

Abstract
Background:
Globular glial tauopathy (GGT) has been associated with frontotemporal dementia syndromes; little is known about the clinical and imaging characteristics of GGT and how they differ from other non-globular glial 4-repeat tauopathies (N4GT) such as progressive supranuclear palsy (PSP) or corticobasal degeneration (CBD).
Methods:
For this case-control study the Mayo Clinic brain banks were queried for all cases with an autopsy-confirmed diagnosis of GGT between 01/01/2011 and 31/10/2021. Fifty patients with N4GT (30 PSP, 20 CBD) were prospectively recruited and followed at Mayo Clinic, Minnesota. MR imaging was used to characterize patterns of gray/white matter atrophy, MR-parkinsonism index, midbrain volume, and white matter hyperintensities.18F-Fluorodeoxyglucose-, 11C Pittsburg compound-, and 18F-flortaucipir-PET scans were reviewed.
Results:
Twelve patients with GGT were identified: 83% were women compared to 42% in NG4T (P=0.02) with median age at death 76.5 years (range: 55–87). The most frequent clinical features were eye movement abnormalities, parkinsonism, behavioral changes, cognitive impairment followed by pyramidal tract signs and speech abnormalities. Lower motor neuron involvement was present in 17% and distinguished GGT from NG4T, P=0.035. Primary progressive apraxia of speech was the most frequent initial diagnosis (25%); 50% had a Parkinson-plus syndrome before death. Most GGT patients had asymmetric frontotemporal atrophy with matching hypometabolism. GGT patients had more gray matter atrophy in temporal lobes, normal MR-parkinsonism index, and larger midbrain volumes.
Conclusions:
Female sex, lower motor neuron involvement in the context of a frontotemporal dementia syndrome and asymmetric brain atrophy with preserved midbrain might be suggestive of underlying GGT.
Keywords: globular glial tauopathy, progressive supranuclear palsy, corticobasal degeneration, neuroimaging, diagnosis
INTRODUCTION
Globular glial tauopathy (GGT) is a relatively newly described neurodegenerative proteinopathy in the tau-positive frontotemporal lobar degeneration (FTLD-tau) spectrum with four-repeat (4R) tau inclusions found primarily in the glia (astrocytes/oligodendrocytes).(1) GGT comprises less than 10% of FTLD-tau cases. Additionally, 30% of patients with a GGT have been found to have mutations in the microtubule associated protein tau (MAPT) gene.(2) Three GGT subtypes are recognized: Type I is characterized by globular oligodendroglial inclusions (GOIs) found predominantly in frontotemporal regions without corticospinal tract (CST) involvement(1, 3); Type II is characterized by motor cortex involvement and CST degeneration(1, 4) whereas Type III is characterized by diffuse involvement of frontotemporal and motor cortices with CST and often lower motor neuron involvement(1). Adding to the complexity of GGT is the diversity of clinical phenotypes making it a diagnostic conundrum. Clinical syndromes associated with GGT have been diverse including behavioral variant frontotemporal dementia (bvFTD) and primary progressive aphasia (PPA) especially in GGT type I.(1, 3, 5–8) In addition, Parkinson-plus syndromes such as progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS) as well as primary lateral sclerosis have been described particularly in GGT type II(1, 4) whereas lower motor neuron disease(1) in combination with parkinsonism and dementia is associated with GGT type III. Comprehensive data remain scarce and there are only a handful of case reports and small series describing frontotemporal and occasionally parietal lobe atrophy on MRI.(9–13) To our knowledge there are only two cases in the literature showing frontotemporal hypometabolism on [18F]-fluorodeoxyglucose-PET(10, 14) and a single case report describing changes on single-photon emission tomography (SPECT)(11) and molecular tau-PET in GGT.(15) Overlap of syndromic and imaging features associated with other neuropathologic entities in FTLD-tau spectrum (Pick’s disease, PSP, corticobasal degeneration [CBD]) may hinder the successful enrollment and interfere with outcome evaluation in clinical trials.
In this study we sought to address some of the above-mentioned knowledge gaps. We describe clinical and multimodal (cross-sectional/longitudinal) neuroimaging features in a series of 12 patients with GGT who were followed and came to autopsy over the past 10 years. We also compare demographic, clinical, and neuroimaging features of GGT patients to prospectively followed cohort of patients who had autopsy confirmed non-GGT 4R tauopathy (NG4T) at death (PSP or CBD).
MATERIALS AND METHODS
Patients
The Mayo Clinic brain banks in Minnesota and Florida were queried for all autopsy-confirmed cases meeting international pathological criteria for GGT(1) between 1/1/2011–10/31/2021 from a cohort of 1658 4-repeat tauopathies. All patient charts were reviewed by a clinician, and both clinical and demographic data were abstracted. Two cases were published as case reports.(8, 16, 17) All patients were screened for mutations in MAPT gene.
Fifty patients with autopsy-proven PSP(18, 19) or CBD(20) were selected as a NG4T group for comparison of demographic, clinical and neuroimaging features, including cerebral and midbrain atrophy, white matter hyperintensity (WMH) burden, and magnetic resonance parkinsonism index (MRPI). All NG4T patients were prospectively recruited and followed at Mayo Clinic, Rochester, Minnesota, USA as part of National Institutes of Health funded studies.
Standard protocol approvals, registration, and patient consents
This study was approved by the Mayo Clinic Institutional Review Board; all patients and/or their proxies signed a written informed consent before taking part in research activities in accordance with the 1964 Declaration of Helsinki and its amendments.
Pathologic diagnosis
All neuropathological diagnoses were established using hematoxylin and eosin staining and phospho-tau immunohistochemistry (CP13; mouse monoclonal; 1:1000; a gift from the late Dr Peter Davies; Feinstein Institute for Medical Research) staining slides. GGT was diagnosed when tau-positive and Gallyas positive globular oligodendroglial inclusions (GOIs), coiled body-like structures and/or non-argyrophilic tau-positive globular astrocytic inclusions (GAIs) were identified in frontotemporal, motor cortices and/or CST.(1) GGT cases were classified as Type I, Type II or Type III according to published criteria (Figure 1).(1) PSP was diagnosed when characteristic tau-positive neuronal (neurofibrillary tangles, neuropil threads) and glial (tufted astrocytes, oligodendroglial coiled bodies) lesions were present in vulnerable cortical and subcortical regions.(18, 19) CBD was diagnosed by the presence of cortical and subcortical neuronal (pretangles) and glial lesions (astrocytic plaques) and threadlike processes in gray and white matter.(20)
Figure 1. Three pathologic types of GGT.
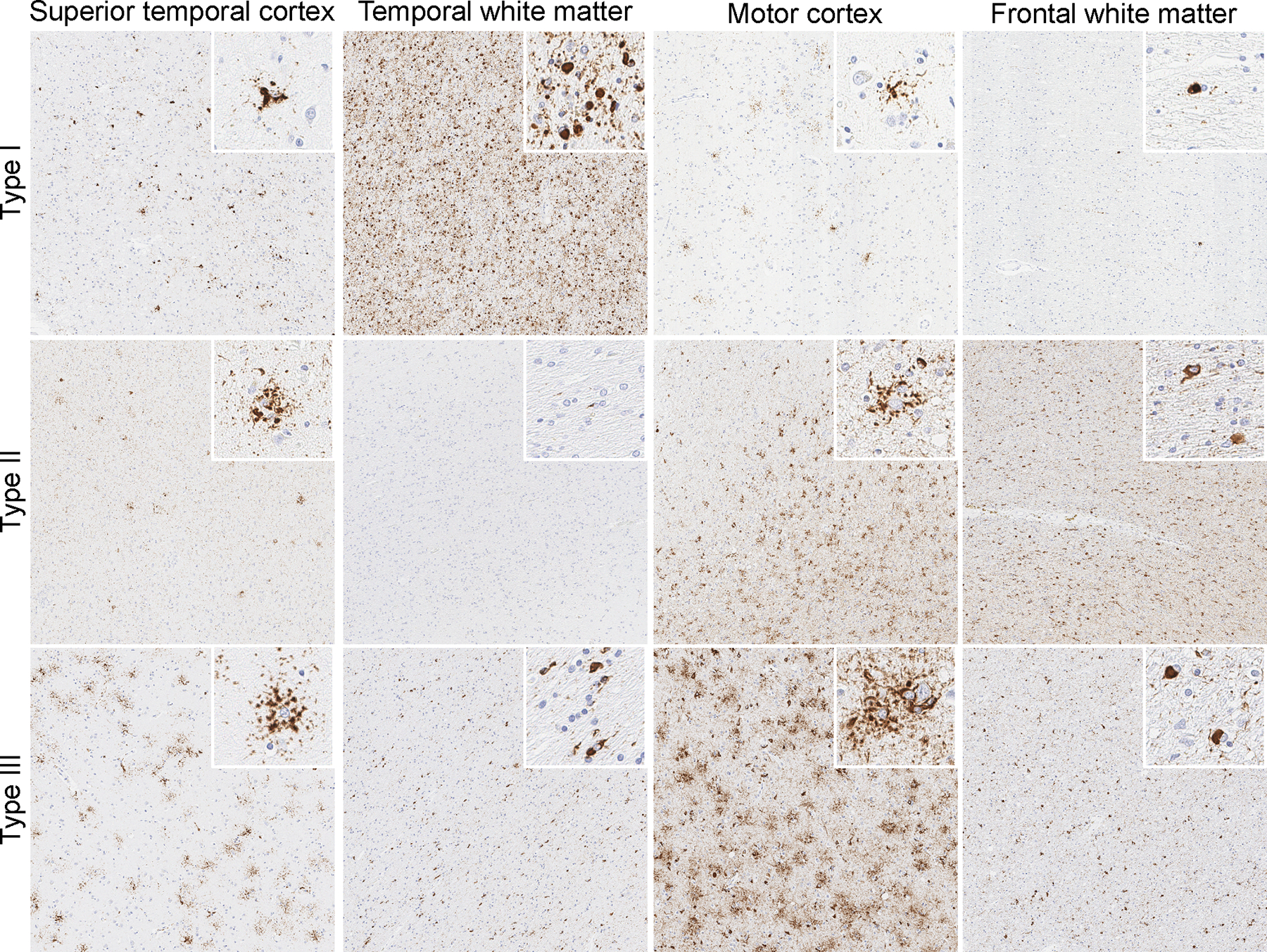
Tau immunohistochemistry with anti-phospho-tau antibody (AT8, Dako, Carpinteria, CA) demonstrating grey and white matter globular astrocytic and oligodendroglial inclusions, respectively, in GGT types I, II and III.
Imaging analysis
MR imaging and analysis
MRI scans were completed and interpreted in all GGT and NG4T patients. Ten GGT patients had images available for review. Seven scans were performed using volumetric research protocol at 3T using a GE scanner, while the remaining three scans were non-volumetric. All NG4T patients had MR scans performed at 3T using a GE scanner with standardized volumetric research protocol.
For the seven GGT patients with volumetric 3T MR imaging, the total intracranial volume (TIV) corrected gray matter volumes for specific regions-of-interest were generated using Statistical Parametric Mapping 12 (SPM12) software and an in-house atlas. The corrected volumes were subsequently utilized in an algorithm template to generate individual z-scores. Atrophic regions with z-scores less than −1.0 were shown in 3D MRIcroGL renders (https://www.mccauslandcenter.sc.edu/crnl).
Using SPM12, voxel-level comparisons of MRI gray and white matter (GM/WM) volumes were performed with voxel-based morphometry (VBM)(21) comparing GGT patients (type I, n = 4; type II, n = 2; type III, n = 1) with volumetric MR scans to the 20 CBD and 30 PSP patients. GGT patients were also compared to 15 healthy controls aged 55 – 79. Analyses were adjusted for age at scan and sex. The average probability maps of GM and WM were smoothed at 6 mm full-width at half maximum.
Details regarding MRPI measurements have been previously published(22) and were performed by a trained imaging analyst using ITK-SNAP software.
All FLAIR images first went through an automated segmentation process using in-house software(23) then were manually edited by trained imaging analysts. Regional and total WMH burden was calculated using an in-house 22-region atlas that categorizes WMH as subcortical or periventricular and corrected for TIV. Logistic regression models, adjusting for the age at scan were used to compare regional WMH burden between GGT and NG4T patients.
PET imaging
Ante mortem 18F-fluorodeoxyglucose (FDG)-PET imaging was available for five patients with GGT. All PET scans were performed using a GE PET/CT scanner (GE Healthcare, Milwaukee, WI) operating in 3D mode. Individual-level patterns of hypometabolism were assessed using 3D stereotactic surface projections(24) using CortexID (GE Healthcare) whereby activity at each voxel is z-scored to an age-segmented normative database.
Three GGT patients had antemortem amyloid-beta PET imaging with Pittsburg Compound B (PiB) and flortaucipir PET imaging performed. Global PiB standardized uptake value ratio (SUVR) was calculated as previously described with a cutoff point of 1.48 used to define PiB-PET positivity.(25) Flortaucipir PET images were divided by uptake in the cerebellar crus gray matter to create SUVR images.
One GGT patient had 123I-FP-CIT scan (dopamine transporter DaT scan, GE Healthcare, Chicago, IL), and three had 99mTc Neurolite SPECT scans performed.
Statistical analysis
All statistical analyses were performed in JMP Pro 14.1.0 (SAS Institute Inc.) software. Two-tailed Fisher’s Exact and Wilcoxon rank sum tests were used for categorical and continuous variables, respectively.
Data availability
Anonymized data are available from the corresponding author upon request from any qualified investigator for purposes of replicating procedures and results.
RESULTS
Demographic and clinical characteristics
Demographic and clinical characteristics of the patients with GGT, as well as those with NG4T are summarized in Table 1. While age at onset did not differ between GGT and NG4T, GGT patients had later symptom onset compared to CBD (P=0.035) but not PSP. Lower motor neuron involvement though infrequent was observed only in GGT patients, significant difference compared to NG4T. Speech abnormalities and parkinsonism were core features of NG4T affecting > 80% of patients but were present in about a half of patients with GGT. Distribution of GGT pathologic subtypes was the following: Type I – 42% (n = 5), Type II – 42% (n = 5), Type III –16% (n = 2), Figure 2. Fifty percent of the GGT patients eventually met clinical criteria for a Parkinson-plus syndrome (PSP/CBS)(26, 27) compared to 86% of patients with NG4T.
Table 1.
Demographic and clinical characteristics of patients with GGT pathologic diagnosis vs non-GGT 4-repeat tauopathy pathology (PSP or CBD)
| Clinical and demographic characteristics | GGT n = 12 |
NG4T‡ n = 50 |
P-value† |
|---|---|---|---|
|
| |||
| Female, n, % | 10 (83) | 21 (42) | 0.022* |
| White, n, % | 11 (92) | 49 (98) | >0.99 |
| Education, median (range), years | 16 (9 – 16) | 16 (12 – 20) | 0.55 |
| Right-handed, n, % | 10 (91) | 42 (84) | 0.64 |
| Family history, n, % | 6 (50)§ | 16 (32) | 0.32 |
| Onset age, median (range), years | 67 (50 – 81) | 63 (47 – 76) | 0.19 |
| Disease duration, median (range), years | 6 (3.4 – 15) | 9 (4.4 – 15) | 0.13 |
| Age at death, median (range), years | 76.5 (55 – 87) | 72 (55 – 88) | 0.37 |
| Age at scan, median (range), years | 71 (54– 82) | 68 (48 – 79) | 0.25 |
| Initial clinical diagnosis, n, % | 0.004** | ||
| Primary progressive AOS | 3 (25) | 17 (34) | |
| Behavioral variant FTD | 2 (17) | 1 (2) | |
| Primary lateral sclerosis | 2 (17) | 0 (0) | |
| Richardson syndrome | 1 (8) | 14 (28) | |
| Progressive supranuclear palsy (non-RS) | 0 (0) | 7 (14) | |
| Corticobasal syndrome | 1 (8) | 2 (4) | |
| Alzheimer’s dementia | 1 (8) | 0 (0) | |
| Semantic variant PPA | 1 (8) | 0 (0) | |
| Nonfluent/agrammatic variant PPA | 0 (0) | 9 (18) | |
| Luria’s dynamic aphasia | 1 (8) | 0 (0) | |
| Final clinical diagnosis, n, % | 0.12 | ||
| Behavioral variant FTD | 2 (17) | 1 (2) | |
| Corticobasal syndrome | 4 (33) | 19 (38) | |
| Richardson syndrome | 2 (17) | 14 (28) | |
| Progressive supranuclear palsy (non-RS) | 0 (0) | 10 (20) | |
| Primary lateral sclerosis | 1 (8) | 0 (0) | |
| Alzheimer’s dementia | 1 (8) | 0 (0) | |
| Primary progressive AOS | 0 (0) | 1 (2) | |
| Semantic variant PPA | 1 (8) | 0 (0) | |
| Nonfluent/agrammatic variant PPA | 1 (8) | 3 (6) | |
| Logopenic variant PPA | 0 (0) | 1 (2) | |
| Progressive auditory agnosia | 0 (0) | 1 (2) | |
| Clinical features, n, % | |||
| Pyramidal tract | 5 (42) | 14 (28) | 0.49 |
| Limb weakness, lower motor neuron | 2 (17) | 0 (0) | 0.035* |
| Speech abnormalities | 5 (42) | 42 (84) | 0.005** |
| Eye movement abnormal | 6 (50) | 31 (62) | 0.52 |
| Parkinsonism | 6 (50) | 48 (96) | 0.003** |
| Language | 4 (33) | 19 (38) | >0.99 |
| Behavior/personality | 6 (50) | 25 (50) | >0.99 |
| Memory/attention/executive function | 6 (50) | 25 (50) | >0.99 |
| Other | 3(25)¶ | 1 (2)¥ | n/a |
P-values are from two-tailed Wilcoxon test for continuous variable and from two-tailed Fisher’s exact test for categorical variables
Non-GGT 4R tauopathy group included autopsy-confirmed PSP (n = 30) and CBD (n = 20)
Mutation in MAPT gene was identified in 2 patients upon death
One patient had hallucinations and delusions; another patient developed voice tremor; third patient had nonverbal orobuccal apraxia and Luria’s dynamic aphasia
One patient had progressive auditory agnosia (verbal and environmental)
Figure 2.

Flow chart representing initial and final clinical diagnoses as well as GGT type for 12 GGT patients.
MR imaging
Asymmetric frontotemporal atrophy was present in most patients with GGT although the severity and focal patterns of atrophy varied greatly among the patients (Figures 3 and Figure 4A–C1). Three-dimensional rendering of gray matter atrophy patterns for the seven GGT patients with volumetric MRI are shown in Figure 3. All seven patients had some degree of temporal lobe atrophy, most frequently asymmetric. Asymmetric frontal lobe involvement was present in all seven patients although it was minimal in patient 1. Patient 4, who presented with primary progressive apraxia of speech (PPAOS), had focal severe involvement of left primary, supplemental motor area, and left marginal gyrus. Both patients with PPAOS evolved to CBS (patients 5 & 7). Patient 6 had non-fluent/agrammatic variant of PPA and severe asymmetric dorsolateral prefrontal cortex atrophy. Patient 7 had significant gray matter atrophy of the premotor cortex which is associated with PPAOS.(28)
Figure 3. Three-dimensional renderings of patterns of gray matter atrophy in seven GGT patients with volumetric MRI.
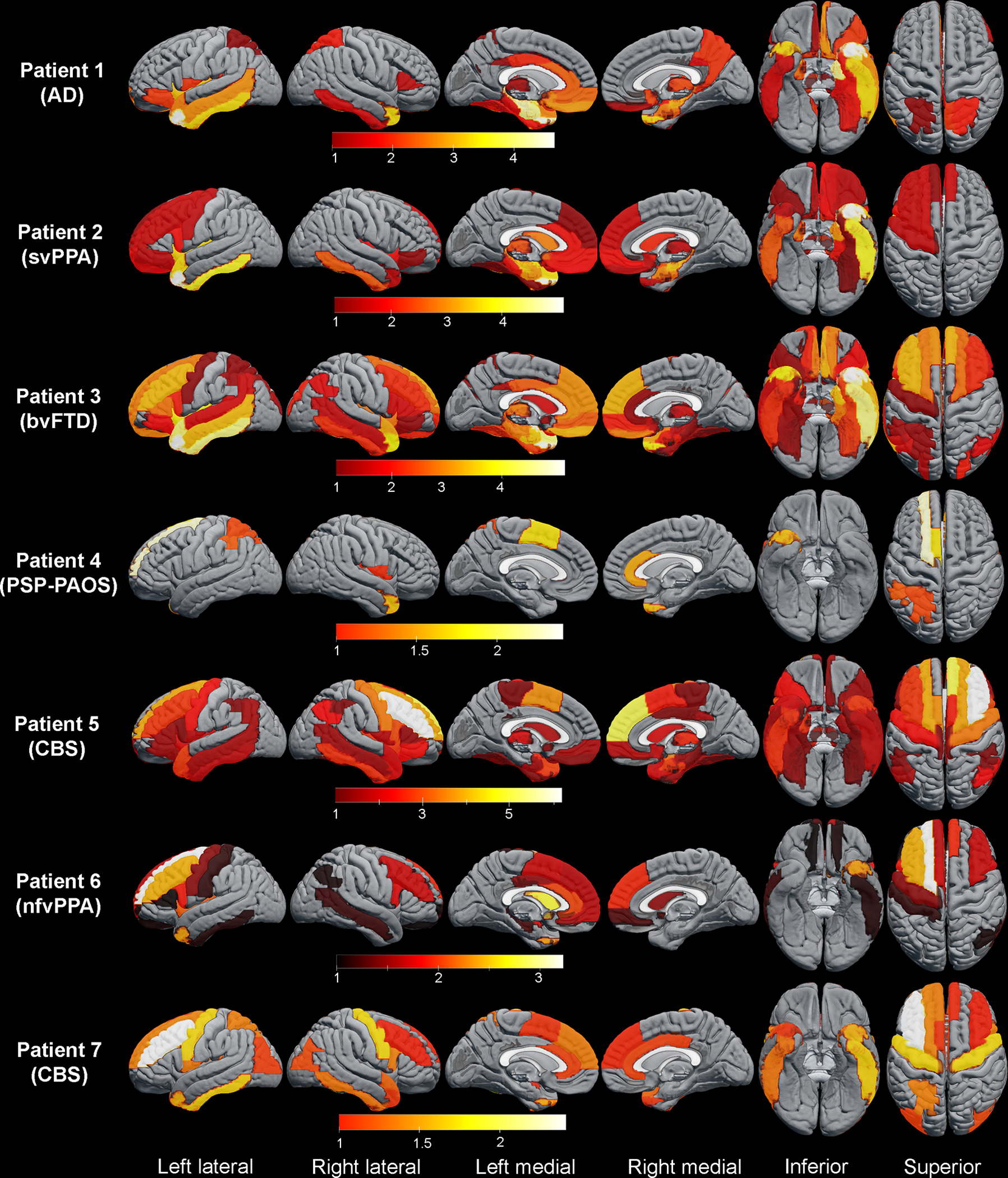
Atrophic regions with z-scores less than −1 are shown. Final clinical diagnoses are indicated in parenthesis for each patient. Lighter color signifies more severe atrophy.
Figure 4. Fluid attenuation inversion recovery (FLAIR) MR, DAT, and PET images of GGT patients.
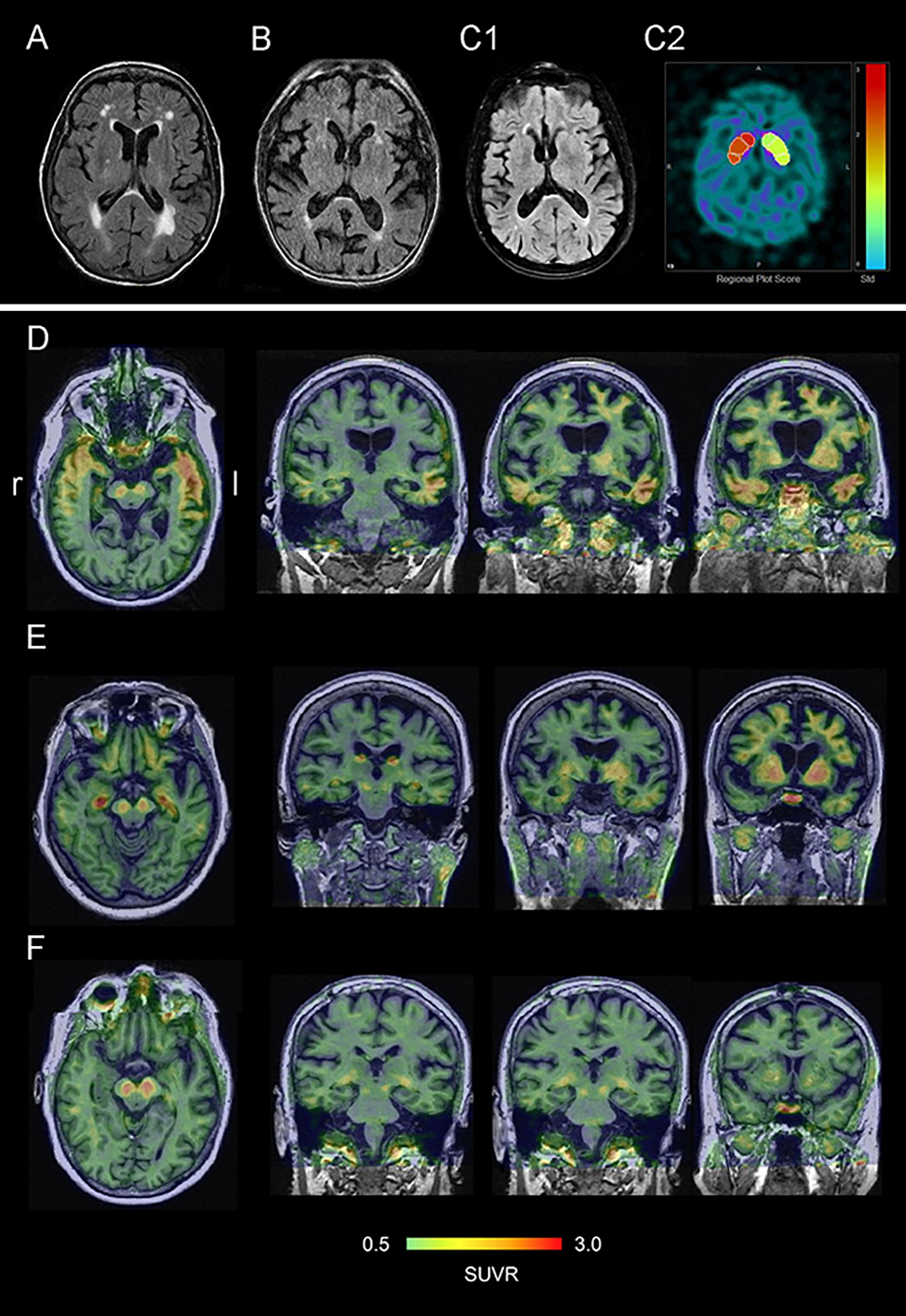
(A) Left temporal atrophy and white mater hyperintensities in patient 8 with CBS phenotype. (B) severe bilateral temporal atrophy in patient 9 with CBS. (C1) atrophy of frontal lobes and right greater than left temporal lobes in patient 10 with PSP-RS phenotype, (C2) decreased radiotracer uptake in right caudate and putamen on DAT scan in patient 10. (D) flortaucipir uptake correlates with pattern of gray matter atrophy on MR and FDG-PET hypometabolism pattern in patient 3. (E) flortaucipir uptake in right greater than left hippocampus, rubrospinal tracts, thalami, caudate and diffusely in frontal cortices in patient 6. (F) flortaucipir uptake mostly in rubrospinal tracts in patient 7. Warmer color correlates with greater flortaucipir uptake.
Sagittal MR scans were available for ten patients, and they showed generalized atrophy of the corpus callosum in 40% (Type I, n=2; Type II, n=1; Type III, n=1) and more severe focal atrophy of the anterior corpus callosum in 30% (all Type I). WMH were moderate in three GGT patients (Type II, n=2 and Type I, n=1) but absent-minimal in the remaining patients.
VBM analysis results comparing GM/WM brain atrophy patterns in GGT to controls, PSP and CBD are shown in Figure 5. PSP had higher MRPI (P=0.002) and smaller midbrain volumes (P=0.002) than GGT patients while there was no difference in MRPI or midbrain volumes between CBD and GGT. No significant difference was detected in regional and total WMH burden between GGT and NG4T patients.
Figure 5. Voxel-based morphometry analysis of MRI gray matter (GM) and white matter (WM) patterns of atrophy between GGT, controls and autopsy-proven PSP and CBD patients.
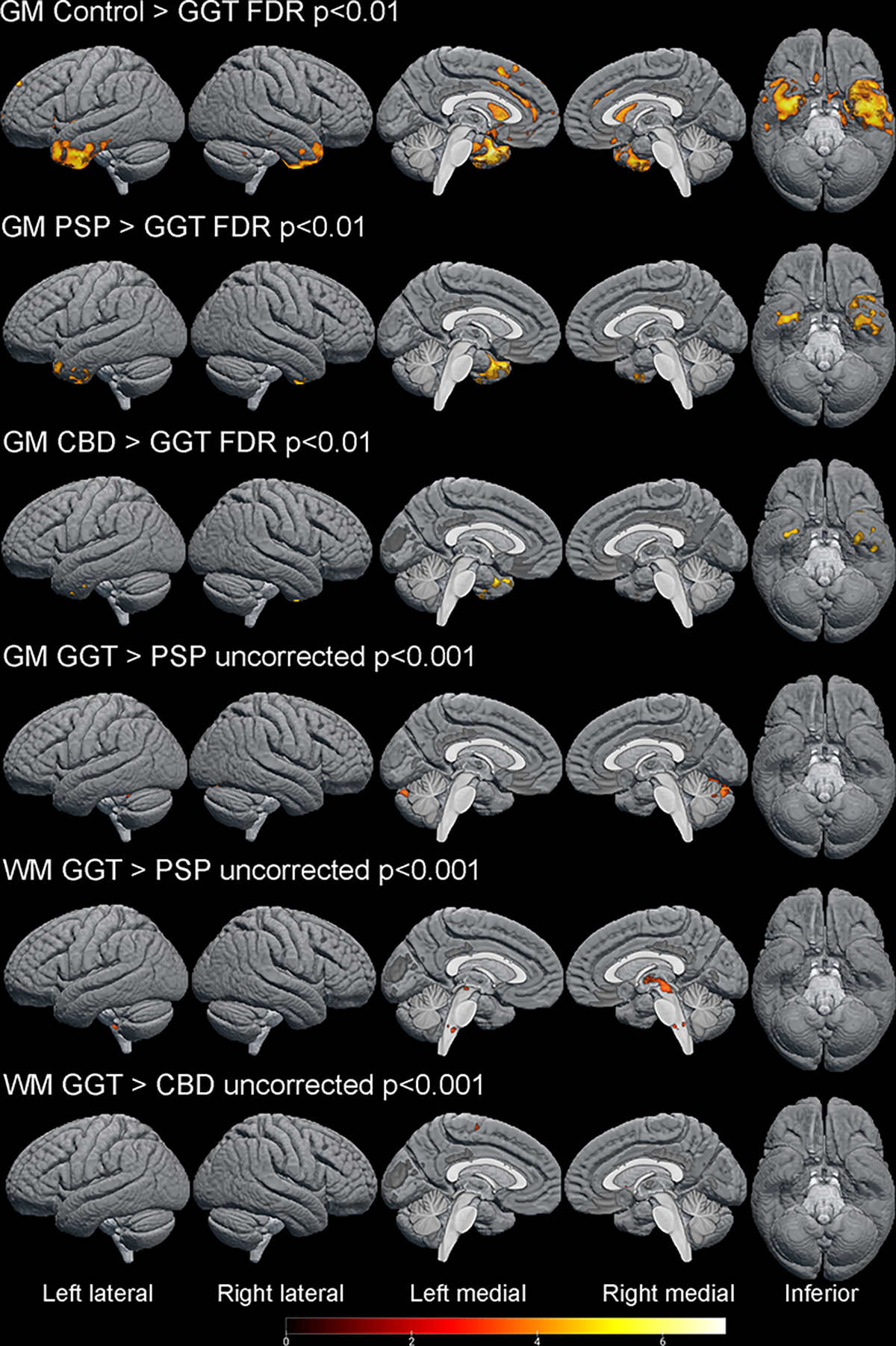
GGT patients have more GM in anterior and medial temporal lobes (left > right), caudate nuclei, anterior cingulate and frontal cortex compared to healthy controls. GGT patients had more GM atrophy in medial temporal lobes compared to PSP and CBD. PSP patients had significantly more WM atrophy in midbrain and medulla oblongata and more GM atrophy in cerebellum. CBD patients had slightly more WM atrophy in motor cortex. Lighter color signifies more atrophy.
PET imaging
Figure 6 demonstrates hypometabolism patterns for a subset of GGT patients. Hypometabolism pattern of patient 3 matches the pattern of GM atrophy on MR scans with severe asymmetric involvement of the temporal lobes as well as the frontal lobe. Patient 4 had mild hypometabolism in primary motor cortex (precentral gyri bilaterally), which was not evident on MR imaging. Patient 5 had severe frontal hypometabolism pattern that matched MR findings, whereas serial FDG-PET scans of patient 7 showed progressive hypometabolism of primary and supplementary motor cortices with overall sparing of temporal and frontal lobes, which were atrophic on MR.
Figure 6. 18F-FDG-PET imaging in 5 patients with GGT.
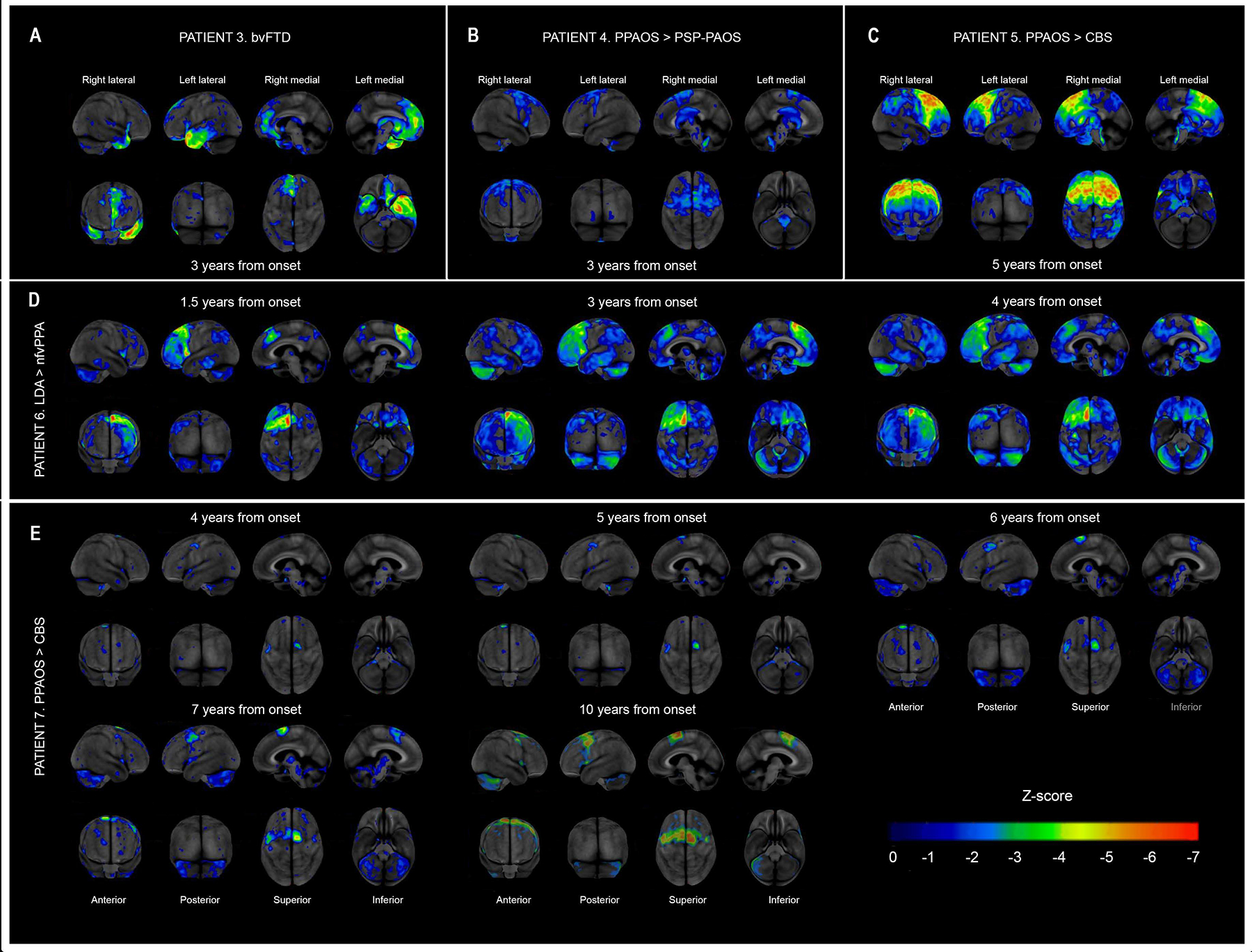
Two diagnoses shown for patients 4, 5, 6, and 7 represent initial clinical diagnosis and final diagnosis. Warmer color signifies more severe hypometabolism. LDA, Luria’s dynamic aphasia.
Two patients with PiB-PET (6 & 7) initially had amyloid-beta negative scans, but patient 7 became PiB-positive (SUVR=1.5) seven years after disease onset. Patient 3 initially had a slightly positive PiB-PET scan (SUVR=1.6) that worsened over two years (SUVR=1.7).
Patient 3 also had flortaucipir-PET imaging that demonstrated increased flortaucipir uptake in the temporal lobes bilaterally and the frontal lobe, which matched the severity of atrophy on MR and hypometabolism on FDG-PET (Figure 4D). In patients 6 flortaucipir uptake correlated less with patterns of atrophy/hypometabolism though showed uptake in frontal cortices (Figure 4E), whereas patient 7 had only mild uptake in temporal regions. All cases had flortaucipir uptake in the rubrospinal tracts whereas patient 6 had also flortaucipir uptake in the hippocampi (Figure 4D–F).
SPECT brain perfusion study was normal in patient 2. It was indeterminate in patient 8, which showed mildly decreased uptake in both posterior frontal regions compatible with early degenerative disease and a small focal defect in the left posterior frontal region likely representing a small infarct. Patient 9 had multiple regions of reduced tracer uptake in both hemispheres, right greater than left, with involvement of the mid and posterior frontal, parietal, temporal lobes bilaterally and with some involvement the left occipital pole and calcarine cortex. Patient 10 had an abnormal DaT scan (Figure 4C2).
DISCUSSION
In this study we examined clinical and neuroimaging features of 12 patients with autopsy proven GGT. PPAOS was the most common initial diagnosis while parkinsonism, eye movement abnormalities, behavioral/personality changes and cognitive impairment were present in the majority by the time of death. An asymmetrical frontotemporal pattern of atrophy/hypometabolism was observed in most patients with variability in severity and focality. FDG-PET studies correlated with clinical presentation, and partially flortaucipir uptake. When compared to NG4T patients, female sex was more frequent among GGT patients. Clinically, the patients with GGT more frequently had lower motor neuron involvement whereas parkinsonism and speech abnormalities were less frequent in GGT. In addition, GGT patients had more severe GM asymmetric temporal atrophy with relative sparing of midbrain.
GGT is known for its phenotypic heterogeneity and can present with bvFTD, svPPA, nfvPPA,(8, 29) CBS, PSP, Alzheimer’s dementia,(30) amyotrophic lateral sclerosis – frontotemporal dementia,(31) primary lateral sclerosis, and Creutzfeldt-Jakob disease.(1, 4, 32) Our case series fit with this variability in clinical presentations of GGT. By the time of death, atypical parkinsonian syndromes were noted in half of the GGT patients with 80% being type II/III suggesting CST involvement. Regardless of the GGT pathologic type (I, II, III), parkinsonism, oculomotor and speech abnormalities were common emphasizing significant overlap with PSP/CBS clinical syndromes though only half of GGT patients clinically evolved to Parkinson-plus syndrome compared to majority of patients with PSP/CBD pathology. Interestingly, the most frequent initial clinical diagnosis was PPAOS(28) suggesting early involvement of the premotor cortex in GGT. Pyramidal signs were also frequent, suggesting involvement of, or progression into the neighboring primary motor cortex.
While most described GGT cases highly resemble established clinical syndromes, quite often they are described as atypical presentations of such (e.g. atypical PSP, CBS, PPA) and do not completely comply with diagnostic criteria.(33) This represents a diagnostic dilemma as many GGT cases may be forced into diagnostic categories for another specific clinical syndrome which may complicate interpretation of clinical trials targeting specific pathologies different from GGT. On the other hand, it raises the question of whether diagnostic criteria for FTLD-spectrum disorders need to be revised and modified to account for GGT.
In this case series we demonstrate that GGT is frequently asymmetric which may aid in differentiating it from Alzheimer’s disease. Furthermore, most GGT cases had frontotemporal involvement as in previous studies.(14, 32) Some involvement of primary motor cortex was observed in most GGT cases, including those with bvFTD and svPPA. Two cases clinically diagnosed as CBS had severe GM atrophy of dorsolateral prefrontal cortex, which was also present, yet less severe, in patient 3 with bvFTD and patient 6 with nfv/agPPA. Asymmetric frontal cortical involvement can be seen with CBS with different areas affected based on underlying pathology with more focal frontal cortices involvement seen in corticobasal degeneration and PSP.(34, 35)
FDG-PET imaging in our patients with GGT correlated with their clinical phenotypes, however, did not always match the GM pattern of atrophy. It is unclear what this dissonance between two imaging modalities represents. In pursuit of identification of potential hallmarks of GGT several studies have assessed patterns of WM involvement, given that GGT is primarily a glial proteinopathy. Corpus callosum abnormalities and WM changes have been described in case reports.(11, 12) In our series, however, only a few patients had moderate periventricular WMH, and we did not observe hypersignal in the corpus callosum on FLAIR in any of the patients; albeit the vast majority had some degree of corpus callosum atrophy. Two out of three patients with anterior corpus callosum atrophy presented with PPA which is consistent with findings reported by Keller et al.(12) As a result, we cannot confirm that corpus collosum involvement is a specific signature feature of GGT that would help distinguish it from other 4R tauopathies.
It is debatable whether tau-PET can aid in ante mortem diagnosis of GGT. Flortaucipir uptake correlated with patterns of atrophy and hypometabolism in one patient but not others, it is unclear whether it also correlates with tau burden. Increased uptake of flortaucipir, the most frequently used PET ligand for Alzheimer’s disease (3R+4R tauopathy) diagnosis, has been reported in 4R tauopathies.(15, 36, 37) In a single case report of flortaucipir-PET in GGT there was more uptake in white matter compared to gray matter which matched the distribution of tau inclusions at autopsy in that case.(15) In our series we found uptake in frontal and temporal regions including in gray matter as well as uptake in midbrain structures. This finding is very interesting given that beta-amyloid was either negative or minimal. This pattern of focal temporal tau uptake in the absence of amyloid is reminiscent of the pathology seen with primary age-related tauopathy.(38) It has also been observed in cases of semantic dementia presumed to be due to underlying TDP-43 pathology.(39) While GGT may be added to the differential diagnosis of elevated temporal lobe tau uptake in the absence of beta-amyloid, one should be cautious with tau-PET results as at least some level of nonspecific flortaucipir binding to 4R has been reported.(36)
When compared to patient with non-GGT 4R tauopathies (CBD/PSP), female sex was more frequent among GGT patients in our series compared to CBD/PSP, finding not reported elsewhere. In the recent review of 88 world-wide reported GGT cases 52% were female.(32) About a half of patients in this series evolved in a Parkinson-plus syndrome, CBS more frequently than PSP, which also is a higher proportion than reported 19% of 88 cases who had parkinsonism or Parkinson-plus syndrome.(32) Whether differences in female sex in this series represent a tissue bank bias is debatable as analyzed cases come from across all of the USA. Higher proportion of Parkinson-plus phenotype might be related to clinical studies focusing on Parkinson-plus syndromes run at Mayo Clinic sites, however, still would not explain female sex predominance as there is no clear sex association with either PSP or CBS.(40) Therefore based on the findings of this study, in the cases of CBS presentation older age of onset, female sex and/or presence of lower motor neuron involvement may be more suggestive of GGT versus CBD pathology.
GGT was associated with the strongest heritability among the frontotemporal tauopathies(2); in our series there was no significant difference in family history of neurodegenerative disease between GGT and NG4T, though higher proportion of GGT patients had positive family history and two had MAPT mutations. Whereas FTLD-MAPT is still classified separately from 4R tauopathies(41) merging these two groups might give a better estimate of genetic risks and heritability of FTLD spectrum tauopathies.(2, 41, 42)
One of the novelties of this study is that we formally compare GGT-associated atrophy patterns with other 4R tauopathies. Voxel-based analysis demonstrated more significant GM volume loss in temporal lobe, particularly in the left hemisphere, in GGT cases compared to controls, PSP, and CBD whereas PSP cases had more severe WM atrophy in the brainstem and GM atrophy in cerebellum. Greater temporal lobe atrophy in GGT compared to PSP/CBD is not surprising given the differences of tau burden distribution between these pathologies(1, 18–20) as well as might explain more frequent bvFTD or semantic dementia phenotype in GGT. Relatively spared midbrain and normal MRPI in GGT patients compared to PSP may explain why eye movement abnormalities and falls are less frequent in GGT cases compared to PSP.(12) Our study was limited, however, by small number of volumetric MR scans available for each of the three GGT types which precluded comparisons across types.
In this study we used current guidelines for establishment of GGT, PSP, and CBD neuropathologic diagnoses. A recent study describing structures of tau filament via cryo-electron microscopy found that among 4R tauopathies tau filament fold is similar between PSP and GGT types I/II and differs from CBD cases.(43) This further supports pathologic differences between GGT and CBD, and might explain clinical overlap between GGT and PSP cases. Whereas tau filament folds in GGT type I and II are similar, it differs from GGT type III.(43) Whether this finding should warrant distinguishing GGT type III as a separate pathologic entity is debatable, phenotypic variability associate with GGT might be due to pathologic variability among the GGT types supported now not only by difference in distribution of the inclusions but also tau filament structure.
Antemortem diagnosis of GGT is challenging due to phenotypic and pathologic heterogeneity, absence of imaging hallmarks and likely underdiagnosis of this pathology due to its relative novelty. Currently, many patients with GGT present as bvFTD, PPA or a Parkinson-plus syndrome diagnoses which may be due to overfitting diagnostic criteria. In certain cases, particularly those that evolve into a Parkinson-plus syndrome, female sex, an early diagnosis of primary progressive apraxia of speech, older age of symptom onset, lower motor neuron involvement, family history of neurodegenerative disorder, and MRI with an asymmetric pattern of atrophy, normal MRPI or minimal midbrain atrophy are features that may help predict GGT.
ACKNOWLEDGEMENTS
We thank AVID Radiopharmaceuticals, Inc., for their support in supplying the AV-1451 precursor, chemistry production advice and oversight, and FDA regulatory cross-filing permission and documentation needed for this work. We would also like to acknowledge The Elsie and Marvin Dekelboum Family Foundation and the Oxley Foundation.
FUNDING
The work was supported by the following National Institutes of Health grants: RF1-NS112153 (KAJ & JLW), R01-DC12519 (JLW), R01-NS89757 (JW & KAJ), R01-DC14942 (KAJ); P30 AG062677 (RCP).
Footnotes
CONFLICTS OF INTEREST
Marina Buciuc, Shunsuke Koga, Nha Trang Thu Pham, Joseph R. Duffy, Aivi Nhuyen, R. Ross Reichard, Farwa Ali, Hugo Botha, Jonathan Graff-Radford, Dennis W. Dickson, Jennifer L. Whitwell, and Keith A. Josephs report no conflicts of interest. David S. Knopman serves on a Data Safety Monitoring Board for the DIAN study; is an investigator in clinical trials sponsored by Biogen, Lilly Pharmaceuticals and the University of Southern California; and receives research support from the NIH. Bradley F. Boeve has served as an investigator for clinical trials sponsored by GE Healthcare, FORUM Pharmaceuticals, C2N Diagnostics and Axovant. He receives publishing royalties Behavioral Neurology of Dementia (Cambridge Medicine, 2009, 2016). He serves on the Scientific Advisory Board of the Tau Consortium. He receives research support from the NIH, the Mayo Clinic Dorothy, Harry T. Mangurian Jr. Lewy Body Dementia Program and the Little Family Foundation. Val J. Lowe consults for Bayer Schering Pharma, Piramal Life Sciences, Life Molecular Imaging, Eisai Inc., AVID Radiopharmaceuticals, and Merck Research and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals and the NIH (NIA, NCI). Ronald C. Petersen serves on data monitoring committees for Janssen Alzheimer Immunotherapy, and is a consultant for Biogen, Roche, Merck, Genentech, Inc; receives publishing royalties from Mild Cognitive Impairment (Oxford University Press, 2003), and receives research support from the NIH/NIA.
REFERENCES
- 1.Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta neuropathologica. 2013;126(4):537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forrest SL, Halliday GM, McCann H, McGeachie AB, McGinley CV, Hodges JR, et al. Heritability in frontotemporal tauopathies. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2019;11:115–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R, et al. White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. Journal of Neuropathology & Experimental Neurology. 2008;67(10):963–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Josephs KA, Katsuse O, Beccano-Kelly DA, Lin W-L, Uitti RJ, Fujino Y, et al. Atypical progressive supranuclear palsy with corticospinal tract degeneration. Journal of Neuropathology & Experimental Neurology. 2006;65(4):396–405. [DOI] [PubMed] [Google Scholar]
- 5.Powers J, Byrne N, Ito M, Takao M, Yankopoulou D, Spillantini M, et al. A novel leukoencephalopathy associated with tau deposits primarily in white matter glia. Acta neuropathologica. 2003;106(2):181–7. [DOI] [PubMed] [Google Scholar]
- 6.Giaccone G, Marcon G, Mangieri M, Morbin M, Rossi G, Fetoni V, et al. Atypical tauopathy with massive involvement of the white matter. Neuropathology and applied neurobiology. 2008;34(4):468–72. [DOI] [PubMed] [Google Scholar]
- 7.Hirano M, Iritani S, Fujishiro H, Torii Y, Kawashima K, Sekiguchi H, et al. Globular glial tauopathy Type I presenting with behavioral variant frontotemporal dementia. Neuropathology. 2020;40(5):515–25. [DOI] [PubMed] [Google Scholar]
- 8.Graff-Radford J, Josephs KA, Parisi JE, Dickson DW, Giannini C, Boeve BF. Globular glial tauopathy presenting as semantic variant primary progressive aphasia. JAMA neurology. 2016;73(1):123–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burrell JR, Forrest S, Bak TH, Hodges JR, Halliday GM, Kril JJ. Expanding the phenotypic associations of globular glial tau subtypes. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2016;4:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erro M, Zelaya M, Mendioroz M, Larumbe R, Ortega-Cubero S, Lanciego J, et al. Globular glial tauopathy caused by MAPT P301T mutation: clinical and neuropathological findings. Journal of neurology. 2019;266(10):2396–405. [DOI] [PubMed] [Google Scholar]
- 11.Ohno Y, Ikeda T, Sakurai K, Yamada K, Tomonari T, Iwasaki Y, et al. Rapid Progression of White Matter Signal Changes and Frontotemporal Atrophy in Globular Glial Tauopathy. Journal of Neuropathology & Experimental Neurology. 2021;80(5):480–3. [DOI] [PubMed] [Google Scholar]
- 12.Keller J, Kavkova A, Matej R, Cséfalvay Z, Rusina R. Corpus callosum hypersignals and focal atrophy: neuroimaging findings in globular glial tauopathy type I. European Journal of Neurology. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rusina R, Csefalvay Z, Kovacs GG, Keller J, Javurkova A, Matej R. Globular glial tauopathy type I presenting as atypical progressive aphasia, with comorbid limbic-predominant age-related TDP-43 encephalopathy. Frontiers in Aging Neuroscience. 2019;11:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zalewski N, Botha H, Whitwell JL, Lowe V, Dickson DW, Josephs KA. FDG-PET in pathologically confirmed spontaneous 4R-tauopathy variants. Journal of neurology. 2014;261(4):710–6. [DOI] [PubMed] [Google Scholar]
- 15.Lowe VJ, Lundt ES, Albertson SM, Min H-K, Fang P, Przybelski SA, et al. Tau-positron emission tomography correlates with neuropathology findings. Alzheimer’s & Dementia. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czarnecki K, Duffy JR, Nehl CR, Cross SA, Molano JR, Jack CR, et al. Very early semantic dementia with progressive temporal lobe atrophy: an 8-year longitudinal study. Archives of neurology. 2008;65(12):1659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tacik P, DeTure M, Lin W-L, Sanchez Contreras M, Wojtas A, Hinkle KM, et al. A novel tau mutation, p. K317N, causes globular glial tauopathy. Acta neuropathologica. 2015;130(2):199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauw J-J, Daniel S, Dickson D, Horoupian D, Jellinger K, Lantos P, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994;44(11):2015-. [DOI] [PubMed] [Google Scholar]
- 19.Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. Journal of neurology. 1999;246(2):II6–II15. [DOI] [PubMed] [Google Scholar]
- 20.Dickson DW, Bergeron C, Chin S, Duyckaerts C, Horoupian D, Ikeda K, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. Journal of Neuropathology & Experimental Neurology. 2002;61(11):935–46. [DOI] [PubMed] [Google Scholar]
- 21.Whitwell JL. Voxel-based morphometry: an automated technique for assessing structural changes in the brain. Journal of Neuroscience. 2009;29(31):9661–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grijalva RM, Pham NTT, Huang Q, Martin PR, Ali F, Clark HM, et al. Brainstem Biomarkers of Clinical Variant and Pathology in Progressive Supranuclear Palsy. Movement Disorders. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graff-Radford J, Arenaza-Urquijo EM, Knopman DS, Schwarz CG, Brown RD Jr, Rabinstein AA, et al. White matter hyperintensities: relationship to amyloid and tau burden. Brain. 2019;142(8):2483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minoshima S, Frey KA, Koeppe RA, Foster NL, Kuhl DE. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. Journal of Nuclear Medicine. 1995;36(7):1238–48. [PubMed] [Google Scholar]
- 25.Jack CR Jr, Wiste HJ, Botha H, Weigand SD, Therneau TM, Knopman DS, et al. The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain. 2019;142(10):3230–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Movement Disorders. 2017;32(6):853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Josephs KA, Duffy JR, Strand EA, Machulda MM, Senjem ML, Master AV, et al. Characterizing a neurodegenerative syndrome: primary progressive apraxia of speech. Brain. 2012;135(5):1522–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim E-J, Lee MJ, Lee J-H, Lee YM, Shin J-H, Shin M-J, et al. Globular glial tauopathy presenting as non-fluent/agrammatic variant primary progressive aphasia with chorea. Parkinsonism & related disorders. 2017;44:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.SantaCruz KS, Rottunda SJ, Meints JP, Bearer EL, Bigio EH, McCarten JR. A case of globular glial tauopathy presenting clinically as alzheimer disease. Alzheimer disease and associated disorders. 2015;29(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasaki R, Mimuro M, Kokubo Y, Imai H, Yoshida M, Tomimoto H. An autopsy case of globular glial tauopathy presenting with amyotrophic lateral sclerosis with dementia. Brain and nerve= Shinkei kenkyu no shinpo. 2016;68(8):945–50. [DOI] [PubMed] [Google Scholar]
- 32.Forrest SL, Kril JJ, Kovacs GG. Association Between Globular Glial Tauopathies and Frontotemporal Dementia—Expanding the Spectrum of Gliocentric Disorders: A Review. JAMA neurology. 2021. [DOI] [PubMed] [Google Scholar]
- 33.Marsili L, Dickson DW, Espay AJ. Globular Glial Tauopathy May be Mistaken for Corticobasal Syndrome—Pointers for the Clinician. Movement disorders clinical practice. 2018;5(4):439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Josephs KA, Whitwell JL, Boeve BF, Knopman DS, Petersen RC, Hu WT, et al. Anatomical differences between CBS-corticobasal degeneration and CBS-Alzheimer’s disease. Movement Disorders. 2010;25(9):1246–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitwell J, Jack C, Boeve B, Parisi J, Ahlskog J, Drubach D, et al. Imaging correlates of pathology in corticobasal syndrome. Neurology. 2010;75(21):1879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghirelli A, Tosakulwong N, Weigand SD, Clark HM, Ali F, Botha H, et al. Sensitivity–Specificity of Tau and Amyloid β Positron Emission Tomography in Frontotemporal Lobar Degeneration. Annals of neurology. 2020;88(5):1009–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schonhaut DR, McMillan CT, Spina S, Dickerson BC, Siderowf A, Devous MD Sr, et al. 18F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and parkinson disease: a multicenter study. Annals of neurology. 2017;82(4):622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta neuropathologica. 2014;128(6):755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makaretz SJ, Quimby M, Collins J, Makris N, McGinnis S, Schultz A, et al. Flortaucipir tau PET imaging in semantic variant primary progressive aphasia. Journal of Neurology, Neurosurgery & Psychiatry. 2018;89(10):1024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swallow DM, Zheng CS, Counsell CE. Systematic review of prevalence studies of progressive supranuclear palsy and corticobasal syndrome. Movement Disorders Clinical Practice. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forrest SL, Kril JJ, Stevens CH, Kwok JB, Hallupp M, Kim WS, et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain. 2018;141(2):521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Josephs KA. Rest in peace FTDP-17. Brain. 2018;141(2):324–31. [DOI] [PubMed] [Google Scholar]
- 43.Shi Y, Zhang W, Yang Y, Murzin AG, Falcon B, Kotecha A, et al. Structure-based classification of tauopathies. Nature. 2021;598(7880):359–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data are available from the corresponding author upon request from any qualified investigator for purposes of replicating procedures and results.