

Abstract
Shedding, loss of expression, or internalization of natural killer group 2, member D (NKG2D) ligands from the tumor cell surface leads to immune evasion, which is associated with poor prognosis in patients with cancer. In many cancers, matrix metalloproteinases cause the proteolytic shedding of NKG2D ligands. However, it remained unclear how to protect NKG2D ligands from shedding. Here, we showed that the shedding of the mouse NKG2D ligand Rae-1 can be prevented by two critical acetyltransferases, GCN5 and PCAF, which acetylate the lysine residues of Rae-1 to avoid shedding both in vitro and in vivo. In contrast, mutations at lysines 80 and 87 of Rae-1 abrogated this acetylation and thereby desensitized tumor cells to NKG2D-dependent immune surveillance. Notably, the protein levels of GCN5 correlated with the expression levels of the human NKG2D ligand ULPB1 in a human tumor tissue microarray and, more importantly, with prolonged overall survival in many cancers. Our results suggest that the acetylation of Rae-1 protein at lysines 80 and 87 by GCN5 and PCAF protects Rae-1 from shedding so as to activate NKG2D-dependent immune surveillance. This discovery may shed light on new targets for NKG2D immunotherapy in cancer treatment.
Keywords: NKG2D ligand stabilization, GCN5, PCAF, acetylation
Introduction
One hallmark of cancer is immune evasion, in which tumor cells modulate the display of cell surface proteins, such as natural killer group 2, member D (NKG2D) ligands, to escape from immune surveillance. During tumor progression, loss of NKG2D ligands from tumor cell surfaces by proteolytic shedding [1, 2], degradation [3], or exosome secretion [4] disables NKG2D-mediated immune surveillance against tumor cells and is often associated with poor prognosis [5]. The shed soluble NKG2D ligands in patients’ sera interfere with the antitumor immune response by downregulating the NKG2D receptor, resulting in tumor progression and reduced overall survival [6].
Histone acetyltransferases covalently modify histone tails to regulate gene transcription and non-histone proteins to regulate cellular signaling and functions [7]. A number of studies have revealed that inhibition of histone deacetylases (HDACs) upregulates expression of the NKG2D ligands MICA and MICB, thereby sensitizing tumor cells to immune cell attack [8]; these studies suggest that NKG2D ligands could be regulated through mechanisms mediated by histone acetyltransferases. The histone acetyltransferases GCN5 (Kat2a) and PCAF (Kat2b), also known as lysine acetyltransferases (KATs) [9], bring acetyl groups to lysines on both histones and non-histone proteins, affecting gene transcription and expression. An alternative way to affect protein stability is to directly acetylate the target protein via a posttranslational mechanism. Acetylation, as an “unconventional” posttranslational modification, has gained increasing interest for its roles in regulating inflammation and immunity [10–13]. In particular, lysine acetylation is conserved among species and plays essential roles in the regulation of enzymes for cellular processes and protein regulation [14–18]. p53, one of the most important tumor suppressors, was found to be acetylated via p300/CBP upon DNA damage [19] to increase protein stability and activity [7, 20]. A number of other proteins, including sterol regulatory element-binding proteins [21], Smad7 [22, 23], HNF [24], AIRE [25], GCPII [26], P27 (Kip1) [27], Cdt1 [28], and beta-catenin [29] in eukaryotes, as well as RNase R [30] in bacteria, can also be acetylated to enhance protein stability.
We previously reported that restoration of the NKG2D ligand Rae-1 on the tumor cell surface was associated with the acetyltransferase function of GCN5 and PCAF [31], but the underlying mechanism remained unknown. The present study revealed that the KATs GCN5 and PCAF do not regulate the transcription of Rae-1. Instead, they directly acetylate Rae-1 at lysine (K) 80 and K87, which protect Rae-1 from matrix metalloproteinase (MMP)-mediated shedding from the tumor cell surface. Furthermore, we found that overexpression of GCN5 and PCAF in solid tumors in vivo boosted immune surveillance and the associated NKG2D-dependent tumor cell death.
Materials and Methods
Animal studies
BLAB/c mice (6–8 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME). A total of 1 × 105 K7M3 tumor cells in 15 µL of phosphate-buffered saline (PBS) was inoculated into BALB/c mice via intraosseous injection. The mouse care and handling procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of The University of Texas MD Anderson Cancer Center.
Cell lines
The cancer cell lines CT26 (mouse colon adenocarcinoma) and LLC (mouse lewis lung carcinoma) and HEK 293T/17 (human embryonic kidney) cell lines were obtained from ATCC (Rockville, MD). The K7M3 (mouse osteosarcoma) cell line was provided by Dr. Eugenie S. Kleinerman’s laboratory (MD Anderson Cancer Center). All cell lines were grown in Dulbecco’s modified Eagle medium (Mediatech, Manassas, VA) supplemented with glutamine and heat-inactivated 10% fetal bovine serum (Life Technologies, Grand Island, NY) and 10 U/mL each of penicillin and streptomycin (Life Technologies). All cell lines were characterized and tested for Mycoplasma contamination by the Cytogenetics and Cell Authentication Core Facility at MD Anderson Cancer Center.
Flow cytometry analysis
Cells were incubated with primary and secondary antibodies for 30 minutes at 4 °C. The stained cells were analyzed on an Attune acoustic focusing cytometer (Applied Biosystems, Carlsbad, CA). Data were analyzed with Attune software (Applied Biosystems) or FlowJo software (BD Biosciences, San Jose, CA).
Immunoblotting assays
Frozen tissue samples were smashed and then homogenized in 0.4 mL of ice-cold lysis buffer with 5 to 8 silicone beads using a Mini-Beadbeater (BioSpec Products, Bartlesville, OK). The protein extract was separated from tissue residues by centrifugation at the maximum speed for 20 minutes at 4 °C. Sixty micrograms of total protein was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes by using the iBlot gel transfer device (Invitrogen, Grand Island, NY). The membranes were blotted with primary and secondary antibodies to detect the proteins of interest.
Immunohistochemistry and immunofluorescence staining
Frozen tumor sections were sequentially fixed with cold acetone, acetone plus chloroform (1:1), and acetone. Tissue sections were blocked with blocking buffer (5% normal horse serum and 1% normal goat serum in PBS) and then incubated with a primary antibody overnight at 4 °C. The next day, the tissues were incubated with a secondary antibody for 1 hour at room temperature. Nuclei were counterstained with hematoxylin (Sigma-Aldrich, St. Louis, MO).
DNA transfection
HEK 293T/17 cells (1 × 106) were transfected with 2 μg of plasmid DNAs (Rae-1WT, Rae-1mut, Rae-1K80R, Rae-1K87R, GCN5 or PCAF) by using X-tremeGENE HP DNA transfection reagent (Roche Diagnostics, Indianapolis, IN).
Intratumoral delivery of GCN5 and PCAF DNA by electroporation
GCN5 and PCAF DNA (5 μg) was injected intratumorally into K7M3 tumor-bearing mice. Electroporation was then performed using the following parameters: 2 20-ms pulses of 150 V cm−1 with a 100-ms interval between pulses.
CT26 cell lysates
To retrieve CT26 cell lysates, the culture medium was removed from CT26 cells, and 200 μL PBS per 106 cells was added. The cells were snap-frozen at −80 °C for 15 minutes and then thawed at room temperature. This freeze/thaw cycle was repeated 3 times. The mixture was collected and sonicated briefly before centrifugation at 16,300g at 4 °C to remove the cell debris. The cell lysates were added to cell culture medium at 1:5 dilution as needed.
RNA isolation and quantitative PCR
RNA was isolated from tumors with TRIzol reagent (Invitrogen, Carlsbad, CA). Real-time quantitative PCR was conducted as described previously [32]. GAPDH and TATA box-binding protein (TBP) were used as reference genes.
Co-immunoprecipitation
Cells were washed with 1 mL of PBS and subjected to lysis in 0.5 mL of lysis buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% NP-40 [pH 8.0]). Twenty-five microliters of protein A/G beads were incubated with 2 μg of the appropriate antibody for 4 hours at 4 °C and then added to 1 mg of cell or tissue lysates and incubated overnight at 4 °C. The beads were washed thrice in 1 mL of lysis buffer, spun down, and boiled in 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis loading buffer. The Rae-1 antibody binds both Rae-1ε and Rae-1β proteins.
In vitro acetylation assay
HEK 293 cells were transiently transfected with wild-type Rae-1 (Rae-1WT) or mutated Rae-1 (Rae-1mut). Rae-1 was immunoprecipitated from 500 μg of cell lysates using an anti–Rae-1 antibody[33] plus protein G-agarose (Sigma) and washed in acetyltransferase assay buffer (50 mM Tris-Cl [pH 8], 10% glycerol, 10 mM butyric acid, 0.1 mM EDTA, 1 mM DTT, 1 mM PMSF). Rae-1 precipitates from 50 μg of cell lysates in 50 μg of assay buffer were incubated with 10 µM acetyl coenzyme A (Sigma) and 500 ng PCAF (Sigma) plus GCN5 recombinant proteins (NOVUS Biologicals) for 45 minutes at 30 °C on a rotating platform, followed by incubation in sodium dodecyl sulfate-sample buffer and immunoblotting assays [34].
Cytotoxic T-lymphocyte assay
K7M3 or HEK 293 cells were labeled with 5 μM CellTracker Violet Dye (ThermoFisher) and incubated for 5 hours at 37 °C with murine splenocytes at effector-to-target ratios of 10:1, 25:1, and 50:1. After incubation, the cells were stained with propidium iodide (1 mg/mL; Sigma Aldrich). Live target cells were identified according to light-scatter parameters and propidium iodide negativity. Survival of the target cells was measured as the normalized percentage of target cells that remained after incubation with CD8+ T cells.
Enzyme-linked immunosorbent assay
The wells of high-binding 96-well plates were coated with anti-mouse Rae-1 polyclonal antibody (ThermoFisher) overnight at 4 °C, washed and blocked using an ELISA Deluxe Set (BioLegend). The supernatant collected from CT26Rae1 cells and positive and negative controls were added to the antibody-coated wells for incubation for 2 hours at room temperature, followed by incubation with biotin-labeled anti–Rae-1 monoclonal antibody [33], avidin-horseradish peroxidase solution, and substrate solution from the ELISA Deluxe Set. The reaction was stopped after 20 minutes, and the absorbance was read at 450 nm.
cBioPortal for cancer genomics
KAT and NKG2D ligands gene expression data and survival data were obtained from The Cancer Genome Atlas (TCGA) portal (https://www.cbioportal.org/).
Statistical analysis
Continuous variables were analyzed by 2-sided Student t-test to compare 2 treatment groups or by 1-way analysis of variance to compare more than 2 treatment groups. Prism software (GraphPad Software, La Jolla, CA) was used for the analyses. We set a P value < 0.05 as the threshold to indicate statistical significance.
Antibody and chemical list
| Antibody | Source | Cat# | |||
|---|---|---|---|---|---|
| Rae-1 | ThermoFisher | PA5–93166 | |||
| GCN5L2 | Cell Signaling Technology | 3305 | |||
| PCAF | Cell Signaling Technology | 3378 | |||
| β-Actin | Cell Signaling Technology | 3700 | |||
| Mouse NKG2D/CD314 | R&D | BAM1437 | |||
| Acetylated-Lysine | Cell Signaling Technology | 9441 | |||
| GAPDH | Cell Signaling Technology | 5174 | |||
| Chemial | Source | Cat# | |||
| panobinostat | Selleckchem | LBH589 | |||
| GM6001 | Sigma | CC10 | |||
| SAHA | Sigma | SML0061 | |||
| MS-275 | Sigma | EPS002 | |||
| EX-527 | Sigma | E7034 | |||
| cycloheximide | Sigma | C4859 | |||
| anacardic acid | Sigma | A7236 | |||
Results
HDAC inhibitors stabilize Rae-1 protein on the tumor cell surface
NKG2D ligands are shed from tumor cells via proteolytic cleavage mediated by MMPs. These ligands are strikingly increased in the sera of patients with high-grade cancers [35]. Elevated levels of soluble NKG2D ligands could neutralize NKG2D function and in turn impair its antitumor cytotoxic function. We noticed that expression of the NKG2D ligand Rae-1 on the surface of Rae-1– overexpressing CT26 (mouse colon adenocarcinoma) cells was reduced during culture and that this reduction could be prevented by the MMP inhibitor GM6001 (Fig. 1A). Additionally, Rae-1 expression on the tumor cell surface was inversely correlated with that in the cell culture supernatant determined via ELISA assay, implying that Rae-1 was shed from the tumor cell surface to the culture medium (Fig. 1B). The next question was how to restore Rae-1 expression to the tumor cells from which Rae-1 was shed. In a previous report, we demonstrated that induced expression of the acetyltransferases GCN5 and PCAF led to Rae-1 restoration on tumor cells [36]. In that vein, we treated K7M3 (mouse osteosarcoma) and LLC (mouse Lewis lung carcinoma) tumor cells with 4 HDAC inhibitors (SAHA, MS-275, panobinostat, and EX-527) that abrogate different families of HDACs. In both cell lines, panobinostat most dramatically induced Rae-1 expression on the tumor cell surface (Fig. 1C).
Figure 1. Histone deacetylase inhibitors stabilize Rae-1 expression on tumor cells.
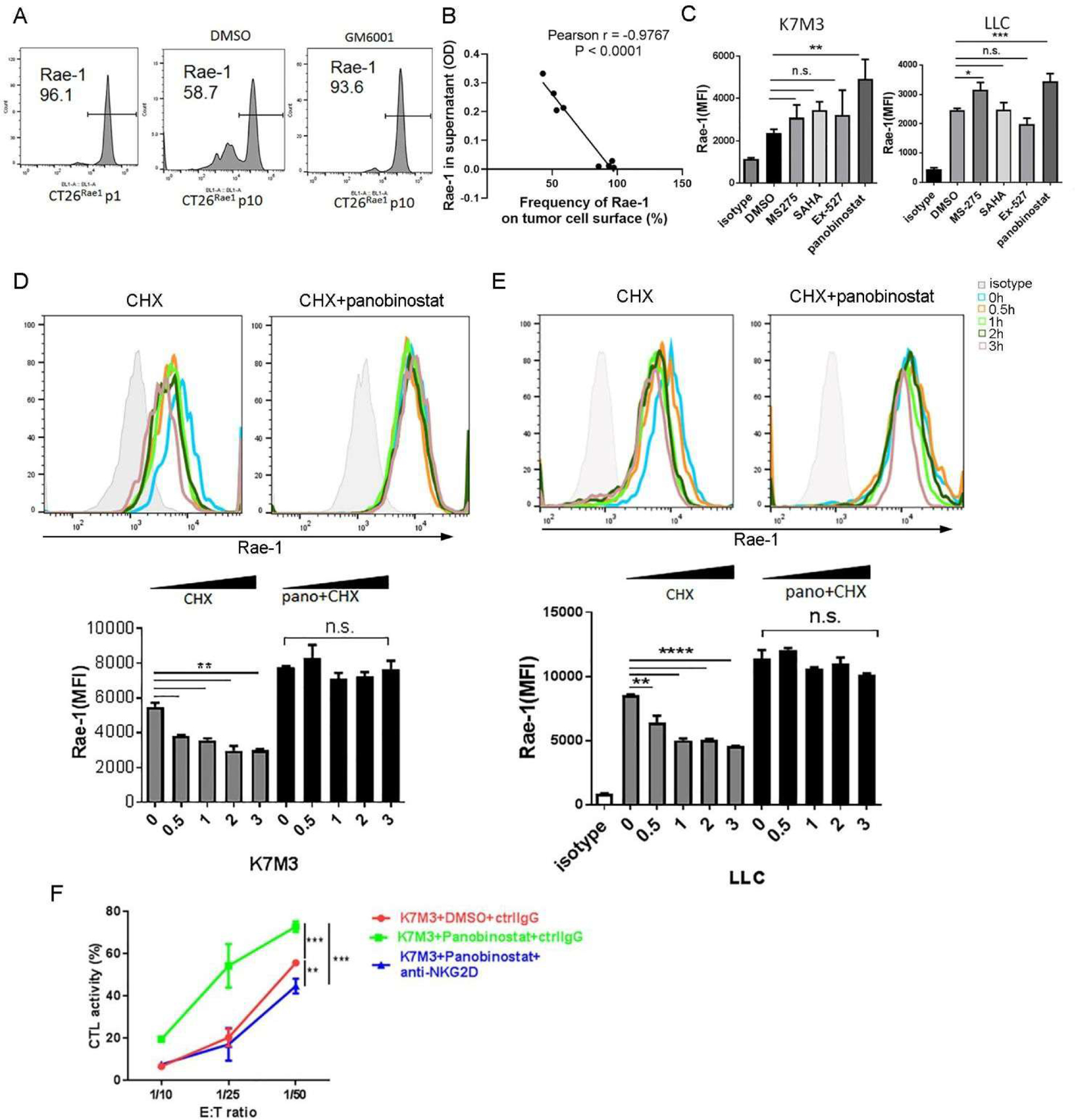
(A) CT26Rae1 cells at passage 10 were treated with DMSO or the MMP inhibitor GM6001 (10 μM) for 24 hours. Rae-1 expression on the surface of CT26Rae1 cells at passage 1 and passage 10 was determined using flow cytometry. (B) Soluble Rae-1 in the cell culture supernatant of CT26Rae1 cells at different passages was determined using ELISA. The frequency of cell surface Rae-1 on CT26Rae1 cells at different passages was determined using flow cytometry. Correlation of cell surface Rae-1 (%) and soluble Rae-1 (OD) was determined by the Pearson’s product moment test. (C) K7M3 and LLC cells were treated with DMSO, MS-275 (5 μM), SAHA (5 μM), EX-527 (5 μM), or panobinostat (100 nM) for 24 hours, and Rae-1 levels were measured using flow cytometry. Bar graphs show the mean fluorescence intensity (MFI) of Rae-1. (D, E) K7M3 (D) and LLC (E) cells were pretreated with DMSO or panobinostat (100 nM) for 1 hour and then treated with cycloheximide (CHX; 1 μg/mL) for 30 minutes or 1, 2, or 3 hours. Rae-1 expression was determined using flow cytometry. Bar graphs show the MFI of Rae-1 on the cell surface. (F) Cytolytic killing activity of splenocytes that were stimulated with CD3/CD28 antibodies against K7M3 cells treated with DMSO or panobinostat (100 nM) in the presence or absence of an anti-NKG2D blocking antibody at the indicated ratios for 5 hours. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; NS, not significant.
To further establish whether panobinostat can prevent Rae-1 from shedding from tumor cells, we treated K7M3 and LLC tumor cells with the translation inhibitor cycloheximide, which dramatically reduced cell surface expression of Rae-1 as early as 30 minutes after treatment. In contrast, pretreatment with panobinostat not only upregulated Rae-1 expression but also stabilized Rae-1 on the surface of tumor cell, suggesting that inhibition of histone or Rae-1 deacetylation protected Rae-1 from shedding (Fig. 1D, 1E). The panobinostat- or vehicle control-treated K7M3 cells were also exposed to stimulated splenocytes isolated from tumor-bearing mice, and panobinostat markedly enhanced the tumor cells’ susceptibility to NKG2D-dependent cytolytic activity (Fig. 1F).
KATs boost Rae-1 expression in vivo independently of transcriptional regulation
Although it is well established that HDAC inhibitors boost NKG2D ligand expression on tumor cells, rendering tumor cells more susceptible to NKG2D-dependent killing in vitro, it is difficult to duplicate this effect in vivo. We discovered the critical roles of the KATs GCN5 and PCAF in Rae-1 upregulation in tumors in vivo [31]. In the present study, instead of using HDAC inhibitors, we directly introduced GCN5 and PCAF into K7MC tumors in vivo via electroporation (Fig. 2). Overexpression of GCN5 and PCAF in tumors was confirmed via immunoblotting (Fig. 2A) and was associated with a significant increase of Rae-1 expression in tumors in vivo (Fig. 2B). Moreover, administration of GCN5 and PCAF recruited NKG2D+ immune cells to tumors, and this effect was completely abrogated by blocking NKG2D (Fig. 2C). These results revealed that GCN5 and PCAF could enhance NKG2D-mediated antitumor immunity by boosting Rae-1 expression in tumors.
Figure 2. Lysine acetyltransferases boost in vivo Rae-1 expression independently of transcriptional regulation.
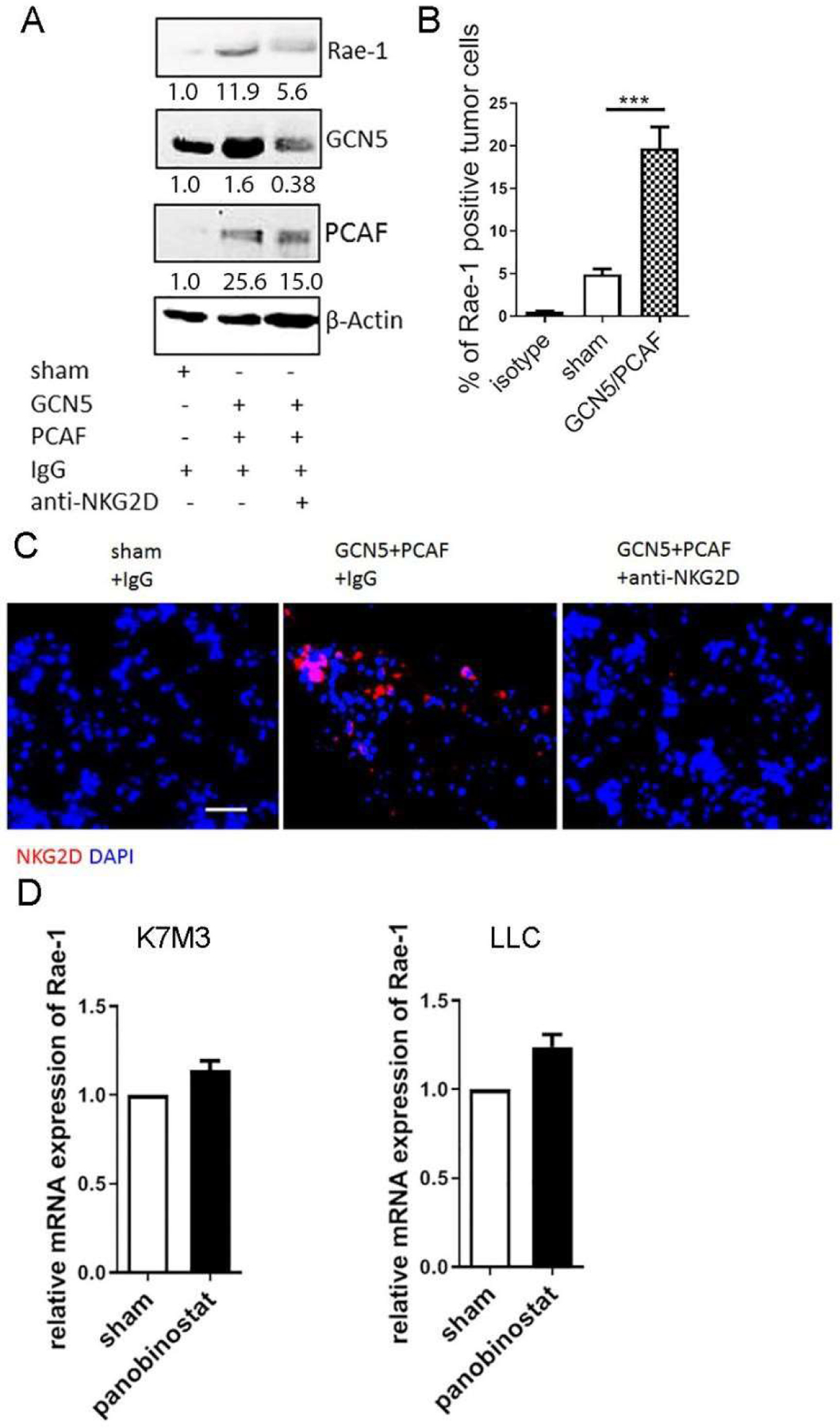
K7M3 orthotopic tumors were established in Balb/c mice (N = 3) and treated with control IgG or anti-NKG2D blocking antibody (250 μg/mouse) twice weekly. Fourteen days after tumor inoculation, GCN5 and PCAF DNA was administered intratumorally, followed by electroporation. Four days later, tumors were harvested for assays. (A) Immunoblotting of K7M3 tumor lysates to detect Rae-1, GCN5, PCAF, and loading control. (B) Rae-1 expression on tumor cell surfaces was determined using flow cytometry. Bar graphs show the percentage of Rae-1–positive cells. (C) Immunofluorescence staining of NKG2D receptor on tumor sections. Scale bar, 50 μm. (D) K7M3 and LLC cells were treated with DMSO or panobinostat (100 nM) for 24 hours. Rae-1 mRNA level was determined using qPCR. Bar graphs show Rae-1 transcription relative to DMSO-treated cells. ***P < 0.001.
As others have emphasized that HDAC inhibitors modify the histone tail to promote NKG2D ligand expression [8, 37], we tested the mRNA expression of Rae-1 in tumors after administration of DMSO or panobinostat. Surprisingly, the increase in Rae-1 mRNA expression was very small compared with the increase in protein expression (Fig. 2D). This discrepancy between Rae-1 transcription and protein upregulation led us to hypothesize that Rae-1 is stabilized through posttranslational acetylation.
Rae-1 protein is directly acetylated by GCN5 and PCAF
To determine whether Rae-1 undergoes acetylation by GCN5 and PCAF, K7M3 tumor cells were transiently transfected with GCN5 or PCAF plasmid DNA for 36 hours (Fig. 3A). Cell lysates from transfected cells were immunoprecipitated with an anti–Rae-1 antibody and immunoblotted with anti–acetyl-lysine (Fig. 3A). These results showed that levels of acetylated Rae-1 were significantly increased upon GCN5 or PCAF transfection (Fig. 3A). To further validate whether GCN5 and PCAF acetylate Rae-1 under physical interaction in vitro, HEK 293 cells were transiently transfected with Rae-1, and transfection was confirmed via flow cytometry (Fig. 3B). Rae-1 was precipitated from cell lysates and used as a substrate of acetyltransferases GCN5 and PCAF in the presence of acetyl-coenzyme A, which provided an essential substrate for Rae-1 acetylation. Immunoblots showed that the yield of acetylated Rae-1 was not changed by GCN5 alone (Fig. 3B). Strikingly, however, the KAT domain of PCAF substantially increased levels of acetylated Rae-1, and GCN5 and PCAF synergistically enhanced Rae-1 acetylation (Fig. 3B).
Figure 3. Rae-1 protein is directly acetylated by GCN5 and PCAF.
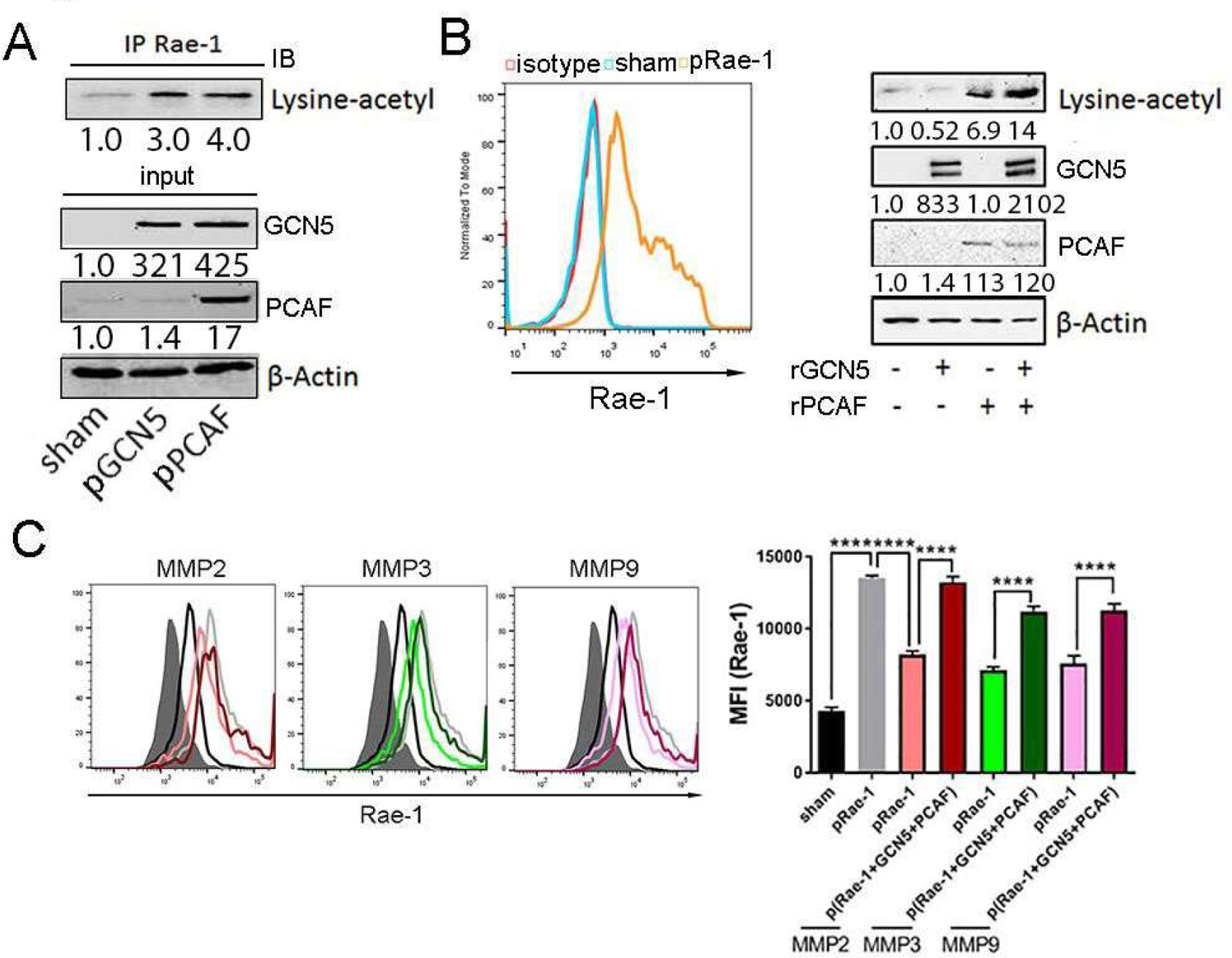
(A) K7M3 cells were transfected with control, GCN5, or PCAF DNA for 36 hours. Rae-1 acetylation was determined using co-immunoprecipitation assays. (B) HEK 293 cells were transfected with Rae-1 DNA for 36 hours, and Rae-1 expression was confirmed with flow cytometry. Rae-1 acetylation was validated using an in vitro acetylation assay. (C) HEK 293 cells were transfected with Rae-1 DNA alone or co-transfected with Rae-1 plus GCN5 and PCAF DNA. After 36 hours, cells were co-cultured with MMP2, MMP3, or MMP9 recombinant protein (10 ng/mL) for 24 hours. Rae-1 expression was determined using flow cytometry. Bar graphs show the MFI of Rae-1 on K7M3 cells. ****P < 0.0001.
We next reasoned that if acetylation of Rae-1 stabilizes Rae-1 on the tumor cell surface, acetylated Rae-1 should be resistant to proteolytic cleavage by MMPs. To test this hypothesis, we transfected HEK 293 cells that lacked MMPs with Rae-1 alone or with Rae-1, GCN5, and PCAF. The cells were then treated with vehicle control or MMPs (Fig. 3C). As expected, tumor cells treated with MMPs shed unacetylated Rae-1 from their surfaces. However, co-expression of Rae-1 with GCN5 and PCAF prevented Rae-1 from cleaving from tumor cells (Fig. 3C). These results validated our hypothesis that acetylated Rae-1 is resistant to enzyme-mediated shedding.
Lysine 80 and lysine 87 are acetylation sites of Rae-1
The bioinformatics tools PAIL [38] (Table 1) and ASEB [39] (Table 2) both predicted K80 and K87 as highly likely acetylation sites of Rae-1. To further define the most critical lysine(s), lysine-to-arginine (R) mutants were generated to determine whether lysine mutation disables Rae-1 acetylation. In vitro acetylation assays were repeated by transfecting HEK 293 cells with control DNA, wild-type Rae-1 (Rae-1WT), or mutant Rae-1 (Rae-1mut) (Fig. 4A). In vitro Rae-1 acetylation assays showed that Rae-1K87R was acetylated at a lower level than was Rae-1WT. In striking contrast, Rae-1K80R almost completely impaired Rae-1 acetylation by GCN5 and PCAF (Fig. 4A). These results demonstrated that K80 and K87 are the target acetylation sites of GCN5 and PCAF. In theory, if K80 and K87 are the true acetylation sites, Rae-1K80RK87R should be susceptible to enzyme-mediated shedding. To test this hypothesis, HEK 293 cells were transfected with control DNA, Rae-1WT, Rae-1WT plus GCN5 and PCAF, or Rae-1mut plus GCN5 and PCAF. Because HEK 293 cells are MMP deficient, transfection of Rae-1WT or Rae-1mut for 36 hours yielded similar levels of Rae-1 expression on the cell surface (Fig. 4B). We next treated these HEK 293 cells with CT26 cell lysates, in which MMPs are abundant [40]. Without GCN5 and PCAF transfection, CT26 lysates dramatically reduced cell surface expression of Rae-1WT at 24 and 48 hours, but with the addition of GCN5 and PCAF, Rae-1WT was resistant to shedding, but Rae-1mut was susceptible to it (Fig. 4C).
Figure 4. Rae-1 can be acetylated at lysine 80 and lysine 87.
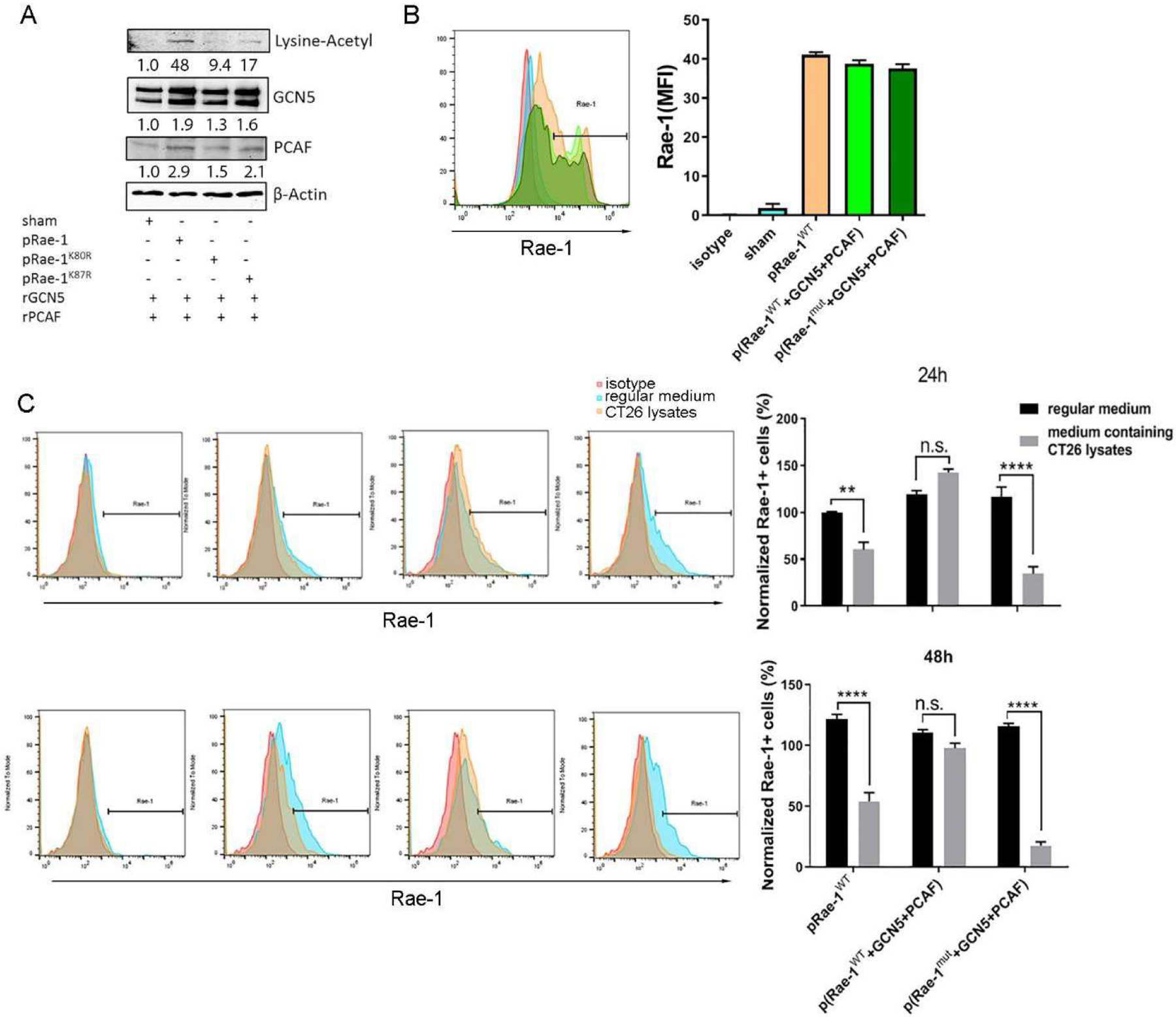
(A) HEK 293 cells were transiently transfected with Rae-1WT, Rae-1K87R, or Rae-1K80R DNA. Rae-1 acetylation was determined by in vitro acetylation assay followed by immunoblotting. (B) HEK 293 cells were transiently transfected with control, Rae-1WT, Rae-1WT plus GCN5 and PCAF, or Rae-1K87RK80R plus GCN5 and PCAF DNAs. Flow cytometry was used to determine Rae-1 levels on the cell surface. Bar graph shows MFI of Rae-1 on HEK 293 cells. (C) HEK 293 cells from (A-B) were cultured in regular cell culture medium or a medium containing CT26 cell lysates for 24 or 48 hours. Rae-1 levels were determined using flow cytometry. Bar graphs show the normalized frequency of Rae-1–expressing cells relative to Rae-1WT–transfected cells at 24 hours. **P < 0.01; ****P < 0.0001; n.s., not significant.
Acetylation of Rae-1 at K80 and K87 stabilizes Rae-1 on the tumor cell surface
To further elucidate how Rae-1 acetylation at K80 and K87 is associated with Rae-1 stability, we first confirmed that downregulation of cell surface Rae-1 was due to proteolytic cleavage by MMPs found in most tumors. HEK 293 cells were transiently transfected with Rae-1WT or Rae-1mut and cultured in a medium containing CT26 cell lysates for 24 hours. Cell surface Rae-1 expression was dramatically decreased, but this downregulation was rescued by treatment with the MMP inhibitor GM6001 or overexpression of GCN5 and PCAF (Fig. 5A, 5B, 4C). These findings suggest that GCN5 and PCAF function in this context as an MMP inhibitor to impair proteolytic cleavage. Moreover, to demonstrate that the acetyltransferase activity of GCN5 and PCAF prevents Rae-1 shedding by MMPs, HEK 293 cells were transiently transfected with GCN5 and PCAF as well as Rae-1WT or Rae-1mut in CT26 lysate-containing medium and then treated with the acetyltransferase inhibitor anacardic acid. As acetyltransferase activity was ablated, GCN5 and PCAF lost their ability to prevent Rae-1 shedding (Fig. 5A, 5B), implying that they rescue Rae-1 from shedding via acetylation.
Figure 5. Acetylation of Rae-1 at K80 and K87 protects the protein from shedding induced by MMPs.
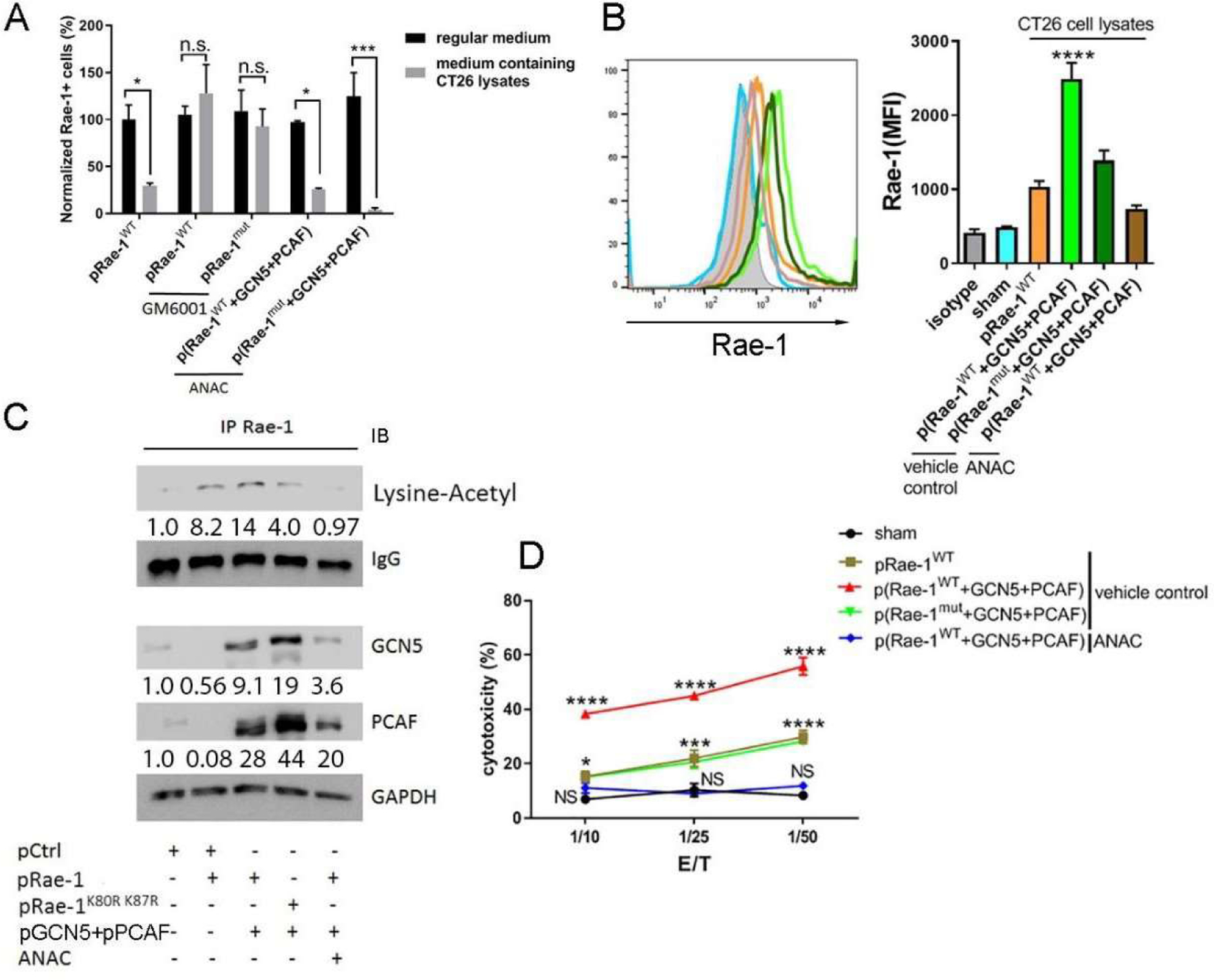
HEK 293 cells were transiently transfected as described in Fig. 4B. (A) The transfected HEK 293 cells were treated with DMSO, GM6001 (10 μM), or anacardic acid (ANAC) (5 μM) in regular cell culture medium or medium containing CT26 cell lysates for 24 hours. Rae-1 levels were determined using flow cytometry. Bar graphs show the normalized frequency of Rae-1–expressing cells relative to Rae-1WT -transfected cells in regular cell culture medium. (B) Transfected cells were cultured in medium containing CT26 cell lysates in the presence or absence of ANAC (5 μM) for 24 hours. Rae-1 levels were determined using flow cytometry. Bar graphs show the MFI of Rae-1 expression on the cell surface. (C) Co-immunoprecipitation assay to determine Rae-1WT and Rae-1K87RK80R acetylation in the presence or absence of ANAC. (D) Cytolytic activity of stimulated splenocytes against Rae-1WT- or Rae-1K87RK80R-transfected HEK 293 cells at the indicated ratios in the presence or absence of ANAC. *P < 0.05; ***P < 0.001; ****P < 0.0001; n.s., not significant.
To analyze the acetylation status of Rae-1, Rae-1 was immunoprecipitated from the transfected cells shown in Fig. 5B and blotted with a lysine-acetyl antibody (Fig. 5C). Input blots showed the transfection of KATs. The levels of acetylated Rae-1WT were increased by overexpression of GCN5 and PCAF, whereas Rae-1mut showed only low basal acetylation levels, and anacardic acid totally abolished Rae-1 acetylation (Fig. 5C). Remarkably, levels of Rae-1 acetylation were correlated with cell surface expression of Rae-1 (Fig. 5B). These results suggested that acetylated Rae-1 is resistant to MMP-mediated proteolytic shedding and is stable on the cell surface.
We next aimed to determine the functional advantage of acetylated Rae-1 in terms of sensitizing tumor cells to immune cell attack—in other words, whether the acetylation of Rae-1 alters its interaction with NKG2D+ immune cells. To address this question, the transfected cells shown in Fig. 5A and 5B were labeled with a violet cell tracker dye. We showed in a previous report that stimulating CD28 signaling strongly induced NKG2D expression on CD8+ T cells [41]. Thus, the Rae-1 transfected cells were incubated with activated mouse splenocytes that highly expressed NKG2D at different ratios in CT26 cell lysate-containing medium for 6 hours, and the NKG2D-dependent cytolytic activity of the T cells was evaluated via flow cytometry (Fig. 5D). Strikingly, co-expression of Rae-1WT and GCN5/PCAF dramatically increased the cytolytic efficacy of T cells, whereas expression of Rae-1mut attenuated cell killing by cytotoxic lymphocytes, even in the presence of GCN5 and PCAF. In contrast, anacardic acid abolished Rae-1 acetylation, rendering tumor cells completely resistant to killing by NKG2D+ splenocytes. These results demonstrated that acetylated Rae-1 is fully functional in rendering tumor cells susceptible to NKG2D+ immune cell cytotoxicity.
High levels of KATs are associated with longer overall survival durations and higher expression of NKG2D ligands
In order to assess whether KATs might upregulate NKG2D ligands in human cancers and extend the survival of cancer patients, we compared the survival times of cancer patients with high versus low expression of KATs using data from The Cancer Genome Atlas database. Notably, we found that in patients with colorectal carcinoma, sarcoma, lung adenocarcinoma, or liver cancer, high expression of KATs in tumors was associated with significant extension of survival times (Fig. 6A). Nevertheless, the gene expression of KATs had no correlation with the mRNA transcription of human NKG2D ligands (Fig. 6B), which clearly showed that KATs had very little effect on the epigenetic transcriptional upregulation of NKG2D ligands in tumors in vivo, even though this effect was very common in vitro. We further assessed the correlation between the protein levels of KATs and NKG2D ligands in human tumors by analyzing expression of GCN5 and the human NKG2D ligand ULBP1 by using immunohistochemical staining of tissue microarrays from patients with liver cancer. Notably, high protein levels of GCN5 were strongly correlated with expression of ULBP1 in liver cancers at various stages (Fig. 6C), showing that KATs play a critical role in stabilizing NKG2D ligands in human cancers.
Figure 6. High levels of lysine acetyltransferases (KATs) are associated with longer overall survival durations in human cancer patients and correlated with higher protein expression of the human NKG2D ligands.
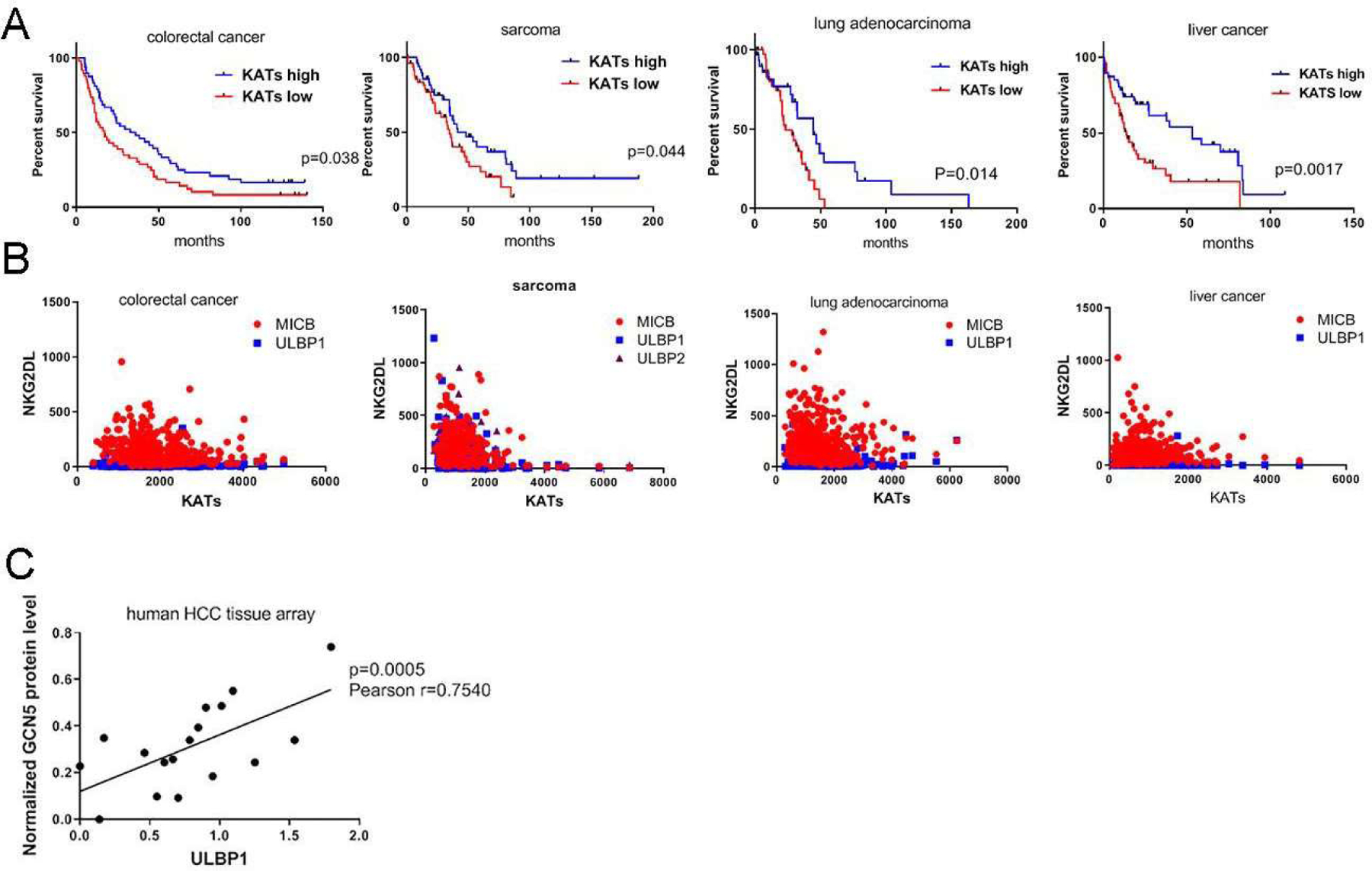
(A) Kaplan-Meier curves showing the overall survival durations of patients with colorectal carcinoma, sarcoma, lung adenocarcinoma, and liver cancer stratified by KAT gene expression levels (www.cbioportal.org). The 25% of patients with the highest KAT levels were classified into the KAT-high group, and the 25% of patients with the lowest KAT levels were classified into the KAT-low group. (B) Correlation of KATs and NKG2D ligand expression (mRNA) in human cancer patient data obtained from TCGA. (C) Correlation of GCN5 and ULBP1 protein levels in human liver cancer tissues. An array of human liver cancer tissues of different stages was subjected to immunohistochemical staining for GCN5 and ULBP1. The dot graph represents the intensity (mean ± SEM) of GCN5 and ULBP1 staining.
Discussion
A number of studies have reported that NKG2D-dependent antitumor immune surveillance effectively eradicates tumor cells at an early stage of tumor development, but as tumors progress, tumor cells tend to evade immune surveillance by shedding NKG2D ligands and releasing soluble products into the serum [42, 43]. These soluble NKG2D ligands not only compromise NKG2D receptor functions on natural killer and effector T cells but also stimulate the expansion of myeloid-derived suppressor cells to augment suppressive immunity. As a result, elevated levels of soluble NKG2D ligands in the serum serve as an indicator of poor prognosis and reduced overall survival in patients with early- and late-stage melanoma [6] and with early-stage breast cancer [44], colorectal cancer, prostate cancer, hepatocellular carcinoma, and neuroblastoma [44–46]. In this study, we demonstrated that posttranslational acetylation of NKG2D ligands obstructs proteolytic shedding and therefore stabilizes the expression of NKG2D ligands on the tumor cell surface.
The expression of NKG2D ligands is regulated at transcriptional, posttranscriptional, and posttranslational levels. The most well-known mechanism is initiated by DNA damage, which stimulates ataxia-telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR) protein kinases and a downstream kinase cascade to stabilize NKG2D ligand transcripts [47]. In addition, 90-kDa heat shock protein (HSP-90) promotes the transcription of NKG2D ligands MICA and MICB [48, 49], and the cell cycle regulator E2F regulates NKG2D ligand transcripts in coordination with stress responses [50]. During oncogenic transformation, NKG2D ligands are induced via a c-myc–dependent mechanism [51], whereas viral infection triggers PI3K-dependent NKG2D ligand upregulation [52]. Additionally, MICA and MICB can be regulated by microRNAs and the EGF receptor pathway at the posttranscriptional level [53–55]. The posttranslational regulation of NKG2D ligands has not been fully characterized, although one study showed that the murine NKG2D ligand Mult1 is expressed in normal cells but absent on the cell surface of cancer cells owing to ubiquitination-dependent degradation [56].
Our data for the first time established that acetylation of the NKG2D ligand Rae-1 protects the protein from proteolytic shedding and subsequently stabilizes Rae-1 expression on tumor cell surfaces. It is noteworthy that posttranslational acetylation functions as a double-edged sword: while it promotes protein activity in certain cases [57–60], it can also suppress proteins through degradation and deactivation [61, 62]. In our case, both the HDAC inhibitor panobinostat and the KATs GCN5 and PCAF sensitized tumor cells to NKG2D-dependent killing by cytotoxic T lymphocytes. Additionally, overexpression of GCN5 and PCAF elevated NKG2D ligand expression in tumors in vivo, resulting in NKG2D+ immune cell accumulation in tumors. These results support our conclusion that posttranslational acetylation of Rae-1 not only stabilizes the protein but also, and more importantly, enhances the effector function of immune surveillance ligands in rendering tumor cells more “visible” to antitumor immunity.
The acetylation sites of Rae-1 were identified using bioinformatics tools and then validated by lysine-to-arginine dominant-negative mutations. Rae-1K80R and Rae-1K87R disabled Rae-1 acetylation in vitro and in vivo, thereby allowing MMP-mediated proteolytic shedding and downregulating Rae-1 expression on the tumor cell surface. It has been suggested that posttranslational modifications, phosphorylation and acetylation for instance, may prevent enzyme-induced proteolytic cleavage of proteins to modulate their biological functions. Hu et al. [63], for example, discovered that phosphorylation of acinus by Akt rendered cells resistant to apoptotic cleavage and served as the mechanism by which the Akt pathway regulates cell survival. Similarly, acetylation of lysine residues of APE1 in tumors reduced proteolysis and maintained the essential role of this protein in tumor cell proliferation [64]. Our data, similarly, suggested that acetylation of Rae-1 at K80 and K87 might block the target amino acids that induce proteolytic shedding to prevent Rae-1 shedding.
In normal cells, acetyltransferases localize in the nucleus. However, others have reported that GCN5 accumulates in the cytoplasm of apoptotic and cancerous cells [65], where it functions as the acetyltransferase of the cytoplasmic marker tubulin [66]. Intriguingly, in our in vivo tumor model, overexpressed GCN5 localized either in the nucleus alone or in both the nucleus and cytoplasm, implying that cytoplasm-localized GCN5 could play a role as a posttranslational acetyltransferase of target proteins. The tumor cells in which GCN5 localized mainly in the nucleus had lower levels of Rae-1 than did the tumor cells with both nuclear and cytoplasmic GCN5 (data not shown).
There has been increasing interest in studying the impact of posttranslational acetylation on cellular and biological functions. Our study clearly showed that instead of transcriptional induction of NKG2D ligands, which is always difficult to achieve in vivo, therapeutics that enable the posttranslational acetylation of NKG2D ligand Rae-1 on K80 and K87 to avoid proteolytic shedding may enhance the protein’s stability on the tumor cell surface in vivo. By these means, tumor cells could be marked with stabilized NKG2D ligands; along with our previously reported strategy to enhance NKG2D receptor expression on T cells [41, 67], this could activate persistent NKG2D antitumor cytotoxicity to eliminate tumors in vivo.
Acknowledgments
This study was supported by the National Institutes of Health through grant R01 CA200574 and Cancer Center Support Grant P30 CA016672. The following Cancer Center Support Grant core resources were used: Genetically Engineered Mouse Facility and Monoclonal Antibody Core Facility. The authors would like to thank Dr. Amy Ninetto from Scientific Publications, Research Medical Library, MD Anderson Cancer Center for editing the manuscript.
Abbreviations:
- NKG2D
natural killer group 2, member D
- HDAC
histone deacetylase
- KAT
lysine acetyltransferase
- MMP
matrix metalloproteinase
Footnotes
Competing interests: The authors have no potential conflicts of interest to declare.
References
- [1].Waldhauer I, Steinle A, Proteolytic release of soluble UL16-binding protein 2 from tumor cells, Cancer Res, 66 (2006) 2520–2526. [DOI] [PubMed] [Google Scholar]
- [2].Boutet P, Aguera-Gonzalez S, Atkinson S, Pennington CJ, Edwards DR, Murphy G, Reyburn HT, Vales-Gomez M, Cutting edge: the metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein, J Immunol, 182 (2009) 49–53. [DOI] [PubMed] [Google Scholar]
- [3].Cerwenka A, New twist on the regulation of NKG2D ligand expression, J Exp Med, 206 (2009) 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, Reyburn HT, Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes, Cancer Res, 70 (2010) 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Champsaur M, Lanier LL, Effect of NKG2D ligand expression on host immune responses, Immunol Rev, 235 (2010) 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Paschen A, Sucker A, Hill B, Moll I, Zapatka M, Nguyen XD, Sim GC, Gutmann I, Hassel J, Becker JC, Steinle A, Schadendorf D, Ugurel S, Differential clinical significance of individual NKG2D ligands in melanoma: soluble ULBP2 as an indicator of poor prognosis superior to S100B, Clin Cancer Res, 15 (2009) 5208–5215. [DOI] [PubMed] [Google Scholar]
- [7].Insinga A, Monestiroli S, Ronzoni S, Carbone R, Pearson M, Pruneri G, Viale G, Appella E, Pelicci P, Minucci S, Impairment of p53 acetylation, stability and function by an oncogenic transcription factor, EMBO J, 23 (2004) 1144–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, Steinle A, Salih HR, Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate, Cancer research, 65 (2005) 6321–6329. [DOI] [PubMed] [Google Scholar]
- [9].Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, Shilatifard A, Workman J, Zhang Y, New nomenclature for chromatin-modifying enzymes, Cell, 131 (2007) 633–636. [DOI] [PubMed] [Google Scholar]
- [10].Biggar KK, Li SS, Non-histone protein methylation as a regulator of cellular signalling and function, Nat Rev Mol Cell Biol, 16 (2015) 5–17. [DOI] [PubMed] [Google Scholar]
- [11].Cao X, Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease, Nat Rev Immunol, 16 (2016) 35–50. [DOI] [PubMed] [Google Scholar]
- [12].Chen T, Dent SY, Chromatin modifiers and remodellers: regulators of cellular differentiation, Nat Rev Genet, 15 (2014) 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, Jiang Y, Wang C, Cao X, Methyltransferase SETD2-Mediated Methylation of STAT1 Is Critical for Interferon Antiviral Activity, Cell, 170 (2017) 492–506 e414. [DOI] [PubMed] [Google Scholar]
- [14].Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M, Lysine acetylation targets protein complexes and co-regulates major cellular functions, Science, 325 (2009) 834–840. [DOI] [PubMed] [Google Scholar]
- [15].Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y, Lei QY, Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth, Mol Cell, 51 (2013) 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Johnsson A, Xue-Franzen Y, Lundin M, Wright AP, Stress-specific role of fission yeast Gcn5 histone acetyltransferase in programming a subset of stress response genes, Eukaryot Cell, 5 (2006) 1337–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Naguleswaran A, Elias EV, McClintick J, Edenberg HJ, Sullivan WJ Jr., Toxoplasma gondii lysine acetyltransferase GCN5-A functions in the cellular response to alkaline stress and expression of cyst genes, PLoS Pathog, 6 (2010) e1001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Xue-Franzen Y, Johnsson A, Brodin D, Henriksson J, Burglin TR, Wright AP, Genome-wide characterisation of the Gcn5 histone acetyltransferase in budding yeast during stress adaptation reveals evolutionarily conserved and diverged roles, BMC Genomics, 11 (2010) 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E, DNA damage activates p53 through a phosphorylation-acetylation cascade, Genes Dev, 12 (1998) 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG, PML regulates p53 acetylation and premature senescence induced by oncogenic Ras, Nature, 406 (2000) 207–210. [DOI] [PubMed] [Google Scholar]
- [21].Giandomenico V, Simonsson M, Gronroos E, Ericsson J, Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors, Mol Cell Biol, 23 (2003) 2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Simonsson M, Heldin CH, Ericsson J, Gronroos E, The balance between acetylation and deacetylation controls Smad7 stability, J Biol Chem, 280 (2005) 21797–21803. [DOI] [PubMed] [Google Scholar]
- [23].Gronroos E, Hellman U, Heldin CH, Ericsson J, Control of Smad7 stability by competition between acetylation and ubiquitination, Mol Cell, 10 (2002) 483–493. [DOI] [PubMed] [Google Scholar]
- [24].Rausa FM 3rd, Hughes DE, Costa RH, Stability of the hepatocyte nuclear factor 6 transcription factor requires acetylation by the CREB-binding protein coactivator, J Biol Chem, 279 (2004) 43070–43076. [DOI] [PubMed] [Google Scholar]
- [25].Incani F, Serra M, Meloni A, Cossu C, Saba L, Cabras T, Messana I, Rosatelli MC, AIRE acetylation and deacetylation: effect on protein stability and transactivation activity, J Biomed Sci, 21 (2014) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Choi JY, Kim JH, Jo SA, Acetylation regulates the stability of glutamate carboxypeptidase II protein in human astrocytes, Biochem Biophys Res Commun, 450 (2014) 372–377. [DOI] [PubMed] [Google Scholar]
- [27].Perez-Luna M, Aguasca M, Perearnau A, Serratosa J, Martinez-Balbas M, Jesus Pujol M, Bachs O, PCAF regulates the stability of the transcriptional regulator and cyclin-dependent kinase inhibitor p27 Kip1, Nucleic Acids Res, 40 (2012) 6520–6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Glozak MA, Seto E, Acetylation/deacetylation modulates the stability of DNA replication licensing factor Cdt1, J Biol Chem, 284 (2009) 11446–11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ge X, Jin Q, Zhang F, Yan T, Zhai Q, PCAF acetylates {beta}-catenin and improves its stability, Mol Biol Cell, 20 (2009) 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liang W, Malhotra A, Deutscher MP, Acetylation regulates the stability of a bacterial protein: growth stage-dependent modification of RNase R, Mol Cell, 44 (2011) 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hu J, Bernatchez C, Zhang L, Xia X, Kleinerman ES, Hung MC, Hwu P, Li S, Induction of NKG2D Ligands on Solid Tumors Requires Tumor-Specific CD8+ T Cells and Histone Acetyltransferases, Cancer Immunol Res, 5 (2017) 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dibra D, Cutrera JJ, Xia X, Birkenbach MP, Li S, Expression of WSX1 in tumors sensitizes IL-27 signaling-independent natural killer cell surveillance, Cancer research, 69 (2009) 5505–5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu J, Vien LT, Xia X, Bover L, Li S, Generation of a monoclonal antibody against the glycosylphosphatidylinositol-linked protein Rae-1 using genetically engineered tumor cells, Biol Proced Online, 16 (2014) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kuninger D, Lundblad J, Semirale A, Rotwein P, A non-isotopic in vitro assay for histone acetylation, J Biotechnol, 131 (2007) 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, Stevanovic S, Rammensee HG, Steinle A, Tumor-associated MICA is shed by ADAM proteases, Cancer Res, 68 (2008) 6368–6376. [DOI] [PubMed] [Google Scholar]
- [36].Hu J, Bernatchez C, Zhang L, Xia X, Kleinerman ES, Hung MC, Hwu P, Li S, Induction of NKG2D Ligands on Solid Tumors Requires Tumor-Specific CD8+ T Cells and Histone Acetyltransferases, Cancer Immunol Res, (2017). [DOI] [PMC free article] [PubMed]
- [37].Schmudde M, Braun A, Pende D, Sonnemann J, Klier U, Beck JF, Moretta L, Broker BM, Histone deacetylase inhibitors sensitize tumour cells for cytotoxic effects of natural killer cells, Cancer letters, 272 (2008) 110–121. [DOI] [PubMed] [Google Scholar]
- [38].Li A, Xue Y, Jin C, Wang M, Yao X, Prediction of Nepsilon-acetylation on internal lysines implemented in Bayesian Discriminant Method, Biochem Biophys Res Commun, 350 (2006) 818–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang L, Du Y, Lu M, Li T, ASEB: a web server for KAT-specific acetylation site prediction, Nucleic Acids Res, 40 (2012) W376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen YJ, Chang WM, Liu YW, Lee CY, Jang YH, Kuo CD, Liao HF, A small-molecule metastasis inhibitor, norcantharidin, downregulates matrix metalloproteinase-9 expression by inhibiting Sp1 transcriptional activity in colorectal cancer cells, Chem Biol Interact, 181 (2009) 440–446. [DOI] [PubMed] [Google Scholar]
- [41].Hu J, Batth IS, Xia X, Li S, Regulation of NKG2D+CD8+ T-cell-mediated antitumor immune surveillance: Identification of a novel CD28 activation-mediated, STAT3 phosphorylation-dependent mechanism, Oncoimmunology, 5 (2016) e1252012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, Weller M, Friese MA, TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells, Brain, 129 (2006) 2416–2425. [DOI] [PubMed] [Google Scholar]
- [43].Schmiedel D, Mandelboim O, NKG2D Ligands-Critical Targets for Cancer Immune Escape and Therapy, Front Immunol, 9 (2018) 2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].de Kruijf EM, Sajet A, van Nes JG, Putter H, Smit VT, Eagle RA, Jafferji I, Trowsdale J, Liefers GJ, van de Velde CJ, Kuppen PJ, NKG2D ligand tumor expression and association with clinical outcome in early breast cancer patients: an observational study, BMC Cancer, 12 (2012) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, Kimura R, Miyagi T, Mochizuki K, Sasaki Y, Hayashi N, Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid, Int J Cancer, 104 (2003) 354–361. [DOI] [PubMed] [Google Scholar]
- [46].Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, Pende D, Steinle A, Ferrone S, Pistoia V, Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma, Neoplasia, 6 (2004) 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gasser S, Orsulic S, Brown EJ, Raulet DH, The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor, Nature, 436 (2005) 1186–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T, Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium, Proc Natl Acad Sci U S A, 93 (1996) 12445–12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fionda C, Soriani A, Malgarini G, Iannitto ML, Santoni A, Cippitelli M, Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation, J Immunol, 183 (2009) 4385–4394. [DOI] [PubMed] [Google Scholar]
- [50].Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH, RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry, J Exp Med, 209 (2012) 2409–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Unni AM, Bondar T, Medzhitov R, Intrinsic sensor of oncogenic transformation induces a signal for innate immunosurveillance, Proc Natl Acad Sci U S A, 105 (2008) 1686–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tokuyama M, Lorin C, Delebecque F, Jung H, Raulet DH, Coscoy L, Expression of the RAE-1 family of stimulatory NK-cell ligands requires activation of the PI3K pathway during viral infection and transformation, PLoS Pathog, 7 (2011) e1002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, Mandelboim M, Mandelboim O, Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D, Nat Immunol, 9 (2008) 1065–1073. [DOI] [PubMed] [Google Scholar]
- [54].Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O, Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells, Cell Host Microbe, 5 (2009) 376–385. [DOI] [PubMed] [Google Scholar]
- [55].Vantourout P, Willcox C, Turner A, Swanson CM, Haque Y, Sobolev O, Grigoriadis A, Tutt A, Hayday A, Immunological visibility: posttranscriptional regulation of human NKG2D ligands by the EGF receptor pathway, Sci Transl Med, 6 (2014) 231ra249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nice TJ, Coscoy L, Raulet DH, Posttranslational regulation of the NKG2D ligand Mult1 in response to cell stress, The Journal of experimental medicine, 206 (2009) 287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kim H, Mendez R, Chen X, Fang D, Zhang K, Lysine Acetylation of CREBH Regulates Fasting-Induced Hepatic Lipid Metabolism, Mol Cell Biol, 35 (2015) 4121–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yang H, Zhou L, Shi Q, Zhao Y, Lin H, Zhang M, Zhao S, Yang Y, Ling ZQ, Guan KL, Xiong Y, Ye D, SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth, EMBO J, 34 (2015) 1110–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fernandes J, Weddle A, Kinter CS, Humphries KM, Mather T, Szweda LI, Kinter M, Lysine Acetylation Activates Mitochondrial Aconitase in the Heart, Biochemistry, 54 (2015) 4008–4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shan C, Elf S, Ji Q, Kang HB, Zhou L, Hitosugi T, Jin L, Lin R, Zhang L, Seo JH, Xie J, Tucker M, Gu TL, Sudderth J, Jiang L, DeBerardinis RJ, Wu S, Li Y, Mao H, Chen PR, Wang D, Chen GZ, Lonial S, Arellano ML, Khoury HJ, Khuri FR, Lee BH, Brat DJ, Ye K, Boggon TJ, He C, Kang S, Fan J, Chen J, Lysine acetylation activates 6-phosphogluconate dehydrogenase to promote tumor growth, Mol Cell, 55 (2014) 552–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sun C, Wang M, Liu X, Luo L, Li K, Zhang S, Wang Y, Yang Y, Ding F, Gu X, PCAF improves glucose homeostasis by suppressing the gluconeogenic activity of PGC-1alpha, Cell Rep, 9 (2014) 2250–2262. [DOI] [PubMed] [Google Scholar]
- [62].Cazzalini O, Sommatis S, Tillhon M, Dutto I, Bachi A, Rapp A, Nardo T, Scovassi AI, Necchi D, Cardoso MC, Stivala LA, Prosperi E, CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis, Nucleic Acids Res, 42 (2014) 8433–8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hu Y, Yao J, Liu Z, Liu X, Fu H, Ye K, Akt phosphorylates acinus and inhibits its proteolytic cleavage, preventing chromatin condensation, EMBO J, 24 (2005) 3543–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bhakat KK, Sengupta S, Adeniyi VF, Roychoudhury S, Nath S, Bellot LJ, Feng D, Mantha AK, Sinha M, Qiu S, Luxon BA, Regulation of limited N-terminal proteolysis of APE1 in tumor via acetylation and its role in cell proliferation, Oncotarget, 7 (2016) 22590–22604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Blanco-Garcia N, Asensio-Juan E, de la Cruz X, Martinez-Balbas MA, Autoacetylation regulates P/CAF nuclear localization, J Biol Chem, 284 (2009) 1343–1352. [DOI] [PubMed] [Google Scholar]
- [66].Fermento ME, Gandini NA, Lang CA, Perez JE, Maturi HV, Curino AC, Facchinetti MM, Intracellular distribution of p300 and its differential recruitment to aggresomes in breast cancer, Exp Mol Pathol, 88 (2010) 256–264. [DOI] [PubMed] [Google Scholar]
- [67].Hu J, Zhu S, Xia X, Zhang L, Kleinerman ES, Li S, CD8+T cell-specific induction of NKG2D receptor by doxorubicin plus interleukin-12 and its contribution to CD8+T cell accumulation in tumors, Mol Cancer, 13 (2014) 34. [DOI] [PMC free article] [PubMed] [Google Scholar]