive (Figures S3B and S3C), and 91.2 ± 1.4% were colocalized with CCK-BRs (Figures S3D and S3E). It showed that much more CCK was in the entorhino-neocortical projections than that in the commissural terminals of pathway L→R. According to our previous studies, cortical neural plasticity could be readily induced by presynaptic and postsynaptic coactivation in the presence of CCK.28 Hence, we proposed that the high-frequency activation of entorhino-neocortical projections could trigger CCK release from their terminals, and the released CCK enabled the interhemispheric cortical plasticity of pathway L→R (Figure 3A). To monitor the dynamics of CCK, we adopted the G protein-coupled receptor (GPCR) activation-based CCK sensor, which is a circular-permutated green fluorescent protein inserted into the intracellular domain of CCK-BR. Once the CCK-BR binds with CCK, the increased fluorescence intensity can be measured by fiber photometry.
Figure 3.

High-frequency activation of the entorhino-neocortical projections could release CCK that enables the interhemispheric cortical plasticity
(A) A schematic diagram illustrated the hypothetic model of the heterosynaptic neuroplasticity for the commissural LTP induction. Left: The activation of the L→R pathway; middle: High-frequency activation of the entorhino-neocortical afferents triggered the CCK release; right: Synaptic plasticity of the interhemispheric cortical pathway enabled by the regulation of CCK.
(B) AAV-syn-CCKsensor was injected into the auditory cortex at site R, and pAAV-Syn-FLEX-ChrimsonR-tdTomato/pAAV-Syn-ChrimsonR-tdTomato into the entorhinal cortex, ipsilateral to site R in CCK-ires-Cre/CCK-KO mice. Enlarged images show the AAV-hSyn-Chrimson-tdTomato injection site (upper right) and co-localized (lower right) entorhino-neocortical projections (lower left) with CCKsensor expression (lower middle, Scale bars: 1 mm and 200 μm).
(C) Upper: The diagram of the experiment design. An optical fiber was attached to the cortex’s surface near site R to activate entorhino-neocortical terminals by red light. Another optical fiber was placed into site R to monitor the fluorescence intensity. Lower: Traces of fluorescence signal of the CCK-sensor before and after the high-frequency laser stimulation (50 Hz red light, HFLS) in CCK-Cre or CCK-KO mice. Averaged ΔF/F0% was shown as the mean value in solid line and SEM in shadow area. The fluorescence increased after HFLS in CCK-Cre mice (red), but the HFLS could not induce the increase in CCK-KO mice (blue).
(D) Bar charts showing the averaged ΔF/F0% from the different groups before and after HFS. (Bonferroni multiple comparisons test: before vs. after HFLS in CCK-Cre: ∗∗, p < 0.0001, n = 21, N = 11; before vs. after HFLS in CCK-KO: ns, p = 1.0, n = 22, N = 11; After HFLS in CCK-Cre vs. CCK-KO: p < 0.0001).
(E) Upper: Electrode and drug-injection pipette placement. ES was applied to site L. CCK or ACSF was injected at the stimulation site R; Lower: Normalized fEPSP slopes before and after CCK (red) or ACSF (blue) injections at site R followed by ES (CCK group: ∗∗, two-way RM ANOVA, p < 0.001; ACSF group: ns, two-way RM ANOVA, p = 0.658; After CCK vs. after ACSF: ∗∗, two-way RM ANOVA, p < 0.001).
(F) Upper: Electrode and drug-injection pipette placement. ES was applied to site L. CCK or ACSF was injected at the recording site L. Lower: Normalized fEPSP slopes before and after CCK (red) or ACSF (blue) injection at site L, followed by ES (CCK group: ∗∗, two-way RM ANOVA, p < 0.001; ACSF group: ns, two-way RM ANOVA, p = 0.784; After CCK vs. after ACSF: ∗∗, two-way RM ANOVA, p < 0.001).
(G) Representative images of CCK-BR expression in CCK-ABRKO and wild-type mice. Upper: Immunolabeling showing CamKll+ and CCK-BR+ neurons in the auditory cortex of wild-type mice (129S, scale bar: 20 μm). Lower: Immunolabeling showing CamKll+ and CCK-BR+ neurons in the auditory cortex of CCK-ABRKO mice (scale bar: 20 μm).
(H) Upper: Positions of recording and stimulating electrodes in the mouse auditory cortex. Lower: Normalized fEPSPs slopes before and after HFS in 129s mice (red) or CCK-ABRKO mice (blue) (two-way RM ANOVA, 129s mice; ∗∗p < 0.001; CCK-ABRKO mice: ns, p = 0.377; After HFES-L in 129s vs. CCK-ABRKO: ∗∗, two-way RM ANOVA, p < 0.001).
We injected the AAV-syn-CCK sensor virus into site R, and pAAV-Syn-FLEX-ChrimsonR-tdTomato/pAAV-Syn-ChrimsonR-tdTomato into the entorhinal cortex, ipsilateral to site R in CCK-Cre/CCK-KO mice (Figure 3B). The AAV-syn-CCK sensor was well expressed six weeks after the virus was injected into the auditory cortex36 (Figure 3B). We then applied a high-frequency red laser (HFLS) at the auditory cortex to activate the entorhino-neocortical terminals. We observed the fluorescence increased significantly in CCK-Cre mice but not in CCK-KO mice (Figures 3C and 3D Averaged ΔF/F0%: two-way ANOVA followed by Bonferroni multiple comparisons test, significant interaction, F [1, 41] = 28.48, p < 0.0001; before vs. after HFLS in CCK-Cre: −0.003 ± 0.004 vs. 0.253 ± 0.040, p < 0.0001; before vs. after HFLS in CCK-KO: 0.004 ± 0. 002 vs. 0.001 ± 0.025, ns, p = 1.0; After HFLS in CCK-Cre vs. CCK-KO: 0.253 ± 0.040 vs. 0.001 ± 0.025, p < 0.0001), suggesting that CCK was released from entorhino-neocortical afferents by the high-frequency activation. We also applied HFLS to the entorhinal cortex directly. Similarly, fluorescence enhanced greatly in CCK-Cre mice but not in CCK-KO mice (Figures S4A and S4B averaged ΔF/F0%: two-way ANOVA followed by Bonferroni multiple comparisons test, significant interaction, F [1, 34] = 36.24, p < 0.0001; before vs. after HFLS in CCK-Cre: 0.004 ± 0.002 vs. 0.271 ± 0.029, p < 0.0001; before vs. after HFLS in CCK-KO: 0.007 ± 0.003 vs. 0.001 ± 0.034, ns, p = 1.0; After HFLS in CCK-Cre vs. CCK-KO: 0.271 ± 0.029 vs. 0.001 ± 0.034, p < 0.0001). Lastly, we confirmed that fluorescence intensity in the auditory cortex increased after the administration of CCK-4 but not after artificial cerebrospinal fluid (ACSF) on the surface of the auditory cortex (Figures S4C and S4D Averaged ΔF/F0% of CCK-Cre mice: two-way ANOVA followed by Bonferroni multiple comparisons test, significant interaction, F [1, 20] = 10.99, p = 0.0035; before vs. after CCK4: 0.068 ± 0.031 vs. 9.00 ± 3.12, ∗∗, p = 0.0014; before vs. after ACSF: −0.062 ± 0.050 vs. −1.11 ± 0.320, ns, p = 1.0; After CCK-4 vs. after ACSF: 9.00 ± 3.12 vs. −1.11 ± 0.320, p < 0.0001; averaged ΔF/F0% of CCK-KO mice: two-way ANOVA followed by Bonferroni multiple comparisons test, significant interaction, significant interaction, F [1, 17] = 12.8, p = 0.0023; before vs. after CCK4: 0.035 ± 0.022 vs. 8.89 ± 2.04, p = 0.0001; before vs. after ACSF: −0.018 ± 0.031 vs. −0.540 ± 0.814, ns, p = 1.0; After CCK-4 vs. after ACSF: 8.89 ± 2.04 vs. −0.540 ± 0.814, p < 0.0001).
We proved that high-frequency activation of entorhino-neocortical CCK projections could trigger the release of CCK. It was reasonable to deduce that besides HFES-R, HFES-L could also induce high-frequency activation of the entorhino-neocortical projections ipsilaterally at site L. Like HFES-R, HFES-L could also trigger CCK release from the entorhino-neocortical terminals and then enable the local neural plasticity around stimulation site L, which formed the local cortical LTP. Hence, we further investigated whether CCK could enable neural plasticity at either site L or site R during the activation of interhemispheric cortical pathway L→R. In this experiment, we infused either CCK or ACSF into the proximity of recording site R after baseline testing (Figure 3E upper panel). After the infusion, LFS (0.1 Hz) was applied to site L to induce both pre- and postsynaptic activation, which was necessary for enabling neural plasticity, as described previously.28 The interhemispheric cortical LTP was induced by the CCK infusion at site R but not the ACSF infusion (two-way RM ANOVA, significant interaction F [1,183] = 139.1, p < 0.001; pairwise comparison, before vs. after CCK, 100.4 ± 0.9% vs. 143.2 ± 2.5%, p < 0.001, n = 20 from 5 mice; whereas before vs. after ACSF, pairwise comparison, 100.6 ± 0.9% vs. 99.4 ± 2.7%, p = 0.658; pairwise comparison, Difference between CCK group and ACSF group at 60 min after the pairing is 43.8 ± 3.7%, p < 0.001, Figure 3E). Subsequently, we infused either CCK or ACSF into the stimulation site L after baseline testing, followed by LFS to site L (Figure 3F upper panel). The recorded fEPSPs were potentiated by the CCK infusion but not by the ACSF infusion (two-way RM ANOVA, significant interaction F [1,203] = 134.8, p < 0.001; pairwise comparison, before vs. after CCK, 100.5 ± 0.6% vs. 137.2 ± 1.8%, p < 0.001, n = 24 from 5 mice, whereas before vs. after ACSF, 99.7 ± 0.7% vs. 100.4 ± 2.2%, p = 0.784, n = 17 from 4 mice. The difference between the CCK group and ACSF group at 60 min after the pairing was 36.6 ± 2.8%, p < 0.001, Figure 3F). Although LTP could be induced by the injection of CCK at either site R (Figure 3E) or site L (Figure 3F), this LTP occurred in different pathways. One is the interhemispheric cortical LTP (Figure 3E), and the other (Figure 3F) is a local LTP.
Lastly, we adopted CCK-ABRKO mice to prove that CCK is needed in the neocortical LTP. Genetic modification has better specificity than antagonists to CCK receptors. Genotyping results indicated that the inserted genes were in our homozygous CCK-ABRKO mice (see Figures S5A–S5C). Immunochemistry staining showed minimal CCK-BR signals in CCK-ABRKO mice in contrast with their wild-type control (129S, Figures 3G and S5D). To reconfirm the critical role of CCK in inducing cortical neural plasticity, we examined whether the knockout of CCK-AR and CCK-BR could result in deficits in HFS-induced neural plasticity. Indeed, the recorded fEPSPs were not potentiated by HFS in the auditory cortex of CCK-ABRKO mice (two-way RM ANOVA, significant interaction F[1,118] = 67.6, p < 0.001; pairwise comparison, 100.3 ± 0.8% vs. 103.8 ± 3.4%, before vs. after HFS, p = 0.377, N = 12 mice; Figure 3H blue dots), whereas significant potentiation was induced in the wild-type 129s mice group (pairwise comparison, 100.7 ± 0.8% vs. 149.4 ± 4.4%, before vs. after HFS, p < 0.001, N = 12 mice; Figure 3H red dots). Altogether, these results shown in Figure 3 confirmed that HFS induced the release of CCK at the stimulation site in the neocortex, enabling neural plasticity to occur in the surrounding areas.
Discussion
In the present study, we had shown that: 1) When we applied the commonly used method (HFES-L) to induce LTP of contralateral cortical pathway L→R, a local cortical LTP was formed. The neural plasticity occurred in site L but not in site R, meaning that it was a local LTP of neural connectivity around the stimulation site, but not the LTP of the interhemispheric cortical pathway L→R. 2) LTP of the interhemispheric cortical pathway could only be induced in a heterosynaptic manner through the modulation of entorhino-neocortical afferents. 3) The entorhino-neocortical projections influenced cortical neural plasticity via CCK release, which occurred once their terminals were activated by high-frequency stimulation. Then, the CCK could enable neural plasticity through the presynaptic and postsynaptic co-activities.
HFES-L could induce local cortical LTP, and the real interhemispheric cortical LTP could only be enabled in a heterosynaptic manner
Neural activity is vital to developing callosal connections in the visual and somatosensory cortex during post-natal periods.12,13,37 In this time period, the formation of callosal synapses is supported by presynaptic and postsynaptic neuronal activity based on the Hebbian rule.12,13 Evidence for the sensitive period has also been found in the development of auditory callosal plasticity.38 Nevertheless, few studies have focused on how an adult brain enables interhemispheric cortical plasticity to adapt to a changing environment. Cortical plasticity has been shown to decline with age, and this often correlates with neurocognitive impairments.2,39 The neuronal degeneration in Alzheimer’s disease has been described as a progressive cortical disconnection syndrome, among which corpus callosum atrophy is one of the indicators.40,41,42 Also, LTP-like cortical plasticity impairment can predict the onset of dementia in patients.21 Understanding the mechanism of cortical plasticity may reveal mechanisms underlying neurodegenerative disease. We investigated these questions through the induction of interhemispheric cortical LTP in the auditory cortex.
We adopted two separate sites in the two hemispheres of the auditory cortex as the stimulation site L and the recording site R, respectively. Optogenetic and electrophysiological experiments verified that LTP of the response to ES could be recorded after the HFES-L, but the response to laser activation of the commissural terminals at the recording site did not exhibit any LTP. This result suggests that the plasticity only occurred at the stimulation site but not at the synapses of terminals at the recording site. The local cortical LTP was formed but not the interhemispheric cortical LTP. Furthermore, high-frequency laser activation of the interhemispheric afferent terminals at the recording site could not induce LTP of the interhemispheric cortical pathway. The widely used experimental method for inducing LTP is to deliver HFS to the presynaptic axon of a synapse or group of synapses.18,22,43 HFS can evoke the activation of both presynaptic fibers and postsynaptic neurons; therefore, HFS-induced LTP is always considered Hebbian LTP, which requires simultaneous pre- and postsynaptic depolarization.14,17,18,19,22,23,24 The current understanding pointed out that LTP occurs at the synapses of the axon terminals that project onto neurons at the recording site.15 Thus, the failure of the interhemispheric cortical LTP induction by HFES-L indicated that the interhemispheric cortical LTP could not be homosynaptic LTP.
In contrast, the HFS at the recording site R readily induced this LTP. Compared to the laser stimulation of the specific projection terminals, the HFS at the recording site could also activate other afferent projections, implying that interhemispheric cortical LTP should be heterosynaptic.
LTP of the interhemispheric cortical pathway could be modulated through the entorhino-neocortical CCK projections
The neocortex and hippocampus are considered important for each stage of declarative or explicit memories, including formation, maintenance, and retrieval phases.44,45,46 The entorhinal cortex in the medial temporal lobe has been shown to form strong reciprocal connections with the neocortex, serving as the gateway from the hippocampus to the neocortex.47,48,49 Neurons in the entorhinal cortex exhibit extensive CCK labeling50,51,52 and project to neocortical areas, including the auditory cortex.28,48,49 CCK is the most abundant among all neuropeptides,53 and the mice lacking the CCK gene exhibit poor performance in passive avoidance tasks and display impaired spatial memory.54 The local infusion of CCK agonist could potentiate neural responses, while the CCK-BR antagonist could prevent the establishment of a cross-modal association.28,29 Our results revealed that optogenetic HFS of the entorhino-neocortical CCK projections pairing with the activation of the interhemispheric cortical pathway readily induced the interhemispheric cortical LTP, while activating the interhemispheric cortical pathway alone induced no LTP. Given that CCK enabled neural plasticity, we believe entorhino-neocortical projections modulated the cortical neuroplasticity in a heterosynaptic manner through CCK release: The activation of CCK-BR increased intracellular calcium concentration of the postsynaptic neurons at site R.55,56,57 We assumed that the calcium elevation could facilitate the calcium-dependent protein kinase II (CaMKII) to be activated by the interhemispheric input, which could trigger the recruitment of AMPARs at the post-synapses58,59,60 (Figure 3A, middle and left panel). A few slice electrophysiology studies demonstrated that the interhemispheric LTP could be induced by delivering HFS to the presynaptic projection.25,26 It seems to contradict our in-vivo results. The explanation is that the difference in calcium concentrations in-vitro slice or in-vivo recording may influence the results of LTP induction.61,62,63 Presynaptic calcium concentration relates to the release probability of CCK from entorhinal projections, and the activation of CCKBr increases intracellular Ca2+ concentration of postsynaptic neuron, which facilitates the occurrence of heterosynaptic plasticity. Inglebert et al.63 indicated that classical Hebbian plasticity induction at rat hippocampal synapses was greatly influenced by calcium. The abnormally higher extracellular calcium concentration in-vitro and physiology calcium concentration in-vivo make the plasticity rules differ greatly.63
Together with earlier publications,28,29,64 we conclude that participation of the entorhino-neocortical CCK projections is necessary to induce interhemispheric cortical LTP at a system level.
High-frequency activation of the entorhino-neocortical projections could release CCK that enables the cortical plasticity
Fiber photometry is well developed to monitor the release of various kinds of neural chemicals.36 Genetically encoded sensors that detect various neurotransmitters and neuropeptides have been adopted in studying neural circuits.65
The CCK sensor detected CCK was released from entorhino-neocortical projections by optogenetic HFS. This result was consistent with previous findings by Chen et al.,29 who found an increased CCK concentration after HFS of the auditory cortex by using microdialysis and ELISA, while the temporal resolution of the CCK sensor, what we employed in this study is much higher (time-resolved vs. 30 min). The results of the current study indicated that CCK was elevated for over a minute. While in our previous brain slice study, CCK was present only during the HFS, and the LTP lasted at least 1 h after CCK was washed away.29 So, we believe CCK can execute its function as plasticity induction at a high rate. The reason for the long-time elevation of the CCK may be: The artificial HFS is too intense for a normal functional brain, resulting in a large amount of CCK release. The released CCK8s could stay for almost half an hour at 37°C before degradation.66 Previous studies have demonstrated that CCK could enable neural plasticity, but they focused on the ipsilateral cortical pathway.28,29 Our last experiment further confirmed that LTP of the interhemispheric cortical pathway could be induced under the application of CCK at the recording site without HFS. Similarly, the application of CCK at the stimulation site could induce the local cortical LTP without HFS. No LTP could be triggered in CCK-ABRKO mice. These data confirmed the importance of CCK in neural plasticity. It is the CCK released from the entorhino-neocortical afferents triggered by HFS at the auditory cortex that modulates cortical plasticity, including interhemispheric cortical plasticity.
The complexity of the cortical network
Most callosal projections are glutamatergic and are derived from neurons in cortical layers II/III and V, targeting layers II/III and layer V neurons.67,68 In this study, we placed the stimulation and recording electrodes into the layer V because entorhinal CCK neurons mainly project to layer V neurons in the auditory cortex.29
As a complicated cellular system, the neocortex consists of six layers in a horizontal view. Neurons in each layer present diverse features in anatomy and function.69 Besides, these cells in the cortex connect tightly, whether within or across layers, mainly through excitatory regulations.70 In the sensory cortex, information propagates from layer IV to layers II/III and then to layers V/VI, since layer IV principal cells send the main output to layer II/III and layer II/III pyramidal neurons project extensively to layers V/VI.71,72 Layer V neurons have the most substantial axonal innervation of subcortical and cortical structures, making up a significant cortex output.72 In the entorhinal cortex, layer V neurons are also the primary origin of entorhinal projections to diverse cortical and subcortical structures.73
Interhemispheric projection can directly activate contralateral pyramidal neurons, as well as inhibitory interneurons, and it is cell-type dependent.7,8 Due to the complexities of the cortical network and the multiple neuronal targets of interhemispheric projection, the response at the recording site is a mix of monosynaptic excitation, polysynaptic excitation, and polysynaptic inhibition. However, in our electrophysiological experiments, we only recorded the slope of fEPSP, which mainly represents the direct excitatory response. The maximum slope of fEPSP appears within 5 ms in most of our optical stimulation recordings (see representative single fEPSP traces of laser activation in Figures 1C, 1F, 1G, 1J, 2F, 2G, and 2H), which is in a monosynaptic time window.74,75 Intracellular recordings in cats confirmed that the contralateral stimulation of A1 elicited excitatory postsynaptic potentials that were often followed by inhibitory postsynaptic potentials.76 Thus, Polysynaptic activity may contribute to the slope of fEPSP but not much.77 The influence of interhemispheric input on a given cortical area is a mix of direct and indirect projections, as well as excitation and inhibition. We have to admit that it is not comprehensive to only consider direct excitatory projections. The fine balance between excitatory projection neurons and local GABAergic interneurons is also crucial for the proper function and plasticity of the cortex. Since there are some CCK-expressing interneurons (CCK-INs) localized in the cortex and previous investigators proposed several mechanisms regarding the role of CCK in the GABAergic microcircuit and interactions with excitatory circuits. For those coexpressing both CCK and vasoactive intestinal peptide interneurons (VIP-CCK-INs), most studies hold the idea that VIP-CCK-INs take the leading control in the disinhibition by inhibiting somatostatin-expressing interneurons or parvalbumin-expressing interneurons in the cortex.78,79 While CCK-INs also project directly to the pyramidal neurons, the function of those CCK-INs remains elusive. Future research in this direction is necessary.
In conclusion, our results explain a CCK-based heterosynaptic mechanism underlying the formation of interhemispheric cortical plasticity, which is modulated by entorhinal efferent. Recently non-invasive brain stimulation are increasingly being applied in many neurological and psychiatric disorders, especially in Alzheimer’s disease, in which LTP impairment has been widely demonstrated as one of the earliest pathological events occurring, correlating with memory impairment and subsequent decline.20,21 Our current study revealed that the reason for the impairment of LTP-like cortical plasticity might be due to the atrophy of entorhinal cortex, as the degeneration of the entorhinal cortex occurs before the onset of overt dementia.80,81 How this specific type of plasticity influences behaviors, and the mechanism of CCK released regulating learning and memory in a behavioral context can be further explored in future studies. Overall, this novel regulation could provide avenues for a comprehensive understanding of cortical connectivity in the future and offer new targets for neurological disorders involving cortical plasticity.
Limitations of the study
In our electrophysiological experiments, we only recorded the slope of fEPSP, which mainly represents the direct excitatory response. The influence of interhemispheric input on a given cortical area is a mix of direct and indirect projections, as well as excitation and inhibition. The fine balance between excitatory projection neurons and local GABAergic interneurons is also crucial for neuronal network organization and information processing. We have to admit that it is not comprehensive to only consider direct excitatory projections.
Entorhinal neurons project to widespread cortical and subcortical areas. These areas may project to the auditory cortex and subsequently, make up an indirect projection from the entorhinal cortex to the auditory cortex. In our experiment, we injected a virus of Chronos into the entorhinal cortex of mice. The laser was only provided in the auditory cortex during the entorhinal projection activation. Thus, only the projections of the entorhinal-auditory were activated. However, there is still a slight chance that entorhinal projection neurons were activated antidromically. We believe the entorhinal cortex is crucial for CCK release since entorhinal cortex CCK knockdown by shRNA could abolish the LTP of auditory evoked potential in the lateral amygdala of mice.82 However, we cannot ensure this is the same case in the auditory cortex. A further shRNA study may solve this question.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CamKII α (Rabbit polyclonal) | Abcam | Cat# ab5683 RRID: AB_305050 |
| Anti-CCK (26–33) (CCK-8) (Rabbit polyclonal) | Sigma | Cat# C2581 RRID: AB_258806 |
| Anti-PSD95 (Mouse monoclonal) | Thermofisher | Cat# MA1-045 RRID: AB_325399 |
| Anti-GAD67 (Mouse monoclonal) | Millipore | Cat# MAB5406 RRID: AB_2278725 |
| Anti-CCK-BR (Goat polyclonal) | Abcam | Cat# ab77077 RRID: AB_1523282 |
| Anti-rabbit IgG Alexa 647 (Donkey polyclonal) |
Jackson ImmunoResearch Labs | Cat# 711-605-152 RRID: AB_2492288 |
| Anti-goat IgG Alexa 647 (Donkey polyclonal) |
Jackson ImmunoResearch Labs | Cat# 705-605-147 RRID:AB_2340437 |
| Anti-mouse IgG Alexa 594 (Donkey polyclonal) |
Jackson ImmunoResearch Labs | Cat# 715-585-150 RRID: AB_2340854 |
| Anti-rabbit IgG Alexa 405 (Donkey polyclonal) |
Jackson ImmunoResearch Labs | Cat# 711-475-152 RRID: AB_2340616 |
| Anti-rabbit IgG Alexa 594 (Goat polyclonal) |
Jackson ImmunoResearch Labs | Cat# 111-584-147 RRID: NA |
| Bacterial and virus strains | ||
| AAV9-Syn-Chronos-GFP | UNC vector core | N/A |
| pAAV-Syn-ChrimsonR-tdT | Addgene | RRID: Addgene_59171 |
| AAV9-Syn-Flex-Chronos-GFP | UNC vector core | N/A |
| AAV-Ef1a-DIO-EYFP | BrainVTA | N/A |
| AAV9-Syn-ChrimsonR-tdTomato | UNC vector core | N/A |
| pAAV-hSyn-CCK2.3 | Vigene Bioscience | N/A |
| pAAV-Syn-FLEX-rc[ChrimsonR-tdTomato] | Addgene | RRID: Addgene_62723 |
| Chemicals, peptides, and recombinant proteins | ||
| DAPI | Santa Cruz Biotechnology | Cat# sc-3598 |
| Pentobarbital sodium (Dorminal 20%) | Alfasan International B.V | N/A |
| Urethane | Sigma-Aldrich | Cat# U2500 |
| Lidocaine | Tokyo Chemical Industry | Cat# L0156 |
| Atropine | Sigma-Aldrich | Cat# A0257 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Laboratory Animal Research Unit, City University of Hong Kong | RRID: IMSR_JAX:000664 |
| Mouse: CCK-ires-Cre | Jackson Laboratories | RRID: IMSR_JAX:019021 |
| Mouse: CCK-CreER | Jackson Laboratories | RRID: IMSR_JAX: 012710 |
| Mouse: CCK-ABKO | Jackson Laboratories | RRID: IMSR_JAX: 006365 |
| Mouse: 129S1/SvImJ | Jackson Laboratories | RRID: IMSR_JAX: 002448 |
| Software and algorithms | ||
| TDT OpenEX | Tucker-Davis Technologies |
https://www.tdt.com/component/openexsoftware-suite/ |
| Matlab | Mathworks | http://www.mathworks.com/products/matlab/ |
| ImageJ | Schindelin et al. | https://imagej.nih.gov/ij/ |
| SPSS | IBM | https://www.ibm.com/products/spss-statistics |
| Other | ||
| Fiber Optic Cannula | Inper | Cat# FOC-W-L-6-20037 |
| Tungsten-wires | California Fine Wire | Cat#100–211 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jufang HE (jufanghe@cityu.edu.hk).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
The Animal Subjects Ethics Sub-Committee of the City University of Hong Kong approved all experimental procedures. Experiments were conducted using adult C57BL/6J, CCK-Cre, CCK-KO, CCK-ABRKO and 129S1/SvImJ mice (male, 2 to 4 months old). Animals were housed at 20–24C with 40–60% humidity under a 12-hour-light/12-hour-dark cycle (lights off from 8:00 am to 8:00 pm) with free access to food and water.
Method details
In-vivo extracellular recordings
Virus injection
Animals were anesthetized with pentobarbital (100 mg/kg) and supplemented with atropine (0.05 mg/kg, Sigma). The anesthetized mice were head-fixed on a stereotaxic device (RWD Life Science). In the experiments shown in Figure 1, C57BL/6 wild-type (C57) mice were adopted as the subjects. AAV9-Syn-Chronos-GFP (300 nL, 4.5E12 gc/mL, Boyden/UNC vector core, for Figures 1A–1C and 1E–1G) or pAAV-Syn-ChrimsonR-tdTomato virus (300 nL, 5.2E12 gc/mL, Addgene for Figures 1H–1J) was injected into one hemisphere of the auditory cortex (AP: −2.7 mm posterior to bregma, ML: −4.2 mm lateral to the midline, DV: 0.7 mm below the pia), at a rate of 30 nL/min (Nanoliter Injector, World Precision Instruments). In the experiments in Figure 2, CCK-ires-Cre (Ccktm1.1(Cre)Zjh/J, C57 background, Jackson Laboratory) mice were adopted as the subjects. AAV9-Syn-Flex-Chronos-eYFP (300 nL, 6.5E12 gc/mL, Boyden/UNC vector core)/AAV-Ef1a-DIO-eYFP (300 nL, 6.4E12 gc/mL, BrainVTA, Wuhan, as control) was injected into the entorhinal cortex (AP: 4.2 mm posterior to bregma; ML: 3.5 mm lateral to the midline; DV: 3.0 mm below the pia), and AAV9-Syn-ChrimsonR-tdTomato (300 nL, 4.1E12 gc/mL, Boyden/UNC vector core) was injected into the contralateral auditory cortex (AP: 2.7 mm posterior to bregma, ML: −4.2 mm lateral to the midline, DV: 0.7 mm below the pia), so that we could separately activate the afferents from the entorhinal and the contralateral auditory cortex with different wavelengths of the laser. We used the wavelength of 473 nm to activate the afferent terminals from the entorhinal cortex with Chronos expression and the wavelength of 635 nm to activate the afferent terminals from the contralateral auditory cortex with ChrimsonR expression. The power of the 473 nm laser and 635 nm laser at the optic fiber end was maintained at 20 mW/mm2 and 30 mW/mm2, respectively.
Surgery
Anesthesia was induced by urethane sodium (2 g/kg, i.p.) and maintained throughout the surgery and during neuronal recordings, with periodic supplements. Atropine sulfate (0.05 mg/kg, s.c.) was administered 15 min before the induction of anesthesia to inhibit tracheal secretions. A local anesthetic (xylocaine, 2%) was liberally applied to the incision site. Animals were mounted in a stereotaxic device, and a midline incision was made in the scalp. A craniotomy was performed (−2 mm to −4 mm posterior and −1.5 mm to −3 mm ventral to sagittal sutur) to access the auditory cortex of each hemisphere, and the dura mater was opened. For all in vivo experiments, electrodes were bilaterally and symmetrically inserted into layer V of the auditory cortex (where the virus was injected and its symmetric site, if applicable). For the electrophysiological experiments without laser stimulation, tungsten electrodes (<500 kΩ, FHC Inc.) could be used for either recording or stimulation. For the optogenetic experiments, glass pipette electrodes (homemade) rather than metal electrodes were used for recording to avoid the photoelectric effect, and an optical fiber was lowered onto the surface of recording site R for the laser stimulation. For all experiments involving drug infusions, the tips of glass pipettes were covered by 0.1 μL silicone oil to avoid leaking. Throughout the experiment, body temperature was maintained at 37–38°C with a heating blanket (homeothermic blanket system, Harvard Apparatus, Holliston, MA, USA).
Recording
The stimulating electrode was inserted into the left auditory cortex virus injection (if applicable) site (site L), and the recording electrodes array was placed symmetrically into the right auditory cortex (site R). Electrode placements were confirmed by neuronal responses to stimulation applied to the contralateral hemisphere, and the locations of these electrodes were confirmed to receive mutual projections. Electrodes were positioned by a stepping-motor micro driver-controlled from outside of the soundproofed chamber. Recording electrodes were attached to a 16-channel head stage, pre-amplified, passed onto an acquisition processor, filtered with a bandwidth of 1–5,000 Hz, and stored using TDT software (OpenEX, TDT, Alachua, FL).
In the experiment shown in Figures 1A–1C, we first recorded fEPSPs in response to ES (ES-L, 0.1 Hz at site L, 50–150 μA) and laser (LS-R, 0.1 Hz at site R, ≈5 mW, 5 ms duration) for 16 min, during which the ES-L and LS-R were alternately delivered with an interval of 5 s. The maximum current recorded was defined as that which could elicit a saturated fEPSP. We adopted a current equal to 40% of the current evoking the maximum fEPSP as the testing current and 80% of that as the HFS current. The fEPSPs were recorded for 16 min before and 60 min after the HFS, which was applied to the stimulation site L (HFES-L).
In the experiment shown in Figures S1E and S1F, a glass pipette was inserted adjacent to the recording site R for lidocaine infusion. After recording baseline fEPSPs in response to ES-L for 16 min, we infused lidocaine (1.0% in saline, 0.5 μL) at a rate of 0.05 μL/min. After the neural activities at site R were silenced, HFS was delivered to site L (HFES-L). No HFS was provided in the control group. fEPSPs were then recorded for 90 min after the neural activities at site R started to recover.
In the experiment shown in Figures 1E–1G, high-frequency laser stimulation (HFLS, 5 pulses at 80 Hz for each burst, 10 bursts at 5 Hz for each train, using 4 trains, with an inter-train-interval of 10 s) was delivered to site R (HFLS-R). fEPSPs in response to ES-L and LS-R were recorded for 16 min before and 60 min after the HFLS-R application.
For the experiment in Figures 1H–1J, fEPSPs in response to ES-L and LS-R were recorded 16 before and 60 minutes after the application of HFES-R.
For the experiment in Figure 2, high-frequency (50 Hz, 5 ms, 10 pulses) blue laser stimulation (HFLS) to the entorhino-neocortical projection terminals was applied to site R followed by the pairing of the low-frequency (1 Hz, 5 pulses) ES-L. The pairing of blue light HFLS and low-frequency ES-L (HFLS-ES) was repeated 10 times with an inter-trial interval of 10s. fEPSPs in response to ES-L and LS-R were recorded 16 min before and 60 min after the HFLS-ES paring.
In the experiment shown in Figures 3E, a glass pipette was inserted adjacent to the recording electrode (site R) to infuse CCK-4 (0.5 μL, 10 μm, Sigma) or artificial cerebrospinal fluid (ACSF, Harvard Apparatus, USA) into the auditory cortex of C57 mice at a rate of 0.1 μL/min. ES (0.1 Hz) was continuously presented during the infusion. fEPSPs in response to ES were recorded 16 min before and 60 min after the injection.
Similarly, in the experiment shown in Figure 3F, a glass pipette was inserted adjacent to the stimulating electrodes (site L) to infuse CCK-4 (0.5 μL, 10 μm, Sigma) or ACSF into the auditory cortex of C57 mice at a rate of 0.1 μL/min. ES (0.1 Hz) was continuously presented during the infusion. fEPSPs in response to ES were recorded for 16 min before and 60 min after the injection.
In the experiment shown in Figures 3G and 3H, we adopted CCK-ABRKO mice (Cckartm1Kpn, Cckbrtm1Kpn; 006365, Jackson Lab) and their wild-type control, 129s mice, as the subjects. Two electrode arrays were inserted symmetrically into both hemispheres of the auditory cortex, one for stimulating (site L) and the other for recording (site R). fEPSPs were recorded for 16 min before and 60 min after the HFES-L.
Data analysis
Mean fEPSP slopes before and after the pairings were normalized, calculated by linear regression and compared using two-way ANOVA with repeated measures (RM) followed by Bonferroni-adjusted pairwise comparison.
Fiberphotometry
Preparation
In all the fiberphotometry experiments, pAAV-hSyn-CCK2.3 (3.5E13 gc/mL, Vigene Bioscience) was injected into the auditory cortex of CCK-Cre or CCK-KO [CCK-CreER, Cck−/−, Ccktm2.1(Cre/ERT2)Zjh/J] mice (AP: −2.6 and −3.2 mm posterior to bregma, ML: −4.2 mm lateral to the midline, DV: −0.90 mm below the pia). This GPCR activation-based CCK sensor, GRABCCK, was developed by inserting a circular-permutated green fluorescent protein (cpEGFP) into the intracellular domain of CCK-BR.83 When the endogenous or exogenous ligand (CCK) bind to the CCK-BR, a conformational change in cpEGFP will be happened as well as an increase in fluorescence intensity, and the CCK activity could be visualized in vivo. After a 6-week recovery, a craniotomy was performed to access the auditory cortex (−2 mm to −4 mm posterior and −1.5 mm to −3 mm ventral to sagittal suture), and the dura mater was opened. An optic fiber (400 μm diameter, 0.22 NA, Thorlabs, Newton, NJ) was lowered into the auditory cortex (100-200 μm from the brain surface) to record the signal of the CCK sensor (Figures 3B–3D and S4A–S4D). Before signal recording, the optic fiber was lowered to the brain surface at different sites to confirm the best site for the recording, where we could capture the strongest fluorescence signal of CCK sensor. This optic fiber cannula was attached to a single fluorescent MiniCube (Doric Lenses, Quebec, QC, Canada) with built-in dichroic mirrors and LED light sources through a fiber patch cord. The excitation light at 470 and 405 nm were released by two fiber-coupled LEDs (M470F3 and M405FP1, Thorlabs) and were sinusoidally modulated at 210 and 330 Hz, respectively. The 473 nm channel is the GRABCCK channel and the 405 nm channel is employed as the isosbestic control channel. An LED driver (LEDD1B, Thorlabs) coupled to the RZ5D processor (TDT, Alachua, FL) managed the excitation light’s intensity through the software Synapse. The emission fluorescence was captured and transmitted by a bandpass filter in the MiniCube. To avoid photobleaching, the excitation light intensity at the tip of the patch cord’s tip was adjusted to less than 30 μW. The fluorescent signal was then detected, amplified, and transformed into an analog signal by the photoreceiver (Doric Lenses). The analog signal was then digitalized by the RZ5D processor and subjected to a 1 kHz low-pass analysis using Synapse software.
Recording
For the laser stimulation experiments in Figures 3B–3D and S4A, CCK-Cre and CCK-KO mice were adopted as the subjects. pAAV-Syn-FLEX-ChrimsonR-tdTomato (400 nL, 8.5E12 gc/mL, Addgene)/pAAV-Syn-ChrimsonR-tdT (400 nL, 6.5E12 gc/mL, Addgene) was infused into the entorhinal cortex of CCK-Cre or CCK-KO mice (−4.2 mm posterior, 3.5 mm lateral, and 3.0 mm ventral to bregma) about 6 weeks before. For the experiment in Figure S4A, an additional craniotomy was performed (−4 to −5 mm posterior and −2.5 to −3.5 lateral to bregma for mice) to access the entorhinal cortex. One optic fiber (100 μm diameter, 0.22 NA, Thorlabs, Newton, NJ) was inserted into the auditory cortex (100-200 μm from the brain surface, Figures 3B–3D), or the entorhinal cortex (Figure S4A ∼2.6 mm ventral to the brain surface) of mice to deliver the high-frequency laser stimulation. The fluorescent signal was recorded 25s before and 75s after the HFLS (5–10 mW, 635 nm) application.
For the experiments in Figures S4C we measured the CCK sensor activities before and after the CCK-4 application in different type of mice. CCK-4 (100 μM, 3 μL, Abcam) or ACSF (3 μL) was applied on the brain surface. The fluorescent signal was recorded 2 min before and 5 min after the drug application.
Fiber photometry analysis
Analysis of the signal was done by the custom-written MATLAB (Mathworks) codes. We first extracted the signal of the 473 nm and 405 nm channels corresponding to the defined periods before and after each stimulus or CCK4/ACSF infusion. A fitted 405 nm signal was created by regressing the 405 nm channel onto a linear fit of its respective 473 channel (MATLAB polyfit function). The fluorescence change (ΔF/F) was then calculated with the formula (473 nm signal − fitted 405 nm signal)/fitted 405 nm signal.84
Immunohistology and image acquisition
Preparation
Mice were anesthetized by an overdose of pentobarbital sodium and transcardially perfused with 30 mL cold phosphate-buffered saline (PBS) and 30 mL 4% (w/v) paraformaldehyde (4% PFA). Brain tissue was removed, post-fixed with 4% PFA, and treated with 30% (w/v) sucrose in 4% PFA at 4°C for 2–3 days. Brain tissue of 30 μm thickness was sectioned on a cryostat (Leica CM3500, Germany) and preserved with antifreeze buffer (20% (v/v) glycerol and 30% (v/v) ethylene glycol diluted in PBS) at −20°C.
Brain slices were washed 3 times per 10 minutes each time before blocking with the buffer (10% (v/v) goat serum in PBS with 0.2% (v/v) Triton X-100) at room temperature. After 2 hours of blocking, the slices were bathed in the rabbit anti-CamKll (Abcam, ab5683, 1:1000, shown in Figures S1A, S2A, S2D, and S2I), rabbit anti-CCK (Sigma, C-2581, 1:3000, shown in the Figures S2B, S2H, S3A, and S3B), mouse anti-PSD95 (Thermofisher, MA1-045, 1:500, shown in the Figures S3A, S3B, and S3D), mouse -anti-GAD67 (Millipore, MAB5406, 1:500, shown in Figures S2A and S2J), goat anti-CCK-BR (Abcam, ab77077, 1:1000, shown in Figures 3G and S3D) which was prepared with blocking buffer shaking for 36 hours at 4°C. After washing 4 times in PBS (each time for 10 min), sections were incubated with the corresponding fluorophore-conjugated secondary antibodies (1:250): donkey anti-rabbit 647 (Jackson Immunostar 711-605-152), donkey anti-goat 647 (Jackson Immunostar 705-605-147, donkey anti-mouse 594 (Jackson Immunostar 715-585-150), donkey anti-rabbit 405 (Jackson Immunostar 711-475-152), or donkey anti-rabbit 594 (Jackson Immunostar 111-584-147) for 2.5 h at 25°C. Sections were then rinsed with PBS three times before DAPI staining (1:5000 (v/v) diluted in PBS) and mounting.
All the sections were mounted with 70% (v/v) glycerol in PBS on slides. Image acquisition (10X, 20X, 63X, and 100X magnification) was performed with Nikon A1HD25 confocal Microscope (Nikon, Japan).
IHC images analysis
The images were analyzed with NIS element (Nikon) or ImageJ (NIH).85 The 8-bit grayscale image was background subtracted for counting immunoreactive signals before applying a threshold to all images. The threshold was adjusted within 10% of the average intensity, and signals at or above the threshold are considered immunopositive.
For quantification of eYFP+/GFP+/tDTomato+ neurons co-expressing CamKll (Figures S1A, S2A, S2D, and S2I), CCK (Figures S2B and S2H), Entorhinal cortex projecting eYFP+ terminals coexpressing CCK (Figures S3A, S3B and S3C), Entorhinal cortex projecting eYFP+ terminals coexpressing CCK-BR (Figures S3D and S3E), 20X images or 100X Z-stack images were acquired for 2–3 fields of view surrounding the AC of each slice. The percentage of eYFP+/GFP+/tDTomato+ cells, as identified by their fluorescent protein expression, overlapping with the CCK, CamKll, or GAD67 was calculated for each field of view. For the quantification of eYFP+ terminals of entorhino-neocortical projections, coexpressing CCK or CCK-BR, terminals were firstly identified by the spot detection with NIS, which recognized the colocalized position of eYFP+ and PSD95 as excitatory terminals. The number of those terminals was counted and summarized. Then those terminals were examined if there is CCK or CCK-BR. Colocalized points were finally recognized as eYFP+ terminals of entorhino-neocortical projections, coexpressing CCK or CCK-BR. The neurons or terminals were counted blindly by others using the NIS element and ImageJ.
Quantification and statistical analysis
All statistical analyses (two-way RM ANOVA) were done in SPSS (IBM, USA). Pairwise comparisons were adjusted by Bonferroni correction. N represents the number of animals and n represents the number of recordings (change to a different stimulation/recording site). Statistical significance was set at p < 0.05.
Acknowledgments
This work was supported by the following:
Hong Kong Research Grants Council, General Research Fund: 11101215M, 11103220M, 11101818M, 11102417M, 11101521M (GRF, JFH); Hong Kong Research Grants Council, Collaborative Research Fund: C1014-15g, C7048-16g (CRF, JFH); Innovation and Technology Fund: MRP/053/18X, MRP/101/17X, GHP_075_19GD (ITF, JFH); Health and Medical Research Fund: 08194106, 03141196, 06172456, 09203656 (HMRF, XL,XC, JFH); National Natural Science Foundation of China: 31671102 (NSFC, JFH); Key Area Research and Development Program of Guangdong Province, 2018B030340001 (XL).
We thank the Wong Chun Hong Endowed Chair Professorship, the Charlie Lee Charitable Foundation, the Fong Shu Fook Tong Foundation, and the Croucher Foundation for generous supports to J.H.; Eduardo Lau, Siu Lung Ng, and Yin Anqi for administrative and technical assistance; and Prof. Lin Xu for reviewing the manuscript.
Author contributions
X.L. and J.H. designed research; X.L., F.H., and E.Y. conducted the electrophysiological experiments; X.L., X.H., and M.C. collected and analyzed the data of fiber photometry experiments; X.L., H.L., F.H., T.C., and X.W. collected and analyzed the anatomy data; P.T., C.C., X.C., and J.Q. prepared some experimental setup; X.L. and J.H. wrote the paper; X.L., L.H., S.B., X.W., X.C., and J.H. edited the paper.
Declaration of interests
The authors declare no competing interests.
Published: March 31, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106542.
Contributor Information
Xiao Li, Email: xli293@cityu.edu.hk.
Jufang He, Email: jufanghe@cityu.edu.hk.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Thiebaut de Schotten M., Forkel S.J. The emergent properties of the connected brain. Science. 2022;378:505–510. doi: 10.1126/science.abq2591. [DOI] [PubMed] [Google Scholar]
- 2.Freitas C., Perez J., Knobel M., Tormos J.M., Oberman L., Eldaief M., Bashir S., Vernet M., Peña-Gómez C., Pascual-Leone A. Changes in cortical plasticity across the lifespan. Front. Aging Neurosci. 2011;3:5. doi: 10.3389/fnagi.2011.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leuner B., Gould E. Structural plasticity and hippocampal function. Annu. Rev. Psychol. 2010;61:111–140. doi: 10.1146/annurev.psych.093008.100359. C1-C3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hensch T.K., Bilimoria P.M. Dana Foundation; 2012. Re-opening Windows: Manipulating Critical Periods for Brain Development. [PMC free article] [PubMed] [Google Scholar]
- 5.Fame R.M., MacDonald J.L., Macklis J.D. Development, specification, and diversity of callosal projection neurons. Trends Neurosci. 2011;34:41–50. doi: 10.1016/j.tins.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aboitiz F., Montiel J. One hundred million years of interhemispheric communication: the history of the corpus callosum. Braz. J. Med. Biol. Res. 2003;36:409–420. doi: 10.1590/s0100-879x2003000400002. [DOI] [PubMed] [Google Scholar]
- 7.Rock C., Apicella A.J. Callosal projections drive neuronal-specific responses in the mouse auditory cortex. J. Neurosci. 2015;35:6703–6713. doi: 10.1523/JNEUROSCI.5049-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bloom J.S., Hynd G.W. The role of the corpus callosum in interhemispheric transfer of information: excitation or inhibition? Neuropsychol. Rev. 2005;15:59–71. doi: 10.1007/s11065-005-6252-y. [DOI] [PubMed] [Google Scholar]
- 9.Pietrasanta M., Restani L., Caleo M. The corpus callosum and the visual cortex: plasticity is a game for two. Neural Plast. 2012;2012:838672. doi: 10.1155/2012/838672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poirier P., Lepore F., Provençal C., Ptito M., Guillemot J.-P. Binaural noise stimulation of auditory callosal fibers of the cat: responses to interaural time delays. Exp. Brain Res. 1995;104:30–40. doi: 10.1007/BF00229853. [DOI] [PubMed] [Google Scholar]
- 11.Poirier P., Miljours S., Lassonde M., Lepore F. Sound localization in acallosal human listeners. Brain. 1993;116:53–69. doi: 10.1093/brain/116.1.53. [DOI] [PubMed] [Google Scholar]
- 12.Mizuno H., Hirano T., Tagawa Y. Pre-synaptic and post-synaptic neuronal activity supports the axon development of callosal projection neurons during different post-natal periods in the mouse cerebral cortex. Eur. J. Neurosci. 2010;31:410–424. doi: 10.1111/j.1460-9568.2009.07070.x. [DOI] [PubMed] [Google Scholar]
- 13.Mizuno H., Hirano T., Tagawa Y. Evidence for activity-dependent cortical wiring: formation of interhemispheric connections in neonatal mouse visual cortex requires projection neuron activity. J. Neurosci. 2007;27:6760–6770. doi: 10.1523/JNEUROSCI.1215-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Artola A., Singer W. Long-term potentiation and NMDA receptors in rat visual cortex. Nature. 1987;330:649–652. doi: 10.1038/330649a0. [DOI] [PubMed] [Google Scholar]
- 15.Bliss T.V., Collingridge G.L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 16.Nabavi S., Fox R., Proulx C.D., Lin J.Y., Tsien R.Y., Malinow R. Engineering a memory with LTD and LTP. Nature. 2014;511:348–352. doi: 10.1038/nature13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lømo T. Patterns of activation in a monosynaptic cortical pathway: the perforant path input to the dentate area of the hippocampal formation. Exp. Brain Res. 1971;12:18–45. [PubMed] [Google Scholar]
- 18.Bliss T.V., Lømo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buonomano D.V., Merzenich M.M. Cortical plasticity: from synapses to maps. Annu. Rev. Neurosci. 1998;21:149–186. doi: 10.1146/annurev.neuro.21.1.149. [DOI] [PubMed] [Google Scholar]
- 20.Di Lorenzo F., Ponzo V., Motta C., Bonnì S., Picazio S., Caltagirone C., Bozzali M., Martorana A., Koch G. Impaired spike timing dependent cortico-cortical plasticity in Alzheimer’s disease patients. J. Alzheimers Dis. 2018;66:983–991. doi: 10.3233/JAD-180503. [DOI] [PubMed] [Google Scholar]
- 21.Di Lorenzo F., Motta C., Casula E.P., Bonnì S., Assogna M., Caltagirone C., Martorana A., Koch G. LTP-like cortical plasticity predicts conversion to dementia in patients with memory impairment. Brain Stimul. 2020;13:1175–1182. doi: 10.1016/j.brs.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 22.Staubli U., Lynch G. Stable hippocampal long-term potentiation elicited by ‘theta’pattern stimulation. Brain Res. 1987;435:227–234. doi: 10.1016/0006-8993(87)91605-2. [DOI] [PubMed] [Google Scholar]
- 23.Hebb D.O. Psychology Press; 2005. The Organization of Behavior: A Neuropsychological Theory. [Google Scholar]
- 24.Löwel S., Singer W. Selection of intrinsic horizontal connections in the visual cortex by correlated neuronal activity. Science. 1992;255:209–212. doi: 10.1126/science.1372754. [DOI] [PubMed] [Google Scholar]
- 25.Sah P., Nicoll R.A. Mechanisms underlying potentiation of synaptic transmission in rat anterior cingulate cortex in vitro. J. Physiol. 1991;433:615–630. doi: 10.1113/jphysiol.1991.sp018446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petrus E., Saar G., Ma Z., Dodd S., Isaac J.T.R., Koretsky A.P. Interhemispheric plasticity is mediated by maximal potentiation of callosal inputs. Proc. Natl. Acad. Sci. USA. 2019;116:6391–6396. doi: 10.1073/pnas.1810132116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J.-Y., Lonjers P., Lee C., Chistiakova M., Volgushev M., Bazhenov M. Heterosynaptic plasticity prevents runaway synaptic dynamics. J. Neurosci. 2013;33:15915–15929. doi: 10.1523/JNEUROSCI.5088-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X., Yu K., Zhang Z., Sun W., Yang Z., Feng J., Chen X., Liu C.-H., Wang H., Guo Y.P., He J. Cholecystokinin from the entorhinal cortex enables neural plasticity in the auditory cortex. Cell Res. 2014;24:307–330. doi: 10.1038/cr.2013.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X., Li X., Wong Y.T., Zheng X., Wang H., Peng Y., Feng H., Feng J., Baibado J.T., Jesky R., et al. Cholecystokinin release triggered by NMDA receptors produces LTP and sound-sound associative memory. Proc. Natl. Acad. Sci. USA. 2019;116:6397–6406. doi: 10.1073/pnas.1816833116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirkwood A., Bear M.F. Hebbian synapses in visual cortex. J. Neurosci. 1994;14:1634–1645. doi: 10.1523/JNEUROSCI.14-03-01634.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iriki A., Pavlides C., Keller A., Asanuma H. Long-term potentiation in the motor cortex. Science. 1989;245:1385–1387. doi: 10.1126/science.2551038. [DOI] [PubMed] [Google Scholar]
- 32.Goulas A., Uylings H.B.M., Hilgetag C.C. Principles of ipsilateral and contralateral cortico-cortical connectivity in the mouse. Brain Struct. Funct. 2017;222:1281–1295. doi: 10.1007/s00429-016-1277-y. [DOI] [PubMed] [Google Scholar]
- 33.Wiesel T.N. Postnatal development of the visual cortex and the influence of environment. Nature. 1982;299:583–591. doi: 10.1038/299583a0. [DOI] [PubMed] [Google Scholar]
- 34.Welsh J.P., Harvey J.A. Pavlovian conditioning in the rabbit during inactivation of the interpositus nucleus. J. Physiol. 1991;444:459–480. doi: 10.1113/jphysiol.1991.sp018888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nordholm A.F., Thompson J.K., Dersarkissian C., Thompson R.F. Lidocaine infusion in a critical region of cerebellum completely prevents learning of the conditioned eyeblink response. Behav. Neurosci. 1993;107:882–886. doi: 10.1037//0735-7044.107.5.882. [DOI] [PubMed] [Google Scholar]
- 36.Sun F., Zeng J., Jing M., Zhou J., Feng J., Owen S.F., Luo Y., Li F., Wang H., Yamaguchi T., et al. A genetically encoded fluorescent sensor enables rapid and specific detection of dopamine in flies, fish, and mice. Cell. 2018;174:481–496.e19. doi: 10.1016/j.cell.2018.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang C.-L., Zhang L., Zhou Y., Zhou J., Yang X.-J., Duan S.-m., Xiong Z.-Q., Ding Y.-Q. Activity-dependent development of callosal projections in the somatosensory cortex. J. Neurosci. 2007;27:11334–11342. doi: 10.1523/JNEUROSCI.3380-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steele C.J., Bailey J.A., Zatorre R.J., Penhune V.B. Early musical training and white-matter plasticity in the corpus callosum: evidence for a sensitive period. J. Neurosci. 2013;33:1282–1290. doi: 10.1523/JNEUROSCI.3578-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koch G., Di Lorenzo F., Bonnì S., Giacobbe V., Bozzali M., Caltagirone C., Martorana A. Dopaminergic modulation of cortical plasticity in Alzheimer’s disease patients. Neuropsychopharmacology. 2014;39:2654–2661. doi: 10.1038/npp.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armstrong R.A. The spatial pattern of discrete β-amyloid deposits in Alzheimers disease reflects synaptic disconnection. Dementia. 1996;7:86–90. doi: 10.1159/000106859. [DOI] [PubMed] [Google Scholar]
- 41.Morrison J., Scherr S., Lewis D.A., Campbell M., Bloom F., Rogers J., Benoit R. In: The Biological Substrates of Alzheimer’s Disease. Scheibel A.B., Wechsler A.F., Brazier M.A., editors. Academic PressOrlando, FL; 1986. The laminar and regional distribution of neocortical somatostatin and neuritic plaques: implications for Alzheimer’s disease as a global neocortical disconnection syndrome; pp. 115–131. [Google Scholar]
- 42.Hampel H., Teipel S.J., Alexander G.E., Horwitz B., Teichberg D., Schapiro M.B., Rapoport S.I. Corpus callosum atrophy is a possible indicator of region–and cell type–specific neuronal degeneration in Alzheimer disease: a magnetic resonance imaging analysis. Arch. Neurol. 1998;55:193–198. doi: 10.1001/archneur.55.2.193. [DOI] [PubMed] [Google Scholar]
- 43.Huang Y.-Y., Kandel E.R. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn. Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- 44.Eichenbaum H., Cohen N.J. Oxford University Press on Demand; 2004. From Conditioning to Conscious Recollection: Memory Systems of the Brain. [Google Scholar]
- 45.Murray E.A., Mishkin M. Object recognition and location memory in monkeys with excitotoxic lesions of the amygdala and hippocampus. J. Neurosci. 1998;18:6568–6582. doi: 10.1523/JNEUROSCI.18-16-06568.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zola S.M., Squire L.R. In: The Oxford Handbook of Memory. Tulving E., Craik F.I.M., editors. Oxford University Press; 2000. The medial temporal lobe and the hippocampus; pp. 485–500. [Google Scholar]
- 47.Brown M.W., Aggleton J.P. Recognition memory: what are the roles of the perirhinal cortex and hippocampus? Nat. Rev. Neurosci. 2001;2:51–61. doi: 10.1038/35049064. [DOI] [PubMed] [Google Scholar]
- 48.Canto C.B., Wouterlood F.G., Witter M.P. What does the anatomical organization of the entorhinal cortex tell us? Neural Plast. 2008;2008:381243. doi: 10.1155/2008/381243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swanson L.W., Köhler C. Anatomical evidence for direct projections from the entorhinal area to the entire cortical mantle in the rat. J. Neurosci. 1986;6:3010–3023. doi: 10.1523/JNEUROSCI.06-10-03010.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenwood R.S., Godar S.E., Reaves T.A., Jr., Hayward J.N. Cholecystokinin in hippocampal pathways. J. Comp. Neurol. 1981;203:335–350. doi: 10.1002/cne.902030303. [DOI] [PubMed] [Google Scholar]
- 51.Innis R.B., Corrêa F.M., Uhl G.R., Schneider B., Snyder S.H. Cholecystokinin octapeptide-like immunoreactivity: histochemical localization in rat brain. Proc. Natl. Acad. Sci. USA. 1979;76:521–525. doi: 10.1073/pnas.76.1.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Köhler C., Chan-Palay V. The distribution of cholecystokinin-like immunoreactive neurons and nerve terminals in the retrohippocampal region in the rat and Guinea pig. J. Comp. Neurol. 1982;210:136–146. doi: 10.1002/cne.902100204. [DOI] [PubMed] [Google Scholar]
- 53.Rehfeld J.F. Immunochemical studies on cholecystokinin. II. Distribution and molecular heterogeneity in the central nervous system and small intestine of man and hog. J. Biol. Chem. 1978;253:4022–4030. [PubMed] [Google Scholar]
- 54.Lo C.-M., Samuelson L.C., Chambers J.B., King A., Heiman J., Jandacek R.J., Sakai R.R., Benoit S.C., Raybould H.E., Woods S.C., Tso P. Characterization of mice lacking the gene for cholecystokinin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008;294:R803–R810. doi: 10.1152/ajpregu.00682.2007. [DOI] [PubMed] [Google Scholar]
- 55.Lee Y.M., Beinborn M., McBride E.W., Lu M., Kolakowski L.F., Jr., Kopin A.S. The human brain cholecystokinin-B/gastrin receptor. Cloning and characterization. J. Biol. Chem. 1993;268:8164–8169. [PubMed] [Google Scholar]
- 56.Simasko S.M., Wiens J., Karpiel A., Covasa M., Ritter R.C. Cholecystokinin increases cytosolic calcium in a subpopulation of cultured vagal afferent neurons. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002;283:R1303–R1313. doi: 10.1152/ajpregu.00050.2002. [DOI] [PubMed] [Google Scholar]
- 57.Miyoshi R., Kito S., Nomoto T. Cholecystokinin increases intracellular Ca2+ concentration in cultured striatal neurons. Neuropeptides. 1991;18:115–119. doi: 10.1016/0143-4179(91)90102-o. [DOI] [PubMed] [Google Scholar]
- 58.Opazo P., Labrecque S., Tigaret C.M., Frouin A., Wiseman P.W., De Koninck P., Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 59.Lisman J., Yasuda R., Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hayashi Y., Shi S.H., Esteban J.A., Piccini A., Poncer J.C., Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 61.Habets R.L.P., Borst J.G.G. Post-tetanic potentiation in the rat calyx of held synapse. J. Physiol. 2005;564:173–187. doi: 10.1113/jphysiol.2004.079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chindemi G., Abdellah M., Amsalem O., Benavides-Piccione R., Delattre V., Doron M., Ecker A., Jaquier A.T., King J., Kumbhar P., et al. A calcium-based plasticity model for predicting long-term potentiation and depression in the neocortex. Nat. Commun. 2022;13:3038. doi: 10.1038/s41467-022-30214-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inglebert Y., Aljadeff J., Brunel N., Debanne D. Synaptic plasticity rules with physiological calcium levels. Proc. Natl. Acad. Sci. USA. 2020;117:33639–33648. doi: 10.1073/pnas.2013663117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Z., Zheng X., Sun W., Peng Y., Guo Y., Lu D., Zheng Y., Li X., Jendrichovsky P., Tang P., et al. Visuoauditory associative memory established with cholecystokinin under anesthesia is retrieved in behavioral contexts. J. Neurosci. 2020;40:2025–2037. doi: 10.1523/jneurosci.1673-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Broussard G.J., Liang R., Tian L. Monitoring activity in neural circuits with genetically encoded indicators. Front. Mol. Neurosci. 2014;7:97. doi: 10.3389/fnmol.2014.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Migaud M., Durieux C., Viereck J., Soroca-Lucas E., Fournié-Zaluski M.C., Roques B.P. The in vivo metabolism of cholecystokinin (CCK-8) is essentially ensured by aminopeptidase A. Peptides. 1996;17:601–607. doi: 10.1016/0196-9781(96)00036-8. [DOI] [PubMed] [Google Scholar]
- 67.Petreanu L., Huber D., Sobczyk A., Svoboda K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat. Neurosci. 2007;10:663–668. doi: 10.1038/nn1891. [DOI] [PubMed] [Google Scholar]
- 68.Kawaguchi Y. Receptor subtypes involved in callosally-induced postsynaptic potentials in rat frontal agranular cortex in vitro. Exp. Brain Res. 1992;88:33–40. doi: 10.1007/BF02259126. [DOI] [PubMed] [Google Scholar]
- 69.Petersen C.C.H. The functional organization of the barrel cortex. Neuron. 2007;56:339–355. doi: 10.1016/j.neuron.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 70.Douglas R.J., Martin K.A.C. Neuronal circuits of the neocortex. Annu. Rev. Neurosci. 2004;27:419–451. doi: 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- 71.Gilbert C.D., Wiesel T.N. Morphology and intracortical projections of functionally characterised neurones in the cat visual cortex. Nature. 1979;280:120–125. doi: 10.1038/280120a0. [DOI] [PubMed] [Google Scholar]
- 72.Feldmeyer D. Excitatory neuronal connectivity in the barrel cortex. Front. Neuroanat. 2012;6:24. doi: 10.3389/fnana.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cappaert N.L., Van Strien N.M., Witter M.P. The Rat Nervous System. Elsevier; 2015. Hippocampal formation; pp. 511–573. [Google Scholar]
- 74.Smith C.C., Kissane R.W.P., Chakrabarty S. Simultaneous assessment of homonymous and heteronymous monosynaptic reflex excitability in the adult rat. Eneuro. 2018;5 doi: 10.1523/ENEURO.0227-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doyle M.W., Andresen M.C. Reliability of monosynaptic sensory transmission in brain stem neurons in vitro. J. Neurophysiol. 2001;85:2213–2223. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- 76.Mitani A., Shimokouchi M. Neuronal connections in the primary auditory cortex: an electrophysiological study in the cat. J. Comp. Neurol. 1985;235:417–429. doi: 10.1002/cne.902350402. [DOI] [PubMed] [Google Scholar]
- 77.Karnup S., Stelzer A. Temporal overlap of excitatory and inhibitory afferent input in Guinea-pig CA1 pyramidal cells. J. Physiol. 1999;516:485–504. doi: 10.1111/j.1469-7793.1999.0485v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pfeffer C.K., Xue M., He M., Huang Z.J., Scanziani M. Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat. Neurosci. 2013;16:1068–1076. doi: 10.1038/nn.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pi H.-J., Hangya B., Kvitsiani D., Sanders J.I., Huang Z.J., Kepecs A. Cortical interneurons that specialize in disinhibitory control. Nature. 2013;503:521–524. doi: 10.1038/nature12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DE TOLEDO-MORRELL L., Goncharova I., Dickerson B., Wilson R.S., Bennett D.A. From healthy aging to early Alzheimer's disease: in vivo detection of entorhinal cortex atrophy. Ann. N. Y. Acad. Sci. 2000;911:240–253. doi: 10.1111/j.1749-6632.2000.tb06730.x. [DOI] [PubMed] [Google Scholar]
- 81.Goldman J.G., Stebbins G.T., Bernard B., Stoub T.R., Goetz C.G., deToledo-Morrell L. Entorhinal cortex atrophy differentiates Parkinson's disease patients with and without dementia. Mov. Disord. 2012;27:727–734. doi: 10.1002/mds.24938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feng H., Su J., Fang W., Chen X., He J. The entorhinal cortex modulates trace fear memory formation and neuroplasticity in the mouse lateral amygdala via cholecystokinin. Elife. 2021;10 doi: 10.7554/eLife.69333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jing M., Zhang Y., Wang H., Li Y. G-protein-coupled receptor-based sensors for imaging neurochemicals with high sensitivity and specificity. J. Neurochem. 2019;151:279–288. doi: 10.1111/jnc.14855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bruno C.A., O'Brien C., Bryant S., Mejaes J.I., Estrin D.J., Pizzano C., Barker D.J. pMAT: an open-source software suite for the analysis of fiber photometry data. Pharmacol. Biochem. Behav. 2021;201 doi: 10.1016/j.pbb.2020.173093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.