

Abstract
Precision oncology informed by genomic information has evolved in leaps and bounds over the last decade. Although non-small cell lung cancer (NSCLC) has moved to center-stage as the poster child of precision oncology, multiple targetable genomic alterations have been identified in various cancer types. RET alterations occur in roughly 2% of all human cancers. The role of RET as oncogenic driver was initially identified in 1985 after the discovery that transfection with human lymphoma DNA transforms NIH-3T3 fibroblasts. Germline RET mutations are causative of multiple endocrine neoplasia type 2 syndrome, and RET fusions are found in 10–20% of papillary thyroid cases and are detected in most patients with advanced sporadic medullary thyroid cancer. RET fusions are oncogenic drivers in 2% of Non-small cell lung cancer. Rapid translation and regulatory approval of selective RET inhibitors, selpercatinib and pralsetinib, have opened up the field of RET precision oncology. This review provides an update on RET precision oncology from bench to bedside and back. We explore the impact of selective RET inhibitor in patients with advanced NSCLC, thyroid cancer, and other cancers in a tissue-agnostic fashion, resistance mechanisms, and future directions.
Keywords: RET, RET fusions, Non-small cell lung cancer, Thyroid cancer, Tissue-agnostic, Clinical trials
1. Introduction
Over the last decade, genome-guided precision oncology has resulted in more effective therapy for patients with many types of cancer (Adashek, Subbiah, & Kurzrock, 2020; Subbiah & Kurzrock, 2016, 2018). Patients with non-small cell lung cancer (NSCLC) have especially benefited from precision oncology, although RET fusions, NTRK fusions, high levels of microsatellite instability, and deficient mismatch repair can be targeted in a tissue-agnostic manner (Adashek, Subbiah, & Kurzrock, 2020). RET alterations occur in approximately 2% of all human cancers (Subbiah & Cote, 2020a, 2020b; Subbiah, Yang, Velcheti, Drilon, & Meric-Bernstam, 2020; Thein, Velcheti, Mooers, Wu, & Subbiah, 2021a). The incidence of RET rearrangement in non small cell lung cancer (NSCLC) seems to be higher in younger, <60 years old, patients with a history of light to never smokers, and no major correlation with the gender or the ethnicity.
As RET fusions have a low prevalence in other cancers, data are lacking pertaining to the clinical characteristics across multiple tumor types.
The role of RET as oncogenic driver was initially discovered in 1985 after the detection that transfection with human lymphoma DNA transforms NIH-3T3 fibroblasts (Subbiah & Cote, 2020a, 2020b). In the 1980s, RET fusions were found in papillary thyroid cancer and germline RET mutations were proved to be responsible of multiple endocrine neoplasia type 2 (MEN2) syndrome. RET alterations are detected in 10–20% patients with papillary thyroid cancers and in almost all patients with medullary thyroid cancer (MTC) (Adashek et al., 2021).
RET is a proto-oncogene located on chromosome 10q11.2. It encodes a transmembrane glycoprotein receptor tyrosine kinase which interacts with ligands from the family of glial-derived neurotrophic factors (GDNF) (Fig. 1-1 and 1-2). RET plays a significant role in the development and function of the enteric nervous system and the renal system.
Fig. 1.

RET mechanisms in normal and oncogenic states. (1) Wild-type RET is a transmembrane protein with extracellular cadherin-like and cysteine-rich domains and an intracellular tyrosine kinase domain and isoform-specific tail. (2) Upon binding of the wild-type RET to a GFRα bound to a GDNF family ligand, RET homodimerizes, is autophosphorylated, and activates signaling through RAS, RAF, MEK, or ERK pathways. Ligands are color-coded (GDNF in red, NRTN in yellow, ARTN in green, and PSPN in blue), and respective GFRα receptors are color-matched. Calcium-binding is represented by bright green crosses and phosphorylation by the letter p in a yellow circle. (3) RET with oncogenic mutations, such as those indicated, is constitutively active. (4) Oncogenic RET with two of the most common fusions, CCDC6 and KIF58, is intracellular, undergoes ligand-independent dimerization, and is constitutively active.
RET activation occurs when a cell-membrane-bound GFRα binds to a GDNF ligand, leading to homodimerization and autophosphorylation (Subbiah & Cote, 2020a, 2020b). This causes the RET kinase domains to trigger the downstream signaling, activating the MAPK, JAK-STAT and PI3K-AKT-mTOR proliferativepathways (Subbiah & Cote, 2020a, 2020b).
RET's role in the enteric nervous system means that loss-of-function mutations, as seen in the hereditary Hirschsprung's disease, can cause bowel dysfunction. Similarly, other RET mutations can lead to urinary tract malformations (Subbiah, Yang, et al., 2020; Thein, Velcheti, Mooers, Wu, & Subbiah, 2021b).
The chromosomal RET rearrangement between the 3′ coding region in the kinase domain on chromosome 10 and a 5′ gene with a coiled-coil or LIS1 homology domain causes constitutive oncogenic RET activation in non-small cell lung cancer (NSCLC) (Subbiah, Yang, et al., 2020) (Fig. 1). Most frequently, the rearrangements, or fusion are intra-chromosomal and occur with partner genes including KIF5B, CCDC6, and NCOA4. Inter-chromosomal, partners have also been identified. Alternatively, inter-chromosomal recombination can occur. It involves the exchange of nucleotide sequences between homologous chromosomes or identical DNA molecules.
RET gene fusions are frequently detected in papillary thyroid carcinoma (Grieco et al., 1990) including those that involve CCDC6 and NCOA4 (Subbiah, Yang, et al., 2020). RET fusions with KIF5B, ROS, and ALK have been reported in lung adenocarcinoma and colorectal cancer (Subbiah, Yang, et al., 2020). RET fusions with BCR and FGFR1OP have been detected in patients with chronic myelomonocytic leukemia (Ballerini et al., 2012). RET fusions partner genes and breakpoints influence the characteristics of the resulting RET protein. The former also determine whether the RET oncoprotein is constitutively active and may result in the formation of multi-kinase signaling hubs.
In thyroid cancer, (Adashek et al., 2021; Subbiah, Yang, et al., 2020), RET predominantly results from point mutations, with those in the extracellular domain leading to the hereditary MEN2A syndrome (Fig. 1-3). The most common of mutation among patients with MEN2A is the M918T alteration (Adashek et al., 2021). MEN2A patients are characterized by marfanoid habitus, the majority develop MTC and pheochromocytomas, and some develop hyperparathyroidism.
Notably RET rearrangements are driver mutations hence mutually exclusive with other driver mutations in NSCLC, though recent data have shown the co-occuring genomic alteration, in particular with the Kirsten rat sarcoma viral oncogene (KRAS) (Skoulidis & Heymach, 2019).
In this review, we will first discuss RET gene and RET protein structure and function. We will describe the current state of RET precision oncology. We explore relevance of RET alterations in patients with advanced NSCLC, thyroid cancer, and other cancers. Finally, we describe resistance mechanisms to RET-targeted therapies and discuss ongoing efforts to more effectively treat patients with RET oncogene-driven cancers.
2. RET structure and function
The RET pre-mRNA is alternatively spliced to generate three isoforms. The transcripts for alternative splicing include the first 19 exons of RET that are then followed by varying sequences (Carter et al., 2001). Each RET isoform is unique and named in accordance with the number of amino acids located at each C-terminus (Fig. 1-1): RET9, RET43, and RET51. The C-terminal isoform tails of these proteins contain 9, 43, and 51 amino acids, respectively (Ishizaka et al., 1989; Myers, Eng, Ponder, & Mulligan, 1995; Tahira, Ishizaka, Itoh, Sugimura, & Nagao, 1990). RET9 and RET51 are highly conserved within vertebrates (Mulligan, 2014) and were first described in 1990 after characterization of RET proto-oncogene cDNA clones obtained from a human neuroblastoma cell line (Tahira et al., 1990). The third isoform, RET43, is expressed at low levels and has a low degree of conservation between mouse, rat, and human (Carter et al., 2001). The RET isoforms have unique binding affinities for factors involved in downstream signaling and likely have distinct signaling functions (Mulligan, 2014; Peterson & Bogenmann, 2004). These alternative splicing patterns at the 3′ end of RET are important for its biological function and differences were previously reported between the RET isoforms studied in vivo (Arighi, Borrello, & Sariola, 2005; Carter et al., 2001; Tahira et al., 1990). These are the result of protein interactions with tyrosine (Y1062) (Arighi et al., 1997), the common and last amino acid found in the three distinct isoforms (RET9, RET43, and RET51)(Arighi et al., 2005; Ibanez, 2013) Phosphorylated tyrosine (Y1062) is located within a docking site for SHC and IRS-1, two adaptor molecules, and the docking protein FRS2 (Kurokawa et al., 2001; Lorenzo et al., 1997) (Lorenzo et al., 1997, Kurokawa et al., 2001). The SHC molecule binds phosphorylated Y1062 through the SH2 or PTB domain depending on the RET isoform (Ishiguro et al., 1999; Melillo et al., 2001). RET 43 does not bind SHC, while binding FRS2 weakly. The PTB domains of the SHC adaptor molecule, IRS-1 and FRS2 are capable of binding RET51 and RET9(Carter et al., 2001). On the other hand, RET9 also has the ability to bind SHC with greater affinity via the SH2 rather than PTB domain (Lorenzo et al., 1997) (Table 1).
Table 1.
Affinities to binding domains following phosphorylation of Y1062.
| Adaptor Molecule |
RET9 | RET43 | RET51 | Binding Domains |
|---|---|---|---|---|
| SHC | □ ◊ ●Efficient binding | ◊ Undetected | ◊ ●Efficient binding | □SH2 |
| IRS-1 | ◊PTB | |||
| FRS2 | ◊ ●Binding | ◊ ●Low binding | ◊ ●Binding |
●Binding Affinity, □SH2, ◊PTB
RET extracellular regions include four cadherin-like and one cysteine-rich domain (Fig. 1-1) (Mulligan, 2014). The cadherin-like domains involve about 110 amino acids, and there is one calcium binding site between cadherin-like domain 2 and cadherin-like domain 3 (Anders, Kjar, & Ibanez, 2001; Ibanez, 2013). The binding of Ca2+ is necessary for interaction of RET protein with a ligand from the GDNF family (Ibanez, 2013).
The cysteine-rich region of RET is cell-membrane adjacent and is essential for protein configuration and ligand binding. Several mutations associated with MEN2A and familial MTC are detected in this domain (Anders et al., 2001; Mulligan, 2014; Pasini et al., 1997; Wang, 2013). A subgroup of cysteine RET variants termed ‘Janus mutations,’ which affect C609, 611, 618 and 620 are associated with RET gain-of-function and loss-of-function diseases (Arighi et al., 2004). We have listed the different possible mutations and their possible phenotype in Tables 1.
RET requires a co-receptor for ligand binding (Airaksinen, Titievsky, & Saarma, 1999), consisting of a soluble GDNF ligand and a GDNF family receptors alpha co-receptor (GFRα). In 1996, two studies characterized the multiple components that comprise the RET receptor(Arighi et al., 1997; Liu et al., 1996). These studies established that GFRα1 is needed to bind GDNF and that the GDNF-GFRα1 complex regulates the dimerization of RET. In the years that followed, three additional GDNF family ligands were discovered including neurturin, artemin, and persephin, which bind to GFRα2, α3, and α4, respectively. The GDNF first binds to GFRα, which is bound to the cell membrane by glycosylphosphatidylinositol. This complex recruits RET, and when two RET molecules are assembled, their cysteine-rich domains are brought together, activating tyrosine kinase function.
3. Targeted RET inhibition in NSCLC and thyroid carcinoma
RET fusions were first identified in NSCLC patients 2012(Subbiah, Yang, et al., 2020). The typical phenotype of patients harboring RET fusions is that of young, non-smokers, with adenocarcinoma histology, though these fusions can be found in any patient population (Subbiah, Yang, et al., 2020). Molecular profiling for RET alterations should not be limited to the typical patient population and should be incorporated into multigene screening. RET was initially targeted with multi-kinase inhibitors (MKIs) in NSCLC. However, MKIs have poor specificity. They are better inhibitors of kinases like VEGFR2 than RET, resulting in side effects due to off-target toxicity (Subbiah & Cote, 2020a, 2020b). Despite this, cabozantinib and vandetanib are two MKIs approved in the treatment of MTC. In addition, lenvatinib and sorafenib are approved therapies for differentiated thyroid cancer. The use of the MKIs are based on histology rather than the presence of RET alterations or any other biomarker.
RET precision oncology has been made possible by the development of novel, highly selective RET inhibitors. Potent and highly selective RET inhibitors, Selpercatinib and Pralsetinib, have recently received regulatory approval based on evidence of safety and efficacy in RET fusion-positive lung, thyroid and medullary thyroid cancers (Drilon et al., 2020; Gainor et al., 2021; Subbiah et al., 2021; Subbiah et al., 2021; Wirth et al., 2020).
3.1. Selpercatinib
Selpercatinib, originally known as LOXO-292 and marketed as Retevmo®, is an oral highly selective RET inhibitor (Subbiah et al., 2018) The phase I/II LIBRETTO-001 trial (NCT03157128) in patients with RET fusion-positive solid tumors led to FDA approval of selpercatinib (Drilon et al., 2018; Subbiah et al., 2021a, 2021b; Wirth et al., 2020).
In the phase I part of the trial, 49 patients with previously treated advanced NSCLC with RET rearrangement were treated with 20 mg selpercatinib once daily up to 240 mg twice daily. The study dose in the 56 patients in the phase II trial was 160 mg twice daily. There was a 64% ORR (95% CI: 54–73%), which was not influenced by previous checkpoint inhibitors or MKIs. Among 39 treatment-naïve patients, there was an 85% ORR. Among 11 previously-treated patients with brain metastases, there was a 91% ORR and 27% complete response rate, supporting high central nervous system (CNS) activity. Overall, the median duration of response (DoR) was 17.5 months (95% CI: 12 months – not reached).
The LIBRETTO-001 trial accrued 55 patients harboring RET-positive MTC, after progression on prior MKIs (Wirth et al., 2020). In this subset of patients, there was a 69% ORR (95% CI: 55–81%), and an 82% one year progression-free survival (PFS) rate (95% CI: 69–90%). Among 88 previously untreated patients, there was a 73% ORR (95% CI: 62–82%) and 92% one year PFS rate (95% CI: 82–97%). The response rate did not differ greatly among 19 previously-treated patients, with a 79% ORR (95% CI: 54–94%) and 64% one year PFS rate (95% CI: 37–82%).
The safety profile of selpercatinib is manageable, with grade 3–4 treatment-emergent adverse effects (TEAEs) in 59% of patients, comprising mainly hypertension (17.1%) and liver enzyme elevation (16.5%). The most common low-grade toxicities included xerostomia (38.9%), diarrhea (36.6%) and hypertension (35.9%).The earliest TEAE was hypersensitivity at 1.7 weeks and the latest, rash, 9.3 weeks after treatment initiation. Dose adjustments, interruptions and discontinuation were more common among patients who received treatment longer.
3.2. Pralsetinib
Pralsetinib, originally known as BLU-667 and now marketed as Gavreto® is an oral selective RET inhibitor (Subbiah et al., 2018). Pralsetinib was FDA approved in December 2020 and EMA approved in November 2021 for patients with advanced NSCLC harboring RET fusions. These approvals are based on the ongoing phase I/II ARROW study (NCT03037385) of pralsetinib in patients with NSCLC, thyroid cancer and other RET-rearranged advanced cancers (Vivek Subbiah, et al., 2021; Subbiah, Hu, Gainor, et al., 2021).
The ARROW trial is a multicenter study in 11 countries (Gainor et al., 2021;Subbiah, Hu, Gainor, et al., 2021; Subbiah, Hu, Wirth, et al., 2021). Patients received pralsetinib at a dose of 400 mg per day. It includes four cohorts: 67 patients with RET-mutant MTC who were treated with prior MKIs, 42 treatment-naïve patients with RET-mutant MTC, 10 previously treated patients MTC who did not receive MKIs, and a cohort of 319 patients with other cancer types who had RET alterations. The endpoints included safety, ORR and DoR.
Among the 92 patients in the RET-mutant MTC cohort, the median age was 59 (19–83) and roughly two-thirds of the patients were male (Subbiah, Hu, Gainor, et al., 2021). About 60% of patients had an Eastern Cooperative Oncology Group performance status of 1 or 2, and 10% had a history of CNS/brain metastases. Various RET alterations were detected: In 61% of patients, the mutation was M918T. Mutations in the cysteine-rich domain were detected in 29% of subjects. A further 3% had V804M or V804L mutations, which are mutations in a gatekeeper residue that modulates interaction of MKIs with the ATP-binding pocket. The remainder of patients had less common alterations. Across the cohorts, the baseline patients characteristics were similar.
At data cut off, the 53 previously treated patients appeared to respond very well to pralsetinib, with a 60% ORR (95% CI: 46–74%) of which 2% had complete responses (Subbiah, Hu, Gainor, et al., 2021). There was a 96% disease control rate. At data cut-off, neither the median DoR nor the median PFS were reached. Seventy-five percent of patients were still on praseltinib and 94% of responders were still on treatment. Among 42 patients who were treatment-naïve and ineligible for standard therapy, there was a 74% ORR and 100% disease control rate. Neither median DoR nor median PFS were reached in this cohort.
Pralsetinib had a manageable safety profile (Subbiah, Hu, Gainor, et al., 2021; Subbiah, Hu, Wirth, et al., 2021). Most TEAEs were low-grade, the most common being increased liver enzymes (57%), anemia (24%), constipation (23%), and hypertension (22%). Regarding grade 3–4 TEAEs, the most common were hypertension (11%), followed by hematologic toxicity, including neutropenia (10%), anemia (8%) and neutropenia (6%). Four percent of patients discontinued praseltinib due to toxicity (Subbiah, Hu, Gainor, et al., 2021; Subbiah, Hu, Wirth, et al., 2021).
3.3. Clinical detection of RET alterations
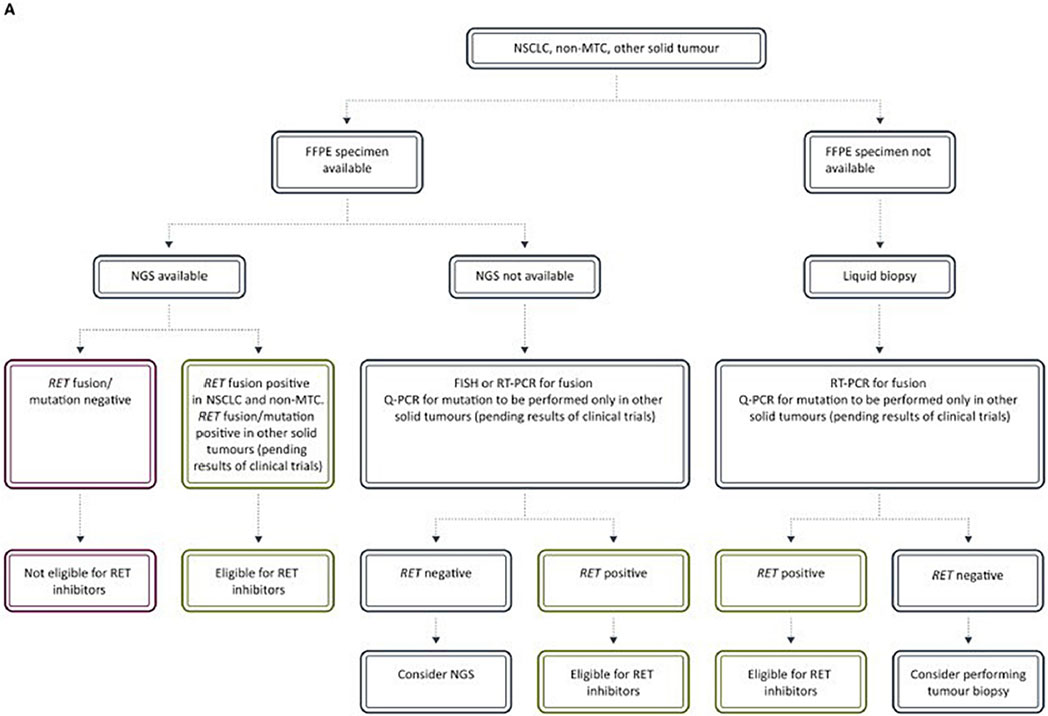
Novel RET inhibitors are dependent on the detection of RET alterations. As with regulatory approval for drugs, diagnostics used in personalized medicine are subject to assessment and approvals. The ESMO Translational Research and Precision Medicine Working Group has published recommendations for the detection of RET rearrangements and mutations (Table 2). The diagnostic approach can include immunohistochemistry (IHC), fluorescent in situ hybridization (FISH), reverse transcription-polymerase chain reaction (RT-PCR), DNA and RNA sequencing. Due to low sensitivity and specificity, IHC alone is not recommended for RET screening. Both novel therapies and diagnostic accuracy are drastically changing the management of RET altered thyroid and lung cancers.
Table 2.
ESMO Translational Research and Precision Medicine Working Group recommendations for routine clinical laboratory detection of targetable RET rearrangements and mutations with their strengths and limitations. Adapted from (Belli et al., 2020; Belli et al., 2021).
| Method | Sensitivity | Specificity | Detection of partner | Detection of expression | Use in screening |
|---|---|---|---|---|---|
| IHC | moderatea | moderateb | no | yes | no |
| FISH | high | high | no/yesc | no | rarely |
| RT-PCR | moderate/highd | high | yes/noe | yes | rarely |
| DNA-seq NGS | moderatef | high/moderateg | yes | no | yes |
| RNA-seq NGS | high | high | yes | yesh | yes |
| |||||
False-positive up to 40%.
False-negative up to 40%.
Requires the use of specific fusion partner probe.
Used in settings with several possible fusion partners, there is risk of lower sensitivity.
Does not allow the detection of novel fusion partners.
False-positive: detected rearrangements by DNA-based assays may not result in fusions, and correlation with RNA-based confirmation of predicted fusion transcript is required.
False-negative: some introns involved in RET rearrangements may be inadequately evaluated for technical reasons.
Indication of the in-frame nature of the fusion, which is predictive of functionality. DNA-seq NGS, DNA sequencing by next-generation sequencing; FISH, fluorescent in situ hybridization; IHC, immunohistochemistry; RNA-seq NGS, RNA sequencing by next-generation sequencing; RT-PCR, reverse transcription-polymerase chain reaction.
4. Intracranial activity of selective RET inhibitors
Patients with NSCLC with intracranial metastases have poor prognosis and impaired quality of life (Subbiah, Gainor, Oxnard, et al., 2021a, 2021b). Before the advent of selective RET inhibitors, a combination of mTOR inhibitor everolimus was shown to improve blood brain penetration of vandetanib pre-clinically and clinically (Minocha, Khurana, Qin, Pal, & Mitra, 2012; Subbiah et al., 2015). Both selective RET inhibitors, selpercatinib and pralsetinib have very high intracranial activity. In the LIBRETTO-001 trial, selpercatinib at the dose of 160 mg twice a day showed 82% response rate, and median duration of intracranial response was not reached (95% CI, 9.3-NE) at a median duration of follow-up of 9.5 months (IQR = 5.7, 12.0) (Drilon et al., 2020; Subbiah, Gainor, Oxnard, et al., 2021a, 2021b). The median intracranial PFS was 13.7 months. Similarly, in the ARROW study, pralsetinib showed intracranial activity in all patients with NSCLC and brain metastases with 56% response rate with a median duration of intracranial response not reached (Gainor et al., 2021). In patients with MTC, selpercatinib was active in leptomeningeal disease as well (Andreev-Drakhlin, Cabanillas, Amini, & Subbiah, 2020).
5. Tissue-agnostic activities of RET inhibitors
Historically, tumors have been diagnosed and treated based on their site of origin and histological subtype. The development of more sophisticated technologies such as next-generation sequencing (NGS) has resulted in a better understanding of the role and impact of molecular features of cancer cells. Evidence suggests that different cancer types can share similar molecular alterations, challenging the dogma that cancer treatment should be driven by site and histology. A shift from treatment based on tumor site and histology to treatment based on genomic aberrations has been triggered by use of NGS techniques and the development of more active and selective tyrosine kinase inhibitors (TKIs). For instance, selpercatinib and pralsetinib treatments resulted in ORRs of over 45% when selective RET inhibitors are administered to patients with cancers in organs other than lung and thyroid who have RET alterations (Subbiah et al., 2022; Vivek Subbiah, et al., 2021; Subbiah et al., 2021; Subbiah et al., 2022). These data validate RET fusions as oncogenic drivers in multiple cancers and demonstrate tissue-agnostic activities of RET inhibitors. Selpercatinib is now US FDA approved in a tissue-agnostic manner for all RET fusion positive cancers.
6. Immunotherapy in NSCLC patients with RET alterations
ICIs are standard front-line therapy, either alone or in combination with a second ICI and/or chemotherapy, in advanced non-oncogene-addicted NSCLC. Not all oncogenic alterations confer resistance to ICIs; for instance, KRAS-driven tumors, in particular, those with the G12C mutation, are sensitive to immunotherapy (Addeo et al., 2021; Torralvo, Friedlaender, Achard, & Addeo, 2019). The impact of RET alterations remains unknown, as patients in previous clinical trials were not evaluate for these mutations. In the context of the recent approval of selective RET inhibitors, it is important to understand how RET alterations influence response to ICIs to best tailor the therapeutic sequence.
So far only retrospective studies have explored the role of ICIs in patients with NSCLC who have RET alterations. NSCLC patients with RET rearrangements were among patients treated with pembrolizumab or nivolumab, alone or with ipilimumab, atezolizumab, or durvalumab. The median tumor mutational burden in patients with RET alterations was 1.75 mutations/Mb, significantly lower than the 5.27 mutations/Mb detected in subjects with wild-type RET. Among the 13 response-evaluable patients who had RET alterations, the median PFS was 3.4 months and was independent of PD-L1 expression and tumor mutational burden (Offin et al., 2019). In the IMMUNOTARGET registry, which comprises the data on the activity of ICIs in oncogene-driven NSCLC, 16 of 551 patients had RET rearrangements (Mazieres et al., 2019). Patients received pembrolizumab or nivolumab. Among patients with RET rearrangements, the ORR was 6%, and disease progression was observed in 75% of patients with a median PFS of 2.1 months. In a retrospective study of 59 NSCLC patients with RET rearrangements, 13 patients received ICIs. The results were similarly disappointing with a 7.7% ORR and 2.1 month median PFS. In a single-center study from the MD Anderson Cancer Center, among 70 patients treated with systemic therapy for cancer with RET alterations, ICI therapy was associated with an increased risk for treatment discontinuation (Hegde et al., 2020). All of these trials suggest that, if available, targeted therapies appear to be a better choice than ICIs for patients who have RET alterations. Additional data about the impact of ICIs in patients with RET alterations are needed. A Chinese trial is currently recruiting NSCLC patients with RET alterations with the goal of comparing chemotherapy to chemoimmunotherapy (NCT04322591). The LIBRETTO-431 trial (NCT04194944) will also compare responses of NSCLC patients with RET alterations to first line treatments with the ICI pembrolizumab.
7. Use of multi-kinase inhibitors in treatment of cancers with RET alterations
The first kinase inhibitors imatinib and gefitinib were approved by the FDA in 2003, and a variety of kinase inhibitors have since been developed to treat cancers associated with RET alterations including chronic myelogenous leukemia, gastrointestinal stromal tumors, MEN2A/2B, familial MTC, NSCLC, and breast cancer (Subbiah & Cote, 2020a, 2020b).
Commonly used kinase inhibitors (e.g., alectinib, cabozantinib, lenvatinib, ponatinib, regorafenib, sorafenib, sunitinib, vandetanib, and RXDX-105) can be classified into types I and II according to their distinct mode of kinase inhibition (Subbiah, Velcheti, et al., 2018; Takahashi, Kawai, & Asai, 2020). Vandetanib and sunitinib are type I inhibitors; these compounds bind the ATP-binding pocket of the activated kinase (Plaza-Menacho, Mologni, & McDonald, 2014). In contrast, sorafenib, cabozantinib, ponatinib and RXDX-105 are type II inhibitors. These bind the ATP-binding pocket while the kinase is in the inactive conformation (Plenker et al., 2017). Efficacies of MKIs have been evaluated in NSCLC and MTC (Thein et al., 2021b). Subsets of patients presented with fusions of RET with various genes including KIF5B, CCDC6, TRIM33, ERC1, CLIP1, NCOA4, and RELCH (Takamori et al., 2021). In advanced MTC, vandetanib was compared to placebo and demonstrated a PFS improvement from 19.3 to 30.5 months. Among patients with more aggressive disease, cabozantinib improved PFS compared to placebo, at 11.2 months versus 4.0 months. Despite these encouraging results, clinical efficacy of MKIs in patients with RET fusions is limited, and significant toxicities are reported. This is likely because MKI inhibitors broadly inhibit kinases including VEGFR, EGFR, MET, and ALK (Thein et al., 2021b).
8. Resistance to selective RET inhibitors
8.1. On-target resistance mechanisms
Resistance to selective RET inhibitors occurs by means of on-target and off-target mechanisms (Fig. 2). Among patients treated with selpercatinib in advanced RET fusion-positive MTC and NSCLC, and who responded initially, circulating tumor DNA was analyzed. The emergence of RET G810 mutations was detected prior to clinical resistance (Solomon et al., 2020). This is a so-called solvent front mutation as it either sterically blocks kinase inhibitor binding and/or destabilizes favorable electrostatic interactions between the inhibitor and the binding site on the kinase. Additionally, analyses of a patient who presented with RET-mutant MTC and a patient with a CCDC6-RET fusion NSCLC who initially responded to selpercatinib but later developed resistance identified selpercatinib-resistant RET mutants that also imparted pralsetinib resistance in cell culture (Subbiah et al., 2021). In the MTC patient, mutations in RET were detected at the solvent front (G810C/S) and in the hinge region (Y806C/N) mutations. The RET G810C mutation was detected in the NSCLC patient. Additionally, five RET kinase domain mutations in three non-gatekeeper residues were found in analyses of 39 selpercatinib-resistant cell lines. All five selpercatinib-resistant RET mutations also resulted in resistance to pralsetinib. No gatekeeper mutations have been detected in RET inhibitor-resistant tumors or cell lines (Subbiah et al., 2021). Selpercatinib susceptibility was maintained in the presence of the RET V804 gatekeeper mutation (Subbiah, Velcheti, et al., 2018).
Fig. 2.

RET inhibitor resistance mechanisms. The RET V804M/L gate-keeper mutations confer resistance to MKIs but are overcome by selective RET inhibitors. Solvent front mutations (e.g., RET G10S/C/R) confer resistance to selective RET inhibitors. Mutations or amplifications of MET or NTRK underlie off-target mechanisms of resistance to selective RET inhibitor therapy. EMT may also play a role in acquisition of RET inhibitor resistance.
8.2. Off-target resistance mechanisms
A recent study identified an acquired KHDRBS1-NTRK3 fusion as a resistance mechanism to ss KIF5B-RET lung cancer that initially responded and then developed resistance to selpercatinib. In post- clinical valiation studies, in BaF3 cells co-expressing KIF5B-RET and KHDRBS1-NTRK3 are resistant to selpercatinib. Co-treatment of these cells with selpercatinib and larotrectinib suppressed induced apoptosis in these cells (Subbiah et al., 2021). This example shows that real-time integration of preclinical studies that apply molecular profiling into clinical practice is critical.
A recent study found MET amplification as a recurrent off-target resistance mechanism in RET rearranged NSCLC (Rosen et al., 2021). Tissue and plasma NGS or FISH analyses among 18 patients treated with selpercatinib and/or pralsetinib detected three (15%) cases of acquired MET amplification, without RET mutations. There was also one case of KRAS amplification. In a case report of selpercatinib resistance in a 48-year-old female never-smoker with KIF5B-RET NSCLC, there was also a novel MET amplification that was not present prior to treatment. A similar case found that MET-driven resistance was overcome by concurrent selpercatinib and crizotinib therapy (Rosen et al., 2021).
These findings suggest that MET amplification is an off-target acquired resistance mechanism to selective RET inhibition. Combination therapies with selective MET inhibitors such as tepotinib, capmatinb and savolitinib, or the use of MKIs may be an effective therapeutic strategy and require further assessment (Lin et al., 2020).
8.3. Resistance due to activation of the epithelial-mesenchymal transition
Although an association between the epithelial-mesenchymal transition (EMT) and RET-targeted therapy has not been experimentally established, activation of EMT has been implicated in development of resistance to various TKIs (Zhuo et al., 2008; Zhuo, Wang, Zhuo, Zhang, & Chen, 2008). EMT induction results in resistance to a MET-specific inhibitor in MET-amplified EGFR inhibitor-resistant NSCLC cells in culture. A preclinical study of lung cancer demonstrated that induction of EMT is associated with resistance to the ALK inhibitors alectinib, ceritinib, and lorlatinib. However, reverting EMT in vitro and in vivo by pre-treating cells with quisinostat, a histone deacetylase inhibitor, was able to block drug resistance. This finding indicates that conditioning cells with a histone deacetylase inhibitor followed by treatment with a kinase inhibitor could be an approach to overcome resistance driven by EMT in the heterogeneous tumor tissue (Fukuda et al., 2019). In a clinical specimen study, Gainor et al. observed that EMT was present in the tumors of NSCLC patients with ALK rearrangements who had resistance to ALK-specific TKIs (Gainor et al., 2016).
EMT also appears to be involved in resistance to other TKIs. NSCLC cells resistant to erlotinib, an EGFR inhibitor, or SU11274, a MET inhibitor, undergo EMT through induction of ZEB-1, which represses the transcription of E-cadherin or by upregulation of β-catenin, respectively (Rastogi et al., 2016). MET amplification and EMT exist as distinct mechanisms of acquired resistance to erlotinib in EGFR-mutated, NSCLC HCC827 cells (Jakobsen et al., 2017). EMT can be both a primary driver of resistance and an acquired mechanism of resistance to EGFR TKIs in lung cancer cell lines (Ware et al., 2013). Thompson et al. demonstrated that the degree of EMT impacts the sensitivity of wild-type EGFR NSCLC cells lines to TKIs. This was proven both in cultures and as xenografts (Thomson et al., 2005). These findings suggest that EMT impacts the sensitivity to TKIs and that tumors that lack an EMT signature (e.g., E-cadherin-positive and vimentin/fibronectin-negative) are most likely to respond to treatment. Further, as treatment of TKI-resistant NSCLC cells with an siRNA that reduces expression of β-catenin or with an miR-200a mimic increases the sensitivity of the TKI-resistant cells to the TKIs (Rastogi et al., 2016), suggesting that a combination of an EMT signaling inhibitor with a TKI might benefit some patients.
Clement et al. recently reported that sequencing results indicated that MET inhibitor resistance was epigenetically arbitrated by EMT and the associated upregulation of FGFR1 rather than resulting from a common genetic alteration (Clement, Gammelgaard, Nielsen, & Sorensen, 2020). Having said that, the association between EMT and RET-targeted therapy has not been experimentally established yet.
Therefore, future studies should also consider epigenetic regulation as an additional parameter in the design of studies and in new treatment discovery.
9. Future perspectives
Data from phase I/II trials are clear: Highly selective RET inhibitors are effective treatments for cancers associated with RET alterations. Two phase III trials are ongoing that are expected to confirm the superiority of RET-targeted therapy compared to current standards of care in NSCLC patients with RET rearrangements. The LIBRETTO-431 trial (NCT04194944) will compared selpercatinib to two standard-of-care arms, one consisting of platinum and pemetrexed chemotherapy, the other chemotherapy and pembrolizumab. The AcceleRET-Lung trial (NCT04222972) will compare pralsetinib to chemotherapy, with or without immunotherapy. In light of the recent approval of adjuvant osimertinib in early NSCLC with EGFR mutations, testing of RET inhibitors in an earlier setting is warranted. The phase III LIBRETTO-432 study (NCT04819100) will compare adjuvant selpercatinib to placebo after definitive therapy for stage IB-IIIA NSCLC, be it surgery or radiotherapy (studies listed in Table 3) (see Table 4).
Table 3.
Ongoing or planned clinical trials with selective RET inhibitors Selpercatinib and Pralsetinib.
| Drug | Name of the trial | NCT Number | Type of cancer | Status | FDA approval |
|---|---|---|---|---|---|
| Selpercatinib | LIBRETTO 001 | NCT03157128 | Advanced solid tumor, NSCLC, MTC | Ongoing (Registration complete) | positive |
| Pralsetinib | ARROW | NCT03037385 | Advanced solid tumor, NSCLC and MTC | Ongoing (Registration complete) | positive |
| Selpercatinib | LIBRETTO-431 | NCT04194944 | NSCLC | Ongoing | – |
| Pralsetinib | AcceleRET-Lung | NCT04222972 | NSCLC | Ongoing | – |
| Selpercatinib | LIBRETTO-531 | NCT04211337 | MTC | Ongoing | – |
| Pralsetinib | AcceleRET-MTC | NCT04760288 | MTC | Planned | – |
Table 4.
Molecular effects of RET mutations in multiple endocrine neoplasia 2.
| Mutation location |
Affected RET Codons |
Putative function of the wild-type residue |
Predicted mutation effects | Phenotype | Recommended intervention |
|---|---|---|---|---|---|
| Extracellular-cysteine rich domain | C609 | Contributes to tertiary structure of RET through the formation of intramolecular disulfide bonds | Weakly activating. Alteration in protein folding and maturation.Formation of mutant RET dimers that are constitutively active in the absence of ligands | MEN 2A and FMTC | Prophylactic thyroid surgery before the age of 5. |
| C611 | |||||
| C618 | |||||
| C620 | Under some conditions may delay beyond 5 years | ||||
| C630 | |||||
| C634 | Role in formation of intramolecular disulfide bonds | Strongly activating. Ligand-independent dimerization of receptor molecules, enhanced phosphorylation of intracellular substrates. | MEN 2A | Surgery <5 years | |
| Intracellular tyrosine kinase domain | L790, Y791 | In the N-terminal lobe of the RET kinase | Moderately activating. Affects ATP binding and inter-lobe flexibility. | MEN 2A and FMTC | May delay surgery beyond 5 years |
| E768 | In close proximity with the ATP binding site | Alters interactions within the region and facilitates the transition to an active conformation | FMTC | ||
| V804 | A gatekeeper residue which regulates access to the ATP binding site | Alters hinge flexibility and positioning of RET helices for catalysis | FMTC | ||
| S891 | C-terminal lobe of the kinase, adjacent to the activation loop of the kinase | Alters activation loop conformation and promotes monomeric RET activation | MEN 2A and FMTC | ||
| A883 | Situated next to activation loop | Strongly activating. Local conformational change which destabilizes the inactive form of the protein and promotes its activation | MEN 2B | As early as possible (within first year of life) | |
| M918 | Lies in the substrate-binding pocket of the kinase and plays a role in stabilizing the receptor–ATP complex | Strongly activating. Alters protein conformation and substrate specificity. The mutant can dimerize and become phosphorylated in the absence of ligand stimulation | MEN 2B |
FMTC, familial medullary thyroid carcinoma; MEN 2, multiple endocrine neoplasia 2; RET, REarranged during Transfection.
There are two further reasons to offer RET-targeted therapy upfront. Given both the risk of CNS progression on standard therapy and high drop-off rates on each subsequent treatment line in NSCLC, opting to keep a TKI for a subsequent line may prevent a patient from receiving this life-prolonging therapy. Currently approved RET inhibitors have good CNS penetration and durable responses. The second reason we would opt for upfront TKIs is toxicity. Highly selective RET inhibitors have a favorable toxicity profile compared to chemotherapy with or without ICIs. Furthermore, although RET inhibitors have not yet been tested in this context, exposing patients to certain TKIs, such as osimertinib, after ICI treatment increases the risk of serious TEAEs.
In conclusion, while we await phase III trial results, our opinion is that front-line RET inhibitors should be offered to those cancer patients with RET alterations. Rapid translation of selective RET inhibitors and global regulatory approvals have opened up the field of RET precision oncology in NSCLC, thyroid cancer, and multiple other cancers in a tissue-agnostic fashion harboring RET fusions. Continued deployment of NGS assays to detect RET alterations are crucial for identification of patients who could benefit the most from such therapy.
Acknowledgments
Vivek Subbiah (VS) is an Andrew Sabin Family Foundation Fellow at The University of Texas MD Anderson Cancer Center. VS acknowledges support of The Jacquelyn A. Brady Fund. JW and VS and are supported by National Institutes of Health grant R01CA242845. H.S.,JW. And VS are supported by R01CA273168. MD Anderson Cancer Center Department of Investigational Cancer Therapeutics is supported by the Cancer Prevention and Research Institute of Texas (RP1100584), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy, 1U01 CA180964, NCATS Grant UL1 TR000371 (Center for Clinical and Translational Sciences), and the MD Anderson Cancer Center Support Grant (P30 CA016672). Figures were created using Biorender.
Abbreviations:
- RET
Rearranged during transfection
Footnotes
Declaration of Competing Interest
V. Subbiah reports grants from Eli Lilly/LOXO Oncology, Blueprint Medicines Corporation, Turning Point Therapeutics, Boston Pharmaceuticals; and grants from Helsinn Pharmaceuticals during the conduct of the study; in addition, V. Subbiah reports a grant and advisory board/consultant position with Eli Lilly/Loxo Oncology during the conduct of the study; research grants from Roche/Genentech, Bayer, GlaxoSmithKline, Nanocarrier, Vegenics, Celgene, Northwest Biotherapeutics, Berghealth, Incyte, Fujifilm, D3, Pfizer, Multivir, Amgen, Abbvie, Alfa-sigma, Agensys, Boston Biomedical, Idera Pharma, Inhibrx, Exelixis, Blueprint Medicines, Altum, Dragonfly Therapeutics, Takeda, National Comprehensive Cancer Network, NCI-CTEP, University of Texas MD Anderson Cancer Center, Turning Point Therapeutics, Boston Pharmaceuticals, Novartis, Pharmamar, Medimmune; an advisory board/consultant position with Helsinn, Incyte, QED Pharma, Daiichi-Sankyo, Signant Health, Novartis, Relay therapeutics, Pfizer, Roche, Medimmune; travel funds from Pharmamar, Incyte, ASCO, ESMO; other support from Medscape; all outside the submitted work.
Data availability
No data was used for the research described in the article.
References
- Adashek JJ, Subbiah V, & Kurzrock R (2020). From tissue-agnostic to N-of-one therapies: (R)evolution of the precision paradigm. Trends Cancer. 7(1), P15–28 JANUARY 01, 2021. [DOI] [PubMed] [Google Scholar]
- Adashek JJ, Desai AP, Andreev-Drakhlin AY, Roszik J, Cote GJ, & Subbiah V (2021). Hallmarks of RET and co-occuring genomic alterations in RET-aberrant cancers. Molecular Cancer Therapeutics 20, 1769–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addeo A, Passaro A, Malapelle U, Luigi Banna G, Subbiah V, & Friedlaender A (2021). Immunotherapy in non-small cell lung cancer harbouring driver mutations. Cancer Treatment Reviews 96, Article 102179. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Titievsky A, & Saarma M (1999). GDNF family neurotrophic factor signaling: Four masters, one servant? Molecular and Cellular Neurosciences 13, 313–325. [DOI] [PubMed] [Google Scholar]
- Anders J, Kjar S, & Ibanez CF (2001). Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin-like domains and a calcium-binding site. The Journal of Biological Chemistry 276, 35808–35817. [DOI] [PubMed] [Google Scholar]
- Andreev-Drakhlin A, Cabanillas M, Amini B, & Subbiah V (2020). Systemic and CNS activity of selective RET inhibition with Selpercatinib (LOXO-292) in a patient with RET-mutant medullary thyroid Cancer with extensive CNS metastases. JCO Precision Oncology 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arighi E, Alberti L, Torriti F, Ghizzoni S, Rizzetti MG, Pelicci G, … Borrello MG (1997). Identification of Shc docking site on ret tyrosine kinase. Oncogene 14, 773–782. [DOI] [PubMed] [Google Scholar]
- Arighi E, Borrello MG, & Sariola H (2005). RET tyrosine kinase signaling in development and cancer. Cytokine & Growth Factor Reviews 16, 441–467. [DOI] [PubMed] [Google Scholar]
- Arighi E, Popsueva A, Degl’Innocenti D, Borrello MG, Carniti C, Perala NM, … Sariola H (2004). Biological effects of the dual phenotypic Janus mutation of ret cosegregating with both multiple endocrine neoplasia type 2 and Hirschsprung’s disease. Molecular Endocrinology 18, 1004–1017. [DOI] [PubMed] [Google Scholar]
- Ballerini P, Struski S, Cresson C, Prade N, Toujani S, Deswarte C, … Delabesse E (2012). RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia 26, 2384–2389. [DOI] [PubMed] [Google Scholar]
- Belli C, Anand S, Gainor JF, Penault-Llorca F, Subbiah V, Drilon A, … Curigliano G (2020. December). Progresses toward precision medicine in RET-altered solid tumors. Clinical Cancer Research 26(231). [DOI] [PubMed] [Google Scholar]
- Belli C, Penault-Llorca F, Ladanyi M, Normanno N, Scoazec JY, Lacroix L, … Curigliano G (2021). ESMO recommendations on the standard methods to detect RET fusions and mutations in daily practice and clinical research. Annals of Oncology 32, 337–350. [DOI] [PubMed] [Google Scholar]
- Carter MT, Yome JL, Marcil MN, Martin CA, Vanhorne JB, & Mulligan LM (2001). Conservation of RET proto-oncogene splicing variants and implications for RET isoform function. Cytogenetics and Cell Genetics 95, 169–176. [DOI] [PubMed] [Google Scholar]
- Clement MS, Gammelgaard KR, Nielsen AL, & Sorensen BS (2020). Epithelial-to-mesenchymal transition is a resistance mechanism to sequential MET-TKI treatment of MET-amplified EGFR-TKI resistant non-small cell lung cancer cells. Translational Lung Cancer Research 9, 1904–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, … Hyman DM (2018). Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. The New England Journal of Medicine 378, 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, … Subbiah V (2020). Efficacy of Selpercatinib in RET fusion-positive non-small-cell lung cancer. The New England Journal of Medicine 383, 813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Takeuchi S, Arai S, Katayama R, Nanjo S, Tanimoto A, Nishiyama A, Nakagawa T, Taniguchi H, Suzuki T, Yamada T, Nishihara H, Ninomiya H, Ishikawa Y, Baba S, Takeuchi K, Horiike A, Yanagitani N, Nishio M, & Yano S (2019). Epithelial-to-mesenchymal transition is a mechanism of ALK inhibitor resistance in lung Cancer independent of ALK mutation status. Cancer Research 79, 1658–1670. [DOI] [PubMed] [Google Scholar]
- Gainor JF, Curigliano G, Kim DW, Lee DH, Besse B, Baik CS, … Subbiah V (2021). Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): A multi-cohort, open-label, phase 1/2 study. The Lancet Oncology 22, 959–969. [DOI] [PubMed] [Google Scholar]
- Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, … Shaw AT (2016). Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discovery 6, 1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, … Vecchio G (1990). PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 60, 557–563. [DOI] [PubMed] [Google Scholar]
- Hegde A, Andreev-Drakhlin AY, Roszik J, Huang L, Liu S, Hess K, … Subbiah V (2020). Responsiveness to immune checkpoint inhibitors versus other systemic therapies in RET-aberrant malignancies. ESMO Open 5, Article e000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez CF (2013). Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb Perspect Biol, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro Y, Iwashita T, Murakami H, Asai N, Iida K, Goto H, Hayakawa T, & Takahashi M (1999). The role of amino acids surrounding tyrosine 1062 in ret in specific binding of the shc phosphotyrosine-binding domain. Endocrinology 140, 3992–3998. [DOI] [PubMed] [Google Scholar]
- Ishizaka Y, Itoh F, Tahira T, Ikeda I, Sugimura T, Tucker J, … Nagao M (1989). Human ret proto-oncogene mapped to chromosome 10q11.2. Oncogene 4, 1519–1521. [PubMed] [Google Scholar]
- Jakobsen KR, Demuth C, Madsen AT, Hussmann D, Vad-Nielsen J, Nielsen AL, & Sorensen BS (2017). MET amplification and epithelial-to-mesenchymal transition exist as parallel resistance mechanisms in erlotinib-resistant, EGFR-mutated, NSCLC HCC827 cells. Oncogenesis 6, Article e307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, Iwashita T, Murakami H, Hayashi H, Kawai K, & Takahashi M (2001). Identification of SNT/FRS2 docking site on RET receptor tyrosine kinase and its role for signal transduction. Oncogene 20, 1929–1938. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Liu SV, McCoach CE, Zhu VW, Tan AC, Yoda S, … Gainor JF (2020). Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Annals of Oncology 31, 1725–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Vega QC, Decker RA, Pandey A, Worby CA, & Dixon JE (1996). Oncogenic RET receptors display different autophosphorylation sites and substrate binding specificities. The Journal of Biological Chemistry 271, 5309–5312. [DOI] [PubMed] [Google Scholar]
- Lorenzo MJ, Gish GD, Houghton C, Stonehouse TJ, Pawson T, Ponder BA, & Smith DP (1997). RET alternate splicing influences the interaction of activated RET with the SH2 and PTB domains of Shc, and the SH2 domain of Grb2. Oncogene 14, 763–771. [DOI] [PubMed] [Google Scholar]
- Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, … Gautschi O (2019). Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Annals of Oncology 30, 1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo RM, Carlomagno F, De Vita G, Formisano P, Vecchio G, Fusco A, … Santoro M (2001). The insulin receptor substrate (IRS)-1 recruits phosphatidylinositol 3-kinase to ret: Evidence for a competition between Shc and IRS-1 for the binding to Ret. Oncogene 20, 209–218. [DOI] [PubMed] [Google Scholar]
- Minocha M, Khurana V, Qin B, Pal D, & Mitra AK (2012). Co-administration strategy to enhance brain accumulation of vandetanib by modulating P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp1/Abcg2) mediated efflux with m-TOR inhibitors. International Journal of Pharmaceutics 434, 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan LM (2014). RET revisited: Expanding the oncogenic portfolio. Nature Reviews. Cancer 14, 173–186. [DOI] [PubMed] [Google Scholar]
- Myers SM, Eng C, Ponder BA, & Mulligan LM (1995). Characterization of RET proto-oncogene 3′ splicing variants and polyadenylation sites: A novel C-terminus for RET. Oncogene 11, 2039–2045. [PubMed] [Google Scholar]
- Offin M, Guo R, Wu SL, Sabari J, Land JD, Ni A, … Drilon A (2019). Immunophenotype and response to immunotherapy of RET-rearranged lung cancers. JCO Precision Oncology 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini A, Geneste O, Legrand P, Schlumberger M, Rossel M, Fournier L, … Billaud M (1997). Oncogenic activation of RET by two distinct FMTC mutations affecting the tyrosine kinase domain. Oncogene 15, 393–402. [DOI] [PubMed] [Google Scholar]
- Peterson S, & Bogenmann E (2004). The RET and TRKA pathways collaborate to regulate neuroblastoma differentiation. Oncogene 23, 213–225. [DOI] [PubMed] [Google Scholar]
- Plaza-Menacho I, Mologni L, & McDonald NQ (2014). Mechanisms of RET signaling in cancer: Current and future implications for targeted therapy. Cellular Signalling 26, 1743–1752. [DOI] [PubMed] [Google Scholar]
- Plenker D, Riedel M, Bragelmann J, Dammert MA, Chauhan R, Knowles PP, … Sos ML (2017). Drugging the catalytically inactive state of RET kinase in RET-rearranged tumors. Science Translational Medicine 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi I, Rajanna S, Webb A, Chhabra G, Foster B, Webb B, & Puri N (2016). Mechanism of c-Met and EGFR tyrosine kinase inhibitor resistance through epithelial mesenchymal transition in non-small cell lung cancer. Biochemical and Biophysical Research Communications 477, 937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen EY, Johnson ML, Clifford SE, Somwar R, Kherani JF, Son J, … Oxnard GR (2021). Overcoming MET-dependent resistance to selective RET inhibition in patients with RET fusion-positive lung cancer by combining selpercatinib with crizotinib. Clinical Cancer Research 27, 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F, & Heymach JV (2019). Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nature Reviews. Cancer 19, 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BJ, Tan L, Lin JJ, Wong SQ, Hollizeck S, Ebata K, … Rothenberg SM (2020). RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. Journal of Thoracic Oncology 15, 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Berry J, Roxas M, Guha-Thakurta N, Subbiah IM, Ali SM, … Heymach JV (2015). Systemic and CNS activity of the RET inhibitor vandetanib combined with the mTOR inhibitor everolimus in KIF5B-RET re-arranged non-small cell lung cancer with brain metastases. Lung Cancer 89, 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Cassier PA, Siena S, Garralda E, Paz-Ares L, Garrido P, … Curigliano G (2022). Pan-cancer efficacy of pralsetinib in patients with RET fusion-positive solid tumors from the phase 1/2 ARROW trial. Nature Medicine 28, 1640–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, & Cote GJ (2020a). Advances in targeting RET-dependent cancers. Cancer Discovery 10, 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, & Cote GJ (2020b). Advances in targeting RET-dependent cancers. Cancer Discovery 10, 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Gainor JF, Oxnard GR, Tan DSW, Owen DH, Cho BC, … Drilon A (2021a). Intracranial efficacy of selpercatinib in RET fusion-positive non-small cell lung cancers on the LIBRETTO-001 trial. Clinical Cancer Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Gainor JF, Oxnard GR, Tan DSW, Owen DH, Cho BC, … Drilon A (2021b). Intracranial efficacy of selpercatinib in RET fusion-positive non-small cell lung cancers on the LIBRETTO-001 trial. Clinical Cancer Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, … Evans EK (2018). Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discovery 8, 836–849. [DOI] [PubMed] [Google Scholar]
- Subbiah V, Hu MI, Wirth LJ, Schuler M, Mansfield AS, Curigliano G, … Taylor MH (2021. AUGUST 01). Pralsetinib for patients with advanced or metastatic RET-altered thyroid cancer (ARROW): A multi-cohort, open-label, registrational, phase 1/2 study. The Lancet Diabetes and Endocrinology 9(8), P491–501. [DOI] [PubMed] [Google Scholar]
- Subbiah V, Hu MI-N, Gainor JF, Mansfield AS, Alonso G, Taylor MH, … Curigliano G (2021). Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion–positive solid tumors. Journal of Clinical Oncology 39, 467.33434059 [Google Scholar]
- Subbiah V, Konda B, Bauer T, McCoach C, Falchook G, Takeda M, Patel J, Weiss J, Peled N, Bazhenova L, Soldatenkova V, French P, Drove N, Gautschi O, & Drilon A (2021). CT011 - Efficacy and safety of selpercatinib in RET fusion-positive cancers other than lung or thyroid cancers. In 2021 American association for cancer research (AACR) annual meeting. Virtual Meeting. [Google Scholar]
- Subbiah V, & Kurzrock R (2016). Universal genomic testing needed to win the war against cancer: Genomics IS the diagnosis. JAMA Oncology 2, 719–720. [DOI] [PubMed] [Google Scholar]
- Subbiah V, & Kurzrock R (2018). Challenging standard-of-care paradigms in the precision oncology era. Trends in Cancer 4, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Shen T, Tetzlaff M, Weissferdt A, Byers LA, Cascone T, … Wu J (2021). Patient-driven discovery and post-clinical validation of NTRK3 fusion as an acquired resistance mechanism to selpercatinib in RET fusion-positive lung cancer. Annals of Oncology 32, 817–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas ME, … Drilon A (2018). Selective RET kinase inhibition for patients with RET-altered cancers. Annals of Oncology 29, 1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Shen T, Terzyan SS, Liu X, Hu X, Patel KP, … Wu J (2021). Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann Oncol 32, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Wolf J, Konda B, Kang H, Spira A, Weiss J, … Drilon A (2022. Oct). Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): A phase 1/2, open-label, basket trial. The Lancet Oncology 23(10), 1261–1273. [DOI] [PubMed] [Google Scholar]
- Subbiah V, Yang D, Velcheti V, Drilon A, & Meric-Bernstam F (2020). State-of-the-art strategies for targeting RET-dependent cancers. Journal of Clinical Oncology 38, 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahira T, Ishizaka Y, Itoh F, Sugimura T, & Nagao M (1990). Characterization of ret proto-oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma cell line. Oncogene 5, 97–102. [PubMed] [Google Scholar]
- Takahashi M, Kawai K, & Asai N (2020). Roles of the RET proto-oncogene in cancer and development. JMA Journal 3, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Matsubara T, Haratake N, Toyokawa G, Fujishita T, Toyozawa R, Ito K, Yamaguchi M, Taguchi K, Okamoto T, & Seto T (2021). Targeted therapy for RET fusion lung cancer: Breakthrough and unresolved issue. Frontiers in Oncology 11, Article 704084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thein KZ, Velcheti V, Mooers BHM, Wu J, & Subbiah V (2021a). Precision therapy for RET-altered cancers with RET inhibitors. Trends Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thein KZ, Velcheti V, Mooers BHM, Wu J, & Subbiah V (2021b). Precision therapy for RET-altered cancers with RET inhibitors. Trends Cancer 7, 1074–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, … Haley JD (2005). Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Research 65, 9455–9462. [DOI] [PubMed] [Google Scholar]
- Torralvo J, Friedlaender A, Achard V, & Addeo A (2019). The activity of immune checkpoint inhibition in KRAS mutated non-small cell lung cancer: A single centre experience. Cancer Genomics Proteomics 16, 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X (2013). Structural studies of GDNF family ligands with their receptors-insights into ligand recognition and activation of receptor tyrosine kinase RET. Biochimica et Biophysica Acta 1834, 2205–2212. [DOI] [PubMed] [Google Scholar]
- Ware KE, Hinz TK, Kleczko E, Singleton KR, Marek LA, Helfrich BA, … Heasley LE (2013). A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis 2, Article e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth LJ, Sherman E, Robinson B, Solomon B, Kang H, Lorch J, … Cabanillas ME (2020). Efficacy of selpercatinib in RET-altered thyroid cancers. The New England Journal of Medicine 383, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo W, Wang Y, Zhuo X, Zhang Y, Ao X, & Chen Z (2008). Knockdown of snail, a novel zinc finger transcription factor, via RNA interference increases A549 cell sensitivity to cisplatin via JNK/mitochondrial pathway. Lung Cancer 62, 8–14. [DOI] [PubMed] [Google Scholar]
- Zhuo WL, Wang Y, Zhuo XL, Zhang YS, & Chen ZT (2008). Short interfering RNA directed against TWIST, a novel zinc finger transcription factor, increases A549 cell sensitivity to cisplatin via MAPK/mitochondrial pathway. Biochemical and Biophysical Research Communications 369, 1098–1102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.