

Abstract
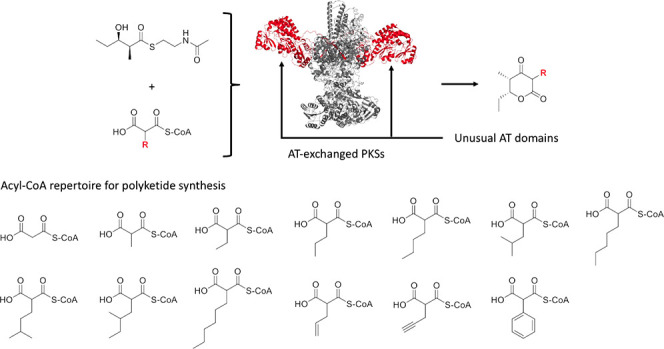
Modular polyketide synthases (PKSs) are polymerases that employ α-carboxyacyl-CoAs as extender substrates. This enzyme family contains several catalytic modules, where each module is responsible for a single round of polyketide chain extension. Although PKS modules typically use malonyl-CoA or methylmalonyl-CoA for chain elongation, many other malonyl-CoA analogues are used to diversify polyketide structures in nature. Previously, we developed a method to alter an extension substrate of a given module by exchanging an acyltransferase (AT) domain while maintaining protein folding. Here, we report in vitro polyketide biosynthesis by 13 PKSs (the wild-type PKS and 12 AT-exchanged PKSs with unusual ATs) and 14 extender substrates. Our ∼200 in vitro reactions resulted in 13 structurally different polyketides, including several polyketides that have not been reported. In some cases, AT-exchanged PKSs produced target polyketides by >100-fold compared to the wild-type PKS. These data also indicate that most unusual AT domains do not incorporate malonyl-CoA and methylmalonyl-CoA but incorporate various rare extender substrates that are equal to in size or slightly larger than natural substrates. We developed a computational workflow to predict the approximate AT substrate range based on active site volumes to support the selection of ATs. These results greatly enhance our understanding of rare AT domains and demonstrate the benefit of using the proposed PKS engineering strategy to produce novel chemicals in vitro.
Introduction
Assembly-line enzymes produce a variety of bioactive molecules that are difficult to chemically synthesize. Modular polyketide synthases (PKSs) are one group of assembly-line enzymes that are composed of several catalytic modules and incorporate an α-carboxyacyl coenzyme A (CoA) at each module.1,2 Each PKS module contains several protein domains, including ketosynthase (KS), acyltransferase (AT), acyl carrier protein (ACP), ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER). Among these domains, the polyketide chain elongation reaction is catalyzed by the KS, AT, and ACP, while the other catalytic domains modify the nascent polyketide chain attached to the ACP. After chain extension and modification reactions, the resulting polyketide intermediate is transferred to the next module for further elongation and processing. A full-length polyketide tethered to an ACP is then offloaded from the assembly line, usually by a thioesterase (TE) domain. In many cases, the resulting polyketide is further decorated by non PKS enzyme(s). Some of these compounds are clinically used as antibacterial (erythromycin, leucomycin), antifungal (nystatin, natamycin), and immunosuppressant agents (FK506, rapamycin).3
AT domains are responsible for extender substrate selection. Although more than 90% of ATs incorporate either malonyl-CoA or methylmalonyl-CoA,4 at least 20 α-carboxyacyl-CoAs, all malonyl-CoA analogues at the C2 position, have been proposed to be incorporated into naturally occurring polyketides. These rare extender substrates include butylmalonyl-CoA, pentylmalonyl-CoA, hexylmalonyl-CoA, allylmalonyl-CoA, and benzylmalonyl-CoA, to name a few.5 Several different PKS engineering techniques have been developed to incorporate unnatural extender substrates.6 One of the most common approaches is the exchange of an entire AT domain for a homologue with different substrate specificity. Although many polyketide analogues have been produced by replacing one of the methylmalonyl-CoA specific ATs in target PKSs using a malonyl-CoA specific AT,7 most of the engineered PKSs showed significantly reduced catalytic activities compared to their wild-type counterparts, probably due to either incompatible domain boundaries used and/or substrate preference of downstream domains of the PKSs. Since then, more precise PKS engineering techniques have been used to exchange ATs.8
Previously, we experimentally analyzed several different domain boundaries to exchange an AT domain using two model PKS systems in vitro, which are module 6 of the erythromycin PKS (DEBS M6+TE) and module 1 of the lipomycin PKS.9 Kinetic parameters obtained from the AT-exchanged mutants revealed compatible and incompatible domain boundaries and suggested that an entire linker between KS and AT domains (KS-AT Linker = KAL) and the first part of a post AT linker (Post AT Linker 1 = PAL1) should be included to exchange an AT domain, which appears to maintain folding of the resulting AT-exchanged PKS module (KAL-AT-PAL1 exchange).9 We also reported on the successful exchange of ATs in another PKS module.10 Furthermore, recent molecular dynamics simulations support that KAL-AT-PAL1 fragments are structurally more stable than those lacking PAL1 or both KAL and PAL1.11 Here, we selected unusual ATs that are predicted to incorporate rare extender substrates and constructed the corresponding AT-exchanged PKSs based on KAL-AT-PAL1 fragments. Twelve AT-exchanged PKSs (=71%) were active when assayed with 14 different extender substrates, including the standard extender substrates, malonyl-CoA and methylmalonyl-CoA. Our ∼200 in vitro reactions resulted in several new-to-nature polyketides that have not been reported. Furthermore, some of the AT-exchanged PKSs produced target polyketide titers >100-fold compared to the wild-type PKS, indicating that we could employ the KAL-AT-PAL1 exchange to incorporate unusual AT domains into PKS modules to produce new-to-nature molecules. We also describe a framework we have developed to predict AT substrates based on three-dimensional (3D) models of AT active sites, which aids the selection of ATs for unnatural polyketide production.
Results and Discussion
Construction of AT-exchanged PKSs
In our previous AT domain exchange efforts, to replace the native KAL-AT-PAL1 fragments with counterparts from other PKS modules, we selected module 4 of the epothilone PKS, module 1 of the borrelidin PKS, module 2 of the rapamycin PKS, module 9 of the indanomycin PKS, module 2 of the spinosad PKS, and module 7 of the curacin PKS, all of which, except for module 4 of the epothilone PKS (see Table 1), exclusively incorporate malonyl-CoA. We then identified KAL-AT-PAL1 fragments by searching highly conserved GTNAH and LPTY(A/P)FQ(H/R)xRYWL sequences (Table S1).9 The average length of KAL-AT-PAL1 used was 442 ± 32 amino acids (the native KAL-AT-PAL1 fragments are 442 and 452 amino acids long, respectively). Approximately 80% of the resulting AT-exchanged PKSs were active to produce the predicted polyketides.
Table 1. AT-Exchanged PKSs Used in This Study.
| PKSs | source bacteria | source genes | source of KAL-AT-PAL1s | natural substrates | purification yields (relative to WT) | abbreviations |
|---|---|---|---|---|---|---|
| DEBS M6 + TE | Saccharopolyspora erythraea NRRL 2338 | eryAI | methylmalonyl-CoA | 1.0 | WT | |
| 1 | Sorangium cellulosum SMP44 | epoD | module 4 | malonyl-CoAmethylmalonyl-CoA | 0.7 | Epo-4 |
| 2 | Streptomyces cinnamonensis ATCC 15413 | monAIV | module 5 | methylmalonyl-CoAethylmalonyl-CoA | 1.6 | Mon-5 |
| 3 | Streptomyces caelestis NRRL 2821 | nidA3 | module 5 | ethylmalonyl-CoA | 1.1 | Nid-5 |
| 4 | Streptomyces sp. SN-593 | revA | module 4 | butylmalonyl-CoAhexylmalonyl-CoA etc. | 0.9 | Rev-4 |
| 5 | Streptomyces sp. W112 | divB | module 4 | ethylmalomyl-CoA | 0.9 | Div-4 |
| 6 | Streptomyces sp. CNH-189 | ansE | module 8 | isobutylmalonyl-CoA | 0.7 | Ans-8 |
| 7 | Streptomyces flaveolus DSM 9954 | sfaI | module 13 | 3-oxobutylmalonyl-CoA | 0.8 | San-13 |
| 8 | Sorangium cellulosum So ce690 | leuA | module 2 | 1-hydroxyisopentylmalonyl-CoA | 0.5 | Leu-2 |
| 9 | Streptomyces sp. W112 | divD | module 6 | isobutenylmalonyl-CoA | 1.1 | Div-6 |
| 10 | Salinispora tropica CNB-476 | salA | module 1 | 2-chloroethylmalonyl-CoA | 0.1 | Sal-1 |
| 11 | Streptomyces ambofaciens ATCC 23877 | pks4 | module 12 | hexylmalonyl-CoAisoheptylmalonyl-CoA etc. | 0.1 | Sta-12 |
| 12 | Streptomyces sp. CNQ431 | spnD | module 3 | benzylmalonyl-CoA | 0.3 | Spl-3 |
In the present work, in addition to module 4 of the epothilone PKS, we selected 16 PKS modules that are proposed to incorporate rare extender substrates to create a second generation of AT-exchanged PKSs using DEBS M6+TE as an acceptor (Figure 1a,b). The average length of KAL-AT-PAL1 fragments used was 454 ± 21 amino acids long. Escherichia coli K207–3,12 which is an engineered strain whose genome encodes the substrate promiscuous phosphopantetheinyl transferase Sfp from Bacillus subtilis that converts the produced apo-PKSs to their corresponding holo forms, was transformed with a plasmid that encodes each PKS variant (Table S2) and used to produce active PKS proteins. These proteins were then purified by metal ion affinity chromatography and anion exchange chromatography. Although five chimeric PKSs could not be purified with acceptable purity, 11 new AT-exchanged PKSs were successfully purified (Figure S1). Approximately 70% of the purified PKSs showed purification yields comparable to the wild-type PKS, which indicates that most AT-exchanged PKSs were structurally stable (Table 1).
Figure 1.
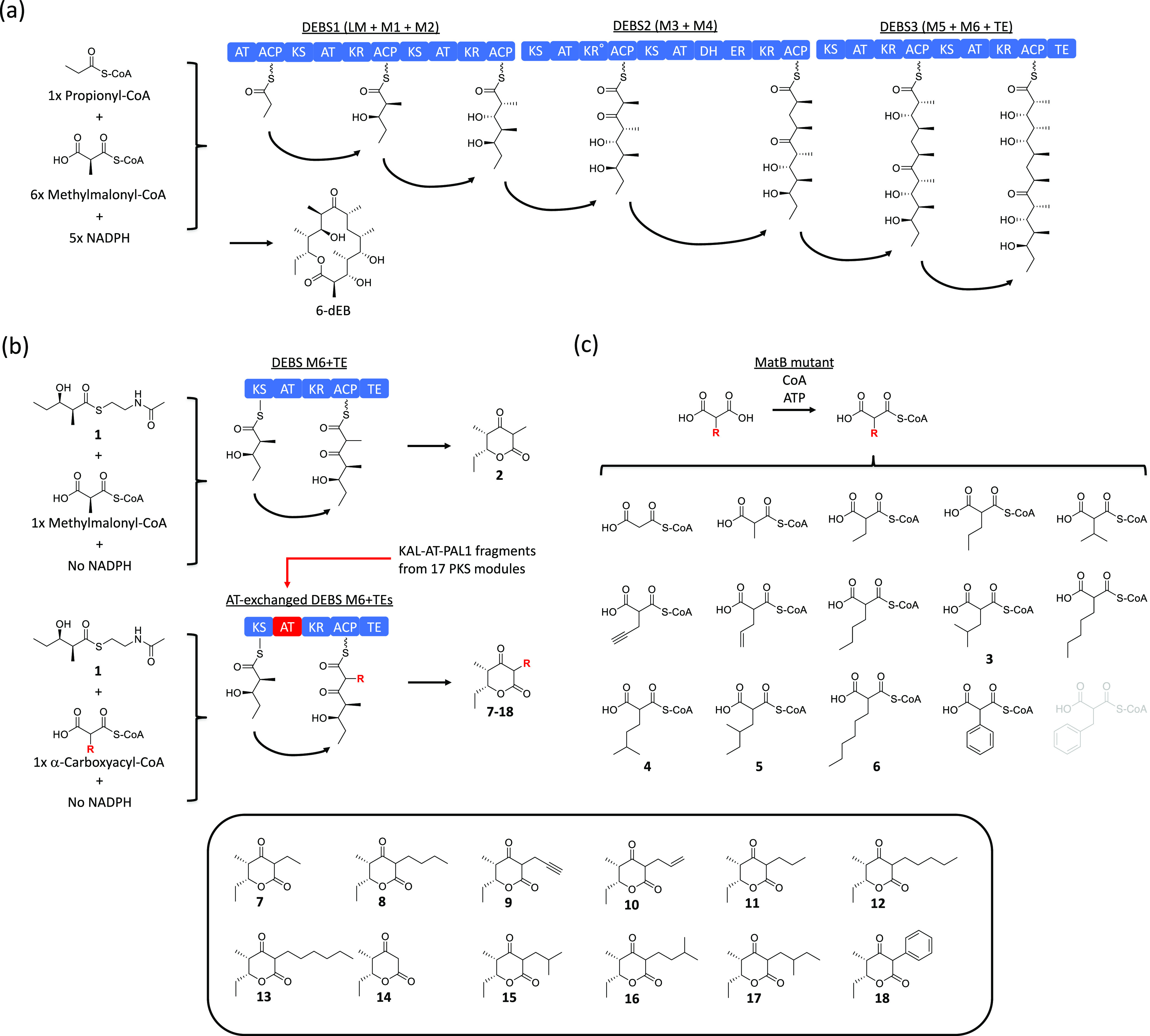
Model PKS systems and extender substrates used in this study. (a) Proposed model for 6-dEB biosynthesis by DEBS. (b) Proposed model for biosynthesis of 2, 7–18 using the truncated PKS system. (c) α-Carboxylacyl-CoAs synthesized using a MatB mutant. Benzylmalonyl-CoA was not synthesized (shown in gray). Abbreviations: 6-dEB, 6-deoxyerythronolide B; ATP, adenosine triphosphate; AT, acyltransferase; ACP, acyl carrier protein; CoA, coenzyme A; DH, dehydratase; DEBS, 6-deoxyerythronolide B synthase; ER, enoyl reductase; KR, ketoreductase; KR°, redox inactive ketoreductase; KS, ketosynthetase; LM, loading module; M, module; PKS, polyketide synthase; TE, thioesterase; and TKL, triketide lactone.
Preparation of Rare Extender Substrates
Except for malonyl-CoA and methylmalonyl-CoA, PKS extender substrates are not commercially available. To synthesize various α-carboxyacyl-CoAs, we employed an engineered acyl-CoA synthetase reported previously (MatB T207G/M306I), which efficiently converts malonic acid or malonic acid analogues at the C2 position into the corresponding acyl-CoAs.13 Fifteen diacids, including malonic acid and methylmalonic acid, were incubated with CoA and MatB T207G/M306I, and production of the corresponding acyl-CoAs and consumption of free CoA were monitored by liquid chromatography–time-of-flight mass spectrometry (LC-TOF MS). All diacids, except for benzylmalonic acid, were successfully converted into the corresponding acyl-CoAs (Figures 1c and S2a). Product yields were approximately ∼90%, except for phenylmalonyl-CoA, which was produced at a 25% yield (Figure S2a). Although production of most of these acyl-CoAs has been reported previously,13,14 we observed four additional products that have not been reported in the literature: isobutylmalonyl-CoA (3), isopentylmalonyl-CoA (4), 2-methylbutylmalonyl-CoA (5), and hexylmalonyl-CoA (6). As shown in Figure S2b,c, the mass errors observed were within a few ppm, except for 2-methylbutylmalonyl-CoA (5). Some ATs we selected are proposed to use 3-oxobutylmalonyl-CoA, 1-hydroxyisopentylmalonyl-CoA, isobutenylmalonyl-CoA, 2-chloroethylmalonyl-CoA, isoheptylmalonyl-CoA, and/or 4-methylhexylmalonyl-CoA. However, these extender substrates were not synthesized in the present study because the corresponding diacids or diesters, which can be converted to diacids by hydrolysis, were either not commercially available or too expensive to have synthesized for the experiment.
Qualitative Analysis of Unnatural Polyketide Production
To test if a panel of α-carboxyacyl-CoAs can be used as extender substrates, a starter substrate (1), which is a thioester derivative of the natural diketide chain elongation intermediate of DEBS module 2, and each PKS were directly added into a MatB reaction mixture, where the resulting triketide lactones only differ from each other in the functionality at the C2 position. Each reaction contained a different extender substrate at approximately 300–400 μM, except for phenylmalonyl-CoA, whose reaction mixture contained approximately 100 μM of the acyl-CoA due to the low yield in the MatB reaction. Because KM values of PKS AT domains are usually in the 1–10 μM range,9 we have assumed that each PKS reaction is saturated with a given extender substrate. To simplify product analysis, we omitted NADPH from the reactions because DEBS M6 + TE has been reported to produce both the reduced (3-hydroxy) and the nonreduced (3-keto) forms of the polyketide products in the presence of NADPH.9 After the overnight reaction, the resulting polyketides were extracted with ethyl acetate, dried, and dissolved in 50% methanol. LC-TOF MS was used to detect and identify the polyketides produced (Figure 2 and Tables S3–S14).
Figure 2.
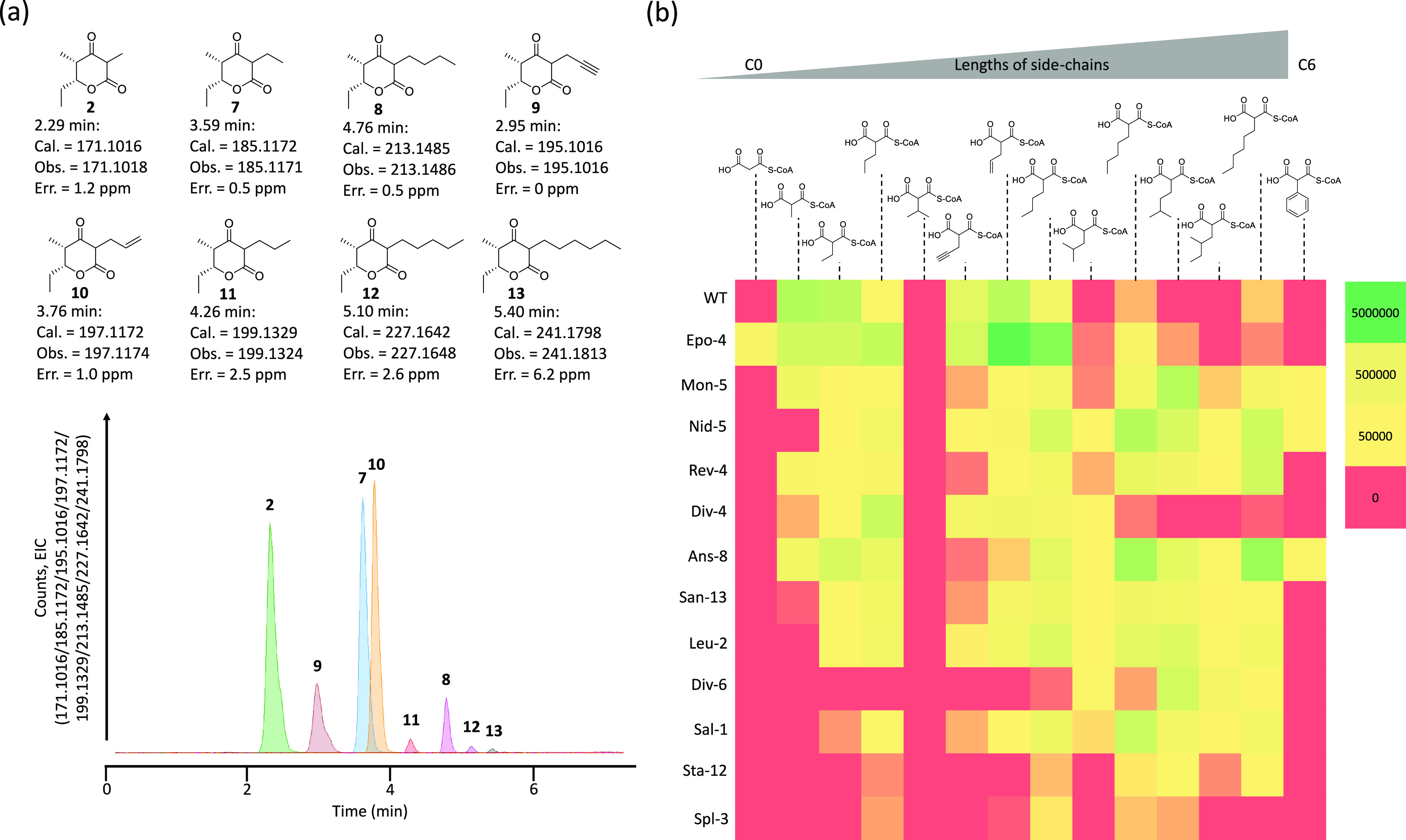
In vitro production of unnatural polyketides from various extender substrates. (a) Production of polyketides 2, and 7–13 by the wild-type PKS. Extracted ion chromatograms (EIC) of 171.1016, 185.1172, 195.1016, 197.1172, 199.1329, 213.1485, 227.1642, and 241.1798 were overlaid. Observed mass errors were within a few ppm, except for polyketide 13 (6.2 ppm). (b) Production of polyketides 2, and 7–18 by the wild-type PKS and 12 AT-exchanged PKSs with 14 extender substrates. Heatmaps show LC-MS peak areas of each reaction (mass counts <5000 were regarded as 0). Each value is an average of at least two experiments.
The wild-type DEBS M6+TE incorporated not only the natural substrate, methylmalonyl-CoA, but also unnatural extender substrates ethylmalonyl-CoA, butylmalonyl-CoA, propargylmalonyl-CoA, and allylmalonyl-CoA (7–10), as determined by LC-TOF MS, where the mass errors were within a few ppm.15 As shown in Figure 2a, we also observed several other polyketide products using propylmalonyl-CoA, pentylmalonyl-CoA, and hexylmalonyl-CoA with reasonable mass errors (11–13), and the elution times were shifted depending on the sizes of the C2 side chains. The mass counts for most products, however, were approximately 100-fold lower than that of 2 (2-methyl), 7 (2-ethyl), and 10 (2-allyl), as shown in Table S3. We did not see any product when isopropylmalonyl-CoA was used as an extender substrate, which is consistent with a previous report.15 It appears that other acyl-CoAs listed in Figure 1c are not processed by the wild-type PKS.
We previously reported that an AT-exchanged PKS, where the KAL-AT-PAL1 of DEBS M6+TE was exchanged with the counterpart in module 4 of the epothilone PKS (Epo-4, see Table 1 for abbreviations of AT-exchanged PKSs used in this study) showed a wild-type level of activity to produce product 2 (2-methyl); the kcat of the engineered PKS was 85% of that of WT. Epo-4 also produced product 14 (2-desmethyl) using malonyl-CoA as an extender substrate (Figure S3), which is not accepted by the native AT.9 In the present study, we investigated whether Epo-4 incorporates another 12 extender substrates in vitro (Figure 2b, second row). While we could not confirm polyketide production with isobutylmalonyl-CoA and isopentylmalonyl-CoA using WT, Epo-4 produced polyketides 15 (2-isobutyl) and 16 (2-isopentyl) using these extender substrates (Figure S4). Interestingly, Epo-4 showed increased production of 11 (2-propyl) by ∼20-fold, 8 (2-butyl) by ∼10-fold, and 12 (2-pentyl) by ∼10-fold compared to WT, while production of 14 (2-hexyl) was similar (Table S3). Our data suggest that Epo-4 AT, when incorporated into the DEBS M6, also accepts substrates that are slightly larger than the substrates it is known to naturally use (Table 1 and Figure S5).
We performed similar polyketide production experiments using the other 11 AT-exchanged PKSs. The production data are summarized in Figure 2b and Tables S4–S14. Dejong et al. previously surveyed AT specificities of PKS modules in silico and reported that most AT domains incorporate either malonyl-CoA (70.8%) or methylmalonyl-CoA (23.7%).4 Because the native metabolisms of many Streptomyces species normally generate the two standard substrates, AT domains that use rare extender substrates must avoid incorporating malonyl-CoA and methylmalonyl-CoA. Indeed, of the 12 AT-exchanged PKSs, only the PKS containing Epo-4 AT, which natively uses malonyl-CoA as a substrate, produced polyketide 14 (2-desmethyl) when malonyl-CoA was used as an extender. Similarly, most of the AT-exchanged mutants did not produce or produced a small amount of 2 (2-methyl). While Mon-5 and Ans-8 produced a low level of polyketide 2 (2-methyl), the monensin PKS is known to naturally incorporate both methylmalonyl-CoA and ethylmalonyl-CoA at module 5 (Table 1). The ansalactam PKS is not known to produce methyl-substituted natural products, to our knowledge. It is possible that the downstream KS domains of the PKS may not extend methyl-substituted products.
We also tested whether these AT-exchanged PKSs produce polyketides with the rare extender substrates (Figure 1c). Overall, as was the case with Epo-4, the AT domains tested were able to incorporate substrates that are equal to in size or larger than their natural substrates by a few carbon atoms (Figures 2b, S5, and Tables S4–S14). Again, since >90% of ATs use malonyl- or methylmalonyl-CoA for extension, and it would be rare to have multiple other malonyl-CoA analogues incorporated into a polyketide, these results may explain how ATs avoid incorporating incorrect (malonyl- or methylmalonyl-CoA) substrates in their natural environment. Using AlphaFold2, we also tested if the structures of the AT domains were perturbed from their native state by incorporation into the DEBS M6.16 The calculated root-mean-square deviation values for ATs in the AT-exchanged DEBS M6 relative to the same ATs to their native PKS modules were less than 0.2 Å (0.09 < Å < 0.20), suggesting that their structures are virtually identical after AT exchange. This hypothesis is supported by our previous AT exchange efforts, where methylmalonyl-CoA incorporation has never been observed when we employed malonyl-CoA-specific ATs in KAL-AT-PAL1 exchange.9 We have also observed that many AT-exchanged PKSs showed increased production of several different polyketides. In some cases, product levels were >100-fold compared to the native DEBS M6 + TE (Tables S5 and S8). It should also be noted that we have observed new polyketides 17 [2-(2-methylbutyl)] and 18 (2-phenyl) with 2-methylbutylmalonyl-CoA and phenylmalonyl-CoA, which were both produced using the Mon-5, Nid-5, and Ans-8 ATs (Figure S6, Tables S4, S5, and S8). Interestingly, there is no known natural PKS that incorporates phenylmalonyl-CoA, to our knowledge. These results indicate that we can diversify the extender substrate portfolio using KAL-AT-PAL1 exchange strategy combined with unusual AT domains in vitro.
Quantitative Analysis of Unnatural Polyketide Production
To further confirm in vitro polyketide production, we purchased authentic standards for 7 (2-ethyl), 8 (2-butyl), 13 (2-hexyl), and 18 (2-phenyl), which were used to reanalyze representative PKS reactions. These synthetic standards have not been used to quantify PKS products in previous studies, to our knowledge, and, importantly, we noticed that our assumption that detection efficiency of the various lactone products by LC-TOF MS is equivalent or increases/decreases linearly depending on sizes of side chains was not correct. As expected, retention times in LC-TOF MS were well matched between authentic standards and polyketides synthesized in vitro (Figure S7), which validates target polyketide production. To simplify polyketide quantification, we selected six PKSs (those with WT, Epo-4, Nid-5, Rev-4, Ans-8, and San-13 ATs) and quantified the production of polyketides 2 (2-methyl), 7 (2-ethyl), 8 (2-butyl), and 13 (2-hexyl) using endpoint assays. Unfortunately, the authentic standard 18 (2-phenyl) was not stable enough to reliably quantify enzymatic products. We have chosen these PKSs based on activities (Figure 2b and Tables S3–S14), protein purification yields (Table 1), and proposed substrate binding motifs of ATs (Figure 3a–c and Table S15). The four binding motifs were selected based on previous literature, except for the first motif.11,17 The first motif is in proximity of the active site serine, according to our structural modeling.
Figure 3.
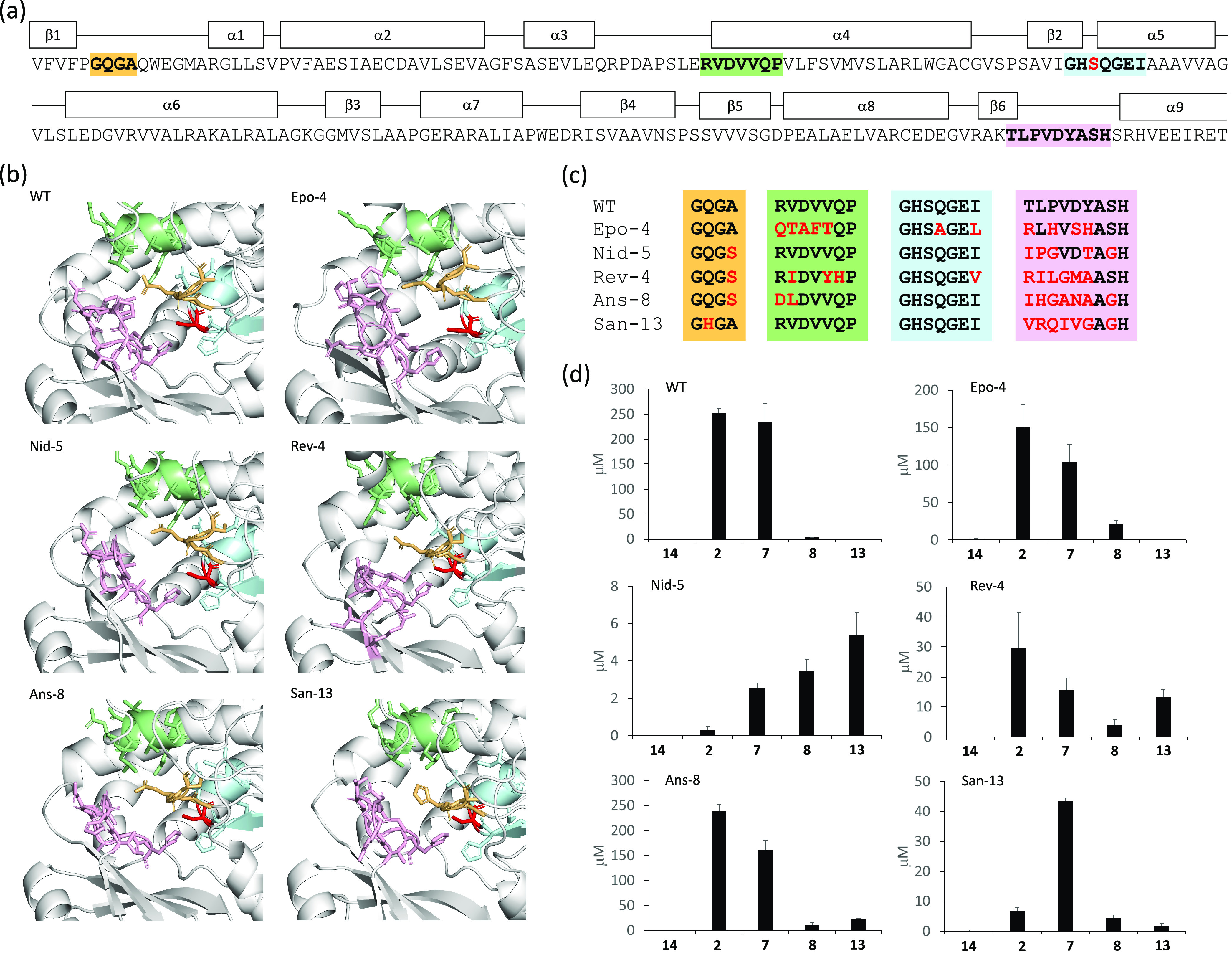
Structural models of ATs and quantitation of unnatural polyketides produced. (a) Amino acid sequence of DEBS M6 AT and secondary structure elements predicted by AlphaFold2. Amino acid residues that may form a substrate binding pocket are shown in orange, green, cyan, and pink. The active site serine residue is shown in red. (b) Tertiary structures of six ATs predicted by AlphaFold2. The same color scheme is used as in panel (a). (c) Sequence alignment of amino acid residues of six ATs that may form a substrate binding pocket. Amino acid residues that are different from DEBS M6 AT are shown in red. (d) Production of polyketides 2 (2-methyl), 7 (2-ethyl), 8 (2-butyl), 13 (2-hexyl), and 14 (2-desmethyl) were quantified at 16 h using the corresponding authentic standards. Each value and error are calculated from three independent experiments. Abbreviations (see Figure 1 and Table 1).
As shown in Figure 3d, LC-TOF MS analysis with authentic standards revealed nearly indistinguishable production levels of polyketides 2 (2-methyl) and 7 (2-ethyl) by WT, indicating that the KS does not discriminate methylmalonyl and ethylmalonyl extension units in condensation reactions, provided that the chain extension is the rate-limiting reaction, as previously suggested.2 On the other hand, other polyketide products were produced at a concentration less than 1% of those of 2 and 7. Based on these observations, one can conclude that potential substrate binding motifs of WT (GQGA, RVDVVQP, GHSQGEI, and TLPVDYASH) may together allow methylmalonyl-CoA and ethylmalonyl-CoA as extender substrates.
The AT of Nid-5 and San-13 have identical second (RVDVVQP) and third (GHSQGEI) substrate binding motifs to those of DEBS module 6 (Figure 3c). However, the product profiles of the three PKSs (WT, Nid-5, and San-13) are very different (Figure 3d), indicating that the second and third binding motifs are not major determinants of substrate incorporation for the ATs. On the other hand, WT, Epo-4, and Ans-8 have different substrate binding motifs (Table S15) but showed somewhat similar product distributions, with the exception that Ans-8 could also incorporate hexylmalonyl-CoA (Figure 4b). These data, if AT structures are not perturbed by KAL-AT-PAL1 exchange as was previously discussed, suggest that the substrate binding motifs are not sufficient to predict AT substrate specificity and that a new analytical algorithm is needed to precisely predict AT substrate specificity to select the desired AT domains.
Figure 4.

Analysis of the active site shape for 1000+ ATs with substrate annotations. (a) Schematic illustrating pipeline for mapping AT active sites. (b) Comparison of active site volume for each AT model grouped by the annotated substrate. The pocket volume is calculated by summing the active site point cloud. The white dots represent the median pocket volume. The ends of the thick black bars represent the first and third quartiles. The ends of the thin black bars represent the minimum and maximum. (c) Active site volumes were calculated from five relaxed structural models predicted by AlphaFold2. (d) Two-dimensional decomposition of active site shape using t-SNE colored by the annotated substrate. Eleven ATs used in this study are scattered within the red box.
AT Active Site Mapping to Predict Substrate Specificity
To develop a workflow for AT substrate prediction, we modeled structures of >1000 AT domains using AlphaFold2 (classified by their documented substrate choice) and then developed a simple algorithm that aligned the structures, detected the active sites, and created 3D clouds to represent the shape of the active sites (Figures 4a and S8). Using this shape model, we compared active site volumes of various AT domains and revealed a loose correlation between the volumes and sizes of the side chains of extender substrates (Figure 4b). Active site volumes of ATs that were used in this study are summarized in Figure 4c. Most ATs appear to have larger active site volumes (>1500 Å3) relative to ATs that natively incorporate methylmalonyl-CoA. Violin plots (Figure 4b) suggest that many of the ATs we tested in vitro prefer extender substrates with C4–C6 side chains, roughly in line with our experimental results, with the exception of Epo-4 AT (Figure 2b). While Epo-4 AT has the smallest active site volume (Figure 4c), it incorporates a wide range of extender substrates. We have also visualized relationships between various AT domains by decomposing the 3D point cloud into two-dimensional feature vectors. AT domains that are predicted to incorporate malonyl-CoA or methylmalonyl-CoA were clearly separated from each other (Figure 4d and Data Set S1). On the other hand, unusual AT domains, except for Epo-4 AT, are scattered mostly throughout AT domains that are proposed to incorporate methylmalonyl-CoA. Phylogenetic analysis of whole AT domains demonstrates a similar trend.18 However, it is clear that the specificity and promiscuity of an enzyme is a spectrum with many determining factors. It is very likely that other active site properties beyond geometry such as electrostatic and hydrophobic interactions are required to precisely predict such a spectrum, which is currently under investigation. Lastly, we have modeled active sites of >6000 AT domains, including nonannotated ATs, which were obtained from UniRef9019 using Epo-4 AT as a query. This analysis showed that the spectrum of AT active site shape is very vast and indicates that the ATs with substrate annotations only partly cover the entire structural space (Figure S9).
KS Active Site Mutation for Unnatural Polyketide Biosynthesis
In hybrid modular PKS systems, KS engineering may play an important role in efficient unnatural polyketide production. Murphy et al. reported that a specific KS active site residue could be changed (A to W) to broaden the substrate specificity of DEBS M3 KS.20 Although the corresponding mutation in DEBS M6 KS appears not to greatly enhance chain extension with unnatural starter substrates,21 we sought to test if the mutation would have some effect on extender substrate incorporation in our engineered PKS systems. To do this, we purified WT, Epo-4, and Rev-4 variants that possess A162W mutation using the same purification protocols. The purification yields were comparable to those of the parental PKSs.
We initially analyzed WT with A162W mutation and observed an increased level of incorporation of methylmalonyl-CoA, albeit with essentially no effect on the use of ethylmalonyl-CoA, butylmalonyl-CoA, and hexylmalonyl-CoA as extender substrates (Figure S10). Next, we investigated whether the mutated KS with Epo-4 AT would produce an increased amount of polyketide 8 (2-butyl). It appears that the A to W mutation does not affect incorporation of a butylmalonyl extension unit, although it slightly changed the yields of the products 2 and 7 (Figure S10). However, when the KS mutation was combined with Rev-4 AT, we observed a different product profile (Figure S10). Although production of polyketide 8 (2-butyl) was not affected, the mutated PKS produced one-third the amount of polyketide 2 (2-methyl) and ∼3-fold more polyketides 7 (2-ethyl) and 13 (2-hexyl) than with the native KS. While these data partly support the results in the mutated DEBS KS3,20 KS engineering strategy should be explored to dramatically increase the incorporation of extender substrates with long side chains.
Conclusions
In summary, the modular nature of modular PKSs not only accounts for the thousands of natural polyketide structures but also provides a wealth of engineering opportunities.22 Among catalytic domains in the PKSs, AT domains are often a primary target of PKS engineering efforts because the AT determines the specific extender substrate(s) for each polymerization reaction. In the present study, we focused on expanding extender substrates of the PKSs using the KAL-AT-PAL1 exchange strategy.9 Using this method combined with unusual AT domains, we successfully produced several new-to-nature polyketides by extending with isobutylmalonyl-CoA, isopentylmalonyl-CoA, 2-methylbutylmalonyl-CoA, and phenylmalonyl-CoA, none of which can be used by the native AT. We also found AT domains that incorporate pentylmalonyl-CoA and/or hexylmalonyl-CoA very efficiently (>100-fold more efficient than those of the native AT). Previously, the entire DEBS PKS was reconstituted in vitro,23 and a few ethyl polyketide analogues were produced when both methylmalonyl-CoA and ethymalonyl-CoA were mixed in the in vitro assay. This result suggests that it is challenging to specify extender substrate incorporation by AT domain exchange alone if the structures of extender substrates are similar to methylmalonyl-CoA. On the other hand, our results indicate that AT domains that naturally incorporate methylmalonyl-CoA cannot incorporate extender substrates where side chain lengths are >C3 (Figures 3d, 4b, and S5). Hence, it is necessary to exchange the AT to incorporate extender substrates with >C4 side chains. Since it is extremely difficult to chemically replace side chains of naturally occurring polyketides, the KAL-AT-PAL1 exchange that we have outlined above is an effective way to selectively produce target unnatural polyketides in vitro and presumably in vivo. In the future, it may be necessary to develop a general strategy to broaden KS specificity as the simple active site mutation in the DEBS M6 KS was not effective in improving production of most targeted polyketides in our study.
Methods
Chemicals
All chemicals were purchased from Sigma-Aldrich (United States) unless otherwise described. (2S,3R)-3-hydroxy-2-methylpentanoic acid-SNAC thioester (1), (4S,5R)-3-oxo-2,4-dimethyl-5-hydroxy-heptanoic acid-d-lactone (2), and (4S,5R)-3-oxo-4-methyl-5-hydroxy-heptanoic acid-d-lactone (14) were synthesized as previously described.24 (4S,5R)-3-Oxo-2-ethyl-4-methyl-5-hydroxy-heptanoic acid-d-lactone (7), (4S,5R)-3-oxo-2-butyl-4-methyl-5-hydroxy-heptanoic acid-d-lactone (8), (4S,5R)-3-oxo-2-hexyl-4-methyl-5-hydroxy-heptanoic acid-d-lactone (13), and (4S,5R)-3-oxo-2-phenyl-4-methyl-5-hydroxy-heptanoic acid-d-lactone (18) were purchased from Acme Bioscience.
Plasmids and Strains
Plasmids and strains used in this study are listed in Supplementary Table 2. The plasmids and strains have been deposited in the public version of JBEI registry (http://public-registry.jbei.org) and are physically available from the corresponding author upon request (keasling@berkeley.edu). KAL-AT-PAL1 genes were codon-optimized for E. coli and synthesized. These genes were subcloned into pSY121 using KpnI and SalI sites.9
Protein Expression and Purification
Each PKS was produced in E. coli K207–312 and purified as described previously.25 Briefly, PKS genes were expressed in the E. coli at 18 °C in the presence of IPTG, and the proteins produced were purified by immobilized metal ion chromatography and anion exchange chromatography at 4 °C. The fractions containing target PKSs were concentrated and stored at −80 °C. Protein concentrations were measured using Bradford assay. MatB T207G/M306I was produced in E. coli BL21(DE3) and purified as described previously.13 Briefly, the MatB gene was expressed in the E. coli at 18 °C in the presence of IPTG, and the proteins produced were purified by immobilized metal ion chromatography at 4 °C. The fractions containing MatB T207G/M306I were concentrated and stored at −80 °C. Protein concentrations were measured using the Bradford assay.
In Vitro α-carboxyacyl-CoA Biosynthesis
Each a-carboxyacyl-CoA was synthesized as described previously.13 Briefly, each a-carboxyacyl-CoA was synthesized in 225 μL of reaction mixture containing 100 mM phosphate (pH 7.2), 2 mM MgCl2, 2 mM DTT, 400 μM ATP, 400 μM CoA, 800 μM malonic acid or the analogues, and 1 μM MatB T207G/M306I at room temperature (∼25 °C). Aliquots were removed at 1 h and 16 h and quenched by adding an equal volume of methanol. After centrifugation, the supernatants were analyzed by LC-TOF-MS (see the Supporting Information for details). For diacids that are not commercially available, which are isobutylmalonic acid, isoamylmalonic acid, (2-methyl)butylmalonic acid, and propargylmalonic acid, we purchased the corresponding diesters and hydrolyzed them using KOH to produce diacids.
In Vitro Polyketide Biosynthesis
Each a-carboxyacyl-CoA was synthesized in 225 μL of the reaction mixture containing 100 mM phosphate (pH 7.2), 2 mM MgCl2, 2 mM DTT, 400 μM ATP, 400 μM CoA, 800 μM malonic acid or the analogues, and 1 μM MatB T207G/M306I at room temperature (∼25 °C). After overnight incubation, each polyketide product was synthesized by adding a PKS and a starter substrate (1) into the reaction mixture at final concentrations of 1 μM and 5 mM, respectively. Aliquots were removed at 1 h and 16 h and quenched by extracting the product with twice the volume of ethyl acetate. After drying the organic phase, 50 mL of 50% methanol was added into each sample tube and well mixed. The resulting solutions were then analyzed by LC-TOF-MS (see the Supporting Information for details).
Creating Active Site Point Clouds
1070 AT sequences with substrate annotations were obtained from the ClusterCAD database,26 which uses antiSMASH2727 determined domain boundaries. 6546 diverse AT sequences with unknown substrates were obtained by first querying the UniRef90 database with jackhmmer28 using the sequence of Epo-4 AT and an e-value threshold of 1E-4. The initial 107 659 hits were filtered down with hhfilter29 by removing sequences with coverage under 75% (-cov = 0.75) and further filtering to the most diverse set of sequences (-diff = 5000). Each protein sequence was then modeled using the AlphaFold216 protein structure prediction algorithm with the following settings: --db_preset = reduced_dbs, --model_preset = monomer_ptm, --relax = true, --max_template_date = 2022-12-31, and --num_models = 5. AlphaFold modeling was separated into two stages: a CPU-focused MSA generation stage was performed on 24-core Intel Xeon E5–2670 v3 nodes and required approximately 22 min of compute time for each protein; a GPU-focused structure generation stage utilized a Nvidia GeForce GTX 1080ti GPU with 2-cores from an Intel Xeon E5–2623 v3 equipped node and required approximately 25 min of compute time to generate five relaxed models for each protein. The resultant 38 145 protein structures, which include models for the 13 ATs analyzed in this study, were structurally aligned in parallel to a reference protein DEBS M6 AT with TM-align30 and GNU parallel.31 The active site for each protein was mapped using fpocket,32 with the minimum spheres per pocket parameter set to 30. Consistent active site boundaries were determined by aligning a bounding box in PyMol33 with the AutoDockTools Plugin.34 Using the bounding box coordinates, a euclidean grid is created to represent the active site coordinates into a consistent matrix representation. These active site matrices are decomposed into 2 features using a t-SNE35 decomposition for visualization.
Acknowledgments
This work was performed as part of the US Department of Energy (DOE) Joint BioEnergy Institute (https://www.jbei.org) supported by DOE, Office of Science, Office of Biological and Environmental Research, under contract DEAC02-05CH11231 between DOE and Lawrence Berkeley National Laboratory and the DOE Distinguished Scientist Fellow Program to J.D.K. E.E. was supported by Formas Mobility Grant No. 2017-00335. S.Y. was supported by research funds from Yamagata Prefectural Government and Tsuruoka City, Japan.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c11027.
The authors declare the following competing financial interest(s): The authors declare the following competing financial inter-est(s): J.D.K has a financial interest in Amyris, Ansa Biotechnol-ogies, Apertor Pharma, Berkeley Yeast, Cyklos Materials, Deme-trix, Lygos, Napigen, ResVita Bio, and Zero Acre Farms.
Notes
The authors declare the following competing financial interest(s): J.D.K. has a financial interest in Amyris, Ansa Biotechnologies, Apertor Pharma, Berkeley Yeast, Cyklos Materials, Demetrix, Lygos, Napigen, ResVita Bio, and Zero Acre Farms.
Supplementary Material
References
- a Cortes J.; Haydock S. F.; Roberts G. A.; Bevitt D. J.; Leadlay P. F. An usually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora-erythraea. Nature 1990, 348, 176–178. 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]; b Donadio S.; M J S.; McAlpine J. B.; Swanson S. J.; Katz L. Modular organization of genes required for complex polyetide biosynthesis. Science 1991, 252, 675–679. 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- Khosla C.; Tang Y.; Chen A. Y.; Schnarr N. A.; Cane D. E. Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu. Rev. Biochem. 2007, 76, 195–221. 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Dejong C. A.; Chen G. M.; Li H.; Johnston C. W.; Edwards M. R.; Rees P. N.; Skinnider M. A.; Webster A. L.; Magarvey N. A. Polyketide and nonribosomal peptide retro-biosynthesis and global gene cluster matching. Nat. Chem. Biol. 2016, 12, 1007–1014. 10.1038/nchembio.2188. [DOI] [PubMed] [Google Scholar]
- Wilson M. C.; Moore B. S. Beyond ethylmalonyl-CoA: the functional role of crotonyl-CoA carboxylase/reductase homologs in expanding polyketide diversity. Nat. Prod. Rep. 2012, 29, 72–86. 10.1039/C1NP00082A. [DOI] [PubMed] [Google Scholar]
- a Oliynyk M.; Brown M. J. B.; Cortes J.; Staunton J.; Leadlay P. F. A hybrid modular polyketide synthase obtained by domain swapping. Chem. Biol. 1996, 3, 833–839. 10.1016/S1074-5521(96)90069-1. [DOI] [PubMed] [Google Scholar]; b Reeves C. D.; Murli S.; Ashley G. W.; Piagentini M.; Hutchinson C. R.; McDaniel R. Alteration of the Substrate Specificity of a Modular Polyketide Synthase Acyltransferase Domain through Site-Specific Mutations. Biochemistry 2001, 40, 15464–15470. 10.1021/bi015864r. [DOI] [PubMed] [Google Scholar]; c Kumar P.; Koppisch A. T.; Cane D. E.; Khosla C. Enhancing the modularity of the modular polyketide synthases: transacylation in modular polyketide synthases catalyzed by malonyl-CoA:ACP transacylase. J. Am. Chem. Soc. 2003, 125, 14307–14312. 10.1021/ja037429l. [DOI] [PubMed] [Google Scholar]
- a McDaniel R.; Thamchaipenet A.; Gustafsson C.; Fu H.; Betlach M.; Betlach M.; Ashley G. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “‘unnatural”’ natural products. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 1846–1851. 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Patel K.; Piagentini M.; Rascher A.; Tian Z. Q.; Buchanan G. O.; Regentin R.; Hu Z.; Hutchinson C. R.; McDaniel R. Engineered biosynthesis of geldanamycin analogs for Hsp90 inhibition. Chem. Biol. 2004, 11, 1625–1633. 10.1016/j.chembiol.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Bayly C. L.; Yadav V. G. Towards Precision Engineering of Canonical Polyketide Synthase Domains: Recent Advances and Future Prospects. Molecules 2017, 22, 235. 10.3390/molecules22020235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzawa S.; Deng K.; Wang G.; Baidoo E. E.; Northen T. R.; Adams P. D.; Katz L.; Keasling J. D. Comprehensive in Vitro Analysis of Acyltransferase Domain Exchanges in Modular Polyketide Synthases and Its Application for Short-Chain Ketone Production. ACS Synth. Biol. 2017, 6, 139–147. 10.1021/acssynbio.6b00176. [DOI] [PubMed] [Google Scholar]
- Curran S. C.; Hagen A.; Poust S.; Chan L. J. G.; Garabedian B. M.; de Rond T.; Baluyot M. J.; Vu J. T.; Lau A. K.; Yuzawa S.; et al. Probing the Flexibility of an Iterative Modular Polyketide Synthase with Non-Native Substrates in Vitro. ACS Chem. Biol. 2018, 13, 2261–2268. 10.1021/acschembio.8b00422. [DOI] [PubMed] [Google Scholar]
- Kalkreuter E.; Bingham K. S.; Keeler A. M.; Lowell A. N.; Schmidt J. J.; Sherman D. H.; Williams G. J. Computationally-guided exchange of substrate selectivity motifs in a modular polyketide synthase acyltransferase. Nat. Commun. 2021, 12, 2193. 10.1038/s41467-021-22497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murli S.; Kennedy J.; Dayem L. C.; Carney J. R.; Kealey J. T. Metabolic engineering of Escherichia coli for improved 6-deoxyerythronolide B production. J. Ind. Microbiol. Biotechnol. 2003, 30, 500–509. 10.1007/s10295-003-0073-x. [DOI] [PubMed] [Google Scholar]
- Koryakina I.; McArthur J.; Randall S.; Draelos M. M.; Musiol E. M.; Muddiman D. C.; Weber T.; Williams G. J. Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases. ACS Chem. Biol. 2013, 8, 200–208. 10.1021/cb3003489. [DOI] [PubMed] [Google Scholar]
- Koryakina I.; Kasey C.; McArthur J. B.; Lowell A. N.; Chemler J. A.; Li S.; Hansen D. A.; Sherman D. H.; Williams G. J. Inversion of Extender Unit Selectivity in the Erythromycin Polyketide Synthase by Acyltransferase Domain Engineering. ACS Chem. Biol. 2017, 12, 114–123. 10.1021/acschembio.6b00732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koryakina I.; McArthur J. B.; Draelos M. M.; Williams G. J. Promiscuity of a modular polyketide synthase towards natural and non-natural extender units. Org. Biomol. Chem. 2013, 11, 4449–4458. 10.1039/c3ob40633d. [DOI] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav G.; Gokhale R. S.; Mohanty D. Computational approach for prediction of domain organization and substrate specificity of modular polyketide synthases. J. Mol. Biol. 2003, 328, 335–363. 10.1016/S0022-2836(03)00232-8. [DOI] [PubMed] [Google Scholar]
- Jenke-Kodama H.; Sandmann A.; Muller R.; Dittmann E. Evolutionary implications of bacterial polyketide synthases. Mol. Biol. Evol. 2005, 22, 2027–2039. 10.1093/molbev/msi193. [DOI] [PubMed] [Google Scholar]
- Suzek B. E.; Wang Y.; Huang H.; McGarvey P. B.; Wu C. H.; the UniProt Consortium UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. 10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy A. C.; Hong H.; Vance S.; Broadhurst R. W.; Leadlay P. F. Broadening substrate specificity of a chain-extending ketosynthase through a single active-site mutation. Chem. Commun. 2016, 52, 8373–8376. 10.1039/C6CC03501A. [DOI] [PubMed] [Google Scholar]
- Klaus M.; Buyachuihan L.; Grininger M. Ketosynthase Domain Constrains the Design of Polyketide Synthases. ACS Chem. Biol. 2020, 15, 2422–2432. 10.1021/acschembio.0c00405. [DOI] [PubMed] [Google Scholar]
- a Yuzawa S.; Mirsiaghi M.; Jocic R.; Fujii T.; Masson F.; Benites V. T.; Baidoo E. E. K.; Sundstrom E.; Tanjore D.; Pray T. R.; et al. Short-chain ketone production by engineered polyketide synthases in Streptomyces albus. Nat. Commun. 2018, 9, 4569. 10.1038/s41467-018-07040-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zargar A.; Valencia L.; Wang J.; Lal R.; Chang S.; Werts M.; Wong A. R.; Hernandez A. C.; Benites V.; Baidoo E. E. K.; et al. A bimodular PKS platform that expands the biological design space. Metab. Eng. 2020, 61, 389–396. 10.1016/j.ymben.2020.07.001. [DOI] [PubMed] [Google Scholar]
- Lowry B.; Robbins T.; Weng C. H.; O’Brien R. V.; Cane D. E.; Khosla C. In vitro reconstitution and analysis of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 2013, 135, 16809–16812. 10.1021/ja409048k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sharma K. K.; Boddy C. N. The thioesterase domain from the pimaricin and erythromycin biosynthetic pathways can catalyze hydrolysis of simple thioester substrates. Bioorg. Med. Chem. Lett. 2007, 17, 3034–3037. 10.1016/j.bmcl.2007.03.060. [DOI] [PubMed] [Google Scholar]; b Castonguay R.; He W.; Chen A. Y.; Khosla C.; Cane D. E. Stereospecificity of ketoreductase domains of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 2007, 129, 13758–13769. 10.1021/ja0753290. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hinterding K.; Singhanat S.; Oberer L. Stereoselective synthesis of polyketide fragments using a novel intramolecular Claisen-like condensation/reduction sequence. Tetrahedron Lett. 2001, 42, 8463–8465. 10.1016/S0040-4039(01)01840-8. [DOI] [Google Scholar]
- Yuzawa S.; Eng C. H.; Katz L.; Keasling J. D. Broad substrate specificity of the loading didomain of the lipomycin polyketide synthase. Biochemistry 2013, 52, 3791–3793. 10.1021/bi400520t. [DOI] [PubMed] [Google Scholar]
- Eng C. H.; Backman T. W. H.; Bailey C. B.; Magnan C.; Garcia Martin H.; Katz L.; Baldi P.; Keasling J. D. ClusterCAD: a computational platform for type I modular polyketide synthase design. Nucleic Acids Res. 2018, 46, D509–D515. 10.1093/nar/gkx893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K.; Shaw S.; Kloosterman A. M.; Charlop-Powers Z.; van Wezel G. P.; Medema M. H.; Weber T. antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. 10.1093/nar/gkab335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L. S.; Eddy S. R.; Portugaly E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinf. 2010, 11, 431. 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M.; Meier M.; Mirdita M.; Vöhringer H.; Haunsberger S. J.; Söding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinf. 2019, 20, 473. 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Luo W.; Veach R. A.; Hickman A. B.; Wilson M. H.; Dyda F. Structural basis of seamless excision and specific targeting by piggyBac transposase. Nat. Commun. 2020, 11, 3446. 10.1038/s41467-020-17128-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V.; Schmidtke P.; Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinf. 2009, 10, 168. 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenik Z.; Gazak R.; Kadlcik S.; Steiningerova L.; Rynd V.; Janata J. C-C bond cleavage in biosynthesis of 4-alkyl-L-proline precursors of lincomycin and anthramycin cannot precede C-methylation. Nat. Commun. 2018, 9, 3167. 10.1038/s41467-018-05455-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobak D.; Berens P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. 2019, 10, 5416. 10.1038/s41467-019-13056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.