

Summary
Loss of function of inhibitory immune checkpoints, unleashing pathogenic immune responses, is a potential risk factor for autoimmune disease. Here, we report that patients with the autoimmune vasculitis giant cell arteritis (GCA) have a defective CD155-CD96 immune checkpoint. Macrophages from patients with GCA retain the checkpoint ligand CD155 in the endoplasmic reticulum (ER) and fail to bring it to the cell surface. CD155low antigen-presenting cells induce expansion of CD4+CD96+ T cells, which become tissue invasive, accumulate in the blood vessel wall, and release the effector cytokine interleukin-9 (IL-9). In a humanized mouse model of GCA, recombinant human IL-9 causes vessel wall destruction, whereas anti-IL-9 antibodies efficiently suppress innate and adaptive immunity in the vasculitic lesions. Thus, defective surface translocation of CD155 creates antigen-presenting cells that deviate T cell differentiation toward Th9 lineage commitment and results in the expansion of vasculitogenic effector T cells.
Keywords: immune checkpoint receptors, CD96, CD155, ER stress, IL-9, autoimmune vasculitis, giant cell arteritis, macrophage, T cell, autoimmunity
Graphical abstract
Highlights
-
•
Patients with giant cell arteritis have a defective CD155-CD96 immune checkpoint
-
•
Antigen-presenting cells (APCs) retain CD155 in the endoplasmic reticulum
-
•
CD155low APCs favor the differentiation of IL-9-producing CD4+CD96+ memory T cells
-
•
IL-9 drives tissue-damaging immunity in vasculitis
Ohtsuki et al. report that patients with the autoimmune vasculitis giant cell arteritis (GCA) have a tolerance defect caused by the loss of an inhibitory immune checkpoint. CD155low-expressing antigen-presenting cells drive expansion of CD4+CD96+ T cells that produce the cytokine IL-9. Blocking IL-9 has therapeutic efficacy in experimental vasculitis.
Introduction
Immune checkpoint receptor/ligand pairs critically control the kinetics, duration, and intensity of immune responses, as exemplified by the high expression of checkpoint ligands on tumor cells that enable tumor immune evasion.1,2,3 Immune checkpoint blockade has revolutionized cancer therapy but also causes immune-related adverse events (irAEs), often mimicking autoimmune disease.4 Accordingly, inherited or acquired deficiency of suppressive checkpoints may have relevance in spontaneous autoimmunity. In support of this concept, patients with the autoimmune vasculitis giant cell arteritis (GCA) have low expression of the checkpoint ligand PD-L1 on antigen-presenting cells (APCs), and blockade of the PD-1/PD-L1 checkpoint exacerbates arteritis.5
GCA is an autoimmune vasculitis of medium and large arteries, frequently involving the aorta. Disease hallmarks include granulomatous infiltrates in the vessel wall, typically associated with multinucleated giant cells.6 Vascular infiltrates contain CD4+ T cells and highly activated macrophages (Mφs),7 and the vessel wall reacts with a maladaptive response to injury, forming lumen-occlusive intimal hyperplasia.8 Aberrantly expressed NOTCH1 receptors on CD4+ T cells enhance co-stimulatory signaling and foster T cell transmigration from vasa vasora into the vascular wall through NOTCH-Jagged interactions.9 Lesional T cells produce an array of effector cytokines, including interferon-γ (IFN-γ), interleukin-2 (IL-2), IL-22, and IL-9.10,11 Loss of self-tolerance has been attributed to the loss of immunoinhibitory T regulatory cells, which release NADPH-containing exosomes to inhibit neighboring CD4+ T cells.12 Age is an established risk factor with disease onset consistently after the age of 50 years13 Based on serial biopsy studies, many patients have refractory vasculitis with persistent inflammation despite immunosuppressive therapy.14
Given the granulomatous character of GCA, Mφs are key contributors to the disease process.15 Circulating monocytes overexpress MMP-9 and digest basal membranes to get access to the adventitial space.16 GCA Mφs produce chemokines and cytokines, have comparably low metabolic activity,17 and express low density of PD-L1, thus functioning as “permissive” APCs.5 Accordingly, the disease process relies on CD80/CD86-dependent co-stimulation to build tissue-damaging infiltrates that trigger neoangiogenesis and wall remodeling.8 Wall-residing Mφs participate in the handling of oxidative stress17 and supply growth and angiogenesis factors18 that mediate immune-mediated vascular restructuring.
Comparative studies between atherosclerosis and GCA have been informative in linking immune cell phenotype to function and disease.17 Glucose is a dominant energy source for Mφs in coronary artery disease, fueling high mitochondrial activity,17 cytokine secretion,19 and high PD-L1 expression.17 Mφs accumulating in atherosclerotic lesions express high amounts of the checkpoint ligand CD155,20 and a hyperactive CD155 immune checkpoint impairs the host’s anti-viral immunity. Overall, atherosclerotic lesions recruit and retain Mφs displaying immunosuppressive functions, contrasting the “permissive” Mφs associated with GCA.
The transmembrane glycoprotein CD155, also known as the poliovirus receptor (PVR), is a member of the immunoglobulin superfamily and regulates natural killer (NK) adhesion and effector functions.21 CD155 is expressed on monocytes, dendritic cells, and tumor cells, where it provides a negative signal and functions as a classical immunoinhibitory ligand. CD155 binds to the CD96, TIGIT, and CD226 surface receptors on T cells and NK cells. CD96 and TIGIT are co-inhibitory receptors, and CD226 may also transmit co-stimulatory signals.22,23,24
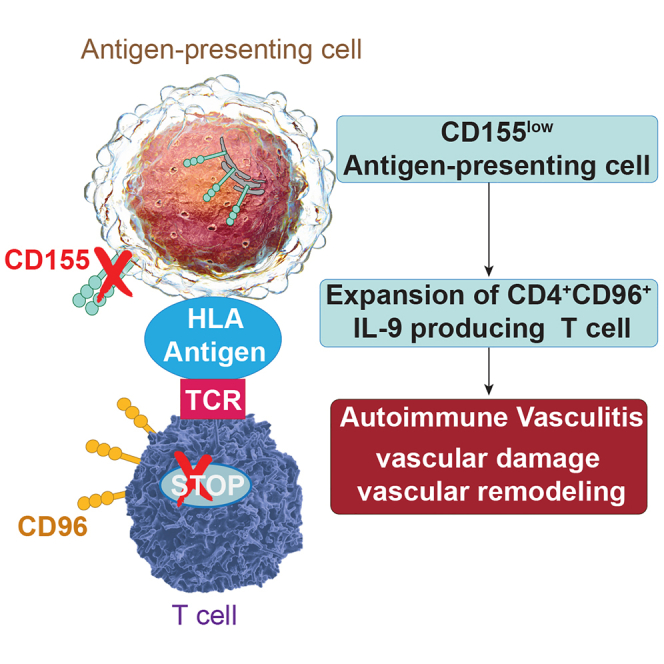
Here, we have identified a defect in GCA Mφs that contributes to the patients’ tolerance loss and amplifies disease activity in the affected arteries. GCA Mφs retained the inhibitory ligand CD155 in the endoplasmic reticulum (ER), endowing them with strong immunostimulatory capacities. Such dysfunctional APCs shifted T cell differentiation, expanding CD96+ T cells within the memory compartment. CD4+ CD96+ T cells were functionally committed to produce the effector cytokine IL-9, and loss-of-function and gain-of-function experiments identified IL-9 as a tissue-damaging molecule in vascular inflammation.
Results
Immunoinhibitory receptors in autoimmune vasculitis
In GCA, disease-relevant tolerance defects have been mapped to co-stimulatory and co-inhibitory checkpoints,5,8 suggesting a key role of T cell receptor signaling strength. Professional APCs express low amounts of PD-L1 and provide strong CD80/86-dependent stimulation.5,8 Most lesional T cells are high expressers for PD-1, but how the peripheral T cell repertoire is affected by dysbalanced APC-T cell interaction is unknown. We designed a panel of 6 immune checkpoint receptors to comprehensively analyze memory CD4+ T cell distributions in the peripheral blood of patients with GCA (n = 8) and age-matched controls (n = 8) (Figures 1A and 1B). CD4+ T cells were classified as memory populations based on CD45RA and CCR7 expression. CD45RA− CCR7+ T cells identified as central memory cells (Tcms) fell into 5 clusters (clusters 1, 2, 3, 5, and 7). CD45RA− CCR7− effector memory T cells (Tems) were composed of three classification clusters (clusters 4, 8, and 9) and CD45RA+CCR7− terminally differentiated effector memory cells (TEMRAs) formed two distinguishable clusters (clusters 6 and 10).
Figure 1.

Enrichment of CD96hi CD4+ T cells in giant cell arteritis (GCA)
(A–D) Freshly harvested peripheral blood mononuclear cells (PBMCs) from patients with GCA and age-matched healthy individuals were immunophenotyped by multiparametric flow cytometry for the expression of 6 immunoinhibitory receptors. CD4+ memory T cell clusters were classified based on CD45RA and CCR7 expression.
(A) Uniform manifold approximation and projection (UMAP) clustering of memory CD4+ T cells, delineating 10 clusters among central memory cells (CD45RA− CCR7+), effector memory cells (CD45RA− CCR7−), and terminally differentiated effector memory cells (TEMRAs; CD45RA+ CCR7−). Data are a mix of samples from 8 patients and 8 controls. Plot of 10 identified PhenoGraph clusters is overlaid on the UMAP projection.
(B) Heatmap demonstrating expression of the 6 checkpoint receptors in the 10 clusters based on mean fluorescence intensity (MFI). z scores are shown.
(C–E) UMAP clustering of memory CD4+ T cells from 8 healthy individuals (C) and 8 patients with GCA (D). Quantified frequencies (%) of each cluster are presented as pie charts. T cell subsets that are enriched or reduced in patients with GCA are indicated (E).
(F) CD96 mRNA transcripts were quantified by RT-PCR. Data from 10 patients and 10 controls.
(G) CD96 expression quantified by flow cytometry in PBMCs from a confirmation cohort of 16 patients and 16 age-matched controls. MFIs are shown, and each dot represents one patient or control.
(H) CD4+ T cells isolated from controls and patients with GCA were stimulated for 0, 3, or 6 days. Kinetics of CD96 expression measured by flow cytometry (n = 4 patients, n = 4 controls).
(I and J) Temporal arteries were collected from diagnostic biopsies of patients with GCA and sinus biopsies from patients with granulomatosis with polyangiitis (GPA).
(I) Targeted tissue transcriptomic analysis of GCA-affected temporal arteries compared with non-inflamed arteries. CD96 transcripts evaluated by RT-PCR. Each lane represents one tissue sample.
(J) Temporal artery (left) and sinal tissue (right) sections were stained for CD3 (red) and CD96 (green). Nuclei marked by DAPI. Representative images from 4 patients. Frequencies of CD3+ CD96high T cells quantified in 50 randomly selected high-power fields. Scale bar: 20 μm. Mean ± SEM with individual values shown.
(F) Mann-Whitney test. (G) Two-tailed unpaired t test. (H) Mann-Whitney test and two-tailed unpaired t test. ∗p < 0.05, ∗∗p < 0.01.
Expression profiles for the 6 immune checkpoint receptors created a total of 10 clusters across the memory T cell landscape (Figures 1A and 1B). The CD4+ Tcm compartment contained 2 subsets of CD96-expressing cells (clusters 1 and 2) and 2 subsets characterized by TIGIT expression (clusters 5 and 7). Tems included CD226-expressing cells, and one subset co-expressed LAG3. End-differentiated TEMRAs mainly had high expression of TIM3 and LAG3, combined with subsets of TIGITlow- and TIM3low-expressing cells. In essence, inhibitory checkpoint receptors were differentially distributed among memory CD4+ T cells, with each memory subpopulation having one “marker” checkpoint receptors: CD96 and TIGIT on Tcms, CD226 on Tems, and TIM3 and LAG3 on TEMRAs. PD-1 was rather selective and appeared on TIGIThi Tcms.
Comparative analysis of circulating memory CD4+ T cells in patients with GCA and controls yielded separable distributions of the 10 classification clusters (Figures 1C and 1D). Most noticeable differences occurred within the central memory compartment. Compared with controls, patients’ CD96hi Tcms expanded from 10.9% to 16.9% and TIGIThi Tcms declined from 24.5% to 17.4%. In addition, patient-derived CD4+ memory T cells had a higher frequency of the small CD226hi LAG3hi subset and had a higher proportion of TIM3hi LAG3hi end-differentiated cells (Figure 1E), commonly considered representing exhausted T cells.
We sought to validate the observation that CD96-expressing Tcms were expanded in GCA (Figures 1F, 1G, and S1). GCA CD4+ T cells had higher gene expression (n = 10 controls, n = 10 patients; Figure 1F) and higher CD96 protein expression (n = 16 controls, n = 16 patients; Figures 1G and S1). Kinetic studies confirmed that resting and activated GCA CD4+ T cells expressed higher CD96 cell surface density (Figure 1H).
To assess the disease relevance of CD4+CD96+ T cells, we analyzed the vasculitic lesions. CD96 mRNA transcripts were abundantly expressed in GCA-affected temporal artery biopsies and were low in non-inflamed arteries (Figure 1I). Multicolor immunofluorescence staining of tissue sections yielded similar results. 90%–95% of the CD3+ T cells in the vasculitic lesions stained positive for CD96 (Figure 1J). Tissues from patients with granulomatosis with polyangiitis (GPA), a small vessel vasculitis affecting sinuses, lungs, and kidneys, contained only a small subset of 5%–10% of CD96+ T cells (Figure 1J).
Together, these data detected a shift of checkpoint receptor-expressing T cells in patients with GCA, localized the shift to the central memory population, and identified the checkpoint receptor CD96 on almost all vasculitic T cells.
CD96 functions as an inhibitory checkpoint receptor in vasculitis
CD96, together with TIGIT and CD226, forms a family of immunoglobulin (Ig) superfamily receptors that interact with nectin and nectin-like molecules.25 TIGIT is an inhibitory receptor on both T cells and NK cells, and CD226 is believed to also provide activating signals. The intracellular domain of human CD96 contains an immunoreceptor tyrosine-based inhibition motif (ITIM), which confers inhibitory potential. Since the GCA tissue lesions were populated by CD96+ T cells, we aimed at defining the functional role of the receptor in the disease process.
We induced vasculitis in chimeric mice that were engrafted with human arteries and immunoreconstituted with T cells, B cells, and APCs from patients with GCA.26 To test the functional impact of CD96 signaling, CD4+ T cells were transfected with control or CD96-specific small interfering RNA (siRNA) prior to the adoptive transfer. siRNA transfection reduced CD96 expression by 60% (Figure S2). Low CD96 expression on CD4+ T cells had profound implications for vasculitogenic immunity. Comparison of disease severity in arteries from mice reconstituted with CD96low T cells versus controls demonstrated exacerbation of vasculitis by several readout parameters (Figure 2): density of the mononuclear cell infiltrate, T cell accumulation in the lesion, and enhanced microangiogenesis. The density of tissue-supplying microvessels was assessed by staining for von Willebrand factor (vWF)+α smooth muscle actin (αSMA)+ lumina (Figure 2E). Adoptive transfer of CD96lowCD4+ T cells increased microvessel density by about half (Figure 2F). Also, targeted transcriptomics revealed significantly higher abundance of T cell receptor (TCR) and pro-inflammatory cytokines. Specifically, quantitative comparison in tissues from CD4+CD96hi- versus CD4+CD96low-reconstituted chimeras demonstrated a major impact on T cell effector cytokine production (IFN-γ, IL-21) and innate immunity (tumor necrosis factor [TNF], IL-6, and IL-1β) (Figure 2G).
Figure 2.

CD96 provides a negative signal in autoimmune vasculitis
CD4+ T cells from patients with GCA were transfected with CD96-specific or control siRNA and adoptively transferred into chimeric NSG mice that had been engrafted with human arteries. Human artery grafts were explanted after 2 weeks and processed for RNA extraction and immunohistochemical analysis.
(A) Tissue sections of explanted arteries were stained with hematoxylin and eosin to assess the invasion of the arterial media by inflammatory cells.
(B) Density of the infiltration was quantified based on nuclear counts in H&E-stained images (n = 7).
(C and D) Tissues were stained for CD3 (red) to estimate the density of tissue infiltrating T cells. Cell numbers were enumerated.
(E and F) Immunostaining of microvessels in the arterial wall. Vascular lumina visualized as vWF+/αSMA+ structures. Enumeration of microvessels shown as bar graphs.
(G) Tissue transcriptomics for T cell receptor (TCR) and cytokines in grafts from mice injected with control or CD96 siRNA-transfected CD4+ T cells. RT-PCR data shown as heatmap.
Scale bars: 100 μm (A) and 20 μm (C and E). Mean ± SEM with individual values shown. (B and D) Two-tailed paired t test. (F) Two-tailed unpaired t test. (G) Two-tailed paired t test or Wilcoxon matched-pairs signed-rank test. ∗p < 0.05, ∗∗p < 0.01, n.s., not significant.
These loss-of-function experiments established that CD96+ T cells promote vasculitis through T cell- and Mφ-dependent responses and that CD96 is an inhibitory checkpoint receptor in this autoimmune disease.
CD155low-expressing APCs in GCA
The identification of CD96 as an immunoinhibitory molecule on vasculitic T cells and the accumulation of CD96+ T cells within the inflamed vessel wall raised the question of whether CD96-dependent signaling is defective and directed attention to the ligand. CD96 interacts with CD155, a receptor expressed on Mφs and dendritic cells27 markedly upregulated upon activation.28,29 The CD155 intracellular domain contains an ITIM, classifying the molecule as an immunoinhibitory ligand.
We profiled CD155 expression on monocyte-derived Mφs (MDMs) from patients with GCA, patients with GPA, and age-matched controls (Figures 3A and 3B). In healthy individuals and in patients with GPA, all Mφs expressed high surface density of CD155. In contrast, the entire population of GCA Mφs had low CD155 surface expression, reaching about 50% of the mean fluorescence intensity (MFI) in controls.
Figure 3.

CD155low-expressing antigen-presenting cells in GCA
CD14+ cells isolated from the peripheral blood of patients with GCA and age-matched controls were differentiated into monocyte-derived Mφs and stimulated with LPS/IFN-γ.
(A and B) Flow cytometric analysis of CD155 on Mφs from a patient with GCA (A; n = 12) or GPA as a disease control (B; n = 5). Representative histograms and MFI are shown.
(C) Comparison of CD155 expression on Mφs from 7 untreated patients with GCA, 6 corticosteroid-treated patients with GCA, and 7 age-matched controls. Individual MFIs are shown.
(D) Dual-color immunofluorescence staining of tissue sections for CD68 (green) and the checkpoint ligand CD155 (red). Nuclei marked with DAPI (blue). Temporal arteries were collected from patients with GCA and sinonasal biopsies from patients with GPA. Frequencies of CD68+ CD155high Mφs were counted in 50 distinct tissue regions. Representative data from 5 tissues. Scale bar: 20 μm.
(E) Immunoblotting of total cell CD155 protein in control and GCA Mφs. β-Actin served as loading control. Bar graph shows protein quantification in individual patients and controls.
(F) Monocyte-derived Mφs were induced from patients with GCA and age-matched controls, and CD155 mRNA transcripts were quantified by RT-PCR. Data from 12 patients and 12 controls.
Mean ± SEM with individual values shown. (A, B, and E) Two-tailed unpaired t test. (C) One-way ANOVA with Tukey’s multiple-comparisons test. (F) Mann-Whitney test. ∗∗∗p < 0.001, n.s., not significant.
To rule out that the CD155low phenotype was a consequence of immunosuppressive therapy, we compared CD155 surface density in untreated and treated patients with GCA (Figure 3C). CD155 expression was unaffected by therapy and was indistinguishably low in both patient cohorts (Figure 3C).
To define the status of CD155 expression in the vasculitic lesions, tissue sections from inflamed temporal arteries were analyzed by dual-color immunofluorescence staining (Figure 3D). Vasculitic lesions were densely populated by CD68+ Mφs, but these tissue-dwelling Mφs had a low signal for CD155 or were entirely negative. Tissue sections from GPA sinusitis served as controls. Again, vasculitic infiltrates were densely populated by CD68+ Mφs, but they displayed a strong signal for CD155, resembling the findings in activated Mφs (Figures 3A–3C).
To begin to understand mechanisms underlying the CD155low phenotype in GCA Mφs, we quantified CD155 mRNA and CD155 protein in patient-derived and healthy Mφs by RT-PCR and immunoblotting (Figures 3E and 3F). Unexpectedly, GCA and control Mφs produced similar amounts of CD155 transcripts and contained similar concentrations of CD155 protein.
Together, these data assigned the breakdown of CD96-dependent signaling to the APCs, which failed to bring the CD96 ligand CD155 to their surface.
CD155 is trapped in the ER of GCA Mφs
Like other transmembrane proteins, CD155 is produced in the membrane of the ER to traffic to the ER-Golgi intermediate compartment (ERGIC) and the Golgi body.30 Dependent on how the protein reaches the plasma membrane and whether it is secreted or recycled, it may also associate with endosomes and multivesicular bodies (MVBs).31,32,33 To localize the CD155 protein in the cell, we applied a screening process relying on the marking of subcellular organelles. GCA MDMs were stimulated with lipopolysaccharide (LPS) and co-stained for CD155 and a series of organelle markers: calnexin for the ER; P58 for the ERGIC; GM130 for the Golgi body; EE1 for the early endosome; CD63 for the late endosome; LC3 for the autophagosome; LAMP2a for the lysosome; and HSP60 for the mitochondria (Figure 4A). We utilized image analysis to estimate co-localization of CD155 with intracellular structures (Figure 4B). Systematic analysis yielded the highest co-localization coefficient with the ER, suggesting that CD155 protein remained at the ER membrane. The signal at the ERGIC was higher than at any of the other organelles, indicating that some of the protein trafficked toward the Golgi and got arrested at the ERGIC. To confirm CD155 retention of CD155 at the ER, we applied immunoblotting of purified ER (Figure 4C). Calnexin served as a control for ER membranes. ER membranes isolated from GCA cells retained about 50% higher amounts of CD155 protein (Figure 4C). CD155-ER co-localization was characteristic for patient Mφs. Direct comparison between healthy and GCA MDMs showed minimal retention of CD155 together with the calnexin signal in controls (Figure S4B).
Figure 4.

CD155 is trapped in the ER of GCA Mφs
Mφs were induced from peripheral blood precursor cells as in Figure 3.
(A) Dual-color immunofluorescence imaging of Mφs stained for CD155 and cellular organelles. Markers represent the following organelles: ER, calnexin; ERGIC, P58; Golgi body, GM130; early endosome, EE1; late endosome, CD63; lysosome, LAMP2a; autophagosome, LC3; mitochondria, HSP60. Cellular stains were analyzed by imaging software to reveal co-localization as white dots (bottom panel).
(B) Co-localization coefficient between CD155 and individual cell organelles in GCA Mφs. At least 25 cells were quantified from each independent experiment.
(C) The ER fraction was isolated from healthy and GCA Mφs and CD155 protein expression analyzed by immunoblotting. Calnexin served as control. Western blot and quantitative data are shown.
(D) ER stress signatures compared in control and patient-derived Mφs. Transcript levels of ER stress markers were quantified by RT-PCR. z scores are shown (n = 12).
(E) Quantification of the ER stress-related protein BiP in control and GCA Mφs. Representative immunoblot and results from 3 independent experiments.
(F) Dual-color immunofluorescence staining of tissue sections for the Mφ marker CD163 (red) and the ER stress protein BiP (green). Nuclei marked with DAPI (blue). Temporal arteries were collected from patients with GCA. Sinonasal biopsies from patients with GPA served as disease control.
(G) Mφs were treated with the ER stress-inducer tunicamycin or DMSO. ER fractions were isolated, and CD155 was quantified by immunoblotting. Calnexin served as a control. Representative immunoblot and data from 4 independent experiments.
Mean ± SEM with individual data shown. Scale bars: 5 μm (A) and 20 μm (F). (B–E) Two-tailed unpaired t test. (G) Two-tailed paired t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Protein synthesis at the ER is majorly affected by the activity status of the organelle related to protein folding and lipid synthesis.34 To evaluate the stress status of the ER in GCA Mφs, we quantified transcripts for 6 genes typically upregulated during the unfolded protein response (UPR) (Figure 4D).35,36 ER stress assessed in control and patient-derived Mφs by transcript quantification of ATF4, ATF6, BiP, CHOP, TRIB3, and XBP. GCA Mφs showed increased transcription of genes encoding ER chaperones and enzymes involved in ER protein degradation, confirming a state of chronic ER stress. Western blotting for BiP in stimulated Mφs from controls and patients showed a 5-fold upregulation of the ER stress marker in GCA (Figure 4E). To verify that increased ER stress was a relevant mechanism in the tissue infiltrates, we stained tissue sections from inflamed temporal arteries for the ER-stress protein BiP (Figure 4F). Tissue lesions from GPA sinusitis served as disease control. The vast majority of CD163+ Mφs in the GCA arteries had a strong signal for BiP. In the control tissues, showing granulomatous infiltrates in GPA sinusitis, a strong BiP signal was exclusively seen in CD163−, non-Mφ cells.
To test whether the UPR is sufficient to prevent CD155 from leaving the ER and traveling to the Golgi body, we induced ER stress with tunicamycin in healthy Mφs (Figure 4G). ER-bound CD155 protein concentrations were significantly higher in tunicamycin-stressed versus control cells.
From these data, we conclude that Mφ in patients with GCA are in a state of chronic ER stress, inducing the retention of the transmembrane protein CD155 on the ER surface and disabling inhibitory signals directed at T cells.
CD155low Mφs induce IL-9-producing effector T cells
To define pathways through which CD155low Mφs shift T cell differentiation and promote vasculitogenic immunity, we explored the functional impact of CD155low APCs. We stimulated healthy CD4+ T cells with healthy or GCA Mφs and evaluated the induction of effector functions. To avoid a bias introduced by alloantigen recognition, we loaded Mφs with anti-CD3 antibodies to provide consistent TCR triggering. Healthy CD4+ T cells were co-cultured with the two types of Mφs, and the impact on T cell differentiation was monitored by quantifying transcripts for 15 effector cytokines, including all currently known T cell effector cytokines. Effector cytokines were barely detectable in Mφ-free cultures. Compared with controls, GCA Mφs induced a distinct pattern of T cell effector molecules (Figure 5A). Specifically, patient-derived Mφs biased CD4+ T cells toward the production of IL-9, IFN-γ, and IL-21. Notably, several of the effector cytokines, including granulocyte macrophage colony-stimulating factor (GM-CSF), IL-2, IL-17, IL-22, and TNF were unaffected by the origin of the APC (Figure 5A). Also, cytokines associated with Th2-ness (IL-4, IL-5) were equally abundant in T cells activated with control or GCA Mφs.
Figure 5.

CD155-CD96 interaction promotes differentiation of IL-9-producing effector T cells
Monocyte-derived Mφs were generated from patients with GCA or healthy controls and used as antigen-presenting cells (APCs) for CD4+ T cells. Induction of T cell effector cytokines was assessed by RT-PCR or flow cytometry.
(A) CD4+ T cells from the Mφ-T cell co-cultures were isolated after 72 h. Transcripts for 15 pro-inflammatory effector cytokines were quantified by RT-PCR. Each column represents Mφs from one patient or one control.
(B–E) Frequencies of IL-9-producing (B and C), IFN-γ-producing (D), or TNF-α-producing (E) CD4+ T cells induced by healthy, GCA, or GPA (C) Mφs. Effector cytokines were measured by flow cytometry. Frequencies of cytokine-producing CD4+ T cells from 6 to 12 independent experiments are presented.
(F) CD4+ T cells were stimulated with healthy Mφs in the absence and presence of anti-CD155 antibody. Intracellular IL-9 protein expression in T cells was analyzed by flow cytometry. Frequencies of IL-9-producing CD4+ T cells from 8 experiments.
(G) CD4+ T cells were stimulated with healthy Mφs in the absence and presence of anti-CD96 antibody. Induction of IL-9-producing T cells was determined by flow cytometry. Frequencies of IL-9-producing CD4+ T cells from 6 experiments.
Mean ± SEM with individual values shown. (A and C–G) Two-tailed paired t test. (B) Wilcoxon matched-pairs signed rank test. ∗∗p < 0.01, ∗∗∗p < 0.001, n.s., not significant.
To verify the differences, we applied flow cytometry staining for intracellular cytokines (Figures 5B–5E). Stimulation by GCA Mφs doubled the frequencies of IL-9-producing CD4+ T cells (Figure 5B), while Mφs from patients with GPA resembled healthy controls (Figure 5C). Similarly, GCA Mφs promoted expansion of IFNγ+ T cells (Th1 cells) (Figure 5D). TNF+ CD4+ T cells appeared with equal frequency when stimulated with healthy or GCA Mφs (Figure 5E).
To explore whether the shift in T cell differentiation, specifically the induction of IL-9-producing T cells, was dependent on CD155 and the interaction of CD155 with CD96, we relied on antibody-blocking studies (Figures 5F–5G). Healthy CD4+ T cells were activated with healthy Mφs in the absence and presence of anti-CD155 antibodies. Frequencies of IL-9-producing CD4+ T cells tripled when the checkpoint was blocked (Figure 5F). Similarly, blocking access to the CD96 receptor resulted in doubling of Th9-comitted effector T cells (Figure 5G).
To summarize, CD155low Mφs from patients with GCA shape the process of T cell differentiation. Lack of CD155-dependent signaling enables the expansion of uncommon, IL-9-producing effector T cells.
IL-9-producing T cells in the vasculitic lesions
To investigate the relevance of Th9 cells in vasculitis, we examined temporal artery biopsies from patients with GCA. Targeted transcriptomics confirmed the presence of IL-9 transcripts exclusively in GCA-affected tissues, with non-inflamed arteries and the non-inflamed aortic wall essentially negative (Figure 6A). Dual-color immunohistochemistry for CD3+ T cells and the IL-9 protein showed clusters of IL-9-producing cells (Figure 6B) in all GCA-affected arteries. These clusters were typically localized outside of the lamina elastic externa. The clustering of the IL-9-producing T cells was suggestive of in situ clonal expansion.
Figure 6.

IL-9+ T cells in the vasculitic lesions of GCA
Arteries affected with GCA were collected by diagnostic biopsy.
(A) Tissue transcriptomics (RT-PCR) for IL-9 in GCA-affected arteries; normal, non-inflamed arteries; and normal, non-inflamed aorta (n = 10 samples each).
(B–D) Tissue sections from GCA arteries were immunostained with anti-CD3 antibody (green) and anti-IL-9 antibody (red). Nuclei marked with DAPI (blue).
(B) Representative images from 6 arteries. Scale bar: 100 μm.
(C) Frequencies of CD3+ IL-9+ cells in CD3+ T cells in independent high-power fields.
(D) Distribution of IL-9-producing T cells in the intimal, medial, and adventitial layers. Data from 6 arteries.
(E) IL-9 receptor transcripts expressed in GCA-affected arteries compared with normal, non-inflamed aortas and arteries (RT-PCR) (n = 8 samples each).
Mean ± SEM with individual values shown. (A and E) One-way ANOVA with Tukey’s multiple-comparisons test was used. ∗∗p < 0.01, ∗∗∗p < 0.001, n.s., not significant.
To provide quantitative data, we enumerated IL-9+ T cells in the lesions by immunohistochemistry (Figure 6C). Th9 frequencies varied between 6% and 26% of the T cell infiltrate and reached an average of 11.1%. In all GCA arteries, Th9 cells were rare in the intima and media and >90% resided in the adventitia (Figure 6D).
We asked whether IL-9 receptors (IL-9Rs) are expressed in the tissue site to allow the cytokine to interfere with inflammatory and remodeling processes (Figure 6E). Healthy arteries yielded negative to very low signals for IL-9R mRNA. All arteries affected by GCA expressed IL-9R transcripts, displaying similar variability as for IL-9 mRNA itself (Figure 6A).
These data identified IL-9 as one of the effector cytokines of vasculitic T cells and sized the fraction of Th9 cells at about 10%. Th9 cells exclusively mapped to the adventitia, suggesting a role in early steps of vasculitis.
IL-9-producing T cells are pathogenic effector cells
The mere presence of an effector T cell subset in vasculitis lesions does not prove a pathogenic role. We designed loss-of-function and gain-of-function experiments to test the relevance of IL-9 as a driver of autoimmune vasculitis. Arterial wall inflammation was induced in human arteries engrafted into NSG mice. Each chimeric mouse was reconstituted with T cells, B cells, and monocytes donated by a patient with active GCA.
We first explored whether Th9 cells are enriched in the inflamed vessel wall. Frequencies of human CD4+ IL-9+ T cells were measured in the spleen, the blood, and the engrafted artery of chimeric mice. In the spleen and the blood, 5% of CD4+ T cells produced IL-9 (Figure 7A). Frequencies were 3-fold higher in the inflamed arteries, strongly suggestive for selective recruitment of Th9 cells (Figure 7A).
Figure 7.

IL-9 promotes tissue damage in autoimmune vasculitis
Immunodeficient NSG mice were engrafted with human arteries and immunoreconstituted with PBMCs from a patient with active GCA.
(A) Human CD4+ T cells were extracted from the mouse spleen, the murine blood, and the explanted artery grafts. Frequencies of IL-9-producing T cells were determined by fluorescence-activated cell sorting (FACS) utilizing intracellular staining.
(B–F) Chimeric mice carrying the same artery and the same PBMC were assigned to one of four treatment arms and were treated for 1 week by intraperitoneal injection on alternative days: vehicle, rhIL-9 (200 ng), control IgG (100 μg/intraperitoneal injection), or anti-IL-9 antibody (100 μg/intraperitoneal injection).
(B) Tissue sections of explanted arteries were stained with H&E. Representative images from one of 8 artery grafts.
(C) PBS- or rhIL-9-treated arteries (n = 6 each) or control or anti-IL-9 treated arteries (n = 7 each) were immunostained with anti-CD3 antibody (green) and DAPI (blue). Representative immunostains are presented.
(D) The density of the T cell infiltrate in arteries form the 4 treatment arms was determined by enumerating CD3+ T cell in tissue sections. Each dot represents one artery.
(E) Gene expression profiling of inflammatory genes in extracts from the explanted arteries. RT-PCR analysis of 8 arteries treated with either vehicle or rhIL-9 shown as heatmap in z scores.
(F) Gene expression profiling for inflammatory cytokines and co-stimulatory ligands in tissue extracts of control- and anti-IL-9-treated blood vessels by RT-PCR.
(G–I) CD4+ T cells from patients with GCA were transfected with CD96-specific or control siRNA and adoptively transferred into artery-engrafted NSG mice as in Figure 2. Chimeras were assigned to two treatment arms: injection of anti-IL-9 antibody or control IgG as in (B)–(F).
(G) Tissue sections of explanted arteries were stained with H&E. Representative images from one of 8 artery grafts.
(H) Tissue sections were stained for CD3 (red) to mark tissue infiltrating T cells and compare the density of tissue infiltrates.
(I) Gene expression profiling for inflammatory cytokines and co-stimulatory ligands in tissue extracts of control-, siCD96-, and siCD96 plus anti-IL-9-treated blood vessels by RT-PCR.
Mean ± SEM with individual values shown. Scale bars: 100 μm (B and G) and 20 μm (C and H). (A and I) One-way ANOVA with Tukey’s multiple-comparisons test. (D– F) Two tailed paired t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n.s., not significant.
In subsequent experiments, chimeric mice were treated with recombinant human IL-9 (rhIL-9) or vehicle for 1 week. Histologic evaluation of explanted artery grafts connected surplus IL-9 to aggressive inflammation, characterized by destruction of wall structures, almost complete loss of the medial layer, and moth-eaten areas in the residual media and intima (Figure 7B). The lamina elastica externa and interna were fragmented throughout. Quantification of tissue-infiltrating T cells revealed 3-fold higher numbers of CD3+ T cells in rhIL-9- versus vehicle-treated grafts (Figures 7C and 7D). Comparative analysis of tissue transcriptomes in arteries explanted from vehicle- and rhIL-9-treated chimeras showed that the cytokine was able to induce robust vasculitis (Figure 7E). Transcripts for the T cell cytokines IFN-γ, IL-17, and IL-21 were all upregulated. In parallel, upregulation of CD80 and CD86 transcripts provided evidence for Mφ activation, supported by markedly higher IL-1β, IL-6, and TNF mRNA.
To validate the pathogenic role of IL-9 in loss-of-function experiments, we used anti-IL-9 antibody (Ab)-blocking studies. Vasculitis was induced in artery-engrafted NSG mice, and chimeras were treated with anti-IL-9 Ab or isotype-control IgG. Dense inflammatory infiltrates penetrated throughout all wall layers in the control IgG-treated grafts, and anti-IL-9-treated tissues had sparse infiltrates (Figure 7B). The density of the T cell infiltrates declined markedly in anti-IL-9-treated grafts (Figure 7C). Blocking of IL-9 resulted in a sharp reduction of tissue-invading T cells (Figure 7D). Anti-IL-9 Ab reduced the number of tissue-infiltrating T cells from 50 to 20 cells/high-power field (HPF). Tissue transcriptomics for a core module of inflammatory markers are presented as a heatmap and show the profound effect of IL-9 blockade on vascular inflammation (Figure 7F). Selective inhibition of IL-9 resulted in suppression of T cell effector cytokines, including IFN-γ, IL-17, and IL-21. A sharp reduction of IL-1β, IL-6, and TNF in the tissue of anti-IL-9-treated chimeras confirmed the inhibitory effect of Ab treatment on innate immunity. CD80 and CD86 transcripts were highly sensitive to anti-IL-9 treatment, confirming IL-9’s role in regulating tissue Mφs (Figure 7F).
To provide evidence that the inflammation resulting from a defective CD155-CD96 checkpoint was dependent on IL-9, we induced vasculitis with intact or CD96 siRNA-transfected CD4+ T cells and combined it with anti-IL-9 Ab treatment (Figures 7G–7I). As in Figure 2, CD96 loss of function in the adoptively transferred CD4+ T cells resulted in aggressive vasculitis, with dense wall infiltrates and wide-spread neoangiogenesis (Figures 7G–7H). Blocking IL-9 alleviated the wall infiltrates and reduced intrawall angiogenesis. Tissue transcriptomic analysis confirmed that anti-IL-9 treatment reversed the exacerbation of vasculitis by blunting the induction of innate and adaptive effector cytokines (Figure 7I).
Together, these data established IL-9 as a driver cytokine of vascular inflammation, endowed with regulatory control over innate and adaptive immune responses in the inflamed arteries.
Discussion
Immune checkpoints control the initiation, the duration, the strength, and the outcome of immune responses, and checkpoint blockade leads to tissue inflammation mimicking autoimmune disease. Current work implicates a defective CD155-CD96 checkpoint in spontaneous autoimmunity manifesting as large vessel vasculitis. Patients with GCA had a shift in their functional T cell repertoire, expanding CD96+ memory CD4 T cells in the blood and in the inflamed arteries. Knockdown experiments assigned the CD96 receptor to an inhibitory pathway, defining vasculitis as the consequence of insufficient CD96 signaling. APCs from patients with vasculitis, including Mφs in the vasculitic lesions, were typically low in the expression of CD155, the ligand interacting with CD96. CD155lo Mφs produced the protein but retained it on the ER membranes. CD155lo Mφs induced a program of T cell differentiation that favored the IL-9-producing lineage. Loss-of-function and gain-of-function experiments confirmed that IL-9 is a key driver of vasculitis, promoting vascular remodeling and tissue-destructive immunity. These data link the CD155-CD96 checkpoint to the differentiation and the expansion of the Th9 lineage and classify GCA as a disease of checkpoint failure that is caused by abnormal intracellular trafficking of the checkpoint ligand CD155.
IL-9 has a pleiotropic range of functions, has been associated with allergic disease and anti-parasitic immune responses,37,38 and aggravates the development of atherosclerosis.39 A variety of cell types can produce IL-9, including mast cells, NKT cells, Th2 cells, Th17 cells, regulatory T cells (Tregs), and group two innate lymphoid cells (ILC2s), but by far the most important source is CD4+ T cells committed to the Th9 lineage.40,41,42,43,44,45 Th9 differentiation requires a complex transcription factor network, including STAT6, IRF4, the transcription factor PU.1, and the basic leucine zipper transcription factor, ATF-like (BATF),46 classifying Th9 cells as a distinct and homogeneous lineage. IL-9 has anti-tumor activity,47,48,49 in line with its tissue-destructive capabilities. High expression of IL-9 mRNA in inflammatory bowel disease (IBD) has identified Th9 cells as disease drivers in ulcerative colitis.50,51 However, dependent on timing and context of inflammatory conditions in vivo, IL-9 may be anti-inflammatory.52 The abundance of IL-9 and IL-9Rs in GCA-affected arteries identifies IL-9 as an inflammatory amplifier in vasculitis. IL-9Rs were broadly expressed in vascular and infiltrating cells. The degree of tissue destruction in arteries exposed to excess IL-9 was unexpected. Usually, inflammatory attack to the vessel wall elicits neoangiogenesis and intimal hyperplasia,18 compatible with a maladaptive tissue healing response. Arteries from IL-9-treated chimeras had essentially lost the medial layer; elastic membranes were fragmented, and the tissue had areas of complete destruction. Th9 cells accounted for about 10% of the T cell infiltrate in vasculitic arteries, raising the question of how this relatively small effector subset can determine disease intensity. Th9 cells sat almost exclusively in the adventitia, arranged in dense T cell clusters, where they may have access to adventitial dendritic cells that control tissue tolerance.53
In current work, IL-9 derived mainly from CD96+ T cells, confirming studies in IBD that have associated low CD96 signal strength with high IL-9 production.54 CD96 is expressed on CD4+ T cells, CD8+ T cells, and NK cells and, together with CD226 and TIGIT, is known to bind to nectins and nectin-like proteins.22 Members of the CD96/TIGIT/CD226 receptor family provide co-stimulatory and co-inhibitory signals. CD226 enhances T cell activation, TIGIT is an established co-inhibitory receptor.23,55,56,57 CD96 appears to transmit both positive as well as negative signals,58,59 but in the setting of large vessel vasculitis, loss of CD96 signaling profoundly exacerbated disease. These data are in line with previous reports in which CD96+ Th9 cells displayed strong pro-inflammatory capacities.54 The tissue-destructive pattern of CD96+ T cells in vasculitis mimicked the profound inflammatory potential of such cells in the murine colon, producing aggressive colitis. Tissue-damaging effector functions were clearly linked to IL-9 release, as documented by Ab-blocking studies and the tissue destruction mediated by recombinant IL-9.
Data support the concept that CD96+ Th9 cells become pathogenic effectors due to insufficient suppression. The peripheral blood and the inflamed tissue of patients with GCA contained expanded numbers of CD4+ CD96+ T cells. In the immunophenotypic studies, CD96 was predominantly expressed on human memory CD4+ T cells, with a small population of effector memory cells positive for the receptor. Remarkably, TIGIT could be placed on a distinct population of memory CD4+ T cells, and PD-1 was co-expressed with TIGIT in a subset of memory cells. Like CD96, TIGIT was absent from most Tems, but unlike CD96, TIGIT was detected on a subpopulation of end-differentiated CD4 T cells (Figure 1). Vasculitis-associated shifts in the functional T cell repertoire included the enrichment of CD96+ CD4+ T cells and the decline of TIGIT+ CD4+ T cells, clearly indicating independent regulation of these memory T cell populations despite their shared ability to interact with CD155. The cytoplasmic tails of CD96 and TIGIT both contain an immunoreceptor tyrosine-based inhibition motif (ITIM) but possibly access differential signaling pathways to direct differentiating T cells toward expansion or contraction.
The pinnacle defect that disabled the CD155-CD96 pathway in GCA related to the processing of CD155. Low surface expression of CD155 prevented APCs from patients with GCA from delivering a CD96-dependent stop signal. Surprisingly, the total cell amount of CD155 was well maintained in such CD155low APCs, and mRNA concentrations for the CD155-encoding gene PVR were indistinguishable in patients and controls. CD155 was produced but retained in the cytosol, specifically in the ER. ER retention of CD155 was associated with an ER stress response and was inducible with the ER stressor tunicamycin. Signals leading to excess ER stress in GCA Mφs are currently unknown, and the precise molecular mechanisms leading to ER retention require further investigation. Several mechanisms of posttranslational processing of CD155 have been described. A recent study connected excess expression and excess inhibitory function of CD155 to alterations in PVR mRNA methylation and stability,20 creating CD155hi inhibitory Mφs in patients with coronary artery disease. As a receptor for poliovirus, the protein has complex subcellular trafficking pathways, partially regulated by posttranslational modifications such as SUMOylation and partially by interacting with the dynein light chain Tctex-1, which transports the protein to distinct subcellular localizations.60,61
The study has important clinical implications and can help pave the way for checkpoint-directed therapy in spontaneous autoimmune disease. In GCA, the CD155-CD96 checkpoint is the second checkpoint of pathogenic relevance, coming in after the PD-L1-PD1 checkpoint.5 For both of these checkpoints, in vivo data have documented that their failing enhanced disease activity. Molecular mechanisms underlying the deficiency of PD-L1/PD-1 signaling in GCA are not understood, but current data have identified a processing defect in the checkpoint ligand CD155 as a steppingstone to autoimmunity. Notably, the loss in negative signals provided by APCs had profound impact on the fate decisions of T cells, guiding them toward the Th9 lineage. CD96 is expressed relatively late in the T cell activation program, predictably affecting contraction of the T cell response more than the induction. This disease mechanism in autoimmune disease provides opportunities for repair, which could begin with restoring CD155 intracellular trafficking but could also extend to blocking the expansion of IL-9-producing, tissue-invasive T cells and their effector cytokines.
Limitations of the study
The current study links hypoactivity of the CD155-CD96 immune checkpoint to GCA, adding a second faulty immune checkpoint to the pathology of this vasculitis. The question arises whether GCA is a syndrome of “lost inhibition,” with multiple flawed immune checkpoints. We did not solve the molecular mechanism causing ER retention of CD155, which may be relevant for an array of inhibitory ligands and could ultimately identify GCA as a syndrome of ER pathology. Finally, we did not clarify how IL-9 causes vascular destruction, how IL-9 communicates with immune and stromal cells in the vessel wall, and whether IL-9 represents a clinically relevant biomarker of vasculitis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse IgG | BioLegend | 400124 |
| mouse IgG | Thermo Fisher Scientific | 02–6502 |
| Alexa Fluor 594-mouse IgG | Thermo Fisher Scientific | A-11032 |
| PE-mouse IgG | Thermo Fisher Scientific | P-852 |
| FITC-rabbit IgG | abcam | ab97050 |
| Alexa Fluor 488-rabbit IgG | Thermo Fisher Scientific | A-11008 |
| CD3 | Dako | A0452 |
| FITC-CD3 | BioLegend | 344804 |
| FITC-CD4 | BioLegend | 300506 |
| FITC-CD4 | BioLegend | 357406 |
| BV480-CD8 | BD Biosciences | 566121 |
| FITC-CD14 | BioLegend | 325604 |
| BV650-CCR7 | BioLegend | 353234 |
| PE-Cy7-CD45 | BioLegend | 368532 |
| BV570-CD45RA | BioLegend | 304132 |
| CD68 | Thermo Fisher Scientific | PA5-32331 |
| CD68 | Cell Signaling Technology | 76437 |
| CD96 | Thermo Fisher Scientific | PA5-97568 |
| CD96 | BioLegend | 338402 |
| PE-CD96 | BioLegend | 338406 |
| BV421-CD96 | BioLegend | 338418 |
| PE-PD-1 | BioLegend | 329906 |
| PE/Dazzle594-TIGIT | BioLegend | 372716 |
| BV711-CD226 | BioLegend | 338334 |
| PE/Cy5-TIM3 | BioLegend | 345052 |
| PE/Cy7-LAG3 | Thermo Fisher Scientific | 25-2239-42 |
| CD155 | Thermo Fisher Scientific | MA5-13493 |
| CD155 | BioLegend | 337502 |
| CD155 | Thermo Fisher Scientific | MA5-29762 |
| PE-CD155 | BioLegend | 337507 |
| CD63 | Santa Cruz Biotechnology | sc-5275 |
| GM130 | abcam | ab52649 |
| ERGIC-53/p58 | Sigma | E1031 |
| LAMP2A | abcam | ab18528 |
| Calnexin | ABclonal | A15631 |
| EEA1 | ABclonal | A0592 |
| LC3 | MBL | PM036 |
| HSP60 | Santa Cruz Biotechnology | sc-13115 |
| PerCP/Cy5.5-IFN-γ | BioLegend | 502526 |
| IL-9 | BioLegend | 507704 |
| IL-9 | proteintech | 66144-1-Ig |
| BV421-IL-9 | BD Biosciences | 564254 |
| Biological samples | ||
| PBMC | Stanford University School of Medicine/Mayo Clinic | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dynabeads Human T-Activator CD3/CD28 | Thermo Fisher Scientific | 11132D |
| Lymphoprep | Cosmo Bio USA | AXS-1114547 |
| M-CSF | BioLegend | 574806 |
| IFN-γ | Sino Biological | 11725 |
| LPS | MilliporeSigma | L2630 |
| Accutase Cell Detachment Solution | Innovative Cell Technologies | AT-104 |
| DAPI | Thermo Fisher Scientific | D1306 |
| Critical commercial assays | ||
| EasySep™ Human Monocyte Enrichment Kit | Stemcell Technologies | 19058 |
| EasySep™ human naive CD4 T cell isolation kit | Stemcell Technologies | 19555 |
| EasySep™ human CD4 T cell isolation kit | Stemcell Technologies | 17952 |
| Experimental models: Organisms/strains | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice | The Jackson Laboratory | IMSR_JAX:005557 |
| Oligonucleotides | ||
| CD96 siRNA | horizon | L-020045-02-0005 |
| control siRNA | horizon | D-001810-01-05 |
| CD96 siRNA | Santa Cruz Biotechnology | sc-45460 |
| control siRNA | Santa Cruz Biotechnology | sc-37007 |
| Software and algorithms | ||
| ZEN Software | Carl Zeiss | RRID:SCR_018163 |
| FlowJo Flow Cytometry Analysis FlowJo | FlowJo | RRID:SCR_00852 |
| ImageJ | ImageJ | RRID:SCR_003070 |
| Prism Software | GraphPad | RRID:SCR_002798 |
Resource availablity
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cornelia M. Weyand (cweyand@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Data: The data that support the plots within this paper and other findings of this study are available from the lead contact upon request.
-
•
Code: This paper does not report original code.
-
•
General Statement: Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and subject details
Patients and tissues
144 GCA patients with typical histological findings of GCA on temporal artery biopsy were enrolled into the study. Demographic characteristics of the patient population are summarized in Table S1. Demographically matched healthy subjects without any history of cancer, autoimmune disease, or chronic viral infection were recruited from the blood banks of Stanford University and Mayo Clinic Rochester (n = 145). Patients with a diagnosis of ANCA-positive granulomatosis with polyangiitis (GPA) were enrolled as disease controls.
Temporal arteries were collected from diagnostic biopsies of individuals suspected to have vasculitis. Serial sections of all biopsies were assessed for the presence of inflammatory cells, fragmentation of elastic laminae, and thickening of the adventitial and intimal layers. A diagnosis of vasculitis required unequivocal evidence for mononuclear cells infiltrating the vessel wall combined with damage to the wall structure. Nasal and sinal tissues were collected from diagnostic biopsies in patients suspected to have GPA. Normal human aorta, axillary and temporal arteries were donated by organ donors and harvested within 12 h postmortem.
The Institutional Review Board approved all bio-specimen collections and all procedures; written informed consent was obtained from all participants as appropriate. The Animal Care and Use Committee reviewed and approved all aspects of the animal protocol.
Cell preparation and culture
Peripheral blood mononuclear cells (PBMCs) were isolated from the peripheral blood of healthy individuals and patients by density gradient centrifugation with Lymphoprep (#AXS-1114547; Cosmo Bio USA, CA, United States). Monocytes isolated from PBMCs were isolated with EasySep Human Monocyte Enrichment Kit (#19058; Stemcell Technologies), or by plastic adherence as previously described.19 Monocytes were differentiated into Mφ by treatment with 20 ng/mL macrophage-CSF (M-CSF) (#574806; BioLegend, CA, United States) for 5-day as reported.19 Mφ were further differentiated by stimulating with 100 U/mL IFN-γ (#11725-HNAS; Sino Biological; PA, United States) and 100 ng/mL LPS (#L2630; MilliporeSigma, MA, United States). Mφ were detached from plates using Accutase Cell Detachment Solution (#AT-104; Innovative Cell Technologies, CA, United States) and Cell Scraper (#3010; Corning, NY, United States). Naive CD4 T cells and total CD4 T cells were purified using the EasySep human naive CD4 T cell and human CD4 T cell isolation kits (#19555, #17952; Stemcell Technologies, Vancouver, Canada). Cells were cultured in RPMI 1640 medium (#11875135; Thermo Fisher Scientific, MA, United States) supplemented with 10% FBS (#100–106; GeminiBio, CA, United States) and were incubated at 37°C with 5% CO2.
Human artery-severe combined immunodeficiency mouse chimeras
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from the Jackson Laboratory (IMSR_JAX:005557; Sacramento, CA) and chimeras were generated as previously described.5,16,62 Randomly selected male and female mice were used in this study at the age of 6–10 weeks. Non-inflamed human temporal or axillary arteries were engrafted subcutaneously into the back of the NSG mice.5,16,62 Seven days after the engraftment, PBMC from GCA patients were adoptively transferred into the chimeras (10–20 million cells per mouse). Chimeras were randomly assigned to 2–3 treatment arms in each experiment. Engrafted arteries were harvested on day 21 and shock-frozen for RNA isolation or embedded into OCT for H&E staining or immunostaining.
For gain-of-function and loss-of-function experiments, anti-human IL-9 (100 μg, #507704, BioLegend), isotype control IgG (100 μg, #400124, BioLegend), human recombinant IL-9 (200 ng, #11844-H08B-5, Sino Biological), or saline were injected intraperitoneally every other day for one week. CD96 was knocked down by siRNA technology. CD4 T cells were isolated from GCA patients and transfected with siRNA using the Amaxa Nucleofector system and the Human Nucleofector kit (Lonza). CD96 siRNA-transfected T cells were recombined with autologous T cell-depleted peripheral mononuclear cells and adoptively transferred into chimeric mice to induce vasculitis (total 10–20 million cells per mouse). To extract T cells from the inflamed arteries, explanted tissues were minced and shaken at 37 οC, 100 rpm, for 1 h with 1.5 mg/mL collagenase I (#LS004196; Worthington Biochemical Corporation, NJ, United States) and 100 μg/mL DNase I (#E1011-A; Zymo Research, CA, United States) in HBSS (#14025076; Thermo Fisher Scientific). Cells were filtered to prepare single-cell suspensions using 35 μm filters.
Method details
Macrophage-T cell co-cultures
PBMC-derived monocytes from patients with GCA or healthy donor were isolated using the EasySep Human Monocyte Enrichment Kit (#19058; Stemcell Technologies). 10,000 cells were seeded with M-CSF, cultured for 5-day in 96-well plates, and stimulated with IFN-γ/LPS for 24-h as described above. After stimulation, monocyte derived Mφ were loaded with 1.5 μg/mL anti-CD3/OKT antibody (BioLegend) as previously desribed19 and co-cultured with 50,000 isolated naive CD4 T cells from healthy individuals for 72 h. Antibody-blocking experiments with anti-CD155 antibody (Thermo Fisher Scientific), anti-CD96 antibody (BioLegend), or isotype control antibodies (Thermo, Biolegend) followed previously described protocols.63
Quantitative reverse transcription polymerase chain reaction (RT-PCR)
Methods for RNA isolation, reverse transcription and quantitative PCR have been published.16,64 Gene transcript numbers were adjusted relative to β-actin transcripts. Primers are listed in Table S2.
Flow cytometry and multiparametric phenotyping
Expression of surface molecules was analyzed using the BD LSRFortessa (BD Biosciences, CA, USA) or the CYTEK Aurora (Cytek Biosciences, CA, United States). Data analysis followed the workflow in FlowJo (Tree Star, OR, United States). Cells were stained with diluted antibodies, and Zombie NIR (#423106, BioLegend) was used to distinguish live/dead cells. Staining was performed in wash buffer (BD Biosciences) for 30 min at 4 οC. For multiparametric assays, after exclusion of dead cell by Zombie NIR staining, memory T cells were gated based on expression of CD45RA and CCR7. Using FlowJo software, dimensionality reduction was performed applying uniform manifold approximation and projection (UMAP). FlowJo plugin Phenograph was used for the identification of unique cell populations (cluster) on the resulting UMAP map. Quantified frequencies of each cluster are presented as pie charts. To evaluate intracellular cytokine production, cells were treated with Cell Activation Cocktail (#423301, BioLegend) for 6 h, permeabilized using the BD Cytofix/Cytoperm Kit (#554714, BD Biosciences), stained with specific antibodies, and analyzed by flow cytometry. GolgiStop (#554724, BD Biosciences) was added at the same time to inhibit protein release to the extracellular space. The antibodies used in flow cytometry experiments are listed in Table S3.
Immunohistochemistry and immunofluorescence
Tissues were shock-frozen in OCT on dry ice and blocks stored in −80°C as described.5,62 Five-micron sections were fixed with pre-cold acetone for 15 min and incubated with 0.3% H2O2 buffer for 15 min at room temperature. For dual-color immunohistochemistry, tissue sections were stained with primary antibodies. Bound antibodies were visualized with anti-rabbit or anti-mouse secondary antibodies. Tissues were counterstained with DAPI (#D1306, Thermo Fisher Scientific) and images were taken with an Olympus fluorescence microscopy system (Olympus, Tokyo, Japan). Tissues stained with anti-CD96 or anti-CD155 antibodies were analyzed by the All-in-One Fluorescence Microscope BZX800E system (Keyence, Kyoto, Japan) and Zeiss LSM780 confocal microscope (Zeiss, Oberkochen, Germany). The antibodies used in immunohistochemistry are listed in Table S2.
Quantification and statistical analysis
All analyses were performed using Prism9 (Version 9.5.0; GraphPad Software, La Jolla, CA). Statistical significance was assessed by unpaired or paired 2-tailed Student’s t test (parametric), Mann-Whitney U test or Wilcoxon test (nonparametric), or one-way analysis of variance (ANOVA) with post hoc Tukey’s multiple comparisons test (3 or more groups). The number of samples used in the analysis, the method of analysis, and the meaning of the bar graphs are described in the legend of each figure.
Acknowledgments
This work was supported by the National Institutes of Health (R01AR042527, R01AI108906, R01HL142068, and R01HL117913 to C.M.W. and R01AI108891, R01AG045779, U19AI057266, and R01AI129191 to J.J.G.).
Author contributions
Conceptualization, C.M.W., J.J.G., and S.O.; formal analysis, S.O. and C.W.; investigation, S.O., C.W., R.W., H.Z., M.A., and G.J.B.; patient recruitment, C.M.W., J.J.G., S.O., G.J.B., M.C.B., J.J.M., and K.J.W.; writing – original draft, C.M.W., J.J.G., and S.O.; supervision, C.M.W., J.J.G., and G.J.B.; funding acquisition, C.M.W. and J.J.G.
Declaration of interests
K.J.W. has received clinical trial support from Eli Lilly, GSK, and Kiniksa; he received consulting fees and honoraria from Chemocentryx.
Published: April 18, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101012.
Supplemental information
References
- 1.Baumeister S.H., Freeman G.J., Dranoff G., Sharpe A.H. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 2.Fritz J.M., Lenardo M.J. Development of immune checkpoint therapy for cancer. J. Exp. Med. 2019;216:1244–1254. doi: 10.1084/jem.20182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okazaki T., Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 4.Ramos-Casals M., Brahmer J.R., Callahan M.K., Flores-Chávez A., Keegan N., Khamashta M.A., Lambotte O., Mariette X., Prat A., Suárez-Almazor M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Prim. 2020;6:38. doi: 10.1038/s41572-020-0160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H., Watanabe R., Berry G.J., Vaglio A., Liao Y.J., Warrington K.J., Goronzy J.J., Weyand C.M. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc. Natl. Acad. Sci. USA. 2017;114:E970–E979. doi: 10.1073/pnas.1616848114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pugh D., Karabayas M., Basu N., Cid M.C., Goel R., Goodyear C.S., Grayson P.C., McAdoo S.P., Mason J.C., Owen C., et al. Large-vessel vasculitis. Nat. Rev. Dis. Prim. 2022;7:93. doi: 10.1038/s41572-021-00327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weyand C.M., Goronzy J.J. Immune mechanisms in medium and large-vessel vasculitis. Nat. Rev. Rheumatol. 2013;9:731–740. doi: 10.1038/nrrheum.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H., Watanabe R., Berry G.J., Nadler S.G., Goronzy J.J., Weyand C.M. CD28 signaling controls metabolic fitness of pathogenic T cells in medium and large vessel vasculitis. J. Am. Coll. Cardiol. 2019;73:1811–1823. doi: 10.1016/j.jacc.2019.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen Z., Shen Y., Berry G., Shahram F., Li Y., Watanabe R., Liao Y.J., Goronzy J.J., Weyand C.M. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci. Transl. Med. 2017;9:eaal3322. doi: 10.1126/scitranslmed.aal3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciccia F., Rizzo A., Guggino G., Cavazza A., Alessandro R., Maugeri R., Cannizzaro A., Boiardi L., Iacopino D.G., Salvarani C., Triolo G. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatology. 2015;54:1596–1604. doi: 10.1093/rheumatology/kev102. [DOI] [PubMed] [Google Scholar]
- 11.Terrier B., Geri G., Chaara W., Allenbach Y., Rosenzwajg M., Costedoat-Chalumeau N., Fouret P., Musset L., Benveniste O., Six A., et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum. 2012;64:2001–2011. doi: 10.1002/art.34327. [DOI] [PubMed] [Google Scholar]
- 12.Jin K., Wen Z., Wu B., Zhang H., Qiu J., Wang Y., Warrington K.J., Berry G.J., Goronzy J.J., Weyand C.M. NOTCH-induced rerouting of endosomal trafficking disables regulatory T cells in vasculitis. J. Clin. Invest. 2021;131:e136042. doi: 10.1172/JCI136042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunder G.G., Bloch D.A., Michel B.A., Stevens M.B., Arend W.P., Calabrese L.H., Edworthy S.M., Fauci A.S., Leavitt R.Y., Lie J.T., et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33:1122–1128. doi: 10.1002/art.1780330810. [DOI] [PubMed] [Google Scholar]
- 14.Maleszewski J.J., Younge B.R., Fritzlen J.T., Hunder G.G., Goronzy J.J., Warrington K.J., Weyand C.M. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod. Pathol. 2017;30:788–796. doi: 10.1038/modpathol.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weyand C.M., Goronzy J.J. Immunology of giant cell arteritis. Circ. Res. 2023;132:238–250. doi: 10.1161/CIRCRESAHA.122.322128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe R., Maeda T., Zhang H., Berry G.J., Zeisbrich M., Brockett R., Greenstein A.E., Tian L., Goronzy J.J., Weyand C.M. MMP (matrix metalloprotease)-9-producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ. Res. 2018;123:700–715. doi: 10.1161/CIRCRESAHA.118.313206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watanabe R., Hilhorst M., Zhang H., Zeisbrich M., Berry G.J., Wallis B.B., Harrison D.G., Giacomini J.C., Goronzy J.J., Weyand C.M. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight. 2018;3:e123047. doi: 10.1172/jci.insight.123047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piggott K., Biousse V., Newman N.J., Goronzy J.J., Weyand C.M. Vascular damage in giant cell arteritis. Autoimmunity. 2009;42:596–604. doi: 10.1080/08916930903002495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirai T., Nazarewicz R.R., Wallis B.B., Yanes R.E., Watanabe R., Hilhorst M., Tian L., Harrison D.G., Giacomini J.C., Assimes T.L., et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016;213:337–354. doi: 10.1084/jem.20150900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao T.V., Hu Z., Ohtsuki S., Jin K., Wu B., Berry G.J., Frye R.L., Goronzy J.J., Weyand C.M. Hyperactivity of the CD155 immune checkpoint suppresses anti-viral immunity in patients with coronary artery disease. Nat. Cardiovasc. Res. 2022;1:634–648. doi: 10.1038/s44161-022-00096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kučan Brlić P., Lenac Roviš T., Cinamon G., Tsukerman P., Mandelboim O., Jonjić S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell. Mol. Immunol. 2019;16:40–52. doi: 10.1038/s41423-018-0168-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Georgiev H., Ravens I., Papadogianni G., Bernhardt G. Coming of age: CD96 emerges as modulator of immune responses. Front. Immunol. 2018;9:1072. doi: 10.3389/fimmu.2018.01072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shibuya A., Campbell D., Hannum C., Yssel H., Franz-Bacon K., McClanahan T., Kitamura T., Nicholl J., Sutherland G.R., Lanier L.L., Phillips J.H. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity. 1996;4:573–581. doi: 10.1016/s1074-7613(00)70060-4. [DOI] [PubMed] [Google Scholar]
- 24.Yu X., Harden K., Gonzalez L.C., Francesco M., Chiang E., Irving B., Tom I., Ivelja S., Refino C.J., Clark H., et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 25.Chan C.J., Andrews D.M., Smyth M.J. Receptors that interact with nectin and nectin-like proteins in the immunosurveillance and immunotherapy of cancer. Curr. Opin. Immunol. 2012;24:246–251. doi: 10.1016/j.coi.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Piggott K., Deng J., Warrington K., Younge B., Kubo J.T., Desai M., Goronzy J.J., Weyand C.M. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. 2011;123:309–318. doi: 10.1161/CIRCULATIONAHA.110.936203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lupo K.B., Matosevic S. CD155 immunoregulation as a target for natural killer cell immunotherapy in glioblastoma. J. Hematol. Oncol. 2020;13:76. doi: 10.1186/s13045-020-00913-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan C.J., Martinet L., Gilfillan S., Souza-Fonseca-Guimaraes F., Chow M.T., Town L., Ritchie D.S., Colonna M., Andrews D.M., Smyth M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014;15:431–438. doi: 10.1038/ni.2850. [DOI] [PubMed] [Google Scholar]
- 29.Kamran N., Takai Y., Miyoshi J., Biswas S.K., Wong J.S.B., Gasser S. Toll-like receptor ligands induce expression of the costimulatory molecule CD155 on antigen-presenting cells. PLoS One. 2013;8:e54406. doi: 10.1371/journal.pone.0054406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shikano S., Li M. Membrane receptor trafficking: evidence of proximal and distal zones conferred by two independent endoplasmic reticulum localization signals. Proc. Natl. Acad. Sci. USA. 2003;100:5783–5788. doi: 10.1073/pnas.1031748100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bourne H.R., Sanders D.A., McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 32.Gordon D.M., Lyver E.R., Lesuisse E., Dancis A., Pain D. GTP in the mitochondrial matrix plays a crucial role in organellar iron homoeostasis. Biochem. J. 2006;400:163–168. doi: 10.1042/BJ20060904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roche P.A. Intracellular protein traffic in lymphocytes: "how do I get THERE from HERE"? Immunity. 1999;11:391–398. doi: 10.1016/s1074-7613(00)80114-4. [DOI] [PubMed] [Google Scholar]
- 34.Hotamisligil G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bommiasamy H., Back S.H., Fagone P., Lee K., Meshinchi S., Vink E., Sriburi R., Frank M., Jackowski S., Kaufman R.J., Brewer J.W. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009;122:1626–1636. doi: 10.1242/jcs.045625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sriburi R., Jackowski S., Mori K., Brewer J.W. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicolaides N.C., Holroyd K.J., Ewart S.L., Eleff S.M., Kiser M.B., Dragwa C.R., Sullivan C.D., Grasso L., Zhang L.Y., Messler C.J., et al. Interleukin 9: a candidate gene for asthma. Proc. Natl. Acad. Sci. USA. 1997;94:13175–13180. doi: 10.1073/pnas.94.24.13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimbara A., Christodoulopoulos P., Soussi-Gounni A., Olivenstein R., Nakamura Y., Levitt R.C., Nicolaides N.C., Holroyd K.J., Tsicopoulos A., Lafitte J.J., et al. IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J. Allergy Clin. Immunol. 2000;105:108–115. doi: 10.1016/s0091-6749(00)90185-4. [DOI] [PubMed] [Google Scholar]
- 39.Zhang W., Tang T., Nie D., Wen S., Jia C., Zhu Z., Xia N., Nie S., Zhou S., Jiao J., et al. IL-9 aggravates the development of atherosclerosis in ApoE-/- mice. Cardiovasc. Res. 2015;106:453–464. doi: 10.1093/cvr/cvv110. [DOI] [PubMed] [Google Scholar]
- 40.Elyaman W., Bradshaw E.M., Uyttenhove C., Dardalhon V., Awasthi A., Imitola J., Bettelli E., Oukka M., van Snick J., Renauld J.C., et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc. Natl. Acad. Sci. USA. 2009;106:12885–12890. doi: 10.1073/pnas.0812530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gounni A.S., Nutku E., Koussih L., Aris F., Louahed J., Levitt R.C., Nicolaides N.C., Hamid Q. IL-9 expression by human eosinophils: regulation by IL-1beta and TNF-alpha. J. Allergy Clin. Immunol. 2000;106:460–466. doi: 10.1067/mai.2000.109172. [DOI] [PubMed] [Google Scholar]
- 42.Hültner L., Kölsch S., Stassen M., Kaspers U., Kremer J.P., Mailhammer R., Moeller J., Broszeit H., Schmitt E. In activated mast cells, IL-1 up-regulates the production of several Th2-related cytokines including IL-9. J. Immunol. 2000;164:5556–5563. doi: 10.4049/jimmunol.164.11.5556. [DOI] [PubMed] [Google Scholar]
- 43.Lu L.F., Lind E.F., Gondek D.C., Bennett K.A., Gleeson M.W., Pino-Lagos K., Scott Z.A., Coyle A.J., Reed J.L., Van Snick J., et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006;442:997–1002. doi: 10.1038/nature05010. [DOI] [PubMed] [Google Scholar]
- 44.Nowak E.C., Weaver C.T., Turner H., Begum-Haque S., Becher B., Schreiner B., Coyle A.J., Kasper L.H., Noelle R.J. IL-9 as a mediator of Th17-driven inflammatory disease. J. Exp. Med. 2009;206:1653–1660. doi: 10.1084/jem.20090246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turner J.E., Morrison P.J., Wilhelm C., Wilson M., Ahlfors H., Renauld J.C., Panzer U., Helmby H., Stockinger B. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 2013;210:2951–2965. doi: 10.1084/jem.20130071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaplan M.H. The transcription factor network in Th9 cells. Semin. Immunopathol. 2017;39:11–20. doi: 10.1007/s00281-016-0600-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Y., Hong S., Li H., Park J., Hong B., Wang L., Zheng Y., Liu Z., Xu J., He J., et al. Th9 cells promote antitumor immune responses in vivo. J. Clin. Invest. 2012;122:4160–4171. doi: 10.1172/JCI65459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu Y., Wang Q., Xue G., Bi E., Ma X., Wang A., Qian J., Dong C., Yi Q. Th9 cells represent a unique subset of CD4(+) T cells endowed with the ability to eradicate advanced tumors. Cancer Cell. 2018;33:1048–1060.e7. doi: 10.1016/j.ccell.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purwar R., Schlapbach C., Xiao S., Kang H.S., Elyaman W., Jiang X., Jetten A.M., Khoury S.J., Fuhlbrigge R.C., Kuchroo V.K., et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat. Med. 2012;18:1248–1253. doi: 10.1038/nm.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerlach K., Hwang Y., Nikolaev A., Atreya R., Dornhoff H., Steiner S., Lehr H.A., Wirtz S., Vieth M., Waisman A., et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat. Immunol. 2014;15:676–686. doi: 10.1038/ni.2920. [DOI] [PubMed] [Google Scholar]
- 51.Nalleweg N., Chiriac M.T., Podstawa E., Lehmann C., Rau T.T., Atreya R., Krauss E., Hundorfean G., Fichtner-Feigl S., Hartmann A., et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut. 2015;64:743–755. doi: 10.1136/gutjnl-2013-305947. [DOI] [PubMed] [Google Scholar]
- 52.Angkasekwinai P., Dong C. IL-9-producing T cells: potential players in allergy and cancer. Nat. Rev. Immunol. 2021;21:37–48. doi: 10.1038/s41577-020-0396-0. [DOI] [PubMed] [Google Scholar]
- 53.Ma-Krupa W., Jeon M.S., Spoerl S., Tedder T.F., Goronzy J.J., Weyand C.M. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J. Exp. Med. 2004;199:173–183. doi: 10.1084/jem.20030850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stanko K., Iwert C., Appelt C., Vogt K., Schumann J., Strunk F.J., Ahrlich S., Schlickeiser S., Romagnani C., Jürchott K., et al. CD96 expression determines the inflammatory potential of IL-9-producing Th9 cells. Proc. Natl. Acad. Sci. USA. 2018;115:E2940–E2949. doi: 10.1073/pnas.1708329115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chauvin J.M., Pagliano O., Fourcade J., Sun Z., Wang H., Sander C., Kirkwood J.M., Chen T.h.T., Maurer M., Korman A.J., Zarour H.M. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Invest. 2015;125:2046–2058. doi: 10.1172/JCI80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joller N., Hafler J.P., Brynedal B., Kassam N., Spoerl S., Levin S.D., Sharpe A.H., Kuchroo V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011;186:1338–1342. doi: 10.4049/jimmunol.1003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stanietsky N., Simic H., Arapovic J., Toporik A., Levy O., Novik A., Levine Z., Beiman M., Dassa L., Achdout H., et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA. 2009;106:17858–17863. doi: 10.1073/pnas.0903474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiang E.Y., de Almeida P.E., de Almeida Nagata D.E., Bowles K.H., Du X., Chitre A.S., Banta K.L., Kwon Y., McKenzie B., Mittman S., et al. CD96 functions as a co-stimulatory receptor to enhance CD8(+) T cell activation and effector responses. Eur. J. Immunol. 2020;50:891–902. doi: 10.1002/eji.201948405. [DOI] [PubMed] [Google Scholar]
- 59.Dougall W.C., Kurtulus S., Smyth M.J., Anderson A.C. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017;276:112–120. doi: 10.1111/imr.12518. [DOI] [PubMed] [Google Scholar]
- 60.Mueller S., Cao X., Welker R., Wimmer E. Interaction of the poliovirus receptor CD155 with the dynein light chain Tctex-1 and its implication for poliovirus pathogenesis. J. Biol. Chem. 2002;277:7897–7904. doi: 10.1074/jbc.M111937200. [DOI] [PubMed] [Google Scholar]
- 61.Zitti B., Molfetta R., Fionda C., Quatrini L., Stabile H., Lecce M., de Turris V., Ricciardi M.R., Petrucci M.T., Cippitelli M., et al. Innate immune activating ligand SUMOylation affects tumor cell recognition by NK cells. Sci. Rep. 2017;7:10445. doi: 10.1038/s41598-017-10403-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang H., Watanabe R., Berry G.J., Tian L., Goronzy J.J., Weyand C.M. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation. 2018;137:1934–1948. doi: 10.1161/CIRCULATIONAHA.117.030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stamm H., Klingler F., Grossjohann E.M., Muschhammer J., Vettorazzi E., Heuser M., Mock U., Thol F., Vohwinkel G., Latuske E., et al. Immune checkpoints PVR and PVRL2 are prognostic markers in AML and their blockade represents a new therapeutic option. Oncogene. 2018;37:5269–5280. doi: 10.1038/s41388-018-0288-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watanabe R., Shirai T., Namkoong H., Zhang H., Berry G.J., Wallis B.B., Schaefgen B., Harrison D.G., Tremmel J.A., Giacomini J.C., et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J. Clin. Invest. 2017;127:2725–2738. doi: 10.1172/JCI92167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: The data that support the plots within this paper and other findings of this study are available from the lead contact upon request.
-
•
Code: This paper does not report original code.
-
•
General Statement: Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.