

Summary
Enzalutamide (ENZA), a second-generation androgen receptor antagonist, has significantly increased progression-free and overall survival of patients with metastatic prostate cancer (PCa). However, resistance remains a prominent obstacle in treatment. Utilizing a kinome-wide CRISPR-Cas9 knockout screen, we identified casein kinase 1α (CK1α) as a therapeutic target to overcome ENZA resistance. Depletion or pharmacologic inhibition of CK1α enhanced ENZA efficacy in ENZA-resistant cells and patient-derived xenografts. Mechanistically, CK1α phosphorylates the serine residue S1270 and modulates the protein abundance of ataxia telangiectasia mutated (ATM), a primary initiator of DNA double-strand break (DSB)-response signaling, which is compromised in ENZA-resistant cells and patients. Inhibition of CK1α stabilizes ATM, resulting in the restoration of DSB signaling, and thus increases ENZA-induced cell death and growth arrest. Our study details a therapeutic approach for ENZA-resistant PCa and characterizes a particular perspective for the function of CK1α in the regulation of DNA-damage response.
Keywords: prostate cancer, enzalutamide, CRISPR screening, CK1α, ATM
Graphical abstract
Highlights
-
•
CRISPR screen identifies CK1α as a target to overcome enzalutamide resistance
-
•
CRISPR screen and RNA-seq illustrate DSB signaling involvement in enzalutamide resistance
-
•
ATM is a prognostic biomarker to predict the clinical outcome of enzalutamide
-
•
CK1α-dependent phosphorylation of ATM primes it for proteasomal degradation
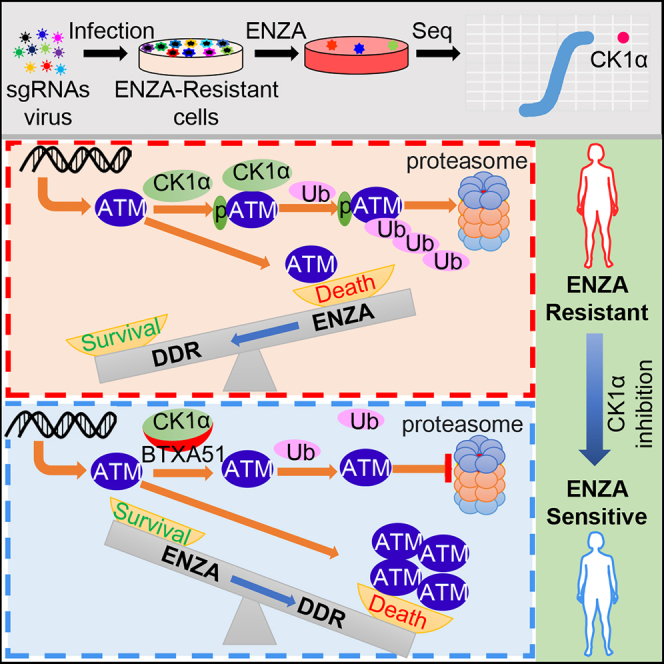
Liu et al. identify CK1α as a target to overcome enzalutamide resistance of prostate cancer. CK1α phosphorylates and modulates the protein abundance of ataxia telangiectasia mutated (ATM). Inhibition of CK1α stabilizes ATM, resulting in the restoration of double-strand break signaling, and thus increases enzalutamide-induced cell death and growth arrest.
Introduction
Prostate cancer (PCa) is the most common non-cutaneous malignancy among males and is also the leading cause of cancer-related death in the world.1 Androgen deprivation therapy is the first line of therapy for metastatic PCa, but most patients will eventually progress to castration-resistant PCa (CRPC). The 5-year survival rate of men with metastatic CRPC is only 31%,2 highlighting the urgent need for development of other therapies. Enzalutamide (ENZA), a potent second-generation androgen receptor (AR) antagonist, has been approved for metastatic CRPC therapy and has shown remarkable improvement in patient survival outcomes.3 Importantly, a recent large-scale randomized trial established that ENZA could delay the time to metastasis and thereby prolong overall survival of patients with non-metastatic CRPC.4 Nevertheless, most patients ultimately progress to ENZA resistance, which has unreliable and limited treatment options. Therefore, it is critical to investigate other treatments for ENZA-resistant PCa.
Various studies have defined characterization of ENZA resistance, which are divided into two general categories: AR-dependent and AR-independent mechanisms. AR-dependent ENZA-resistant mechanisms include restoration of AR activity due to AR amplification, AR point mutations that convert ENZA from an antagonist to an agonist, and expression of AR splice variants that are constitutively active because of the lack of the ligand-binding domain.5 AR-independent ENZA-resistant mechanisms are defined as bypassing AR signaling via other hormone nuclear receptors, such as glucocorticoid receptor,6 or acquiring lineage plasticity characteristics by expressing neuroendocrine and stem cell-associated genes through epigenetic mechanisms.7,8 However, few therapeutic targets have moved forward to clinical trial or treatment.
Clustered regularly interspaced short palindrome repeats associated protein 9 (CRISPR-Cas9) technology is an efficient approach for gene editing, whereby Cas9, an endonuclease, cuts DNA at a location specified by a guide RNA. Loss-of-function pooled library screens using CRISPR-Cas9 have displayed a powerful ability to identify genes related to drug response.9,10 By use of an unbiased kinome-wide CRISPR-Cas9 library screen, we discovered casein kinase 1α (CK1α) was a top candidate whose knockout sensitized cells to ENZA treatment. CK1α participates in extensive regulatory roles in numerous cellular processes including cell metabolism and differentiation in development, autophagy, chromosome segregation, cell cycle, apoptosis, immune response, and neurodegeneration.11 Mutations and alterations of CK1α are detected in various tumor entities including acute myeloid leukemia (AML), lung, esophageal, urothelial, kidney, breast, pancreas, and ovarian cancers.11,12,13 Moreover, CK1α expression is associated with poorer overall survival in cancer patients.14 Additionally, CK1α has also been reported to be implicated in acquired drug resistance to erlotinib in non-small cell lung cancer.15 These findings suggest that CK1α is a conditionally essential oncogene and a potential target for cancer therapy. However, whether and how CK1α is involved in PCa development and ENZA treatment response is ambiguous.
Results
A kinome-wide CRISPR-Cas9 knockout screen identifies CK1α as a therapeutic target to overcome ENZA resistance
Protein kinases regulate nearly all aspects of cell life, and their alterations or mutations contribute to cancer initiation and progression. Consequently, kinases have been major targets for cancer therapy. So far, 76 kinase inhibitors have been approved for use in the clinic and many others are in clinical trials.16 To identify potential kinases whose inhibition might overcome ENZA resistance in PCa treatment, we performed a pooled kinome-wide CRISPR-Cas9 knockout screen in an intrinsic ENZA-resistant PCa cell line 22Rv1 cells using a lentiviral single guide RNA (sgRNA) library (Figure 1A) that contained 3,052 unique sgRNAs targeting 763 human kinases.
Figure 1.

A kinome-wide CRISPR-Cas9 knockout screen identifies CK1α as a therapeutic target to overcome ENZA resistance
(A) Workflow of the kinome-wide CRISPR-Cas9 screen. MOI, multiplicity of infection; 22Rv1-Cas9, 22Rv1 cell line stably expressing Cas9.
(B) Workflow of CRISPR screen data analysis with MAGeCK.
(C) Scatterplot showing gene differential beta score (subtracting the control beta score from the treatment beta score) versus gene beta score in control samples. CSNK1A1, MTOR, and PLK1 are the top three negatively selected genes that mediated sensitivity to ENZA. Genes in the kinome library that overlap with essential genes and non-essential genes, reported previously, are highlighted in yellow and blue, respectively.
First, we generated a stable cell line expressing Cas9 in 22Rv1 cells (22Rv1-Cas9) (Figure S1A) and then validated its efficacy of gene knockout by targeting AR-full-length with two sgRNAs (Figure S1B). We also compared the response of 22Rv1-Cas9 and parental 22Rv1 cells (22Rv1-wild type [WT]) to ENZA to ensure that expression of Cas9 did not change the sensitivity of cells to ENZA (Figure S1C). After transduction at a low multiplicity of infection (MOI = 0.3) followed by selection with puromycin, one-third of the infected cells (500× coverage) were harvested as the baseline for sequencing, and the rest of the cells were continuously cultured for additional 2 weeks to allow gene editing (Figure 1A). Cells were then randomly split and treated with ENZA or DMSO for an additional week. Next, the sgRNAs incorporated in the cells were amplified from genomic DNA and subjected to sequencing, followed by model-based analysis of genome-wide CRISPR-Cas9 knockout (MAGeCK)17 (Figure 1B). Consistent with previous CRISPR screening,17,18 multiple essential and non-essential genes identified in previous screens were also identified in our analysis (Figure 1C), validating our approach. Among the genes we identified, CSNK1A1 (coding CK1α) was the number-one candidate whose knockout enhanced the efficacy of ENZA in 22Rv1 cells (Figure 1C and Table S1). Of note, the top second and third candidates, MTOR and PLK1, respectively, have been reported to be involved in ENZA resistance in previous studies.19,20
Validation of CK1α as a target to treat ENZA-resistant PCa
To validate CK1α as an effective target to overcome ENZA resistance in PCa, we compared the cellular response to ENZA upon CK1α depletion. As indicated, CK1α depletion significantly decreased cell viability and colony formation of 22Rv1 cells upon ENZA treatment (Figures 2A and S2A). Similarly, knockdown of CK1α also dramatically enhanced the efficacy of ENZA treatment in two other acquired ENZA-resistant PCa cell lines, C4-2R (Figures 2B, S2B, and S2C) and MR49F (Figures 2C, S2B, and S2D), indicating that this finding is not cell line specific. In addition, consistent with these observations, ENZA treatment induced a significant increase of cleaved poly(ADP-ribose) polymerase (c-PARP), a marker of apoptosis, in 22Rv1 cells with CK1α depletion, but not 22Rv1-WT cells, in a dose-dependent manner (Figure 2D). Finally, overexpression of CK1α attenuated ENZA-induced elevation of c-PARP in ENZA-sensitive PCa cell line, C4-2 (Figure 2E), and induced ENZA resistance (Figures S2E and S2F).
Figure 2.

Validation of CK1α as a target to treat ENZA-resistant PCa
(A) In vitro proliferation of 22Rv1 cells expressing sgCtrl or two sgRNAs against CK1α was determined by AquaBluer assay after treatment for 72 h with the indicated concentrations of ENZA. Inset: immunoblot (IB) analysis of whole-cell lysates (WCLs) of 22Rv1 cells expressing sgCtrl or two sgRNAs against CK1α.
(B and C) C4-2R (B) or MR49F (C) cells expressing short hairpin control (shCtrl) or two shRNAs targeting CK1α were pre-treated with 100 ng mL−1 doxycycline (Dox) for 48 h to induce CK1α knockdown and then treated with ENZA for 72 h, followed by AquaBluer assay to determine cell viability. Insets: IB analysis of WCL of C4-2R (B) or MR49F (C) cells expressing shCtrl or shCK1α treated with 100 ng mL−1 Dox for 48 h.
(D) 22Rv1 cells expressing sgCtrl or sgRNA targeting CK1α, treated with different concentrations (10, 20, 40, and 80 μM) of ENZA for 24 h and subjected to IB.
(E) IB analysis of WCL derived from C4-2 cells stably expressing empty vector (EV) or FLAG-CK1α after treatment with different concentrations (10, 20, 40, and 80 μM) of ENZA for 24 h.
(F–H) In vivo 22Rv1 xenograft assay. (F) A schema showing the experiment design of in vivo 22Rv1 xenograft and treatment strategy. 22Rv1 (sgCtrl or sgCK1α#1) cells were injected into the flanks of pre-castrated male nude mice. When the tumors reached 100 mm3, the mice (n = 5 for sgCtrl, n = 6 for sgCK1α#1) were treated with vehicle or ENZA (20 mg kg−1 by oral gavage daily, 5 days on and 2 days off for 5 weeks). Tumor volume (G) and tumor weight (H) are shown as mean ± SEM.
(I–K) In vivo LuCaP 35CR PDX assay. (I) A schema showing the experimental design of in vivo LuCaP 35CR PDX. LuCaP 35CR tumor bits were implanted into the flanks of pre-castrated NSG mice. When the tumors reached 50 mm3, the mice were pre-treated with ENZA (50 mg kg−1, by oral gavage daily, 5 days on and 2 days off for 1 week) and then treated with vehicle (n = 11) or BTX-A51 (5 mg kg−1, by oral gavage, two days on and one day off, n = 10) or ENZA (20 mg kg−1, by oral gavage every 3 days, n = 11) or the combination (n = 10) for the indicated time. Tumor volume (J) is shown as mean ± SEM, and survival percentage is shown in (K).
Next, we tested these observations in an in vivo setting. Accordingly, castrated nude mice carrying xenograft tumors derived from WT 22Rv1 or 22Rv1 with CK1α depletion were subjected to treatment with ENZA (Figure 2F), followed by measurement of tumor growth. As expected, CK1α depletion (Figures S3B and S3F) increased the sensitivity of 22Rv1 xenografts to ENZA treatment compared with 22Rv1-WT, as indicated by limited tumor growth (Figures 2G and S3D) and reduced tumor weight (Figures 2H, S3A, S3E, and S3G). Furthermore, with respect to clinical relevance, inhibition of CK1α with BTX-A51, a compound targeting CK1α currently in a clinical trial (NCT04243785) to treat AML, significantly enhanced the efficacy of ENZA in ENZA-resistant patient-derived xenograft (PDX) models, LuCaP 35CR and LuCaP 77CR,21 and improved survival of the mice without apparent toxicity (Figures 2I–2K and S3I–S3N). Collectively, these results demonstrate that CK1α is a promising target to treat ENZA-resistant PCa.
CK1α regulates double-strand break (DSB)-response signaling
To dissect the underlying mechanisms of targeting CK1α to sensitize cancer cells to ENZA, we performed RNA sequencing (RNA-seq) of 22Rv1 cells upon CK1α knockout or overexpression. With gene set enrichment analysis (GSEA), we found that 287 gene sets were significantly enriched in CK1α-overexpressing cells compared with the control, 253 (206 plus 47) pathways downregulated (Figure 3A) and 34 (30 plus 4) pathways upregulated (Figure 3B). In contrast, 140 gene sets were significantly enriched in CK1α-knockout cells compared with the control, 63 (47 plus 16) upregulated (Figure 3A) and 77 (73 plus 4) downregulated pathways (Figure 3B). Further analysis indicated that 51 (47 plus 4, Figures 3A and 3B; Table S2) gene sets were significantly enriched in both CK1α-knockout and CK1α-overexpressing cells but with the opposite enriched trend. Interestingly, of these 51 pathways, nearly 80% (40/51) were related to DNA-damage response (DDR) (Figures 3C, S4A, and S4B), and, in particular, DNA DSB response by homologous recombination (HR) (Figures 3D and 3E), which includes a series of pathways that monitor DNA integrity and activate cell-cycle checkpoints and DNA repair. Specifically, knockout of CK1α activated the DSB response, as indicated by the hyperactivity of DSB-signaling-related gene sets, such as G2/M DNA-damage checkpoint (Figure 3F), and DNA DSB repair pathways (Figures 3G and S4C–S4H), whereas CK1α overexpression inhibited DSB-response signaling, validated by downregulation of these pathways (Figures 3H, 3I, and S4I–S4N). In addition, we also confirmed these findings with The Cancer Genome Atlas (TCGA) dataset of PCa patients. As indicated, patients with a higher level of CK1α (Figure 3J) exhibited lower activity of G2/M DNA-damage checkpoint and DNA DSB repair pathways (Figures 3K and 3L), indicating that the correlation of CK1α and DSB response was not just a phenomenon in cultured cells. Taken together, these results suggest that CK1α negatively regulates DSB-response signaling, a possible mechanism for ENZA resistance.
Figure 3.

CK1α regulates DSB-response signaling
(A) Venn diagram showing gene sets from Hallmarks, Reactome, BioCarta, and KEGG database that are downregulated in 22Rv1 cells after CK1α overexpression (OE) compared with the control (Ctrl) or gene sets that are upregulated in 22Rv1 cells with CK1α knockout (KO) compared with the Ctrl. Heatmap shows the overlapped gene sets.
(B) Venn diagram showing gene sets from Hallmarks, Reactome, BioCarta, and KEGG database that are downregulated in 22Rv1 cells with CK1α KO or gene sets that are upregulated in 22Rv1 cells after CK1α OE. Heatmap shows the overlapped gene sets.
(C) Pie chart showing the portion of the DNA-damage response (DDR)-related gene sets in the overlapped gene sets in (A) and (B).
(D and E) Bar plots showing that gene sets related to DNA double-strand break (DSB) response are positively regulated in CK1α KO versus Ctrl (D), but negatively regulated in CK1α OE versus Ctrl (E). p = 0.001, if p value is less than or equal to 0.001. NES, normalized enrichment score.
(F–I, K, and L) Gene set enrichment analysis (GSEA) showing the enrichment of G2/M checkpoint and DNA DSB repair in the indicated groups: CK1α KO versus Ctrl in 22Rv1 cells (F and G), CK1α OE versus Ctrl (H and I) in 22Rv1 cells, and CK1α-high versus CK1α-low in prostate cancer patients from TCGA database (K and L).
(J) A histogram of the gene expression levels of CK1α of TCGA-PRAD tumor samples was plotted to demonstrate the distribution of CK1α level.
DSB signaling is involved in ENZA resistance
PCa cells often harbor germline or somatic alterations in DDR genes, which are associated with disease outcome and treatment response.22 Interestingly, when reviewing the results of our CRISPR screen, we discovered that the depletion of kinases (CHEK2, ATM, ATMIN, HUS1, NEK9, and TP53RK) in DSB-response signaling rendered 22Rv1 cells more resistant to ENZA (Figures 4A and S5A; Table S1). To validate this observation, we selected the top two candidates, ATM (coding ATM) and CHEK2 (coding CHK2), then devised knockout for these genes in 22Rv1 and C4-2 cells, respectively. As indicated, knockout of CHK2 and ATM immensely improved cell survival upon ENZA treatment in 22Rv1 (Figures 4B, 4C, S5B, and S5C) and C4-2 cells (Figures 4D, 4E, S5D, and S5E). In addition, improvement of the cell survival was more significant in ENZA-sensitive C4-2 cells than in ENZA-resistant 22Rv1 cells.
Figure 4.

DSB signaling is involved in ENZA resistance
(A) Scatterplot showing gene beta score of ENZA treatment versus gene beta score in control samples. Resistance or essentiality related genes are highlighted as indicated.
(B–E) In vitro proliferation of 22Rv1 cells (B and C) or C4-2 cells (D and E) expressing sgCtrl or two sgRNAs against ATM (B and D) or CHK2 (C and E) was determined by AquaBluer assay after treatment for 72 h with the indicated concentrations of ENZA. Insets are IB results of WCL of 22Rv1 cells (B and C) or C4-2 cells (D and E) expressing sgCtrl or two sgRNAs against ATM (B and D) or CHK2 (C and E).
(F–I) GSEA showing the enrichment of G2/M checkpoint and DNA DSB repair gene sets in the indicated groups, C4-2R versus C4-2 (F and G), non-responder (NR) to ENZA versus responder (R) (dataset from the study described by Alumkal et al.7) (H and I).
(J) Venn diagram showing genes that are downregulated in 22Rv1 cells after CK1α overexpression (OE) compared with the control or genes that are upregulated in 22Rv1 cells with CK1α knockout (KO) compared with the control. Heatmap showing expression of the overlapped genes.
(K) GSEA showing the enrichment of “CK1α negative signature” in NR to ENZA versus R (dataset from the study described by Alumkal et al.7).
Recently, genomic alteration of TP53 (coding p53), a key effector of the DSB response, was reported to be associated with a shorter time on treatment with the AR signaling inhibitors (ARSIs) in clinical outcome.23 Consistent with this study, we also observed that depletion of TP53RK (coding TP53RK), a key upstream kinase that phosphorylates and activates p53, could induce ENZA resistance (Figures 4A and S5A). To further confirm the role of DSB signaling in the ENZA response, we also examined the effect of p53 abundance in the ENZA response. As expected, knockout of p53 significantly induced resistance to ENZA in two ENZA-sensitive cell lines, LNCaP and C4-2 (Figures S5F–S5I). Also, p53 knockout attenuated CK1α-depletion-induced sensitivity to ENZA (Figure S5J).
Additionally, our findings are supported by RNA-seq data from both cell lines and human patients (Figures 4F–4I). We performed RNA-seq with ENZA-sensitive C4-2 cells in comparison with ENZA-resistant C4-2R cells. As indicated, GSEAs revealed that the DSB-related gene sets, DNA DSB repair response, and G2/M DNA-damage checkpoint pathways have lower activity in ENZA-resistant C4-2R cells compared with ENZA-sensitive C4-2 cells (Figures 4F and 4G). Furthermore, Alumkal et al. recently published a study7 that characterized the genomic landscape of patients with de novo resistance to ENZA. This provides the opportunity to test the association of DSB signaling and ENZA response with a clinical dataset. Consistent with our findings in vitro, DNA DSB repair and G2/M DNA-damage checkpoint pathways were also inactivated in patients who showed no response to ENZA compared with responsive patients (Figures 4H and 4I). This suggests that DSB signaling is compromised in ENZA non-responders. Moreover, the “CK1α negative signature,” generated by overlapping the genes downregulated by CK1α overexpression and upregulated by CK1α knockout in 22Rv1 cells (Figure 4J and Table S3), was also negatively enriched in ENZA non-responders compared with responders (Figure 4K).
ATM is involved in CK1α-associated regulation of DSB signaling
Next, to uncover the underlying mechanism of CK1α in the regulation of DSB signaling, we examined the indicator of DSB response, γH2AX, and the regulator of the G2/M checkpoint, CHK2, upon manipulation of CK1α. As expected, DSB signaling was activated upon CK1α knockout, as indicated by increased phosphorylation of H2AX (γH2AX) and CHK2, but decreased upon CK1α overexpression (Figure 5A). Interestingly, the abundance of ATM, the primary initiator of the DSB response, was increased upon CK1α knockout but decreased upon CK1α overexpression (Figure 5A). Consistent with this observation, CK1α knockdown (Figures 5B and 5C) or inhibition with two different selective inhibitors of CK1α kinase, BTX-A51 or D4476 (Figures 5D–5H), in 22Rv1 and C4-2 cells dramatically upregulated the protein level of ATM as well as the phosphorylation of H2AX and CHK2. Moreover, this result was supported by in vivo experiments, as BTX-A51 treatment caused significant accumulation of ATM protein and γH2AX in PDXs (Figures 6E and 6F).
Figure 5.

ATM is involved in regulation of DSB signaling by CK1α
(A) IB analysis of WCL from 22Rv1 cells with Ctrl, sgCK1α, or FLAG-CK1α.
(B and C) IB analysis of WCL from 22Rv1 cells (B) or C4-2 cells (C) with shCtrl or shCK1α treated with 100 ng mL−1 Dox for 48 h to induce CK1α knockdown.
(D and E) IB analysis of WCL from 22Rv1 cells (D) or C4-2 cells (E) treated with 100 nM BTX-A51 or 20 μM D4476 for 24 h.
(F–H) Immunofluorescence staining of ATM (F), γH2AX (G), and p-CHK2 (H) in 22Rv1 cells after 100 nM BTX-A51 or 20 μM D4476 treatment for 24 h. Scale bars, 10 μm (top) and 50 μm (bottom).
(I and J) Representative images of CK1α and ATM immunohistochemistry (IHC) staining in a tissue microarray (TMA) of primary human prostate cancer samples. Scale bar, 500 μm.
(K) Spearman’s correlation coefficient analysis of IHC staining for ATM and CK1α in TMA of prostate cancer patient specimens. A linear regression line helps visualize the negative correlation between the two genes.
(L and M) GSEA showing enrichment of the ATM pathway in the indicated groups, CK1α-high versus CK1α-low in prostate cancer patients from TCGA database (L); CK1α OE versus Ctrl in 22Rv1 cells (M).
(N and O) IB analysis of WCL from 22Rv1 (N) or C4-2 (O) cells expressing sgCtrl or two sgRNAs against ATM after treatment with 100 nM BTX-A51 (N) or 20 μM D4476 (O) for 24 h.
Figure 6.

ATM-initiated DSB signaling is involved in modulation of the ENZA response by CK1α
(A) Scatterplot together with Spearman’s correlation was applied to evaluate the correlation strength between the duration of ARSI treatment and ATM mRNA expression based on the “SU2C PNAS2019” datasets. A linear regression line helps visualize the correlation. The analysis concerns only those patients whose ARSI treatment status is recorded as “off treatment” = TRUE (n = 50).
(B) Histogram of the FPKM-normalized gene level of ATM of the “SU2C PNAS-2019” samples were plotted to demonstrate the distribution of ATM levels (n = 55). The dashed line indicates the median expression of ATM.
(C) Kaplan-Meier curve together with the log-rank p value was generated to evaluate the association between the possibility of patients continuing on the line of ARSI treatment and the ATM expression level (dichotomized as high versus low with median chosen as the cutoff) (n = 55).
(D) GSEA showing the enrichment of the ATM pathway in non-responsive (NR) patients to ENZA compared with patients responsive (R) to ENZA (dataset from the study reported by Alumkal et al.7).
(E–G) Representative images of ATM (E), γh2AX (F), and cleaved caspase 3 (G) IHC staining in LuCaP 35CR PDXs with the indicated treatments. Scale bars, 100 μm (top) and 20 μm (bottom).
(H) In vitro proliferation of 22Rv1 cells expressing sgCtrl or sgCK1α or sgCK1α and sgATM was determined by AquaBluer assay after treatment for 72 h with the indicated concentrations of ENZA. Inset: IB analysis of whole-cell lysates from 22Rv1 cells expressing sgCtrl or sgCK1α or sgCK1α and sgATM.
(I and J) In vitro proliferation of 22Rv1 cells expressing sgCtrl or two sgRNAs against ATM was detected by AquaBluer assay after combined treatment with the indicated concentrations of ENZA and 50 nM BTX-A51 (I) or 10 μM D4476 (J) for 72 h.
Subsequently, we investigated whether CK1α regulates the level of ATM in human PCa. To address this issue, we examined CK1α and ATM expression in patient samples by performing immunohistochemistry (IHC) on a tissue microarray containing a cohort of primary PCa specimens (Table S4). IHC staining was evaluated by measuring both staining intensity and percentage of positive cells. Representative IHC images display low or high staining of CK1α as well as the signal of ATM in the same case (Figures 5I and 5J). Further analysis showed that ATM protein expression is inversely correlated with the level of CK1α among the patients (Figure 5K). This finding was further supported by GSEA of RNA-seq datasets from TCGA PCa patients and cell lines; patients with a higher level of CK1α (Figure 5L) or cells with overexpressed CK1α (Figure 5M) displayed lower activity of the ATM pathway.
To further solidify these findings, we determined whether ATM was involved in CK1α-associated regulation of DSB signaling. Accordingly, ATM was knocked out utilizing two sgRNAs in 22Rv1 and C4-2 cells, followed by manipulating CK1α activity with the inhibitors BTX-A51 and D4476. CK1α inhibition significantly increased ATM abundance, as well as phosphorylation of CHK2 and H2AX in WT cells, whereas depletion of ATM dramatically attenuated CK1α-inhibition-induced phosphorylation of CHK2 and H2AX (Figures 5N and 5O). This finding revealed that ATM does participate in CK1α-mediated regulation of the DSB response. Furthermore, it has been reported that DNA-dependent protein kinase catalytic subunits (DNA-PKcs) also regulated DSB signaling by phosphorylating H2AX and CHK2. We thus depleted DNA-PKcs in 22Rv1 and C4-2 cells, followed by CK1α inhibition. As indicated, depletion of DNA-PKcs did not attenuate CK1α-inhibition-induced DSB signaling (Figures S6A and S6B). Overall, these results indicate that CK1α regulates DSB signaling by modulating the abundance of ATM.
ATM-initiated DSB signaling is involved in the modulation of the ENZA response by CK1α
As described above, ATM and DSB-response-related signaling are correlated with the ENZA response (Figure 4). A previous study,23 focusing on the genomic landscape of metastatic CRPC with clinical outcomes, showed no significant correlation between genomic alterations of ATM and the treatment probability on first-line ARSIs, ENZA, and abiraterone. In contrast, with the transcriptome dataset of this study, we found that the ATM mRNA level was significantly associated with the time on treatment with ARSIs (Figure 6A). Patients with a higher level of ATM expression had a significantly longer time to progress on ARSIs treatment compared with patients with a lower expression of ATM (Figures 6B and 6C). This finding is further supported by Cox hazards ratio analysis, which showed a significant decreased hazard in patients with a higher level of ATM compared with patients with a lower level of ATM (Figure S6C). This notion is also supported by the dataset from the study of Alumkal et al.,7 which showed that the ATM pathway was inactivated in patients who were categorized as non-responsive to ENZA (Figure 6D). Of note, during the preparation of this manuscript, another clinical study reported that patients with circulating tumor cells (CTCs) harboring genomic gains of ATM had an enhanced clinical benefit to ARSIs.24 Overall, these findings indicate that either ATM level or activity could be a prognostic biomarker for the clinical outcome of ARSIs.
Since CK1α regulates DSB signaling by modulating ATM abundance (Figure 5), we sought to determine whether ATM-initiated DSB response was involved in regulation of the ENZA response by CK1α. A previous study showed that ENZA could induce a BRCA-deficient state, “BRCAness.”25 Consistent with this finding, in ENZA-sensitive C4-2 cells ENZA treatment activated DSB signaling and induced cell apoptosis, indicated by phosphorylation of H2AX and CHK2 and increased c-PARP (Figure 2E). Overexpression of CK1α reversed these phenotypes through reducing ATM abundance (Figure 2E). Also, ATM depletion attenuated ENZA inducing DSB response (Figure S6D). Conversely, in ENZA-resistant 22Rv1 cells, CK1α knockout stabilized ATM protein and sensitized cells to ENZA treatment, indicated by increased DSB response and apoptosis (Figure 2D). These observations were further validated in vivo via a PDX treatment regime. Monotreatment with ENZA in the ENZA-resistant PDX group did not cause obvious apoptosis, but significant cell death was observed when ENZA was combined with CK1α inhibition, accompanied by elevated levels of ATM and γH2AX (Figures 6E–6G and S6E). Consistent with these findings, ATM knockout rendered cells resistant to the selective inhibitors of CK1α, BTX-A51 (Figure S6F) and D4476 (Figure S6G). Furthermore, ATM knockout significantly attenuated CK1α depletion or inhibition-associated enhancement of ENZA efficacy in 22Rv1 cells (Figures 6H–6J). Collectively, these findings suggest that ATM and ATM-associated DSB response signaling are essential for modulation of the ENZA response by CK1α.
CK1α regulates ATM stability by phosphorylating ATM at S1270
To gain mechanistic insight into how CK1α regulates ATM abundance, we first checked our RNA-seq data. However, neither CK1α knockout nor CK1α overexpression significantly affected the level of ATM mRNA (Figures S7A and S7B), suggesting that CK1α does not regulate the transcription level of ATM. Ectopic expression of CK1α significantly reduced ATM abundance (Figures 7A and 7B) and induced ATM polyubiquitination (Figure 7C), and MG132 could reverse CK1α-induced ATM degradation (Figures 7B and S7C). Collectively, these observations suggest that CK1α regulates ATM stability in a ubiquitin-dependent manner. Consistent with this notion, the half-life of ATM protein was significantly shorter in CK1α-overexpressing cells compared with WT cells (Figure 7D).
Figure 7.

CK1α regulates ATM stability by phosphorylating ATM
(A) HEK293T cells were co-transfected with FLAG-ATM and Myc-CK1α or EV and harvested for IB.
(B) 22Rv1 cells stably expressing EV or FLAG-CK1α were treated with 20 μM MG132 or DMSO for 6 h and harvested for IB.
(C) HEK293T cells co-transfected with FLAG-ATM, HA-CK1α, and Myc-ubiquitin (ub) were treated with 20 μM MG132 for 12 h and harvested for anti-FLAG immunoprecipitation (IP), followed by IB.
(D) 22Rv1 cells stably expressing EV or FLAG-CK1α were treated with 20 μg mL−1 cycloheximide (CHX) for different times and harvested for IB. The protein abundance of ATM was quantified using Image lab (Bio-Rad).
(E) HEK293T cells were co-transfected with HA-CK1α and FLAG-ATM or EV, and harvested for anti-FLAG IP, followed by IB.
(F) IB analysis of anti-ATM immunoprecipitates and WCL derived from 22Rv1 cells.
(G and H) in vitro kinase assay. After the recombinant human full-length ATM (G) or different fragments (H) were incubated with purified CK1α in the presence of [γ-32P]ATP, the reaction mixtures were resolved by SDS-PAGE followed by autoradiography.
(I) Mass spectrometry (MS) analysis of ATM phosphorylation by CK1α after the in vitro kinase assay of recombinant human ATM fragment 3 with purified CK1α. MS/MS of phosphorylated and intact peptide DEVKSIANQIQEDW for S1270. For clarity, only b+ ions are shown (highlighted in black). The presence of phosphorylation on S1270 was confirmed by the ladders of b3+-b13+ ions in the MS spectrum.
(J) The recombinant human ATM fragment 3 with the indicated mutations were incubated with purified CK1α in the presence of [γ-32P]ATP. The reaction mixtures were resolved by SDS-PAGE followed by autoradiography.
(K) The recombinant human ATM fragment 3 (WT or S1270A) was incubated with purified CK1α in the presence of ATP. The reaction mixtures were resolved by SDS-PAGE followed by IB.
(L and N) IB analysis of WCL derived from 22Rv1 (L) or C4-2 (N) cells expressing sgCtrl or two sgRNAs against CK1α.
(M and O) IB analysis of WCL derived from 22Rv1 (M) or C4-2 (O) cells treated with 20 μM D4476 or 100 nM BTX-A51 for 24 h.
(P) 22Rv1 cells stably expressing EV or FLAG-CK1α were treated with 20 μM MG132 or 100 nM BTX-A51 for 12 h and harvested for IB.
(Q) HEK293T cells were co-transfected with FLAG-ATM (WT or S1270A) and Myc-CK1α, and harvested for IB.
(R) HEK293T cells were co-transfected with FLAG-ATM (WT or S1270), HA-CK1α, and Myc-ub, treated with 20 μM MG132 for 12 h, and harvested for anti-FLAG IP, followed by IB.
(S) HEK293T cells were co-transfected with FLAG-ATM (WT or S1270A) and Myc-CK1α, then treated with 20 μg mL−1 CHX for the indicated times, followed by IB and quantification of protein turnover with Image Lab (Bio-Rad).
Because CK1α is a kinase, the idea of whether ATM is a substrate of CK1α was explored. To support this hypothesis, we detected a physical interaction between ectopically expressed or endogenous CK1α and ATM proteins in HEK293T and 22Rv1 cells (Figures 7E, 7F, S7D, and S7E). Furthermore, an in vitro kinase assay showed that CK1α directly phosphorylated ATM (Figure 7G) on a site(s) located in fragment 3 (amino acids 830–1,290) (Figure 7H). Next, two sites, S1270 identified by mass spectrometry (Figure 7I) and S1281 predicted by Group-based Prediction System (version 5.0), were considered as potential CK1α phosphorylation sites. We further showed that mutation of S1270 to alanine (S1270A), but not S1281, largely diminished CK1α-mediated phosphorylation of ATM (Figure 7J). Subsequently, this finding was verified by a specific antibody recognizing ATM when phosphorylated at S1270 (p-ATM S1270), which was validated by an in vitro kinase assay. As indicated (Figure 7K), only the incubation of CK1α kinase rendered ATM fragment 3 (ATM-F3) be recognized by anti-p-ATM S1270 antibody, whereas ATM-F3 itself, ATM-F3-S1270A, or ATM-F3-S1270A incubating with CK1α kinase was undetectable by the antibody. Depletion or pharmacologic inhibition of CK1α dramatically decreased the phosphorylation of ATM at S1270 in 22Rv1 (Figures 7L and 7M) and C4-2 (Figures 7N and 7O) cells. Overexpression of CK1α induced ATM phosphorylation at S1270, and the CK1a kinase inhibitor BTX-A51 significantly abolished this induction (Figure 7P). Lastly, mutation of S1270 to alanine rendered ATM resistant to CK1α-mediated polyubiquitination (Figure 7R) and degradation (Figure 7Q), and prolonged the half-life of ATM protein upon CK1α overexpression (Figure 7S).
Discussion
Recently, the mechanism of lenalidomide’s high efficiency in pre-leukemic myelodysplastic syndrome (MDS) with deletion of chromosome 5q (del(5q)) was addressed by identification of CK1α as a lenalidomide-regulated CRL4CRBN substrate.13 CSNK1A1 (coding CK1α) is located in the deleted region of del(5q) MDS, and the resultant haploinsufficient expression of CK1α sensitizes cells to lenalidomide treatment, although this is limited to patients with one active allele of CK1a. Recent identification of BTX-A51, a selective CK1α inhibitor,26 provides an opportunity to realize the full therapeutic potential of targeting CK1a in cancer therapy. In this study, we identify CK1α as a therapeutic target to overcome ENZA resistance with a kinome-scale CRISPR-Cas9 knockout screen. Of note, BTX-A51 has been approved by the US Food and Drug Administration in clinical trials for patients with AML. In addition, BTX-A51 has shown the ability to block superenhancer targets (CDK7/CDK9) and prevent transcription of the key oncogene MYC, another established target for PCa therapy.26,27 Therefore, our findings will have a strong translational potential and will be beneficial to patients suffering with ENZA-resistant CRPC.
CK1α is a well-established regulator of β-catenin protein stability. CK1α phosphorylates β-catenin at S45, which is subsequently phosphorylated by GSK3β, leading to its degradation.28,29 Thus, CK1α inhibition would likely increase β-catenin abundance and activate Wnt signaling, while an increase of β-catenin has been linked to gain of ENZA resistance.30 This paradox has also been documented in colon cancer models. Stabilizing β-catenin, a key driver of colon cancer, by homologous deletion of CK1α does not induce tumorigenesis.31 The underlying mechanism of this observation is that CK1α-loss-induced activation of β-catenin and the Wnt pathway is distinguished from other models of Wnt hyperactivation, such as adenomatous polyposis coli ablation. CK1α loss would induce p53-dependent growth arrest and apoptosis, which counteracts the pro-tumorigenic effects of Wnt hyperactivation.31,32 Thus, CK1α is a double-edged sword. We propose that cells resistant to ENZA maintain the abundance of CK1α at a certain level in order to balance the Wnt-β-catenin pathway and DSB-response signaling to survive under ENZA treatment. This partially explains the observation of no significant correlation between the level of CK1α and ARSI treatment probability (data not shown). However, fully inhibiting CK1α with BTX-A51 tips this balance toward cell death. Upon combination treatment of BTX-A51 with ENZA, cells will undergo apoptosis due to ATM-stabilization-initiated extensive DSB-response signaling, including the p53 pathway, regardless of the level of β-catenin or Wnt activity.
Previous studies showed that AR and AR variants were involved in DNA-repair progression in PCa cells by driving expression of a cohort of DNA-repair and DDR genes.33,34,35 ENZA treatment could suppress the expression of HR genes and induce HR deficiency and “BRCAness,”25 a phenotypic trait that cells share with characteristics of BRCA1/2 mutation carriers, which subsequently promote DNA-damage-induced cell death. In congruence with these findings, we observed that ENZA treatment induced DSB response and cell apoptosis in ENZA-sensitive cells. Patients or cells categorized as non-responsive to ENZA showed a lower activity of DSB-response signaling, but restoration of DSB-response signaling by inhibition of CK1α rendered otherwise resistant cells sensitive to ENZA. In addition, CK1α inhibition decreased AR activity (Figures S7I–S7L), which will further stimulate the DSB response and cell death. Our findings further solidify DDR signaling as an essential player in the ENZA response and identify a therapeutic target to halt disease progression.
Kinases ATM, ataxia telangiectasia and Rad3-related protein (ATR), and DNA-PKcs play pivotal roles in DDR by phosphorylating a variety of effectors to control DNA repair, cell-cycle arrest, or cell death in response to DNA damage.36 In contrast to ATR and DNA-PKcs, ATM serves as a central node in response to DNA DSB.37 ATM must be tightly regulated, as aberrant activation of ATM could lead to toxic DNA repair, cell-cycle arrest, senescence, or apoptosis.36 We showed that ATM was a direct substrate of CK1α in regulation of DNA DSB signaling and the ENZA response. It has been well established that p53 abundance is regulated by CK1α, whereas ATM is an important regulator of p53 activity and stability in a direct or indirect manner.36,38 Therefore, our findings expand our knowledge of the scale of CK1α in the regulation of DDR and offer another mechanism of the regulation of p53 by CK1α through the modulation of the abundance of ATM.
As described above, DSB signaling is important in ENZA-induced cell death. As the primary initiator of the DSB response, ATM is significant in this process. A previous study has demonstrated that CHK1 inhibitor induced ATM activation and DDR, which enhanced the efficacy of ENZA.35 Patients with a high expression level of ATM showed a significantly increased timeframe to progression on ARSIs treatment compared with patients with a lower level of ATM. A more recent clinical study reported that ATM and BRCA2 were surprisingly differentially gained in ENZA-resistant versus-sensitive CTCs DNA, with BRCA2 gain enriched in resistant patients and ATM gain enriched in sensitive patients.24 ENZA could induce “BRCAness,”25 which would subsequently lead to ATM-dependent cell death, whereas BRCA2 gain would attenuate ENZA-induced “BRCAness” and cell death. Therefore, ATM and BRCA2 seem to play opposite roles in the ENZA response. This also explains why alterations of DNA-damage genes (including ATM and BRCA1/BRCA2) are not correlated with ENZA treatment probability,23 whereas the expression level of ATM itself is.
Lineage plasticity has been implicated in drug resistance, particularly molecularly targeted therapies such as ARSI. In PCa, loss of p53, RB1, and androgen-dependent signaling are the hallmarks of lineage plasticity or neuroendocrine PCa, which has shown strong resistance to ARSIs.39,40,41 At present there is no established therapeutic approach for these patients. As a well-established negative regulator of p53,31 inhibition of CK1α will increase p53 abundance. Here we showed that CK1α was also involved in the regulation of DDR signaling through the modulation of ATM level in a phosphorylation-dependent manner. These findings suggest that CK1α might be a target to slow the progression of lineage plasticity PCa due to ARSI treatment. Further experiments need to be conducted to validate and establish an accurate model.
Limitations of the study
With a CRISPR-Cas9 library screen, we identified CK1α was the number-one candidate gene that is involved in ENZA response. Although we focused on CK1α in this study, there are also other interested targets in the candidate list that need to be explored in future studies. In addition to regulating ATM stability and DDR signaling, we also found that CK1α could modulate AR signaling and cell migration (Figures S7I–S7M). Whether and how AR signaling and cell migration were involved in CK1α and ENZA response also need to be addressed in future studies. Furthermore, protein datasets with ENZA response from clinical samples could be used to solidify our findings of CK1α and ATM in ENZA response and their correlations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Cas9 | Cell Signaling Technology | Cat# 14697; RRID:AB_2750916 |
| Rabbit monoclonal anti-AR | Cell Signaling Technology | 5153Cat# 5153; RRID:AB_10691711 |
| Rabbit monoclonal anti-β-Actin | Cell Signaling Technology | Cat#4970; RRID:AB_2223172 |
| Rabbit polyclonal anti-CK1α | Cell Signaling Technology | Cat# 2655; RRID:AB_2283593 |
| Rabbit monoclonal anti-ATM | Cell Signaling Technology | Cat# 2873; RRID:AB_2062659 |
| Rabbit polyclonal anti-CHK2 | Cell Signaling Technology | Cat# 2662; RRID:AB_2080793 |
| Rabbit monoclonal anti-phospho-CHK2 (T68) | Cell Signaling Technology | Cat# 2197; RRID:AB_2080501 |
| Rabbit monoclonal anti-γH2AX | Cell Signaling Technology | Cat# 9718; RRID:AB_2118009 |
| Rabbit monoclonal anti-H2AX | Cell Signaling Technology | Cat# 7631, RRID:AB_10860771 |
| Rabbit monoclonal anti-DNA-PKcs | Cell Signaling Technology | Cat# 38168; RRID:AB_2799128 |
| Rabbit monoclonal anti-Cleaved PARP | Cell Signaling Technology | Cat# 5625; RRID:AB_10699459 |
| Rabbit monoclonal anti-Myc-Tag | Cell Signaling Technology | Cat# 2278, RRID:AB_490778 |
| Rabbit monoclonal anti-HA-tag | Cell Signaling Technology | Cat# 3724; RRID:AB_1549585 |
| Rabbit polyclonal anti-Cleaved Caspase-3 | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| Goat polyclonal anti-rabbit IgG, HRP-linked | Cell Signaling Technology | Cat# 7074; RRID:AB_2099233 |
| Goat polyclonal anti-mouse IgG, HRP-linked | Cell Signaling Technology | Cat# 7076; RRID:AB_330924 |
| Mouse monoclonal anti-alpha-Tubulin | Cell Signaling Technology | Cat# 3873; RRID:AB_1904178 |
| Mouse monoclonal anti-vinculin | Sigma-Aldrich | Cat# V4505; RRID:AB_477617 |
| Rabbit polyclonal anti-FLAG | Sigma-Aldrich | Cat# F7425; RRID:AB_439687 |
| Mouse monoclonal anti-CK1α | Santa Cruz Biotechnology | Cat# sc-74582; RRID:AB_2084662 |
| Mouse monoclonal anti-p53 | Santa Cruz Biotechnology | Cat# sc-126; RRID:AB_628082 |
| Mouse monoclonal anti-HA Magnetic Beads | Thermo Fisher Scientific | Cat# 88836; RRID:AB_2749815 |
| Mouse monoclonal anti-FLAG Magnetic Beads | Sigma-Aldrich | Cat# M8823; RRID:AB_2637089 |
| Rabbit monoclonal anti-ATM | Abcam | Cat# ab201022 |
| Rabbit monoclonal anti-ATM | Abcam | Cat# ab32420; RRID:AB_725574 |
| Rabbit monoclonal anti-CK1α | Abcam | Cat# ab223144 |
| Rabbit polyclonal anti-CK1α | OriGene | Cat# TA313698 |
| Rabbit polyclonal anti-phospho-ATM (S1270) | This paper | N/A |
| Goat polyclonal anti-Rabbit IgG | Thermo Fisher Scientific | Cat# A-11034; RRID:AB_2576217 |
| Bacterial and virus strains | ||
| lentiCas9-Blast | Sanjana et al.42 | Addgene Cat# 52962 |
| lentiGuide-Puro | Sanjana et al.42 | Addgene Cat# 52963 |
| LentiGuidPuro-hTP53_sgRNA-1 | Wang et al.43 | Addgene Cat# 88853 |
| sgTP53_3 | Hong et al.44 | Addgene Cat# 78164 |
| pcDNA3.1(+) FLAG-His-ATM wt | Canman et al.45 | Addgene Cat# 31985 |
| PRKDC gRNA1 | Doench et al.46 | Addgene Cat# 77861 |
| PRKDC gRNA2 | Doench et al.46 | Addgene Cat# 77862 |
| ATM gRNA1 | Doench et al.46 | Addgene Cat# 77530 |
| ATM gRNA2 | Doench et al.46 | Addgene Cat# 77531 |
| CHEK2 gRNA1 | Doench et al.46 | Addgene Cat# 76487 |
| CHEK2 gRNA2 | Doench et al.46 | Addgene Cat# 76488 |
| CSNK1A1 gRNA1 | Doench et al.46 | Addgene Cat# 76188 |
| CSNK1A1 gRNA2 | Doench et al.46 | Addgene Cat# 76189 |
| Tet-plko-puro | Wiederschain et al.47 | Addgene Cat# 21915 |
| pLC-FLAG-CSNK1A1-WT-Puro | Patil et al.48 | Addgene Cat# 123319 |
| pcDNA3.1-HA | Oskar Laur | Addgene Cat# 128034 |
| psPAX2 | Didier Trono | Addgene Cat# 12260 |
| pCMV-VSV-G | Stewart et al.49 | Addgene Cat# 8454 |
| GST-ATM fragments | Dr. Zhenkun Lou Lab (Mayo Clinic, Rochester, USA) | N/A |
| Endura Competent Cells | Lucigens | Cat# 60242 |
| Biological samples | ||
| Patient-derived xenografts (PDX) | University of Washington | LuCaP 35CR |
| Patient-derived xenografts (PDX) | University of Washington | LuCaP 77CR |
| Chemicals, peptides, and recombinant proteins | ||
| Enzalutamide | Selleck Chemicals | Cat# S1250 |
| doxycycline | Selleck Chemicals | Cat# S5159 |
| cycloheximide | Selleck Chemicals | Cat# S7418 |
| MG132 | Selleck Chemicals | Cat# S2619 |
| D4476 | Selleck Chemicals | Cat# S7642 |
| BTX-A51 | DC Chemicals | Cat# DC11250 |
| CK1α | Thermo Fisher Scientific | Cat# PV3850 |
| ATM | Sigma-Aldrich | Cat# 14-933 |
| Polybrene | Sigma-Aldrich | Cat# TR-1003-G |
| Hoechst | Sigma-Aldrich | Cat# B2261 |
| Kinase Buffer | Cell Signaling Technology | Cat# 9802 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| DMSO | Sigma-Aldrich | Cat# D2650 |
| carboxymethyl cellulose | Selleck Chemicals | Cat# S6703 |
| Tween 80 | Sigma-Aldrich | Cat# P4780 |
| Solutol | MedChemExpress | Cat# HY-Y1893 |
| 2-hydroxy prolyl-b-cyclodextrin | MedChemExpress | Cat# HY-101103 |
| AquaBluer Solution | MultiTarget | Cat# 6015 |
| Critical commercial assays | ||
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | Cat# E0554 |
| RNeasy Mini kit | Qiagene | Cat# 74104 |
| Plasmid Maxi Kit | Qiagene | Cat# 12162 |
| Deposited data | ||
| Raw and analyzed data | This paper | GSE203362 |
| Experimental models: Cell lines | ||
| 22Rv1 | ATCC | CRL-2505 |
| C4-2 | ATCC | CRL-3314 |
| LNCaP | ATCC | CRL-1740 |
| MR49F | Dr. Amina Zoubeidi Lab (University of British Columbia, Vancouver, BC, Canada) | N/A |
| C4-2R | Dr. Allen Gao Lab (University of California, Davis, CA, USA) | N/A |
| C4-2B | Dr. Ka Wing Fong (University of Kentucky, USA) | N/A |
| HEK293T | Dr. Andrea Kasinski Lab (Purdue University, USA) | N/A |
| Experimental models: Organisms/strains | ||
| NU/J mice | The Jackson Laboratory | RRID:IMSR_JAX:002019 |
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice | The Jackson Laboratory | RRID:IMSR_JAX:005557 |
| Oligonucleotides | ||
| sgAR-full#1 5′GTTACACGTGGACGACCAGA-3′ | This paper | N/A |
| sgAR-full#2 5′GTGTCCAGCACACACTACACC-3′ | This paper | N/A |
| shCK1α#1 5′GCCACAGTTGTGATGGTTGTT-3′ | This paper | N/A |
| shCK1α#2 5′GCAAGCTCTATAAGATTCTTC-3′ | This paper | N/A |
| Recombinant DNA | ||
| Myc-DDK-CSNK1A1 (#RC217936) | OriGene | Cat# RC217936 |
| PcDNA3.1-HA-CK1α | This paper | N/A |
| Tet-plko-puro-CK1α | This paper | N/A |
| sgAR-full length | This paper | N/A |
| Kinome CRISPR Knockout Library | Doench et al.46 | Addgene Cat# 75314 |
| Software and algorithms | ||
| MAGeCK | Li et al.50 | https://sourceforge.net/p/mageck/wiki/Home/ |
| R package limma | Ritchie et al.51 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| GSEA4.1.0 | Subramanian et al.52 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ImageJ | Schneider et al.53 | https://imagej.nih.gov/ij/ |
| GraphPad Prism 8.0 | GraphPad | https://www.graphpad.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be direct to and will be fulfilled by the lead contact, Dr. Xiaoqi Liu (xiaoqi.liu@uky.edu).
Materials availability
All reagents generated in this study are accessible from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell culture
Human PCa cell lines LNCaP, C4-2, MR49F, C4-2R, C4-2B and 22Rv1 were cultured in RPMI-1640 medium (Sigma-Aldrich). Among these cell lines, LNCaP is androgen-dependent, whereas C4-2 is an androgen-independent cell line derived from LNCaP. MR49F is an ENZA-resistant derivative of LNCaP obtained from Dr. Amina Zoubeidi (University of British Columbia, Vancouver, BC, Canada), whereas C4-2R is derived from C4-2 and kindly provided by Dr. Allen Gao (University of California, Davis, CA, USA). The concentration of ENZA for ENZA-resistance maintenance of MR49F and C4-2R was 10 or 20 μM, respectively. ENZA was removed for at least 48 hrs before any experiment. 22Rv1 is an established intrinsic ENZA resistant cell line. C4-2B cell line was a gift from Dr. Ka Wing Fong (University of Kentucky, USA). LNCaP, C4-2, and 22Rv1 cells were purchased from American Type Culture Collection. HEK293T cell was a gift from Dr. Andrea Kasinski (Purdue University, USA) and cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich). All medium contained 10% fetal bovine serum (Atlanta Biologicals, GA, USA), 100 U ml-1 of penicillin and 100 μg ml−1 streptomycin. Cells were cultured in a humidified atmosphere at 37 °C, with 5% CO2.
Mouse models and treatment
All animal experiments were approved by the Institutional Animal Care and Use Committee at University of Kentucky (KY, USA). Patient derived xenograft (PDX) experiments were conducted as previous described.21 Male NSG (NOD scid gamma) mice (age 6-8 weeks) were castrated and then implanted subcutaneously with LuCaP 35CR or LuCaP 77CR tumor bits two weeks later. When the tumor volume reached 50 mm3, mice were pre-treated with ENZA (50 mg kg-1 in PBS containing 1% carboxymethyl cellulose, 0.1% Tween 80, 5% DMSO) by oral gavage daily, for a routine of 5 days on, 2 days off. After one week, ENZA resistance would have developed.21 Mice were randomly assigned into four groups: Control (with vehicle); ENZA (20 mg kg-1 in PBS containing 1% carboxymethyl cellulose, 0.1% Tween 80, 5% DMSO, by oral gavage every 3 days); BTX-A51 (5 mg kg-1 in ddH2O containing 10% DMSO, 10% Solutol and 10% 2-hydroxy prolyl-b-cyclodextrin, by oral gavage, two days on and one day off); or combination with ENZA and BTX-A51 (Figures 2I and S3J). Tumor growth was monitored twice weekly by measuring the length and width of tumors. Tumor volume was evaluated using the formula: (length × width2)/2. For survival studies, the mice were sacrificed when tumor volume exceeded 1,500 mm3, when the diameter exceeded 20 mm, when the animals became compromised, or after 12 weeks, whichever developed first for the end point of the study. After humane euthanization, the tumors were harvested for paraffin embedding or frozen for subsequent analyses.
22Rv1 in vivo xenograft experiments were conducted by subcutaneous injection of 2 × 106 22Rv1 cells (wild-type or CK1α-knockout) (100 μl in 50% PBS and 50% Matrigel, Corning) into the flanks of pre-castrated male nude mice (Jackson Lab, MI, USA). When the tumor volume reached 100 mm3, daily gavage treatment with 20 mg kg-1 ENZA or vehicle (PBS containing 1% carboxymethyl cellulose, 0.1% Tween 80, 5% DMSO) was started (5 days on, 2 days off). Tumor growth was monitored twice weekly by measuring the length and width of tumors. Tumor volume was evaluated using the formula: (length × width2)/2. After humane euthanization, tumors were harvested for paraffin embedding or frozen for subsequent analyses.
Method details
Kinome-wide CRISPR-Cas9 screen
22Rv1 cells were transduced with lentiCas9-Blast and then selected with 10 μg ml-1 blasticidin for 3 weeks to generate a stable cell line expressing Cas9 (22Rv1-Cas9). Single clones were picked and further determined Cas9 expression and ability to edit genes knockout.
Human Kinome CRISPR pooled library (Brunello)46 was a gift from Drs. John Doench and David Root. The library was amplified as previously described.46 Briefly, the Kinome library was diluted to 50 ng ul-1 in water and then electroporated using Endura electrocompetent cells. The transformations were plated onto pre-warmed agar plates and incubated for 14 hrs at 32 °C. Colonies were harvested and the library plasmids were purified with a maxi scale plasmid prep. To make lentivirus, the library plasmids were co-transfected with packaging plasmids pCMV-VSV-G and psPAX2 into HEK293T cells with Lipofectamine 2000. After 6 hrs, the medium was changed and further cultured for an additional 60 hrs, then viral supernatants were harvested and centrifuged at 2,000 rpm at 4 °C for 10 min to pellet cell debris. The supernatant was then filtered through a 0.45 μm membrane and viruses were concentrated with a high-speed centrifuge (24, 000 rpm, 2 hrs). The titer of the lentiviruses was determined with 22Rv1 cells in a functional assay by measuring puromycin resistance after transduction.
Infections were set up at 1500-fold coverage of the library. 22Rv1-Cas9 cells were infected at a low multiplicity of infection (MOI = 0.3) with the above prepared kinome library viruses in the presence of 8 μg ml−1 polybrene, followed by selection with puromycin for 3 days, then 1/3 of the infected cells (500 × coverage) were harvested as the baseline for deep sequencing and the remaining cells were continuously cultured for an additional two weeks waiting for genes edition. Then the cells were randomly split and treated with 20 μM ENZA or DMSO for an additional one week. The sgRNAs incorporated into the cells were amplified from genomic DNA and sequenced on Illumina HiSeq 2500.
Analysis of CRISPR-Cas9 screen data
Screen data analysis was performed by MAGeCK50 software. Raw sequencing reads were processed by FastQC for quality control, 21-27 bp were trimmed from the 5’ end until the protospacer sequence was reached, then mapped to the expected sgRNA sequence with zero mismatches tolerated. The read counts of sgRNAs were normalized by non-targeting control sgRNAs (Table S5). Using the MAGeCK MLE algorithm, the normalized sgRNA read counts in ENZA and DMSO samples were compared to the initial baseline sgRNA distribution and negative control sgRNAs to estimate a beta score for each targeted gene in the treatment and control groups separately. The beta scores of all genes were normalized by the median beta score of the essential genes to make the cell proliferation rates comparable between treatment and control samples and to reduce false-positive hits.17 The top negatively selected genes were those with the smallest negative values in the differential beta score, which was calculated by subtracting the control beta score from the treatment beta score. We defined 8 classes of genes with respect to resistance and essentiality: 1) Resistance related = beta score is negative in control, it becomes positive after ENZA treatment; 2) Resistance gain = beta score is around 0 in control, beta score becomes positive after ENZA treatment; 3) Resistance increased = beta score is positive in control, it becomes more positive after ENZA treatment; 4) Essentiality lost= beta score is negative in control, it becomes around 0 after ENZA treatment; 5) Essentiality reduced = beta score is negative in control, it becomes less negative after ENZA treatment; 6) Essentiality increased = beta score is negative in control, it becomes more negative after ENZA treatment; 7) Essentiality gain = beta score is around 0 in control, it becomes negative after ENZA treatment; 8) Others = genes not in the above mentioned 7 classes. “Gain” refers to the beta score is 0 in control, and it becomes positive (resistance gain) or negative (essentiality gain) after ENZA treatment. “Increased” means the beta score is already positive or negative, and it becomes more positive (resistance increased) or more negative (essentiality increased) after ENZA treatment. “Related” indicates a more complicated situation that the gene changes from an essential gene in control to a resistance gene in ENZA treatment.
RNA sequencing and analysis
Total RNA derived from 22Rv1, 22Rv1 with CK1α knockout and 22Rv1 cells with CK1α overexpression was purified using a RNeasy Mini kit. The RNA sequencing was run as a paired-end 150bp in length with 30 million reads, on the Illumina HiSeq 2500 (Novogene). Raw sequences were mapped via HISAT254 (v2.1.0) to the human reference genome GRCh38. Read counting was performed using featureCounts55 from the subread package (v1.5.1). Differential gene expression was analyzed by R package limma51 (v3.42.2). All p-values were corrected for multiplicity by the Benjamini-Hochberg method. Gene Set Enrichment Analysis (GSEA) statistical analysis was carried out with publicly available software from the Broad Institute (http://www.broadinstitute.org/gsea/index.jsp). Gene expression results were ranked and used to conduct GSEA. GSEA was performed to evaluate enrichment of differential expression patterns curated from Hallmarks, Reactome, BioCarta, and KEGG database within the MSigDBv7.4.
RNA sequencing of C4-2 and C4-2R cells has been described.56 Briefly, extracted total RNA from C4-2 and C4-2R cells was sequenced as a paired-end 100bp in length with 30 million reads, on the Illumina HiSeq 2500 (Purdue Genomics Facility). Tophat2 was used to align reads to the Ensembl Homo sapiens genome database version GRCh38.p5. The htseq-count script in HTSeq v.0.6.1 was run to count the number of reads mapping to each gene. DESeq2, edgeR and Cufflinks2 were used for differential expression analysis. Gene expression results were ranked and used to conduct GSEA. GSEA was performed accordingly.
Tissue microarray (TMA) construction and IHC staining
For TMA, formalin-fixed paraffin-embedded tissue blocks from prostate cancer patients who underwent radical prostatectomy at the University of Kentucky (KY, USA) were cored to construct a TMA through the Markey Cancer Center Biospecimen Procurement and Translational Pathology Shared Resource Facility. Approval for use of human prostate tissue was obtained from the University of Kentucky Institutional Review Board. Cancerous, adjacent benign epithelial, or benign prostatic hyperplasia tissue cores (2 mm) were used, with duplicate cores from each patient. Four-micron thick sections of the TMA were cut for IHC staining. For regular xenograft tumor, four-micron thick sections of formalin-fixed paraffin-embedded tumor tissue were cut for IHC staining. Staining was carried out on Ventana Discover Ultra. Antigen retrieval was performed using CC1 (Roche) (for ATM, γH2AX and Cleaved Caspase-3) or CC2 (Roche) (for CK1α) with standard conditions. Slides were incubated with primary antibodies (ATM at 1:50, γH2AX at 1:100, Cleaved Caspase-3 at 1:150 and CK1α at 1:50) for 1 hr at 37 °C, incubated with anti-Rabbit-HQ (Roche), followed by anti-HQ-HRP (Roche) and visualized with DAB prior to light counterstain with Mayer’s hematoxylin. The TMA staining was evaluated and scored by a pathologist. The regular staining of tumor xenografts was also evaluated by a pathologist by comparison to the positive control. Images were taken with a Nikon microscopic camera or scanned with the Aperio Digital Pathology Slide Scanner.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde for 15 min at room temperature (RT), permeabilized in 0.2% Triton X-100 for 10 min and blocked with 10% FBS/PBS (v/v) for 30 min. Afterwards, the cells were incubated with appropriate primary antibodies overnight at 4°C and secondary antibodies for 1 hr at RT. All antibodies were diluted in PBS containing 1% BSA and 1% normal goat serum. The dilution ratios of the primary antibodies were 1:500 for ATM and γH2AX, 1:100 for p-CHK2. The secondary antibody was used at 1: 500. The nucleus was stained with Hoechst. Images were captured with a confocal microscope (Nikon).
Immunoblot (IB) and immunoprecipitation (IP) analyses
Cell lysates were prepared as previously described.57 Briefly, cultured cells were harvested and lysed with the lysis buffer (50 mM Tris, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.2% SDS, protease inhibitors, and phosphatase inhibitors). For tumor tissue, the lysates were prepared by homogenization using the above lysis buffer followed by sonication with a probe sonicator. After quantification with BCA assay, equal amounts of protein were loaded on SDS-PAGE gel and then transferred to nitrocellulose membranes, followed by IB with the indicated antibodies. For IP analysis, 1 mg protein collected with the lysis buffer (50 mM Tris, 150 mM NaCl, 0.5% sodium deoxycholate,1% NP-40, protease inhibitors) were incubated with the indicated primary antibodies overnight at 4°C followed by the protein A/G magnetic beads for 3 hrs at RT or antibody-conjugated beads overnight at 4 °C. The immunocomplexes were washed three times with the lysis buffer and analyzed by IB.
AquaBluer assay to determine cell viability
AquaBluer assay was performed according to the manufacturer’s instructions. Cells were seeded at 3000-8000 (dependent on the cell size) per well in 96-well culture plates for cells grow overnight. Cells were then treated as indicated and further cultured for 72 hrs. Dilute AquaBluer™ (1: 100) in the culture medium and mix well. The medium was then removed from the cell culture and 100 μl of the diluted AquaBluer™ was added to each well. The plate was returned to the incubator for another 4 hrs. Once placed in a fluorescence plate reader, the fluorescence intensity was read at 540ex/590em.
Colony formation assay
Cells were seeded at 500-2000 (dependent on cell size) per well in 6-well or 12-well culture plates. After growing for 24 hrs, cells were treated as indicated and further cultured for 3 weeks. The medium was then removed and the cells were rinsed carefully with PBS. Cells were fixed with 4% paraformaldehyde for 15 min at RT before they were stained with 0.5% crystal violet for 30 min at RT. After staining, the crystal violet solution was removed and rinsed carefully with tap water. Colonies were left to dry on the plates at RT. The images were taken with the ChemiDoc Imaging System (Bio-Rad).
In vitro kinase assay
In vitro kinase assays were performed with a kinase buffer (25 mM Tris-HCl (pH 7.5), 5 mM beta-glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM Na3VO4 and 10 mM MgCl2) supplemented with 125 μM ATP and 10 μCi of [γ- 32P] ATP at 30 °C for 30 min in the presence of CK1α kinase and GST-ATM proteins. After the reaction, mixtures were resolved by SDS-PAGE, the gels were stained with Coomassie brilliant blue, dried, and subjected to autoradiography. In vitro kinase assays for mass spectrometry analysis were performed with the kinase buffer above supplemented with 200 μM ATP at 30 °C for 30 minutes in the presence of CK1α kinase and GST-ATM proteins. After the reaction, proteins were denatured, resolved on SDS–PAGE, the gels were stained with Coomassie brilliant blue, the protein bands were cut and subjected to mass spectrometry analysis.
sgRNA or shRNA clones
To clone the sgRNA or shRNA to the vectors, lentiGuide-Puro or Tet-plko-puro was cut by restriction enzymes and dephosporylated with FastAP (Thermo Fisher). Oligonucleotides for the sgRNAs or shRNAs sequence (sgAR-full#1 5′GTTACACGTGGACGACCAGA-3′ sgAR-full#2 5′GTGTCCAGCACACACTACACC-3′, shCK1α#1 5′GCCACAGTTGTGATGGTTGTT-3′, shCK1α#2 5′GCAAGCTCTATAAGATTCTTC-3′) were phosphorylated using polynucleotide kinase and then annealed by heating to 95 °C for 5 minutes and cooling to 25 °C at 1.5 °C minute-1. Annealed oligos were then ligated into lentiGuide-Puro or Tet-plko-puro above using T4 ligase (New England Biolabs) at 16 °C overnight.
Wound scratch assay
A scratch was introduced into the confluent cell monolayer by using a pipette tip. Cells were then washed gently with phosphate-buffered saline (PBS) twice. Cells were further cultured with complete medium contained 2% Fetal Bovine Serum in a humidified atmosphere at 37 °C, with 5% CO2 for 72 hrs. The migration of cells into the wounded area was recorded by a microscope (Nikon).
Bioinformatic analysis
Correlation analysis with Spearman’s correlation coefficient
The Spearman’s Correlation coefficient and its associated 95% confidence interval (CI) were used to quantify the correlation between ATM and CK1α expression in TMA IHC staining. A linear regression line helps visualize the negative correlation between the two genes.
Analysis of “SU2C PNAS2019” dataset
Scatter plot together with Spearman’s rank correlation coefficient was applied to evaluate the correlation strength between the duration of ARSI treatment and FPKM-normalized ATM mRNA expression based on the “SU2C PNAS2019” datasets.23 A linear regression line helped to visualize the correlation. The analysis concerns only those patients whose ARSI treatment status is recorded as “Off treatment” =TRUE. SU2C PNAS-2019 mRNA FPKM expression values were downloaded from the cBioportal website (https://www.cbioportal.org/). The histogram of the FPKM-normalized gene levels of ATM of the SU2C PNAS-2019 samples was plotted to demonstrate the distribution of ATM levels. A dashed line indicates the median expression of ATM. Kaplan-Meier curve, together with the logrank p-value, was generated to evaluate the association between the possibility of patients staying on the line of ARSI treatment and the ATM expression levels (dichotomized as high versus low with median chosen as the cutoff). Forest plot associated with the statistics based on Cox proportional hazards model was generated to evaluate the association between the ARSI-treatment duration length and the ATM mRNA levels (dichotomized as high versus low with median chosen as the cutoff). A Hazard Ratio being less than 1 indicates that patients in the ATM-high group tended to remain on the line of ARSI treatment longer than those in the ATM-low group.
GSEA analysis with TCGA dataset (https://www.cancer.gov/tcga)
TCGA-PRAD mRNA FPKM expression values were downloaded from the GDC data portal and manually converted to TPM-normalized gene levels. The histogram of the interested gene level from the TCGA-PRAD tumor samples was plotted to demonstrate the distribution of gene expression. The dashed lines indicate the median or quartile expression of the interested genes. Differential comparison analysis between the indicated gene expression high (>= cut off, indicated in the figure legend) and the low (< cut off) subgroups was performed based on the quasi-likelihood F-test in R package edgeR. Genes were then ranked based on p values together with the corresponding signs of log2-foldchange. Pre-ranked GSEA was then performed accordingly.
Analysis of “Dr. Alumkal PNAS2020” dataset
Dr. Joshi Alumkal kindly provided the raw count table from their RNA-seq dataset.7 Raw read counts were log transformed and analyzed by limma51 to generate log2 fold change for non-responders versus responders, which was used to pre-rank the genes as the input for GSEA. A CK1α gene signature composed of 188 genes was generated by overlapping up regulated genes (adjusted p<0.05, knockout/control ratio >1) in CK1α knockout samples and down regulated genes (adjusted p<0.05, overexpression/control ratio <1) in CK1α overexpression samples. Heatmap was generated by R package pheatmap.
Quantification and statistical analysis
For comparisons between two groups of independent datasets, unpaired t-test was performed, and p-value and standard error of the mean (SEM) were reported. For comparisons among more than two groups, one-way ANOVA was performed, p-values and SEM were reported. For drug response curves, IC50 was calculated by non-linear regression and then t-test or one-way ANOVA was performed based on the groups for comparisons. For all figures, ∗ represents p<0.05, ∗∗ represents p<0.01, ∗∗∗ represents p<0.001.
Acknowledgments
This work was supported by NIH R01 CA157429 (X.L.), R01 CA196634 (X.L.), R01 CA264652 (X.L.), and R01 CA256893 (X.L.). This study was aided by grant #IRG 19-140-31 from the American Cancer Society (J.L.). This research was also supported by the Biospecimen Procurement & Translational Pathology, Biostatistics and Bioinformatics, and Flow Cytometry and Immune Monitoring Shared Resources of the University of Kentucky Markey Cancer Center (P30CA177558). We acknowledge Dr. Joshi Alumkal (University of Michigan, Ann Arbor, MI, USA) for sharing the RNA-seq database from the patients with ENZA treatment. We thank Heather Russell-Simmons at the Markey Cancer Center Research Communications Office and Eleanor Erikson for proofreading the paper.
Author contributions
J.L. designed and performed experiments, analyzed the data, and wrote the manuscript. X.L. designed the research and supervised the project. Y. Zhao, D.H., S.T., C.L., C.W., and L.L. performed the analysis of CRISPR screening, RNA-seq, and other bioinformatics. K.M.J. conducted manuscript editing. D.B.A. evaluated and scored the IHC staining. Y. Zhang conducted the amplification of the CRISPR library. J.C. performed the mass spectrometry assay and analyzed the data. K.M.J., Q.Z., and X.W. conducted xenograft tumor harvesting and some data analysis.
Declaration of interests
The authors declare no competing interests.
Published: April 18, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101015.
Supplemental information
Data and code availability
-
•
The datasets of CRISPR Screen and RNA-seq are available at NCBI (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE203362), referring to GSE203362.
-
•
This paper does not generate the original code.
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of Incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Swami U., McFarland T.R., Nussenzveig R., Agarwal N. Advanced prostate cancer: treatment advances and future directions. Trends Cancer. 2020;6:702–715. doi: 10.1016/j.trecan.2020.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Davis I.D., Martin A.J., Stockler M.R., Begbie S., Chi K.N., Chowdhury S., Coskinas X., Frydenberg M., Hague W.E., Horvath L.G., et al. Enzalutamide with standard first-line therapy in metastatic prostate cancer. N. Engl. J. Med. 2019;381:121–131. doi: 10.1056/NEJMoa1903835. [DOI] [PubMed] [Google Scholar]
- 4.Hussain M., Fizazi K., Saad F., Rathenborg P., Shore N., Ferreira U., Ivashchenko P., Demirhan E., Modelska K., Phung D., et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med. 2018;378:2465–2474. doi: 10.1056/NEJMoa1800536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson P.A., Arora V.K., Sawyers C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer. 2015;15:701–711. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arora V.K., Schenkein E., Murali R., Subudhi S.K., Wongvipat J., Balbas M.D., Shah N., Cai L., Efstathiou E., Logothetis C., et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alumkal J.J., Sun D., Lu E., Beer T.M., Thomas G.V., Latour E., Aggarwal R., Cetnar J., Ryan C.J., Tabatabaei S., et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc. Natl. Acad. Sci. USA. 2020;117:12315–12323. doi: 10.1073/pnas.1922207117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beltran H., Prandi D., Mosquera J.M., Benelli M., Puca L., Cyrta J., Marotz C., Giannopoulou E., Chakravarthi B.V.S.K., Varambally S., et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu G., Chhangawala S., Cocco E., Razavi P., Cai Y., Otto J.E., Ferrando L., Selenica P., Ladewig E., Chan C., et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 2020;52:198–207. doi: 10.1038/s41588-019-0554-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang S., Zhang M., Sun J., Yang X. Casein kinase 1alpha: biological mechanisms and theranostic potential. Cell Commun. Signal. 2018;16:23. doi: 10.1186/s12964-018-0236-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knippschild U., Krüger M., Richter J., Xu P., García-Reyes B., Peifer C., Halekotte J., Bakulev V., Bischof J. The CK1 family: contribution to cellular stress response and its role in carcinogenesis. Front. Oncol. 2014;4:96. doi: 10.3389/fonc.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krönke J., Fink E.C., Hollenbach P.W., MacBeth K.J., Hurst S.N., Udeshi N.D., Chamberlain P.P., Mani D.R., Man H.W., Gandhi A.K., et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523:183–188. doi: 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richter J., Kretz A.L., Lemke J., Fauler M., Werner J.U., Paschke S., Leithäuser F., Henne-Bruns D., Hillenbrand A., Knippschild U. CK1alpha overexpression correlates with poor survival in colorectal cancer. BMC Cancer. 2018;18:140. doi: 10.1186/s12885-018-4019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lantermann A.B., Chen D., McCutcheon K., Hoffman G., Frias E., Ruddy D., Rakiec D., Korn J., McAllister G., Stegmeier F., et al. Inhibition of casein kinase 1 alpha prevents acquired drug resistance to erlotinib in EGFR-mutant non-small cell lung cancer. Cancer Res. 2015;75:4937–4948. doi: 10.1158/0008-5472.CAN-15-1113. [DOI] [PubMed] [Google Scholar]
- 16.Cohen P., Cross D., Jänne P.A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat. Rev. Drug Discov. 2021;20:551–569. doi: 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B., Wang M., Zhang W., Xiao T., Chen C.H., Wu A., Wu F., Traugh N., Wang X., Li Z., et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 2019;14:756–780. doi: 10.1038/s41596-018-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S., et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z., Hou X., Shao C., Li J., Cheng J.X., Kuang S., Ahmad N., Ratliff T., Liu X. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res. 2014;74:6635–6647. doi: 10.1158/0008-5472.CAN-14-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato M., Banuelos C.A., Imamura Y., Leung J.K., Caley D.P., Wang J., Mawji N.R., Sadar M.D. Cotargeting androgen receptor splice variants and mTOR signaling pathway for the treatment of castration-resistant prostate cancer. Clin. Cancer Res. 2016;22:2744–2754. doi: 10.1158/1078-0432.CCR-15-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam H.M., Nguyen H.M., Labrecque M.P., Brown L.G., Coleman I.M., Gulati R., Lakely B., Sondheim D., Chatterjee P., Marck B.T., et al. Durable response of enzalutamide-resistant prostate cancer to supraphysiological testosterone is associated with a multifaceted growth suppression and Impaired DNA damage response transcriptomic program in patient-derived xenografts. Eur. Urol. 2020;77:144–155. doi: 10.1016/j.eururo.2019.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wengner A.M., Scholz A., Haendler B. Targeting DNA damage response in prostate and breast cancer. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21218273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abida W., Cyrta J., Heller G., Prandi D., Armenia J., Coleman I., Cieslik M., Benelli M., Robinson D., Van Allen E.M., et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA. 2019;116:11428–11436. doi: 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta S., Halabi S., Kemeny G., Anand M., Giannakakou P., Nanus D.M., George D.J., Gregory S.G., Armstrong A.J. Circulating tumor cell genomic evolution and hormone therapy outcomes in men with metastatic castration-resistant prostate cancer. Mol. Cancer Res. 2021;19:1040–1050. doi: 10.1158/1541-7786.MCR-20-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L., Karanika S., Yang G., Wang J., Park S., Broom B.M., Manyam G.C., Wu W., Luo Y., Basourakos S., et al. Androgen receptor inhibitor-induced "BRCAness" and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aam7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minzel W., Venkatachalam A., Fink A., Hung E., Brachya G., Burstain I., Shaham M., Rivlin A., Omer I., Zinger A., et al. Small molecules Co-targeting CKIalpha and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell. 2018;175:171–185.e25. doi: 10.1016/j.cell.2018.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koh C.M., Bieberich C.J., Dang C.V., Nelson W.G., Yegnasubramanian S., De Marzo A.M. MYC and prostate cancer. Genes Cancer. 2010;1:617–628. doi: 10.1177/1947601910379132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amit S., Hatzubai A., Birman Y., Andersen J.S., Ben-Shushan E., Mann M., Ben-Neriah Y., Alkalay I. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002;16:1066–1076. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu C., Li Y., Semenov M., Han C., Baeg G.H., Tan Y., Zhang Z., Lin X., He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z., Cheng L., Li J., Farah E., Atallah N.M., Pascuzzi P.E., Gupta S., Liu X. Inhibition of the wnt/beta-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res. 2018;78:3147–3162. doi: 10.1158/0008-5472.CAN-17-3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elyada E., Pribluda A., Goldstein R.E., Morgenstern Y., Brachya G., Cojocaru G., Snir-Alkalay I., Burstain I., Haffner-Krausz R., Jung S., et al. CKIalpha ablation highlights a critical role for p53 in invasiveness control. Nature. 2011;470:409–413. doi: 10.1038/nature09673. [DOI] [PubMed] [Google Scholar]
- 32.Pribluda A., Elyada E., Wiener Z., Hamza H., Goldstein R.E., Biton M., Burstain I., Morgenstern Y., Brachya G., Billauer H., et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell. 2013;24:242–256. doi: 10.1016/j.ccr.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Kounatidou E., Nakjang S., McCracken S.R.C., Dehm S.M., Robson C.N., Jones D., Gaughan L. A novel CRISPR-engineered prostate cancer cell line defines the AR-V transcriptome and identifies PARP inhibitor sensitivities. Nucleic Acids Res. 2019;47:5634–5647. doi: 10.1093/nar/gkz286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polkinghorn W.R., Parker J.S., Lee M.X., Kass E.M., Spratt D.E., Iaquinta P.J., Arora V.K., Yen W.F., Cai L., Zheng D., et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3:1245–1253. doi: 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karanika S., Karantanos T., Li L., Wang J., Park S., Yang G., Zuo X., Song J.H., Maity S.N., Manyam G.C., et al. Targeting DNA damage response in prostate cancer by inhibiting androgen receptor-CDC6-ATR-chk1 signaling. Cell Rep. 2017;18:1970–1981. doi: 10.1016/j.celrep.2017.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blackford A.N., Jackson S.P. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 37.Vítor A.C., Huertas P., Legube G., de Almeida S.F. Studying DNA double-strand break repair: an ever-growing toolbox. Front. Mol. Biosci. 2020;7:24. doi: 10.3389/fmolb.2020.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shieh S.Y., Ikeda M., Taya Y., Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L., Goodrich D.W. RB1, cancer lineage plasticity, and therapeutic resistance. Annu. Rev. Cell Biol. 2022;6:201–221. doi: 10.1146/annurev-cancerbio-070120-092840. [DOI] [Google Scholar]
- 40.Mu P., Zhang Z., Benelli M., Karthaus W.R., Hoover E., Chen C.C., Wongvipat J., Ku S.Y., Gao D., Cao Z., et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rickman D.S., Beltran H., Demichelis F., Rubin M.A. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat. Med. 2017;23:1–10. doi: 10.1038/nm.4341. [DOI] [PubMed] [Google Scholar]
- 42.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Q., Zou Y., Nowotschin S., Kim S.Y., Li Q.V., Soh C.L., Su J., Zhang C., Shu W., Xi Q., et al. The p53 family coordinates Wnt and nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell. 2017;20:70–86. doi: 10.1016/j.stem.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong A.L., Tseng Y.Y., Cowley G.S., Jonas O., Cheah J.H., Kynnap B.D., Doshi M.B., Oh C., Meyer S.C., Church A.J., et al. Integrated genetic and pharmacologic interrogation of rare cancers. Nat. Commun. 2016;7 doi: 10.1038/ncomms11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Canman C.E., Lim D.S., Cimprich K.A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M.B., Siliciano J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 46.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiederschain D., Wee S., Chen L., Loo A., Yang G., Huang A., Chen Y., Caponigro G., Yao Y.M., Lengauer C., et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8:498–504. doi: 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- 48.Patil A., Manzano M., Gottwein E. CK1alpha and IRF4 are essential and independent effectors of immunomodulatory drugs in primary effusion lymphoma. Blood. 2018;132:577–586. doi: 10.1182/blood-2018-01-828418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stewart S.A., Dykxhoorn D.M., Palliser D., Mizuno H., Yu E.Y., An D.S., Sabatini D.M., Chen I.S.Y., Hahn W.C., Sharp P.A., et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W., Köster J., Xu H., Chen C.H., Xiao T., Liu J.S., Brown M., Liu X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015;16:281. doi: 10.1186/s13059-015-0843-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 56.Farah E., Li C., Cheng L., Kong Y., Lanman N.A., Pascuzzi P., Lorenz G.R., Zhang Y., Ahmad N., Li L., et al. NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. J. Biol. Chem. 2019;294:8543–8554. doi: 10.1074/jbc.RA118.006983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J., He D., Cheng L., Huang C., Zhang Y., Rao X., Kong Y., Li C., Zhang Z., Liu J., et al. p300/CBP inhibition enhances the efficacy of programmed death-ligand 1 blockade treatment in prostate cancer. Oncogene. 2020;39:3939–3951. doi: 10.1038/s41388-020-1270-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The datasets of CRISPR Screen and RNA-seq are available at NCBI (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE203362), referring to GSE203362.
-
•
This paper does not generate the original code.
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.