

Summary
Immune checkpoint-inhibitor combinations are the best therapeutic option for advanced hepatocellular carcinoma (HCC) patients, but improvements in efficacy are needed to improve response rates. We develop a multifocal HCC model to test immunotherapies by introducing c-myc using hydrodynamic gene transfer along with CRISPR-Cas9-mediated disruption of p53 in mouse hepatocytes. Additionally, induced co-expression of luciferase, EGFP, and the melanosomal antigen gp100 facilitates studies on the underlying immunological mechanisms. We show that treatment of the mice with a combination of anti-CTLA-4 + anti-PD1 mAbs results in partial clearance of the tumor with an improvement in survival. However, the addition of either recombinant IL-2 or an anti-CD137 mAb markedly improves both outcomes in these mice. Combining tumor-specific adoptive T cell therapy to the aCTLA-4/aPD1/rIL2 or aCTLA-4/aPD1/aCD137 regimens enhances efficacy in a synergistic manner. As shown by multiplex tissue immunofluorescence and intravital microscopy, combined immunotherapy treatments enhance T cell infiltration and the intratumoral performance of T lymphocytes.
Keywords: hepatocellular carcinoma, immunotherapy, hydrodynamic gene transfer, PD-1, CTLA-4, CD137, IL-2
Graphical abstract
Highlights
-
•
Hydrodynamic gene cotransfer provides an antigenic model of hepatocellular carcinoma
-
•
EGFP and luciferase expression allows the tracking of tumor cells in vivo
-
•
Anti-PD-1, anti-CTLA-4, anti-CD137, IL-2, and adoptive T cell therapy synergize
-
•
Intravital microscopy imaging shows changes in T cell behavior upon immunotherapy
Ochoa et al. show the combination of immunotherapy strategies for multifocal hepatocellular carcinoma in vivo via oncogenic hydrodynamic gene transfer. Immunotherapy synergistic regimens combine checkpoint inhibitors, interleukin-2, agonist anti-CD137 monoclonal antibodies, and adoptive T cell therapy.
Introduction
Immunotherapy with checkpoint inhibitors has become the backbone of first-line treatments for advanced hepatocellular carcinoma (HCC).1,2 Formerly, sorafenib was considered the first treatment choice. However, randomized clinical trial results have conclusively shown that combinations of the anti-PDL1 mAbs (monoclonal antibodies) atezolizumab or durvalumab with either the anti-VEGF blocking mAb bevacizumab3 or with the anti-CTLA-4 mAb tremelimumab,4 respectively, surpassed the overall survival benefit of sorafenib. The ongoing revolution of immunotherapy for HCC started demonstrating clinical activity in second-line advanced HCC patients with the anti-PD-1 mAbs nivolumab5 and pembrolizumab.6 The clinical activity of the combination of nivolumab with the anti-CTLA-4 mAb ipilimumab for second-line treatment of sorafenib-refractory or intolerant patients was also conducive to FDA fast-track approval.7
Although a remarkable improvement in patient survival has been achieved, progress is needed to further enhance efficacy and to discover predictive and pharmacodynamic biomarkers. Hence, reliable mouse models for HCC immunotherapy are considered a key goal. This is especially so as, despite considerable efforts, no reliable clinically useful biomarkers have been identified.8,9
Dr. A. Lujambio’s group reported on an interesting mouse model based on the hydrodynamic gene transfer of genetic elements into adult hepatocytes. This genetic intervention gave rise to multifocal HCC subjected to immune surveillance when the driving oncogene, MYC, was directly linked to expression of strong foreign antigens. However, this model presents a strong immune surveillance response that limits the utility of this model as a platform for testing combination immunotherapies.10 More recently, the model has been used to address the contribution of several oncogenes to the carcinogenesis process.11 Due to the previous paucity of reliable animal HCC models for therapeutic developments, this methodology holds much potential to advance the field.12
The area of cancer immunotherapy as a whole is very actively seeking synergistic combinations to advance the field addressing resistance mechanisms or acting on more than one mechanism of action.13 Animal models in which immunotherapy exerts suboptimal effects become crucial to evaluate new therapeutic combinations and compare the efficacy of the approaches. Otherwise, clinical experimentation on combinations including triplets and higher order combinations in the absence of conclusive preclinical evidence poses serious difficulties for physicians, patients, and the pharmaceutical industry.
The model based on hydrodynamic gene intervention inserting the c-myc oncogene and deleting p53 is also featured by some level of heterogeneity among the multiple HCC foci. This is considered relevant and advantageous since heterogeneity in cancer is considered a serious obstacle14,15,16 that needs to be overcome by cancer therapeutics.
In this study, a hydrodynamic gene transfer approach was used to co-engineer c-myc expression, p53 disruption, and the expression of EGFP, luciferase, and the human gp100 melanosomal antigen. These multigenic interventions were possible since in the hydrodynamic gene transfer experimental setting, hepatocytes can simultaneously get transfected with multiple constructs.10 In this experimental setting of multifocal HCC, immunotherapy interventions using checkpoint inhibitors, cytokines, and adoptive T cell therapy could be tested to improve the outcome of immunotherapy combination regimens as well as to interrogate the underlying immune mechanisms.
Results
A multifocal HCC mouse model suitable for testing immunotherapy interventions
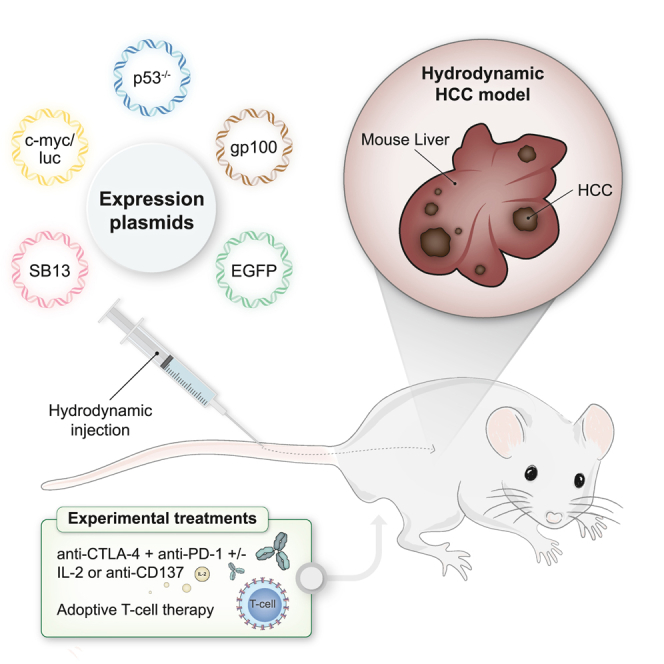
Hydrodynamic gene transfer is achieved by rapid injection through the tail vein of mice using a solution containing gene constructs.17 These techniques could be used to transfer gene expression cassettes or to disrupt loci with CRISPR-Cas9 approaches.10 Based on this property, we co-injected five gene constructs encoding (1) human c-myc, luciferase, (2) EGFP, (3) the human gp100 melanosomal antigen, and (4) a gRNA guide for p53 and the CAS9 nuclease (Figure 1A). To favor genome integration of the transgene cassettes, a sleeping beauty transposase expression cassette (5) was included, and all the other gene expression constructs were flanked by sleeping beauty recognized sites.18
Figure 1.

A multifocal HCC model to test immunotherapy strategies
(A and B) Collection of expression plasmids used to transfer hepatocytes by hydrodynamic injection into the lateral tail vein (A) of mice to attain hepatocyte co-expression of the indicated genes and CRISPR-Cas9 knock-out of p53 (B).
(C) Images of representative resected livers at the indicated days following hydrodynamic injection and a microphotograph of an H&E staining of a liver tissue section from a representative case on day 28 post gene transfer.
(D) Survival of the mice following c-myc, luciferase transfer, and p53 elimination compared with the same procedure with the addition of the plasmids encoding EGFP and the human gp100 melanosomal antigen. This experiment has been replicated twice with similar results (n = 13 per group).
(E) Quantitative RT-PCR assessment of gp100 expression in RNA extracted from the liver of the gene-transferred tumor-bearing mice and a control mouse. Each point represents data from a different dissected tumor nodule.
(F) A described antigenic peptide in the sequence of gp100 was added to splenocytes derived from the indicated groups of mice, and IFNγ release into the supernatant was measured following a 72-h culture. Each symbol corresponds to a single mouse. Means and SEM are shown.
(G) A representative case of fluorescent tumors that were observed under white light illumination and epifluorescence detection.
(H) EGFP fluorescence in tumor cells from cell suspensions of terminal mouse livers following fluorescence-activated cell sorting-gating on live cells. See also Figures S1–S3.
The five constructs were co-injected into mice (Figures 1A and 1B), and expression of all these constructs is expected in a variable fraction of hepatocytes (ranging from 5% to 15%).17 Mice receiving these combinations developed easily observable multifocal neoplastic disease in their livers (Figure 1C), which was histologically indistinguishable from human HCC (Figure 1C). Such tumors could also be followed by ultrasound examination (Figure S1). To ensure that the transformed cells were amenable to immunotherapy experimentation, we introduced the human gp100 melanosomal antigen, cognizant that its dominant H2-Kb-restricted epitope is recognized by the TCR transgene of pmel-1 transgenic mice.
Although the foreign human sequences in c-myc and gp100 were expected to raise antigenicity together with luciferase and EGFP, we considered such an increase in antigenicity to be very convenient features to model immunotherapy, since the expression of the c-myc oncogene and the disruption of p53 would be immunologically silent. However, it is already known that introducing other strong foreign antigens such as ovalbumin results in rejection by immunosurveillance.10
Penetrance of the carcinogenesis phenomena was 95% in the model. Compared with c-myc + p53 knockdown, the addition of the luciferase, EGFP, and gp100 transgenes only slightly delayed time to death (Figure 1D), with 80% of the mice already dead 70 days following the hydrodynamic gene transfer procedure in most experiments.
In tumor-bearing mice, dissection of individual tumors indicated variable levels of expression of the gene encoding gp100, thereby reflecting some degree of heterogeneity (Figure 1E). This finding dovetails with the observations that splenocytes in such gene-transferred mice responded to the human gp100 epitope sequence presented by H-2Kb by releasing IFNɣ into the culture supernatants (Figure 1F), although with some degree of variability. Moreover, some antigenicity was similarly substantiated for the reported antigenic peptides of human c-myc,19 luciferase,20 and EGFP21 following intensive vaccination (Figures S2A–S2C).
Figures 1G and 1H show that tumors are fluorescent upon UV light exposure and that fluorescent tumor cells in cell suspensions were detectable by flow cytometry (Figures 1G and 1H). Moreover, there was heterogeneity in gp100 and EGFP co-expression, indicating that perhaps only some of the hepatocytes take up and express all the plasmids in the mixture. As can be seen in Figures S3A–S3C, an important fraction of gene-transferred hepatocytes following hydrodynamics gene transfer expressed either EGFP or gp100 but not both transgenes. This indicates that the model is likely to reflect tumor heterogeneity.
Efficacy of checkpoint inhibitor combinations can be enhanced by CD137 agonists and IL-2
HCC treatment with PD(L)-1 blockade + CTLA-4 blockade is one of the first line choices for patients with advanced disease.7 Hence, we modeled this regimen in mice starting treatment when animals bore microscopic tumors on day +7 of the post-hydrodynamic multi-gene transfer (Figure 2A). In this setting, either anti-PD-1 of anti-CTLA-4 mAbs showed very limited efficacy in terms of luciferase bioluminescence reduction in the hepatic region and overall survival (Figures 2B and 2C). However, mice treated with the anti-PD-1 + anti-CTLA-4 mAbs combination showed an advantage in terms of tumor burden and survival, even if tumors in most animals eventually progressed and mice succumbed to the disease (Figures 2B and 2C).
Figure 2.

Effects of combinations of immunotherapy agents on the multifocal HCC model and bioluminescence follow-up of the tumors
(A) Mice bearing multifocal HCC following hydrodynamic injection were treated at weekly intervals with anti-PD-1, anti-CTLA-4, or a combination of the two checkpoint inhibitors.
(B) Bioluminescence images on days 32 and 64 and their quantification. Medians and 95% CI are shown.
(C) Survival of the experimental groups of mice.
(D) Similar scheme of treatments but testing triplet combinations including anti-PD-1 + anti-CTLA-4 + anti-CD137 and anti-PD-1 + anti-CTLA-4 + IL-2 starting on day 7. IL-2 was given in 3-day cycles dosed every 12 h as indicated.
(E) Bioluminescence images on day 32 and 64 to monitor tumor burden. Medians and 95% CI are shown
(F) Survival of the experimental groups of mice.
(G) Similar experiments postponing treatment onset to day 21.
(H) Bioluminescence imaging. Images with a crossed rectangle represent dead mice at the time of bioluminescence imaging.
(I) Overall survival. Number of mice and median overall survival are provided.
(J) Individual tumor size follow-up of the groups of mice given the indicated treatments. Statistical comparisons were made using Kruskal-Wallis, repeated measures ANOVA, followed by Tukey’s test calculated with logarithmic data transformation and log rank (Mantel-Cox) test. All experiments were repeated at least twice. See also Figures S4 and S5.
In this same setting, we studied triplet regimens adding recombinant IL-2 or agonist anti-CD137 mAb since both treatments have been reported to be synergistic with double checkpoint blockade.22,23 As shown in Figure 2B, both triplets were therapeutically active since most mice had virtually undetectable tumors on day +32 and a dramatic survival advantage was observed (Figures 2D–2F).
To study whether this treatment scheme could be efficacious in more advanced stages of this aggressive multifocal HCC disease model, we postponed the treatment onset to day +21 and observed limited but observable efficacy of the anti-PD1 + anti-CTLA-4 + IL-2 triplet in terms of tumor burden reduction and survival advantage. These experiments illustrate the suitability of this model to iteratively test and improve therapeutic outcomes with experimental treatments (Figures 2G–2J). The identity of the antigens is difficult to address beyond the observations on gp100 and human c-myc shown in Figures 1F and S2B. EGFP seems to be also recognized as an antigen upon combined treatments, while anti-luciferase IFNγ-producing T cells are not detectable upon combined immunotherapy regimens (Figure S4).
To confirm results on the efficacy of the combinations, we used mice subcutaneously engrafted with the HCC cell line PN-299-L, generated from HCC arising upon hydrodynamic gene transfer by Dr. Lujambio’s group.24 Tumor-bearing syngeneic mice were treated, as shown in Figure S5A, either with single agents or with the combinations. As shown in Figures S5B–S5D, the triple combinations were more efficacious and achieved in some cases complete eradications of the tumors: six out of 11 in the case of anti-PD1 + anti-CTLA4 + antiCD137 and nine out of 11 in anti-PD1 + anti-CTLA4 + IL2.
T cell infiltrates correlate with efficacy of the therapeutic regimens that are mediated by CD8 and CD4 T cells
Next, we performed treatment experiments as described in Figure 3A and sacrificed mice on day +32 to retrieve and study their livers (Figure 3A). Paraffin tissue sections were subjected to a multiplex immunofluorescence panel. As expected, tumors were smaller in the therapy group (Figure 3B). The multiplex immunofluorescence panel yielded evidence for enhanced T cell infiltrates upon anti-PD-1 + anti-CTLA-4 treatment. Importantly, the infiltrates were much more abundant when anti-CD137 mAb or IL-2 were added to the regimens. Both CD8 and CD4 T cells were increased including Foxp3+ Tregs (Figure 3C). Notably, a fraction of all such T cell subsets were actively proliferating as denoted by Ki-67 co-expression.
Figure 3.

Immunotherapy combinations enhance T lymphocyte infiltration in the multifocal HCC model
(A) Schematic timeline of the experiments.
(B) Representative images for the excised livers and multiplex immunofluorescence staining of tumor sections.
(C) Quantification in liver tissue sections of the densities of the indicated lymphocyte subsets in groups of mice treated as depicted in (A). Each group included four to five mice. Means and SEM are shown.
Our most efficacious treatment was the triplet with IL-2, and in animals treated with this combination, we performed CD4 and CD8 depletions with monoclonal antibodies that showed a necessary requirement for T cells (Figures 4A–4D). Single depletions of either CD4 or CD8 T cells seemed to result in shorter survival even if differences did not reach statistical significance (Figure 4D). CD4 + CD8 double depletions completely abrogated the efficacy of treatment, thus suggesting a prominent role for CD4+ T cells.
Figure 4.

CD4 and CD8 T cells are required for the efficacy of the anti-PD-1 + anti-CTLA-4 + IL-2 triple immunotherapy regimen
(A) Scheme of the timeline of the experiments selectively depleting CD4, CD8 T cells, or both T cell subsets.
(B) Bioluminescence images of the groups of mice at the three indicated time points.
(C) Quantification and statistical comparison (Kruskal-Wallis test) of the images in (B). Each symbol represents an individual mouse. Medians and 95% CI are shown.
(D) Overall survival of the experimental groups of mice.
Modeling adoptive T cell therapy
Expression of gp100 and pmel-1 TCR transgenic mice permitted adoptive T cell therapy regimens to be tested in the hydrodynamic gene-transferred model. As a proof of concept, we treated mice as early as day +2 after oncogenic hydrodynamic gene transfer (Figure 5A). Following in vivo luciferase activity over time, we observed a clear decrease in the signal that was frequently curative in mice receiving pmel-1 but not in those mice that had not been gene-transferred with the human gp100 cognate antigen as a specificity control (Figures 5B–5D). Hence, adoptive T cell therapy is highly efficacious in this very early stage of the malignancy model.
Figure 5.

Adoptive T cell therapy with anti-gp100 CD8+ TCR transgenic T cells in the HCC model
(A) The HCC model was treated as early as 2 days after hydrodynamic injection of the plasmids with preactivated pmel-1 T cells as indicated. Mice were monitored for tumor burden by bioluminescence, and survival was recorded.
(B) Time course of bioluminescence (median ± 95% CI) of the indicated groups of mice. As a specificity control, a group of mice received pmel-1 transfer without prior gp100 in the plasmid mixture for hydrodynamic gene transfer.
(C) Corresponding bioluminescence images.
(D) Overall survival of the experimental treatment groups of mice.
(E) Similar experimental setting as in (A) but postponing T cell transfer until day 14 post-hydrodynamic injection. In this case, a group of mice received control TCR transgenic T cells that recognize an irrelevant antigen in this model (OT1 T cells that recognize an ovalbumin H2-Kb-presented epitope).
(F and G) Bioluminescence quantification (median ± 95% CI) and images recorded at the indicated time points.
(H) Survival follow-up of the treated mice (number of mice and survival median are provided for each group of mice).
However, if treatment onset is postponed until day +14 (Figure 5E), the therapy is much less efficacious, with only some objective reduction of the burden as assessed by luciferase expression and a marginal benefit in survival. Importantly, if similarly preactivated and transferred TCR transgenic cells recognizing other irrelevant antigens were used (OT-I lymphocytes), no effect was substantiated (Figures 5F–5H).
Given the fact that both tumor antigen-specific adoptive T cell therapy and immunomodulation offered room for improvement, we tested on a 21-day established HCC a combination of the anti-PD-1 + anti-CTLA-4 + IL-2 regimen with adoptive transfer of pmel-1 TCR transgenic cells. In this difficult-to-treat setting, clear evidence for a beneficial effect was found in the groups treated with combined immunomodulation and adoptive T cell therapy in terms of reducing tumor burden over time (Figures 6A–6D).
Figure 6.

Efficacy of the combination of triple immunomodulatory therapy plus adoptive T cell therapy
(A) Scheme of treatments including groups of mice receiving only pmel-1 T cell transfer, triple (anti-PD-1 + anti-CTLA-4 + IL-2) immunomodulatory therapy with either OT1 T cells or pmel-1 T cells as quadruple combined immunotherapy regimens.
(B) Serial bioluminescence imaging at representative time points.
(C) Individual bioluminescence growth curves for each group. Statistical differences were calculated by repeated measures ANOVA tests with subsequent Tukey’s test and calculated upon logarithmic transformation of the data (p values are provided under the graphs).
(D) Overall survival of the indicated groups of mice for which the median survival time and numbers of mice are provided.
Intravital microscopy experiments show improved performance of antitumor T cells upon immunotherapy combination
Experiments as above suggested that the efficacy of triplet immunomodulation was dependent on T cells and resulted in prominent T cell infiltrates. Combinations encompassing transfer of resting pmel-1 lymphocytes and triplet immunomodulation allowed the tumor-specific T cells to be traced by means of multiphoton confocal microscopy on surgically exposed livers of living mice. In these experimental settings, time-lapse imaging showed EGFP+ malignant cells and RFP+ T cells, either endogenous in CD2-RFP transgenic mice (Figure 7A and Video S1) or when the mice had been transferred with RFP+ pmel-1 cells (Figure 7B). Figure 7A shows the infiltration of tumors and surrounding parenchyma by red fluorescent endogenous lymphocytes. Of note, the areas around blood vessels showed more prominent T cell infiltration. Figure 7B shows the intratumoral presence of red fluorescent T cells following the adoptive transfer of RFP+ pmel-1 lymphocytes. Of note, triplet treatments resulted in the induction of apoptosis in EGFP+ malignant cells as ascertained by flow cytometry on EGFP+ sorted cell suspensions or by immunochemistry on excised liver serial sections, as shown in Figures S6A–S6E.
Figure 7.

Intravital multiphoton confocal microscopy imaging of multifocal HCC-bearing livers in mice undergoing immunotherapy
CD2-RFP transgenic mice were subjected to multifocal HCC induction by the five-plasmid hydrodynamic gene transfer and subjected to intravital microscopy of surgically exposed livers following the treatment with RatIgG (control) or the triple immunomodulatory combination (anti-PD-1 + anti-CTLA-4 + IL-2) started on day 35.
(A) Representative microphotographs of non-tumor and tumor liver on day 43 showing endogenous CD2-RFP T cells and the corresponding quantifications according to experimental treatments.
(B) Quantification of the density of the red fluorescent cells corresponding to transferred RFP+ pmel-1 T cells and images from the indicated experimental treatments groups.
(C) Experiments as in (B) in non-transgenic C57Bl/6 mice that had undergone hydrodynamic HCC induction and that were transferred with resting RFP+ pmel-1 CD8+ T cells on day 32. Mice were given control or triple immunomodulatory treatment on day 35, and intravital microscopy images were acquired on day 43. Representative images are shown as well as quantifications pertaining to motility, migration speed, and meandering index from four imaged cases are represented. These images correspond to Video S2. See also Figure S6.
Multifocal HCC tumors were generated by hydrodynamic injection in CD2-RFP mice. Mice were treated with anti-PD-1+ anti-CTLA-4 + IL-2 according to the treatment scheme represented in Figure 2 starting on day 35. Livers were observed by intravital imaging on day 43. Mice were injected with anti-CD31 fluorescent antibodies right before surgery to visualize blood vessels (blue). Videos show endogenous T cell infiltration (RFP, red) into tumor lesions (EGFP, green). Video is 600× accelerated. Related to Figure 7.
Our intravital imaging experiments showed, in conditions either visualizing pmel-1 cells or endogenous T cells, that immunotherapy triplet with IL-2 enhanced T cell infiltration. Moreover, time-lapse intravital microscopy showed enhanced mobility and directional migration of the tracked T cells visualized in the tumors upon immunomodulatory therapy instigation (Figure 7C). A representative video shows such behavior changes that were contingent on checkpoint inhibition + IL-2 treatment 7 days prior to intravital imaging, which also showed indirect signs of clear disruption of EGFP+ tumor cells upon immunotherapy (Video S2).
Multifocal HCC tumors were generated by hydrodynamic injection in wild-type mice, and 4 weeks later, mice were transferred with 5 × 106 pmel-1 RFP+ CD8 T cells (RFP, red). Mice were treated with anti-PD-1 + anti-CTLA-4 + IL-2 according to the treatment scheme represented in Figure 2 starting on day 35. Intravital microscopy was performed at 6 weeks after hydrodynamic injection. Mice were injected with anti-CD31 fluorescent antibodies right before surgery to visualize blood vessels (blue). Videos show the dynamics of anti-gp100 CD8+ T cells (RFP, red) into tumor lesions (EGFP, green). Videos are 600× accelerated. Related to Figure 7.
Discussion
Reliable animal modeling of HCC for immunotherapy aims to preclinically define and predict improvements in efficacy and to elucidate the underlying mechanisms of action. Definition of the antigens and multifocal heterogeneous disease are considered advantageous features of the models. Gp100 and pmel-1 cells were chosen because of relatively modest TCR affinity that would more faithfully mimic clinically relevant immune responses.25 Previous attempts from our group modeled multipronged immunotherapy in inducible c-myc transgenic mice in which OVA as a surrogate tumor antigen was co-expressed with c-myc. However, this GEMM model had serious limitations since it only works in neonatal mice.26 Despite this caveat, we were able to establish that immunotherapy regimens encompassing PD-1 blockade, CD137, and OX40 agonists synergized for efficacy.27 In that model, we also reported synergistic efficacy if adoptive T cell therapy with antigen-specific CD8 and CD4 T lymphocytes was included in the treatment strategy.27
The hydrodynamic oncogenesis model originally published by the group of A. Lujambio is of exceptional value to study carcinogenesis and combination of genetic elements.10,11,28 However, it was inefficient to study combination immunotherapies. Luciferase and EGFP as reporter genes are very useful, while also weakly conveying antigenicity to the tumors. Reportedly, if strong antigenic genes were co-transferred, animals rejected their nascent tumors because of immunosurveillance mechanisms.10 In this study, we conferred moderate levels of antigenicity that permitted comparative preclinical evaluations of immunotherapy regimens.
The hydrodynamic model offers several advantages over engrafted syngeneic cell lines since it probably reflects more accurately the coevolution of cancer and immune responses either at baseline or following pressures elicited by treatment. It also reflects the heterogeneity of a tumor model in human HCC. Heterogeneity of response to treatment among mice reflects the human situation of variable response to immunotherapy and might be useful to discover predictive and pharmacodynamics biomarkers.
The nature of the antigens in the mouse system is likely related to the reporter transgenes and more clearly in our hands to the melanosomal antigen gp100 with some contribution of human c-myc and EGFP. In humans, antigenicity will likely be variable and heterogeneous, and these immunobiology features are recapitulated by the mouse model as well.
In our hands, the modified model is suitable for comparative immunotherapy experimentation, and indeed, we were able to study the efficacy of anti-PD-1 + anti-CTLA-4 combinations, which are currently used in clinical practice.7 Furthermore, we clearly observed improved tumor control by adding agents to form triplets when incorporating recombinant IL-2 or anti-CD137 agonist mAbs.29 The choice of these agents was made because a new wave of IL-2 and CD137 agents are undergoing clinical trials.22,30 The efficacy observed in our experiments was truly remarkable, showing intense intratumor T cell infiltrates. However, the model still offers room to test further improvements with experimental agents.
The triplet resulting from the addition of IL-2 to the anti-PD-1 + anti-CTLA-4 doublet achieved a very significant increase in efficacy even against well-established tumors. This synergy is reminiscent of work by Dr. R. Ahmed’s group on IL-2 combined with checkpoint inhibition in a murine model of chronic viral infections.31 In the clinic, new formats of IL-2 are being developed that reduce or ablate binding to CD25 to avoid preferential Treg stimulation. Such mutant or pegylated forms of IL-2 would be the most suitable in our opinion.32,33 Adoptive T cell transfer in liver cancer has reportedly benefited HCC patients in the early stages of disease1 and is being developed with CAR T cells targeting glypican-3.34 Thus, we have experimentally modeled adoptive transfer with pmel-1 CD8+ TCR transgenic lymphocytes that showed evidence for certain levels of efficacy, and this approach synergized with immunomodulatory combinations.
In our hands, CD4 T cells were critical for the efficacious effect of combined immunotherapy as previously observed in other mouse tumor models,35 and CD4+ T lymphocytes prominently infiltrated the HCC tumors upon immunotherapy. These observations have prompted ongoing investigations to address the antitumor mechanisms mediated by CD4 T cells in HCC immunotherapy and to study the role of CD4 T cells in adoptive immunotherapy approaches.27
Intravital microscopy offered evidence for potentiated T cell activities. Time-lapse intravital multiphoton confocal microscopy imaging indicates that immunotherapy does not only result in more numerous T cell infiltrates but also in changes regarding the behavior and motility of intratumor T lymphocytes when mice had received immunotherapy.
The multifocal HCC model that we present offers the opportunity for further sophistication, testing other regimens including those checkpoint inhibitors with VEGF or VEGFR targeted therapies as well as modeling coexisting fibrotic liver damage36 and steatosis.37 Recently, the synergy of an anti-VEGF + anti-PDL1 combination has been reported in a similar preclinical model to be more efficacious than the single-agent treatments.8 Other combinations are to be explored, and the model offers opportunities to study the role of known targets for other existing immunotherapy agents (i.e., LAG3, TIM3, and TIGIT).1,38
It must be realized that the liver is considered a highly tolerogenic organ for viral, transplantation, and cancer immunology.39 This applies not only to experimental HCC expressing the SV40 T antigen40 but also to metastasis of other malignancies nesting in the liver.41 The adoptive transfer setting will enable us to study T cell exhaustion and myeloid suppression in ongoing mechanistic research (M.C.O., unpublished data).
In summary, we have made improvements in a relatively simple, reproducible, and reliable spontaneous and heterogeneous HCC mouse model that enabled us to test various immunotherapies. With it, we have investigated clinically feasible combinatorial regimens including feasible triplets and intravitally visualized the antitumor immune response occurring in vivo performing multiphoton confocal microscopy of intrahepatic HCC lesions.
Limitations of the study
The clinical reality of the complex immunobiology of human HCC and the exploitation of immunotherapy combinations against this deadly disease are only partially simulated by the hydrodynamic gene transfer mouse models because of the following reasons: first, the oncogenesis and immunoediting processes usually take many months in human patients to occur as opposed to the 2- to 3-week intervals described in this study. Second, HCC is often accompanied by underlying liver diseases such as cirrhosis, chronic viral hepatitis, autoimmunity, or NASH that are yet to be accounted for in these mouse models. Third, the level of tumor heterogeneity in the much larger burdens of disease observed in humans is conceivably much higher than that accounted for in the model.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CTLA-4 (9D9) for in vivo use | BioXcell | BE0164; RRID:AB_10949609 |
| Anti-PD-1 (RMP1-14) for in vivo use | BioXcell | BE0273; RRID:AB_2687796 |
| Anti-CD137 (3H3) for in vivo use | BioXcell | BE0239; RRID:AB_2687721 |
| Anti-CD4 (GK1.5) for in vivo use, depletion | BioXcell | BE0003-1; RRID:AB_1107636 |
| Anti-CD8β (53-5.8) for in vivo use, depletion | BioXcell | BE0223; RRID:AB_2687706 |
| RatIgG for in vivo use | BioXcell | BE0094; RRID:AB_1107795 |
| Anti-CD3 (SP7) | Abcam | Ab16669; RRID:AB_443425 |
| Anti-CD8 (D4W2Z) | Cell Signaling Technology | 98941; RRID:AB_2756376 |
| Anti-Foxp3 (D6O8R) | Cell Signaling Technology | 12653; RRID:AB_2797979 |
| Anti-CD4 (D7D2Z) | Cell Signaling Technology | 25229; RRID:AB_2798898 |
| Anti-CD11b (6H12) | Nous Biologicals | NB110-89474; RRID:AB_1216361 |
| Anti-KI-67 (rabbit polyclonal) | Abcam | Ab15580; RRID:AB_443209 |
| Anti-cleaved Caspase-3 (Asp175) | Cell Signaling Technology | 9661; RRID:AB_2341188 |
| Anti-Melanoma gp100 antibody [EP4863(2)] | Abcam | ab137078; RRID:AB_2732921 |
| Anti-GFP (Green Fluorescent Protein) pAb (Polyclonal Antibody) | Novus biologicals | NB100-1770; RRID:AB_523903 |
| Rabbit anti-Goat IgG (H + L) Secondary Antibody, Biotin | Invitrogen | 31732; RRID:AB_228393 |
| PE Rabbit Anti- Active Caspase-3 | BD Biosciences | 51-68655X; RRID:AB_393957 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human IL-2 (Proleukin) | Novartis | CN70389 |
| EGFP 118–126 (DTLVNRIEL) | Genscript | Han et al.21 |
| Luciferase 264–272 (LMYRFEEEL) | Genscript | Limberis et al.20 |
| Human gp100 petide KVPRNQDWL | Genscript | RP20344 |
| c-Myc 229–238 (SSPQGSPEPL) | Thermo-Scientific | Helm et al.19 |
| c-Myc 201–240 (DSSSPKSCASQDSSAFSPSSDSLL SSTESSPQGSPEPLVL) |
Thermo-Scientific | Helm et al.19 |
| OVA 257–264 (SIINFEKL) | Invitrogen | Vac-sin |
| D-luciferine | Xenogen | XR-1001 |
| DAPI | Akoya-Biosciences | FP1490 |
| Faramount aqueous mounting medium | Dako | S3025 |
| Citrate unmasking solution | Cell Signaling Technology | 14746 |
| Diaminobenzidine | Dako | K3468 |
| Collagenase I | Sigma-Aldrich | CO130 |
| DNAse I | Calbiochem | 260913 |
| Critical commercial assays | ||
| Maxwell RSC simplyRNA extraction kit | Promega | AS1340 |
| Endo-free maxiprep | Quiagen | 12362 |
| M-MLV enzyme | Invitrogen | 28025013 |
| IFNγ ELISA KIT | BD OptEIA | 551866 |
| CD8 + T cell Isolation Kit, human | Miltenyi | 130-096-495 |
| Experimental moles: Cell lines | ||
| PN-299-L | Dr. Amaia Lujambio Icahn School of Medicien at Mount Sinai10 |
N/A |
| Experimental models: Organisms/strains | ||
| Mouse WT: C57BL/6 | Envigo | C57BL/6NHsd |
| Mouse Pmel1 (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J) | Jackson Laboratory | Strain: 00502324 |
| Mouse (OT-1 C57BL/6-Tg(TcraTcrb)1100Mjb/J) www.jax.org/strain/003831"/> | Jackson Laboratory | Strain: 003831 |
| CD2-RFP | Dr. Mark Coles University of York24 |
N/A |
| CD2-RFP x Pmel-1 | In house | N/A |
| Oligonucleotides | ||
| Primers for c-myc (FW 5′GGATTCTCTGCTCTCCT CGAC- and RV 5′-GATGTGTGGAGACGTGGCAC) |
Sigma-Aldrich | N/A |
| Primers for human gp100 (FW 5′AGTGAGCGAAG TGATGGGAA- and RV 5′- TCTCTAGCGGTTGTC TCCAC) |
Sigma-Aldrich | N/A |
| Recombinant DNA | ||
| pT3-EFIa-MYC-IRES-luciferase | Dr. Amaia Lujambio Icahn School of Medicien at Mount Sinai10 |
RRID:Addgene_129775 |
| pT3-c-myc | Dr. Amaia Lujambio Icahn School of Medicien at Mount Sinai10 |
N/A |
| pCMV-SB13 | Dr. Amaia Lujambio Icahn School of Medicien at Mount Sinai10 |
N/A |
| Px330-sg-p53 | Dr. Amaia Lujambio Icahn School of Medicien at Mount Sinai10 |
N/A |
| pT4-EGFP | This paper | N/A |
| pT4-gp100 | This paper | N/A |
| Software and algorithms | ||
| Prism 8 | GraphPad Prism | https://www.graphpad.com; RRID:SCR_002798 |
| Imaris 9 | Bitplane |
http://www.bitplane.com/imaris/imaris; RRID:SCR_007370 |
| CytExpert Software | Beckman Coulter |
https://www.mybeckman.com.br/flow-cytometry/research-flow-cytometers/cytoflex/software; RRID:SCR_017217 |
| M3 Vision | Biospace labs | https://biospacelab.com/products/#software |
| In-Form software (2.4.8) | Akoya Biosciences | https://www.akoyabio.com/phenoimager/software/inform-tissue-finder/; RRID:SCR_019155 |
| QuPath | https://qupath.github.io/; RRID: SCR_018257 | |
| Fiji | ImageJ | https://imagej.net/; RRID:SCR_002285 |
Resource availability
Lead contact
Correspondence and request for materials should be addressed to the lead contact for resources, Dr. Ignacio Melero (imelero@unav.es).
Materials availability
Plasmids generated in this study will be made available from the lead contact upon request.
Experimental model and subject details
Animal models
Mice were group (6 animals per cage) open-system housed under pathogen-free conditions at the animal facility of the Center for Applied Medical Research (CIMA, Pamplona, Spain). Five to seven-week-old male and female mice were used for all the in vivo experiments. C57BL/6N were purchased from Envigo (Barcelona, Spain). hCD2dsRed (named CD2-RFP throughout the paper) mice were a kindly gifted from Mark Coles from the University of York,42 OT1 (C57BL/6-Tg(TcraTcrb)1100Mjb/J), pmel-1 (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J)25 and pmel-1x CD2RFP mice were bred in our facilities (CIMA, Pamplona, Spain) in a closed system, one male and three female per cage. Animal were moved to a new cage separated from their parents when they were three weeks old and are placed in an open system, 6 animals per cage. All animal procedures were approved by the animal experimentation ethics committee of the regional government of Navarra (protocol 108-19 and 014-21).
Cell lines
The cell line PN-299-L (kindly provided by A. Lujambio, Mount Sinai, New York) was used for a subcutaneous tumor model.This cell line was derived from an HCC induced by the hydrodynamic transfection of the liver with the plasmids pT3-EF1a-MYC-IRES-luciferase-OS (MYC-lucOS), pT3-N90-CTNNB1 (CTNNB1) and the SB13 transposase–encoding plasmid as described by Ruiz de Galarreta et al.10 The cell line was cultured in RPMI 1640 medium with Glutamax (Gibco 61870044) and 10% decomplemented fetal bovine serum. Cells were cultured at 37°C and 5% of CO.2
Method details
In vivo treatments and depletions
Anti-CTLA-4 (9D9), anti-PD-1 (RMP1-14), anti-CD137 (3H3), anti-CD4 (GK1.5) and anti CD8β (53-5.8) were obtained from BioXcell. RatIgG was used as a control and was also obtained from BioXcell (BE0094). Human recombinant IL-2 (Aldesleukin, Clinigen) was used for treatments.
Antibodies were administered to the mice intraperitoneally, at a dose of 100μg per mouse in 100 μL of PBS solution at the indicated points. IL-2 was administered intraperitoneally in cycles of 3 days, at a dose of 105IU/mouse every 12h (6 doses each cycle).
To induce subcutaneous tumors in mice 2 × 106 cells in 100μL of PBS were injected subcutaneously to anesthetized mice. Treatments started on day 7 and tumors were measured with and electronic caliper twice a week. Area of the tumors was calculated assuming a square fit, multiplying two perpendicular dimensions. Mice were sacrificed when tumors reach 225mm2 area.
Plasmids and hydrodynamic tail-vein injection
The pT3-c-myc-luc, (pT3-EFIa-MYC-IRES-luciferase, Plasmid #129775 Addgene), pT3-c-myc, pCMV-SB13 and pgRNAp53-Cas9 (px330-sg-p53) plasmids were kindly provided by Dr. Lujambio (Icahn School of Medicine at Mount Sinai, New York, NY).10 Briefly, the pT3-EF1a-MYC-IRES-luciferase (MYC-luc) vector was constructed inserting the the “IRES-luciferase” sequence PCR-amplified from pMSCV-IRESluciferase into the linearized pT3-EF1a-MYC vector opened with PmeI restriction enzyme. For the px330-sg-p53 plasmid, the px330 vector (Addgene plasmid # 42230) was digested with BbsI and ligated with annealed oligonucleotides of the p53 guide.
TpT4-EGFP was cloned using the vector pT4/HB (Plasmid #108352 Addgene18) and the EGFP insert was included in the HindIII/XbaI site. The EGFP insert was formed by a CMV Enhacer/CAG Promotor region, a 5 ‘ UTR Polylinker, a KOZAK sequence (GCCGCCACC), the EGFP ORF, a 3’ UTR Polylinker, an h-betaGlobin 3′ UTR (x2) sequence and an SV40 polyA signal. The pT4-gp100 design was similar, including the human melanosomal protein ORF between the SpeI/BsiWI targets. The insert synthesis and the cloning were performed by Genescript. Plasmid productions were made using Endo-Free Maxi or Mega kits from Qiagen following the manufacturer's instructions. A volume of 2 mL of saline dilution containing the appropriate plasmid concentration was hydrodynamically injected through the vein tail with a 27-G needle at a rate of 0.4 mL/s.17 Each mouse received 12μg of pT3-c-myc-luc, 2.5μg of pCMV-SB13 and 10μg of pgRNAp53-Cas9, pT4-EGFP and pT4-gp100.
RNA isolation and quantitative RT-PCR
Total RNA was purified from livers using the Maxwell RSC simplyRNA extraction kit (Promega), according to the manufacturer’s instructions and subsequently retrotranscribed into cDNA using M-MLV enzyme (Invitrogen). Real-time PCR reactions were performed in a Bio-Rad CFX qPCR system with customized primers for c-myc (FW 5′GGATTCTCTGCTCTCCTCGAC- and RV 5′-GATGTGTGGAGACGTGGCAC) and gp100 (FW 5′AGTGAGCGAAGTGATGGGAA- and RV 5′- TCTCTAGCGGTTGTCTCCAC). c-human myc gene expression was used as housekeeping.
Immunization and IFNγ determination
Customized peptides for immunization of EGFP 118–126 (DTLVNRIEL)21 and luciferase 264–272 (LMYRFEEEL)20 were purchased from Genescript. Human gp100 peptide KVPRNQDWL was obtained also from Genescript (Cat. No. RP20344). SSPQGSPEPL 229–23819 for “short” c-MYC and 201–240 DSSSPKSCASQDSSAFSPSSDSLLSSTESSPQGSPEPLVL for “long” c-MYC were custom purchased from Thermo-Scientific.
To induce the immunization, mice were inoculated subcutaneously with 80μg of the different peptides along with 100μL of incomplete Freund adjuvant (BD) and 50 μg of poly I:C (Sigma) in a total volume of 200μL, once a week for three weeks. 21 days after the first inoculation, spleens were harvested and splenocytes were cultured at a concentration of 4 × 106 cells/mL with 10 μg/mL of the corresponding peptide for 72h. Then, supernatants were collected and stored at −20°C until the IFNɣ quantification that was performed by ELISA using the BD OptEIA 551866 kit following the manufacturer instructions. ex vivo restimulation of splenocytes from mice bearing tumors was performed also at a concentration of 4 × 106 cells/mL with 10 μg/mL of human gp100 peptide for 72h.
EGFP visualization
EGFP expression in tumors was verified by epifluorescence. Tumor were illuminated with an SFA Light Base Nightsea lamp that emits 440–460 nm led light and visualized through a 500 nm filter. Also, tumors were mechanically disrupted to obtain a cellular suspension and expression of EGFP was verified by flow cytometry with a Cytoflex S (Beckman Coulter) cytometer.
Bioluminescence
Luciferase was quantified by bioluminescence at different time points. For this purpose, mice were anesthetized using ketamine (Richter pharma) and xylazine (Rompun, Bayer) and injected with 100μl of D-luciferine i.p. (Xenogen) at a concentration of 30 mg/mL. Mice were placed in the imaging chamber of a PhotonIMAGER Optima system (Biospace labs). A color-scale photograph of the animals was acquired, followed by a bioluminescent acquisition. Regions of interest were drawn over the animal image, as well as over regions of no signal, which were used as background readings. Light intensity was quantified using photons/s. The color-scale photograph and data images from all studies were superimposed using M3 Vision software (Biospace labs).
Activation of pmel-1 and OT-I lymphocytes
Spleens and lymph nodes from pmel-1 and OT-1 transgenic mice were mechanically disrupted to obtain a cell suspension. OT-1 splenocytes and lymph node cells were activated during 48 h with 1 ng/mL of OVA 257–264 peptide (Invivogen). The pmel-1 splenocytes and lymph nodes cells were activated for 48 h with 100 ng/mL of hgp100 peptide (Genscript). After activation, 6 × 106 lymphocytes were injected per mouse in a PBS solution intravenously.
Immunohistochemistry
Immunohistochemistry was applied using the following primary antibodies: goat anti-GFP (Novus Biologicals, NB100-1770), rabbit anti-melanoma gp100 (Abcam, ab137078) and rabbit anti-cleaved caspase-3 (Cell Signaling, 9661). Antigen retrieval was applied for 30 min at 95°C in 0.01 M Tris-1 mM EDTA, pH = 9. For goat primary antibody (GFP), sections were first incubated with rabbit anti-goat (ThermoFisher, 31732), and then the EnVision+ System (Dako, K400311-2) was used in all cases according to the manufacturer instructions.
Detection and quantification of EGFP+ and GP100+ cells in brightfield images of serial histological sections were carried out using an in-house plugin developed for Fiji/ImageJ, an open-source Java-based image processing software. The plugin was developed by the Imaging Platform of the Center for Applied Medical Research (CIMA). The two images were first aligned by image registration,43 hematoxylin and DAB channels were then separated by means of a color deconvolution plugin,44 and positive cells for each marker were then detected by segmenting nuclei in hematoxylin and looking for positive DAB areas in each segmented cell. Double-positive cells were finally retrieved by using an AND logical operation between the two segmentation masks.
Assessment of apoptosis in malignant cells
Single cell suspensions from excised livers containing tumors from mice receiving treatments or control antibodies were obtained by enzymatic digestion (Collagenase I CO130, Sigma-Aldrich and DNAse I 260913, Calbiochem) and mechanic disaggregation. EGFP+ cells were sorted to over 99% purity using BD FACSAria sorter. Sorted cells were intracellularly stained with a mAb that recognizes cleaved caspase 3 (clone C92-605, Bioscience Pharmingen) and analyzed in a Cytoflex S flow cytometer (Beckman Coulter). Percentages of EGFP+ cells stained for activated caspase 3 were estimated.
Multiplexed immunofluorescence
A seven-color multiplex immunofluorescence panel based on tyramide signal amplification was used for simultaneous detection of CD3 (T cells), CD8 (cytotoxic T lymphocytes), CD4 and Foxp3 (Regulatory T cells, Tregs), CD11b (myeloid cells), Ki67 (cell proliferative activity), and diamidino-2-phenylindole (DAPI) on tumor sections from formalin-fixed paraffin-embedded (FFPE) samples. The validation pipeline for the multiplex immunofluorescence protocol has been previously described by our group.45 At the end of the protocol, nuclei were counterstained with spectral DAPI (Akoya Biosciences) and sections were mounted with Faramount Aqueous Mounting Medium (Dako).
Imaging, spectral Unmixing and Phenotyping
Multiplexed immunofluorescence slides were scanned on a Vectra-Polaris Automated Quantitative Pathology Imaging System (Akoya Biosciences) as described previously.46,47 Whole tissue present in a single FFPE tissue section was imaged, spectrally unmixed and exported as a component TIF image tile using Akoya Biosciences’ In-form software (version 2.4.8). Component TIF image tiles were then imported into the open-source digital pathology software QuPath version 0.2.0-m9 and stitched together using the x-y coordinates to create a new pyramidal TIF file for image analysis. Image analysis was performed on the whole tissue sections.
Cell segmentation was performed in the whole multispectral image using QuPath software version 0.2.0-m9.48 Nuclear detection was carried out on the DAPI channel using a custom, unsupervised watershed algorithm as described previously.46 A random trees algorithm classifier was generated to further subclassify the cells as CD3+, CD8+, CD4+ Foxp3+, CD11b+, and Ki67+. Cells close to the border of the images were removed to reduce the risk of artifacts. CD4+ T-cells were defined as CD3+ CD8−. Cells negative for these markers were defined as “other cell types”. Measurements were calculated as cell densities (cells/mm2).
Intravital microscopy
For the intravital microscopy of tumors in the liver, CD2-RFP mice or WT mice were used to generate liver tumors by hydrodynamic injection of the following plasmids (pT3-c-myc, pT4-EGFP, pT4-gp100, pgRNAp53-Cas9 and pCMV-SB13). WT mice were transferred with resting 5 × 106 RFP+ pmel-1immunomagnetically isolated CD8+ T cells (Miltenyi kit ref. 130-096-495) i.v. transferred on day 32. On day 35 treatment of anti-PD-1+ anti-CTLA-4 mAbs or RatIgG control was administered, followed by a 3-day cycle of IL-2 on days 39–41. Intravital confocal microscopy was performed on day 43–44 after hydrodynamic plasmid injection as previously described.49 Briefly, the liver was surgically exposed and isolated from respiratory movements by separating the rib cage by pulling with a thread and keeping the liver tissue adhered to the coverslip with the help of wet pieces of paper. The temperature of the mice was maintained with a rectal probe connected to a heating blanket (Kemp) and mice were kept asleep under Isoflurane anesthesia (2%). Mice were placed on a custom-built stage and imaged with an LSM880 inverted microscope (Zeiss) equipped with a 25X water immersion objective (NA, 0.8). Imaging sessions took from 2 to 4 h per mice and time-lapse acquisitions lasted from 30 min to 2 h with frames taken every 2 min. Several tumor areas were imaged per session and mice. Time-lapse videos were analyzed using the IMARIS (Bitplane) software. RFP+ T cells were segmented using the spots tool and tracked manually over time. Individual cell motility parameters were generated from such tracks. Speed (track length/time), chemotactic index or straightness (Track displacement length/track length) were measured. Time-lapse videos were generated in IMARIS software and edited with Final Cut Pro (Apple software).
Quantification and statistical analysis
GraphPad Prism V.8 (LA Jolla, California, USA) was used for appropriate statistical analysis. Survival data were analyzed with log rank (Mantel-Cox) test and differences between means were analyzed using the U-Mann Whitney test, Kruskal-Wallis test followed by an uncorrected Dunn's test or repeated measures ANOVA followed by Tukey's test. For statistical comparisons of bioluminescence data, logarithmic transformations were used. p-values are indicated in the figures.
Acknowledgments
We are grateful to the support provided by Dr. Diego Alignani (flow cytometry unit), Dr. Xabier Morales and Dr. Mikel Ariz (imaging facility), Dr. Laura Guembe (morphology platform), and Eneko Elizalde and Elena Ciordia (animal experimentation service). Dr. Belen Palencia and Esther Guirado are acknowledged for project management. This project has been supported by PID2020-112892RB funded by MCIN/AEI/10.13039/501100011033 and SAF2017-83267-C2-1-R funded by MCIN/AEI/10.13039/501100011033 and by FEDER “Una manera de hacer Europa” to I.M. as well as Mark Foundation and Fundación Fero. This project has also received funding from Fundación de la Asociación Española Contra el Cáncer (AECC) (HR21-00083), Fundació la Marató de TV3 (488/C/2019), Fundación La Caixa, and Fundación BBVA. A.T. is supported by a Ramon y Cajal Contract (RYC2019-026406-I and PID2020-113174RA100 funded by MCIN/AEI/10.13039/501100011033 and EI FSE “invierte en tu futuro”).
Author contributions
Conception or design of the work: M.C.O., P.B., A.T., and I.M. Data collection: M.C.O., C.A., S.S.G., S.G., and I.E. Data analysis and interpretation: M.C.O., C.A., S.S.G., S.G., M.A., I.O., J.G.V., I.E., C.L., A.C., P.B., A.A., A.T., and I.M. Drafting the article: M.C.O., C.A., A.T., and I.M. Critical revision of the article: M.C.O., S.S.G., J.A., B.S., and A.T. Final approval of the version to be published: all authors.
Declaration of interests
I.M. acknowledges grants from Roche, Alligator, Genmab, BMS, AstraZeneca, Pharmamar, and Bioncotech, as well as consultancy fees from BMS, Roche, Genmab, Numab, F-Star, Biolinerx, Pierre Fabre, Sanofi, Gossamer, Alligator, AstraZeneca, and Pharmamar. B.S. received consulting fees from Adaptimmune, AstraZeneca, Bayer, BMS, BTG, Eisai, Exelixis, Eli-Lilly, IPSEN, Merck, Onxeo, Roche, and Sirtex; lecture fees from AstraZeneca, Bayer, BMS, Eisai, Eli-Lilly, Incyte, IPSEN, Roche, and Sirtex; and institutional research grants from BMS and Sirtex.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: April 10, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101009.
Supplemental information
Data and code availability
-
•
Data statement: This study did not generate any unique datasets.
-
•
Code Statement: This study did not generate any unique code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon reasonable request.
References
- 1.Sangro B., Sarobe P., Hervás-Stubbs S., Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021;18:525–543. doi: 10.1038/s41575-021-00438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greten T.F., Abou-Alfa G.K., Cheng A.L., Duffy A.G., El-Khoueiry A.B., Finn R.S., Galle P.R., Goyal L., He A.R., Kaseb A.O., et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immunotherapy for the treatment of hepatocellular carcinoma. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finn R.S., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., Kudo M., Breder V., Merle P., Kaseb A.O., et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 4.Abou-Alfa G.K., Lau G., Kudo M., Chan S.L., Kelley R.K., Furuse J., Sukeepaisarnjaroen W., Kang Y.-K., Van Dao T., De Toni E.N., et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid. 2022;1 doi: 10.1056/EVIDoa2100070. [DOI] [PubMed] [Google Scholar]
- 5.El-Khoueiry A.B., Sangro B., Yau T., Crocenzi T.S., Kudo M., Hsu C., Kim T.-Y., Choo S.-P., Trojan J., Welling T.H., et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finn R.S., Ryoo B.Y., Merle P., Kudo M., Bouattour M., Lim H.Y., Breder V., Edeline J., Chao Y., Ogasawara S., et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J. Clin. Oncol. 2020;38:193–202. doi: 10.1200/JCO.19.01307. [DOI] [PubMed] [Google Scholar]
- 7.Yau T., Kang Y.K., Kim T.Y., El-Khoueiry A.B., Santoro A., Sangro B., Melero I., Kudo M., Hou M.-M., Matilla A., et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol. 2020;6 doi: 10.1001/jamaoncol.2020.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu A.X., Abbas A.R., de Galarreta M.R., Guan Y., Lu S., Koeppen H., Zhang W., Hsu C.-H., He A.R., Ryoo B.Y., et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022;28:1599–1611. doi: 10.1038/s41591-022-01868-2. [DOI] [PubMed] [Google Scholar]
- 9.Sangro B., Melero I., Wadhawan S., Finn R.S., Abou-Alfa G.K., Cheng A.-L., Yau T., Furuse J., Park J.-W., Boyd Z., et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020;73:1460–1469. doi: 10.1016/j.jhep.2020.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruiz de Galarreta M., Bresnahan E., Molina-Sánchez P., Lindblad K.E., Maier B., Sia D., Puigvehi M., Miguela V., Casanova-Acebes M., Dhainaut M., et al. β-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9:1124–1141. doi: 10.1158/2159-8290.CD-19-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molina-Sánchez P., Ruiz de Galarreta M., Yao M.A., Lindblad K.E., Bresnahan E., Bitterman E., Martin T.C., Rubenstein T., Nie K., Golas J., et al. Cooperation between distinct cancer driver genes underlies intertumor heterogeneity in hepatocellular carcinoma. Gastroenterology. 2020;159:2203–2220.e14. doi: 10.1053/j.gastro.2020.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berraondo P., Ochoa M.C., Olivera I., Melero I. Immune desertic landscapes in hepatocellular carcinoma shaped by β-catenin activation. Cancer Discov. 2019;9:1003–1005. doi: 10.1158/2159-8290.CD-19-0696. [DOI] [PubMed] [Google Scholar]
- 13.Sanmamed M.F., Berraondo P., Rodriguez-Ruiz M.E., Melero I. Charting roadmaps towards novel and safe synergistic immunotherapy combinations. Nat. Cancer. 2022;3:665–680. doi: 10.1038/s43018-022-00401-1. [DOI] [PubMed] [Google Scholar]
- 14.Hill W., Caswell D.R., Swanton C. Capturing cancer evolution using genetically engineered mouse models (GEMMs) Trends Cell Biol. 2021;31:1007–1018. doi: 10.1016/j.tcb.2021.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Bailey C., Black J.R.M., Reading J.L., Litchfield K., Turajlic S., McGranahan N., Jamal-Hanjani M., Swanton C. Tracking cancer evolution through the disease course. Cancer Discov. 2021;11:916–932. doi: 10.1158/2159-8290.CD-20-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong L.-Q., Peng L.-H., Ma L.-J., Liu D.-B., Zhang S., Luo S.-Z., Rao J.-H., Zhu H.-W., Yang S.-X., Xi S.-J., et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J. Hepatol. 2020;72:896–908. doi: 10.1016/j.jhep.2019.12.014. [DOI] [PubMed] [Google Scholar]
- 17.Liu F., Song Y., Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y., Pryputniewicz-Dobrinska D., Nagy E.É., Kaufman C.D., Singh M., Yant S., Wang J., Dalda A., Kay M.A., Ivics Z., Izsvák Z. Regulated complex assembly safeguards the fidelity of Sleeping Beauty transposition. Nucleic Acids Res. 2017;45:311–326. doi: 10.1093/nar/gkw1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Helm F., Kammertoens T., Lehmann F.M., Wilke A., Bruns H., Mautner J., Bornkamm G.W., Gerbitz A. Targeting c-MYC with T-cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0077375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Limberis M.P., Bell C.L., Wilson J.M. Identification of the murine firefly luciferase-specific CD8 T-cell epitopes. Gene Ther. 2009;16:441–447. doi: 10.1038/gt.2008.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han W.G.H., Unger W.W.J., Wauben M.H.M. Identification of the immunodominant CTL epitope of EGFP in C57BL/6 mice. Gene Ther. 2008;15:700–701. doi: 10.1038/sj.gt.3303104. [DOI] [PubMed] [Google Scholar]
- 22.Etxeberria I., Glez-Vaz J., Teijeira Á., Melero I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open. 2020;4 doi: 10.1136/esmoopen-2020-000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wrangle J.M., Patterson A., Johnson C.B., Neitzke D.J., Mehrotra S., Denlinger C.E., Paulos C.M., Li Z., Cole D.J., Rubinstein M.P. IL-2 and beyond in cancer immunotherapy. J. Interferon Cytokine Res. 2018;38:45–68. doi: 10.1089/jir.2017.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lasarte-Cia A., Lozano T., Cano D., Martín-Otal C., Navarro F., Gorraiz M., Casares N., Vivas I., Lasarte J.J. Intratumoral STING agonist injection combined with irreversible electroporation delays tumor growth in a model of hepatocarcinoma. BioMed Res. Int. 2021;2021 doi: 10.1155/2021/8852233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Overwijk W.W., Theoret M.R., Finkelstein S.E., Surman D.R., de Jong L.A., Vyth-Dreese F.A., Dellemijn T.A., Antony P.A., Spiess P.J., Palmer D.C., et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ney J.T., Schmidt T., Kurts C., Zhou Q., Eckert D., Felsher D.W., Schorle H., Knolle P., Tüting T., Barchet W., et al. Autochthonous liver tumors induce systemic T cell tolerance associated with T cell receptor down-modulation. Hepatology. 2009;49:471–481. doi: 10.1002/hep.22652. [DOI] [PubMed] [Google Scholar]
- 27.Morales-Kastresana A., Sanmamed M.F., Rodriguez I., Palazon A., Martinez-Forero I., Labiano S., Hervas-Stubbs S., Sangro B., Ochoa C., Rouzaut A., et al. Combined immunostimulatory monoclonal antibodies extend survival in an aggressive transgenic hepatocellular carcinoma mouse model. Clin. Cancer Res. 2013;19:6151–6162. doi: 10.1158/1078-0432.CCR-13-1189. [DOI] [PubMed] [Google Scholar]
- 28.Moon H., Park H., Ro S.W. c-Myc-driven Hepatocarcinogenesis. Anticancer Res. 2021;41:4937–4946. doi: 10.21873/anticanres.15307. [DOI] [PubMed] [Google Scholar]
- 29.Melero I., Shuford W.W., Newby S.A., Aruffo A., Ledbetter J.A., Hellström K.E., Mittler R.S., Chen L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997;3:682–685. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 30.Hernandez R., Põder J., LaPorte K.M., Malek T.R. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat. Rev. Immunol. 2022;22:614–628. doi: 10.1038/s41577-022-00680-w. [DOI] [PubMed] [Google Scholar]
- 31.Blattman J.N., Grayson J.M., Wherry E.J., Kaech S.M., Smith K.A., Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 2003;9:540–547. doi: 10.1038/nm866. [DOI] [PubMed] [Google Scholar]
- 32.Diab A., Tannir N.M., Bentebibel S.E., Hwu P., Papadimitrakopoulou V., Haymaker C., Kluger H.M., Gettinger S.N., Sznol M., Tykodi S.S., et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced solid tumors: phase I dose-escalation study of safety, efficacy, and immune activation (PIVOT-02) Cancer Discov. 2020;10:1158–1173. doi: 10.1158/2159-8290.CD-19-1510. [DOI] [PubMed] [Google Scholar]
- 33.Rosen D.B., Kvarnhammar A.M., Laufer B., Knappe T., Karlsson J.J., Hong E., Lee Y.-C., Thakar D., Zúñiga L.A., Bang K., et al. TransCon IL-2 β/γ: a novel long-acting prodrug with sustained release of an IL-2Rβ/γ-selective IL-2 variant with improved pharmacokinetics and potent activation of cytotoxic immune cells for the treatment of cancer. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2022-004991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li D., Li N., Zhang Y.F., Fu H., Feng M., Schneider D., Su L., Wu X., Zhou J., Mackay S., et al. Persistent polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology. 2020;158:2250–2265.e20. doi: 10.1053/j.gastro.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alspach E., Lussier D.M., Miceli A.P., Kizhvatov I., DuPage M., Luoma A.M., Meng W., Lichti C.F., Esaulova E., Vomund A.N., et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696–701. doi: 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reiberger T., Chen Y., Ramjiawan R.R., Hato T., Fan C., Samuel R., Roberge S., Huang P., Lauwers G.Y., Zhu A.X., et al. An orthotopic mouse model of hepatocellular carcinoma with underlying liver cirrhosis. Nat. Protoc. 2015;10:1264–1274. doi: 10.1038/nprot.2015.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfister D., Núñez N.G., Pinyol R., Govaere O., Pinter M., Szydlowska M., Gupta R., Qiu M., Deczkowska A., Weiner A., et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592:450–456. doi: 10.1038/s41586-021-03362-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiu D.K.-C., Yuen V.W.-H., Cheu J.W.-S., Wei L.L., Ting V., Fehlings M., Sumatoh H., Nardin A., Newell E.W., Ng I.O.-L., et al. Hepatocellular carcinoma cells up-regulate PVRL1, stabilizing PVR and inhibiting the cytotoxic T-cell response via TIGIT to mediate tumor resistance to PD1 inhibitors in mice. Gastroenterology. 2020;159:609–623. doi: 10.1053/j.gastro.2020.03.074. [DOI] [PubMed] [Google Scholar]
- 39.Crispe I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009;27:147–163. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 40.Buschow C., Charo J., Anders K., Loddenkemper C., Jukica A., Alsamah W., Perez C., Willimsky G., Blankenstein T. In vivo imaging of an inducible oncogenic tumor antigen visualizes tumor progression and predicts CTL tolerance. J. Immunol. 2010;184:2930–2938. doi: 10.4049/jimmunol.0900893. [DOI] [PubMed] [Google Scholar]
- 41.Yu J., Green M.D., Li S., Sun Y., Journey S.N., Choi J.E., Rizvi S.M., Qin A., Waninger J.J., Lang X., et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 2021;27:152–164. doi: 10.1038/s41591-020-1131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veiga-Fernandes H., Coles M.C., Foster K.E., Patel A., Williams A., Natarajan D., Barlow A., Pachnis V., Kioussis D. Tyrosine kinase receptor RET is a key regulator of Peyer’s patch organogenesis. Nature. 2007;446:547–551. doi: 10.1038/nature05597. [DOI] [PubMed] [Google Scholar]
- 43.Lowe D.G. Distinctive image features from scale-invariant keypoints. Int. J. Comput. Vis. 2004;60:91–110. doi: 10.1023/b:visi.0000029664.99615.94. [DOI] [Google Scholar]
- 44.Ruifrok A.C., Johnston D.A. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 2001;23:291–299. doi: 10.1038/nature05597. [DOI] [PubMed] [Google Scholar]
- 45.Alvarez M., Molina C., De Andrea C.E., Fernandez-Sendin M., Villalba M., Gonzalez-Gomariz J., Ochoa M.C., Teijeira A., Glez-Vaz J., Aranda F., et al. Intratumoral co-injection of the poly I:C-derivative BO-112 and a STING agonist synergize to achieve local and distant anti-tumor efficacy. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-002953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abengozar-Muela M., Esparza M.V., Garcia-Ros D., Vásquez C.E., Echeveste J.I., Idoate M.A., Lozano M.D., Melero I., de Andrea C.E. Diverse immune environments in human lung tuberculosis granulomas assessed by quantitative multiplexed immunofluorescence. Mod. Pathol. 2020;33:2507–2519. doi: 10.1038/s41379-020-0600-6. [DOI] [PubMed] [Google Scholar]
- 47.Martinez-Valbuena I., Valenti-Azcarate R., Amat-Villegas I., Riverol M., Marcilla I., de Andrea C.E., Sánchez-Arias J.A., Del Mar Carmona-Abellan M., Marti G., Erro M.-E., et al. Amylin as a potential link between type 2 diabetes and alzheimer disease. Ann. Neurol. 2019;86:539–551. doi: 10.1002/ana.25570. [DOI] [PubMed] [Google Scholar]
- 48.Bankhead P., Fernández J.A., McArt D.G., Boyle D.P., Li G., Loughrey M.B., Irwin G.W., Harkin D.P., James J.A., McQuaid S., et al. Integrated tumor identification and automated scoring minimizes pathologist involvement and provides new insights to key biomarkers in breast cancer. Lab. Invest. 2018;98:15–26. doi: 10.1038/labinvest.2017.131. [DOI] [PubMed] [Google Scholar]
- 49.Teijeira Á., Garasa S., Gato M., Alfaro C., Migueliz I., Cirella A., de Andrea C., Ochoa M.C., Otano I., Etxeberria I., et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52:856–871.e8. doi: 10.1016/j.immuni.2020.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Multifocal HCC tumors were generated by hydrodynamic injection in CD2-RFP mice. Mice were treated with anti-PD-1+ anti-CTLA-4 + IL-2 according to the treatment scheme represented in Figure 2 starting on day 35. Livers were observed by intravital imaging on day 43. Mice were injected with anti-CD31 fluorescent antibodies right before surgery to visualize blood vessels (blue). Videos show endogenous T cell infiltration (RFP, red) into tumor lesions (EGFP, green). Video is 600× accelerated. Related to Figure 7.
Multifocal HCC tumors were generated by hydrodynamic injection in wild-type mice, and 4 weeks later, mice were transferred with 5 × 106 pmel-1 RFP+ CD8 T cells (RFP, red). Mice were treated with anti-PD-1 + anti-CTLA-4 + IL-2 according to the treatment scheme represented in Figure 2 starting on day 35. Intravital microscopy was performed at 6 weeks after hydrodynamic injection. Mice were injected with anti-CD31 fluorescent antibodies right before surgery to visualize blood vessels (blue). Videos show the dynamics of anti-gp100 CD8+ T cells (RFP, red) into tumor lesions (EGFP, green). Videos are 600× accelerated. Related to Figure 7.
Data Availability Statement
-
•
Data statement: This study did not generate any unique datasets.
-
•
Code Statement: This study did not generate any unique code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon reasonable request.