

Abstract
Combining two pharmacophores in a molecule can lead to useful synergistic effects. Herein, we show hybrid systems that combine sterically hindered phenols with dinitrobenzofuroxan fragments exhibit a broad range of biological activities. The modular assembly of such phenol/benzofuroxan hybrids allows variations in the phenol/benzofuroxan ratio. Interestingly, the antimicrobial activity only appears when at least two benzofuroxan moieties are introduced per phenol. The most potent of the synthesized compounds exhibit high cytotoxicity against human duodenal adenocarcinoma (HuTu 80), human breast adenocarcinoma (MCF-7), and human cervical carcinoma cell lines. This toxicity is associated with the induction of apoptosis via the internal mitochondrial pathway and an increase in ROS production. Encouragingly, the index of selectivity relative to healthy tissues exceeds that for the reference drugs Doxorubicin and Sorafenib. The biostability of the leading compounds in whole mice blood is sufficiently high for their future quantification in biological matrices.
Keywords: benzofuroxan, sterically hindered phenol, anti-cancer activity, cytotoxicity, apoptosis, ROS production
1. Introduction
Cancer continues to be one of the most serious challenges facing modern science and medicine. Oncological diseases steadily occupy the second place in the list of causes of human death. According to the World Health Organization, by 2030, 15 million people will be dying from this pathology in the world every year [1].
In our search for new highly effective antitumor drugs, we chose a molecular design that can achieve two main goals: (1) to reduce toxicity in a healthy microenvironment and to provide a targeted effect on tumor tissues; (2) to enhance the antitumor effect by combining in one molecule several pharmacophores with different but complementary mechanisms of action.
The inclusion of a sterically hindered phenol (SHP) fragment helps to address the first goal. In healthy cells, where there is a redox balance, phenols will behave as antioxidants, protecting a normal cell from the harmful effects of other pharmacophoric fragments of the structure. The situation changes in a state of oxidative stress observed in tumor cells when numerous ROS (Reactive Oxygen Species) are formed and various metals, mainly iron and copper, accumulate in the unbound state [2,3,4,5,6]. Under these conditions, the phenols are transformed into highly reactive methylene quinones, which have destructive effects on lipids, proteins and DNA, thereby leading to the tumor cells death (Figure 1). The effect of quinones can be multi-fold. Non-hindered quinones are potent electrophiles that can bind to thiol, amine and hydroxyl groups. More generally, the fast redox cycling between phenols and quinones can lead to the formation of intermediates that can transfer an electron to molecular oxygen, transforming it into a superoxide radical anion and, thus, serving themselves as a potentially catalytic source of ROS.
Figure 1.

(A) General approach to the creation of new types of hybrid antitumor agents based on sterically hindered phenols (SHP) and benzofuroxans. (B) Redox cycling between phenols and quinone methides creates radical anions that can reduce molecular oxygen to a superoxide anion. (C) Phosphonate group greatly activates benzylic C-H bonds and facilitates quinone methide radical-anion formation. (D) Generation of NO and ROS from furoxans. ROS = Reactive Oxygen Species.
Such phenol-mediated redox reorientation was shown to be promising for the development of targeted antitumor agents, initiating the mitochondrial pathway of cancer cell apoptosis. The spectrum of biological antitumor activity includes inhibiting metastasis of melanoma and Lewis lung carcinoma [7,8,9], leukemia, colon, liver, ovarian, breast cancers [10], sarcoma 37 and carcinoma [11].
The presence of a benzylic phosphonate group is likely to be important based on the earlier indications [12,13,14] that the introduction of phosphate ester or bisphosphonate ester moieties into anticancer agents (e.g., Cisplatin, Camptothecin and Doxorubicin) can improve drug solubility and antitumor activity. An intriguing possibility is that it can also assist in the formation of quinone methides and their radical anions by activating the C-H bond (Figure 1). It is well known that the phosphonate group can increase C-H acidity by >13 orders of magnitude [15].
Considering the redox activity and ROS generation with SHPs, a logical choice for the second pharmacophore is provided by the benzofuroxan moiety. Benzofuroxans represent an important class of lipophilic thiol-dependent NO donors [16,17]. After the discovery of the key role of nitric oxide as an intracellular regulator of metabolism, benzofuroxans have been playing an increasingly important role in the development of anticancer agents that can generate high levels of NO and inhibit tumor growth in vivo [18,19]. Interestingly, these heterocycles can produce not only nitric oxide (NO•) but also its two redox forms, nitroxyl (HNO) and nitrosonium ion (NO+). From this point of view, the synergy between SHP and furoxan moieties in hybrid systems seem to be an attainable goal.
Since the first reports of the antitumor properties of benzofuroxans [20,21,22,23], cytotoxicity against tumor cell lines of many of their derivatives has been tested [24,25,26,27,28]. A number of benzofuroxan derivatives inhibit the growth of M-HeLa cells in vitro. Furthermore, several of them showed significant activity against P388 lymphatic leukemia and Ehrlich ascitic carcinoma in mice [29]. Interestingly, the antitumor effect of these compounds is due to their dual ability to inhibit DNA synthesis and to cause DNA destruction through a series of single- and double-strand breaks [26]. It is well known that single DNA damage can be fairly easily repaired by the numerous intercellular DNA repair systems. In contrast, multiple damages are much harder to repair and, hence, it is much more likely to lead to cell death. It is noteworthy that the same study [26] emphasizes the positive contribution of electron-withdrawing nitro groups in the benzofuroxan core to cytotoxicity.
In addition, NO is a necessary component of a non-specific defense mechanism against many pathogens, including bacteria, viruses and fungi. A number of publications describe the high fungicidal activity of the so-called “hybrid” benzofuroxan derivatives, which include, in addition to the benzofuroxan moiety, known pharmacophore fragments (amino acids, amino alcohol nitrates, phenols and polyene antibiotics) [30,31]. The activity of a 4,6-dinitro-5,7-dichlorobenzofuroxan derivative against Trichophyton mentagrophytes was four times higher than the activity of the antifungal drug Nystatin [32]. Benzofuroxan derivatives were found to be highly active against phytopathogenic fungi (Rhizoctonia solani, Sclerotinia sclerotiorum, Fusarium graminearum and Phytophthora capsici) [33] and against antibiotic-resistant Staphylococcus bacteria [34].
In summary, our aim is to develop agents with a broad spectrum of biological action. Our basic molecular platform is a substituted sterically hindered phenol, which can be reversibly oxidized to the corresponding methylene quinone in cancer cells but exhibits antioxidant activity in healthy cells. The auxiliary components include a diaminopyridine or diaminophenyl linker for targeted modification, a phosphorus-containing fragment for increasing bioavailability, and benzofuroxan as an additional pharmacophore with the potential to be both the NO-donor and apoptosis-inducing agent (Figure 1).
2. Results and Discussion
2.1. Chemistry
Phosphorylated sterically hindered phenols (SHPs) 2 containing diaminopyridine or diaminophenyl fragments were used as a starting point due to their already identified high and selective cytotoxicity [35]. The synthesis of the starting SHPs was carried out in accordance with the original method developed by us earlier (Scheme 1, [35]). The main stages of this method include preparation of the corresponding phosphorylated phenol, its oxidation to methylene quinone 1, and, as the last step, reaction of the methylene quinone with C-nucleophile, i.e., 2,6-diaminopyridine or 1,3-diaminobenzene. This sequence produces the key compounds 2, that possess two amino groups suitable for further modifications with heterocyclic fragments.
Scheme 1.

The synthesis of the starting SHPs 2 containing diaminopyridine or diaminophenyl fragments.
Finally, the amino groups of the two hindered phosphorylated phenols 2 were used in reactions with nitrochlorobenzofuroxans. This highly reactive heterocycle is superelectrophilic and readily reacts with the appended aniline moieties. The phenol OH group is unreactive under these conditions due to the steric protection provided by the two tert-butyl groups. The final products of this synthetic sequence result are previously unknown “hybrid” phosphorus-containing SHPs containing a nitrobenzofuroxan fragment linked to the SHP fragment through a (hetero)aromatic “linker” (Scheme 2).
Scheme 2.

Synthesis of SHP/furoxan hybrids of varying composition.
Depending on the initial ratio of reagents, it is possible to vary the composition of the final products, leading to the formation of compounds with a composition of 2:1 when two molecules of benzofuroxan interact with one molecule of sterically hindered phenol and compounds of the composition of 1:1, when one molecule of benzofuroxan reacts with one molecule of sterically hindered phenol. The excess phosphorus-containing SHP in both cases was used to neutralize the hydrogen chloride formed during the reaction.
As is known in the literature, benzofuroxan rapidly interconverts in solution between two non-symmetric bicyclic structures through the open dinitroso form (so-called N-1-oxide/N-3-oxide tautomerism, Scheme 3) [36,37]. As a result, the NMR 1H and 13C spectra of benzofuroxan derivatives taken at room temperatures are often broadened as they correspond to a fast mutual transition of equivalent non-symmetric forms, while the spectra taken at low temperature are non-symmetric forms [38]. In our study, we also observed our products in the spectra, not as individual compounds but as a mixture of tautomers. In some cases, they were separated into the pure form (as in the case of compounds 4e–g). In other cases, where the content of the second tautomer was insignificant, we limited ourselves to describing only the main structure.
Scheme 3.

N-1-oxide/N-3-oxide tautomerism in benzofuroxan derivatives.
The complete assignment of signals in 1H and 13C spectra (Figures S1–S45) was accomplished by using the 2D NMR techniques (COSY, 1H-13C HSQC and 1H-13C HMBC). Examples of 2D spectra for compounds 4d and 4e are presented (Figures S12–S14 and S18–S20). For compounds within the series, the assignment of peaks was conducted by analogy, taking into account the effects of neighboring substituents. Some difficulty is that benzofuroxans exist as two tautomers due to oxygen migration. As we showed earlier [39], the presence of only one proton in the tri-substituted benzofuroxan moiety makes it difficult to use two-dimensional NMR experiments. At the same time, the chemical shifts of carbons in these tautomeric forms do not differ significantly. Nevertheless, the difference between C3a and C7a makes it possible to quite unambiguously attribute the observed shape of the benzofuroxan ring.
The exception is the interaction of benzofuroxan 3 with diaminophenyl derivative 2f containing diethylphosphonate fragment. We found that regardless of the ratio of reagents employed, there is only a formation of a mono-substitution product 4f. The 2:1 product cannot be obtained, probably because of a sterically hindered environment for the NH2 group located in the ortho-position relative to the methyldiethylphosphonate fragment.
2.2. Biological Evaluation
For the synthesized compounds, diverse biological properties were studied: antitumor potential with the determination of the mechanisms of action, hemolytic activity, antibacterial properties and biostability.
2.2.1. Anticancer Activity
To start, a new series of synthesized compounds proposed as potential anticancer agents were tested for cytotoxicity against cancer and normal cell lines (Table 1). Data on cytotoxic activity are represented by IC50 values (the concentration of compound that causes 50% cell death in the test population).
Table 1.
In vitro cytotoxic effects for the tested SHP-benzofuroxan hybrids.
| Compounds | IC50, µM | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cancer Cell Lines | Normal Cell Lines | |||||||||
| HuTu 80 | PC3 | PANC-1 | MCF-7 | M-HeLa | T98G | A549 | A375 | WI38 | Chang Liver | |
| 3 | >100 | nd | >100 | nd | 94.1 ± 8.6 | nd | nd | nd | nd | >100 |
| 2a | > 100 | nd | nd | 31.0 ± 2.6 | 16.1 ± 1.3 | nd | nd | nd | nd | >100 |
| 2b | 86.6 ± 6.8 | nd | nd | 34.2 ± 3.0 | 17.1 ± 1.3 | nd | nd | nd | nd | >100 |
| 2c | >100 | nd | nd | 21.8 ± 1.7 | 38.0 ± 2.8 | nd | nd | nd | nd | >100 |
| 2d | 63.2 ± 5.6 | nd | nd | 16.0 ± 1.2 | 7.4 ± 0.7 | nd | nd | nd | nd | 52 ± 3.5 |
| 2e | nd | nd | nd | >100 | 45.3 ± 3.2 | nd | nd | nd | nd | >100 |
| 4a | 5.2 ± 0.4 | 14.0 ± 1.2 | 20.0 ± 1.7 | 7.6 ± 0.6 | 4.2 ± 0.3 | 12.4 ± 1.2 | 13.5 ± 1.2 | 13.5 ± 1.2 | 7.2 ± 0.6 | 12.0 ± 1.2 |
| 4b | 5.4 ± 0.3 | 11.0 ± 0.8 | 5.3 ± 0.4 | 3.1 ± 0.2 | 2.9 ± 0.2 | 8.3 ± 0.7 | 9.3 ± 0.8 | 11.4 ± 0.9 | 5.3 ± 0.4 | 7.0 ± 1.5 |
| 4c | 0.9 ± 0.07 | 1.2 ± 1.0 | 11.2 ± 0.9 | 1.1 ± 0.1 | 0.9 ± 0.07 | 2.2 ± 0.1 | 0.9 ± 0.08 | 2.9 ± 0.2 | 2.0 ± 0.1 | 2.1 ± 0.7 |
| 4d | 13.1 ± 1.1 | 11.3 ± 0.9 | 11.9 ± 0.9 | 9.0 ± 0.7 | 6.3 ± 0.5 | 7.9 ± 0.7 | 9.0 ± 0.7 | 17.0 ± 1.4 | 11.3 ± 1.1 | 21.0 ± 2.4 |
| 4e | 17.1 ± 1.3 | 12.3 ± 1.1 | 17.4 ± 1.3 | 21.5 ± 1.8 | 12.0 ± 1.0 | 40.2 ± 3.1 | 48.3 ± 3.7 | 53.1 ± 4.1 | 25.7 ± 2.2 | 14.3 ± 1.3 |
| 5a | 11.0 ± 0.9 | 7.7 ± 0.6 | 13.5 ± 1.1 | 12.1 ± 1.1 | 7.7 ± 0.6 | 26.8 ± 2.2 | 48.4 ± 3.5 | 21.6 ± 1.8 | 11.4 ± 0.9 | 15.1 ± 1.7 |
| 5c | 3.9 ± 0.3 | 6.8 ± 0.5 | 5.0 ± 0.3 | 4.5 ± 0.4 | 2.4 ± 0.2 | 7.7 ± 0.6 | 7.7 ± 0.6 | 9.0 ± 0.8 | 5.0 ± 0.4 | 4.1 ± 0.8 |
| 5d | 4.9 ± 0.3 | 5.9 ± 0.4 | 2.8 ± 0.2 (SI = 3.3) |
2.1 ± 0.1 (SI = 4.4) |
2.0 ± 0.1 (SI = 4.6) |
4.8 ± 0.4 | 5.2 ± 0.4 | 5.9 ± 0.5 | 8.4 ± 0.7 | 9.2 ± 1.9 |
| DOX | 0.2 ± 0.01 | 1.4 ± 0.1 | 2.2 ± 0.1 | 0.4 ± 0.03 | 2.1 ± 0.2 | 2.0 ± 0.1 | 0.7 ± 0.05 | 0.3 ± 0.02 | 0.4 ± 0.02 | 0.5 ± 0.04 |
| SF | 6.2 ± 0.5 | 11.3 ± 0.9 | 12.0 ± 1.1 | 27.5 ± 2.3 | 25.0 ± 1.9 | 8.6 ± 0.7 | 25.2 ± 2.2 | 6.8 ± 0.5 | 6.6 ± 0.5 | 21.7 ± 1.7 |
HuTu-80 is a duodenal adenocarcinoma; PC3 is a human prostate adenocarcinoma; PANC-1 is a human pancreatic cancer cell line; MCF-7 is a human breast adenocarcinoma (pleural fluid); M-HeLa is a human cervix epitheloid carcinoma; T98G is a glioblastoma cell line; A549 is an adenocarcinomic human alveolar basal epithelial cell line; A375 is human amelanotic melanoma cell line; WI 38 is an human embryonic lung; Chang liver is a line of human liver cells; DOX—doxorubicin, SF—sorafenib. The experiments were repeated three times. SI—selectivity index values relative to Chang liver; nd—not determined. Cells were incubated with substances for 48 h.
It is interesting to compare the activity of hybrid molecules 4–5 to their building blocks, SHPs 2 and chlorobenzofuroxan 3. As can be seen from the data given in Table 1, the simple benzofuroxan 3 does not show cytotoxicity at these concentrations. At the same time, the initial SHPs 2 have cytotoxic activity, which, as we noted earlier, motivated us to choose them as a starting point of our design. Gratifying, the hybrid compounds 4–5 showed relatively high activity against all cancer lines used in the experiments. In addition, the lead compounds have moderate cytotoxicity against the normal Chang liver cells.
When compared with the starting compounds, the SHP/benzofuroxan hybrid 4c is 40 times more active than SHP 2c with respect to M-HeLa and 20 times more active with respect to MCF-7. The activity of compound 5d exceeds the activity of compound 2d by 3.7 times with respect to M-HeLa and 7.6 times with respect to MCF-7. However, it should be noted that the cytotoxicity towards normal cells of these hybrid leader compounds also increases, which makes these compounds more toxic. These data suggest that finding the proper balance between efficacy and selectivity remains a challenge. From these data, we have identified two lead compounds, 4c and 5d, that show high cytotoxicity against human duodenal adenocarcinoma (HuTu 80), human breast adenocarcinoma (MCF-7) and human cervical carcinoma cell lines. The IC50 values of compounds 4c and 5d for these lines were either comparable to or exceeded the activity of the reference drugs Doxorubicin and Sorafenib.
The key indicator for evaluating perspective antitumor drugs is the selectivity index (SI), which was calculated as the ratio between the IC50 value for normal cells and the IC50 value for cancer cells. The selectivity index values for 5d are shown in Table 1. According to the literature guidelines [40], compounds are considered selective at SI ≥ 3. Therefore, the lead compound 5d can be considered selective for MCF-7 and M-HeLa cell lines at SI = 4. Note that the reference drugs Doxorubicin and Sorafenib are significantly inferior in selectivity compared to the lead compound.
Apoptosis is one of the preferred mechanisms of cytotoxic action for the development of new anticancer agents. Apoptosis-inducing properties of the leading compounds, 4c and 5d, were evaluated by flow cytometry at IC50/2 and IC50 concentrations on the M-HeLa cell line (Figure 2a,b). This assay is convenient for detecting apoptosis and for differentiating its stages. In particular, the viable cells are negative for both PI and Annexin V- Alexa Fluor 647 binding; non-viable, necrotic cells are negative for Annexin V- Alexa Fluor 647 binding and positive for PI uptake; cells in early apoptosis are Annexin V- Alexa Fluor 647 positive and PI negative; cells in late apoptosis are positive for both Annexin V- Alexa Fluor 647 binding and PI uptake.
Figure 2.

Induction of apoptosis in M-HeLa cancer cells by compounds 4c and 5d, evaluated by Flow Cytometry Assay, after cell labeling with propidium iodide (PI) and Annexin V- Alexa Fluor 647 (a). L, D, La and Ea labels in the top right control plot correspond to Live, Dead, Late apoptosis and Early apoptosis, respectively. Cells were either unlabeled and untreated (Control) or treated with IC50 and IC50/2 doses of test compounds for 48 h, as indicated. The quantitative analysis of early and late apoptotic cells after drug exposure is represented in (b). Data are presented as mean ± error of the mean (n = 3). **** p ≤ 0.0001 compared to control (one-way ANOVA). Concentration for 4c was (IC50/2- 0.5 µM and IC50—1.0 µM); for 5d (IC50/2—1.0 µM and IC50—2.0 µM).
It can be seen that after 48 h of incubation, the test compounds begin to induce apoptosis in M-HeLa cells. The apoptotic effects are most prominent in the case of the leading compound 4c, especially at the stage of late apoptosis (Figure 2b).
The possibility of apoptosis through the mitochondrial pathway was assessed by flow cytometry using the JC-10 fluorescent dye (in the Mitochondria Membrane Potential Kit). In normal cells, JC-10 accumulates in the mitochondrial matrix, where it forms aggregates identified via their red fluorescence. However, in apoptotic cells, JC-10 diffuses out of the mitochondria, converts to its monomeric form, and emits green fluorescence, which is recorded by a flow cytometer. After treatment with compounds 4c and 5d at concentrations of IC50/2 and IC50, we observed the dissipation of the mitochondrial membrane potential of M-HeLa cells, which became more pronounced in the case of compound 4c (Figure 3a). Figure 3b shows that the intensity of green fluorescence significantly increased relative to the control. These results suggest that the mechanism of action of the studied compounds is associated with the induction of apoptosis, which occurs along the internal mitochondrial pathway.
Figure 3.

(a) Flow cytometry analysis of M-Hela cells treated with compounds 4c and 5d. (b) Quantitative determination of % cells with red aggregates and green monomers. Data are presented as mean ± error of the mean (n = 3). **** p ≤ 0.0001 compared to control (one-way ANOVA). Concentration for 4c was (IC50/2—0.5 µM and IC50—1.0 µM); for 5d (IC50/2—1.0 µM and IC50—2.0 µM).
Apoptosis can be induced in various ways, including an increase in the production of reactive oxygen species (ROS) in the cell, with subsequent oxidative stress and destruction of membrane lipids, proteins and nucleic acids. Thus, chemical compounds that disturb the redox balance and lead to the production and accumulation of ROS are potential agents for targeting the transformed cells. To understand the possible synergy between the different components of these hybrid agents, we compared the ROS production for compounds 4c, 5d, starting phenols 2c, 2d and furoxan 3 at the concentration of IC50 cytotoxicity on M-HeLa cells (Figure 4). A significant increase in CellROX® Deep Red fluorescence intensity indicates an increase in ROS production. This increase is especially pronounced in the presence of compound 4c where ROS production significantly exceeds that for the original phenol 2c and furoxan 3. Hence, it is reasonable to suggest that activity of compound 4c benefits from the synergy between the methylene quinones (as producers of superoxide) and furoxans (as the NO donors).
Figure 4.

Induction of ROS production by compounds 2c, 2d, 3, 4c and 5d. The data were obtained with flow cytometry and the CellROX® Deep Red kit. Concentration for compounds was equivalent to IC50 cytotoxicity on M-HeLa cells. Data are presented as mean ± SD (n = 3). ** p < 0.01, **** p < 0.0001 compared to control; #### p ≤ 0.0001, compared to 2c; $$$$ p ≤ 0.0001 and compared to 3 (one-way ANOVA).
Thus, the phenol-benzofuroxan hybrids exhibit high cytotoxicity. This biological activity is primarily associated with the induction of apoptosis, occurring via the internal mitochondrial pathway and an increase in ROS production.
Additionally, we evaluated the hemolytic activity (i.e., the ability to destroy human erythrocytes) [41,42,43]and biostability in whole mice blood for the leader compounds [44]. The tested compounds do not show hemolytic activity (HC50 >100 µM) and are metabolized relatively slowly (data are presented in supporting information, Figure S46).
2.2.2. Antimicrobial Activity
The synthesized compounds were also tested for antibacterial activity against a number of gram-positive Staphylococcus aureus ATCC 6538P FDA 209P (Sa), Bacillus cereus ATCC 10702 (Bc), Enterococcus faecalis ATCC 29212 (Ef), gram-negative bacteria Escherichia coli ATCC 25922 (Ec) and Pseudomonas aeruginosa ATCC 9027 (Pa), including against methicillin-resistant strains of Staphylococcus aureus MRSA-1 and MRSA-2. Methicillin-resistant strains of S. aureus were provided to us by the Republican Clinical Hospital (Kazan, Russia) from patients with chronic tonsillitis and sinusitis and were highly resistant: MRSA-1—to β-lactams and fluoroquinolones and MRSA-2—only to β-lactams. Antifungal activity was studied on Candida albicans 10231. Neither sterically hindered phenols 2 and benzofuroxan 3 nor their 1:1 hybrids 4 show activity against fungi and bacteria. On the other hand, the introduction of the second benzofuroxan fragment leads to the rise of antimicrobial activity (Table 2). Compounds 5a, 5c and 5d display selective antimicrobial activity against gram-positive bacteria S. aureus, B. cereus, E. faecalis (at the level of the reference drug Chloramphenicol) and strain MRSA 1. These compounds are less active against MRSA-2. All studied compounds were inactive toward gram-negative bacteria and the yeast Candida albicans 10231.
Table 2.
Antimicrobial activity of studied compounds.
| Compounds | Minimum Inhibitory Concentration (MIC), µg/mL | ||||
|---|---|---|---|---|---|
| Sa | Bc | Ef | MRSA-1 | MRSA-2 | |
| 2a | - | - | - | - | - |
| 2c | - | - | - | - | - |
| 2d | - | - | - | - | - |
| 3 | - | - | - | - | - |
| 4a | - | - | - | - | - |
| 4c | - | - | - | - | - |
| 4d | - | - | - | - | - |
| 5a | 125 ± 11 | 62.5 ± 5.3 | 125 ± 10 | 250 ± 20 | 250 ± 20 |
| 5c | 31.3 ± 2.3 | 31.3 ± 2.5 | 62.5 ± 5.3 | 62.5 ± 5.5 | 250 ± 20 |
| 5d | 15.6 ± 1.2 | 62.5 ± 5.4 | 62.5 ± 5.2 | 62.5 ± 5.3 | 250 ± 19 |
| Chloramphenicol | 31.3 ± 2.2 | 62.5 ± 5.4 | 62.5 ± 5.2 | nd | nd |
| Ketoconazole | - | - | - | - | - |
| Minimum bactericidal and fungicidal concentrations (MBC), (MFC) µg/ml | |||||
| 5a | 250 ± 19 | - | - | 250 ± 21 | - |
| 5c | - | 250 ± 20 | - | - | - |
| 5d | 125 ± 10 | - | - | 250 ± 20 | 250 ± 19 |
| Chloramphenicol | - | - | - | - | - |
| Ketoconazole | - | - | - | - | - |
Average of three values measured; ± standard deviation (SD);—means non-active; nd—not determined.
3. Materials and Methods
3.1. Chemistry
IR spectra were recorded on an IR Fourier spectrometer Tensor 37 (Bruker Optik GmbH, Ettlingen, Germany) in the 400–3600 cm−1 range in KBr. The 1H- and 13C-NMR spectra were recorded on a Bruker AVANCE 400 spectrometer (Bruker BioSpin, Rheinstetten, Germany) operating at 400 MHz (for 1H NMR), 101 MHz (for 13C NMR) and 162 MHz (for 31P NMR) and Brucker spectrometers AVANCEIII-500 (Bruker Corporation, Rheinstetten, Germany) operating at 500 MHz (for 1H NMR) and 126 MHz (for 13C MMR). Chemical shifts were measured in δ (ppm) with reference to the solvent (δ = 7.27 ppm and 77.00 ppm for CDCl3; δ = 2.06 ppm and 28.94 ppm for (CD3)2CO, δ = 2.56 ppm and 39.52 ppm for DMSO-d6 for 1H and 13C NMR, respectively). Elemental analysis was performed on a CHNS-O Elemental Analyser EuroEA3028-HT-OM (EuroVector S.p.A., Milan, Italy). ESI-TOF-MS spectra were recorded on a Bruker AmazonX instrument (Bruker Daltonix GmbH, Bremen, Germany). The melting points were determined on JK-MAM-4 Melting-point Apparatus with Microscope (JINGKE SCIENTIFIC INSTRUMENT CO, Shanghai, China). The progress of reactions and the purity of products were monitored by TLC on Sorbfil UV-254 plates (Sorbpolimer, Krasnodar, Russia); the chromatograms were developed under UV light.
7-Chloro-4,6-dinitrobenzofuroxan 3 was synthesized according to the literature [45].
Reaction between sterically hindered phenols 2a–g and 7-chloro-4,6-dinitrobenzofuroxan 3. To a solution of 7-chloro-4,6-dinitrobenzofuroxan 3 (0.8 mmol) in 5 mL of CHCl3 at room temperature was added a solution of sterically hindered phenols 2 (1.6 mmol (for compounds 4) or 1.2 mmol (for compounds 5)) in 5 mL of CHCl3. The reaction was carried out at room temperature and under magnetic stirring, and the conversion was monitored through TLC analysis (eluent: toluene/ethyl acetate, 2:1). The mixture was stirred at room temperature overnight; the crude mixture was precipitated in hexane (10 mL), the obtained solid was filtered off, washed with cold water (100 mL) and dried under vacuum (0.06 mm Hg) at 40 °C temperature to constant weight. In any case, a mixture of compounds 4 and 5 was obtained, depending on the conditions, with a high content of one of them. The crude product was purified by column chromatography (eluent in each case was selected individually) to give the target compound (the second product in this case was isolated in an insignificant amount).
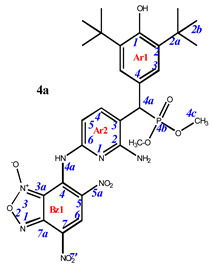
7-((6-amino-5-((3,5-di-tert-butyl-4-hydroxyphenyl)(dimethoxyphosphoryl)methyl)pyridin-2-yl)amino)-4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide (4a). Purple powder, yield 85%. M.p.: 270–271 °C. IR (ν, cm–1): 694 (P–C), 1228 (P=O), 1359 (NO2 symm), 1563 (NO2 asymm), 1623 (furoxan ring), 3448 (NH2) and 3621 (OH). 1H NMR (500 MHz, Acetone-d6): δ 9.10 (s, 1H, H6-Bz1), 8.13 (dd, J = 8.1 Hz, J(PH) = 1.5 Hz, 1H, H4-Ar2), 7.44 (d, J(PH) = 1.8 Hz, 2H, H3-Ar1), 6.73 (brs, 1H, H5-Ar2), 6.11 (s, 1H, OH-Ar1), 4.67 (d, J(PH) = 26.8 Hz, 1H, H4a-Ar1), 3.70 and 3.61 (d, J = 10.7 Hz, 6H, CH3) and 1.47 (s, 18H, H2b-Ar1). 13C{1H} NMR (126 MHz, Acetone-d6) δ 156.7 (C2-Ar2), 154.9 (C1-Ar1), 151.2 (C6-Ar2), 148.1 (C4-Bz), 143.7 (C4-Ar2), 139.1 (C2-Ar1), 139.0 (C7a-Bz), 134.4 (C6-Bz), 128.3 (C7-Bz), 127.7 (d, J(PC) = 7.3 Hz, C3-Ar1), 127.4 (C3-Ar2), 125.3 (C5-Bz), 115.0 (C3a-Bz), 114.0 (C4-Ar1), 104.9 (C5-Ar2), 54.5 and 54.2 (d, J(PC) = 7.2 Hz, CH3), 44.9 (d, J(PC) = 138.7 Hz, C4a-Ar1), 36.0 (C2a-Ar1) and 31.4 (C2b-Ar1). 31P NMR (162 MHz, Acetone-d6) δ 27.30. Found: C, 51.15; H, 5.24; N, 14.83; P, 4.77. Anal. calcd (%) for C28H34N7O10P: C, 50.99; H, 5.20; N, 14.87; P, 4.70. HRMS (ESI) m/z for C28H34N7O10P: calc. 659.21 [M]+, found 658.15 [M-H]+.
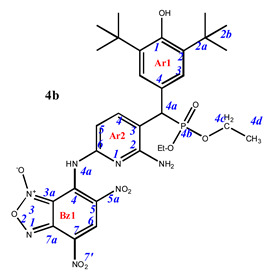
7-((6-amino-5-((3,5-di-tert-butyl-4-hydroxyphenyl)(diethoxyphosphoryl)methyl)pyridin-2-yl)amino)-4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide (4b). Purple powder, yield 78%. M.p.: 142–143 °C. IR (ν, cm–1): 703 (P–C), 1226 (P=O), 1365 (NO2 symm), 1563 (NO2 asymm), 1620 (furoxan ring) and 3440 (NH2); 3616 (OH). Minor amounts of the other tautomer are present in the spectra. 1H NMR (500 MHz, Acetone-d6): δ 9.08 (s, 1H, H6-Bz1), 8.14 (dd, J = 8.1 Hz, J(PH) = 1.5 Hz, 1H, H4-Ar2), 7.44 (d, J(PH) = 1.8 Hz, 2H, H3-Ar1), 6.66 (brs, 1H, H5-Ar2), 4.63 (d, J(PH) = 26.9 Hz, 1H, H4a-Ar1), 4.06 (m, 4H, CH2), 1.47 (s, 18H, H2b-Ar1), 1.24 and 1.17 (tr, J = 7.2 Hz, 6H, CH3). 13C{1H} NMR (126 MHz, Acetone-d6) δ 156.1 (C2-Ar2), 154.7 (C1-Ar1), 151.8 (C6-Ar2), 148.1 (C4-Bz), 144.1 (C4-Ar2), 140.2 (C7a-Bz), 139.0 (C2/C6-Ar1), 133.9 (C6-Bz), 127.9 (C7-Bz), 127.8 (d, J(PC) = 7.3 Hz, C3-Ar1), 127.4 (C3-Ar2), 124.2 (C5-Bz), 115.0 (C3a-Bz), 114.0 (C4-Ar1), 104.9 (C5-Ar2), 64.2 and 53.9 (m, CH2), 45.0 (d, J(PC) = 138.7 Hz, C4a-Ar1), 35.9 (C2a-Ar1), 31.3 (C2b-Ar1) and 17.4 and 17.3 (m, CH3). 31P NMR (162 MHz, Acetone-d6) δ 25.01. Found: C, 52.45; H, 5.52; N, 14.33; P, 4.57. Anal. calcd (%) for C30H38N7O10P: C, 52.40; H, 5.57; N, 14.26; P, 4.50. HRMS (ESI) m/z for C30H38N7O10P: calc. 687.24 [M]+, found 686.18 [M-H]+.
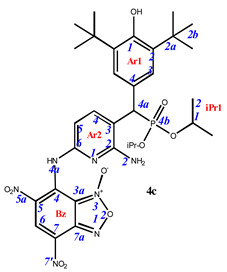
7-((6-amino-5-((3,5-di-tert-butyl-4-hydroxyphenyl)(diisopropoxyphosphoryl)methyl)pyridin-2-yl)amino)-4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide (4c). Claret powder, yield 69%. M.p.: 119–120 °C. IR (ν, cm–1): 705 (P–C), 1206 (P=O), 1364 (NO2 symm), 1569 (NO2 asymm), 1621 (furoxan ring) and 3444 (NH2); 3620 (OH). 1H NMR (500 MHz, Acetone-d6): δ 11.34 (s, 1H, NH- or H4a-Bz), 9.08 (s, 1H, H6-Bz), 8.12 (dd, J = 8.2 Hz, J(PH) = 1.7 Hz, 1H, H4-Ar2), 7.46 (d, J(PH) = 1.8 Hz, 2H, H3-Ar1), 6.67 (d, J = 7.9 Hz, 1H, H5-Ar2), 6.12 (s, 1H, OH-Ar1), 4.76 (brs, 2H, NH2-Ar2), 4.68 and 4.57 (m, 8H, H1-iPr), 4.56 (d, J(PH) = 26.9 Hz, 1H, H4a-Ar1), 1.47 (s, 18H, H2b-Ar1), 1.30, 1.29, 1.16 and 0.99 (d, J = 6.2 Hz, 12H, H2-iPr). 13C{1H} NMR (126 MHz, Acetone-d6) δ 156.3 (d, J(PC) = 10.1 Hz, C2-Ar2), 154.8 (d, J(PC) = 14.3 Hz, C1-Ar1), 151.5 (C6-Ar2), 148.2 (C4-Bz), 143.8 (d, J(PC) = 6.9 Hz, C4-Ar2), 139.9 (C7a-Bz), 137.0 (d, J(PC) = 15.6 Hz, C2-Ar1), 133.7 (C6-Bz), 128.2 (d, J(PC) = 5.6 Hz, C4-Ar1), 128.0 (d, J(PC) = 7.5 Hz, C3-Ar1), 127.7 (C7-Bz), 124.1 (C5-Bz), 115.5 (C3-Ar2), 113.9 (C3a-Bz), 104.4 (C5-Ar2), 73.0 and 72.5 (d, J(PC) = 7.2 Hz, C1-iPr), 46.0 (d, J(PC) = 140.4 Hz, C4a-Ar1), 35.9 (C2a-Ar1), 31.3 (C2b-Ar1), 25.2, 25.0 24.6 and 24.4 (d, J(PC) = 5.4 Hz, C1-iPr). 31P NMR (162 MHz, Acetone-d6) δ 23.69. Found: C, 53.75; H, 5.86; N, 13.64; P, 4.22. Anal. calcd (%) for C32H42N7O10P: C, 53.70; H, 5.92; N, 13.70; P, 4.33. HRMS (ESI) m/z for C32H42N7O10P: calc. 715.27 [M]+, found 716.27 [M+H]+, 714.25 [M-H]+.
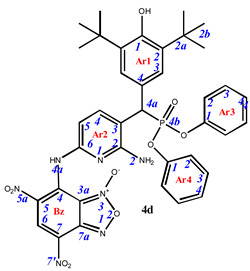
7-((6-amino-5-((3,5-di-tert-butyl-4-hydroxyphenyl)(diphenoxyphosphoryl)methyl)pyridin-2-yl)amino)-4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide (4d). Dark cherry powder, yield 76%. M.p.: 156–157 °C. IR (ν, cm–1): 690 (P–C), 1211 (P=O), 1378 (NO2 symm), 1565 (NO2 asymm), 1592 (C=Carom), 1622 (furoxan ring), 3437 (NH2). 1H NMR (500 MHz, CDCl3): δ 10.69 (s, 1H, NH- or H4a-Bz), 9.18 (s, 1H, H6-Bz), 8.03 (d, J = 7.9 Hz, 1H, H4-Ar2), 7.27 (tr, J = 8.5 Hz, 2H, H3/H5-Ar4), 7.27 (s, 2H, H3-Ar1), 7.21 (tr, J = 8.5 Hz, 2H, H3/H5-Ar3), 7.16 (tr, J = 8.5 Hz, 1H, H4-Ar4), 7.10 (tr, J = 8.5 Hz, 1H, H4-Ar3), 6.95 (d, J = 8.5 Hz, 2H, H2/H6-Ar4 [Ar4 cis- to 4a of Ar1]), 6.80 (d, J = 8.5 Hz, 2H, H2/H6-Ar3), 6.63 (d, J = 7.9 Hz, 1H, H5-Ar2), 5.28 (s, 1H, OH-Ar1), 4.84 (s, 2H, NH2-Ar21), 4.61 (d, J(PH) = 26.7 Hz 1H, H4a-Ar1) and 1.40 (s, 18H, H2b-Ar1). 13C{1H} NMR (126 MHz, CDCl3) δ 156.5 (d, J(PC) = 10.1 Hz, C2-Ar2), 154.0 (C1-Ar1), 150.8 (d, J(PC) = 9.7 Hz, C1-Ar3), 150.3 (d, J(PC) = 9.7 Hz, C1-Ar4 [Ar4 cis- to 4a of Ar1] ), 148.2 (C6-Ar2), 145.8 (C4-Bz), 141.5 (d, J(PC) = 6.9 Hz, C4-Ar2), 137.0 (C2-Ar1), 136.1 (C7a-Bz), 130.8 (C6-Bz), 129.9 (C3-Ar3), 129.8 (C3-Ar4), 127.11 (C7-Bz), 126.5 (C5-Bz), 126.7 (d, J(PC) = 7.5 Hz, C3-Ar1), 125.9 (C4-Ar3), 125.3 (C4-Ar4), 123.3 (d, J(PC) = 5.6 Hz, C4-Ar1), 120.7 (d, J(PC) = 4.1 Hz, C2-Ar3), 120.3 (d, J(PC) = 4.1 Hz, C2-Ar4), 114.9 (C3-Ar2), 111.7 (C3a-Bz), 106.1 (C5-Ar2), 46.1 (d, J(PC) = 140.4 Hz, C4a-Ar1) and 34.6 (C2a-Ar1), 30.4 (C2b-Ar1). 31P NMR (162 MHz, CDCl3) δ 19.09. Found: C, 58.29; H, 4.81; N, 12.45; P, 3.89. Anal. calcd (%) for C38H38N7O10P: C, 58.24; H, 4.89; N, 12.51; P, 3.95. HRMS (ESI) m/z for C38H38N7O10P: calc. 783.24 [M]+, found 782.20 [M-H]+.
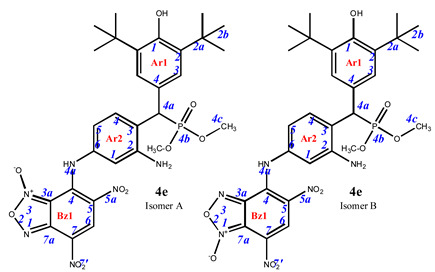
7-((3-amino-4-((3,5-di-tert-butyl-4-hydroxyphenyl)(dimethoxyphosphoryl)methyl)phenyl)amino)-4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide (4e). Maroon powder, yield 72%. M.p.: 138–139 °C. IR (ν, cm–1): 681 (P–C), 1237 (P=O), 1376 (NO2 symm), 1569 (NO2 asymm), 1590 (C=Carom), 1621 (furoxan ring), 3415 (NH2) and 3625 (OH). Exists in solution as two isomers. Major isomer A (85%): 1H NMR (500 MHz, CDCl3): δ 11.17 (brs, NH-Ar2), 9.22 (s, 1H, H6-Bz1), 7.44 (dd, J = 8.2 Hz, J(PH) = 1.6 Hz, 1H, H4-Ar2), 7.22 (d, J(PH) = 1.5 Hz, 2H, H3-Ar1), 6.54 (d, J = 1.7 Hz, 1H, H1-Ar2), 6.50 (dd, J = 8.2 Hz, J = 1.7 Hz, 1H, H5-Ar2), 5.22 (s, 1H, OH-Ar1), 4.56 (d, J(PH) = 26.5 Hz, 1H, H4a-Ar1), 3.62 and 3.66 (d, J = 10.7 Hz, 6H, CH3) and 1.43 (s, 18H, H2b-Ar1). 13C{1H} NMR (126 MHz, CDCl3) δ 153.5 (C1-Ar1), 146.8 (C2-Ar2), 146.0 (C4-Bz), 138.1 (C7a-Bz), 137.8 (C6-Ar2), 136.4 (C2-Ar1), 132.3 (d, J(PC) = 6.3 Hz, C4-Ar2), 131.4 (C6-Bz), 126.5 (d, J(PC) = 7.6 Hz, C3-Ar1), 125.3 (C7-Bz), 125.0 (d, J(PC) = 5.2 Hz, C4-Ar1), 124.5 (C5-Bz), 123.6 (C3-Ar2), 112.0 (C5-Ar2), 110.2 (C1-Ar2), 109.9 (C3a-Bz), 54.0 and 53.5 (d, J(PC) = 7.1 Hz, CH3), 45.6 (d, J(PC) = 139.2 Hz, C4a-Ar1), 34.6 (C2a-Ar1) and 30.5 (C2b-Ar1). 31P NMR (162 MHz, CDCl3) δ 27.28. Isomer B (15%), since the concentration is low, only part of the peaks can be correlated: 1H NMR (500 MHz, CDCl3): δ 11.49 (s, NH-Ar2), 8.93 (s, 1H, H6-Bz1), 7.51 (dd, J = 8.2 Hz, J(PH) = 1.6 Hz, 1H, H4-Ar2), 7.25 (d, J(PH) = 1.5 Hz, 2H, H3-Ar1), 6.76 (d, J = 1.7 Hz, 1H, H1-Ar2), 6.73 (dd, J = 8.2 Hz, J = 1.7 Hz, 1H, H5-Ar2), 5.22 (s, 1H, OH-Ar1), 4.61 (d, J(PH) = 26.5 Hz, 1H, H4a-Ar1), 3.66 and 3.62 (d, J = 10.7 Hz, 6H, CH3) and 1.43 (s, 18H, H2b-Ar1). 31P NMR (162 MHz, CDCl3) δ 28.27. Found: C, 52.95; H, 5.29; N, 12.68; P, 4.65. Anal. calcd (%) for C29H35N6O10P: C, 52.89; H, 5.36; N, 12.76; P, 4.70. HRMS (ESI) m/z for C29H35N6O10P: calc. 658.22 [M]+ and found 657.16 [M-H]+.
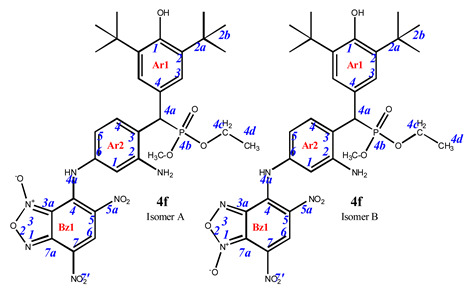
7-((3-amino-4-((3,5-di-tert-butyl-4-hydroxyphenyl)(diethoxyphosphoryl)methyl)phenyl)amino)-4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide (4f). Dark cherry powder, yield 78%. M.p.: 201–202 °C. IR (ν, cm–1): 695 (P–C), 1239 (P=O), 1376 (NO2 symm), 1570 (NO2 asymm), 1621 (furoxan ring), 3250 (NH2) and 3439 (OH). Exists in solution as two isomers. Major isomer A (94%): 1H NMR (400 MHz, DMSO-d6): δ 8.91 (s, 1H, H6-Bz1), 7.54 (dd, J = 8.4 Hz, J(PH) = 1.4 Hz, 1H, H4-Ar2), 7.24 (d, J(PH) = 1.8 Hz, 2H, H3-Ar1), 6.61 (s, 1H, H1-Ar2), 6.55 (dd, J = 8.3 Hz, J = 1.7 Hz, 1H, H5-Ar2), 4.63 (d, J(PH) = 26.7 Hz, 1H, H4a-Ar1), 3.86 (m, 6H, CH3), 1.35 (s, 18H, H2b-Ar1) and 1.09 (m, 4H, CH2). 13C NMR (101 MHz, DMSO-d6) δ 153.6, 147.9, 139.6, 139.0, 138.3, 133.9, 131.4 (d, J(PC) = 5.5 Hz), 129.9, 129.2, 127.6 (d, J(PC) = 5.3 Hz), 127.0 (d, J(PC) = 7.0 Hz), 126.4 (d, J(PC) = 13.4 Hz), 121.4, 112.8, 110.6, 109.8, 62.9 (dd, J(PC) = 20.3, 6.8 Hz), 35.6, 31.4, 22.0 and 17.1 (d, J(PC) = 3.5 Hz). 31P NMR (162 MHz, DMSO-d6) δ 27.89. Found: C, 53.62; H, 5.49; N, 12.45; P, 4.69. Anal. calcd (%) for C30H37N6O10P: C, 53.57; H, 5.54; N, 12.49; P, 4.60.
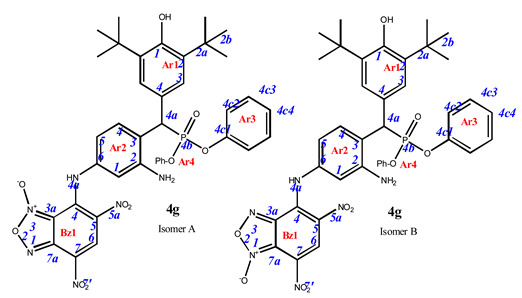
7-((3-amino-4-((3,5-di-tert-butyl-4-hydroxyphenyl)(diphenoxyphosphoryl)methyl)phenyl)amino)-4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide (4g). Purple powder, yield 81%. M.p.: 139–140 °C. IR (ν, cm–1): 689 (P–C), 1238 (P=O), 1377 (NO2 symm), 1567 (NO2 asymm), 1590 (C=Carom), 1620 (furoxan ring), 3437 (NH2) and 3625 (OH). Exists in solution as two isomers. Isomer A (80%): 1H NMR (500 MHz, CDCl3): δ 11.12 (br.s, NH-Ar2), 9.22 (s, 1H, H6-Bz1), 7.67 (dd, J = 8.2 Hz, J(PH) = 1.6 Hz, 1H, H4-Ar2), 7.31 (d, J(PH) = 1.5 Hz, 2H, H3-Ar1), 7.26 and 7.22 (tr, J = 8.0 Hz, 4H, H3/H5 -Ar3 and Ar4), 7.10 and 7.15 (tr, J = 8.0 Hz, 2H, H2/H6-Ar3 and Ar4), 6.92 and 6.82 (d, J = 8.0 Hz, 4H, H2/H6-Ar3 and Ar4), 6.54 (d, J = 1.7 Hz, 1H, H1-Ar2), 6.53 (dd, J = 8.2 Hz, J = 1.7 Hz, 1H, H5-Ar2), 5.25 (s, 1H, OH-Ar1), 4.84 (d, J(PH) = 26.4 Hz, 1H, H4a-Ar1), 1.40 (s, 18H, H2b-Ar1). 13C{1H} NMR (126 MHz, CDCl3) δ 153.8 (C1-Ar1), 150.7 and 150.4 (d, J(PC) = 9.7 Hz, C4c1 -Ar3 and Ar4), 147.1 (C2-Ar2), 146.0 (C4-Bz), 138.0 (C7a-Bz), 138.0 (C6-Ar2), 136.7 (C2-Ar1), 132.2 (d, J(PC) = 6.4 Hz, C4-Ar2), 131.4 (C6-Bz), 129.9 and 129.8 (C4c3/C4c5 -Ar3 and Ar4), 127.1 (d, J(PC) = 9.5 Hz, C3-Ar1), 125.5 and 125.2 (C4c4 -Ar3 and Ar4), 125.3 (C7-Bz), 125.0 (C5-Bz),124.1 (d, J(PC) = 5.2 Hz, C4-Ar1), 122.9 (C3-Ar2), 120.8 and 120.4 (d, J(PC) = 4.2 Hz, C4c2/C4c6 -Ar3 and Ar4), 112.0 (C5-Ar2), 110.2 (C1-Ar2), 110.0 (C3a-Bz), 45.9 (d, J(PC) = 139.2 Hz, C4a-Ar1), 34.6 (C2a-Ar1) and 30.4 (C2b-Ar1). 31P NMR (162 MHz, CDCl3) δ 18.50. Isomer B (20%), since the concentration is low, only part of the peaks can be correlated: 1H NMR (500 MHz, CDCl3): δ 11.48 (brs, NH-Ar2), 8.93 (s, 1H, H6-Bz1), 7.76 (dd, J = 8.2 Hz, J(PH) = 1.6 Hz, 1H, H4-Ar2), 7.33 (d, J(PH) = 1.5 Hz, 2H, H3-Ar1), 7.26 and 7.22 (tr, J = 8.0 Hz, 4H, H3/H5 -Ar3 and Ar4), 7.15 and 7.10 (tr, J = 8.0 Hz, 2H, H2/H6-Ar3 and Ar4), 6.92 and 6.82 (d, J = 8.0 Hz, 4H, H2/H6-Ar3 and Ar4), 6.79 (dd, J = 8.2 Hz, J = 1.7 Hz, 1H, H5-Ar2), 6.74 (d, J = 1.7 Hz, 1H, H1-Ar2), 5.19 (s, 1H, OH-Ar1), 4.88 (d, J(PH) = 26.4 Hz, 1H, H4a-Ar1) and 1.40 (s, 18H, H2b-Ar1). 31P NMR (162 MHz, CDCl3) δ 20.09. Found: C, 59.91; H, 4.95; N, 10.69; P, 4.01. Anal. calcd (%) for C39H39N6O10P: C, 59.84; H, 5.02; N, 10.74; P, 3.96. HRMS (ESI) m/z for C39H39N6O10P: calc. 782.25 [M]+ and found 781.21 [M-H]+.
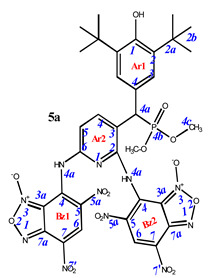
7,7’-((3-((3,5-di-tert-butyl-4-hydroxyphenyl)(dimethoxyphosphoryl)methyl)pyridine-2,6-diyl)bis(azanediyl))bis(4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide) (5a). Bright red powder, yield 91%. M.p.: 160–161 °C. IR (ν, cm–1): 689 (P–C), 1213 (P=O), 1378 (NO2 symm), 1575 (NO2 asymm), 1594 (C=Carom), 1626 (furoxan ring), 3437 (NH2), 3632 (OH). 1H NMR (500 MHz, Acetone-d6): δ 11.05 (s, 2H, NH or H4a -Bz1 and -Bz2), 9.08 (s, 1H, H6-Bz2), 8.61 (s, 1H, H6-Bz1), 8.36 (d, J = 8.4 Hz, 1H, H4-Ar2), 7.58 (2H, H3-Ar1), 7.10 (brs, 1H, H5-Ar2), 5.94 (s, 1H, OH-Ar1), 5.65 (d, J(PH) = 12.4 Hz, 1H, H4a-Ar1), 3.66 and 3.61 (d, J = 10.3 Hz, 6H, CH3), 1.46 (s, 18H, H2b-Ar1). 13C{1H} NMR (Acetone-d6, 126 MHz) δ 157.4 (C2-Ar2), 157.3 (C6-Ar2), 154.5 (C1-Ar1), 149.5 (C4-Bz2), 148.1 (C4-Bz1), 141.6 (C4-Ar2), 141.5 (C7a-Bz1), 138.8 (C2-Ar1), 138.7 (C7a-Bz1), 134.4 (C6-Bz1), 132.8 (C6-Bz2), 130.0 (C5-Bz1), 129.8 (C4-Ar1), 129.5 (C5-Bz2), 128.6 (C7-Bz1), 128.0 (C7-Bz2), 127.9 (d, J(PC) = 7.5 Hz, C3-Ar1), 115.7 (C5-Ar2), 114.4 (C3a-Bz1), 113.7 (C3a-Bz2), 111.0 (C3-Ar2), 54.4 (CH3), 43.2 (d, J(PC) = 140.0 Hz, C4a-Ar1), 35.9 (C2a-Ar1), 31.4 (C2b-Ar1). 31P NMR (162 MHz, CDCl3) δ 27.44. Found: C, 46.27; H, 3.84; N, 17.48; P, 3.49. Anal. calcd (%) for C34H34N11O16P: C, 46.21; H, 3.88; N, 17.44; P, 3.51. HRMS (ESI) m/z for C34H34N11O16P: calc. 883.19 [M]+, found 882.12 [M-H]+.
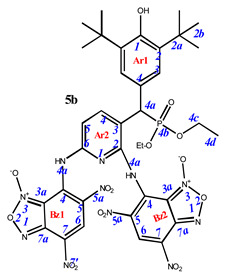
7,7’-((3-((3,5-di-tert-butyl-4-hydroxyphenyl)(diethoxyphosphoryl)methyl)pyridine-2,6-diyl)bis(azanediyl))bis(4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide) (5b). Red powder, yield 88%. M.p.: >250 °C. IR (ν, cm–1): 696 (P–C), 1211 (P=O), 1378 (NO2 symm), 1571 (NO2 asymm), 1624 (furoxan ring) and 3436 (OH). Minor amounts of the other tautomer are present in the spectra. 1H NMR (500 MHz, Acetone-d6): δ 9.05 (s, 1H, H6-Bz2), 8.63 (brs, 1H, H6-Bz1), 8.40 (m, 1H, H4-Ar2), 7.54 (d, J(PH) = 0.6 Hz, 2H, H3-Ar1), 7.07 (d, J = 8.4 Hz, 1H, H5-Ar2), 5.95 (s, 1H, OH-Ar1), 5.53 (d, J(PH) = 25.3 Hz, 1H, H4a-Ar1), 4.02 (m, 4H, H4c-Ar1), 1.45 (s, 18H, H2b-Ar1) and 1.33 and 1.20 (d, J = 6.6 Hz, 6H, CH3 or H4d-Ar1). 13C{1H} NMR (Acetone-d6, 126 MHz) δ 154.9 (C1-Ar1), 149.6 (C2-Ar2), 149.5 (C6-Ar2), 148.2 (C4-Bz2 and C4-Bz1), 142.5 (d, J(PC) = 4.7 Hz, C4-Ar2), 141.8 (C7a-Bz1), 138.7 (C2-Ar1), 138.6 (C7a-Bz1), 134.5 (C6-Bz2), 1330 (C6-Bz1), 129.4 (C5-Bz1), 128.4 (C5-Bz2), 128.3 (C7-Bz1), 128.2 (C7-Bz2), 128.0 (d, J(PC) = 8.0 Hz, C3-Ar1), 127.1 (d, J(PC) = 4.3 Hz, C4-Ar1), 115.7 (d, J(PC) = 5.7 Hz, C3-Ar2), 114.3 (C3a-Bz1), 113.6 (C3a-Bz2), 111.0 (C5-Ar2), 63.7 (m, C4c-Ar1), 44.0 (d, J(PC) = 140.3 Hz, C4a-Ar1), 35.9 (C2a-Ar1), 31.4 (C2b-Ar1) and 17.3 (m, C4d-Ar1). 31P NMR (162 MHz, Acetone-d6) δ 25.97. Found: C, 47.48; H, 4.27; N, 16.88; P, 3.45. Anal. calcd (%) for C36H38N11O16P: C, 47.43; H, 4.20; N, 16.90; P, 3.40. HRMS (ESI) m/z for C36H38N11O16P: calc. 911.22 [M]+ and found 910.16 [M-H]+.
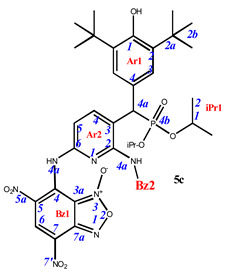
7,7’-((3-((3,5-di-tert-butyl-4-hydroxyphenyl)(diisopropoxyphosphoryl)methyl)pyridine-2,6-diyl)bis(azanediyl))bis(4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide) (5c). Brick red powder, yield 78%. M.p.: >250 °C. IR (ν, cm–1): 695 (P–C), 1210 (P=O), 1360 (NO2 symm), 1565 (NO2 asymm), 1625 (furoxan ring) and 3444 (OH). 1H NMR (500 MHz, Acetone-d6): δ 11.30 (s, 1H, NH- or H4a-Bz2), 10.64 (s, 1H, NH- or H4a-Bz1), 9.07 (s, 1H, H6-Bz2), 8.90 (s, 1H, H6-Bz1), 8.15 (dd, J = 8.4 Hz, J(PH) = 1.0 Hz, 1H, H4-Ar2), 7.56 (d, J(PH) = 0.6 Hz, 2H, H3-Ar1), 7.53 (d, J = 8.4 Hz, 1H, H5-Ar2), 6.22 (s, 1H, OH-Ar1), 4.98 (d, J(PH) = 25.3 Hz, 1H, H4a-Ar1), 4.83 and 4.68 (sept, J(PH) = 6.3 Hz, 8H, H1-iPr), 1.49 (s, 18H, H2b-Ar1), 1.40, 1.38, 1.18 and 1.09 (d, J = 6.2 Hz, 12H, H2-iPr). 13C{1H} NMR (126 MHz, Acetone-d6,) δ 155.0 (C1-Ar1), 149.9 (C2-Ar2), 149.8 (C6-Ar2), 147.4 (C4-Bz2), 147.3 (C4-Bz1), 144.0 (d, J(PC) = 4.7 Hz, C4-Ar2), 139.5 (C7a-Bz1), 139.3 (C2-Ar1), 138.3 (C7a-Bz1), 132.1 (C6-Bz2), 132.0 (C6-Bz1), 130.9 (C5-Bz1), 130.0 (C5-Bz2), 129.7 (C7-Bz1), 128.4 (d, J(PC) = 8.0 Hz, C3-Ar1), 128.2 (C7-Bz2), 127.1 (d, J(PC) = 4.3 Hz, C4-Ar1), 125.9 (d, J(PC) = 5.7 Hz, C3-Ar2), 115.2 (C5-Ar2), 114.2 (C3a-Bz1), 113.6 (C3a-Bz2), 73.5 and 73.0 (d, J(PC) = 7.0 Hz, C1-iPr), 46.5 (d, J(PC) = 140.2 Hz, C4a-Ar1), 36.0 (C2a-Ar1), 31.3 (C2b-Ar1), 25.2, 25.0, 24.7 and 24.4 (d, J(PC) = 5.3 Hz, C1-iPr). 31P NMR (162 MHz, Acetone-d6) δ 24.34. Found: 48.54; H, 4.54; N, 16.32; P, 3.35. Anal. calcd (%) for C38H42N11O16P: C, 48.57; H, 4.50; N, 16.39; P, 3.30. HRMS (ESI) m/z for C38H42N11O16P: calc. 939.25 [M]+ and found 938.24 [M-H]+.
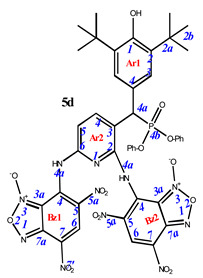
7,7’-((3-((3,5-di-tert-butyl-4-hydroxyphenyl)(diphenoxyphosphoryl)methyl)pyridine-2,6-diyl)bis(azanediyl))bis(4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide) (5d). Brick red powder, yield 75%. M.p.: >250 °C. IR (ν, cm–1): 695 (P–C), 1209 (P=O), 1378 (NO2 symm), 1569 (NO2 asymm), 1622 (furoxan ring) and 3444 (OH). 1H NMR (400 MHz, DMSO-d6) δ 8.87 (s, 1H), 8.67 (s, 1H), 8.34 (d, J = 8.4 Hz, 1H), 8.28 (s, 1H), 7.30 (d, J = 8.1 Hz, 2H), 7.17 (m, 5H), 7.07 (m, 1H), 6.99 (d, J = 8.3 Hz, 2H), 6.87 (br.s, 1H), 6.64 (d, J = 8.2 Hz, 2H), 5.26 (m, 1H) and 1.24 (s, 18H). 13C NMR (126 MHz, DMSO-d6) δ 162.8, 161.7, 153.8, 150.4, 148.4, 147.7, 139.6, 135.2, 134.6, 130.4, 130.2, 130.2, 130.1, 129.9, 129.8, 127.6, 126.6, 125.6, 125.3, 120.9, 120.8, 120.4, 115.1, 111.9, 111.4, 36.2, 34.9 and 30.5.31P NMR (162 MHz, DMSO-d6) δ 18.80. Found: %: C, 52.49; H, 3.77; N, 15.35; P, 3.01. Anal. calcd (%) for C44H38N11O16P: C, 52.44; H, 3.80; N, 15.29; P, 3.07.
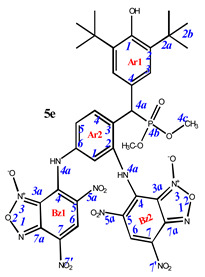
7,7’-((4-((3,5-di-tert-butyl-4-hydroxyphenyl)(dimethoxyphosphoryl)methyl)-1,3-phenylene)bis(azanediyl))bis(4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide) (5e). Red powder, yield 68%. M.p.: >250 °C. IR (ν, cm–1): 689 (P–C), 1212 (P=O), 1378 (NO2 symm), 1578 (NO2 asymm), 1597 (C=Carom), 1624 (furoxan ring) and 3449 (OH). 1H NMR (400 MHz, DMSO-d6) δ 9.00 (s, 1H, H6-Bz2), 8.91 (s, 1H, H6-Bz1), 7.32 (s, 2H, H3-Ar1), 7.18 (s, 1H, Ar2), 7.09 (s, 1H, Ar2), 6.90 (s, 1H, Ar2), 5.07 (d, J(PH) = 24.4 Hz, 1H, H4a-Ar1), 3.62 and 3.58 (d, J = 10.4 Hz, 6H, CH3) and 1.35 (s, 18H, H2b-Ar1). 13C{1H} NMR (DMSO-d6, 126 MHz) δ 161.8, 153.6, 153.3, 148.5, 147.3, 147.3, 139.7, 139.5, 139.4 (d, J(PC) = 7.7 Hz), 138.1, 135.3, 132.9, 131.1, 127.7, 126.9, 126.9, 126.8, 126.6, 126.4, 115.2, 112.6, 111.5, 53.5 (d, J(PC) = 35.4 Hz), 35.1, 30.9 (d, J(PC) = 8.1 Hz) and 30.6. 31P NMR (162 MHz, DMSO-d6) δ 28.65. Found: C, 47.69; H, 4.07; N, 15.82; P, 3.56. Anal. calcd (%) for C35H35N10O16P: C, 47.63; H, 4.00; N, 15.87; P, 3.51. HRMS (ESI) m/z for C35H35N10O16P: calc. 882.20 [M]+ and found 881.12 [M-H]+.
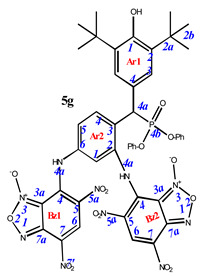
7,7’-((4-((3,5-di-tert-butyl-4-hydroxyphenyl)(diphenoxyphosphoryl)methyl)-1,3-phenylene)bis(azanediyl))bis(4,6-dinitrobenzo[c][1,2,5]oxadiazole 1-oxide) (5g). Red powder, yield 55%. M.p.: 196–197 °C. Minor amounts of the other tautomer are present in the spectra. 1H NMR (600 MHz, DMSO-d6) δ 9.06 (s, 2H), 7.83 (dd, J = 8.3, J (PH) = 1.5 Hz, 1H), 7.39 (s, 2H), 7.32 (m, 2H), 7.19 (m, 4H), 7.10 (m, 1H), 6.93 (d, J = 8.5 Hz, 2H), 6.63 (m, 2H), 6.59 (d, J = 8.3 Hz, 1H), 5.18 (d, J(PH) = 27.9 Hz, 1H) and 1.31 (s, 18H). 13C NMR (101 MHz, DMSO-d6) δ 162.2, 154.3, 151.3 (d, J = 9.7 Hz), 150.9 (d, J = 10.3 Hz), 148.9, 140.1, 135.8, 131.8 (d, J = 5.2 Hz), 130.7, 130.5, 130.4, 130.3, 128.1, 127.3 (d, J = 7.8 Hz), 126.2, 126.1, 125.8, 121.4 (d, J = 3.7 Hz), 121.3, 121.1 (d, J = 3.7 Hz), 120.5, 116.2, 115.7, 111.9, 111.5, 110.4, 35.5 and 31.2. 31P NMR (162 MHz, DMSO-d6) δ 20.38. Found: C, 53.59; H, 3.88; N, 13.90; P, 3.05. Anal. calcd (%) for C45H39N10O16P: C, 53.68; H, 3.90; N, 13.91; P, 3.08. HRMS (ESI) m/z for C45H39N10O16P: calc. 1006.23 [M]+ and found 1005.19 [M-H]+.
3.2. Biology
3.2.1. Cells and Materials
For the experiments, we used tumor cell cultures: M-HeLa clone 11 (epithelioid carcinoma of the cervix, subline HeLa., clone M-HeLa); T 98G—human glioblastoma; PANC-1, human pancreatic carcinoma; HuTu 80, human duodenal adenocarcinoma; MCF7—human breast adenocarcinoma (pleural fluid); A549, human lung carcinoma; WI38, VA 13 subline 2RA, human embryonic lung from the collection of the Institute of Cytology, Russian Academy of Sciences (St. Petersburg); PC3—prostate adenocarcinoma cell line from ATCC (American Type Cell Collection, USA; CRL 1435; human liver cells (Chang liver) from the collection and the Research Institute of Virology of the Russian Academy of Medical Sciences (Moscow). The cells were cultured in a standard Eagle’s nutrient medium manufactured at the Chumakov Institute of Poliomyelitis and Virus Encephalitis (PanEco company, Moscow, Russia), and supplemented with 10% fetal calf serum (Biosera, France) and 1% nonessential amino acids (PanEco company, Russia).
3.2.2. Cytotoxicity Assay
The cytotoxic effect on cells was determined using the colorimetric method of cell proliferation—the MTT test. NADP-H-dependent cellular oxidoreductase enzymes can, under certain conditions, reflect the number of viable cells. These enzymes are able to reduce the tetrazolium dye, (MTT)—3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide, to insoluble blue-violet formazan, which crystallizes inside the cell. The amount of formazan formed is proportional to the number of cells with active metabolism [46]. Cells were seeded on a 96-well Nunc plate at a concentration of 5 × 103 cells per well in a volume of 100 μL of medium and cultured in a CO2 incubator at 37 °C until a monolayer was formed. The process of cell monolayer formation took 24 h. Then, the nutrient medium was removed, and 100 µL of solutions of the test drug in the given dilutions were added to the wells, which were prepared directly in the nutrient medium with the addition of 5% DMSO to improve solubility. After 48 h of incubation of the cells with the tested compounds, the nutrient medium was removed from the plates, and 100 µL of the nutrient medium without serum with MTT at a concentration of 0.5 mg/mL was added and incubated for 4 h at 37 °C. Formazan crystals were added 100 µL of DMSO to each well. Optical density was recorded at 540 nm on an Invitrologic microplate reader (Russia). The experiments for all compounds were repeated three times.
3.2.3. Flow Cytometry Assay
Cell Culture. M-HeLa cells at 1 × 106 cells/well in a final volume of 2 mL were seeded into six-well plates. After 48 h of incubation, various concentrations of compounds 4c and 5d were added to wells.
Cell Apoptosis Analysis. The cells were harvested at 2000 rpm for 5 min and, then washed twice with ice-cold PBS, followed by resuspension in binding buffer. Next, the samples were incubated with 5 μL of annexin V- Alexa Fluor 647 (Sigma-Aldrich, St. Louis, MO, USA) and 5 μL of propidium iodide for 15 min at room temperature in the dark. Finally, the cells were analyzed by flow cytometry (Guava easy Cyte, Merck, Rahway, NJ, USA) within 1 h. The experiments were repeated three times.
Mitochondrial Membrane Potential. Cells were harvested at 2000 rpm for 5 min and then washed twice with ice-cold PBS, followed by resuspension in JC-10 (10 µg/mL) and incubation at 37 °C for 10 min. After the cells were rinsed three times and suspended in PBS, the JC-10 fluorescence was observed by flow cytometry (Guava easy Cyte, Merck, Rahway, NJ, USA).
Detection of Intracellular ROS. M-HeLa cells were incubated with compounds at concentrations of IC50 for 48 h. ROS generation was investigated using flow cytometry assay and CellROX® Deep Red flow cytometry kit. For this M-HeLa cells were harvested at 2000 rpm for 5 min and then washed twice with ice-cold PBS, followed by resuspension in 0.1 mL of medium without FBS, to which was added 0.2 μL of CellROX® Deep Red and incubated at 37 °C for 30 min. After three times washing the cells and suspending them in PBS, the production of ROS in the cells was immediately monitored using flow cytometer (Guava easy Cyte, Merck, Rahway, NJ, USA).
3.2.4. Antimicrobial Activity
Antimicrobial activity of test compounds was determined by serial micro dilutions in 96-well plates using Mueller-Hinton broth for bacterial culture and Sabouraud broth for yeast culture [47]. Cultures of gram-positive bacteria were used in the experiment: Staphylococcus aureus ATCC 6538 P FDA 209P, Bacillus cereus ATCC 10702 NCTC 8035, Enterococcus faecalis ATCC 29212; Gram-negative bacteria: Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 9027 and yeast: Candida albicans ATCC 10231, purchased from the State Collection of Pathogenic Microorganisms and Cell Cultures “SCPM-Obolensk”. Methicillin-resistant strains of S. aureus (MRSA) were isolated from patients with chronic tonsillitis (MRSA-1) and sinusitis (MRSA-2) in the bacteriological laboratory of the Republican Clinical Hospital (Kazan, Russia). The experiments were carried out in triplicate.
3.2.5. Statistical Analysis
IC50 values were estimated using the Quest Graph IC50 Calculator (AAT Bioquest, Inc., Sunnyvale, CA, USA) (Version 2022) (accessed on 25 June 2022) [48].
3.3. Biostability Studies
3.3.1. Preparation of Spikes and Samples
A substance of 1 mg was dissolved in methanol to achieve the solution of 1 mg/mL. By a series of further dilutions, a sample containing 5000 ng/mL in the same solvent was prepared. A total of 50 μL of the prepared solution was added to 450 μL of whole blood, resulting in a spike containing 500 ng/mL of the compound in the matrix.
Aliquot of 10 μL was taken from the spike and added to a 100 μL of the precipitation solution consisting of a mixture of 0.2 M ZnSO4 in water and methanol (2:8, v/v). The sample was then vortex mixed for 20–30 s, incubated for 10–15 min, vortex mixed again and centrifuged for 10 min at 13,400 rpm (Eppendorf MiniSpin). Supernatant (100 μL) was transferred into a vial and analyzed.
3.3.2. Apparatus and LC-MS/MS Conditions
Analyses were carried out using a Shimadzu LC-20AD Prominence chromatograph (Shimadzu, Tokyo, Japan) equipped with a binary gradient pump, cooled autosampler SIL-20AC and column oven. A column packed with a reversed-phase sorbent ProntoSil 120-AQC18 (2 × 75 mm, 5 µm, EcoNova, Novosibirsk, Russia) was used for chromatographic separations. Mobile phase was water (eluent A) and MeOH (eluent B). The following gradient was used: 0 min—10% B; 1 min—90% B; 4.6 min—90%; 4.7 min—100% B; 6.0 min—100% B, followed by the equilibration of the column. Flow rate was 330 µL/min; injection volume was 10 µL. Mass spectrometric detection was performed on an ABSCIEX 6500 QTRAP mass spectrometer (AB SCIEX, Framingham, MA, USA) using negative electrospray ionization. The following parameters were set for the detection: scan mode—MRM, curtain gas (CUR) = 30 psi, collision-induced dissociation gas (CAD) = Medium, ion source voltage (IS) = 5500 V, gas drier temperature (TEM) = 250 °C, sprayer gas (GS1) = 15 psi, drier gas (GS2) = 20 psi, entrance potential (EP) = 10 V and dwell time = 80 msec. Detection parameters for agents 4c and 5d are shown in Table S1 (Supporting Information). The instruments were controlled, and the data were collected using Analyst 1.6.3 software (AB SCIEX); data processing was performed using MultiQuant 2.1 software (AB SCIEX).
3.4. Computations
Computations were calculated using Gaussian16 [49] with a M06-2X/6-311++G** level of theory [50]. Grimme’s dispersion correction (D3) was included [51]. Solvation was modeled using the solvation model based on density (SMD = H2O) [52].
4. Conclusions
The synthesis of novel sterically hindered phenols containing benzofuroxan fragments obtained via aromatic nucleophilic substitution reaction of 7-chloro-4,6-dinitrobenzofuroxan is presented. Depending on the initial ratio of reagents, it is possible to vary the composition of the final products, leading to the formation of compounds with a composition of 2:1 or compounds 1:1. Antimicrobial activity and antitumor potential were studied for the phenols/benzofuroxan hybrids. Most substances exhibit high cytotoxicity against human duodenal adenocarcinoma (HuTu 80), human breast adenocarcinoma (MCF-7) and human cervical carcinoma cell lines. The IC50 values of compounds 4c and 5d for these lines ranged from 0.9 to 5.9 µM and were either comparable to or exceeded the activity of Doxorubicin and Sorafenib. Moreover, the selectivity indices for healthy cells for the compound 5d also exceed those for the reference drugs. A study of the mechanisms of cytotoxicity suggests that the latter can be associated with the induction of apoptosis along the internal mitochondrial pathway and an increase in ROS production. Encouragingly, all tested compounds do not show hemolytic activity (HC50 >100 µM). The biostability of the leading compounds was evaluated in the whole blood of mice, where the substances remained unchanged for two h. This is a positive sign for their future quantitative determination in biological matrices. When studying antimicrobial activity, we note an interesting trend that the effect only appears when at least two benzofuroxan moieties are introduced per phenol.
Overall, the combination of sterically hindered phenol and benzofuroxan in one molecule leads to a number of positive effects, including increased ROS production and greater cytotoxicity. Compounds 4c and 5d can be considered a promising basis for the development of antitumor drugs.
Acknowledgments
The authors are grateful to the Assigned Spectral-Analytical Center of FRC Kazan Scientific Center of RAS for technical assistance in research.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph16040499/s1, Figures S1–S45 (p. 2–46)—copies of NMR spectra of all synthesized compounds; Figure S46 Dependence of substances 4c and 5d peak area on the chromatograms of the initial value (5 min); Table S1 (p. 47–48)—biostability studies; p. 49–51—computational data.
Author Contributions
E.C. and E.G.—investigation (chemistry), writing—original draft preparation, supervision (chemistry), K.M., B.B., N.A. (Nurgali Akylbekov), H.B.T.N. and M.Z.—investigation (chemistry), M.N., A.V., A.L. and S.A.—investigation (in vitro study), N.A. (Nurbol Appazov)—funding acquisition, O.S. and A.B.—project administration, V.S.—investigation (NMR study), K.V. and A.R.—investigation (biostability studies), L.K.—investigation (quantum-chemical computations), I.A.—writing—original draft preparation, project administration. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of all described compounds are available from the author E. Chugunova.
Funding Statement
The synthesis, the study of anticancer and antimicrobial activities were carried out at the Arbuzov Institute of Organic and Physical Chemistry and were supported by the Ministry of Science and Higher Education of the Russian Federation at FRC Kazan Scientific Center (grant No. 075-15-2022-1128). Two-dimensional NMR experiments were funded by the government assignment for the FRC Kazan Scientific Center of RAS. The study of hemolytic activity was funded by the financial support for research from the Ministry of Agriculture of the Republic of Kazakhstan (BR10764960). L.K. acknowledges support by the National Science Foundation Graduate Research Fellowship under Grant No. 1449440 for quantum-chemical computations realization.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Wild C.P., Weiderpass E., Stewart B.W., editors. World Cancer Report: Cancer Research for Cancer Prevention. International Agency for Research on Cancer; Lyon, France: 2020. [Google Scholar]
- 2.Gonzalez F.J. Role of cytochromes P450 in chemical toxicity and oxidative stress: Studies with CYP2E1. Mutat. Res. Mol. Mech. Mutagen. 2005;569:101–110. doi: 10.1016/j.mrfmmm.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 3.Halliwell B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007;401:1–11. doi: 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- 4.Policastro L.L., Ibañez I.L., Notcovich C., Duran H.A., Podhajcer O.L. The Tumor Microenvironment: Characterization, Redox Considerations, and Novel Approaches for Reactive Oxygen Species-Targeted Gene Therapy. Antioxid. Redox Signal. 2013;19:854–895. doi: 10.1089/ars.2011.4367. [DOI] [PubMed] [Google Scholar]
- 5.Sotgia F., Martinez-Outschoorn U.E., Lisanti M.P. Mitochondrial oxidative stress drives tumor progression and metastasis: Should we use antioxidants as a key component of cancer treatment and prevention? BMC Med. 2011;9:62. doi: 10.1186/1741-7015-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Popovici V., Musuc A.M., Matei E., Karampelas O., Ozon E.A., Cozaru G.C., Schröder V., Bucur L., Aricov L., Anastasescu M., et al. ROS-Induced DNA-Damage and Autophagy in Oral Squamous Cell Carcinoma by Usnea barbata Oil Extract—An In Vitro Study. Int. J. Mol. Sci. 2022;23:14836. doi: 10.3390/ijms232314836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milaeva E.R., Shpakovsky D.B., Gracheva Y.A., Antonenko T.A., Osolodkin D.I., Palyulin V.A., Shevtsov P.N., Neganova M.E., Vinogradova D.V., Shevtsova E.F. Some insight into the mode of cytotoxic action of organotin compounds with protective 2,6-di-tert-butylphenol fragments. J. Organomet. Chem. 2015;782:96–102. doi: 10.1016/j.jorganchem.2014.12.013. [DOI] [Google Scholar]
- 8.Milaeva E.R., Shpakovsky D.B., Dodokhova M.A., Kotieva I.M., Safronenko A.V., Alkhusein-Kulyaginova M.S. Means for inhibiting metastasis in the lungs. Patent Ru 2762730. 2021 December 22;
- 9.Milaeva E.R., Shpakovsky D.B., Dodokhova M.A., Kotieva I.M., Safronenko A.V., Alkhusein-Kulyaginova M.S. Means for inhibiting metastasis in the lungs. Patent Ru 2765955. 2022 February 7;
- 10.Edwards C.M., Mueller G., Roelofs A.J., Chantry A., Perry M., Russell R.G.G., Van Camp B., Guyon-Gellin Y., Niesor E.J., Bentzen C.L., et al. ApomineTM, an inhibitor of HMG-CoA-reductase, promotes apoptosis of myeloma cells in vitro and is associated with a modulation of myeloma in vivo. Int. J. Cancer. 2007;120:1657–1663. doi: 10.1002/ijc.22478. [DOI] [PubMed] [Google Scholar]
- 11.Mil’ E.M., Erokhin V.N., Binyukov V.I., Albantova A.A., Volod’kin A.A., Goloshchapov A.N. Apoptotic effect of the anphen sodium antioxidant in combination with H2O2 on Lewis carcinoma cells. Russ. Chem. Bull. 2019;68:2359–2364. doi: 10.1007/s11172-019-2712-z. [DOI] [Google Scholar]
- 12.Hochdörffer K., Abu Ajaj K., Schäfer-Obodozie C., Kratz F. Development of novel bisphosphonate prodrugs of doxorubicin for targeting bone metastases that are cleaved pH dependently or by cathepsin B: Synthesis, cleavage properties, and binding properties to hydroxyapatite as well as bone matrix. J. Med. Chem. 2012;55:7502–7515. doi: 10.1021/jm300493m. [DOI] [PubMed] [Google Scholar]
- 13.Orsini F., Sello G., Sisti M. Aminophosphonic Acids and Derivatives. Synthesis and Biological Applications. Curr. Med. Chem. 2010;17:264–289. doi: 10.2174/092986710790149729. [DOI] [PubMed] [Google Scholar]
- 14.Huang X., Huang R., Gou S., Wang Z., Wang H. Anticancer Platinum(IV) Prodrugs Containing Monoaminophosphonate Ester as a Targeting Group Inhibit Matrix Metalloproteinases and Reverse Multidrug Resistance. Bioconjug. Chem. 2017;28:1305–1323. doi: 10.1021/acs.bioconjchem.7b00117. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X.M., Bordwell F.G. Equilibrium acidities and homolytic bond dissociation energies of the acidic carbon-hydrogen bonds in P-substituted triphenylphosphonium cations. J. Am. Chem. Soc. 1994;116:968–972. doi: 10.1021/ja00082a018. [DOI] [Google Scholar]
- 16.Li X., Wang X., Xu C., Huang J., Wang C., Wang X., He L., Ling Y. Synthesis and biological evaluation of nitric oxide-releasing hybrids from gemcitabine and phenylsulfonyl furoxans as anti-tumor agents. Medchemcomm. 2015;6:1130–1136. doi: 10.1039/C5MD00158G. [DOI] [Google Scholar]
- 17.Fershtat L.L., Makhova N.N. Molecular Hybridization Tools in the Development of Furoxan-Based NO-Donor Prodrugs. ChemMedChem. 2017;12:622–638. doi: 10.1002/cmdc.201700113. [DOI] [PubMed] [Google Scholar]
- 18.Aguirre G., Boiani M., Cerecetto H., Fernández M., González M., León E., Pintos C., Raymondo S., Arredondo C., Pacheco J.P., et al. Furoxan derivatives as cytotoxic agents: Preliminary in vivo antitumoral activity studies. Pharmazie. 2006;61:54–59. [PubMed] [Google Scholar]
- 19.Medana C., Ermondi G., Fruttero R., Di Stilo A., Ferretti C., Gasco A. Furoxans as Nitric Oxide Donors. 4-Phenyl-3-furoxancarbonitrile: Thiol-Mediated Nitric Oxide Release and Biological Evaluation. J. Med. Chem. 1994;37:4412–4416. doi: 10.1021/jm00051a020. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh P.B., Whitehouse M.W. Potential antileukemic and immunosuppressive drugs. Preparation and in vitro pharmacological acitivity of some benzo-2,1,3-oxadiazoles (benzofurazans) and their N-oxides (benzofuroxans) J. Med. Chem. 1968;11:305–311. doi: 10.1021/jm00308a027. [DOI] [PubMed] [Google Scholar]
- 21.Whitehouse M.W., Ghosh P.B. 4-nitrobenzofurazans and 4-nitrobenzofuroxans: A new class of thiol-neutralising agents and potent inhibitors of nucleic acid synthesis in leucocytes. Biochem. Pharmacol. 1968;17:158–161. doi: 10.1016/0006-2952(68)90169-X. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh P., Whitehouse M. Potential Antileukemic and Immunosuppressive Drugs. II. Further Studies with Benzo-2,1,3-oxadiazoles (Benzofurazans) and Their N-Oxides (Benzofuroxans) J. Med. Chem. 1969;12:505–507. doi: 10.1021/jm00303a606. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh P.B., Ternai B., Whitehouse M.W. Potential antileukemic and immunosuppressive drugs. 3. Effects of homocyclic ring substitution on the in vitro drug activity of 4-nitrobenzo-2,1,3-oxadiazoles (4-nitrobenzofurazans) and their N-oxides (4-nitrobenzofuroxans) J. Med. Chem. 1972;15:255–260. doi: 10.1021/jm00273a012. [DOI] [PubMed] [Google Scholar]
- 24.Hay M.P., Wilson W.R., Moselen J.W., Palmer B.D., Denny W.A. Hypoxia-selective antitumor agents. 8. Bis(nitroimidazolyl)alkanecarboxamides: A new class of hypoxia-selective cytotoxins and hypoxic cell radiosensitisers. J. Med. Chem. 1994;37:381–391. doi: 10.1021/jm00029a010. [DOI] [PubMed] [Google Scholar]
- 25.Wink D.A., Kasprzak K.S., Maragos C.M., Elespuru R.K., Misra M., Dunams T.M., Cebula T.A., Koch W.H., Andrews A.W., Allen J.S., et al. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 26.Kessel D., Belton J.G. Effects of 4-nitrobenzofurazans and their N-oxides on synthesis of protein and nucleic acid by murine leukemia cells. Cancer Res. 1975;35:3735–3740. [PubMed] [Google Scholar]
- 27.Thompson S., Kellicutt L. Mutagenicity of anti-cancer nitrobenzofuroxans. Mutat. Res. Mol. Mech. Mutagen. 1977;48:145–153. doi: 10.1016/0027-5107(77)90154-3. [DOI] [PubMed] [Google Scholar]
- 28.Macphee D.G., Robert G.P., Ternai B., Ghosh P., Stephens R. Mutagenesis by 4-nitrobenzofurazans and furoxans. Chem. Biol. Interact. 1977;19:77–90. doi: 10.1016/0009-2797(77)90043-6. [DOI] [PubMed] [Google Scholar]
- 29.Belton J.G., Conalty M.L., O’Sullivan J.F. Anticancer agents—XI. Antitumour activity of 4-amino-7-nitrobenzofuroxans and related compounds. Proc. R. Ir. Acad. B. 1976;76:133–149. [PubMed] [Google Scholar]
- 30.Cerecetto H., Porcal W. Pharmacological properties of furoxans and benzofuroxans: Recent developments. Mini Rev. Med. Chem. 2005;5:57–71. doi: 10.2174/1389557053402864. [DOI] [PubMed] [Google Scholar]
- 31.Chugunova E.A.A., Gazizov A.S.S., Burilov A.R.R., Yusupova L.M.M., Pudovik M.A.A., Sinyashin O.G.G. Benzofuroxans: Their synthesis, properties, and biological activity. Russ. Chem. Bull. 2019;68:887–910. doi: 10.1007/s11172-019-2503-6. [DOI] [Google Scholar]
- 32.Chugunova E.A., Mukhamatdinova R.E., Sazykina M.A., Sazykin I.S., Khammami M.I., Akylbekov N.I., Burilov A.R., Kulik N.V., Zobov V.V. Synthesis and biological activity of new hybrids compounds derived from benzofuroxanes and polyene antibiotics. Russ. J. Gen. Chem. 2016;86:1037–1040. doi: 10.1134/S1070363216050091. [DOI] [Google Scholar]
- 33.Wang L., Li C., Zhang Y., Qiao C. Synthesis and Biological Evaluation of Benzofuroxan Derivatives as Fungicides against Phytopathogenic Fungi. J. Agric. Food Chem. 2013;61:8632–8640. doi: 10.1021/jf402388x. [DOI] [PubMed] [Google Scholar]
- 34.Jorge S.D., Masunari A., Rangel-Yagui C.O., Pasqualoto K.F.M., Tavares L.C. Design, synthesis, antimicrobial activity and molecular modeling studies of novel benzofuroxan derivatives against Staphylococcus aureus. Bioorg. Med. Chem. 2009;17:3028–3036. doi: 10.1016/j.bmc.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 35.Gibadullina E., Nguyen T.T., Strelnik A., Sapunova A., Voloshina A., Sudakov I., Vyshtakalyuk A., Voronina J., Pudovik M., Burilov A. New 2,6-diaminopyridines containing a sterically hindered benzylphosphonate moiety in the aromatic core as potential antioxidant and anti-cancer drugs. Eur. J. Med. Chem. 2019;184:111735. doi: 10.1016/j.ejmech.2019.111735. [DOI] [PubMed] [Google Scholar]
- 36.Hammick D.L., Edwakdes W.A.M., Steiner E.R. The constitution of benzfurazan and benzfurazan oxide. J. Chem. Soc. 1931;0:3308–3313. doi: 10.1039/JR9310003308. [DOI] [Google Scholar]
- 37.Bailey A.S., Case J.R. 4:6-dinitrobenzofuroxan, nitrobenzodifuroxan and benzotrifuroxan: A new series of complex-forming reagents for aromatic hydrocarbons. Tetrahedron. 1958;3:113–131. doi: 10.1016/0040-4020(58)80003-4. [DOI] [Google Scholar]
- 38.Botlton A.J., Ghosh P.B. Benzofuroxans. In: Katritzky A.R., Boulton A.J., editors. Advances in Heterocyclic Chemistry. Volume 10. Academic Press; London, UK: 1969. pp. 1–41. [Google Scholar]
- 39.Chugunova E., Akylbekov N., Shakirova L., Dobrynin A., Syakaev V., Latypov S., Bukharov S., Burilov A. Synthesis of hybrids of benzofuroxan and N-, S-containing sterically hindered phenols derivatives. Tautomerism. Tetrahedron. 2016;72:6415–6420. doi: 10.1016/j.tet.2016.08.047. [DOI] [Google Scholar]
- 40.Salah Ayoup M., Wahby Y., Abdel-Hamid H., Ramadan E.S., Teleb M., Abu-Serie M.M., Noby A. Design, synthesis and biological evaluation of novel α-acyloxy carboxamides via Passerini reaction as caspase 3/7 activators. Eur. J. Med. Chem. 2019;168:340–356. doi: 10.1016/j.ejmech.2019.02.051. [DOI] [PubMed] [Google Scholar]
- 41.Tramer F., Da Ros T., Passamonti S. Screening of fullerene toxicity by hemolysis assay. Methods Mol. Biol. 2012;926:203–217. doi: 10.1007/978-1-62703-002-1_15. [DOI] [PubMed] [Google Scholar]
- 42.Voloshina A.D., Gumerova S.K., Sapunova А.S., Kulik N.V., Mirgorodskaya A.B., Kotenko A.A., Prokopyeva T.M., Mikhailov V.A., Zakharova L.Y., Sinyashin O.G. The structure—Activity correlation in the family of dicationic imidazolium surfactants: Antimicrobial properties and cytotoxic effect. Biochim. Biophys. Acta Gen. Subj. 2020;1864:129728. doi: 10.1016/j.bbagen.2020.129728. [DOI] [PubMed] [Google Scholar]
- 43.Voloshina A.D., Sapunova A.S., Kulik N.V., Belenok M.G., Strobykina I.Y., Lyubina A.P., Gumerova S.K., Kataev V.E. Antimicrobial and cytotoxic effects of ammonium derivatives of diterpenoids steviol and isosteviol. Bioorg. Med. Chem. 2021;32:115974. doi: 10.1016/j.bmc.2020.115974. [DOI] [PubMed] [Google Scholar]
- 44.Neganova M., Aleksandrova Y., Suslov E., Mozhaitsev E., Munkuev A., Tsypyshev D., Chicheva M., Rogachev A., Sukocheva O., Volcho K., et al. Novel Multitarget Hydroxamic Acids with a Natural Origin CAP Group against Alzheimer’s Disease: Synthesis, Docking and Biological Evaluation. Pharmaceutics. 2021;13:1893. doi: 10.3390/pharmaceutics13111893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Norris W.P., Chafin A., Spear R.J., Read R.W. Synthesis and thermal rearrangement of 5-chloro-4,6-dinitrobenzofuroxan. Heterocycles. 1984;22:271–274. doi: 10.3987/R-1984-02-0271. [DOI] [Google Scholar]
- 46.Smolobochkin A.V., Gazizov A.S., Yakhshilikova L.J., Bekrenev D.D., Burilov A.R., Pudovik M.A., Lyubina A.P., Amerhanova S.K., Voloshina A.D. Synthesis and Biological Evaluation of Taurine-Derived Diarylmethane and Dibenzoxanthene Derivatives as Possible Cytotoxic and Antimicrobial Agents. Chem. Biodivers. 2022;19:e202100970. doi: 10.1002/cbdv.202100970. [DOI] [PubMed] [Google Scholar]
- 47.Yu Strobykina I., Voloshina A.D., Andreeva O.V., Sapunova A.S., Lyubina A.P., Amerhanova S.K., Belenok M.G., Saifina L.F., Semenov V.E., Kataev V.E. Synthesis, antimicrobial activity and cytotoxicity of triphenylphosphonium (TPP) conjugates of 1,2,3-triazolyl nucleoside analogues. Bioorg. Chem. 2021;116:105328. doi: 10.1016/j.bioorg.2021.105328. [DOI] [PubMed] [Google Scholar]
- 48.AAT Bioquest Inc Quest GraphTM IC50 Calculator. 2022. [(accessed on 25 June 2022)]. (Version 2022) Available online: https://www.aatbio.com/tools/ic50-calculator.
- 49. [(accessed on 31 January 2023)]. Available online: https://gaussian.com/citation/
- 50.Zhao Y., Truhlar D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008;120:215–241. doi: 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- 51.Grimme S., Antony J., Ehrlich S., Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 52.Marenich A.V., Cramer C.J., Truhlar D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is contained within the article and supplementary material.