

SUMMARY
We report that increasing inhibition from the basal ganglia (BG) to the superior colliculus (SC) through the substantia nigra pars reticulata (nigra) using in vivo optogenetic activation of GABAergic terminals in mice produces contralateral orienting movements. These movements are unexpected because decreases, and not increases, in nigral activity are generally associated with the initiation of orienting movements. We found that, in slice recordings, the same optogenetic stimulation of nigral terminals producing movements in vivo evokes post-inhibitory rebound depolarization followed by Na+ spikes in SC output neurons. Moreover, blocking T-type Ca2+ channels in slices prevent post-inhibitory rebound and subsequent Na+ spiking in SC output neurons and also reduce the likelihood of contralateral orienting in vivo. On the basis of these results, we propose that, in addition to the permissive role, the BG may play an active role in the generation of orienting movements in mice by driving post-inhibitory rebound depolarization in SC output neurons.
Graphical Abstract
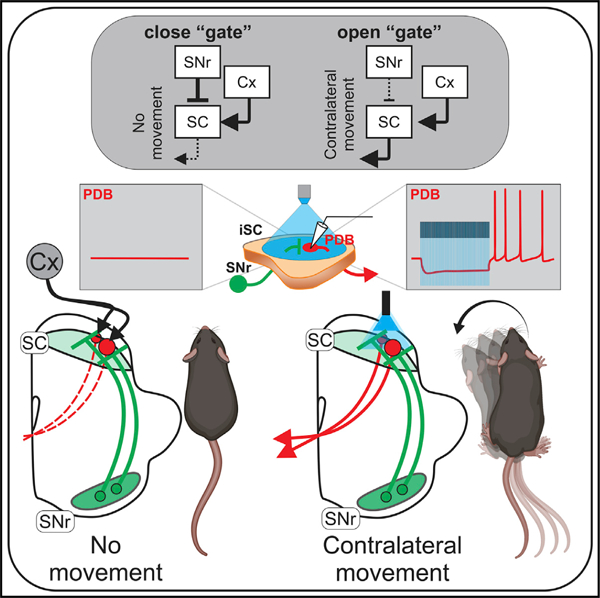
In brief
Villalobos and Basso show that, in addition to the known inhibition from the nigro-collicular circuit, thought to suppress movement, optogenetic activation of the inhibitory inputs to the superior colliculus from the substantia nigra pars reticulata in the mouse evokes contralateral orienting movements and post-inhibitory rebound depolarization in collicular output neurons.
INTRODUCTION
Stemming from experiments performed in monkeys, cats, and rodents in the late 1980s and using traditional electrophysiological and anatomical approaches (Hikosaka and Wurtz, 1985a, 1985b) a model of the role of the basal ganglia (BG) in movement generation in health and disease emerged (DeLong, 1983; Penney and Young, 1983; Albin et al., 1989; DeLong et al., 1990). The model, currently described in all major neuroscience textbooks, proposes that the BG plays a permissive role in the generation of movement through modulation of the amount of inhibition on BG target structures such as the thalamus and superior colliculus (SC). For orienting movements specifically, one of two output nuclei of the BG, the substantia nigra pars reticulata (nigra) contains GABAergic neurons that project primarily to the ipsilateral SC, a key structure involved in the control of orienting (Sato and Hikosaka, 2002; Deniau et al., 2007; Liu and Basso, 2008). The removal of nigral inhibition on the SC, combined with an excitatory drive from the cerebral cortex to the SC, produces contralateral orienting movements. The cascade of disinhibition to the SC is mediated by the direct BG pathway and is offset by activation of the indirect BG pathway, which acts to suppress movements by increasing the inhibitory output of the nigra on the SC (Hikosaka et al., 2000). The disinhibitory nature of BG function enjoys extensive experimental support in a variety of species including monkeys, cats, rodents, and even lampreys (Boussaoud et al., 1985; Reiner et al., 1998; Stephenson-Jones et al., 2012; Grillner and Robertson, 2016). Disinhibition as a mode of BG action is often referred to as the rate model, as it is the rate of spiking in the inhibitory output neurons of the BG on target structures that determines whether or not a movement commences; high rates and more inhibition result in no movement whereas low rates and less inhibition result in movement (Albin et al., 1989; Nelson and Kreitzer, 2014).
Testing models of BG function at the circuit level is now possible with optogenetics. Recent work in mice by use of causal manipulations of the direct and indirect pathways suggests that the BG plays a supportive rather than a permissive role in reaching movements and that the direct and indirect pathways of the striatum operate in concert to produce movement, wherein one disinhibits and the other inhibits movement (Tecuapetla et al., 2016; Yttri and Dudman, 2016; Klaus et al., 2019). We reasoned that we could use optogenetics to provide a causal test of the permissive disinhibition model of orienting at the level of the output via the nigro-collicular circuit. The prediction from the permissive disinhibition model is that increasing the activity of the nigro-collicular circuit unilaterally should suppress contralateral movements. To test this prediction, we used AAV injections to express Chronos in the nigral-inhibitory terminals and activated them in the SC. Surprisingly, we found that unilateral optogenetic activation of nigral afferents in the SC evoked, rather than suppressed, contralateral movements, contrary to the prediction of a model based on permissive disinhibition. To understand the mechanism of this result, we performed ex vivo slice experiments in the SC and found that the same optogenetic stimulation of nigral terminals used in vivo evoked post-inhibitory rebound depolarization (RD) followed by RD-evoked Na+ spikes in the SC output neurons through activation of T-type Ca2+ channels. Consistent with a causal role for T-type Ca2+ channels triggering post-inhibitory RD and RD-evoked Na+ spikes in orienting, we found that in vivo unilateral injection of a T-type Ca2+ channel blocker reduced the likelihood of spontaneous contralateral movements. These combined results suggest that the control of orienting movements by the inhibitory nigro-collicular circuit may involve biophysical mechanisms that drive SC output neurons through inhibition and RD in addition to the well-known role in permissive disinhibition, similar to cerebellar circuits (Person and Raman, 2012). These results point toward a need to revisit our understanding of the role of BG in orienting movements.
RESULTS
Optogenetic activation of nigral terminals in the SC evokes contralateral orienting movements in mice
The idea that more inhibition from the BG produces less movement and less inhibition produces more movement enjoys considerable experimental support, much of which is circumstantial or correlational (Hikosaka and Wurtz, 1983a, 1983b; Hikosaka et al., 2000; Basso and Sommer, 2011; Freeze et al., 2013; Schmidt et al., 2013; Nelson and Kreitzer, 2014; Klaus et al., 2019). We reasoned that we could provide a direct causal test of the rate model by manipulating the inhibition onto the SC by optogenetically activating the nigral afferents. Figure 1A shows three possible outcomes of this experiment. Unilateral activation of nigral afferents increases the suppression of ipsilateral SC neurons, preventing contralateral movements (Figure 1A1). Activation may also disinhibit the contralateral SC through inhibitory commissural connections, resulting in ipsilateral movements (Figure 1A2). A third, previously unconsidered possibility is that activation of the nigral afferents generates spiking in SC output neurons, evoking contralateral movements (Figure 1A3). To arbitrate among these possibilities, we performed unilateral viral injections of AAV9-Syn-Chronos-GFP or AAV9-Flex-Chronos-GFP into the nigra of wild-type or GAD2-Cre mice, respectively, and later used light activation of the terminals located in the SC while the mice moved about freely (Figures 1B and S1).
Figure 1. Optogenetic activation of the nigro-collicular circuit evokes contralateral rotations in mice.
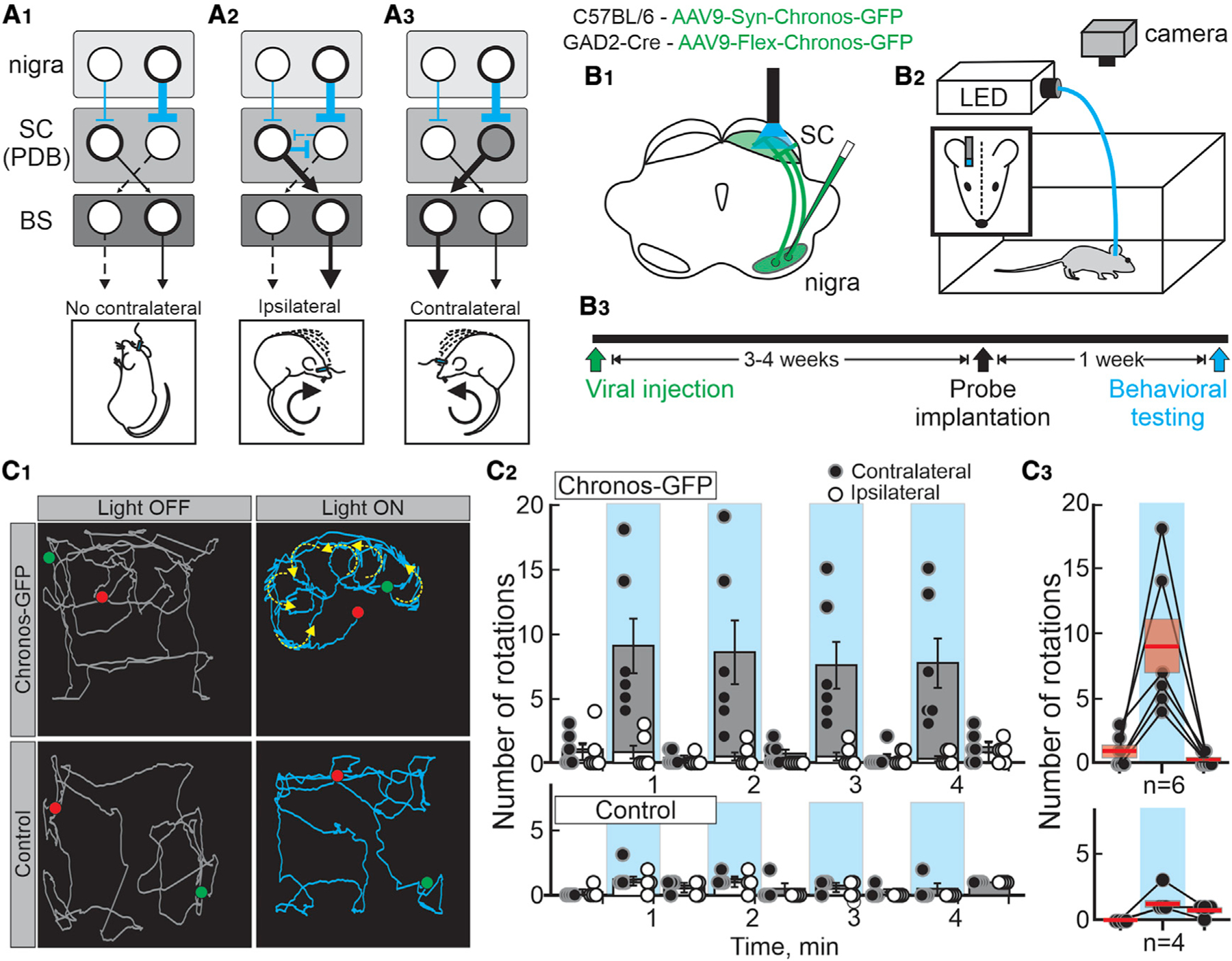
(A1–3) Schematic depiction of the known synaptic connection between the bilateral substantia nigra pars reticulata (nigra), the superior colliculus (SC) pre-dorsal bundle neurons (PDB), and the brainstem- and spinal cord-orienting centers (BS) targeted by the PDB neurons. Cyan and black lines show inhibitory and excitatory connections, respectively. The thickness of the lines depicts the putative level of activity of the circuit, and dashed lines indicate reduced activity. The images below show the predicted outcomes upon unilateral optogenetic activation of the nigro-collicular circuit.
(B1) Schematic of a mouse brain coronal section showing the injection of either AAV9-Syn-Choronos-GFP or AAV9-Flex-Chronos-GFP into the nigra of either wild type (C57BL/6) or GAD2-Cre mice (green) for optogenetic activation of nigro-collicular terminals. The cyan shows the location of the light stimulation into the lateral intermediate/deep layers of the SC, ipsilateral to the virus injection.
(B2) Schematic of the behavioral procedure for unilateral stimulation of freely moving mice in an open-field apparatus.
(B3) Timeline of experimental procedures.
(C1) Traces (collapsed over a 30-s epoch) showing the tracked head movements of a Chronos-GFP and a control mouse (top and bottom, respectively) before (gray traces - OFF) and during (cyan traces - ON) light stimulation. Green and red dots depict starting and endpoints, respectively. Yellow arrows show the movement direction calculated from 5-s epochs.
(C2) Quantification of the number of contralateral (●) and ipsilateral (○) rotations relative to the stimulation site for mice expressing Chronos-GFP and control mice (top and bottom, respectively). Gray and white bars show the mean ± SEM number of rotations recorded in 30-s epochs, with (100 Hz, cyan) and without stimulation (no shading) for all mice. Each circle shows the total number of rotations recorded over four sessions/mouse. Chronos-GFP; n = 6; ON[contra versus ipsi]: 2 × 2 repeated-measures ANOVA f(5,36) = 27.18, p < 0.001; OFF[contra versus ipsi]: (2 × 2 repeated-measures ANOVA f(5,36) = 1.592, p = 0.1871). Control; n = 4; (3 × 2 repeated-measures ANOVA f(9,48) = 0.40, p = 0.930).
(C3) Mean number of contralateral rotations recorded for each of the mice during the first stimulation epoch (Chronos-GFP and control mice, top and bottom, respectively). Lines link the number of rotations recorded for each mouse.
Activation of the nigro-collicular terminals in mice expressing Chronos unexpectedly evoked reliable contralateral rotations, whereas mice injected with control virus showed no discernible pattern of movement with stimulation (Figure 1C1–2 and Video S1). During light-ON epochs, we found a significantly higher number of contralateral than ipsilateral rotations, whereas no differences were observed between rotations during the light-OFF periods. In control mice, there were no differences between the number of contralateral or ipsilateral rotations for any of the epochs (Figure 1C2, bottom). Although variable, all mice expressing Chronos increased the number of contralateral rotations upon light stimulation compared with control mice (Figure 1C3, top:Chronos: OFF, 1.00 ± 0.47, ON, 9.00 ± 2.11; OFF, 0.33 ± 0.19; bottom:Control: OFF, 0.00 ± 0.00, ON, 1.25 ± 0.22, OFF, 0.75 ± 0.22). Thus, surprisingly, 100-Hz optogenetic activation of the inhibitory nigro-collicular circuit in mice produced contralateral orienting movements rather than suppressing them, as expected from a model of permissive disinhibition.
Pre-dorsal bundle neurons show post-inhibitory RD and RD-evoked Na+ channel-mediated spiking
It is unknown how activation of nigral-inhibitory afferents in the SC could lead to a contralateral orienting movement. A possible hypothesis is that pre-dorsal bundle (PDB) output neurons of the SC are excited through post-inhibitory RD evoked by GABA release from nigral terminals innervating the SC. Work in rodents and birds indicates that pallidal (area X) activation evokes post-inhibitory RD and spiking in thalamic (DML) neurons (Person and Perkel, 2005; Kim et al., 2017), but see (Edgerton and Jaeger, 2014). We reasoned that a similar mechanism might be at play in the nigro-collicular circuit of mice.
We first assessed whether PDB neurons could exhibit post-inhibitory RD and RD-evoked spiking. We retrogradely labeled PDB neurons with an injection of retroAAV2-CAG-tdTomato into the pontine reticular nucleus (PnC) and performed patch-clamp recordings of visually identified PDB neurons from ex vivo slices (Figure 2A1–2). A 100-pA current step induced membrane depolarizations and triggered a train of action potentials (Figure 2B1, black trace). A –100-pA step of negative current induced a sustained hyperpolarization, at the end of which the membrane voltage (Vm) repolarized, and post-inhibitory Na+ spikes appeared (Figure 2B1, red trace). 85% (136/160) of PDB neurons showed post-inhibitory spikes upon similar negative current steps, demonstrating that spikes induced by hyperpolarization are common in PDB neurons (Figure 2B2). As in many other neuronal types (Aizenman and Linden, 1999; Sun and Wu, 2008b; Wang et al., 2016; Kurowski et al., 2018), post-inhibitory spiking is commonly associated with RD, usually masked by post-inhibitory spikes. Inhibition of Na+ spikes with TTX (1 µM), unmasks the post-inhibitory RD (red traces, Figure 2C1). Despite the short duration of the RD, PDB neurons can show persistent post-inhibitory Na+ spikes. A possible mechanism for the persistent post-inhibitory spikes, as observed in the SC optic layer neurons (Lo et al., 1998), is a pacemaker activity created by the balance between the afterhyperpolarization (AHP) and the afterdepolarization (ADP) triggered after each action potential. Bath applications of apamin, inhibiting the AHP, thus unmasking the ADP, increased the number of post-inhibitory Na+ spikes (Figure 2C2–3; mean #spikes: ctrl, 2.5 ± 0.6, ap, 7.5 ± 1.3). Despite these results, the mechanism underlying the persistent post-inhibitory spikes is not yet fully understood. To test the relationship between hyperpolarization amplitude, RD, and post-inhibitory spikes in PDB neurons, we used TTX to isolate the RD and found that the amplitude of the RD correlated linearly with the magnitude of the negative-current step and the hyperpolarization when we injected negative step currents under –200 pA (Wang et al., 2016) (Figure 2D1–2). If the hyperpolarization-induced RD underlies the post-inhibitory spikes, increasing the current step amplitudes should increase the number of post-inhibitory spikes. As expected, in the absence of TTX, we found that the amplitude of the hyperpolarizing step also correlated linearly with the number of RD-evoked Na+ spikes (Figure 2E1–2). These results demonstrate that both post-inhibitory RD and RD-evoked Na+ spikes are common features of PDB neurons and that both the RD amplitude and the number of post-inhibitory spikes are proportional to the magnitude of the preceding hyperpolarization, consistent with reports from other neuronal cell types (Lo et al., 1998; Aizenman and Linden, 1999; Wang et al., 2016).
Figure 2. PDB of the SC show robust post-inhibitory rebound depolarization (RD) and spiking.
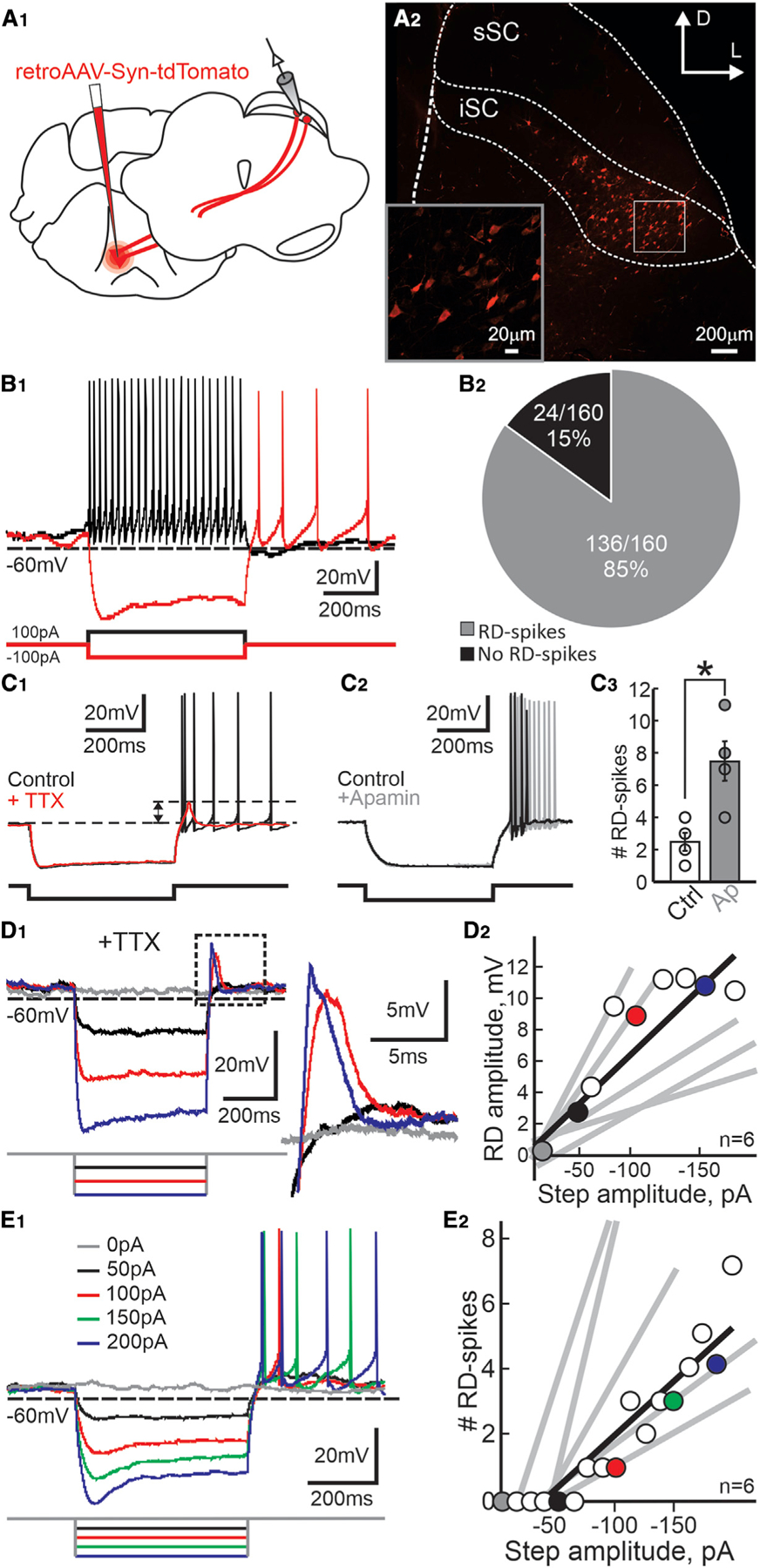
(A1) Schematic of coronal sections of a mouse brain showing the location of viral injections (red) and recordings.
(A2) Confocal image showing the distribution of PDB neurons in the SC (sSC, superficial SC; iSC, intermediate/deep SC). Scale bar, 200 mm. Inset: high magnification of the region indicated by the box. Scale bar, 20 mm.
(B1) Current-clamp traces recorded from a PDB neuron upon 500 ms current injections of 100 pA and –100 pA (back and red traces, respectively).
(B2) Pie chart showing the proportion of PDB neurons showing post-inhibitory spikes in response to 500 ms, –100 pA current injection.
(C1) Current-clamp traces from a single PDB neuron before and after application of TTX (black and red traces, respectively). Dotted lines show the post-inhibitory RD amplitude. Current injection, 500 ms, –100 pA.
(C2) Current-clamp traces recorded from a single PDB neuron before and after bath application of apamin (black and gray traces, respectively). Current injection, 500 ms, –100 pA.
(C3) Quantification of the number of post-inhibitory spikes in PDB neurons before and after application of apamin (white and gray circles and bars, respectively). Current injection, 500 ms, –100 pA. Circles indicate values obtained from individual PDB neurons. Bars, mean ± SE; n = 4, paired t test(3) = 3.54, p = 0.039. Error bars denote SEM. *p < 0.05.
(D1) Current-clamp traces in response to varying amplitude, 500 ms current steps (gray 0 pA, black –50 pA, red –100 pA, blue –150 pA) in the presence of TTX. The area in the dotted black square is magnified on the right.
(D2) Peak amplitude of the RD plotted against the amplitude of the negative step current (mean R2 = 0.82 ± 0.064). Circles and black line correspond to RD amplitude values and linear regression calculated from the neuron in (D1). Colored circles correspond to values from the traces shown in (D1). Gray lines show the linear regressions from five other PDB neurons.
(E1) Current-clamp traces in response to varying amplitude, 500 ms current steps in the absence of TTX showing RD-evoked spikes in PDB neurons.
(E2) The number of RD-evoked spikes plotted against the amplitude of the negative step current (mean R2 = 0.90 ± 0.028). Colored circles correspond to the traces shown in (E1). The black line shows the linear regression calculated from the data obtained from the neuron in (E1). Gray lines show the linear regression calculated for five other PDB neurons.
Optogenetic activation of nigral-inhibitory terminals in the SC induces RD and RD-evoked spiking in PDB neurons
We next sought to investigate whether inhibitory inputs from the nigra into the SC were capable of evoking similar responses in PDB neurons as seen with current injection. We light-activated nigral terminals expressing Chronos opsin in the SC while recording from visually identified PDB neurons (Figures 3A1–3 and S2). Optogenetic stimulation of nigral neurons expressing Chronos increased their spike frequency above spontaneous levels, thus mimicking increases in the nigro-collicular inhibitory drive shown in previous in vivo experiments (Meyer-Luehmann et al., 2002) (Figure S1B1–2). Using patch-clamp recording in ex vivo mouse brain slices, we found that 10-Hz light pulses produced reliable monosynaptic IPSPs in PDB neurons (Figure 3B1); however, 10-Hz light pulses did not evoke RD or spiking in PDB neurons (Figure 3B1–2). On the other hand, 100-Hz stimulation induced a sustained hyperpolarization, often accompanied by robust RDs (Figure 3B3, red trace, and Figure S3). In some PDB neurons, light stimulation produced RD but no spiking (11/29 PDB neurons), perhaps due to the opto-induced hyperpolarization triggering RDs that did not reach the threshold voltage for spiking. As shown in Figure 2E, the likelihood of producing RD-evoked spikes in PDB neurons was proportional to the amplitude of the preceding hyperpolarization. Thus, we reasoned that increasing the amplitude of the opto-induced hyperpolarization should produce reliable RD-evoked spikes. We increased the amplitude of the opto-induced hyperpolarizations by holding the neurons at slightly depolarized Vms, thus increasing the drive of the ions underlying the hyperpolarizations. Injection of small, slow, positive currents inducing subthreshold depolarizations paired with 100-Hz light stimulation of nigral terminals produced reliable post-inhibitory spikes in PDB neurons (Figure 3C1). Note that at more hyperpolarized Vms the light pulses induced a reversal of the opto-induced hyperpolarization (black trace), consistent with an increase in Cl– conductance as a result of GABAA receptor activation by the GABA released from the nigro-collicular terminals (reversal potential, –67.79 ± 0.88 mV; Figure 3C2). The linear correlation between the amplitude of the opto-mediated hyperpolarization and the Vm demonstrates that the same-intensity light pulses produce larger hyperpolarizations at more depolarized Vms (Figure 3C2). Furthermore, as we observed with current injections, the number of post-inhibitory spikes was proportional to the amplitude of the opto-mediated hyperpolarization (Figure 3C3). The currents used to depolarize the Vm were subthreshold and did not evoke spikes before the light pulses (Figure 3D). These results demonstrate that PDB neurons receive monosynaptic inhibitory nigral inputs and that 100-Hz light activation of nigral terminals produces hyperpolarization followed by RD and RD-evoked action potentials in PDB neurons. These findings point toward a previously undescribed mechanism in the SC explaining how activation of inhibitory nigral terminals evokes contralateral movements as observed in vivo.
Figure 3. Optogenetic activation of GABAergic nigral inputs to the SC generates RD-evoked spikes in PDB neurons.
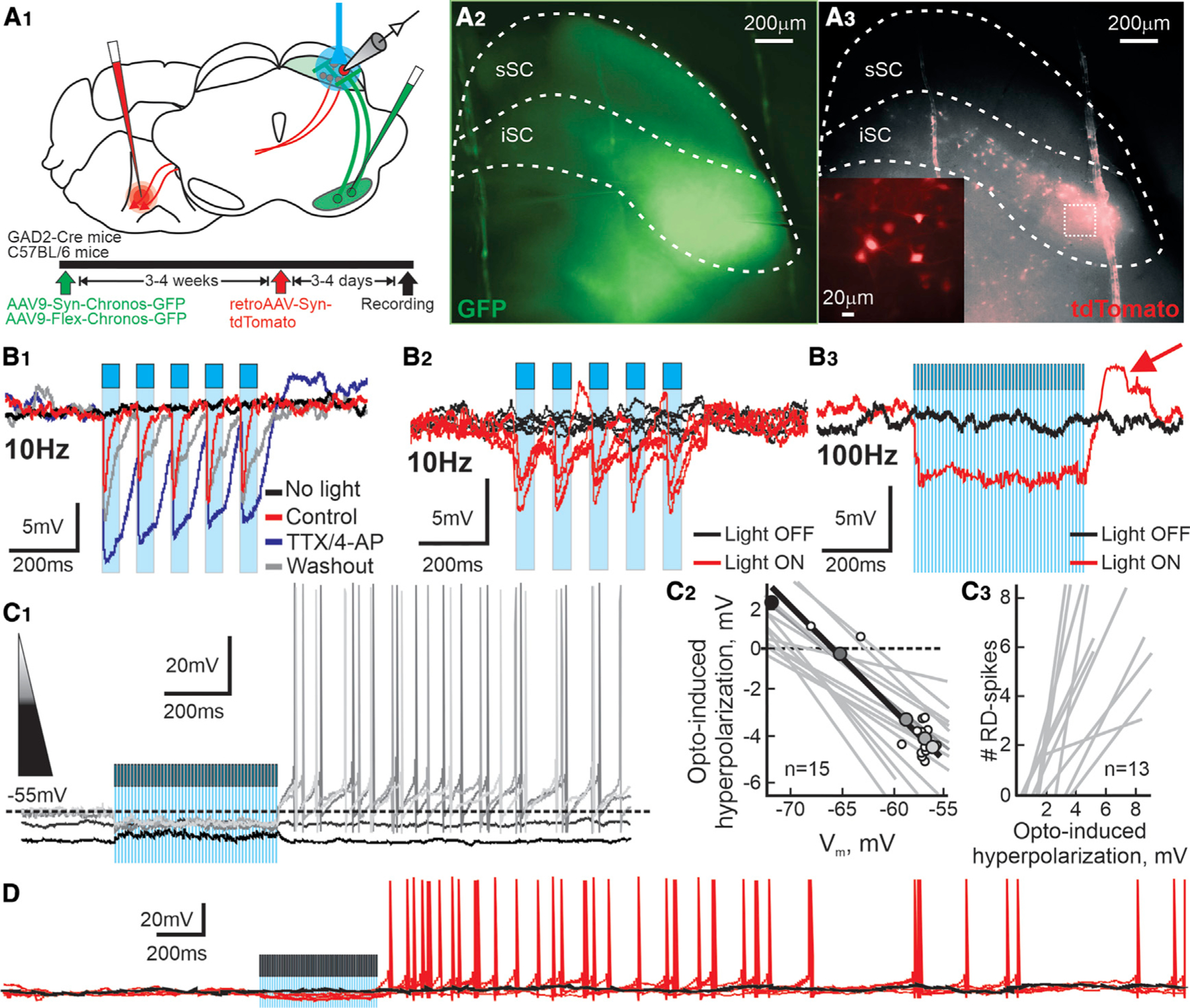
(A1) Schematic coronal sections showing the location of viral injections and recording. Bottom: timeline of experimental procedures.
(A2) Micrograph image of the nigral fibers (GFP, pseudocolored green) in the iSC of a slice used to record PDB neurons. Scale bar, 200 µm.
(A3) Micrograph image of the PDB neurons (tdTomato, pseudocolored red) in the same slice as (A2). Scale bar, 200 µm. Inset: magnification of the area in the white dashed square. Scale bar, 20 µm.
(B1) Current-clamp traces from a PDB neuron showing IPSPs evoked by light pulses (10 Hz) before (red traces, cyan bars), after bath application of TTX/4-AP (blue traces), and after 5 min washout (gray traces).
(B2) Consecutive traces from a single PBD neuron recorded with and without stimulation (red and black traces, respectively; cyan bars, 10-Hz light pulses).
(B3) Traces recorded from the same neuron in (B2) showing voltage response with and without 100-Hz light stimulation (red and black traces, respectively; cyan shading, 100-Hz pulses). The red arrow shows a post-inhibitory RD at the end of the light stimulation.
(C1) Five current-clamp traces recorded from a single PDB neuron at different Vms, indicated by the shaded triangle. Darker traces indicate more hyperpolarized Vms. The cyan shading shows the 500-ms, 100-Hz optogenetic stimulation.
(C2) Plot showing the value of the opto-induced hyperpolarization against the Vm. Shaded circles correspond to traces shown in (C1), and smaller white circles show the rest of the values obtained from the same neuron. The black line shows the calculated linear regression of the neuron in (C1). Gray lines show the linear regressions obtained from 14 other PDB neurons.
(C3) Number of RD-evoked spikes plotted against the amplitude of the opto-induced hyperpolarization in PDB neurons. Gray lines show the linear regressions calculated for 13 PDB neurons.
(D) Extended current-clamp recording from a PDB neuron showing the current before (~1.0 s) and after (~3.0 s) the optogenetic activation of nigral terminals. The figure shows four consecutive traces recorded with and without light stimulation (red and black traces, respectively; cyan shading, 100-Hz pulses).
Features of RD and RD-evoked spiking ex vivo predict features of orienting movements in vivo
We showed that 100-Hz optogenetic stimulation of inhibitory nigral terminals in the SC produced contralateral orienting movements in mice in vivo and post-inhibitory spiking in PDB neurons in brain slices. If post-inhibitory spiking in PDB neurons is the physiological mechanism underlying the in vivo contralateral orienting movements, we reasoned that they should show similar features. First, in slices, 100-Hz but not 10-Hz frequency optogenetic stimulation of nigral terminals induced robust RD and RD spikes (Figure 3B2–3, S3A, and S3B). We predicted that 100-Hz but not 10-Hz unilateral optogenetic activation of nigral terminals of mice expressing Chronos would evoke contralateral orienting movements. As already described, 100-Hz stimulation evoked consistent contralateral rotations, whereas 10-Hz stimulation failed to evoke statistically significant contralateral rotations. Moreover, none of these stimulation patterns evoked ipsilateral rotations (Figure 4A). Thus, only 100-Hz optogenetic stimulation of the nigral terminals in the SC, which evokes robust post-inhibitory RD and RD-evoked spiking in slices ex vivo, triggered contralateral rotations in vivo. 10-Hz stimulation, which induced small RD and did not evoke post-inhibitory spikes ex vivo, was ineffective at producing rotations in vivo.
Figure 4. Features of RD and RD-evoked spiking ex vivo predict features of orienting movements in vivo.
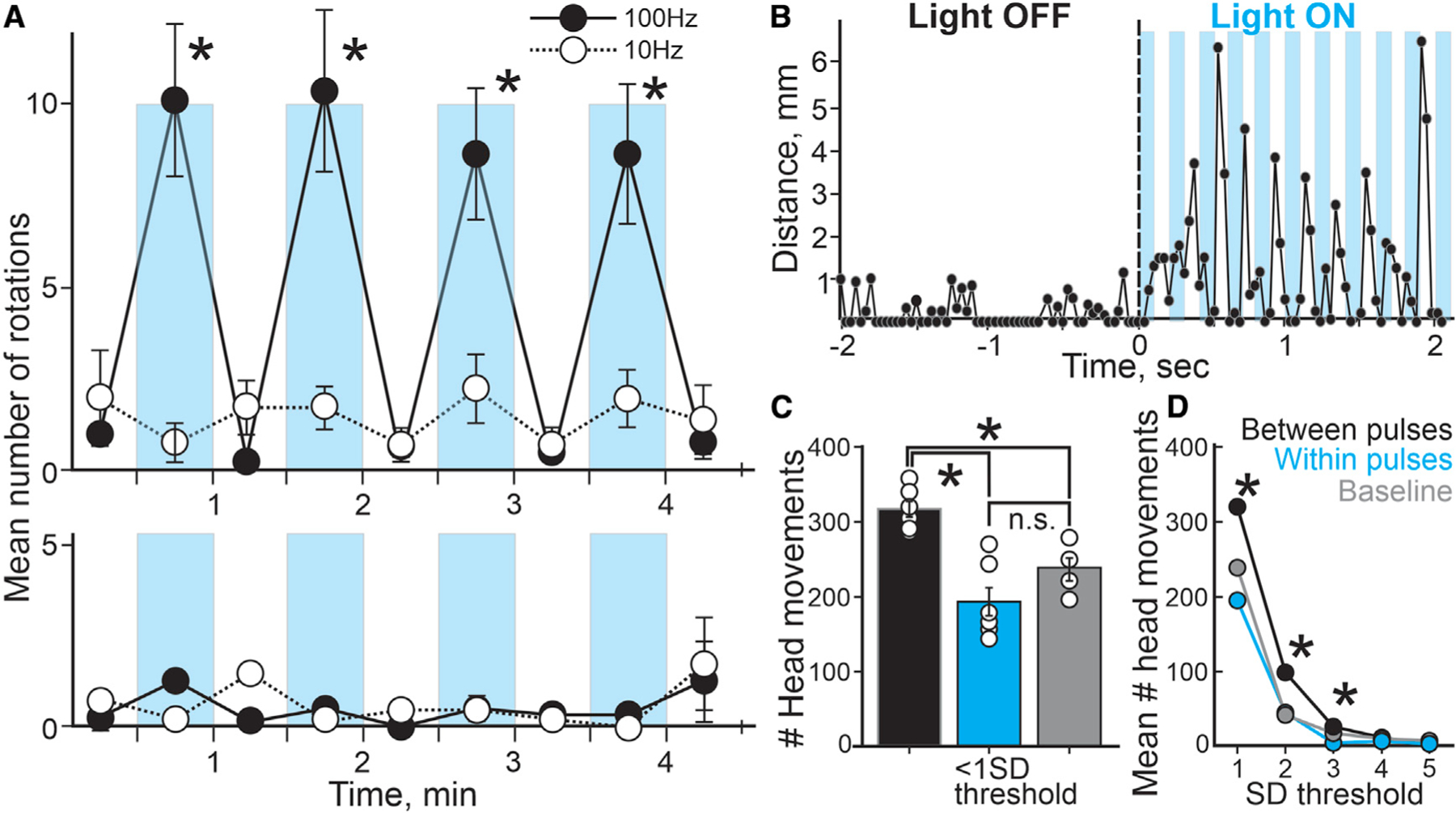
(A) Mean number of rotations (contralateral and ipsilateral; top and bottom, respectively) evoked with different stimulation frequencies (100 Hz [●], 10 Hz [○]) counted in 30-s epochs during the behavioral testing plotted over time. Cyan bars and white spaces represent the LIGHT ON/OFF epochs, respectively (n = 4; 100 Hz: 2 3 2 repeated-measures ANOVA f(3,24) = 39.00, p < 0.0001; 10 Hz: 2 × 2 repeated-measures ANOVA 10 Hz: f(3,24) = 2.88, p = 0.0603).
(B) Distance of the head position of the mouse recorded between consecutive video frames during a 4-s sample before (LIGHT OFF) and during 100-Hz optogenetic stimulation of nigral terminals (LIGHT ON). Each cyan bars represent the 100-ms light pulses during the stimulation.
(C) Plot showing the mean number of head movements (<mean±1SD) measured for each epoch (between pulses, within pulses, and baseline; black, cyan, and gray bars, respectively) recorded during the behavioral sessions. White circles indicate the mean number of head movements recorded for each mouse (n = 6; between versus within versus baseline: One-way ANOVA (2,12) = 12.62, p = 0.0011, post hoc Turkey HSD; within versus baseline: ANOVA, post hoc Turkey HSD: 1SD, p = 0.288; 2SD, p = 0.999; 3SD = p = 0.391.
(D) Mean of the number of head movements of different lengths recorded between video frames for each epoch (n = 6; One-way ANOVA, post-hoc Turkey HSD: 1SD, p = 0.00095; 2SD, p = 0.00104; 3SD, p = 0.0315). Error bars denote SEM throughout. *p < 0.05.
Another feature of the post-inhibitory spikes in ex vivo slices is that they appear at the end of the hyperpolarization of PDB neurons. If the orienting movements evoked after the light activation of the nigro-collicular circuit are underlain by RD-evoked spiking in PDB neurons, we predict they should present similar onsets with respect to the light pulses. A slowed-down video clip of ~1 s before and after the initiation of 100-Hz light stimulus shows a delay in the initiation of the orienting movements with respect to the light stimulus (Video S2). To confirm this, we tracked the head position of the mouse in the video recordings frame by frame and tested when the movements occurred with respect to the light pulses (Figure S4; STAR Methods). We reasoned that if the initiation of the movement occurred synchronously with the onset of the light pulses, most of the head movements should appear in the video frames recorded within the light pulses. On the other hand, if the initiation of the movements occurred at the end of the light pulses, consistent with post-inhibitory spiking, most of the head movements should occur in the video frames in between the light pulses. We found that most of the movements during the light-ON epoch occurred in between the light pulses compared with both within the light pulses and during baseline (Figures 4B and 4C; between pulses, 316.8 ± 10.5, within pulses, 194.0 ± 19.0, baseline, 236.5 ± 15.4). Even though there were a considerable number of movements recorded within the light pulses, they were not statistically different from the number of movements recorded during the baseline, when mice performed random exploratory movements without the light stimulation. We found similar results for larger movements (up to mean ± 3SD), thus indicating that most of the mouse head movements measured during the light-ON stimulation period occurred in between the light pulses, demonstrating a delay in the initiation of the light-evoked movements with respect to the onset of the light pulses.
Taking these results together, we conclude that optogenetic activation of inhibitory nigral terminals in the SC results in contralateral head movements with onsets aligned to the offset of the light stimulation, consistent with the hypothesis that activation of the inhibitory nigro-collicular circuit evokes contralateral orienting movements mediated by post-inhibitory spiking in PDB neurons of the SC.
T-type Ca2+ channels underlie the post-inhibitory RD in PDB neurons of the SC
Having shown that unilateral optogenetic activation of the mouse nigro-collicular circuit generates contralateral rotations in vivo, likely due to post-inhibitory RD and spiking in PDB neurons, we next aimed to investigate the ionic mechanism underlying the post-inhibitory RD and spiking. Previous studies showed that two different channels, the hyperpolarization-activated cyclic nucleotide-gated channel (HCN) and the T-type Ca2+ channels, contribute to generating post-inhibitory RD and spiking in many neuronal types (Kim et al., 2001; Perez-Reyes, 2003; Wang et al., 2016). We sought to investigate whether these same channels were involved in triggering the post-inhibitory RD and spiking in PDB neurons. We first assessed whether PDB neurons exhibit a voltage sag upon negative current injections, a known trademark of the presence of Ih, an inward current generated by HCN channels (McCormick and Pape, 1990; Maccaferri and McBain, 1996; Pape, 1996). We found that only a small fraction of PDB neurons showed significant sags and that the sag amplitude was smaller than that recorded in wide-field vertical (WFV) neurons (Figures S5A–S5D; PDB, 21.6% [8/37]; WFV, 89% [33/37]) (Endo et al., 2008). Furthermore, in PDB neurons showing sags, the HCN channel blocker ZD7288 (ZD, 50 µM) completely reduced its amplitude (Figures 5A and 5B; mean sag amplitude: control, 3.32 ± 0.46 mV; ZD, –0.17 ± 0.66 mV) but failed to inhibit both the post-inhibitory RD (Figure 5C; mean RD amplitude: control, 4.057 ± 0.54 mV, ZD, 4.54 ± 0.80 mV) and the post-inhibitory spikes (Figure 5D). We conclude that HCN channels are unlikely to significantly contribute to the post-inhibitory RD and spiking in PDB neurons.
Figure 5. HCN channels do not mediate the post-inhibitory RD or the RD-evoked spikes in PDB neurons.
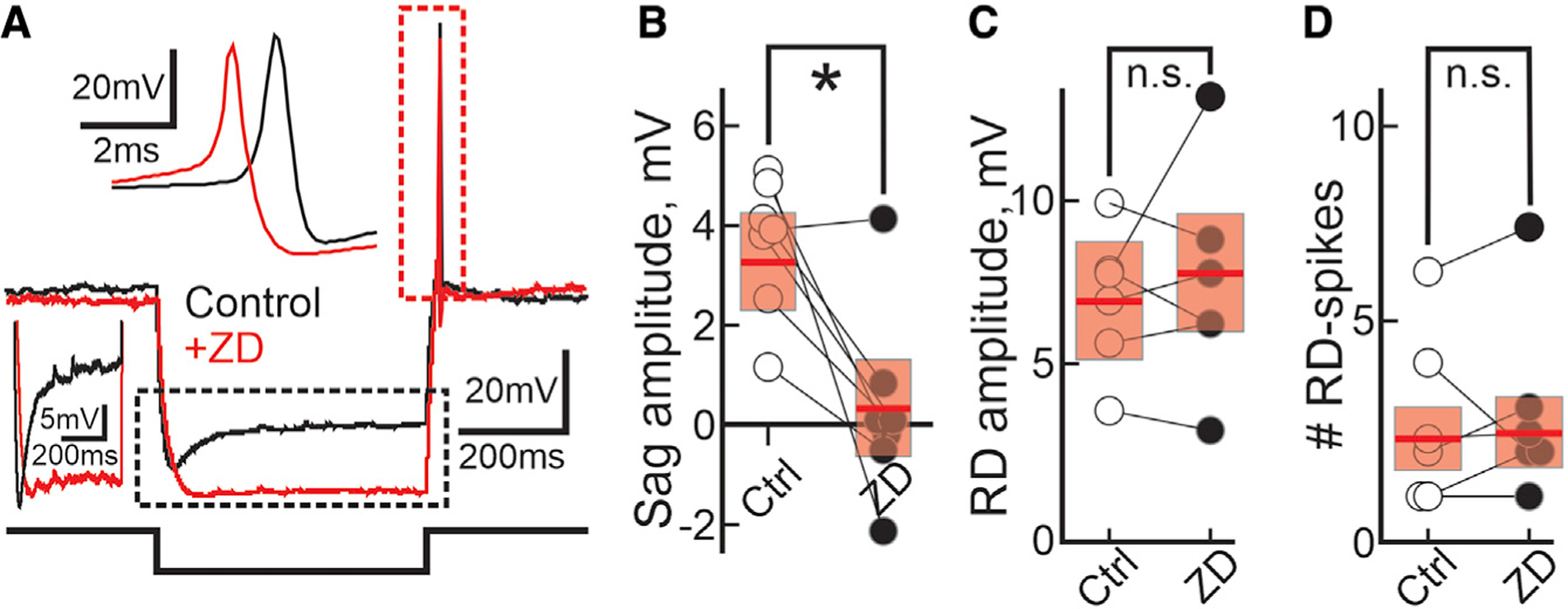
(A) Current-clamp traces from a single PDB neuron showing the changes in voltage before and after bath application of the HCN channel blocker ZD 7288 (black and red traces, respectively). Current injection, 500 ms, –100 pA. The part of the trace outlined by the dotted black rectangle is compressed to the left. The top inset expands the part of the trace outlined by the red dotted rectangle.
(B and C) Plot depicting the amplitude of the sag (n = 7; paired t test(6) = 3.57; p = 0.011) and the RD (n = 6; paired t test(5) = –0.69; p = 0.53) recorded from PDB neurons before and after bath application of ZD (white and black circles, respectively). Current injection, 500 ms, –100 pA. Red bars, mean ± SEM; *p < 0.05; n.s., not significant.
(D) Plot showing the number of RD-evoked spikes before and after ZD application (white and black circles, respectively). Current injection, 500 ms, –100 pA. Red bars, mean ± SE. n = 6; paired t test(5) = −0.35; p = 0.37.
We next asked whether T-type Ca2+ channels underlie the RD leading to post-inhibitory Na+ spiking in PDB neurons as they appear to do in other neuronal cell types (Llinás and Jahnsen, 1982; Jahnsen and Llinás, 1984a, 1984b; Lo et al., 1998; Person and Raman, 2012; Wang et al., 2016). After T-type Ca2+ channel de-inactivation during the post-inhibitory repolarization of the membrane, Ca2+ currents appear at Vms above –60 mV (Perez-Reyes, 2003). If T-type Ca2+ channels underlie the appearance of RD and RD-evoked Na+ spiking in PDB neurons, they should be modulated by changes in the membrane voltage in a manner consistent with the de-inactivation and activation patterns of T-type Ca2+ channels. We first recorded from PDB neurons at different Vms, by applying slow constant current. When held at hyperpolarized Vm (–67 mV), a negative-current step (500 ms, –100pA), normally sufficient to evoke post-inhibitory RD and RD-evoked spiking in PDB neurons, evoked a sustained hyperpolarization, a small RD, and no RD-evoked spiking (Figure 6A, bottom, black trace). At this Vm, the membrane repolarization did not reach a potential capable of de-inactivating/activating a significant number of T-type Ca2+ channels, hence the low amplitude of the RD and the lack of RD-evoked spikes. At more depolarized Vms, the same negative-current step induced the triggering of RD-evoked spikes, likely due to the activation of a larger number of T-type Ca2+ channels (Figure 6A, lighter traces). We then tested how different features of RD and RD-evoked spikes changed at different Vms. We found that in PDB neurons all the parameters tested (except for the hyperpolarization amplitude) correlated linearly with changes in Vm (Figure 6; Hyper, R2 = 0.13 ± 0.048; first RD-spike latency, R2 = 0.69 ± 0.12; RD, R2 = 0.73 ± 0.076; # of RD spikes, R2 = 0.66 ± 0.096). These results show that, although the amplitude of the hyperpolarization remained constant, depolarizing Vms increased the RD amplitude, reduced the latency of the first RD-evoked spike, and increased the number of spikes elicited by the negative-current step, consistent with increased activation of T-type Ca2+ channels (Aizenman and Linden, 1999).
Figure 6. Features of the post-inhibitory RD in PBD neurons.
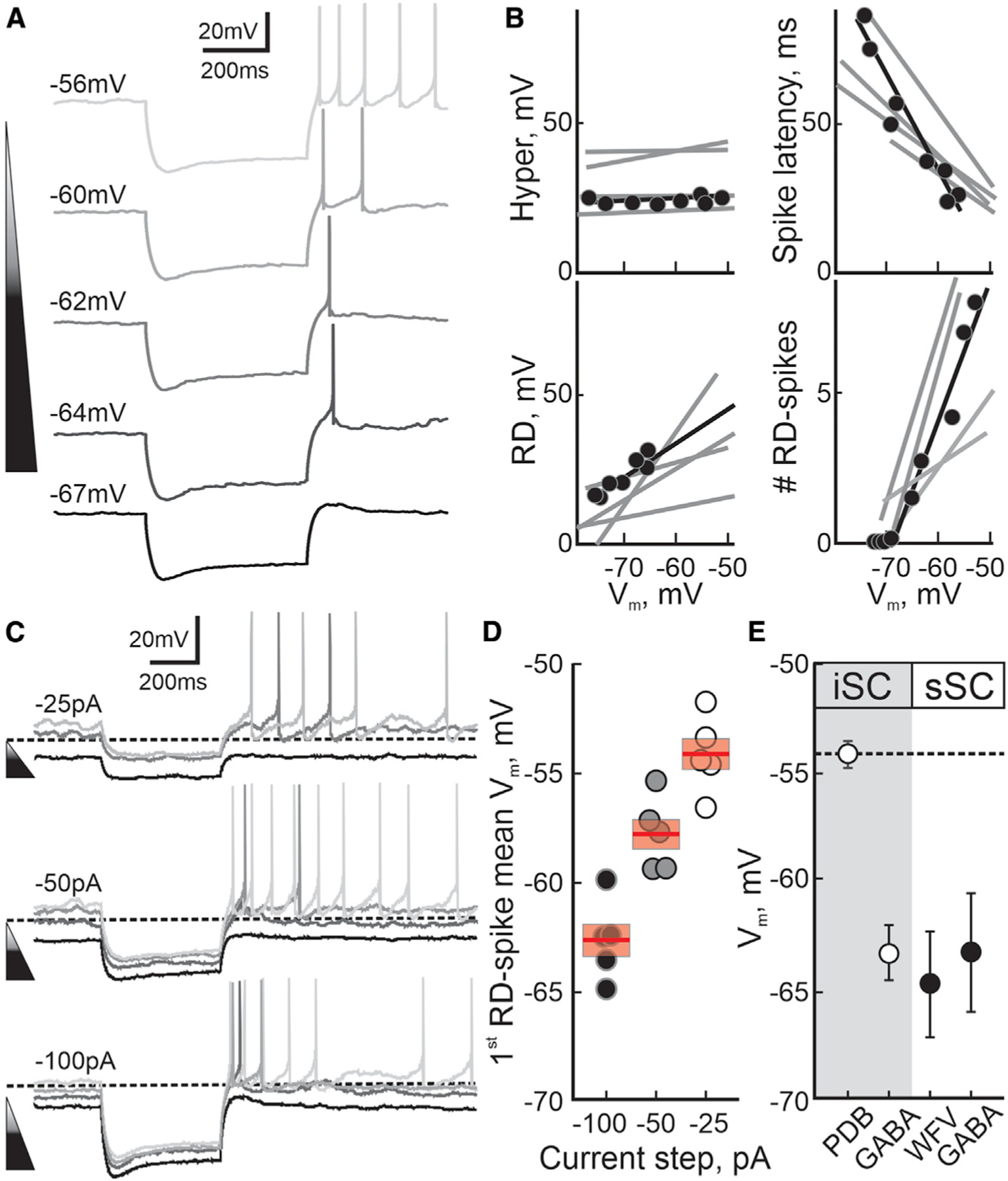
(A) Current-clamp traces from a single PDB neuron showing the voltage response to 500 ms, –100 pA negative-current steps at different Vms. The shade of triangle and traces indicate the Vm from hyperpolarized, –67 mV (darker) to more depolarized –56 mV (lighter).
(B) Electrophysiological parameters of RD in PDB neurons. The black line and circles represent the data and linear regression obtained from the PDB neuron shown in (A). The gray lines show the linear regressions calculated for four other PDB neurons. The top left and right: magnitude of the hyperpolarization induced by the negative current injections (Hyper; t test(5) = 2.51; p = 0.054) and the latency of the first RD spike (spike latency; t test(5) = 6.22, p = 0.01). The bottom left and right panels show the RD amplitude (t test(5) = 9.80, p = 0.001) and the number of RD-evoked spikes (t test(5) = 6.28, p = 0.002).
(C) Current-clamp traces for a PDB neuron at different Vms in response to 500 ms negative-current steps of varying amplitudes –25 pA, –50 pA, and –100 pA (top, middle, and bottom traces, respectively). The shade of traces represents Vms from hyperpolarized (darker) to depolarized (lighter). The dotted line is –60 mV for calibration.
(D) Plot depicting the Vm at which the first RD-evoke spike appears at the end of the hyperpolarization induced by either 500 ms –100 pA ( ), –50 pA (
), –50 pA ( ), or –25 pA (
), or –25 pA ( ) negative-current steps (n = 5). Red bar, mean ± SE.
) negative-current steps (n = 5). Red bar, mean ± SE.
(E) Plot depicting the mean ± SEM values of the resting Vms for different neuronal types in the SC (iSC, white circles: PDB, n = 161; GABA, n = 30. sSC, black circles: WFV, n = 6; GABA, n = 10).
Next, to assess how both the Vm and the hyperpolarization amplitude modulated the RD-evoked spikes, we recorded from PDB neurons at different Vms while applying varying negative current steps. We found that a 500-ms, –25-pA step current induced a small hyperpolarization, RD, and no RD-evoked spikes (Figure 6C, top, black traces). The same current step, at more depolarized Vms, increased the RD amplitude and eventually generated RD-evoked spikes (Figure 6C, top, gray traces). We found similar results with a –50-pA current step (Figure 6C, middle traces). A larger step current of –100 pA resulted in a larger hyperpolarization and RD-evoked spikes even at hyperpolarized Vms compared with the two previous step currents (Figure 6C, bottom traces). These results show that smaller hyperpolarizations can evoke RD-evoked spikes at depolarized Vms, whereas larger hyperpolarizations triggered RD-evoked spikes even at hyperpolarized Vms, consistent with both the hyperpolarization amplitude and the Vm modulating the number of T-type Ca2+ channels deactivated/activated in PDB neurons (–100 pA = –62.41 ± 0.81 mV, –50 pA = –58.69 ± 0.39, –25 pA = –54.67 ± 2.052 mV).
Although present in other neuronal types, T-type Ca2+ channel activation producing the RD is likely to be relevant in PDB neurons (Figures S5E and S5F), given the more depolarized resting Vm, making these neurons more likely to evoke post-inhibitory spikes compared with other SC neuronal cell types (Figure 6E; mean Vm: iSC- PDB, –54.85 ± mV, GABA, –63.74 ± 1.38; sSC- WFV, –65.27 ± 2.60 mV, GABA, –63.72 ± 2.90 mV).
To directly test whether T-type Ca2+ channels underlie the RD and RD-evoked spiking in PDB neurons, we recorded the voltage responses of PDB neurons in ex vivo brain slices after the application of either NiCl2 (Lee et al., 1999; Obejero-Paz et al., 2008; Kim et al., 2017; Toda et al., 2017) or NNC 55–0396, a broad spectrum, and a selective T-type Ca2+ channel blocker. Negative-current injections induced hyperpolarizations followed by RD and RD-evoked spikes, both of which were selectively suppressed by bath application of NiCl2, without affecting the Ih (Figures 7A1–3, S5G, and S5H; mean RD: control, 11.30 ± 1.90 mV, + NiCl2, 1.55 ± 0.67 mV; mean # RD spikes: control, 3.6 ± 1, + NiCl2, 0). Application of the selective T-type Ca2+ channel blocker NNC 55–0396 also suppressed both the post-inhibitory RD and the RD-evoked spikes in PDB neurons (Figure 7B1–3; mean RD: control, 15.10 ± 2.56, + NNC, 3.23 ± 0.91; mean #RD spikes, control, 2.78 ± 0.54, + NNC, 0). On the basis of these results, we conclude that T-type Ca2+ channels underlie the post-inhibitory RD that promotes post-inhibitory Na+ spikes in PDB neurons.
Figure 7. T-type Ca2+ channels underlie the post-inhibitory RD and spiking in PDB neurons and mediate contralateral orienting movements.
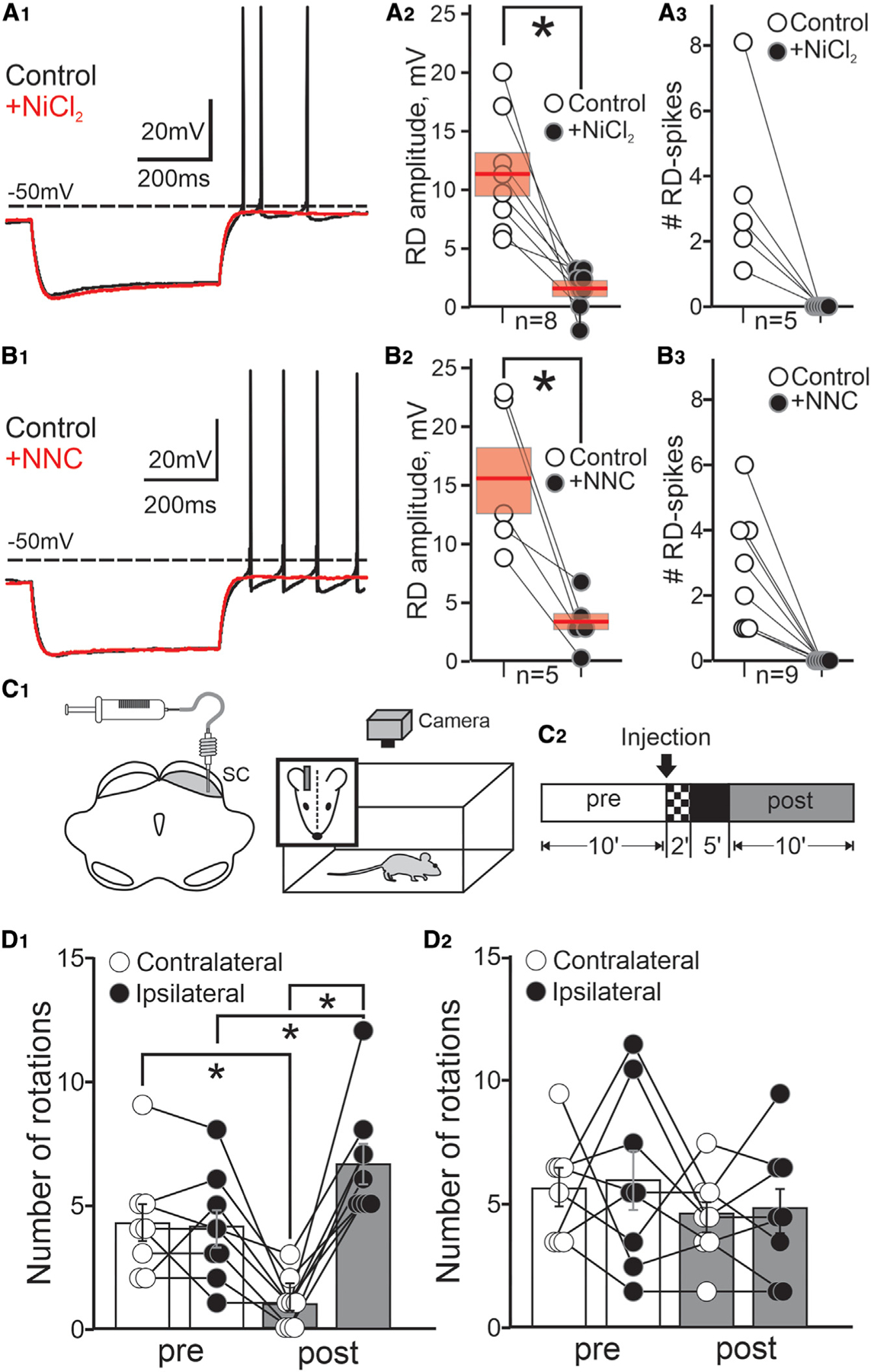
(A1) Current-clamp traces showing the changes in voltage before and after application of 500 mm NiCl2 (black and red traces, respectively). Current injection, 500 ms, –100 pA.
(A2) Plot showing the RD amplitude induced by 500 ms, –100 pA step currents in PDB neurons before ( ) and after application of NiCl2 (
) and after application of NiCl2 ( ). Red bars, mean ± SE; n = 8; paired t test(7) = 3.82, p = 0.0087.
). Red bars, mean ± SE; n = 8; paired t test(7) = 3.82, p = 0.0087.
(A3) Plot showing the number of RD-evoked spikes after 500 ms, –100 pA current step in PDB neurons before ( ) and after bath application of NiCl2 (
) and after bath application of NiCl2 ( ; n = 5; t test(4) = 0.98, p = 0.020).
; n = 5; t test(4) = 0.98, p = 0.020).
(B) Same as in (A) but using NNC 55–0396 (+NNC; 1 mM; red traces and black circles). RD amplitude, n = 6; t test(5) = 3.414, p = 0.042; #RD spikes, n = 10, t test(9) = 4.856, p = 0.0013.
(C1) Schematic of a mouse brain coronal section showing the injection site targeting the SC intermediate/deep layers. Right:schematic of the behavioral procedure.
(C2) Time course of the behavioral experiment (STAR Methods).
(D1) Plot showing the number of contralateral and ipsilateral rotations (white and black circles, respectively) relative to the injection side recorded pre- and post-injection (white and gray bars, mean ± SE) of the specific T-type Ca2+ blocker NNC 55–0396; n = 8; contra: t test(7) = 4.33, p = 0.0017; ipsi: t test(7) = −2.38, p = 0.025.
(D2) same as in (D1) for saline control injections; n = 8; contra: t test(7) = 1.31, p = 0.22; ipsi: t test(7) = 1.67, p = 0.14. Error bars denote SEM, throughout. *p < 0.05.
Finally, to provide direct and causal evidence that T-type Ca2+ channels, responsible for the post-inhibitory RD in PDB neurons, play a role in the generation of contralateral orienting movements in vivo, we unilaterally blocked the activity of T-type Ca2+ channels by injecting NNC 55–0396 in the SC of mice while tracking their behavior in an open field (Figure 7C1–2 and S6A). We reasoned that if the generation of contralateral orienting movements depended on the activation of T-type Ca2+ channels and the resulting RD-evoked spiking of PDB neurons, then unilaterally blocking these channels should decrease the occurrence of contralateral orienting movements. Unilateral injections of NNC 55–0396 into the intermediate/deep layers of the SC significantly reduced the number of spontaneous contralateral rotations and increased the number of ipsilateral rotations compared with similar recording times before the injection (Figure 7D1 and Video S3; # contra rotations: pre, 4.25 ± 0.74, post, 1.00 ± 0.35; ipsi rotations: pre, 4.13 ± 0.74, post, 6.63 ± 0.81). Meanwhile, unilateral saline injections had no discernable effect on post-injection rotations (Figure 7D2). As a positive control, we injected muscimol, a GABAA receptor agonist. As expected, muscimol injections significantly reduced the number of spontaneous contralateral rotations and increased the number of spontaneous ipsilateral rotations (Figure S6B). Interestingly, muscimol increased the number of ipsilateral rotations to a greater extent than did injection of NNC 55–0396 (Figure S6C). We suspect that this effect may result from muscimol affecting multiple neuronal cell types within the SC in addition to the PDB neurons, likely the inhibitory commissural neurons. In contrast, NNC 55–0396 appears to preferentially impact post-inhibitory RD, a feature of PDB neurons. Two independent human raters, blinded to the conditions, obtained similar scores, thus validating the results obtained by the automatic tracking system (STAR Methods and Figures S6D and S6E).
On the basis of these combined findings, we conclude that activation of T-type Ca2+ channels plays a causal role in mediating contralateral orienting movements in mice, and thus, in addition to BG disinhibition, post-inhibitory RD and RD-evoked Na+ spiking may also play an important role in the control of orienting behavior.
DISCUSSION
We used targeted optogenetics to activate the inhibitory afferents arising from the nigra and terminating in the ipsilateral SC of mice. Unexpectedly, we found that unilateral activation of the inhibitory nigral afferents in the SC in vivo produced contralateral orienting movements rather than suppressing them, as the model of BG function based on disinhibition predicts. Ex vivo experiments in brain slices showed that the same stimulus evoking contralateral rotations in vivo (100 Hz) produces a strong hyperpolarization followed by post-inhibitory RD mediated by T-type Ca2+ channels and subsequent Na+ spikes in PDB neurons. Lower-frequency stimulations (10 Hz) failed to evoke orienting movements in vivo as well as post-inhibitory RD and RD spiking in ex vivo PDB neurons. Unilateral pharmacological blockade of T-type Ca2+ channels in the SC in vivo reduced spontaneous contralateral rotations and biased the rotations toward the ipsilateral side. Taking all that together, we propose that, in addition to disinhibition from the nigro-collicular circuit, a second mechanism for producing orienting movements may be the direct activation of PDB neurons through inhibitory nigral terminals via post-inhibitory RD and RD-evoked spiking.
Relationship to previous studies
Our use of the GAD2-Cre mouse and the opsin Chronos allowed us to specifically activate the nigral inhibitory projections in the SC—avoiding spurious activation of the adjacent dopaminergic neurons of the substantia nigra pars compacta—with tight temporal precision, thus, driving the activity of nigral axonal projections at frequencies matching the in vivo discharge rates of the nigral neurons (Meyer-Luehmann et al., 2002). Other areas such as the GABAergic zona incerta (ZI) also project to the SC (Kim et al., 1992; Watson et al., 2015; May et al., 2018; Doykos et al., 2020; Hormigo et al., 2020). Our results, however, are unlikely attributable to the ZI projection, located ~1.0 mm anterior and ~2 mm dorsal to and at a shallower angle than our injection locations. Furthermore, despite the abundant inhibitory projections from the ZI to the SC, the projection strength to PDB neurons in mice appears to be minimal (Doykos et al., 2020). Finally, because we stimulated axons located within the SC, we can also rule out interpretations based on the spread of the virus to other midbrain neurons that do not project to the SC, as would be a concern if we stimulated nigral neuronal cell bodies directly.
To our surprise, we found that light activation of the inhibitory nigro-collicular circuit led to movement generation. According to the current model of BG function, activation of the nigro-collicular circuit should suppress contralateral movements, as seen with local injections of muscimol into the SC (Hikosaka and Wurtz, 1983a, 1983b, 1985a, 1985b; Boussaoud et al., 1985). Optogenetic stimulation of nigral afferents offers several advantages to test the function of the nigro-collicular circuit compared with muscimol injections. First, optogenetic activation is a direct causal test of the role of the nigro-collicular circuit, whereas muscimol injections provide circumstantial evidence, since the SC receives inhibitory inputs from other brain areas in addition to the nigra; so muscimol effects could be as well explained by these other projections. Second, the SC contains inhibitory interneurons that may be affected by muscimol injections but are avoided by targeted activation of nigral afferents. In fact, our observation that muscimol affected ipsilateral movement more than the T-type Ca2+ channel blocker (Figure S6C) is consistent with the idea that muscimol impacts neuronal elements beyond PDB neurons. Third, muscimol is a potent GABAA agonist with long-lasting effects, leaving neurons deeply hyperpolarized for hours (Hikosaka and Wurtz, 1985a, 1985b; Martin and Ghez, 1999; Edeline et al., 2002). Optogenetic activation, in contrast, allows for tighter temporal control of the stimulation and GABA release, which, as our results showed, is needed for the triggering of post-inhibitory RD, and RD-evoked spiking in PDB neurons. The slow effect of muscimol injections in the SC would not provide the temporal resolution to activate the T-type Ca2+ channels and unveil the post-inhibitory RD and RD-evoked spiking in PDB neurons.
Post-inhibitory RD and RD-evoked spiking are found in many neuronal cell types including cerebellar nuclei neurons, thalamic neurons, and WFV neurons of the superficial SC (Person and Perkel, 2005; Mitra and Miller, 2007; Endo et al., 2008; Sun and Wu, 2008a; Hirano and Kawaguchi, 2014; Wang et al., 2016; Chang et al., 2018). Electrical activation of the avian homolog of the mammalian pallidal nucleus, Area X, results in post-inhibitory RD and spiking in DLM neurons, the avian homolog of the mammalian motor thalamus (Person and Perkel, 2005, 2007). Follow-up experiments, however, suggest that rebound spiking in the avian thalamus may stem from coincident excitation from the cerebral cortex (Goldberg and Fee, 2012; Goldberg et al., 2012, 2013). In birds, the coincident activation observed using simultaneous neuronal recordings in Area X and DLM appeared during bouts of singing (Goldberg and Fee, 2012; Goldberg et al., 2012). In our experiments, optogenetic activation of nigral afferents occurred randomly, not correlated with any specific behavior associated with coincident cortical drive. Moreover, the stimulations often occurred at rest, presumably when cortical drives would also be minimal. Our results, however, do not exclude the possibility that excitatory cortical inputs may also trigger orienting movements during silencing of nigral inhibition. Our results do, however, provide strong evidence that nigral inhibition is sufficient to drive spiking in PDB neurons and produce orienting movements. Thus, both disinhibition and active post-inhibitory rebound mediated by T-type Ca2+ channels are likely to play a role in driving orienting movements.
Post-inhibitory RD and RD-evoked spiking, dependent on activation of T-type Ca2+ channels, appear in neurons of the mouse ventrolateral thalamus and are associated with neck and limb muscle contractions (Kim et al., 2017). These experiments suggest that RD-evoked spikes resulting from BG inhibitory input to the thalamus play an active role in the control of movement and are not just associated with abnormal movement (Edgerton and Jaeger, 2014; Kim et al., 2017). Our results are consistent with this and show that the inhibitory input from the BG to the SC can play an active role in normal movement generation. Indeed, a role for nigral inhibition and post-inhibitory spiking in PDB neurons helps explain a puzzling result in the monkey, using electrical stimulation of the nigra. Electrical stimulation of the nigra in monkeys does not suppress visually guided saccades but rather results in a reduced and less variable saccadic reaction time, consistent with the synchronization of a large population of PBD neurons (Basso and Liu, 2007; Liu and Basso, 2008). Moreover, simultaneous recordings of SC neurons, together with electrical activation of the nigra in monkeys, reveal a transient pause in SC saccade-related neurons followed by a rapid activity rebound (Liu and Basso, 2008), consistent with a hyperpolarization followed by RD-evoked spiking reported here.
Expanding the role of the nigro-collicular circuit in orienting behavior
The correlation between nigral pauses and SC bursts, together with findings from reversible inactivation studies, led to a model in which the pause of nigral neuronal activity served as a gate, allowing descending cortical excitatory inputs to the SC to drive the choice of a saccade (Sato and Hikosaka, 2002). Since those original experiments, recordings from nigral neurons, as well as pallidal neurons of the BG, reveal that the activity of BG output neurons is much more nuanced during saccades than the original gating model predicts (reviewed in Basso and Sommer, 2011). For example, some nigral neurons pause their activity as soon as a visual stimulus appears until the end of a saccade (Handel and Glimcher, 1999) while some show graded decreases in activity as saccade probability changes (Basso and Wurtz, 2002). Other nigral neurons show increases in activity around the time of a saccade (Handel and Glimcher, 1999). It is unclear how these varied response profiles from BG neurons would fit into the traditional disinhibition model of orienting movement and, together with recent studies in mice, indicate that nigral influences on the SC are much more complicated than the original model posed (Antal et al., 2014; Essig et al., 2021; Sans-Dublanc et al., 2021). For example, recent work shows that nigral neurons in rodents can discharge transiently at rates >200 spikes/s in association with grooming behavior (Meyer-Luehmann et al., 2002). How these increases in activity fit in with models of movement based on disinhibition is only beginning to be explored.
Recent work from the limb movement system is also changing the way we view the role of the inhibitory output of the BG. Rather than working in a push-pull antagonist framework, the increases and decreases in BG output nuclei activity are thought to work cooperatively to generate actions (Tecuapetla et al., 2016) and even to control the vigor of movement (Yttri and Dudman, 2016). Our results suggest that similar cooperative mechanisms may be at play in the orienting movement system controlled by the nigro-collicular circuit. One possibility is that the nigral neurons showing transient increases in activity work together with neurons that show pausing in activity. The coordinated action of these two neuronal types could lead to enhanced spiking and synchronization of a large population of SC neurons driving an orienting movement through RD as well gating of excitatory inputs. A second possibility is that the pause in nigral activity combined with excitatory cortical (and/or cerebellar) inputs would generate subthreshold depolarizations that create a temporal window in which the likelihood of spiking is increased, similar to what we observed ex vivo with changes in Vm. The precise timing of the inhibition and excitation would then provide a mechanism for enhancing the signal-to-noise ratio in the transformation of sensory signals to motor output. A third, not mutually exclusive possibility, is that the end of the pause in nigral neurons provides an important signal to SC neurons that also serves to synchronize the SC population activity to ensure a fast, coordinated orienting movement, similar to that seen in deep cerebellar nuclei in response to coordinated and synchronized input from inhibitory Purkinje neurons (Person and Raman, 2012).
Limitations of the study
The results presented here are robust and employ multiple experimental methods, all with results consistent with the hypothesis that activation of nigral afferents produces RD and RD-evoked spiking in PDB output neurons of the SC. These experimental results, however, do not rule out a role for the excitatory drive from the cerebral cortex to PDB neurons directly in the control of orienting; they show only that disinhibition is not the only mechanism. The experimental results provide information about the input from the nigra to the PDB neurons of the SC, when we know that nigral afferents also target inhibitory interneurons in SC (Kaneda et al., 2008). Thus, to show definitively that the mechanism of RD plays a causal role in the generation of orienting movements, additional in vivo experiments in which we record from molecularly characterized neuronal cell types within the SC while activating nigral afferents will be crucial.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Michele A. Basso (mbasso@uw.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
Custom MATLAB scripts developed by our laboratory to analyze mouse data are available at (https://zenodo.org/badge/latestdoi/472946729).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals and surgery
We used C57BL6 wild-type mice (The Jackson Laboratory, stock no. 000664) and GAD2-IRES-Cre knock-in mice (The Jackson Laboratory, stock no. 010802). Forty-two (10–12-week-old) mice were used in the slice experiments and 12 (10-week-old) mice were used in the behavioral experiments. Both female and male mice were used throughout the study. All mice were maintained on a reverse circadian 12-h light/12-h dark cycle with food and water provided ad libitum. All surgical procedures were performed using aseptic techniques and were performed using general anesthesia. Post-surgical analgesia was provided, and the animal recovered from surgery for at least 5 days before experiments commenced. All experimental protocols involving mice were approved by the UCLA Chancellor’s Animal Research Committee and complied with standards set by the Public Health Service policy on the humane care and use of laboratory animals, as well as all state and local guidelines.
METHOD DETAILS
AAV vectors
For optogenetic activation of the nigral terminals in the SC (nigro-collicular), AAV9-Syn-Chronos-GFP (UNC, AV6102C), AAV9-Flex-Chronos-GFP (UNC, AV65553) were injected into the nigra of C57BL6 wild type or GAD2 mice. We used the latter mice to target the GABAergic nigral neurons specifically. We used Chronos to excite the nigral afferents in the SC due to its fast kinetic activity (Klapoetke et al., 2014) which allowed us to modulate the levels of nigral inhibition in the SC precisely, mimicking the wide range of activity of nigral neurons (Meyer-Luehmann et al., 2002). We used AAV9-Syn-EGFP (Addgene, 50465-AAV9) as expression control (blank virus). For retrograde labeling of PDB neurons, we used AAV2-retro-CAG-tdTomato (UNC, AV7643B).
Stereotaxic injections
Mice were anesthetized with isoflurane (5.0% induction, 1.5–2.0% maintenance) and placed in a stereotaxic apparatus (Kopf Instruments, CA USA). A midline incision exposed the skull, and the periosteum was removed. The skull was leveled using a digital display console (Kopf Instruments, CA, USA). Bregma and lambda were placed at the same z-position to ensure leveling in the pitch axis and the points AP:−3.20 and ML:+1.5/−1.5 mm were placed at the same z-position to ensure leveling in the roll axis. We made a burr hole in the skull using a dental drill with a 0.5 mm bit at the following coordinates AP:−3.4; ML: +3.2; DV:−4.3. Once the burr hole was made, the virus was loaded into a thin glass pipette connected to a 2.5 µL Hamilton syringe with a compression fitting set (cat# 55750–01, Hamilton, NV USA) and primed with mineral oil (VWR, cat# 9706–128). Aliquots of virus (2.0 µL) were stored at –80°C and thawed at room temperature right before loading into the pipette. The injecting pipette was lowered to the calculated depth. Once in the target location, 200–300 nL of the virus was injected at a speed of 20–30 nL/min using a QSI Stoelting microinjector (cat# 53311, Stoelting Co, IL, USA). The injection was performed at a 30° angle from vertical in the sagittal plane to reach the nigra and avoid the SC. The pipette remained at depth for 10 min following the injection before slowly retracting the pipette. After the injection, the skin was sutured and supplemental fluid, as well as the analgesic carprofen, were provided. The mice recovered in a controlled temperature recovery cage and were then returned to their home cage.
For the slice recording experiments, mice received a viral injection into the nigra as described above and then a second injection of retroAAV2-CAG-tdTomato into the caudal pons (PnC) 3–4 weeks later to label retrogradely the output neurons of the SC. The coordinates used for the PnC injections were, AP:−5.45; ML:−1.3; DV:−4.35. A 10° angle from vertical in the sagittal plane was used for the injection. Mice recovery and post-surgical treatments were completed as described above. Three to four days after the PnC injections, mice were anesthetized with isoflurane, euthanized and the brains were extracted for patch-clamp slice electrophysiological recording experiments as described below. To identify wide-field vertical (WFV) neurons of the superficial SC, we injected cholera-toxin b conjugated with Alexa Fluor 555 (ThermoFisher, cat#: C34776) into the pulvinar (Bickford et al., 2015). Visually identified WFV neurons in the ipsilateral SC labeled retrogradely were recorded using patch-clamp.
Slice electrophysiology
Preparation of acute brain slices was performed according to previous work (Villalobos et al., 2018). Brain slices containing the SC were prepared from adult mice (P30-P50) anesthetized with isoflurane before decapitation. The brains were quickly removed and cooled (4°C) in high sucrose cutting solution (in mM: 240 sucrose, 7 D-glucose, 7 MgCl2, 1.25 NaH2PO4, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2 bubbled to saturation with 95% O2 – 5% CO2). Coronal brain slices (250–300 mm) were cut using a vibratome (Leica VT 1200 S) and subsequently transferred to a recovery chamber containing artificial cerebrospinal fluid (ACSF: 126 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 2 MgCl2, and 10 glucose; ~305 mOsm, pH 7.4) at 35°C for at least an hour before recording. Slices were then transferred to a submerged recording chamber on the stage of an upright Zeiss (Axio Examiner D1) microscope. Slices were superfused (2–3 mL/min) with standard ACSF and maintained at ~30°C using an in-line heater controller (TC-324C, Warner Instruments). Individual PBD neurons were identified under infrared differential interference contrast (IR-DIC) and fluorescence using a Hamamatsu Cooled CCD camera (C11440–42U). Recordings were obtained using 3–5 MU electrodes filled with intracellular solution (150 K-Gluconate, 20 KCl, 0.2 EGTA, 2 MgCl2, 10 HEPES, 2 Na-ATP, and 0.5 Na-GTP; 290 mOsm; pH 7.3). Signals were amplified using a Multiclamp 700B amplifier (Molecular Devices, San Jose, CA, United States), digitized, and stored on a PC. Series resistance and whole-cell capacitance were automatically compensated. To record PnC neurons at different Vms we applied slow constant current (~5.0 s) through the recording electrode. Stable recordings of post-inhibitory RD and spikes were achieved within 1–2 min after breaking into the neuron and were maintained stable for ~30–35 min. Drugs were applied after stable recording within the first 3–5 min after breaking into the neuron. Electrophysiological data were analyzed using the Clampfit 10 software. To activate the nigro-collicular circuit optogenetically, we obtained slices from mice receiving nigral injections with Chronos-GFP. We used Chronos to activate nigral terminals, due to its fast kinetics (Klapoetke et al., 2014) and our desire to more closely match the in vivo discharge rates of nigral neurons which range from 50 to 125 spikes/sec (reviewed in (Basso and Sommer, 2011)). We illuminated the SC in slices through the 40X objective with a 470 nm LED light (~5 mW light output, Mightex). To produce light-evoked postsynaptic potentials in PDB neurons we presented light pulses at different frequencies, 100 Hz and 10 Hz using a 500 ms train duration. The pulse duration for the 100 Hz stimulation was 5 ms and for the 10 Hz stimulation was 50 ms. Similarly, as with the negative current steps, we assessed the RD amplitude and presence of RD-spikes at the end of the stimulation train. All chemicals were purchased from Sigma-Aldrich. To confirm the monosynaptic nature of the PSPs, we performed recordings in the presence of 4-aminopyridine (4-AP, 20 µM) and tetrodotoxin (TTX, 1 µM). To calculate the amplitude of the post-inhibitory RD, we blocked the post-inhibitory Na+ spikes induced after the hyperpolarization by bath applications of TTX. All the sag amplitude of membrane voltage responses to negative step currents was measured as in (Tateno and Robinson, 2011). To test the ionic mechanism of the sag and the RD, we superfused ZD 7288 (ZD, 50 µM), NiCl2 (500 µM), or NNC 55–0396 (1µM) with ACSF. To test the mechanism underlying the persistent post-inhibitory spiking in PDB neurons, we applied the small-conductance Ca2+ activated K+ channel blocker, apamin (300 nM). All recorded neurons were deemed healthy by assessing Vm magnitude and stability, and maximum spike voltage (e.g., < –50 mV and >0 mV, respectively).
Optical fiber implantation
Three to four weeks after the nigral AAV injections, a ceramic ferrule with an optical fiber (0.5 mm in diameter, 3.0 mm in length) was implanted with the fiber tip on top of the lateral portion of the intermediate/deep SC using stereotaxic coordinates (AP:−3.8, ML:+1.6, DV:−2.2), ipsilateral to the side of the nigral injection. After five minutes to allow the probe to settle, the probe was affixed to the skull with consecutive layers of dental acrylic. Once the acrylic dried, the incision was closed with silk sutures. Recovery and post-surgical care were the same as described above. Behavioral testing commenced 5–7 days after the optical fiber implantation. The right SC was implanted in 5 mice and the left SC was implanted in 1 mouse.
Cannula implantation
An internal cannula (26 gauge, 4 mm length, 5 mm pedestal) was implanted in GAD2 mice into the lateral portion of the intermediate/ deep SC using stereotaxic coordinates (AP:−3.8, ML:+1.6, DV:−2.2) on either side of the brain (n = 3 left and n = 5 right). After five minutes to allow the probe to settle, the probe was affixed to the skull with consecutive layers of dental acrylic. Once the acrylic dried, we closed the incision with silk sutures and placed a dummy cannula inside the internal cannula. Behavioral testing commenced 5–7 days after the cannula implantation.
Optogenetic behavioral measurement
Mice were handled daily by the experimenter for at least 2–3 days before making behavioral measurements. Once habituated to handling, mice were transferred to the behavioral room and were habituated to the room conditions for 15–20 min before starting the experiment. Before each session, the apparatus was cleaned with 70% ethanol to eliminate odor cues. All behavioral measurements were made during the same circadian period (13:00–17:00) and were performed at the Behavioral Testing Core at UCLA (BTC-UCLA). Mice were habituated to the open field environment for 10–15 min before the start of each trial. The open field environment consisted of a transparent acrylic box (30 × 30 × 18 cm) and a zenithal video camera (DMK 22AUC03 ImagingSource, NC, USA) with a varifocal lens (Stoelting, UK), connected to a computer. We acquired the videos at 30fps. The light pulses for optogenetic stimulation were controlled by an Ami-2 digital interface (Stoelting, UK) and powered by a Dual-Optogenetics-LED (Prizmatix, Israel) which provided a ~25 mW blue light output power (~2.5–3 mW at the tip of the optic fiber). For every mouse, a behavioral session comprised of a 5 min recording, consisting of 30-s of baseline recordings with no stimulation followed by consecutive 30-s bouts of stimulation and no-stimulation intervals (light ON and light OFF, respectively), repeated four times in a full session (Figure S1C). In each set of experiments, we aimed to test the ability of varying frequencies of stimulation light to induce orienting movements in vivo. The stimulation protocols were selected based on the results of the ex vivo stimulation and consisted of a series of light ON/light OFF transitions, which correspond to the cessation of the inhibitory stimulus, where post-inhibitory spikes would be most likely to occur. Furthermore, these stimulation protocols allow us to rule out any other type of excitatory stimulation that could evoke orienting movements in our experiments, which would be reflected in movements induced by light stimulation regardless of the frequency. We presented light pulses at 100 Hz and 10 Hz. Although nigral neurons show spontaneous activity within 10–40 Hz, nigral neurons can transiently discharge at rates as high as 200 Hz in vivo, and these increases in discharge are associated with the initiation of specific behaviors (Meyer-Luehmann et al., 2002). Although caveats apply to this and other experiments using optogenetic stimulation, the in vivo results demonstrate that the frequencies of stimulation we used, and higher, are seen in normal physiological conditions. The pulse duration for the 100 Hz and 10 Hz stimulation was 5 ms and 50 ms, respectively (Figure S1C1–2). For the 100 Hz, the stimulation trains were presented in 100 ms intervals, so in each 100 ms interval, there were ten light pulses with 5 ms duration. Each 100 ms stimulation interval was followed by 100 ms with no stimulation, thereby producing ~150 light ON/light OFF transitions during a single 30-s bout of stimulation. For the 10 Hz stimulation protocol, we used 1-s stimulation trains intervals, where each interval consisted of ten 50 ms duration light pulses. In the 1-s interval, there was the same number of pulses as with the 100 Hz protocol but at a lower frequency. Each 1 s stimulation interval was followed by a 1-s duration light OFF interval, producing 15 light ON/light OFF transitions during the 30-s bout of stimulation. The 30-s stimulation bouts were repeated four times during the testing sessions for all the stimulation protocols. The order of the stimulation protocol was randomized across all sessions.
SC injection - behavioral measurement
Mice movements were tracked using a zenithal video camera (ELP 2.8 m wide-angle USB camera) and the rotations were scored offline either automatically with the ANY-maze video tracking system (Stoelting Co, IL, USA) or by two independent raters blinded to the experimental condition. Mice were individually housed, and each behavioral session was performed in their cages (dimension: 31 × 15 × 15 cm). A single session consisted of 10 min of pre-injection recording, followed by 2 min of drug infusion and 5 min of diffusion time. Post-injection behavior was recorded for 10 min thereafter. After the session, mice were returned to the vivarium. In the same set of mice, we injected the GABAA agonist muscimol (1µL, 2 mg/–L), the specific T-type Ca2+ channel blocker NNC 55–0396 (1µL, 500µM), and 9.0% physiological saline (1µL) unilaterally through the implanted cannula. We performed muscimol injection experiments first to confirm the location of the cannula in the SC. If an animal showed rotation in this condition, we then used it to test the NNC 55–0396 and saline control. If an animal did not show rotation in response to muscimol, we removed the animal from the study and confirmed its cannula was misplaced (1 out of 9 mice).
Histological procedures
Mice were anesthetized with isoflurane and perfused with phosphate buffered saline (PBS) and PBS containing 4% paraformaldehyde (PFA). Brains were removed and incubated in PFA overnight. After rinsing with PBS, the brains were cryoprotected in 30% sucrose in PBS until sinking. Brains were then embedded in Tissue-Plus O.C.T. Compound (cat# 23–730-571, Fisher Scientific), frozen on dry ice, and cryo-sectioned coronally. 30–60 mm thick sections were mounted on slides and cover-slipped with Fluoro-Gel mounting medium with TES buffer (EMS, 17985–30). Sections were scanned with an LSM 800 (Carl Zeiss) for confocal images or a Leica DMI8 (Leica Microsystems).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details for each experiment can be found in the corresponding figure legends. All the data are expressed as means and standard errors of the mean (SEM). We assessed the statistical significance using parametric tests (t-tests and ANOVA) when the data passed tests for normality (Kolmogorov-Smirnov test of normality) and significance was considered for test statistics with a (*) p value < 0.05.
Quantification of mouse rotations
The measurement of the mouse rotations during the optogenetic behavioral sessions was performed offline using the automated video tracking system ANY-maze (Stoelting Co, IL, USA). A single rotation was defined as the angular position of the mouse head once it reached 360°, either clockwise or counterclockwise from the initial position. For the optogenetic experiments, we counted the total number of full rotations during each of the 30 s bouts (light ON/light OFF) on a session, for four different sessions. For the SC injections experiments, we counted the number of rotations during 10 min recordings pre- and post-injection in four separate sessions. For both optogenetic and SC injection experiments, we linearized the distance traveled by the mouse head on each full rotation and calculated the total distance traveled by the mouse head. To avoid counting rotations resulting from movements of the mouse around the open field (i.e., moving around the perimeter of the space), we defined rotations as the movements comprising 360° with a linearized total distance <50 cm, a distance smaller than the open field perimeter.
Quantification of mouse head movements
To determine whether the orienting movement evoked by optogenetic stimulation occurred synchronously to the onset or offset of light pulses (Figure 4) we measured the distance traveled by the mouse head on a frame-by-frame basis, before (baseline) and during the stimulation epoch. During the stimulation, we divided the measurement epochs into video frames within light pulses or between the light pulses. We measured the position of the mouse head between consecutive video frames offline using an in-house MATLAB routine (code available upon request). The distance traveled by the mouse’s head between consecutive video frames (d) was calculated using the following equation:, where x1 and y1 represent the horizontal and vertical position of the head in the open field during a set frame and x2 and y2 represent the horizontal and vertical position of the head recorded during the following video frame. We obtained the timestamps at which light pulses were applied and aligned those timestamps with their corresponding video frames. We measured the movements between consecutive video frames 15 s before the first light stimulation and 30 s during the light ON periods. These times would allow us to compare a similar number of video frames (~500 frames) for all three conditions (baseline, within, and between light pulses). The mouse head movements were then separated by the length of the movement. For each pair of consecutive frames, the head movements were grouped by whether the traveled distance of the head between frames was smaller than the mean+1SD (<1SD, Figure 4C). The next group corresponds to the distances between mean+1SD and mean+2SD (2SD threshold, Figure 4D). We grouped all the distances in this manner with values up to the mean+5SD.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
|
| ||
| AAV9-Syn-Chronos-GFP | University of North Carolina at Chapel Hill | AV6102C |
| AAV9-Flex-Chronos-GFP | University of North Carolina at Chapel Hill | AV65553 |
| AAV9-Syn-EGFP | Addgene | 50465-AAV9 |
| AAV2-retro-CAG-tdTomato | University of North Carolina at Chapel Hill | AV7643B |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Cholera Toxin Subunit B, Alexa Fluor 555 | ThermoFisher | C34776 |
| 4-Aminopyridine | Sigma-Aldrich | 275875 |
| Muscimol | Sigma-Aldrich | M1523 |
| Tetrodotoxin | Sigma-Aldrich | 554412 |
| ZD 7288 | Tocris | 1000 |
| Nickel(II)chloride | Sigma-Aldrich | 339350 |
| NNC 55–0396 | Tocris | 2268 |
| Apamin | Sigma-Aldrich | A9459 |
| Tissue-Plus O.C.T. Compound | Fisher Scientific | 23–730-571 |
| Fluoro-Gel Mounting Medium with TES Buffer | EMS | 17985–30 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX stock # 000664 |
| Mouse: GAD2-IRES-Cre | The Jackson Laboratory | JAX stock # 010802 |
|
| ||
| Software and algorithms | ||
|
| ||
| pClamp | Molecular Devices | RRID: SCR_011323 |
| Zeiss ZEN 2 (blue edition) | Zeiss | RRID: SCR_013672 |
| Leica Application Suite X | Leica | RRID: SCR_013673 |
| ANY-maze | https://www.any-maze.com/ | RRID: SCR_014289 |
| CorelDRAW Graphics Suite | Corel | RRID: SCR_014235 |
| MATLAB 2020b | MathWorks | RRID:SCR_001622 |
| Custom code | This paper | https://doi.org/10.5281/zenodo.6378006 |
|
| ||
| Other | ||
|
| ||
| 2.5 µL Hamilton syringe with a compression fitting set | Hamilton | Cat # 55750–01 |
| QSI microinjector | Stoelting | Cat # 53311 |
| Video camera | ImagingSource | DMK 22AUCO3 |
| Ami-2 Digital Interface | Stoelting | 60064 |
Highlights.
Activation of the nigro-collicular circuit evokes contralateral rotations in mice
Nigro-collicular activation evokes rebound depolarization in the SC output neurons
T-type Ca2+ blockers inhibit rebound depolarization in SC output neurons
Blockage of T-type Ca2+ channels reduces the number of contralateral movements in mice
ACKNOWLEDGMENTS
We are grateful to Drs. Martha Bickford, Craig Evinger, and Paul May for critical comments on previous versions of the manuscript. We are grateful to Dr. Martha Bickford for assistance with the initial optogenetic experiments and Ms. Psyche Lee for assistance with the initial injections, Mr. Eduardo Alvarez and Ms. McKenna Lah for animal care, Ms. Kelly Zhang for technical support, and Mr. Vaibhav Thakur for statistical assistance. We are also grateful to Ms. Irina Zhuravka, Dr. Lindsay Lueptow, and the Behavioral Training Core at UCLA for help with the in vivo experiments. This work was supported by a Marion Bowen neurobiology postdoctoral fellowship grant to C.A.V. and EY019663 and EY024153 to M.A.B.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110699.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aizenman CD, and Linden DJ (1999). Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J. Neurophysiol 82, 1697–1709. 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, and Penney JB (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci 12, 366–375. 10.1016/0166-2236(89)90074-X. [DOI] [PubMed] [Google Scholar]
- Antal M, Beneduce BM, and Regehr WG (2014). The substantia nigra conveys target-dependent excitatory and inhibitory outputs from the basal ganglia to the thalamus. J. Neurosci 34, 8032–8042. 10.1523/JNEUR-OSCI.0236-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso MA, and Liu P (2007). Context-dependent effects of substantia nigra stimulation on eye movements. J. Neurophysiol 97, 4129–4142. 10.1152/jn.00094.2007. [DOI] [PubMed] [Google Scholar]
- Basso MA, and Sommer MA (2011). Exploring the role of the substantia nigra pars reticulata in eye movements. Neuroscience 198, 205–212. 10.1016/j.neuroscience.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso MA, and Wurtz RH (2002). Neuronal activity in substantia nigra pars reticulata during target selection. J. Neurosci 22, 1883–1894. 10.1523/jneurosci.22-05-01883.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickford ME, Zhou N, Krahe TE, Govindaiah G, and Guido W (2015). Retinal and tectal ‘‘Driver-Like’’ inputs converge in the shell of the mouse dorsal lateral geniculate nucleus. J. Neurosci 35, 10523–10534. 10.1523/JNEUROSCI.3375-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussaoud D, Joseph JP, and Boussaoud D (1985). Role of the cat substantia nigra pars reticulata in eye and head movements. I. Neural activity. Exp. Brain Res 57, 286–296. 10.1007/BF00236534. [DOI] [PubMed] [Google Scholar]
- Chang M, Dian JA, Dufour S, Wang L, Moradi Chameh H, Ramani M, Zhang L, Carlen PL, Womelsdorf T, and Valiante TA (2018). Brief activation of GABAergic interneurons initiates the transition to ictal events through post-inhibitory rebound excitation. Neurobiol. Dis 109, 102–116. 10.1016/j.nbd.2017.10.007. [DOI] [PubMed] [Google Scholar]
- DeLong MR (1983). The neurophysiologic basis of abnormal movements in basal ganglia disorders. Neurobehav. Toxicol. Teratol 5, 611–616. http://www.ncbi.nlm.nih.gov/pubmed/6422317. [PubMed] [Google Scholar]
- DeLong MR, Johns T, and Street NW (1990). Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13, 281–285. 10.1016/0166-2236(90)90110-V. [DOI] [PubMed] [Google Scholar]
- Deniau JM, Mailly P, Maurice N, and Charpier S (2007). The pars reticulata of the substantia nigra: a window to basal ganglia output. Prog. Brain Res 160, 151–172. 10.1016/S0079-6123(06)60009-5. [DOI] [PubMed] [Google Scholar]
- Doykos TK, Gilmer JI, Person AL, and Felsen G (2020). Monosynaptic inputs to specific cell types of the intermediate and deep layers of the superior colliculus. J. Comp. Neurol 528, 2254–2268. 10.1002/cne.24888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edeline JM, Hars B, Hennevin E, and Cotillon N (2002). Muscimol diffusion after intracerebral microinjections : a reevaluation based on electrophysiological and autoradiographic quantifications. Neurobiol. Learn Mem 78, 100–124. 10.1006/nlme.2001.4035. [DOI] [PubMed] [Google Scholar]
- Edgerton JR, and Jaeger D (2014). Optogenetic activation of nigral inhibitory inputs to motor thalamus in the mouse reveals classic inhibition with little potential for rebound activation. Front. Cell. Neurosci 8, 36. 10.3389/fncel.2014.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo T, Tarusawa E, Notomi T, Kaneda K, Hirabayashi M, Shigemoto R, and Isa T (2008). Dendritic Ih ensures high-fidelity dendritic spike responses of motion-sensitive neurons in rat superior colliculus. J. Neurophysiol 99, 2066–2076. 10.1152/jn.00556.2007. [DOI] [PubMed] [Google Scholar]
- Essig J, Hunt JB, and Felsen G (2021). Inhibitory neurons in the superior colliculus mediate selection of spatially-directed movements. Commun. Biol 4, 719. 10.1038/s42003-021-02248-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze BS, Kravitz AV, Hammack N, Berke JD, and Kreitzer AC (2013). Control of basal ganglia output by direct and indirect pathway projection neurons. J. Neurosci 33, 18531–18539. 10.1523/JNEUR-OSCI.1278-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JH, Farries MA, and Fee MS (2012). Integration of cortical and pallidal inputs in the basal ganglia-recipient thalamus of singing birds. J. Neurophysiol 108, 1403–1429. 10.1152/jn.00056.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JH, Farries MA, and Fee MS (2013). Basal ganglia output to the thalamus: still a paradox. Trends Neurosci 36, 695–705. 10.1016/j.tins.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JH, and Fee MS (2012). A cortical motor nucleus drives the basal ganglia-recipient thalamus in singing birds. Nat. Neurosci 15, 620–627. 10.1038/nn.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillner S, and Robertson B (2016). The basal ganglia over 500 million years. Curr. Biol 26, R1088–R1100. 10.1016/j.cub.2016.06.041. [DOI] [PubMed] [Google Scholar]
- Handel A, and Glimcher PW (1999). Quantitative analysis of substantia nigra pars reticulata activity during a visually guided saccade task. J. Neurophysiol 82, 3458–3475. 10.1152/jn.1999.82.6.3458. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Takikawa Y, and Kawagoe R (2000). Role of the basal ganglia in the control of purposive saccadic eye movements. Physiol. Rev 80, 953–978. 10.1152/physrev.2000.80.3.953. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, and Wurtz RH (1983a). Effects on eye movements of a GABA agonist and antagonist injected into monkey superior colliculus. Brain Res 272, 368–372. 10.1016/0006-8993(83)90586-3. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, and Wurtz RH (1983b). Visual and oculomotor functions of monkey substantia nigra pars reticulata. IV. Relation of substantia nigra to superior colliculus. J. Neurophysiol 49, 1285–1301. 10.1152/jn.1983.49.5.1285. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, and Wurtz RH (1985a). Modification of saccadic eye movements by GABA-related substances. I. Effect of muscimol and bicuculline in monkey superior colliculus. J. Neurophysiol 53, 266–291. 10.1152/jn.1985.53.1.266. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, and Wurtz RH (1985b). Modification of saccadic eye movements by GABA-related substances. II. Effects of muscimol in monkey substantia nigra pars reticulata. J. Neurophysiol 53, 292–308. 10.1152/jn.1985.53.1.292. [DOI] [PubMed] [Google Scholar]
- Hirano T, and Kawaguchi SY (2014). Regulation and functional roles of rebound potentiation at cerebellar stellate cell-Purkinje cell synapses. Front. Cell. Neurosci 8, 42–48. 10.3389/fncel.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormigo S, Zhou J, and Castro-Alamancos MA (2020). Zona incerta GABAergic output controls a signaled locomotor action in the midbrain tegmentum. eNeuro 7, 1–16. 10.1523/ENEURO.0390-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, and Llinás R (1984a). Electrophysiological properties of Guinea-pig thalamic neurones: an in vitro study. J. Physiol 349, 205–226. 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, and Llinás R (1984b). Ionic basis for the electro-responsiveness and oscillatory properties of Guinea-pig thalamic neurones in vitro. J. Physiol 349, 227–247. 10.1113/jphysiol.1984.sp015154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda K, Isa K, Yanagawa Y, and Isa T (2008). Nigral inhibition of GABAergic neurons in mouse superior colliculus. J. Neurosci 28, 11071–11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, and Shin HS (2001). Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron 31, 35–45. 10.1016/s0896-6273(01)00343-9. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim Y, Nakajima R, Shin A, Jeong M, Park AH, Jeong Y, Jo S, Yang S, Park H, et al. (2017). Inhibitory basal ganglia inputs induce excitatory motor signals in the thalamus. Neuron 95, 1181–1196.e8. 10.1016/j.neuron.2017.08.028. [DOI] [PubMed] [Google Scholar]
- Kim U, Gregory E, and Hall WC (1992). Pathway from the zona incerta to the superior colliculus in the rat. J. Comp. Neurol 321, 555–575. 10.1002/cne.903210405. [DOI] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346. 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Alves da Silva J, and Costa RM (2019). What, if, and when to move: basal ganglia circuits and self-paced action initiation. Annu. Rev. Neurosci 42, 459–483. 10.1146/annurev-neuro-072116-031033. [DOI] [PubMed] [Google Scholar]
- Kurowski P, Grzelka K, and Szulczyk P (2018). Ionic mechanism underlying rebound depolarization in medial prefrontal cortex pyramidal neurons. Front. Cell. Neurosci 12, 93. 10.3389/fncel.2018.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, and Perez-Reyes E (1999). Nickel block of three cloned T-type calcium channels: Low concentrations selectively block α1H. Biophys. J 77, 3034–3042. 10.1016/S0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, and Basso MA (2008). Substantia nigra stimulation influences monkey superior colliculus neuronal activity bilaterally. J. Neurophysiol 100, 1098–1112. 10.1152/jn.01043.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, and Jahnsen H (1982). Electrophysiology of mammalian thalamic neurones in vitro. Nature 297, 406–408. 10.1038/297406a0. [DOI] [PubMed] [Google Scholar]
- Lo FS, Cork RJ, and Mize RR (1998). Physiological properties of neurons in the optic layer of the rat’s superior colliculus. J. Neurophysiol 80, 331–343. 10.1152/jn.1998.80.1.331. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, and McBain CJ (1996). The hyperpolarization-activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens-alveus interneurones. J. Physiol 497, 119–130. 10.1113/jphysiol.1996.sp021754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JH, and Ghez C (1999). Pharmacological inactivation in the analysis of the central control of movement. J. Neurosci. Methods 86, 145–159. 10.1016/S0165-0270(98)00163-0. [DOI] [PubMed] [Google Scholar]
- May PJ, Basso MA, and Basso MA (2018). Connections between the zona incerta and superior colliculus in the monkey and squirrel. Brain Struct. Funct 223, 371–390. 10.1007/s00429-017-1503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, and Pape HC (1990). Noradrenergic and serotonergic modulation of a hyperpolarization-activated cation current in thalamic relay neurones. J. Physiol 431, 319–342. 10.1113/jphysiol.1990.sp018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Thompson JF, Berridge KC, and Aldridge JW (2002). Substantia nigra pars reticulata neurons code initiation of a serial pattern: implications for natural action sequences and sequential disorders. Eur. J. Neurosci 16, 1599–1608. 10.1046/j.1460-9568.2002.02210.x. [DOI] [PubMed] [Google Scholar]
- Mitra P, and Miller RF (2007). Normal and rebound impulse firing in retinal ganglion cells. Vis. Neurosci 24, 79–90. 10.1017/S0952523807070101. [DOI] [PubMed] [Google Scholar]
- Nelson AB, and Kreitzer AC (2014). Reassessing models of basal ganglia function and dysfunction. Annu. Rev. Neurosci 37, 117–135. 10.1146/annurev-neuro-071013-013916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obejero-Paz CA, Gray IP, and Jones SW (2008). Ni2+ block of Cav3.1 (α1G) T-type calcium channels. J. Gen. Physiol 132, 239–250. 10.1085/jgp.200809988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC (1996). Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu. Rev. Physiol 58, 299–327. 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Penney JB, and Young AB (1983). Speculations on the functional anatomy of basal ganglia disorders. Annu. Rev. Neurosci 6, 73–94. 10.1146/annurev.ne.06.030183.000445. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E (2003). Molecular physiology of low-voltage-activated T-type calcium channels. Physiol. Rev 83, 117–161. 10.1152/phys-rev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Person AL, and Perkel DJ (2005). Unitary IPSPs drive precise thalamic spiking in a circuit required for learning. Neuron 46, 129–140. 10.1016/j.neuron.2004.12.057. [DOI] [PubMed] [Google Scholar]
- Person AL, and Perkel DJ (2007). Pallidal neuron activity increases during sensory relay through thalamus in a songbird circuit essential for learning. J. Neurosci 27, 8687–8698. 10.1523/JNEUROSCI.2045-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person AL, and Raman IM (2012). Synchrony and neural coding in cerebellar circuits. Front. Neural Circuits 6, 97. 10.3389/fncir.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Medina L, and Veenman CL (1998). Structural and functional evolution of the basal ganglia in vertebrates. Brain Res. Brain Res. Rev 28, 235–285. 10.1016/S0165-0173(98)00016-2. [DOI] [PubMed] [Google Scholar]
- Sans-Dublanc A, Chrzanowska A, Reinhard K, Lemmon D, Nuttin B, Lambert T, Montaldo G, Urban A, and Farrow K (2021). Optogenetic fUSI for brain-wide mapping of neural activity mediating collicular-dependent behaviors. Neuron 109, 1888–1905.e10. 10.1016/j.neuron.2021.04.008. [DOI] [PubMed] [Google Scholar]
- Sato M, and Hikosaka O (2002). Role of primate substantia nigra pars reticulata in reward-oriented saccadic eye movement. J. Neurosci 22, 2363–2373. 10.1523/JNEUROSCI.22-06-02363.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, Leventhal DK, Mallet N, Chen F, and Berke JD (2013). Canceling actions involves a race between basal ganglia pathways. Nat. Neurosci 16, 1118–1124. 10.1038/nn.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson-Jones M, Ericsson J, Robertson B, and Grillner S (2012). Evolution of the basal ganglia: dual-output pathways conserved throughout vertebrate phylogeny. J. Comp. Neurol 520, 2957–2973. 10.1002/cne.23087. [DOI] [PubMed] [Google Scholar]
- Sun H, and Wu SH (2008a). Modification of membrane excitability of neurons in the rat’s dorsal cortex of the inferior colliculus by preceding hyperpolarization. Neuroscience 154, 257–272. 10.1016/j.neuroscience.2007.10.055. [DOI] [PubMed] [Google Scholar]
- Sun H, and Wu SH (2008b). Physiological characteristics of postinhibitory rebound depolarization in neurons of the rat’s dorsal cortex of the inferior colliculus studied in vitro. Brain Res 1226, 70–81. 10.1016/j.brainres.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Tateno T, and Robinson HP (2011). The mechanism of ethanol action on midbrain dopaminergic neuron firing: a dynamic-clamp study of the role of Ih and GABAergic synaptic integration. J. Neurophysiol 106, 1901–1922. 10.1152/jn.00162.2011. [DOI] [PubMed] [Google Scholar]
- Tecuapetla F, Jin X, Lima SQ, and Costa RM (2016). Complementary contributions of striatal projection pathways to action initiation and execution. Cell 166, 703–715. 10.1016/j.cell.2016.06.032. [DOI] [PubMed] [Google Scholar]
- Toda K, Lusk NA, Watson GDR, Kim N, Lu D, Li HE, Meck WH, and Yin HH (2017). Nigrotectal stimulation stops interval timing in mice. Curr. Biol 27, 3763–3770.e3. 10.1016/j.cub.2017.11.003. [DOI] [PubMed] [Google Scholar]
- Villalobos CA, Wu Q, Lee PH, May PJ, and Basso MA (2018). Parvalbumin and GABA microcircuits in the mouse superior colliculus. Front. Neural Circuits 12, 35. 10.3389/fncir.2018.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XX, Jin Y, Sun H, Ma C, Zhang J, Wang M, and Chen L (2016). Characterization of rebound depolarization in neurons of the rat medial geniculate body in vitro. Neurosci. Bull 32, 16–26. 10.1007/s12264-015-0006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GD, Smith JB, and Alloway KD (2015). The zona incerta regulates communication between the superior colliculus and the posteromedial Thalamus: implications for thalamic interactions with the dorsolateral striatum. J. Neurosci 35, 9463–9476. 10.1523/JNEUROSCI.1606-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yttri EA, and Dudman JT (2016). Opponent and bidirectional control of movement velocity in the basal ganglia. Nature 533, 402–406. 10.1038/nature17639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
Custom MATLAB scripts developed by our laboratory to analyze mouse data are available at (https://zenodo.org/badge/latestdoi/472946729).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.