

Abstract
We aimed to probe the functions and possible mechanisms of empagliflozin in doxorubicin (Dox)-caused cardiotoxicity. First, a cardiotoxicity rat model was built by continuously injecting Dox intraperitoneally. Then, empagliflozin (30 mg/kg) was gavaged into the rats. Next, echocardiography was utilized for checking the cardiac function of rats, and H&E staining for observing pathological alterations of the myocardial tissues. Besides, biochemical assays and Enzyme-linked Immunosorbent Assay were adopted to detect the creatine kinase isoenzyme (CK-MB), N-terminal pro-brain natriuretic peptide (NT-proBNP), adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine monophosphate (AMP) levels in rat serum and superoxide dismutase (SOD), malondialdehyde acid (MDA), and catalase (CAT) in myocardial tissue, respectively. Furthermore, the expression of AMPK/SIRT-1/PGC-1α signaling pathway-related proteins in the myocardial tissues was tested by Western blot. Continuous intraperitoneal injection of Dox greatly elevated left ventricular end-systolic diameter (LVESD) and left ventricular end-diastolic diameter (LVEDD), reduced fractional shortening (FS) and ejection fraction (EF), and notably up-regulated CK-MB and NT-proBNP level in rats’ serum, thus impairing cardiac function. Empagliflozin treatment could ameliorate myocardial histopathological damage and alleviate cardiac function and tissue damage by down-regulating LVEDD and LVESD, up-regulating EF and FS, and inhibiting CK-MB and NT-proBNP level in serum. Additionally, empagliflozin improved Dox-induced excessive oxidative stress and dysregulation of energy metabolism. Furthermore, empagliflozin activated the AMPK/SIRT-1/PGC-1α signaling pathway in Dox-caused cardiotoxicity rats. In conclusion, in addition to bettering the cardiac tissue and function injury caused by Dox, empagliflozin also improves excessive oxidative stress and energy metabolism. Notably, empagliflozin may exert cardioprotective effects through activating the AMPK/SIRT-1/PGC-1α pathway.
Keywords: cardiotoxicity, doxorubicin (Dox), empagliflozin, AMPK/SIRT-1/PGC-1α, mitochondria
Introduction
Despite the continued decline in cancer-related mortality over the past 20 years, cancer is still the major reason of death globally. Thanks to the application of anticancer drugs, patients with cancer are allowed to improve their survival remarkably now.1 However, it is followed by the adverse reactions caused by many chemotherapeutic drugs to patients. Thereinto, cardiotoxicity is one of the most common toxic reactions of anticancer drugs and seriously endangers the patient’s life.2 Anthracyclines, as one of chemotherapeutic agents used in pediatric and adult sufferers, are often applied clinically to treat malignant tumors such as sarcoma, breast cancer and lymphoma.3 Among them, doxorubicin (Dox) is the commonest anthracycline applied in clinical practice. In spite of the satisfactory therapeutic effect of Dox, it has a limited clinical application due to its dose dependence as well as Dox-induced cumulative cardiotoxicity to one fourth of sufferers.4 Clinically, Dox-caused cardiotoxicity is characterized by decreased left ventricular ejection fraction (EF), increased thickness of ventricular wall, arrhythmias, and heart failure (HF); some patients with severe symptoms may die if they are not treated promptly.5 Studies have reported that the mechanisms of Dox inducing cardiotoxicity appear to be multifactorial, mainly including oxidative stress, raised lipid peroxidation, autophagy suppression, endoplasmic reticulum-mediated apoptosis, mitochondrial damage, and DNA/RNA damage.6 Though the mechanism that Dox induces cardiotoxicity has been extensively researched, the molecular pathogenesis of Dox remains incompletely understood.7 Therefore, it is still a significant challenge to excavate new treatment regimens to reduce Dox-induced cardiotoxicity without altering its anticancer efficacy.
Empagliflozin, as sodium-glucose cotransporter 2 inhibitors, is a novel kind of antidiabetic agent. On basis of previous clinical studies, empagliflozin is surprisingly beneficial to patients with diabetes (type 2) or HF.8 Another study showed that the improvement of empagliflozin on remodeling and cardiac function in rats without diabetes but with LV dysfunction upon myocardial infarction (MI) is related to substantially improved cardiac ATP generation and cardiac metabolism.9 Furthermore, the EMPA-REG OUTCOME trial has revealed that empagliflozin conduces greatly to cardiovascular system via reducing HF hospitalization by 35%, cardiovascular mortality by 38%, and cardiovascular events by 14%.10 Additionally, several clinical trials demonstrated that empagliflozin was markedly helpful to non-diabetic patients. Specifically, empagliflozin further inhibited the worsening of HF and decreased the cardiovascular mortality in HF patients with declined EF, regardless of suffering or not suffering type 2 diabetes.11 An in vitro study pointed out that empagliflozin exerts an antioxidant effect in cardiac myocytes by decreasing Dox-induced lipid peroxidation in cardiac myocytes.10 Besides, Quagliariello et al. discovered in vivo that empagliflozin inhibited inflammation, apoptosis, fibrosis, and ferroptosis in Dox-induced mice via the NLRP3- and MyD88-related pathways, thereby greatly improving cardiac functions.12 Hence, it could be concluded from the above that empagliflozin may have the effect of alleviating Dox-induced cardiotoxicity. Interestingly, Chintan et al. revealed that empagliflozin protected the heart from lipopolysaccharide-induced hyperinflammation and energy expenditure through activating adenosine 5′ monophosphate-activated protein kinase (AMPK).13 So we hypothesize that there are additional potential mechanisms by which empagliflozin may ameliorate Dox-induced cardiotoxicity. In this study, a model of rats with Dox-induced cardiotoxicity was created in vivo by continuous intraperitoneal injection of Dox. Then, the effects of empagliflozin on the rats were assessed by measuring their oxidative stress response, tissue damage and cardiac function in each group. Furthermore, the possible mechanisms of empagliflozin were preliminarily explored, so as to provide basic research support for empagliflozin combined with Dox for clinical treatment to alleviate the cardiotoxicity of Dox.
Materials and methods
Establishment and treatment of the rat model
The Ethics Committee of South China University of Technology (2022039) approved this research. Besides, animals used in the trial were housed according to the program approved through the Institutional Animal Care and Use Committee. Specifically, South China University of Technology provided 24 male rats (adult), and all rats were grouped randomly as the Dox, Control, Dox + Emp (empagliflozin), and Dox + Met (metoprolol) groups (each group for six rats). Dox (2.5 mg/kg) was intraperitoneally injected into SD rats, twice a week, for 4 weeks (cumulative dose 20 mg/kg), to establish the rat model of Dox-induced cardiotoxicity.14 The dose of empagliflozin (30 mg/kg/day) was set based on the previous studies.15,16 After the completion of rat models, the rats in each group received following different treatments next day. Briefly speaking, the Dox group rats were given 3-mL distilled water intragastrically; the Dox + Emp group rats were administrated with 30 mg/kg empagliflozin intragastrically; and the Dox + Met group rats were gavaged with 25 mg/kg metoprolol. The above treatments were implemented once a day, for a total of 4 weeks. As for rats in the Control group, normal saline (the same doses as Dox) was intraperitoneally injected, twice a week, for four consecutive weeks; after that, rats were gavaged with 3-mL distilled water once a day, for a total of 4 weeks. Subsequently, echocardiography was performed in each group on the day after the last dosage of treatment. Finally, the rats were euthanized for the collection of the heart and blood samples.
Echocardiography
Ultrasound imaging and cardiac function measurements in rats were conducted using a high-frequency ultrasound device (Vevo 770, VisualSonics Inc., Toronto, Canada) carrying an MS-250S probe (13–24 MHz). M-mode Teicholz based on two-dimension was used to measure ventricular parameters including EF, fractional shortening (FS), left ventricular end-systolic diameter (LVESD) and left ventricular end-diastolic diameter (LVEDD). Before operation, all rats were weighed, followed by anesthesia via 2–3% isoflurane. Subsequently, their hair in the abdomen, chest, and neck was removed with depilatory creams; their body temperature was controlled at 37°C by fixing them supine on a heating pad. Afterwards, limb electrodes placement was operated on rats to continuously monitor their respiratory rate, body temperature, and heart rate (HR). LVESD, EF, FS, and LVEDD were also recorded. By the way, all electrocardiographic measurements were the average of three cardiac cycles.17
HE staining
After fixing with paraformaldehyde (4%) for 12 h, the rat heart tissues were embedded and sectioned. The sections were washed with PBS three times for 10 min, followed by mounting. After slide preparation, staining was performed based on the instructions of HE (Hematoxylin & Eosin) staining kit (Solarbio, China) and pathological changes of heart tissues were observed under a biomicroscope.
Biochemistry tests
The collected blood of rats in each group was allowed to stand in a centrifuge tube at 4°C. After 20 min, the blood was centrifuged at 3,000 r/min for 20 min at 4°C, and the supernatant (serum) was collected and divided into aliquots for testing. Later, 50 mg of myocardial tissue from rats was collected into a homogenization tube, and homogenization beads were added to the homogenizer to fully homogenize for 3 min until no tissue block was observed. Afterwards, the homogenized tissues were centrifuged at 4°C, 12,000 r/min, and after 10 min, the supernatant was acquired. The levels of creatine kinase isoenzyme (CK-MB) in serum and malondialdehyde acid (MDA), catalase (CAT), and superoxide dismutase (SOD) in myocardial tissues were checked in the light of the corresponding biochemical assay kit (Nanjing Jiancheng Bioengineering Institute, China) manuals.
Enzyme-linked Immunosorbent Assay
Serum from each group of rats isolated in Section 1.4 was utilized to detect the adenosine monophosphate (AMP), adenosine diphosphate (ADP), adenosine triphosphate (ATP), and N-terminal pro-brain natriuretic peptide (NT-proBNP) levels in the serum. The ATP/ADP ratio was calculated as referring to the corresponding ELISA (Enzyme-linked Immunosorbent Assay) kit (Nanjing Jiancheng Bioengineering Institute, China) instructions.
Western blot
Myocardial tissues were loaded with RIPA cell lysate (Beyotime, China) and homogenized in a tissue homogenizer. After 30 min of lysis on ice, the tissues were centrifuged at 10,000 rpm at 4°C; 20 min later, the total protein was extracted, and its concentration was tested with a BCA kit (Beyotime, China). Subsequently, the total protein (20 μg) was separated via SDS-PAGE then transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were washed clean, followed by 1–2 h of blocking with 5% non-fat dry milk. Next, TBST was adopted to wash the membranes for three times, and the diluted primary antibodies (CST, USA) were added for the incubation overnight at 4°C. Later, after washing with TBST for three times, the fixtures were supplemented with the secondary antibodies (CST, USA) for another 1 h of incubation at ambient temperature. Again, the membranes were washed with TBST for three times, and ECL chemiluminescence reagent (Beyotime, China) was dripped for protein development and image acquisition using a gel imaging system. Eventually, Image J software was employed to analyze the gray level of the protein bands, and the relative expression of the protein was calculated with β-actin as an internal control.
Statistical analysis
All results were expressed as mean ± standard deviation (SD) and statistical analysis was performed with SPSS 26.0. Comparisons among multiple groups were performed via one-way analysis of variance. P < .05 indicated significantly different.
Results
Empagliflozin improves doxorubicin-induced impairment of cardiac function in SD rats
To determine the role of empagliflozin in Dox-induced cardiac function, SD rats were housed to establish a Dox-induced cardiotoxicity model then grouped in line with different treatments. Compared with the Control group, Dox group rats exhibited a noticeable climb in LVEDD and LVESD while a marked decline in EF (56.45 ± 1.766% vs. 74.15 ± 2.037%) and FS (24.56 ± 1.439% vs. 47.71 ± 1.418%), indicating the successful construction of a Dox-induced cardiotoxicity rat model. Furthermore, relative to the Dox group, the Dox + Emp and Dox + Met group rats showed a remarkable decrease in LVEDD and LVESD, whereas a great increase in EF (65.88 ± 1.372% and 67.17 ± 2.826% vs. 56.45 ± 1.766%) and FS (35.69 ± 2.096% and 36.15 ± 1.87% vs. 24.56 ± 1.439%) (Fig. 1A–D). All in all, empagliflozin considerably bettered the cardiac function of rats with Dox-caused cardiotoxicity.
Fig. 1.
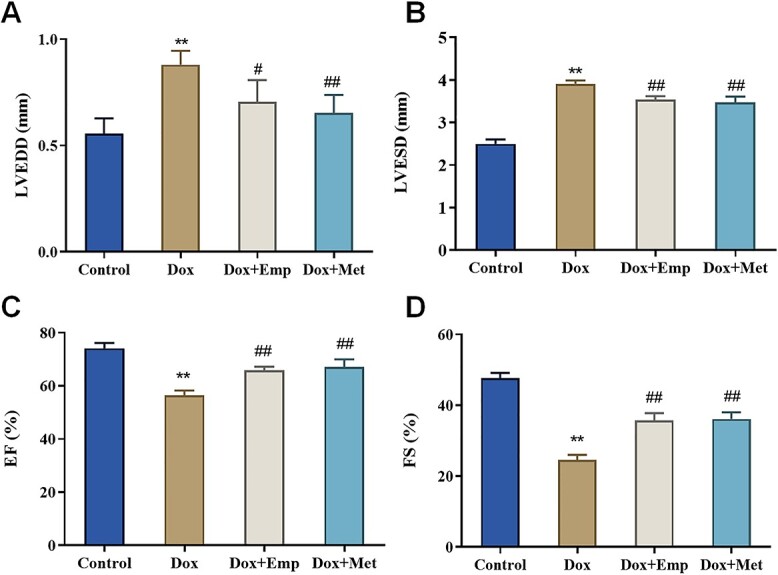
Empagliflozin (Emp) improved doxorubicin (Dox)-induced impairment of cardiac function in SD rats. (A)–(D): echocardiography to evaluate LVEDD (A), LVESD (B), EF (C), and percentage of FS (D). **P < .01 vs. Control group, #P < .05 and ##P < .01 vs. Dox group.
Empagliflozin attenuates doxorubicin-induced myocardial injury in SD rats
To investigate the functions of empagliflozin on Dox-induced myocardial injury, the pathological changes of myocardial tissue in SD rats were assessed by HE staining. Shortly speaking, the Dox group rats displayed some obvious histological changes, such as vacuolation, myocardial fiber disarrangement, myocardial cell degeneration and necrosis, and inflammatory cell infiltration, in the myocardial tissues. Whereas the myocardial tissues in the Dox + Emp group and Dox + Met group rats exhibited moderate vacuolization, irregular arrangement of myocardial fibers, and reduced inflammatory cell infiltration; the histological changes in the two groups were improved to a certain extent compared with those in the Dox group (Fig. 2A). In addition, the serum levels of NT-proBNP and CK-MB were measured in each group. The outcomes indicated that the CK-MB and NT-proBNP levels in the Dox group were remarkably up-regulated relative to those in the Control group (P < .01) while markedly down-regulated in the Dox + Emp and Dox + Met groups compared with those in the Dox group (Fig. 2B/C). Consequently, empagliflozin considerably alleviated Dox-induced myocardial tissue injury and decreased level of myocardial damage-associated factors in rats.
Fig. 2.
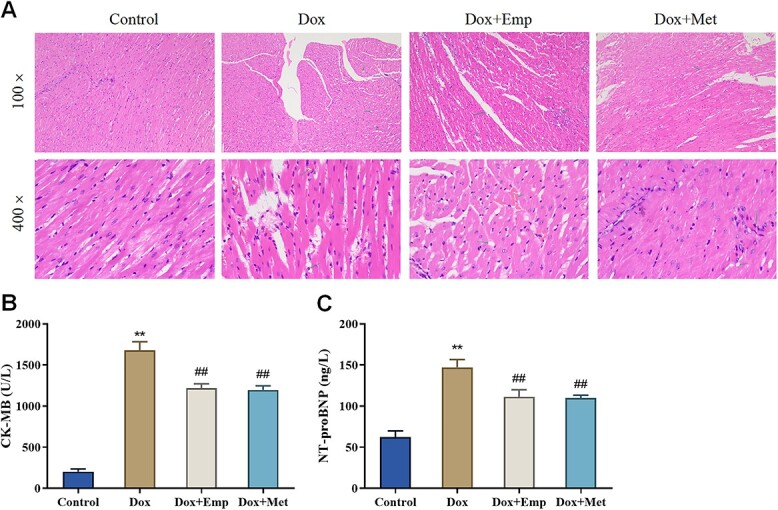
Empagliflozin (Emp) attenuates doxorubicin (Dox)-induced myocardial injury in SD rats. (A): HE staining to observe the pathological changes of myocardial tissue in rats of each group; (B): biochemical detection of creatine kinase isoenzyme (CK-MB) levels in the blood of rats of each group; (C): ELISA to test N-terminal pro-brain natriuretic peptide (NT-proBNP) levels in the blood of rats of each group, **P < .01 vs. Control group, ##P < .01 vs. Dox group.
Empagliflozin inhibits doxorubicin-induced oxidative stress in SD rats
For assessing the effect of empagliflozin on Dox-induced oxidative stress, the levels of oxidative stress-related substances (MDA, SOD, and CAT) in the myocardial tissues of rats were further measured. In brief, relative to the controls, the Dox group presented greatly climbed MDA levels and evidently decreased levels of CAT and SOD. Besides, the MDA levels were markedly lowered, whereas the SOD and CAT levels were obviously elevated in the Dox + Emp and Dox + Met groups matched with those in the Dox group (Fig. 3A–C). In conclusion, empagliflozin significantly inhibited oxidative stress in myocardial tissues of rats with Dox-induced cardiotoxicity.
Fig. 3.

Empagliflozin (Emp) inhibits doxorubicin (Dox)-induced oxidative stress in myocardial tissues of SD rats. (A)–(C): the levels of oxidative stress-related substances MDA (malondialdehyde acid, A), SOD (superoxide dismutase, B), and CAT (catalase, C) in myocardial tissue of rats in each group were detected biochemically. **P < .01 vs. Control group, #P < .05, and ##P < .01 vs. Dox group.
Empagliflozin improves doxorubicin-induced cardiac energy metabolism impairment in SD rats
The effect of empagliflozin on Dox-induced cardiometabolic impairment was further evaluated by observing the changes of ATP, ADP, and AMP in serum and the value of ATP/ADP. Researches manifested that empagliflozin modulated Dox-induced cardiac energy metabolism in rats. Specifically, compared with the Control group, the Dox group exhibited notably higher ADP levels while much lower ATP levels and ATP/ADP value. Additionally, in Dox + Emp and Dox + Met groups, the ADP levels in the serum of rats were significantly lower, whereas the ATP levels and the ATP/ADP value were obviously higher than those in the Dox group. Moreover, there was no significant difference in the changes of AMP levels in the serum of rats (Fig. 4A–D). Thus, it could be concluded that empagliflozin greatly ameliorated mitochondrial energy metabolism disturbances in Dox-induced cardiotoxicity rats.
Fig. 4.
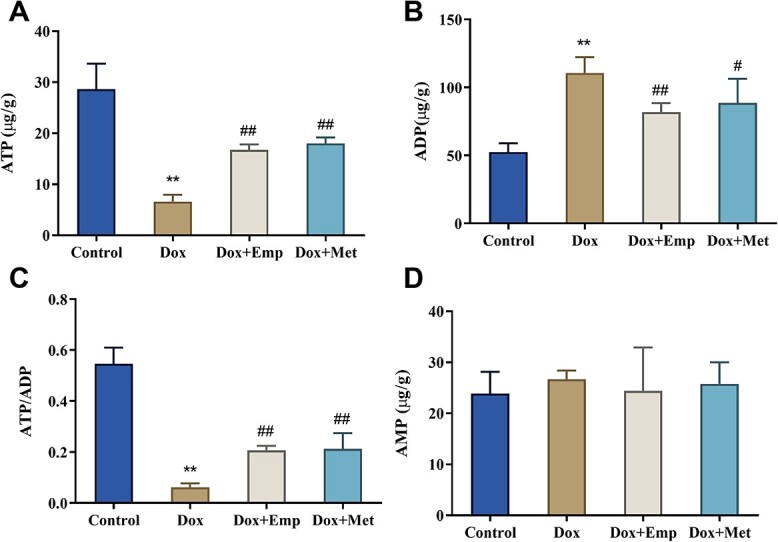
Empagliflozin (Emp) improves doxorubicin (Dox)-induced impairment of cardiac energy metabolism in SD rats. (A)–(D): the levels of ATP (adenosine triphosphate, A), ADP (adenosine diphosphate, B), and AMP (adenosine monophosphate, C) in serum were measured by ELISA and the ATP/ADP ratio (D) was calculated. **P < .01 vs. Control group, #P < .05, and ##P < .01 vs. Dox group.
Empagliflozin activates doxorubicin-induced AMPK/SIRT-1/PGC-1α signaling pathway in SD rats
Studies have demonstrated that AMPK/SIRT-1/PGC-1α signaling pathway is an energy sensing network that significantly regulates mitochondrial biogenesis, energy metabolism, as well as oxidative stress.18 To further determine whether empagliflozin exerts some of the above effects in Dox-induced cardiotoxicity rats through the same pathway, we detected the expression of AMPK/SIRT-1/PGC-1α related pathway proteins (AMPK, p-AMPK, PGC-1α, and SIRT-1) in myocardial tissues of rats. It was shown that the p-AMPK, SIRT-1, PGC-1α protein levels, and the p-AMPK/AMPK value in myocardial tissues of rats in the Dox group were notably decreased relative to those in the Control group, whereas the protein levels of p-AMPK, SIRT-1, PGC-1α and the p-AMPK/AMPK value in myocardial tissues of rats in the Dox + Emp and Dox + Met groups were considerably raised in comparison with those in the Dox group (Fig. 5A/B). From the above, empagliflozin was revealed to activate the AMPK/SIRT-1/PGC-1α signaling pathway in Dox-induced myocardial tissue from SD rats.
Fig. 5.
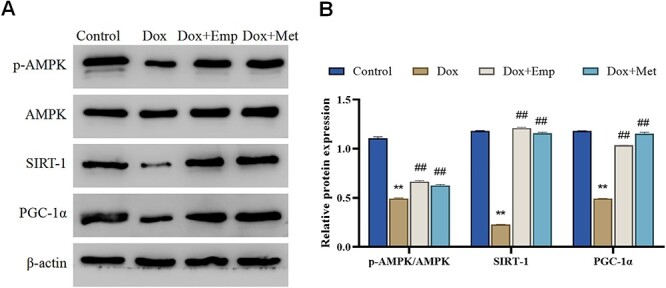
Empagliflozin (Emp) activates doxorubicin (Dox)-induced AMPK/SIRT-1/PGC-1α signaling in SD rats. (A)/(B): Western blot to detect the protein expressions of AMPK/SIRT-1/PGC-1α signaling pathway-related proteins AMPK, p-AMPK, SIRT-1, and PGC-1α in the myocardial tissues of rats in each group (A) and the relative expression levels of the proteins were calculated by Image J (B), **P < .01 vs. Control group, ##P < .01 vs. Dox group.
Discussion
Cardiotoxicity caused by exposure to Dox is a result attributed to multiple mechanisms. Pro-inflammatory cytokine storm, a rise in iROS and [Ca2+]i, mitochondrial dysfunctions, apoptosis, and necrosis are all responsible for this cardiotoxicity.12 Current research shows that empagliflozin might relieve the cardiac disease, independent of the presence or absence of diabetes and regardless of the cause; and such benefits to cardiorenal function have failed to be explained by the function of empagliflozin in lowering blood glucose because some other antidiabetic drugs with stronger antihyperglycemic functions have not presented the similar effects.19 Mustroph et al. also proposed the cardioprotective properties of empagliflozin on account of its role as a calmodulin inhibitor.20 Santos-Gallego pointed out that empagliflozin treatment betters cardiac function and myocardial energetics by increasing myocardial ATP content [(conversion of myocardial fuel metabolism away from glucose toward ketone bodies (KB)], branched-chain amino acid (BCAA) and free fatty acid (FFA), the expression of AMPK and PGC-1α.21 Besides, several studies showed that empagliflozin alleviated cardiotoxicity of anticancer drugs in cancer patients by inhibiting oxidative stress and pro-inflammatory heart microenvironment in our and previous research.12,22,23 Clinically, empagliflozin plays a vital role in protecting cardiac functions.24 In order to comprehensively elucidate the effect and possible mechanism of empagliflozin, we constructed a rat model of Dox-induced cardiotoxicity. In brief, compared with the Control group, the cardiac indices LVEDD and LVESD were markedly elevated, whereas EF and FS were remarkably declined in the Dox group. The HE staining images showed some obvious histological changes in the myocardial tissue of Dox-induced rats, such as vacuolation, myocardial fiber disarrangement, myocardial cell degeneration and necrosis, inflammatory cell infiltration. These ventricular parameters and myocardial tissue pathological changes were consistent with that in Dox-induced rats of Baris et al’s study.25 Thus, it was suggested that the Dox-induced cardiotoxicity rat model was successfully constructed. Additionally, after treatment of empagliflozin, noticeable decreased LVEDD and LVESD while increased EF and FS were observed in rats with Dox-induced cardiotoxicity. This result was consistent with the findings of Baris et al. that empagliflozin improved Dox-induced impairment of cardiac function.26
Furthermore, researches have manifested that CK-MB is one of the isoenzymes of CK and the detection of CK-MB level in serum contributes to determining the myocardial injury degree.27 To be specific, the increase of CK-MB level in serum indicates that CK-MB leaks from the damaged myocardial cell membrane into the circulation, and thus serves as an indicator of cardiotoxicity in clinical practice.28 The synthesis and secreted of NT-proBNP were completed via the ventricular myocardium, and the elevated NT-proBNP level in acute coronary syndrome patients is a reminder of impaired cardiac function.29 Recently, several articles have claimed that the NT-proBNP level in the serum of Dox-induced rats is significantly higher than that of the corresponding normal rats.30–32 In this study, up-regulated CK-MB and NT-proBNP levels were observed in the serum of rats with Dox-induced cardiotoxicity. Fortunately, empagliflozin treatment greatly reduced the CK-MB and NT-proBNP levels in the serum. Moreover, HE staining displayed that empagliflozin highly improved Dox-induced myocardial vacuolation, myocardial fiber disarrangement, myocardial cell degeneration and necrosis, and inflammatory cell infiltration. From the above, it could be seen that empagliflozin markedly alleviated Dox-induced cardiac tissue injury and cardiomyocyte injury.
A study by Zhang et al. implicated that doxorubicin activated the pathways related to DNA response and apoptosis and markedly altered the transcriptome selectively influencing mitochondrial biogenesis and oxidative phosphorylation in cardiomyocytes in the presence of Top2β (an important driving factor of the cardiotoxicity induced by doxorubicin).33 Cappetta et al. also considered mitochondrial oxidative stress as one of vital factors causing Dox-induced cardiotoxicity.34 A low oxidant level is essential for normal signal transduction processes, and excessive oxidant level has been shown to be associated with a variety of pathological conditions.35 The cumulative dose of Dox exceeding 500 mg/m2 will lead to increased oxidative stress,36 and reduced redox cycling of electron transport chain complex I. As a result, large amounts of ROS were produced and interruption of ATP synthesis was thus induced, and meanwhile the generated ROS interfered with normal mitochondrial function by reacting with nearby mitochondrial biomolecules.37 Mitochondrial numbers, the main subcellular target of Dox in cardiomyocytes, increased by 35–40% compared with its numbers in other tissues, possibly being one of the reasons why cardiomyocytes were vulnerable to injury.38 We found that Dox remarkably up-regulated MDA and down-regulated SOD and CAT levels in rat myocardial tissue, whereas empagliflozin treatment greatly lowered down MDA and elevated SOD and CAT levels in myocardial tissue. In conclusion, empagliflozin remarkably declined Dox-induced excessive oxidative stress in cardiac tissue.
Moreover, previous studies have revealed that sirtuins (SIRT) 1 in the heart activates the autophagic process of cardiomyocytes, inhibits apoptosis, and protects the heart from cardiovascular injuries like atherosclerosis and ischemia-reperfusion injury.39 However, treatment with Dox downregulates SIRT1 and affects PGC-1α, thus leading to excessive cell damage, mitochondrial dysfunction, apoptosis, and oxidative stress.39 Besides, it is reported that cardiac damage caused by Dox is linked to AMPK, and AMPK regulates mitochondrial biogenesis, function, autophagy, and fibrosis.40 At the same time, many researches have manifested that Dox is responsible for down-regulation of AMPK activity, which may be the mechanism leading to down-regulation of SIRT1 expression.41 Dox was also found to result in decreased activity of the AMPK/SIRT-1/PGC-1α signaling pathway in this study, and empagliflozin treatment activated AMPK/SIRT-1/PGC-1α signaling pathway activity in rats with Dox-induced cardiotoxicity. What is more, a great many papers state that empagliflozin can activate AMPK. For instance, Zhou et al. found that empagliflozin trigger AMPK activation by restoring the AMP-to-ATP value, inhibited the phosphorylation of Drp1S616, promoted the phosphorylation of Drp1S637, suppressed mitochondrial fission, thereby relieving diabetic myocardial microvascular injury.42 Santos-Gallego et al. also revealed that empagliflozin treatment improved myocardial energetics and cardiac function in nondiabetic porcine model of HF by increasing the expression of AMPK and PGC-1α.21 All the above suggested that empagliflozin may prevent Dox-induced cardiac damage by activating the AMPK/SIRT-1/PGC-1α signaling pathway.
As is mentioned above, empagliflozin improved Dox-induced cardiac tissue, functional impairment, oxidative stress, and energy metabolism probably via activating the AMPK/SIRT-1/PGC-1α signaling pathway. Nevertheless, as the molecular mechanisms underlying its specific effects were not explored in this study, the specific mode of regulation of empagliflozin is uncertain. Besides, since this study was only performed in the body of rats, a large number of clinical trials are required to fully verify the effect of empagliflozin for further clinical use.
Conclusion
To sum up, empagliflozin prevents Dox-induced cardiotoxicity by improving Dox-induced cardiac tissue and function impairment, inhibiting oxidative stress in myocardial tissue, and modulating mitochondrial energy metabolism. Notably, the protective effect of empagliflozin on heart may be correlated with the activation of the AMPK/SIRT-1/PGC-1α signaling pathway. In a word, empagliflozin alleviates Dox-induced adverse effects.
Acknowledgments
Not applicable.
Funding
None.
Conflict of Interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
M.C. conceptualized and designed the study, carried out the initial analyses, reviewed, and revised the manuscript.
Consent for publication
Not applicable.
Consent to participate
Not applicable.
Ethical Approval
The study was approved by Ethics Committee of South China University of Technology (2022039) and animals used were housed according to the protocol approved by the Institutional Animal Care and Use Committee.
List of abbreviations
doxorubicin (Dox); creatine kinase isoenzyme (CK-MB); N-terminal pro-brain natriuretic peptide (NT-proBNP); adenosine triphosphate (ATP); adenosine diphosphate (ADP); adenosine monophosphate (AMP); malondialdehyde acid (MDA); superoxide dismutase (SOD); catalase (CAT); left ventricular end-diastolic diameter (LVEDD); left ventricular end-systolic diameter (LVESD); fractional shortening (FS); ejection fraction (EF); myocardial infarction (MI); Enzyme-linked Immunosorbent Assay (ELISA); Hematoxylin & Eosin (HE); heart rate (HR); polyvinylidene fluoride (PVDF); adenosine 5′ monophosphate-activated protein kinase (AMPK); heart failure (HF); ketone bodies (KB); free fatty acid (FFA); branched-chain amino acid (BCAA).
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018:68(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2. Parkin DM. The global burden of cancer. Semin Cancer Biol. 1998:8(4):219–235. [DOI] [PubMed] [Google Scholar]
- 3. Chmielewska M, Symonowicz K, Pula B, Owczarek T, Podhorska-Okolow M, Ugorski M, Dziegiel P. Expression of metallothioneins I and II in kidney of doxorubicin-treated rats. Exp Toxicol Pathol. 2015:67(4):297–303. [DOI] [PubMed] [Google Scholar]
- 4. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003:97(11):2869–2879. [DOI] [PubMed] [Google Scholar]
- 5. Hekmat AS, Navabi Z, Alipanah H, Javanmardi K. Alamandine significantly reduces doxorubicin-induced cardiotoxicity in rats. Hum Exp Toxicol. 2021:40(10):1781–1795. [DOI] [PubMed] [Google Scholar]
- 6. Tallaj JA, Franco V, Rayburn BK, Pinderski L, Benza RL, Pamboukian S, Foley B, Bourge RC. Response of doxorubicin-induced cardiomyopathy to the current management strategy of heart failure. J Heart Lung Transplant. 2005:24(12):2196–2201. [DOI] [PubMed] [Google Scholar]
- 7. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Prasad SVN, Mutharasan RK, Naik TJ, Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014:124(2):617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019:381(21):1995–2008. [DOI] [PubMed] [Google Scholar]
- 9. Yurista SR, Silljé HHW, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, van Goor H, van Veldhuisen DJ, de Boer RA, Westenbrink BD. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. 2019:21(7):862–873. [DOI] [PubMed] [Google Scholar]
- 10. Kaku K, Lee J, Mattheus M, Kaspers S, George J, Woerle HJ, on behalf of the EMPA-REG OUTCOME® Investigators. Empagliflozin and cardiovascular outcomes in Asian patients with type 2 diabetes and established cardiovascular disease—results from EMPA-REG OUTCOME((R)). Circ J. 2017:81(2):227–234. [DOI] [PubMed] [Google Scholar]
- 11. Heerspink HJL, Sjostrom CD, Jongs N, Chertow GM, Kosiborod M, Hou FF, McMurray JJV, Rossing P, Correa-Rotter R, Kurlyandskaya R, et al. Effects of dapagliflozin on mortality in patients with chronic kidney disease: a pre-specified analysis from the DAPA-CKD randomized controlled trial. Eur Heart J. 2021:42:1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quagliariello V, de Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A, Paccone A, Altucci L, Conte M, Canale ML, et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. 2021:20(1):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sabatino J, de Rosa S, Tammè L, Iaconetti C, Sorrentino S, Polimeni A, Mignogna C, Amorosi A, Spaccarotella C, Yasuda M, et al. Empagliflozin prevents doxorubicin-induced myocardial dysfunction. Cardiovasc Diabetol. 2020:19(1):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qi W, Boliang W, Xiaoxi T, Guoqiang F, Jianbo X, Gang W. Cardamonin protects against doxorubicin-induced cardiotoxicity in mice by restraining oxidative stress and inflammation associated with Nrf2 signaling. Biomed Pharmacother. 2020:122:109547. [DOI] [PubMed] [Google Scholar]
- 15. Daud E, Ertracht O, Bandel N, Moady G, Shehadeh M, Reuveni T, Atar S. The impact of empagliflozin on cardiac physiology and fibrosis early after myocardial infarction in non-diabetic rats. Cardiovasc Diabetol. 2021:20(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shao Q, Meng L, Lee S, Tse G, Gong M, Zhang Z, Zhao J, Zhao Y, Li G, Liu T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc Diabetol. 2019:18(1):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu G, Liu Y, Wang R, Hou T, Chen C, Zheng S, Dong Z. Spironolactone attenuates doxorubicin-induced cardiotoxicity in rats. Cardiovasc Ther. 2016:34(4):216–224. [DOI] [PubMed] [Google Scholar]
- 18. Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009:20(2):98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Packer M. SGLT2 inhibitors produce cardiorenal benefits by promoting adaptive cellular reprogramming to induce a state of fasting mimicry: a paradigm shift in understanding their mechanism of action. Diabetes Care. 2020:43(3):508–511. [DOI] [PubMed] [Google Scholar]
- 20. Mustroph J, Wagemann O, Lücht CM, Trum M, Hammer KP, Sag CM, Lebek S, Tarnowski D, Reinders J, Perbellini F, et al. Empagliflozin reduces ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018:5(4):642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, Flores E, Garcia-Ropero A, Sanz J, Hajjar RJ, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. 2019:73(15):1931–1944. [DOI] [PubMed] [Google Scholar]
- 22. Oh CM, Cho S, Jang JY, Kim H, Chun S, Choi M, Park S, Ko YG. Cardioprotective potential of an SGLT2 inhibitor against doxorubicin-induced heart failure. Korean Circ J. 2019:49(12):1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang CC, Chen YT, Wallace CG, Chen KH, Cheng BC, Sung PH, Li YC, Ko SF, Chang HW, Yip HK. Early administration of empagliflozin preserved heart function in cardiorenal syndrome in rat. Biomed Pharmacother. 2019:109:658–670. [DOI] [PubMed] [Google Scholar]
- 24. Anker SD, Butler J, Filippatos G, Khan MS, Marx N, Lam CSP, Schnaidt S, Ofstad AP, Brueckmann M, Jamal W, et al. Effect of Empagliflozin on cardiovascular and renal outcomes in patients with heart failure by baseline diabetes status: results from the EMPEROR-reduced trial. Circulation. 2021:143(4):337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barış VÖ, Gedikli E, Yersal N, Müftüoğlu S, Erdem A. Protective effect of taurine against doxorubicin-induced cardiotoxicity in rats: echocardiographical and histological findings. Amino Acids. 2019:51(10–12):1649–1655. [DOI] [PubMed] [Google Scholar]
- 26. Barış VÖ, Dinçsoy AB, Gedikli E, Zırh S, Müftüoğlu S, Erdem A. Empagliflozin significantly prevents the doxorubicin-induced acute cardiotoxicity via non-antioxidant pathways. Cardiovasc Toxicol. 2021:21(9):747–758. [DOI] [PubMed] [Google Scholar]
- 27. Liu XH, Zhang ZY, Sun S, Wu XD. Ischemic postconditioning protects myocardium from ischemia/reperfusion injury through attenuating endoplasmic reticulum stress. Shock. 2008:30(4):422–427. [DOI] [PubMed] [Google Scholar]
- 28. Wu S, Zhou Y, Xuan Z, Xiong L, Ge X, Ye J, Liu Y, Yuan L, Xu Y, Ding G, et al. Repeated use of SSRIs potentially associated with an increase on serum CK and CK-MB in patients with major depressive disorder: a retrospective study. Sci Rep. 2021:11(1):13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zdravkovic V, Mladenovic V, Colic M, Bankovic D, Lazic Z, Petrovic M, Simic I, Knezevic S, Pantovic S, Djukic A, et al. NT-proBNP for prognostic and diagnostic evaluation in patients with acute coronary syndromes. Kardiol Pol. 2013:71(5):472–479. [DOI] [PubMed] [Google Scholar]
- 30. Pajović V, Kovácsházi C, Kosić M, Vasić M, Đukić L, Brenner GB, Giricz Z, Bajić D, Ferdinandy P, Japundžić-Žigon N. Phenomapping for classification of doxorubicin-induced cardiomyopathy in rats. Toxicol Appl Pharmacol. 2021:423:115579. [DOI] [PubMed] [Google Scholar]
- 31. Dindaş F, Güngör H, Ekici M, Akokay P, Erhan F, Doğduş M, Yılmaz MB. Angiotensin receptor-neprilysin inhibition by sacubitril/valsartan attenuates doxorubicin-induced cardiotoxicity in a pretreatment mice model by interfering with oxidative stress, inflammation, and caspase 3 apoptotic pathway. Anatol J Cardiol. 2021:25(11):821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adıyaman MŞ, Adıyaman ÖA, Dağlı AF, Karahan MZ, Dağlı MN. Prevention of doxorubicin-induced experimental cardiotoxicity by Nigella sativa in rats. Rev Port Cardiol. 2022:41(2):99–105. [DOI] [PubMed] [Google Scholar]
- 33. Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ETH. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012:18(11):1639–1642. [DOI] [PubMed] [Google Scholar]
- 34. Cappetta D, de Angelis A, Sapio L, Prezioso L, Illiano M, Quaini F, Rossi F, Berrino L, Naviglio S, Urbanek K. Oxidative stress and cellular response to doxorubicin: a common factor in the complex milieu of anthracycline cardiotoxicity. Oxidative Med Cell Longev. 2017:2017:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonomini F, Rodella LF, Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015:6(2):109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wenningmann N, Knapp M, Ande A, Vaidya TR, Ait-Oudhia S. Insights into doxorubicin-induced cardiotoxicity: molecular mechanisms, preventive strategies, and early monitoring. Mol Pharmacol. 2019:96(2):219–232. [DOI] [PubMed] [Google Scholar]
- 37. Eder AR, Arriaga EA. Capillary electrophoresis monitors enhancement in subcellular reactive oxygen species production upon treatment with doxorubicin. Chem Res Toxicol. 2006:19(9):1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goffart S, Kleist-Retzow JC, Wiesner RJ. Regulation of mitochondrial proliferation in the heart: power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res. 2004:64(2):198–207. [DOI] [PubMed] [Google Scholar]
- 39. Ruan Y, Dong C, Patel J, Duan C, Wang X, Wu X, Cao Y, Pu L, Lu D, Shen T, et al. SIRT1 suppresses doxorubicin-induced cardiotoxicity by regulating the oxidative stress and p38MAPK pathways. Cell Physiol Biochem. 2015:35(3):1116–1124. [DOI] [PubMed] [Google Scholar]
- 40. Timm KN, Tyler DJ. The role of AMPK activation for cardioprotection in doxorubicin-induced cardiotoxicity. Cardiovasc Drugs Ther. 2020:34(2):255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bartlett JJ, Trivedi PC, Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol. 2017:104:1–8. [DOI] [PubMed] [Google Scholar]
- 42. Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018:15:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.