

Summary:
Autophagy is an adaptive response to metabolic and therapeutic stress, especially in treatment-refractory cancers such as pancreatic cancer. In this issue of Cancer Discovery, two groups establish ferritinophagy, a selective autophagy program that could become a drug target, as the mechanism that pumps iron into mitochondria via the lysosome, enabling survival and therapy resistance in pancreas cancer.
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most lethal cancers requiring new therapeutic approaches. Autophagy is a process that allows the degradation and recycling of cellular cargo, including proteins and organelles, serving both a catabolic and an anabolic function in cancer cells. Although the role of bulk autophagy (macroautophagy) in the survival, progression, and therapy resistance of pancreatic cancer has been well established (1), there is growing appreciation for the role of selective autophagy as well as the degradation and recycling of specific cargos (e.g., MHC class I; ref. 2). In this issue, Santana-Codina and colleagues (3) and Ravichandran and colleagues (4) report that ferritinophagy, the specific degradation of ferritin through autophagy, regulated by the autophagy cargo receptor nuclear receptor coactivator 4 (NCOA4) promotes survival and therapy resistance in pancreatic cancer.
Autophagy and lysosome function have previously been shown to be increased and essential for pancreatic cancer survival and growth. This pathway provides essential metabolic substrates to cancer cells that are coping with a harsh tumor microenvironment and the oncogenic stress imposed by KRAS mutations that are common in pancreas cancer (5). However, the mechanism for how autophagy sustains tumor growth is quite complex, involving many possible metabolic pathways. Identifying a selective autophagy program that is necessary and sufficient for sustained pancreatic cancer growth could not only shed light on a key vulnerability for cancer cells but lead to a novel therapeutic approach for pancreas cancer. Ferritinophagy is a recently appreciated example of selective autophagy, whereby the autophagy cargo receptor NCOA4 recruits ferritin as cargo into forming autophagosomes. Ferritin is a protein complex that transports iron intracellularly. Once ferritin is delivered to the lysosome through autophagy, ferritin is degraded, releasing iron in a form that becomes available for cellular use (Fig. 1).
Figure 1.
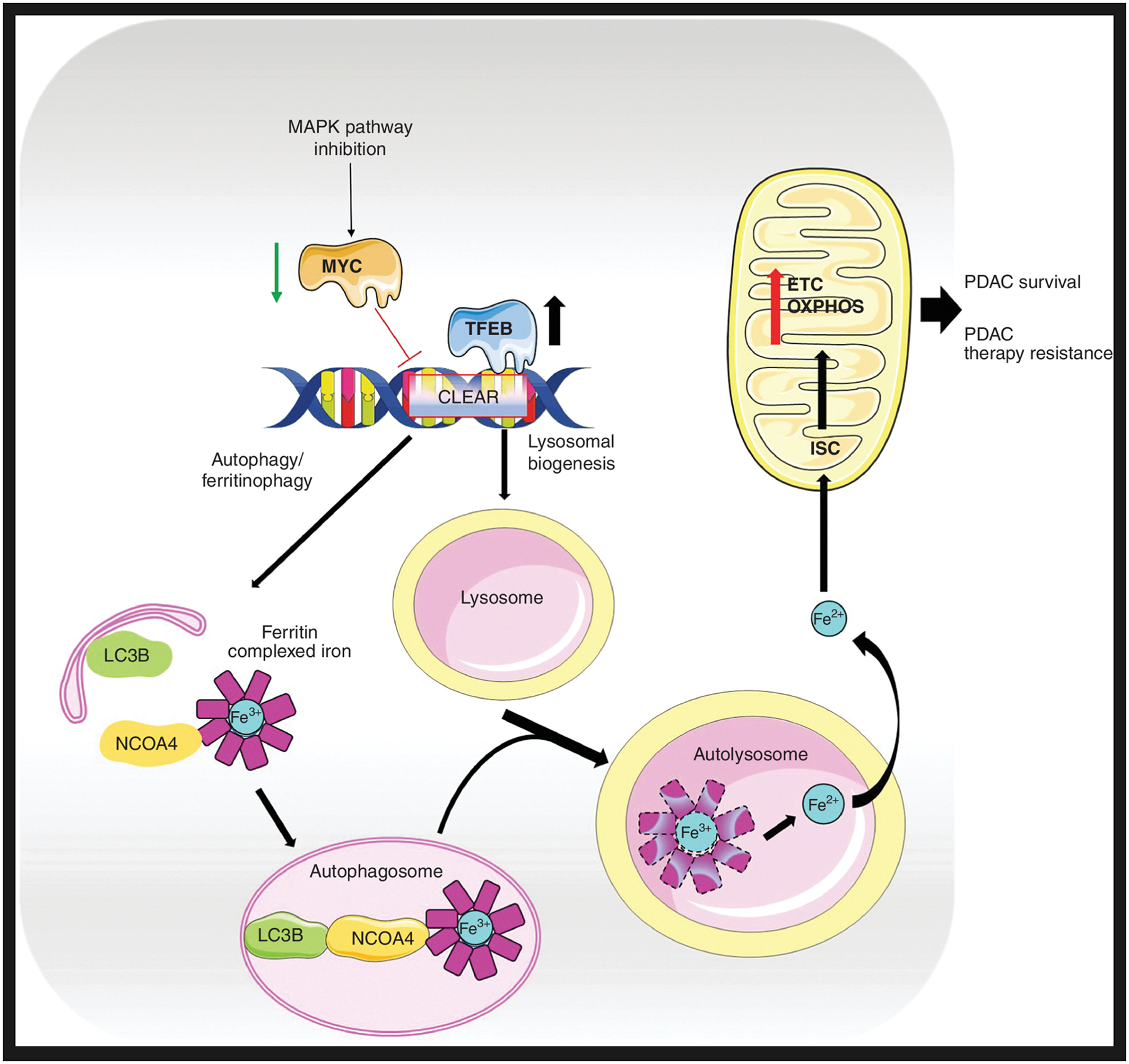
Ferritinophagy drives survival and resistance in pancreas cancer. Ferritinophagy is increased in pancreas cancer. TFEB, a master transcription factor, drives a specific iron metabolism program in addition to lysosomal biogenesis and autophagy. The autophagy cargo receptor NCOA4 binds ferritin, which stores ferric iron (Fe3+). NCOA4–ferritin is processed by the autophagy pathway to release bioactive ferrous iron (Fe2+) in autolysosomes. The ferritinophagy-dependent excess of cellular iron in PDAC increases iron–sulfur cluster (ISC) protein levels in the mitochondria, enhancing the electron transport chain (ETC), oxidative phosphorylation (OXPHOS), and mitochondrial health, thereby supporting pancreatic cancer growth. MAPK pathway inhibition in PDAC leads to reduced MYC competition for TFEB binding sites on CLEAR promoter elements, enabling induction of ferritinophagy, lysosomal biogenesis, and autophagy genes.
Although the mechanism for how ferritinophagy is executed in cells was well understood (6), a functional role of ferritinophagy in cancer cells was not fully established until the report by Santana-Codina and colleagues (3). This group found that NCOA4 levels were strikingly high in PDAC cell lines and patient samples. NCOA4 was found to be a DepMap dependency gene. NCOA4 knockout in cancer cells decreased iron availability and reduced proliferation in vitro and delayed tumor growth in xenografts (3). Using two genetically engineered mouse models of PDAC in which Ncoa4 could be conditionally knocked out or overexpressed, the authors convincingly demonstrated that Ncoa4-driven ferritinophagy was a key driver of pancreatic cancer growth and survival. In the KrasLSL-G12D/+; Trp53fl/+; Pft1a-Cre; Ncoa4fl/fl (KPCN) mouse model, the authors have found that Ncoa4 deletion in pancreatic cancers retards tumor progression and significantly prolongs survival of KPCN compared with KPC mice. Meanwhile, in KPC-Ncoa4Tg/+ mice in which Ncoa4 is overexpressed in pancreatic cancer cells, pancreatic cancer tumors arose at a strikingly rapid rate, significantly reducing overall survival in KPC-Ncoa4Tg/+ compared with KPC mice. To better understand the functional role of ferritinophagy in cancer cells, the investigators performed quantitative proteomics and found that NCOA4 depletion leads to a reduction in iron–sulfur cluster (ISC) protein synthesis—proteins essential for mitochondrial health. Notably, this work further established the paradigm that lysosome-derived iron is essential for the production of ISC proteins in the mitochondria.
In addition to promoting the survival of pancreatic cancer, the autophagy/lysosome pathway promotes therapeutic resistance to pancreas cancer. Three independent studies in 2019 showed that MAPK inhibition in RAS-mutant cancers, with pancreas cancer serving as the main model, leads to a striking induction of autophagy. This was true when any of the major kinases of the MAPK pathway, such as RAF (7), MEK (8), or ERK (9), were targeted. These articles implicated a metabolic crisis imposed by MAPK inhibition, which activated AMPK, a gatekeeper for cytoprotective autophagy. When ERK was targeted, Bryant and colleagues (9) also observed impaired mitochondrial activity, which they proposed as a stress mechanism inducing autophagy, and another selective form of autophagy, mitophagy. However, further work was needed to understand how autophagy was sustaining RAS-mutant cancer cells facing MAPK inhibition.
Ravichandran and colleagues determined that ferritinophagy is activated by MAPK inhibition and is essential to support the mitochondrial health of cancer cells leading to therapy resistance (4). Using pancreatic cancer cell lines and patient-derived organoids, the investigators demonstrated a transcriptional program whereby the microphthalmia/transcription factor E (MiT/TFE) family of master transcription factors (MITF, TFE3, and TFEB) compete with c-MYC for their binding to the promoters of the coordinated lysosomal expression and regulation (CLEAR) network genes. Inhibition of MAPK signaling decreases the stability of c-MYC and allows increased access of TFEB/TFE3 onto CLEAR elements to upregulate the expression of lysosomal biogenesis and autophagy genes.
When MAPK signaling was inhibited in PDAC cells, not only were autophagy and lysosome genes increased, but a coordinated transcriptional program to enable an “iron-replete” state was found with the upregulation of ferritin heavy and light chains (FTH1 and FLH) and downregulation of iron importers and exporters—transferrin receptor and ferroportin, respectively. Leveraging an approach to study lysosomes isolated from MEK inhibitor (MEKi)–treated cells, the investigators found that ferritin and iron levels were increased in MEKi-treated cells. Like the Santana-Codina and colleagues study, upregulation of ferritinophagy also increased the production of ISC proteins, but in the context of MEKi, the functional consequence of ferritinophagy-enabled ISC production was enhanced mitochondrial respiratory chain activity and tricarboxylic acid cycling. The enhanced activity of mitochondria following MAPK inhibition was completely reversed by iron chelation, blockade of NCOA4, or lysosomal inhibition, demonstrating that ferritinophagy was a major component of the autophagy/lysosome program that promoted survival of pancreatic cancer cells with MAPK inhibition.
Together, these two studies identified a novel mechanistic link between ferritinophagy, lysosomal iron liberation, and mitochondrial health (Fig. 1). In contrast to the previous study (8), no mitophagy involvement was reported in these articles following MAPK inhibition. When autophagy is activated, mitophagy is also typically increased; therefore, there may be a regulated dynamic between enhancing mitochondrial function through ferritinophagy while simultaneously degrading mitochondria to recycle nutrients. This work nominates NCOA4 as an interesting new drug target in cancer. Other iron metabolism proteins such as the transferrin receptor have been considered as targets for antibody–drug conjugates, but the feasibility of such drug targets raises concerns due to their widespread role in tissue homeostasis. In addition to the electron transport chain, ISC proteins are also involved in DNA repair and DNA/RNA metabolism, which can also contribute to therapy resistance but could also lead to toxicity if NCOA4 is therapeutically targeted. Developing a chemical inhibitor to NCOA4 may prove challenging, but in the absence of a chemical inhibitor, the effects of systemic Ncoa4 deletion could be modeled in mice with concurrent tumors. Finally, although the inhibition of ferritinophagy produced antitumor effects in the short term, long-term inhibition showed a compensatory increase in iron uptake and heme breakdown, suggesting that there may be tachyphylaxis to a therapeutic NCOA4 inhibitor.
Overall, these two studies advance the concept that selective autophagy programs could be developed into drug targets that may have advantages over targeting bulk autophagosome production or lysosomal function (10). Targeting selective autophagy programs that may be more important for cancer cell survival than normal cell survival could increase the therapeutic index of targeting autophagy. Whether this is the case for ferritinophagy remains to be seen. Moreover, although it is clear that autophagy clears damaged organelles and serves a catabolic homeostasis function, the growing appreciation for the anabolic roles of autophagy, exemplified by the ferritinophagy–lysosome–mitochondria axis, calls for a renewed focus on targeting autophagy in cancer.
Acknowledgments
This work was supported by HHS/NIH/NCI grants P30 CA016520-45, PO1 CA114046, and RO1 CA266404 P50 CA174523.
Authors’ Disclosures
R.K. Amaravadi reports grants and other support from Pinpoint Therapeutics, personal fees from Deciphera, and grants from Novartis and Bristol Myers Squibb outside the submitted work, as well as a patent for dimeric chloroquine licensed to Pinpoint Therapeutics. No disclosures were reported by the other author.
REFERENCES
- 1.Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov 2019; 9:1167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamamoto K, Venida A, Perera RM, Kimmelman AC. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy 2020;16:1524–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santana-Codina N, Quiles Del Rey M, Kapner KS, Zhang H, Gikandi A, Malcolm C, et al. NCOA4-mediated ferritinophagy is a pancreatic cancer dependency via maintenance of iron bioavailability for iron–sulfur cluster proteins. Cancer Discov 2022;12:2180–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravichandran M, Hu J, Cai C, Ward NP, Venida A, Foakes C, et al. Coordinated transcriptional and catabolic programs support iron-dependent adaptation to RAS–MAPK pathway inhibition in pancreatic cancer. Cancer Discov 2022;12:2198–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamamoto K, Iwadate D, Kato H, Nakai Y, Tateishi K, Fujishiro M. Targeting autophagy as a therapeutic strategy against pancreatic cancer. J Gastroenterol 2022. Jun 21 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014;509:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A 2019;116:4508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 2019;25:620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 2019;25:628–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JJ, Jain V, Amaravadi RK. Clinical translation of combined mapk and autophagy inhibition in ras mutant cancer. Int J Mol Sci 2021; 22:12402. [DOI] [PMC free article] [PubMed] [Google Scholar]