

Abstract
Next-generation sequencing technologies have revolutionized our ability to catalog the landscape of somatic mutations in tumor genomes. These mutations can sometimes create so-called neoantigens, which allow the immune system to detect and eliminate tumor cells. However, efforts that stimulate the immune system to eliminate tumors based on their molecular differences have had less success than has been hoped for, and there are conflicting reports about the role of neoantigens in the success of this approach. Here we review some of the conflicting evidence in the literature and highlight key aspects of the tumor–immune interface that are emerging as major determinants of whether mutation-derived neoantigens will contribute to an immunotherapy response. Accounting for these factors is expected to improve success rates of future immunotherapy approaches.
Keywords: neoantigens, immunotherapy, precision medicine, tumor evolution
1. INTRODUCTION
Immunotherapy co-opts an individual’s own immune system to eliminate tumors. This approach to cancer therapy has sometimes resulted in remarkable responses, motivating significant investment into immunotherapy research and development. The last decade has seen rapid advances toward novel immunotherapies that excite immune cells against the molecular differences in tumors, informed by tumor profiling using next-generation sequencing (NGS) technologies. However, overall response rates have been disappointing, and the field is racing to understand why immunotherapies succeed and why they fail. Such efforts are revealing the ways in which the complex and dynamic interplay between tumors and the immune system can lead to short-lived or ineffective immune responses. Some of the emerging pitfalls that can limit the effectiveness of therapy are not yet widely appreciated but are critical to improving outcomes. In this review, we focus on key factors to be considered in the development of immunotherapies that target alterations in the tumor genome in an informed way. Many relevant aspects of mutant peptide selection and neoantigen identification are placed in the context of the major histocompatibility complex (MHC) molecule, tumor evolution, and fundamental variables such as age and sex.
2. A HISTORICAL PERSPECTIVE
The idea that the immune system responds to cancer cells is not new. At the turn of the twentieth century, Georg Schöne in his remarkable book Heteroplastic and Homoplastic Transplantation established the general rules governing the acceptance or rejection of tumor grafts (1). His “laws of transplantation” showed that whereas autografts almost invariably succeed, some degree of foreignness is necessary for rejection to occur, and that a second graft in a recipient that had previously rejected a graft from the same donor undergoes accelerated rejection (cited in 2). Following World War II, George D. Snell and Peter Gorer discovered a genetic locus intimately related to the rejection of tumor grafts that they labeled “H” for histocompatibility (3, 4). This was the first hint that tumor rejection was dependent on the histocompatibility locus. Whereas this observation solidified the concept that tumor rejection and histocompatibility antigens are linked, the observation was more important for experimental biology, as the field of cancer immunology would not exist for many years. This discovery forms the basis of our understanding of cancer immunity and its relationship to tumor rejection, and it is also the subject of this review and its arguments.
By definition, cancer immunity is restricted to phenomena that occur either in the autologous host or in syngeneic animals through specific resistance to malignant cells. Whereas immunological resistance to cancer cells can occasionally occur as the autochthonous reaction to sporadic cancer, stronger resistance develops as a result of prior exposure to the same cancer cells, as predicted by Schöne’s laws of transplantation.
Cancer immunity, much like transplantation immunity, is fundamentally a cell-mediated response similar to delayed type hypersensitivity (5). This is not to say that humoral immunity by antibodies does not play a role, but antibodies must be sufficiently specific to discriminate between cancer cells and normal tissues—something T cells do more efficiently. Thus, T cell responses against cancer are subject to the rules of the MHC with respect to antigen presentation, T cell activation, and recognition by antigen-specific effector T cells.
The first conundrum facing immunologists was how T cells recognize tumor cells and tumor antigens as T cells with high-affinity receptors, for these essentially self-antigens are expected to be deleted or pruned during thymic selection (6, 7). How then can the immune system mount an effective resistance to cancer cells?
2.1. Natural Immune Protection Against Cancer
The concept of natural immune protection against cancer is generally termed immune surveillance and is attributed to F.M. Burnet (8). Burnet defined cancer immunity as the ability of T cells to recognize new antigenic material at the cell surface of cancer cells. Despite his lack of proof, Burnet must be credited for proposing a concept that predicted contemporary thinking on cancer immunity. In his words:
The thesis is that when aberrant cells with proliferative potential arise in the body, they will carry new antigenic determinants on their cell surfaces. When a significant amount of new antigen has developed, a thymus-dependent immunological response will be initiated and eventually eliminates the aberrant cells in essentially the same way as an allograft is destroyed. (Ref. 8, p. 9)
One may argue that the new antigen could be either a conserved antigen not usually expressed in normal tissues or a truly novel antigen created by mutations in the cancer genome.
Adaptive responses are mediated by CD8+ and CD4+ T lymphocytes and are based on the recognition of antigenic peptides expressed in complex with the MHC on the tumor cell surface. In theory, tumor antigens (endogenous antigens) can be processed and presented through the MHC of the host cell and serve as targets of T cells. In humans, there is collective evidence that T cells against characterized tumor antigens can be identified as part of a natural response to the tumor.
Tumor antigens recognized by T cells range from conserved antigens, including antigens coded by cancer-germline genes and antigens overexpressed in cancer cells and cancer testis, to onco-fetal antigens and differentiation antigens (9–11).
However, these classical tumor antigens may not be the ones Burnet predicted to drive immune surveillance. Implicit in his hypothesis was the suggestion that T cells would prevalently recognize peptides from mutated antigens, although he had no proof of this. In contrast to the abundance of conserved antigens T cells can recognize (see the sidebar titled Cancer Antigens Can Be Derived from a Variety of Sources), prior to systematic use of genomic interrogation, the autochthonous recognition of mutated peptides at the surface of cancer cells has only occasionally been shown (Table 1).
Table 1.
Older examples of T cells recognizing mutated epitopes in cancer cells
| CD4+ T cells | ||
|---|---|---|
| Source | Type | References |
| Triosephosphate isomerase | Nonsynonymous mutation | 7, 153 |
| Low-density lipid receptor/GDP-l-fucose:β-d-galactoside-2-α-l-fucosyltransferase | Gene fusion | 9, 154 |
| Triosephosphate isomerase | Nonsynonymous mutation | 11, 155 |
| CD8+ T cells | ||
| Source | Type | References |
| Chimeric P210 BCR-ABL | Gene fusion | 8, 156 |
| Chimeric ETV6-AML1 | Gene fusion | 10, 157 |
CANCER ANTIGENS CAN BE DERIVED FROM A VARIETY OF SOURCES
Cancer antigens can be encoded by cancer-germline genes (e.g., MAGE, NY-ESO1, LAGE1, SAGE1) and differentiation genes (e.g., MART1, TYR, TRP1, PSA, PAP, PSMA), as well as by viruses (e.g., Epstein–Barr virus, hepatitis B and C viruses, human papilloma virus, HIV) and mutated oncogenes (e.g., TP53, KRAS). Cancer antigens can also derive from overexpressed proteins in cancer cells (e.g., TERT, MUC1, Survivin, CEA, ERBB2, and glycoproteins such as T and Tn antigens).
2.2. A Revision of the Theory of Immune Surveillance: Immunoediting
Possibly because the original immune surveillance hypothesis paid little attention to the complex dynamics between cancer cells and immune cells during cancer evolution, Schreiber and colleagues proposed the cancer immunoediting hypothesis (12, 13). This hypothesis predicts that while the human immune system can potentially protect from cancer, it also drives the development of tumors that undergo so-called immunogenic sculpting, rendering them resistant to further attack by T cells. The immunoediting hypothesis describes three phases: elimination, equilibrium, and escape. While the first and second phases are extensions of Burnet’s hypothesis, the third phase (escape) predicts that over time, the immune response stops being effective and the tumor progresses. In evolutionary terms, this is equivalent to the already established concept of immune selection in response to pathogens (14).
The basis for both hypotheses is that antigens displayed on cancer cells as peptides associated with MHC molecules are able to direct a cellular immune response against tumor cells. However, whereas Burnet’s hypothesis was concerned with what T cells might recognize, the immunoediting hypothesis is concerned with a time-based dimension of immunity during cancer evolution. Since most cancers inevitably progress, it is difficult to deny that tumor cells adopt ways to escape immune surveillance. Thus, the dynamics of the tumor–immune cell interplay depend on how tumor antigens shape the immune response over the course of cancer evolution and on the fundamental rules of this process.
2.3. Neoantigens
Cancer is a genetic disease (15) and somatic mutations account for variation in cancer risk (16). This forms the essence of the contemporary paradigm in cancer. Bioinformatic developments aimed at detecting cancer-causing driver mutations from whole-exome, -genome, and -transcriptome sequencing also support the rapid and systematic interrogation of the tumor antigenic landscape by cataloging tumor-specific mutant peptides (neopeptides) deriving from nonsynonymous mutations, frameshift mutations, and gene rearrangements. This allows researchers to probe the relationship between the MHC and genomic mutations at a level of resolution hitherto unachievable.
As discussed elsewhere, not all neopeptides are immunogenic, which implies that not all neopeptides can be rightly called neoantigens (17). High-throughput selection of neopeptides is a bioinformatic process following the basic rule that for any peptide to be immunogenic, binding to the MHC molecule with some degree of thermodynamic stability is necessary. But binding is not sufficient to predict immunogenicity, as this depends on the ability of the peptide–MHC (pMHC) complex to engage a T cell, and predicting the probability that a neopeptide binds with a T cell receptor (TCR) is more difficult given the polymorphic nature of the TCR. We have previously suggested that immunogenicity needs to be validated at an empirical level in the context of an individual’s variability with genetic and environmental influences, which affect the available and expandable T cell repertoire (17).
2.4. A Genomic-Bioinformatic Analysis of the Cancer Immune Interface
As we enter a new phase in the study of cancer immunity, new questions are possible, and their answers may impact therapeutic choices at a time when cancer immunotherapy is enjoying unprecedented attention. What can genomic interrogation and bioinformatic analyses tell us about natural immune protection against cancer? Are mutated peptides the antigens that drive it? How does the mutational process affect immunity in cancer? How does immunogenicity vary during cancer evolution and across heterogeneous cancer cell populations?
3. MECHANISM
MHC class I (MHC-I) provides the adaptive immune system a window into the intracellular health of all nucleated cells. It is a protein complex that consists of an invariant beta-2 microglobulin (B2M) scaffold protein and a variable HLA (classically, HLA-A, HLA-B, and HLA-C) protein that binds and presents short (~9-mer) intracellular peptides to CD8+ T cells. It is through this pathway that virally infected and malignant cells can be identified (Figure 1). Although less discussed, nonclassical HLA proteins (HLA-E, HLA-F, and HLA-G) can be exploited or affected by the tumor (see 18, 19) and can serve as biomarkers for tumor immune evasion (20, 21).
Figure 1.
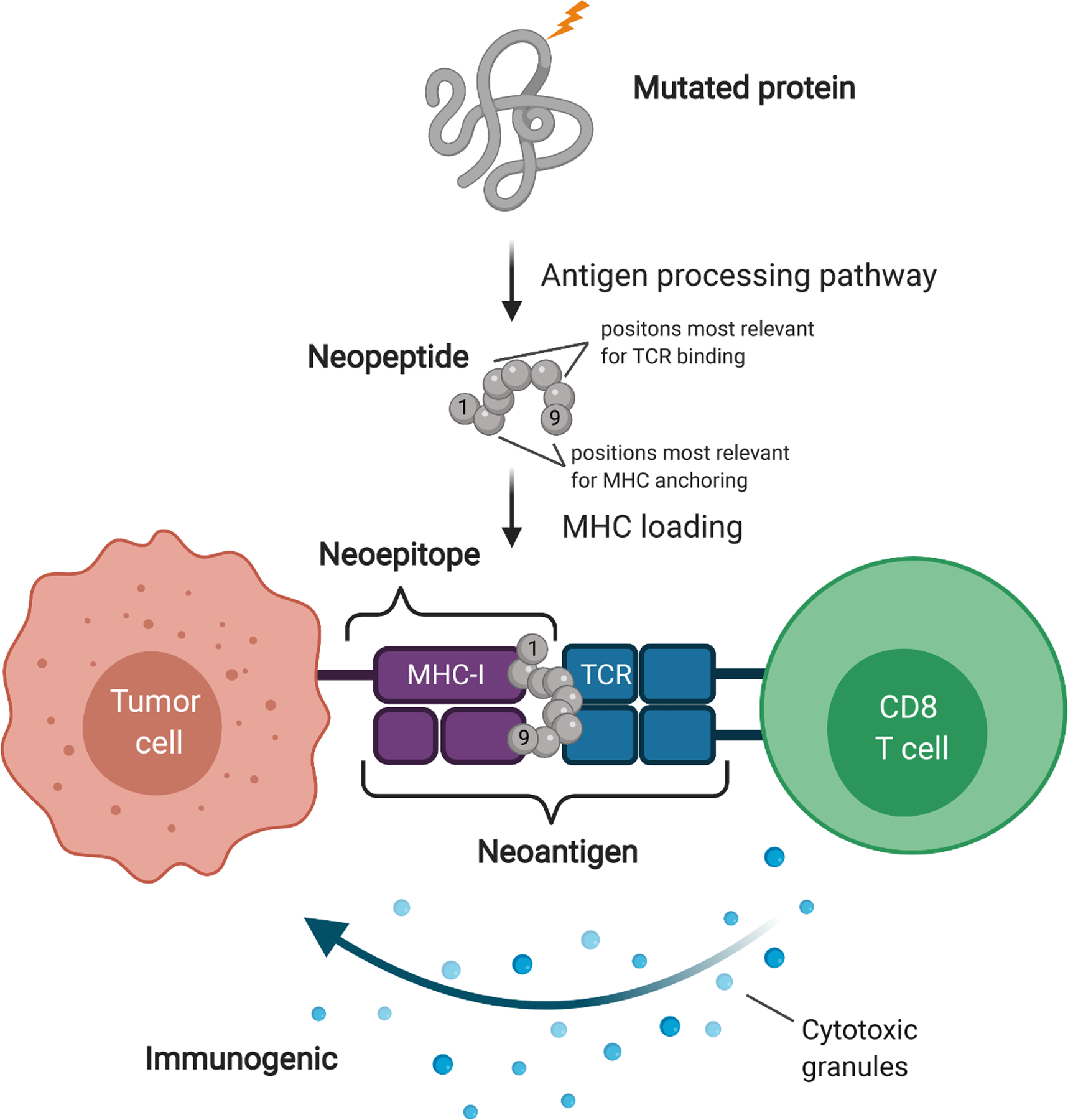
Neoantigens are displayed via MHC-I on tumor cells and recognized by CD8+ T cells. Amino acids in positions 2 and 9 are typically primary MHC-I anchor residues, and amino acids in positions 4–7 are typically TCR-binding residues. Figure adapted from images created with BioRender.com. Abbreviations: MHC, major histocompatibility complex; MHC-I, MHC class I; TCR, T cell receptor.
In contrast, MHC class II (MHC-II) is specifically expressed on professional antigen-presenting cells (APCs), including dendritic cells, B cells, and macrophages, although it has been observed on some tumor cells, likely due to upregulated interferon-gamma (IFNγ) (22). MHC-II is a heterodimer composed of an α chain and more variable β chain (classically DRA1/DRB1, DQA1/DQB1, and DPA1/DPB1), which bind longer (optimally 15-mer) peptides and presenting them to CD4+ T cells.
Antigen-processing pathways differ for MHC-I and MHC-II; peptides presented via MHC-I derive predominantly from endogenous peptides, while those presented via MHC-II are predominantly exogenous. Both peptide types are cleaved: intracellular peptides by the proteasome and exogenous peptides by lysosomal proteases. TAP mediates transport of class I peptides into the endoplasmic reticulum for loading, whereas class II peptides are transported to the antigen-processing compartment via endosomes. Loading of both types onto respective MHC complexes is aided by chaperone proteins: tapasin in class I and HLA-DM in class II (see 23). However, these are not hard rules; exogenous peptides have been found to be presented via MHC-I on APCs via cross-presentation (24), which has implications for tumor immunity and opportunities for enhanced treatment (25).
4. APPLICATION AND CONTROVERSY
One hypothesis is that cancer arises when the immune system fails to identify and eliminate the tumor. The genomic era and neoantigens’ rapid surge to prominence as problem solvers have provided a major boost to cancer immunotherapy. Traditional passive antibody therapy against surface antigens (e.g., HER2, CD20, VEGF), which are now standard of care in certain cancer types (see 26), and vaccination against conserved antigens that produced below-expectation results have been replaced by neoantigen vaccination, passive treatment with antibodies for immune checkpoint blockade (ICB), and adoptive T cell therapies (27) (Figure 2).
Figure 2.
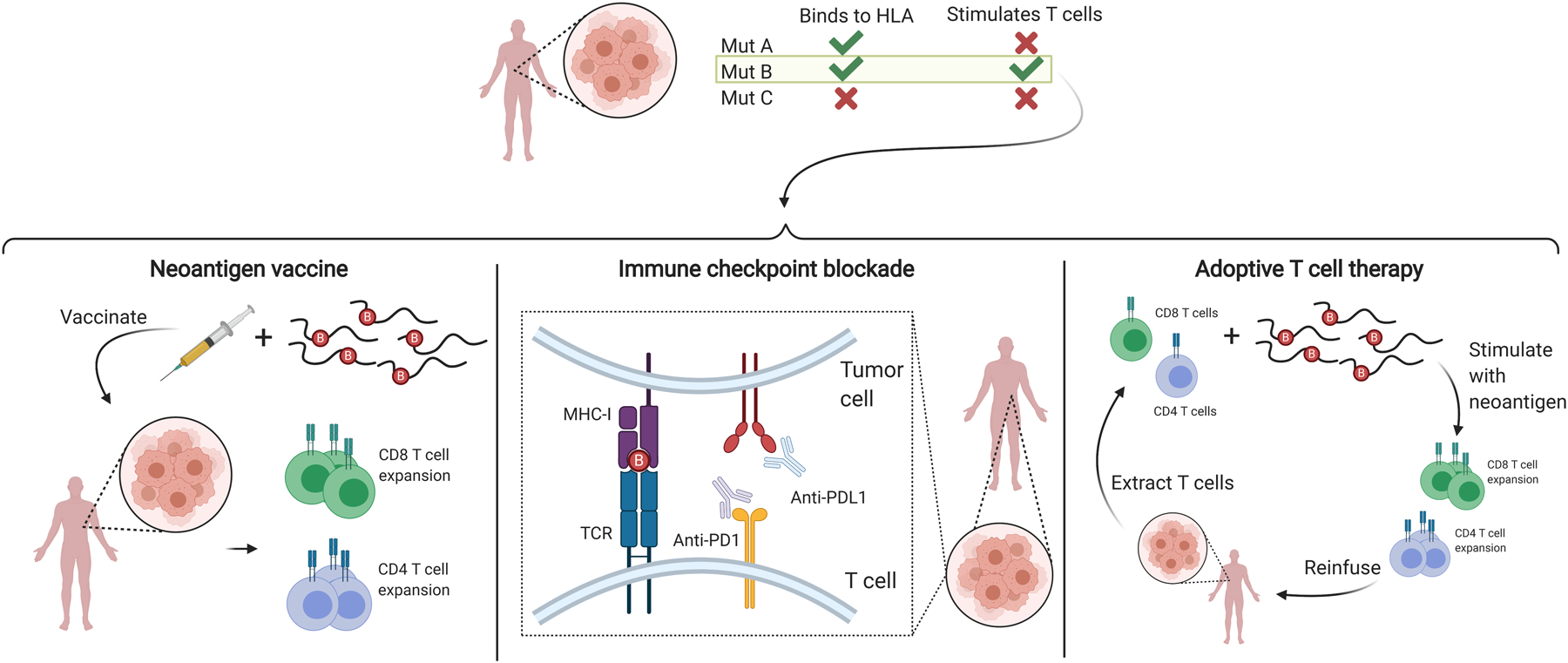
Overview of neoantigen use in common cancer immunotherapies. Appropriate patient-specific neoantigens (e.g., those derived from mutation B in this example) are selected and used to stimulate T cells in neoantigen vaccines and adoptive T cell therapies. Presence of appropriate neoantigens is advantageous for successful immune checkpoint blockade. Figure adapted from images created with BioRender.com. Abbreviations: HLA, human leukocyte antigen; MHC-I, major histocompatibility complex class I; TCR, T cell receptor.
Personalized immunotherapy uses the immune system to attack cancer cells. The process involves identifying and targeting patient-specific nonsynonymous somatic mutations. While all three approaches are part of a concerted effort to offer solutions to the same cause, in our opinion neoantigen vaccination is the truest embodiment of the new revolution: it is personalized, precise, and requires active participation of the patient’s immune system.
4.1. Neoantigen Vaccine Trials: Mixed Results and Uncertain Clinical Benefit
The goal of a neoantigen vaccine is to induce a T cell response exploiting the foreignness characteristic of a neoantigen to elicit a T cell response. This response is theoretically free of the constraints imposed by thymic selection and the purging of T cells with high a affinity for self-antigens (an obstacle for vaccines targeting conserved tumor antigens). In principle, the available T cell repertoire against neoantigens should be larger and of higher affinity than that for conserved antigens. This recapitulates the essence of Schöne’s and later Burnet’s rationale of the optimal condition for tumor rejection by T cells.
To date, only a handful of neoantigen vaccine trials have been reported (Table 2), with overall mixed results and many questions raised. The first report was by Carreno et al. in three melanoma patients (28). The authors selected 21 neopeptides with HLA-A*02:01-binding affinities under 500 nM and performed proteomic analysis to verify tumor-specific expression. Of the 21 peptides tested, only 9 induced a T cell response. Collectively, only 7 peptides (30%) elicited a de novo T cell response in vivo. No long-term follow-up has been reported and the actual clinical benefit of vaccination remains unknown.
Table 2.
Neoantigen vaccine phase I clinical trials
| Study | Patients | Neopeptide length | Total IR neopeptides | Pre-existing IR | CD4+ T cells | CD8+ T cells |
|---|---|---|---|---|---|---|
| Carreno et al. 2015 (28); melanoma | n = 3 | 9-aa peptide (short) | 9/21 (42.8%) | 3/9 | ND | 42.8% |
| Ott et al. 2017 (29); melanoma | n = 6 | 15- to 30-aa IMP (long) | 58/97 (60%) | ND | 60% | 16% |
| 10% | ||||||
| Sahin et al. 2017 (30); melanoma | n = 13 | Short (mRNA) | 75/125 (60%) | 27c/125 | 32.8%d | 9.6%d |
| 15.2%d | ||||||
| Hilf et al. 2019 (31); glioblastoma | n = 15a | 18- to 19-aa peptide (long) | 12/13 (92.3%) | [**AU: ND?**] | 11/13 (84.7%) | 0/13 (0%) |
| 5/13 (38.4%) | ||||||
| Keskin et al. 2019 (32); glioblastoma | n = 10b | 20- to 30-aa IMP (long) | ND | ND | 3/7 (42.8%) poolse | Unclear |
Abbreviations: aa, amino acids; IMP, immunizing long peptide; ND, not determined.
11 patients received APVAC2, the neopeptide vaccine.
Eight patients completed first vaccine, and five patients had at least one booster.
Includes one putative pre-existing response. Data from table S2 in Reference 30.
Values calculated from table S2 in Reference 30.
For the two patients that generated IFNγ responses of five that received at least one booster vaccine.
A second and larger study also in melanoma patients has been reported by Ott et al. (29). In total, 97 neopeptides were identified from 6 patients on the basis of HLA-A and HLA-B binding. Of the 97 neopeptides, a response occurred in 58 instances (60%). Oddly, only 16% of responses were by CD8+ T cells (MHC-I restricted), whereas the 60% were by CD4+ T cells (MHC-II restricted). A minority of responses (10%) were mixed (Table 2). Only 3 out of 97 neopeptide-induced T cell responses were able to recognize autologous melanoma cells. Of note, these T cell responses developed in two out of six patients that had received anti-PD1 antibody treatment after recurrence of disease. While the study showed little clinical benefit, it revealed an intriguing aspect of neoantigen immunobiology: the propensity to preferentially activate CD4+ T cells even though the initial selection was based on MHC-I-binding affinity.
A third study (30), also in melanoma patients, used an RNA-based poly-neoepitope vaccine engineered with both MHC-I (HLA-A, HLA-B) and MHC-II (DRB1, DQB1) predicted neoantigens. Thirteen patients received multiple immunizations and all developed T cell responses against multiple vaccine neoepitopes. Of 125 neopeptides used for immunization, 75 (60%) induced a T cell response. However, whereas only 20 of the 69 (29%) predicted high-affinity MHC-I binders triggered a response, the CD4+ T cell response was more frequent (range of 70–34%) and correlated with binding affinity. A mixed CD4+ and CD8+ T cell response was observed in 15% of instances. Oddly, 20% of the responses were raised against neopeptides with poor HLA-binding scores. Postvaccination biopsies from two patients confirmed the infiltration of vaccine-induced neoantigen-specific T cells and specific killing of autologous melanoma cells. Overall the study reported a significantly reduced rate of metastasis after vaccination and sustained progression-free survival.
Glioblastoma, a brain tumor with limited immune infiltration and low tumor mutational burden (TMB) [except for instances with microsatellite instability–high (MSI-H) status], has been the subject of two recent trials. Hilf et al. (31) targeted both conserved tumor antigens and neopeptides to maximally exploit a response by the available T cell repertoire. Fifteen HLA-A*2:01- or HLA-A*24:02-positive glioblastoma patients were vaccinated, first with a mixture of unmutated antigens, and then with a mixture of neopeptides. This half-personalized vaccination approach showed that unmutated antigens elicited sustained responses of central memory CD8+ T cells in 13 patients, whereas neopeptides induced predominantly CD4+ T cell responses in 8 out of 10 patients. Despite a promising T cell response, the majority of enrolled patients died from their cancer. Keskin et al. reported on personalized neoantigen vaccination in 10 patients newly diagnosed with glioblastoma (32). Patients received a vaccine containing up to 20 putative neoantigens expressed in the tumor. Only the two patients who had not received dexamethasone (a potent immunosuppressant) mounted a neoantigen-specific T cell response consisting predominantly of CD4+ T cells. Despite a systemic and intratumoral T cell response, all patients ultimately died of progressive disease.
4.2. Immune Checkpoint Inhibitors
While neoantigen vaccines are ideal for evaluating in silico prediction of putative neoantigens, ICB has received more attention, as it has shown immediate success in some patients. This success has been associated with TMB, which serves as a proxy for the availability of immunogenic neopeptides to direct an immune response. TMB was first associated with long-term benefit in melanoma (33) and has been applied as a biomarker in many ICB trials across different tumor types with varying degrees of success (34). While typically quantified as the number of nonsynonymous single-nucleotide variants per megabase (35), TMB can also be approximated for smaller gene panels routinely used in clinical settings (36, 37).
While most ICB studies associate high TMB with clinical benefit (33, 35, 38–41), recent studies of ICB response in non-small-cell lung cancer (NSCLC) did not (42–44), and the extent of the correlation varies across studies. However, there were also differences in how the high-TMB threshold was defined. Different TMB thresholds in different tumors may be required (35). Overall, discrepant ICB findings suggest that TMB provides a rough estimate of neoantigen load, and its correlation with outcomes may depend on other factors in a tumor-specific manner (45). What then are the main factors that must be considered to evaluate the potential immunogenicity of the somatic landscape of a tumor?
5. THREE REQUIREMENTS FOR NEOANTIGEN-DIRECTED IMMUNE RESPONSES
Improvements in molecular profiling capabilities and studies of how the tumor genome and tumor microenvironment (TME) relate to immunotherapy success have provided valuable insights into determinants of effective neoantigen-dependent responses. Three areas in particular are worth consideration, namely, MHC functionality, the availability of immunogenic neopeptides, and whether the TME is capable of mounting a response. Many other factors also influence the effectiveness of neoantigen-directed immune responses (see 46–48), but these three factors appear to be necessary for immunotherapies such as ICB and vaccines to be effective.
5.1. Functionality of the MHC
The MHC is essential for antigen-driven immunity; without it, there can be no immune surveillance. Accordingly, MHC integrity and function directly impact neoantigen potential to drive immune responses. Impairment of MHC function is emerging as a relatively frequent occurrence in tumors. Thus, it is increasingly common for studies assessing neopeptides to also evaluate the condition of the MHC.
5.1.1. Tools for assessing MHC integrity.
Assessing MHC integrity entails profiling the alleles an individual carries and determining whether they are present and functional on tumor cells. Variation affecting the MHC can be profiled from DNA, and allele-specific expression can be evaluated from RNA sequencing data (49), although not all gene panels include the HLA locus. Most methods infer HLA alleles from NGS DNA sequencing data by aligning reads, usually from whole-exome sequencing (WXS) (50) [but some tools also accommodate whole-genome sequencing (51) and RNA (52, 53)], to a reference panel of human HLA allele sequences. Following initial alignment, tools employ different optimization and classification methods to identify the most probable set of HLA alleles.
While HLA mutations are called by standard somatic mutation calling pipelines, the region’s high variability makes accurate calling difficult. More accurate calls can be obtained after realignment to a patient-specific HLA reference. PolySolver was the first tool to realign tumor reads to a patient-specific allele set called from normal WXS data, followed by detection of somatic mutations using MuTect and Strelka (50). As with mutation calling, HLA allele–specific copy number analysis first requires aligning of sequencing reads to patient-specific HLA alleles. Tumor coverage is inferred relative to germline coverage using B allele frequencies from identified polymorphic sites between homologous alleles. Lastly, HLA allele–specific copy number is estimated from tumor sequencing coverage depth and used to assess loss of heterozygosity (LOH) of specific alleles. For example, the computational tool LOHHLA classifies copy number < 0.5 as LOH (54).
5.1.2. Somatic variation affecting the MHC.
Common mechanisms of immune evasion can involve somatic impairment of the MHC. These variants include somatic mutations directly impairing protein function, loss of protein products, and altered transcription of necessary pathway genes. Such variation can affect all MHC molecules, or only specific alleles.
5.1.2.1. MHC mutation and loss of heterozygosity.
LOH at the HLA locus affecting specific class I alleles is well established (55), but genomic profiling of larger cohorts has more recently revealed a high frequency of HLA LOH across several tumor types (54, 56). Interestingly, HLA LOH was infrequent in melanoma, which could factor into its higher response rates to ICB. In contrast, HLA LOH was frequent subclonally across multiple tumor types, including clear cell renal cell carcinoma (ccRCC), breast cancer, bladder urothelial carcinoma, endometrial carcinoma, and esophageal adenocarcinoma (56). Even subclonal loss of HLA could shield some tumor cells from immune elimination, promoting disease progression posttherapy. Loss of B2M affects the cell surface expression of all MHC molecules and has been identified as a likely resistance mechanism emerging after ICB therapy (57). Although expected to occur less frequently because cell surface pMHC is an inhibitory receptor for cytolytic natural killer (NK) cells, other factors can inhibit NK cells (58), potentially providing a more conducive environment for MHC loss.
Somatic mutations affecting HLA and B2M were observed in around 5% of TCGA (The Cancer Genome Atlas) patients in pan-cancer analyses (50, 59) and were associated with characteristics of tumor somatic mutation profiles. This points to a model wherein allele-specific HLA mutations bias which neopeptides are presented, while B2M loss-of-function mutations decrease overall pMHC levels (59). Interestingly, despite the association of MSI-H tumors with response to ICB (60), HLA and B2M mutations occur frequently in MSI-H tumors (61), where they can promote immune evasion or escape upon ICB treatment (62).
5.1.2.2. MHC downregulation whether by decreased expression or upstream antigen presentation pathway genes.
Transcriptional downregulation of specific HLA alleles following relapse postimmunotherapy has also been reported and is associated with resistance to immunotherapy (63, 64). Notably, there are multiple documented mechanisms of transcriptional downregulation, including likely hypermethylation of HLA genes (63), HLA mRNA instability due to 3′ UTR (untranslated region) binding by MEX3B (64), NMYC amplification (65), and likely others. Transcriptional repression differs from HLA LOH in that inhibiting the cause of repression, such as treating with hypomethylating agents (63), can restore HLA transcription. Similarly, somatic mutations and alterations can affect the class I antigen presentation pathway (APP) upstream of pMHC binding. Bivalent histone modifications to MHC-I APP gene promoters can repress the pathway and inhibit T cell killing in small-cell lung cancer (66).
5.1.3. MHC dynamics at the cell surface.
Following antigen processing and binding, the amount and duration of pMHC cell surface expression influence T cell recognition. Interestingly, oncogenic BRAFV600E mutations, frequent in melanoma, have been reported to specifically and constitutively internalize MHC-I from the cell surface (67). Similarly, intrinsic DQA1/DQB1 stability has been associated with variation in cell surface density of MHC molecules in engineered fibroblasts, suggesting that inefficient complex assembly influences the amount and duration of cell surface expression (68). Interestingly, less stable alleles have been more often associated with autoimmunity, which is consistent with weak affinity epitopes generating less stable complexes, allowing self-reactive T cells to escape central tolerance (69). Counterintuitively, this could suggest that in some cases, lower-affinity alleles have more potential to generate immune responses than higher-affinity alleles, although this may be more relevant to MHC-II. Better assays for quantifying peptide-specific cell surface pMHC numbers, ratios, and turnover rates will be helpful for understanding the thresholds required for robust T cell responses.
Immunodominance is a phenomenon whereby the cytotoxic T cell population has a hierarchical and focused response toward only certain pMHC complexes despite the availability of other possible pMHC targets (70). This concept is potentially related to variation in immunogenicity of pMHC molecules on the cell surface, although many factors may contribute (71). Although more extensively studied in viral and vaccine studies, immunodominance is also relevant in cancer (72) and presents yet another barrier to predicting immunogenic responses in tumors since it dictates that even when many immunogenic neoantigens are available, responses may be driven by only a minority thereof (71). Further exploration is necessary to determine whether immunodominance could be associated with epitope spreading and the generation of T cell reactivity with alternate antigen specificity.
5.1.4. Germline variation in the MHC.
The frequent somatic loss of MHC-I integrity as a mechanism of immune evasion raises questions about whether heritable variability affecting these alleles could introduce cancer-relevant interindividual differences in immune surveillance. The set of HLA alleles carried in an individual’s genome is a determinant of the diversity of foreign antigens that can be presented to T cells (73). Recent findings suggest this diversity may extend to the potential of an individual’s MHC to present neoepitopes from a greater proportion of the somatic mutations present. Chowell et al. first described an association of heterozygosity with better response to ICB treatment and found that melanoma patients with HLA-B44 supertype alleles in two cohorts received more benefit (74). They subsequently formulated the HLA-I evolutionary divergence score to quantify dissimilarity between HLA alleles, describing the potential of an individual’s alleles to present a more diverse set of neoepitopes (75). In contrast, Negrao et al. (76) did not find an association of HLA zygosity or supertype in ICB-treated NSCLC patients, including those evaluated by Chowell et al. Nonetheless, a recent study by Cummings et al. once again reported a B44 advantage and suggested that differences in B44 carrier outcomes for melanoma versus NSCLC could result from characteristics of mutations generated by the mutational processes underlying those diseases (77). Further studies are needed to help determine the extent to which inherited combinations of HLA alleles shape the individual potential to respond to ICB.
5.2. Availability of Neoantigens
Studies of the so-called immunome suggest that neoantigens capable of driving immune responses are relatively rare (78); thus, neoantigen load might provide a better measure than TMB. Counterintuitively, studies have reported that predicted neoantigen load fails to predict outcomes better than TMB (45, 79). As it does not seem possible that neoantigens are not important for responses (80), this discrepancy points to difficulties with accurately determining what mutations truly have the potential to drive immune responses. What then are the characteristics of neopeptides that lead to immunogenic neoantigens?
5.2.1. The association of tumor mutational burden with neoantigens.
Observations from studies of TMB are helpful for understanding the relationship between mutation quantity and immunogenicity. The general observation that high TMB is more often associated with ICB benefit may be related to higher numbers of neoantigens. However, responses have been observed in low-TMB tumors and failed in high-TMB tumors. Such conflicting reports suggest that TMB itself is an imperfect proxy for the presence of effective neoantigens. Riaz et al. found that the change in TMB early in the course of treatment was a more effective predictor than pretreatment TMB, but this approach requires on-treatment biopsy (81). High TMB associated with DNA mismatch repair deficiency (MMRd) has proven to be highly predictive of patient response to immunotherapy (60, 82). Notably, MMRd/MSI-H tumors often display a high number of indel and frameshift mutations that create more potent neoantigens (83–85), although even among MSI-H metastatic colorectal tumors, higher TMB was associated with better outcomes (83).
Some high-TMB tumors including melanoma and NSCLC have shown different levels of correlation between TMB and ICB response. While higher TMB generally correlates with better outcomes in melanoma (33, 38), in NSCLC there is less evidence supporting benefit (42–44). In melanoma, patients carrying the HLA-B44 supertype responded particularly well, and this was linked to an excess of amino acid substitutions to glutamate (74). Cummings et al. found differences in the rate at which radical glutamic acid substitutions occurred in melanoma and NSCLC, corresponding to differences in UV and tobacco carcinogen–induced mutagenesis (77). In NSCLC patients carrying the B44 allele, a higher burden of radical glutamic acid substitutions in B44 neoepitopes was associated with better outcomes. These reports hint that neopeptide characteristics beyond MHC affinity could determine the potential to drive ICB response and point to a previously unappreciated role for mutational exposures in shaping immune responses.
Altogether these studies and others suggest that quality rather than quantity of neoantigens is important for ICB response (85, 86). Accordingly, the absence of high-affinity MHC-I neoepitopes in patients with high TMB was associated with lower response rates to ICB monotherapy in two distinct cohorts (87). Interestingly, a recent study suggested that extremely high TMB is associated with a more dysfunctional T cell landscape in NSCLC (88). In addition, controlling for HLA LOH improved correlation between TMB and response in NSCLC (89) where HLA LOH is fairly common (54), highlighting that effective antigen presentation is necessary for the relationship with TMB to hold. These findings further underscore the importance of assessing both MHC integrity and the state of the TME in addition to neoantigen availability.
5.2.2. Determinants of neoantigen quality.
To drive effective antitumor immune responses, neoantigens must at a minimum be presented by MHC and recognized by T cells in a manner that generates robust and sustained T cell activity. Because cell surface presentation does not guarantee recognition by a TCR and recognition by a TCR does not guarantee immunogenicity, there must be additional requirements beyond MHC binding to render neopeptides immunogenic. Here we discuss factors that have been implicated as determinants of immunogenicity.
5.2.2.1. Relevance to tumors.
Even if a neoantigen is highly immunogenic, it is unlikely to drive an effective immunotherapy response if it is present in only a minority of tumor cells. Indeed, it has been reported that clonal neoantigens, present in a high proportion of tumor cells, are associated with better responses (90). Tumors may also be more susceptible to immune responses targeted at mutations that represent a dependency, such as driver mutations or nondriver mutations in tumor-essential genes (85). The fact that DNA copy loss affecting clonal neoantigens can act as a mechanism of immune evasion upon ICB treatment supports the thesis that cancer dependency may promote more effective immune responses (91).
5.2.2.2. Binding affinity.
Although pMHC binding affinity may not be sufficient for immunogenicity, it is nonetheless informative since formation of a pMHC complex is necessary for presentation. Some studies suggest that higher affinity is associated with more immunogenicity or better outcomes (30, 87). Many methods predict binding affinity given a peptide and an HLA allele, usually as either an IC50 (half-maximal inhibitory concentration) or a percentile rank, although there is still debate about which has greater predictive power (92). Affinity was found to correspond to cell surface presentation by MHC based on mass spectrometry studies of eluted pMHC complexes (93–95).
5.2.2.3. Stability.
Two peptides might have the same affinity (measured in nM) but differing stability (measured as half-life); stability has been proposed to be more predictive of immunogenicity (96), with anchor position two playing an important role. Harndahl et al. found that 30% of nonimmunogenic binders formed unstable pMHC complexes, providing a possible explanation for their nonimmunogenicity. A recent study found that MHC-I stability is influenced by cell surface sialylation in dendritic cells, with doubled MHC-I half-life when cell surfaces were desialylated (97).
5.2.2.4. Abundance.
Perhaps not surprisingly, the amount of mutant peptide has also proven a useful measure. Mutations that do not get translated to protein cannot generate neoantigens. Expression can be approximated by RNA read support for the mutant allele. Interestingly, high expression appears to compensate for lower binding affinity in some cases, making it less clear how to establish affinity thresholds for predicting cell surface display (98).
5.2.2.5. Agretopicity.
Agretopicity refers to the idea that the mutant peptide should bind the MHC with higher affinity than the wild-type peptide (99). Tolerance should ensure that TCRs capable of initiating self-peptide responses are eliminated from the repertoire; thus, there should be a dearth of T cells available for recognizing mutant peptides with qualities too similar to the wild type (Figure 3a). Agretopicity is usually quantified as the ratio of (100, 101) or difference between (99, 102) mutant binding affinity to wild-type binding affinity.
Figure 3.
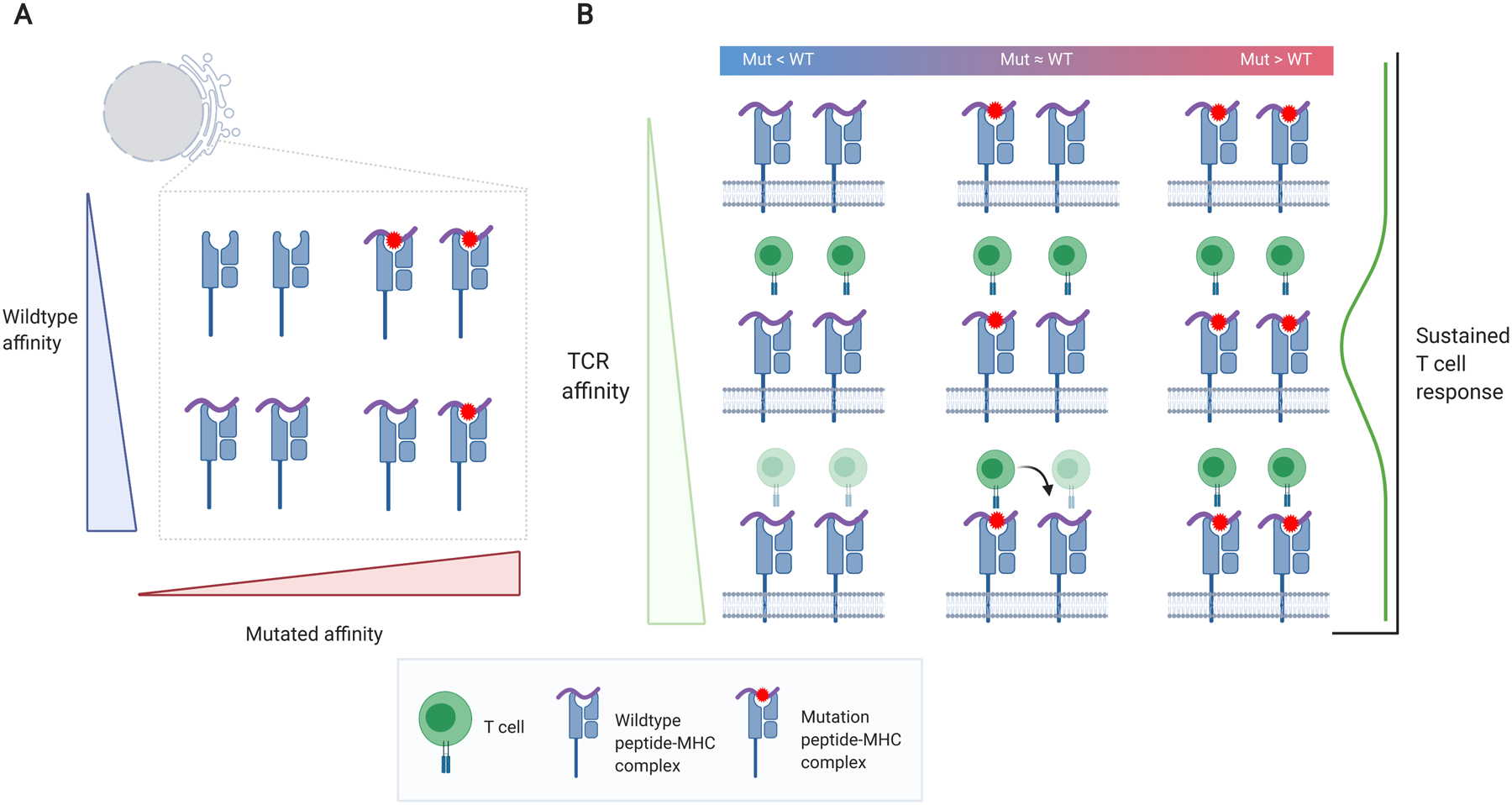
Relations among neoantigens, MHC, TCR binding, and immunogenicity. (a) Neopeptide versus WT peptide affinity. A high differential agretopic index (i.e., the neopeptide has a greater affinity than the WT) will result in greater presentation of the mutation (top right). (b) Effects of peptide affinity in conjunction with TCR affinity on sustained T cell response. High TCR affinity to the WT pMHC complex (bottom left and bottom middle) should result in thymic elimination of T cell clones (faded T cells). However, high TCR affinity to the neoepitope could result in cross-reactivity, also targeting the WT pMHC (bottom middle). Very high TCR affinity may promote short-lived T cell responses (113). High neoepitope–MHC affinity but lower WT-MHC affinity and intermediate TCR affinity may be the optimal combination to promote a sustained T cell response (middle right). Figure adapted from images created with BioRender.com. Abbreviations: MHC, major histocompatibility complex; pMHC, peptide–MHC; TCR, T cell receptor; WT, wild-type.
5.2.2.6. Cross-reactivity.
The best neoantigens may be those that result in the expansion of T cell clones that recognize not only the MHC-bound neoantigen itself, but also the wild-type sequence or another similar self-sequence that would not have otherwise generated an immune response (103) (Figure 3b). However, such neoantigens may also pose the risk of potentially generating off-target immune recognition of normal tissues.
5.2.2.7. Foreignness.
Because the adaptive immune system has evolved to protect us from external threats such as pathogens, it is possible that it is optimized to recognize more foreign-appearing neoantigens (33, 100). Indeed, somatic indel and frameshift mutations have been more likely to bind the MHC and associate with response to ICB (84).
5.2.2.8. Amino acid substitution qualities.
Other evidence suggests a more complex relationship between binding affinity and immunogenicity. Cummings et al. observed that the existence of a high number of mutations that generated HLA-B44 supertype–specific so-called motif neoepitopes, which included a radical glutamic acid substitution at the 5′ anchor position and a sequence match to the HLA-B44 motif otherwise, was associated with better outcomes in patients carrying one or more HLA-B44 alleles (77). Among HLA-B supertypes, only B27 showed a similar effect. Notably, B44 and B27 are the only charged HLA-B supertypes, and motif neoepitopes were those that generated a peptide with the complementary charge to those alleles. Patients carrying B44/B27 alleles without such motif neoepitopes had worse responses to ICB (77), suggesting that the specific characteristic of the 5′ anchor position substitution was a determinant of immunogenicity.
5.2.2.9. Peptide characteristics.
While peptide hydrophobicity and mutation position are likely important for determining antigen presentation (104), amino acids at positions 4–6 of MHC-I-bound peptides may be particularly important for immunogenicity (101, 105–107). High hydrophobicity could actually be a hindrance because it implies that peptide side chains are directed toward the MHC rather than upward, where they could contribute to TCR binding. Accordingly, a recent experiment by TESLA (Tumor Neoantigen Selection Alliance) found that peptide hydrophobicity, while associated with better binding to MHC, was significantly lower in immunogenic neoantigens, which also tended to have overall higher MHC affinity and to be more abundant in tumor cells (101). Interestingly, of the 37 immunogenic neopeptides in their dataset, none had the mutation at the position two anchor residue.
5.2.2.10. Restriction by MHC-I versus MHC-II.
While most studies have focused on neoantigens specific to MHC-I, a 2019 study found that effective immunotherapy responses require neoantigens restricted by both MHC-I and MHC-II (108). The burden of MHC-II specific neoepitopes was among features significantly associated with immunotherapy response in a human melanoma cohort, and maximal class II germline heterozygosity was associated with longer progression free survival (109). It is possible that in the absence of neoantigens capable of stimulating CD4+ T cells, even highly immunogenic class I neoantigens will not generate effective responses.
While less relevant to immunogenicity, mutated peptides must be successfully processed through various aspects of the APP. MHC binding appears to be the largest bottleneck, but alterations to other key aspects of the APP have been suggested to impact the availability and landscape of antigens, including but not limited to proteasomal dysregulation (110), TAP loss (111), and tapasin loss (112). This may further constrain the potential immunogenicity of the tumor genome.
Ultimately, recognition and robust activation by T cells will depend on both neoantigen quality and MHC integrity (Figure 4), and responses could further depend on cell surface pMHC dynamics. Is it advantageous to have a more diverse set of pMHCs on the cell surface or a higher concentration of a particular pMHC? Characteristics of the pMHC-–TCR interaction may also be important. Extremely high-affinity pMHC–TCR interactions have been suggested to result in short-lived and less effective responses (113), and small differences in affinity were found to have large effects on T cell fate (114).
Figure 4.
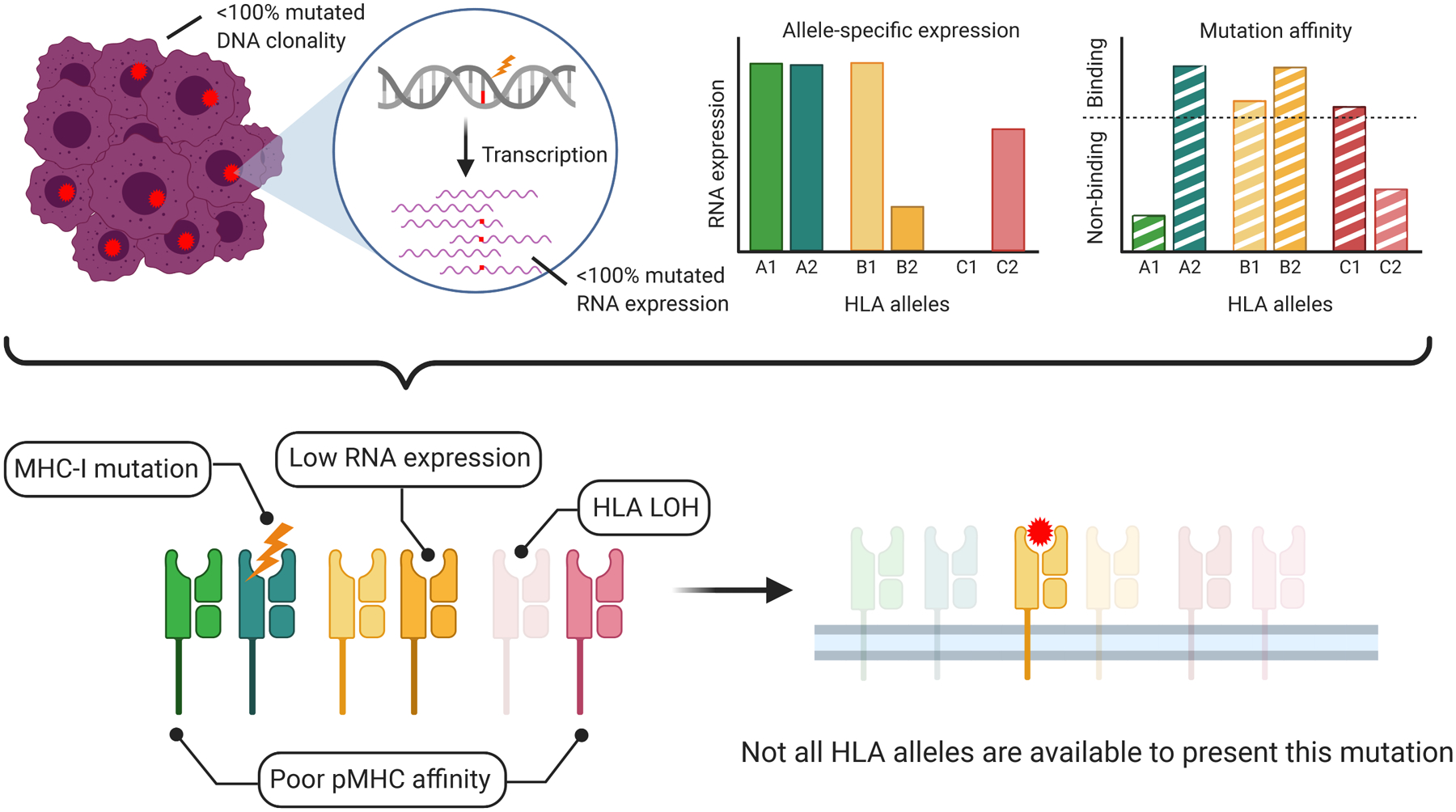
Multiple independent factors influence the potential of a neopeptide to create an effective neoepitope. A mutation with high clonality and robust expression, with the potential to be presented by multiple HLA alleles, may still be ineffective if the MHC alleles to which it can bind are somatically altered or only weakly expressed. Loss of MHC integrity is indicated by faded color. In this way, the landscape of potentially immunogenic neoepitopes may be considerably smaller than what might be predicted based on HLA affinities for neopeptides. Figure adapted from images created with BioRender.com. Abbreviations: HLA, human leukocyte antigen; LOH, loss of heterozygosity; MHC, major histocompatibility complex; MHC-I, MHC class I; pMHC, peptide–MHC.
5.2.3. Effects of immunoediting on neoantigen availability.
Tumors adapt to a dynamic microenvironment that entails nutrient deprivation, hypoxia, and increasing cellular crowding. The immune system is an aspect of the microenvironment with significant potential to impose selective forces on tumor cell populations. Schreiber’s hypothesis of immunoediting suggests that as a result, tumors evolve to be more resistant to elimination by T cells. This has been confirmed by the evolution of resistance to immune elimination posttreatment with immunotherapy, but evidence suggests that immunoediting may render some tumors less responsive even before treatment begins.
Beyond loss of MHC-I integrity, surveying the landscape of neopeptides suggests that immunoediting may enrich for cancer cell populations with less obvious targets for T cells. Lack of presentable neoantigens would impair immune surveillance even with a fully intact APP. While there is general agreement that there exists positive selection for driver mutations even in normal tissue (115), the existence of negative selection is more controversial. Tumor cells with somatic mutations conferring selective advantages (e.g., gain of function mutations in oncogenes or loss of function mutations in tumor suppressors) clearly undergo clonal expansion (116). Negative, or so-called purifying, selection acting on tumor genomes is harder to quantify. The dN/dS ratio, a metric designed for population studies (117, 118), evaluates the ratio of nonsynonymous to synonymous mutations and has been applied to quantify selection and identify driver genes (119). However, in the setting of cancer, dN/dS requires additional refinement to avoid erroneous inference, including taking into account germline contamination, other mutation types, and mutation rate (119, 120). Weak in silico signals of negative selection have been reported (119–121), although these studies do not explicitly account for clonal heterogeneity (122). In contrast, a spatial analysis of ovarian tumor populations identified stronger purifying clonal selection in regions with greater immune infiltration, with depletion of subclonal neoantigens in patients with high CD8+ tumor-infiltrating lymphocyte (TIL) levels (123). Furthermore, a spatiotemporal analysis of tumor heterogeneity in mice revealed a restriction of clonal heterogeneity by the immune system with selective elimination of immunogenic cells (124).
A pan-cancer analysis of patient-specific HLA binding to neopeptides derived from recurrent driver mutations has highlighted a bias whereby poorly presented mutations are more often observed in tumors, providing evidence that driver mutations are also subject to immunoediting via MHC-I and MHC-II (93, 94). Notably, MHC-II appeared to exert stronger selective pressure over MHC-I, highlighting an important role for CD4+ T cells. Effects of immunoediting were more pronounced in female and younger individuals (125), who generally have stronger immune responses (126). While meta-analyses of immunotherapy studies provide conflicting evidence of sex differences in response (127, 128), Ye et al. reported sex-associated differences in relevant immune characteristics of multiple tumor types (129). This necessitates drawing attention to sex and age in the context of immune responses in different tumor types.
The depletion of better-presented nonsynonymous mutations was observed independent of driver status by comparing HLA-neopeptide affinities between expressed and nonexpressed regions of tumor genomes (130). Notably, there was no such depletion of synonymous mutations, suggesting that the effect is not related to differences in somatic mutation rates between expressed and nonexpressed genes. Depletion was also stronger when individuals carried two copies of the displaying allele, suggesting that this might interfere with certain mechanisms of immune evasion such as HLA LOH. Comparing the ratio of expressed to predicted neoepitopes over two time points further suggested downregulation of MHC binding clonal mutations (131). Interestingly, samples with a consistent expressed-to-predicted neoepitope ratio had decreased antitumor immune responses and immune infiltration associated with reduced MHC-I expression. Similarly, mutated-gene-specific promoter hypermethylation was observed, affecting neopeptide expression for nearly one in four neopeptides studied (132). Altogether, these studies suggest an evolutionary advantage of tumors in limiting neoantigen availability and implicate reduced neoantigen expression as a mechanism of immune evasion in tumor subclones facing selective pressure.
5.3. A Supportive Tumor Microenvironment
Even if many neoantigens are expressed and the MHC is fully functional, if there are no immune cells capable of recognizing and responding to neoantigens, immunotherapies will fail to improve outcomes. In hepatocellular carcinoma, TMB and neoantigen quality have failed to predict responses in general, but have been predictive among patients with high levels of GZMA (133), suggesting that high TMB without high-level infiltration of functional T cells is insufficient for a protective antitumor response. Current means to predict a supportive role by the TME involve quantifying lymphocyte infiltration and inhibitory checkpoint molecule expression. Bulk RNA sequencing can also be used to evaluate the condition of the TME. RNA-based measures have proven useful for studying immune activity at the tumor site, although the presence of local heterogeneity in the TME and mechanisms of immune evasion may somewhat limit their utility (132).
5.3.1. Tumor immune infiltrates.
The absence of TILs, has generally been associated with worse immunotherapy outcomes (134). Whether a tumor is immunologically hot (containing high levels of TILs) or cold (containing few TILs) could have bearing on the existence of neoantigen-specific T cell clones that could be reactivated upon ICB. Furthermore, immune cells can play both supporting and suppressive roles at the tumor site, making it desirable to understand which cells are present and in what quantities. Several approaches have been developed to meet this need. While single-cell sequencing would be ideal, deconvolution-based approaches such as CibersortX (135), Epic (136), or xCell (137) can provide an estimate of relative immune cell abundances from bulk RNA sequencing. These methods take advantage of expression patterns specific to immune cell type via a signature matrix and estimate weights for each cell-type signature consistent with the expression of their constituent genes. However it remains unclear what cell types and states to include in signature matrices, as well as whether they should be constructed based on healthy immune cell populations or from immune cells in tumors, as these can have distinct expression profiles (135). In any case, profiling immune cell infiltrates from tumor RNA may provide further information about barriers to generating effective T cell responses against neoantigens.
5.3.2. Functional aspects of T cells.
A variety of T cell types and states have been found in immune-infiltrated tumors, and the TIL landscape may influence the potential for therapeutic response (138–140). If T cells are predominantly in a dysfunctional state, infiltration levels may not correlate with immunotherapy outcomes (141). Several measures have been developed to assess the capacity of infiltrating effector T cells to mount an immune response against tumor cells. Rooney et al. proposed the CYT score, which derives from expression levels of GZMA and PRF1 and is a proxy for TIL cytolytic activity (142). The TIDE score evaluates T cell dysfunction based on 770 genes with expression levels that have been correlated with survival outcomes in the presence of cytolytic TILs (141). The Immunophenoscore integrates expression information for effector T cells with expression information for MHC molecules, immunomodulators, and suppressor cells to capture various factors capable of modifying T cell responses (143). In the absence of RNA sequencing, the Immunoscore, which evaluates the density of beneficial TILs in the tumor core and margins, can be evaluated by immunohistochemistry as part of diagnostic pathology (144).
While CD8+ T cells are classically thought of as mediating neoantigen-directed responses, there is a growing appreciation that CD4+ T cell responses to MHC-II-restricted neoantigens play an important role in effective antitumor immunity. Higher pretreatment CD4+ T cell clonality has been associated with better outcomes in advanced melanoma (145). Tumor vaccine studies designed to generate CD8+ T cell responses reportedly result in more CD4+ T cell responses (29–31, 145). The necessity of both CD4+ and CD8+ T cell activity for effective ICB responses (108) is consistent with the requirement for CD4+ T cell help to generate robust and sustained CD8+ T cell activity. While MHC-II is predominantly expressed on APCs, it can be expressed by tumor cells, usually under conditions of IFNγ signaling. Interestingly, expression of MHC-II by tumor cells has been associated with better outcomes; however, it remains unclear if this is due to MHC-II presentation of neoantigens on tumor cells or reflects some other benefit of IFNγ associated MHC-II expression (22). In general, the level of MHC-II expression on tumor cells is significantly lower than that observed on APCs (98).
6. DISCUSSION
Rapid technological advances and accumulating clinical trial and tumor genomic data provide new insights into the complex dynamics at the tumor–immune cell interface. Understanding this relationship is critical to advance personalized cancer immunotherapy. While it is clear that neoantigens drive tumor–immune interaction dynamics throughout tumor development, tumor progression is inevitable. Thus far, studies evaluating high TMB and neopeptide load in patients that did not undergo immunotherapy found no survival advantage (45, 133), suggesting that TMB and neopeptide load do not alone guarantee effective immune surveillance. Ultimately, the immune system requires help/stimulation to effectively eliminate tumor cells. Neoantigens are central to this effort, but relatively few mutations in tumors generate truly immunogenic neoantigens. We highlight here that the evaluation of neoantigens requires attention to at least three key factors: MHC integrity, neoantigen availability, and a supportive tumor–immune microenvironment.
Mapping the landscape of somatic alterations in tumors to effective neoantigens and immunotherapy responses at scale will inevitably require in silico workflows (Figure 5) (147). Effective algorithms have been developed to predict neopeptides, but identifying neoantigens and correlating them with immunogenicity remain difficult (80). In this review, we have discussed how WXS plus RNA sequencing can be used not only to identify neopeptides but also to disclose potential mechanisms that alone or in combination could hinder natural antitumor immunity and immunotherapy. In our own work, we have identified such factors, including patient genotype–dependent immune visibility of driver mutations, MHC-II presentation, HLA and B2M mutations, the effect of immunoediting over the course of tumor development, and the effect of age and sex on tumor–immune interactions. It is obvious that immunotherapy interventions in the personalized era must take into consideration each and every one of these factors to guarantee a better-informed decision-making process on the suitability of immunotherapy and improve chances of success.
Figure 5.
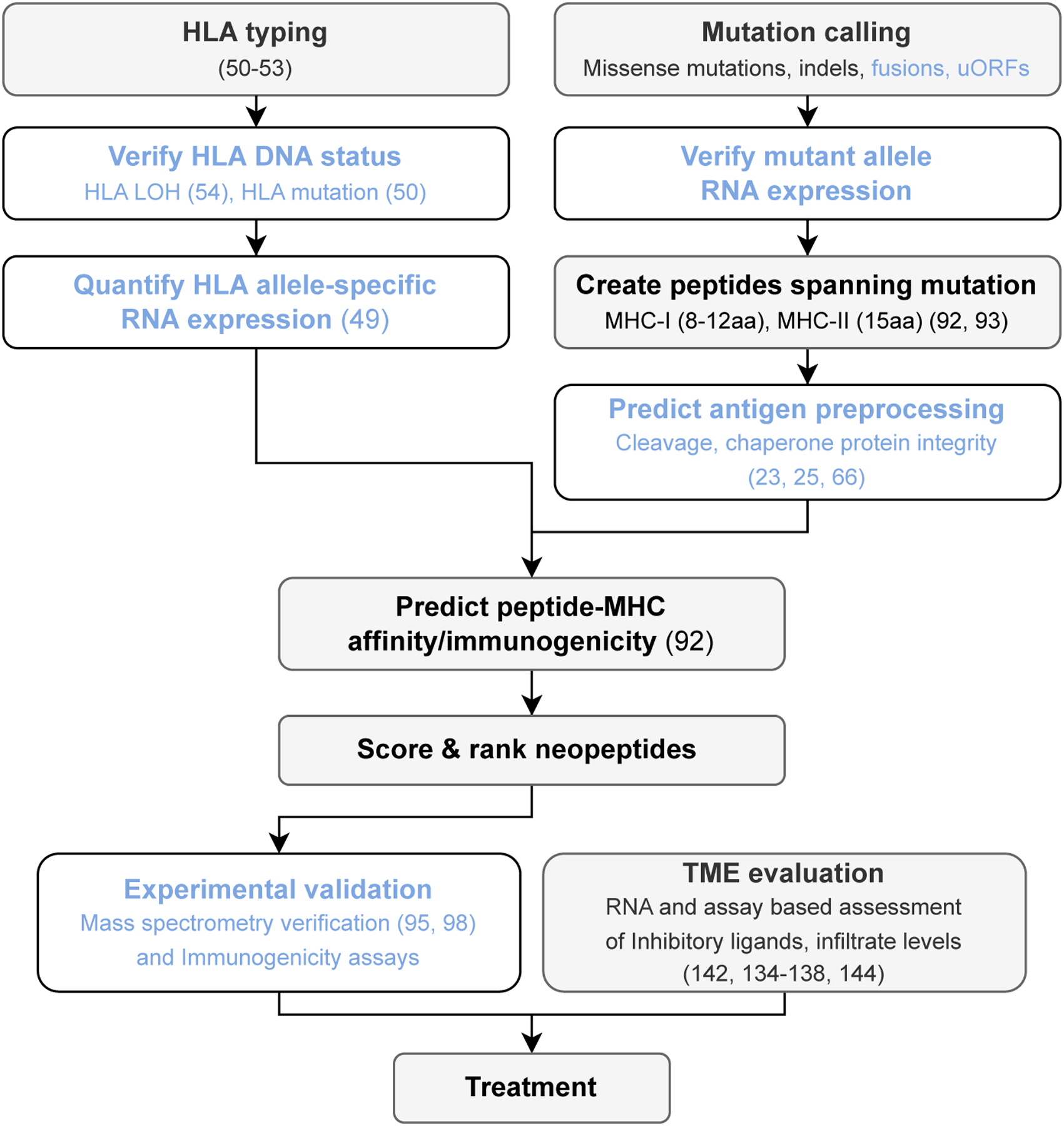
Workflow for in silico mapping of the somatic landscape of tumors to putative immunogenic neoantigens for clinical applications. Blue text indicates steps that are less commonly performed. References for further reading are included in parentheses. Abbreviations: aa, amino acids; HLA, human leukocyte antigen; LOH, loss of heterozygosity; MHC, major histocompatibility complex; MHC-I, MHC class I; MHC-II, MHC class II; uORF, upstream open reading frame.
Although much of the information generated up to this point comes from bulk tissue analyses, we believe that single-cell applications may add new information but may not scale equally well, and we argue that the added benefit may not be sufficient to justify the cost over bulk methods. Going forward, experimental validation of in silico prediction is necessary in clinical settings and should include mass spectrometry to ensure that antigens are processed, as well as in vitro assays of T cell activation to demonstrate the presence of responsive T cells. Whenever feasible, attempts should be made to demonstrate that T cells expanded by a neoantigen of interest recognize and actually kill autologous tumor cells. It is worth mentioning that, based on data from clinical trials, T cell responses against mutant peptides appear biased toward CD4+ T cells. Thus, efforts should evaluate neopeptides for both classes of MHC. Only these systematic analyses may improve the efficacy of personalized cancer immunotherapies.
While functional MHC, neoantigen availability, and a supportive TME are likely to be universal requirements for neoantigen-directed immune responses, characteristics of each may vary by tumor type. There is variation in the efficiency of antigen processing and presentation across different tumors, and consequently different tumors may adopt different mechanisms of immune evasion (148). In ccRCC, where TMB and neoantigen loads tend to be low, expression of the MHC machinery and ratios of specific T cell populations stratified patient responses where TMB and neoantigen predictions could not (149). Specific somatic alterations have been reported to drive immunotherapy resistance and may occur at different rates in different tumor types. For example, PTEN and LKB1 loss in some settings are associated with decreased T cell infiltration and suppressive cytokines, while oncogenic WNT/β-catenin and TGF-β activity are associated with immune cell exclusion from the TME (46, 47). Effectively predicting outcomes may require accounting for these as resistance mechanisms.
Consideration of tumor heterogeneity, spatial organization, and signaling networks should also be part of the decision-making process (150, 151). For instance, higher expression of the MHC machinery and the presence of effective T cells have been associated with lower levels of clonal heterogeneity in ccRCC (149). Other studies have suggested that lower clonal heterogeneity may be associated with more effective host antitumor immunity (124) and that, conversely, heterogeneity contributes to the development of resistance by facilitating cross talk between different cell populations, potentially in a manner that depends on the expression of markers such as PD-L1 and IFNγ that would otherwise be expected to predict better outcomes (152). Longitudinal studies of tumor genomes undergoing immunotherapy may be helpful for understanding the relationship of clonal heterogeneity to immunotherapy resistance.
7. CONCLUSION AND FUTURE DIRECTIONS
Identifying immunogenic neopeptides is crucial for a complete and durable response to immunotherapy but is only the beginning of a validation process we only now begin to appreciate in its complexity. While considerable progress has been made, there remain areas of knowledge that definitely need improvement. In this review we discussed those that, based on data, have been identified and, to the extent possible, quantified. Areas that still need further exploration include the MHC-II processing pathway and what exactly determines binding of the TCR to the pMHC. For the time being, the areas we have identified in this review should provide new guidance to the collective effort to use immunotherapy to treat cancer efficiently and with greater accuracy.
ACKNOWLEDGMENTS
This work was supported by an NIH (National Institutes of Health) National Library of Medicine training grant (T15LM011271) to A.C., an Emerging Leader Award from The Mark Foundation for Cancer Research (18-022-ELA) and a CIFAR (Canadian Institute for Advanced Research) fellowship to H.C., and an RO1 CA220009 grant to M.Z. and H.C.
TERMS AND DEFINITIONS
- IFNγ
interferon-γ is a cytokine that activates the class II pathway and macrophages
- indel
an insertion or deletion mutation
- GZMA
granzyme A protein is a protease released by cytotoxic T and NK cells
- PRF1
perforin-1 is a cytolytic protein produced by cytotoxic T and NK cells that forms membrane pores
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Schöne G 1919. Die Heteroplastische und Homöoplastische Transplantation. Berlin: Springer-Verlag [Google Scholar]
- 2.Silverstein AM. 1989. Transplantation and immunogenetics. In A History of Immunology, pp. 275–304. San Diego, CA: Academic [Google Scholar]
- 3.Gorer PA, Null N, Lyman S, Null N, Snell GD, et al. 1948. Studies on the genetic and antigenic basis of tumour transplantation: linkage between a histocompatibility gene and “fused” in mice. Proc. R. Soc. B 135(881):499–505 [Google Scholar]
- 4.Snell GD, Higgins GF. 1951. Alleles at the histocompatibility-2 locus in the mouse as determined by tumor transplantation. Genetics 36(3):306–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burnet M 1969. Self and Not-Self: Cellular Immunology Book One. Cambridge, UK: Cambridge Univ. Press [Google Scholar]
- 6.Kappler JW, Roehm N, Marrack P. 1987. T cell tolerance by clonal elimination in the thymus. Cell 49(2):273–80 [DOI] [PubMed] [Google Scholar]
- 7.Yu W, Jiang N, Ebert PJR, Kidd BA, Müller S, et al. 2015. Clonal deletion prunes but does not eliminate self-specific αβ CD8+ T lymphocytes. Immunity 42(5):929–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burnet FM. 1970. The concept of immunological surveillance. Prog. Exp. Tumor Res 13:1–27 [DOI] [PubMed] [Google Scholar]
- 9.Old LJ. 2001. Cancer/testis (CT) antigens: a new link between gametogenesis and cancer. Cancer Immun. 1:1. [PubMed] [Google Scholar]
- 10.Finn OJ. 2017. Human tumor antigens yesterday, today, and tomorrow. Cancer Immunol. Res 5(5):347–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. 2014. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14(2):135–46 [DOI] [PubMed] [Google Scholar]
- 12.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. 2002. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol 3(11):991–98 [DOI] [PubMed] [Google Scholar]
- 13.Schreiber RD, Old LJ, Smyth MJ. 2011. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331(6024):1565–70 [DOI] [PubMed] [Google Scholar]
- 14.Karlsson EK, Kwiatkowski DP, Sabeti PC. 2014. Natural selection and infectious disease in human populations. Nat. Rev. Genet 15(6):379–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogelstein B, Kinzler KW. 2004. Cancer genes and the pathways they control. Nat. Med 10(8):789–99 [DOI] [PubMed] [Google Scholar]
- 16.Tomasetti C, Vogelstein B. 2015. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347(6217):78–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vitiello A, Zanetti M. 2017. Neoantigen prediction and the need for validation. Nat. Biotechnol 35(9):815–17 [DOI] [PubMed] [Google Scholar]
- 18.Kochan G, Escors D, Breckpot K, Guerrero-Setas D. 2013. Role of non-classical MHC class I molecules in cancer immunosuppression. OncoImmunology 2(11):e26491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Souza MP, Adams E, Altman JD, Birnbaum ME, Boggiano C, et al. 2019. Casting a wider net: immunosurveillance by nonclassical MHC molecules. PLOS Pathog. 15(2):e1007567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scarabel L, Garziera M, Fortuna S, Asaro F, Toffoli G, Geremia S. 2020. Soluble HLA-G expression levels and HLA-G/irinotecan association in metastatic colorectal cancer treated with irinotecan-based strategy. Sci. Rep 10:8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin A, Yan W-H. 2018. Heterogeneity of HLA-G expression in cancers: facing the challenges. Front. Immunol 9:2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Axelrod ML, Cook RS, Johnson DB, Balko JM. 2019. Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res 25(8):2392–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rock KL, Reits E, Neefjes J. 2016. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 37(11):724–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colbert JD, Cruz FM, Rock KL. 2020. Cross-presentation of exogenous antigens on MHC I molecules. Curr. Opin. Immunol 64:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sánchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Pérez-Gracia JL, et al. 2017. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol 28(Suppl. 12):44–55 [DOI] [PubMed] [Google Scholar]
- 26.Sliwkowski MX, Mellman I. 2013. Antibody therapeutics in cancer. Science 341(6151):1192–98 [DOI] [PubMed] [Google Scholar]
- 27.Rosenberg SA, Restifo NP. 2015. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348(6230):62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, et al. 2015. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348(6236):803–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, et al. 2017. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547(7662):217–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, et al. 2017. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547(7662):222–26 [DOI] [PubMed] [Google Scholar]
- 31.Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanović S, et al. 2019. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565(7738):240–45 [DOI] [PubMed] [Google Scholar]
- 32.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, et al. 2019. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565(7738):234–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, et al. 2014. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med 371(23):2189–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, et al. 2019. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol 30(1):44–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, et al. 2019. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet 51(2):202–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, et al. 2017. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, et al. 2018. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol 36(7):633–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, et al. 2015. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350(6257):207–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, et al. 2015. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348(6230):124–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, et al. 2016. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387(10031):1909–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, et al. 2018. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362(6411):eaar3593. Erratum. 2019. Science 363(6430):eaax1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim S-W, et al. 2019. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N. Engl. J. Med 381(21):2020–31 [DOI] [PubMed] [Google Scholar]
- 43.Garassino M, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, et al. 2019. Evaluation of TMB in KEYNOTE-189: pembrolizumab plus chemotherapy versus placebo plus chemotherapy for nonsquamous NSCLC. J. Thorac. Oncol 14(10):S216–17 (Oral Abstr. 04.06) [Google Scholar]
- 44.Langer C, Gadgeel S, Borghaei H, Patnaik A, Powell S, et al. 2019. KEYNOTE-021: TMB and outcomes for carboplatin and pemetrexed with or without pembrolizumab for nonsquamous NSCLC. J. Thorac. Oncol 14(10):S216 (Oral Abstr. 04.05) [Google Scholar]
- 45.Wood MA, Weeder BR, David JK, Nellore A, Thompson RF. 2020. Burden of tumor mutations, neoepitopes, and other variants are weak predictors of cancer immunotherapy response and overall survival. Genome Med. 12:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keenan TE, Burke KP, Van Allen EM. 2019. Genomic correlates of response to immune checkpoint blockade. Nat. Med 25(3):389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Havel JJ, Chowell D, Chan TA. 2019. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 19(3):133–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yi M, Jiao D, Xu H, Liu Q, Zhao W, et al. 2018. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 17:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aguiar VRC, César J, Delaneau O, Dermitzakis ET, Meyer D. 2019. Expression estimation and eQTL mapping for HLA genes with a personalized pipeline. PLOS Genet. 15(4):e1008091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, et al. 2015. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol 33(11):1152–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie C, Yeo ZX, Wong M, Piper J, Long T, et al. 2017. Fast and accurate HLA typing from short-read next-generation sequence data with xHLA. PNAS 114(30):8059–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. 2014. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30(23):3310–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawaguchi S, Higasa K, Shimizu M, Yamada R, Matsuda F. 2017. HLA-HD: an accurate HLA typing algorithm for next-generation sequencing data. Hum. Mutat 38(7):788–97 [DOI] [PubMed] [Google Scholar]
- 54.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, et al. 2017. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171(6):1259–71.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cabrera T, Maleno I, Lopez-Nevot MA, Redondo M, Fernandez MA, et al. 2003. High frequency of HLA-B44 allelic losses in human solid tumors. Hum. Immunol 64(10):941–50 [DOI] [PubMed] [Google Scholar]
- 56.Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, et al. 2020. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 587:126–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, et al. 2017. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun 8:1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ben-Shmuel A, Biber G, Barda-Saad M. 2020. Unleashing natural killer cells in the tumor microenvironment—the next generation of immunotherapy? Front. Immunol 11:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Castro A, Ozturk K, Pyke RM, Xian S, Zanetti M, Carter H. 2019. Elevated neoantigen levels in tumors with somatic mutations in the HLA-A, HLA-B, HLA-C and B2M genes. BMC Med. Genom 12(Suppl. 6):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, et al. 2017. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357(6349):409–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, et al. 2018. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 8(6):730–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. 1996. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst 88(2):100–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, et al. 2018. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun 9:3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang L, Malu S, McKenzie JA, Andrews MC, Talukder AH, et al. 2018. The RNA-binding protein MEX3B mediates resistance to cancer immunotherapy by downregulating HLA-A expression. Clin. Cancer Res 24(14):3366–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bernards R, Dessain SK, Weinberg RA. 1986. N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell 47(5):667–74 [DOI] [PubMed] [Google Scholar]
- 66.Burr ML, Sparbier CE, Chan KL, Chan Y-C, Kersbergen A, et al. 2019. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 36(4):385–401.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, et al. 2015. BRAFV600E co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol. Res 3(6):602–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miyadera H, Ohashi J, Lernmark Å, Kitamura T, Tokunaga K. 2015. Cell-surface MHC density profiling reveals instability of autoimmunity-associated HLA. J. Clin. Investig 125(1):275–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu GY, Fairchild PJ, Smith RM, Prowle JR, Kioussis D, Wraith DC. 1995. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity 3(4):407–15 [DOI] [PubMed] [Google Scholar]
- 70.Sercarz EE, Lehmann PV, Ametani A, Benichou G, Miller A, Moudgil K. 1993. Dominance and crypticity of T cell antigenic determinants. Annu. Rev. Immunol 11:729–66 [DOI] [PubMed] [Google Scholar]
- 71.Zamora AE, Crawford JC, Thomas PG. 2018. Hitting the target: how T cells detect and eliminate tumors. J. Immunol 200(2):392–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schreiber H, Wu TH, Nachman J, Kast WM. 2002. Immunodominance and tumor escape. Semin. Cancer Biol 12(1):25–31 [DOI] [PubMed] [Google Scholar]
- 73.Messaoudi I, Guevara Patiño JA, Dyall R, LeMaoult J, Nikolich-Zugich J. 2002. Direct link between mhc polymorphism, T cell avidity, and diversity in immune defense. Science 298(5599):1797–800 [DOI] [PubMed] [Google Scholar]
- 74.Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, et al. 2018. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 359(6375):582–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chowell D, Krishna C, Pierini F, Makarov V, Rizvi NA, et al. 2019. Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat. Med 25(11):1715–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Negrao MV, Lam VK, Reuben A, Rubin ML, Landry LL, et al. 2019. PD-L1 expression, tumor mutational burden, and cancer gene mutations are stronger predictors of benefit from immune checkpoint blockade than HLA class I genotype in non-small cell lung cancer. J. Thorac. Oncol 14(6):1021–31 [DOI] [PubMed] [Google Scholar]
- 77.Cummings AL, Gukasyan J, Lu HY, Grogan T, Sunga G, et al. 2020. Mutational landscape influences immunotherapy outcomes among patients with non-small-cell lung cancer with human leukocyte antigen supertype B44. Nat. Cancer In press. 10.1038/s43018-020-00140-1 [DOI] [PubMed] [Google Scholar]
- 78.Nielsen JS, Chang AR, Wick DA, Sedgwick CG, Zong Z, et al. 2017. Mapping the human T cell repertoire to recurrent driver mutations in MYD88 and EZH2 in lymphoma. OncoImmunology 6(7):e1321184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, et al. 2018. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33(5):843–52.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schumacher TN, Schreiber RD. 2015. Neoantigens in cancer immunotherapy. Science 348(6230):69–74 [DOI] [PubMed] [Google Scholar]
- 81.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, et al. 2017. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171(4):934–49.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamashita H, Nakayama K, Ishikawa M, Nakamura K, Ishibashi T, et al. 2018. Microsatellite instability is a biomarker for immune checkpoint inhibitors in endometrial cancer. Oncotarget 9(5):5652–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schrock AB, Ouyang C, Sandhu J, Sokol E, Jin D, et al. 2019. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol 30(7):1096–103 [DOI] [PubMed] [Google Scholar]
- 84.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, et al. 2017. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 18(8):1009–21 [DOI] [PubMed] [Google Scholar]
- 85.McGranahan N, Swanton C. 2019. Neoantigen quality, not quantity. Sci. Transl. Med 11(506):eaax7918. [DOI] [PubMed] [Google Scholar]
- 86.Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, et al. 2017. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551(7681):512–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goodman AM, Castro A, Pyke RM, Okamura R, Kato S, et al. 2020. MHC-I genotype and tumor mutational burden predict response to immunotherapy. Genome Med. 12:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ghorani E, Reading JL, Henry JY, de Massy MR, Rosenthal R, et al. 2020. The T cell differentiation landscape is shaped by tumour mutations in lung cancer. Nat. Cancer 1(5):546–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shim JH, Kim HS, Cha H, Kim S, Kim TM, et al. 2020. HLA-corrected tumor mutation burden and homologous recombination deficiency for the prediction of response to PD-(L)1 blockade in advanced non-small-cell lung cancer patients. Ann. Oncol 31(7):902–11 [DOI] [PubMed] [Google Scholar]
- 90.McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, et al. 2016. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351(6280):1463–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, et al. 2017. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 7(3):264–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nielsen M, Andreatta M, Peters B, Buus S. 2020. Immunoinformatics: predicting peptide–MHC binding. Annu. Rev. Biomed. Data Sci 3:191–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, et al. 2017. MHC-I genotype restricts the oncogenic mutational landscape. Cell 171(6):1272–83.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marty Pyke R, Thompson WK, Salem RM, Font-Burgada J, Zanetti M, Carter H. 2018. Evolutionary pressure against MHC class II binding cancer mutations. Cell 175(2):416–28.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, et al. 2017. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 46(2):315–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Harndahl M, Rasmussen M, Roder G, Dalgaard Pedersen I, Sørensen M, et al. 2012. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur. J. Immunol 42(6):1405–16 [DOI] [PubMed] [Google Scholar]
- 97.Silva Z, Ferro T, Almeida D, Soares H, Ferreira JA, et al. 2020. MHC class I stability is modulated by cell surface sialylation in human dendritic cells. Pharmaceutics 12(3):249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Abelin JG, Harjanto D, Malloy M, Suri P, Colson T, et al. 2019. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity 51(4):766–79.e17 [DOI] [PubMed] [Google Scholar]
- 99.Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, et al. 2014. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med 211(11):2231–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, et al. 2017. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551(7681):517–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, et al. 2020. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell 183(3):818–34.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ghorani E, Rosenthal R, McGranahan N, Reading JL, Lynch M, et al. 2018. Differential binding affinity of mutated peptides for MHC class I is a predictor of survival in advanced lung cancer and melanoma. Ann. Oncol 29(1):271–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, et al. 2014. Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157(5):1073–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chowell D, Krishna S, Becker PD, Cocita C, Shu J, et al. 2015. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. PNAS 112(14):E1754–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Calis JJA, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, et al. 2013. Properties of MHC class I presented peptides that enhance immunogenicity. PLOS Comput. Biol 9(10):e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Capietto A-H, Jhunjhunwala S, Pollock SB, Lupardus P, Wong J, et al. 2020. Mutation position is an important determinant for predicting cancer neoantigens. J. Exp. Med 217(4):e20190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Glanville J, Huang H, Nau A, Hatton O, Wagar LE, et al. 2017. Identifying specificity groups in the T cell receptor repertoire. Nature 547(7661):94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, et al. 2019. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574(7780):696–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. .Anagnostou V, Bruhm DC, Niknafs N, White JR, Shao XM, et al. 2020. Integrative tumor and immune cell multi-omic analyses predict response to immune checkpoint blockade in melanoma. Cell Rep. Med 1(8):100139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen Y, Zhang Y, Guo X. 2017. Proteasome dysregulation in human cancer: implications for clinical therapies. Cancer Metastasis Rev. 36(4):703–16 [DOI] [PubMed] [Google Scholar]
- 111.Ling A, Löfgren-Burström A, Larsson P, Li X, Wikberg ML, et al. 2017. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. OncoImmunology 6(11):e1356143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sokol L, Koelzer VH, Rau TT, Karamitopoulou E, Zlobec I, Lugli A. 2015. Loss of tapasin correlates with diminished CD8+ T-cell immunity and prognosis in colorectal cancer. J. Transl. Med 13:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Presotto D, Erdes E, Duong MN, Allard M, Regamey P-O, et al. 2017. Fine-tuning of optimal TCR signaling in tumor-redirected CD8 T cells by distinct TCR affinity-mediated mechanisms. Front. Immunol 8:1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Conley JM, Gallagher MP, Berg LJ. 2016. T cells and gene regulation: the switching on and turning up of genes after T cell receptor stimulation in CD8 T cells. Front. Immunol 7:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, et al. 2015. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348(6237):880–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, et al. 2020. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 20:555–72 [DOI] [PubMed] [Google Scholar]
- 117.Kryazhimskiy S, Plotkin JB. 2008. The population genetics of dN/dS. PLOS Genet. 4(12):e1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cvijović I, Good BH, Desai MM. 2018. The effect of strong purifying selection on genetic diversity. Genetics 209(4):1235–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, et al. 2017. Universal patterns of selection in cancer and somatic tissues. Cell 171(5):1029–41.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Van den Eynden J, Jiménez-Sánchez A, Miller ML, Larsson E. 2019. Lack of detectable neoantigen depletion signals in the untreated cancer genome. Nat. Genet 51(12):1741–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Weghorn D, Sunyaev S. 2017. Bayesian inference of negative and positive selection in human cancers. Nat. Genet 49(12):1785–88 [DOI] [PubMed] [Google Scholar]
- 122.Bakhoum SF, Landau DA. 2017. Cancer evolution: no room for negative selection. Cell 171(5):987–89 [DOI] [PubMed] [Google Scholar]
- 123.Zhang AW, McPherson A, Milne K, Kroeger DR, Hamilton PT, et al. 2018. Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell 173(7):1755–69.e22 [DOI] [PubMed] [Google Scholar]
- 124.Milo I, Bedora-Faure M, Garcia Z, Thibaut R, Périé L, et al. 2018. The immune system profoundly restricts intratumor genetic heterogeneity. Sci. Immunol 3(29):eaat1435. [DOI] [PubMed] [Google Scholar]
- 125.Castro A, Pyke RM, Zhang X, Thompson WK, Day C-P, et al. 2020. Strength of immune selection in tumors varies with sex and age. Nat. Commun 11:4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Klein SL, Flanagan KL. 2016. Sex differences in immune responses. Nat. Rev. Immunol 16(10):626–38 [DOI] [PubMed] [Google Scholar]
- 127.Conforti F, Pala L, Bagnardi V, De Pas T, Martinetti M, et al. 2018. Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. Lancet Oncol. 19(6):737–46 [DOI] [PubMed] [Google Scholar]
- 128.Zhang T, Jia H, Wu Z. 2018. Sex as a predictor of response to cancer immunotherapy. Lancet Oncol. 19(8):e374. [DOI] [PubMed] [Google Scholar]
- 129.Ye Y, Jing Y, Li L, Mills GB, Diao L, et al. 2020. Sex-associated molecular differences for cancer immunotherapy. Nat. Commun 11:1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang F, Kim D-K, Nakagawa H, Hayashi S, Imoto S, et al. 2019. Quantifying immune-based counterselection of somatic mutations. PLOS Genet. 15(7):e1008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nejo T, Matsushita H, Karasaki T, Nomura M, Saito K, et al. 2019. Reduced neoantigen expression revealed by longitudinal multiomics as a possible immune evasion mechanism in glioma. Cancer Immunol. Res 7(7):1148–61 [DOI] [PubMed] [Google Scholar]
- 132.Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, et al. 2019. Neoantigen-directed immune escape in lung cancer evolution. Nature 567(7749):479–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mauriello A, Zeuli R, Cavalluzzo B, Petrizzo A, Tornesello ML, et al. 2019. High somatic mutation and neoantigen burden do not correlate with decreased progression-free survival in HCC patients not undergoing immunotherapy. Cancers 11(12):1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Badalamenti G, Fanale D, Incorvaia L, Barraco N, Listì A, et al. 2019. Role of tumor-infiltrating lymphocytes in patients with solid tumors: Can a drop dig a stone? Cell Immunol. 343:103753 [DOI] [PubMed] [Google Scholar]
- 135.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, et al. 2019. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol 37(7):773–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. 2017. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 6:e26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Aran D, Hu Z, Butte AJ. 2017. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.van der Leun AM, Thommen DS, Schumacher TN. 2020. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20(4):218–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Thommen DS, Schumacher TN. 2018. T cell dysfunction in cancer. Cancer Cell 33(4):547–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Plesca I, Tunger A, Müller L, Wehner R, Lai X, et al. 2020. Characteristics of tumor-infiltrating lymphocytes prior to and during immune checkpoint inhibitor therapy. Front. Immunol 11:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jiang P, Gu S, Pan D, Fu J, Sahu A, et al. 2018. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med 24(10):1550–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. 2015. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160(1–2):48–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, et al. 2017. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18(1):248–62 [DOI] [PubMed] [Google Scholar]
- 144.Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, et al. 2014. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol 232(2):199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Arakawa A, Vollmer S, Tietze J, Galinski A, Heppt MV, et al. 2019. Clonality of CD4+ blood T cells predicts longer survival with CTLA4 or PD-1 checkpoint inhibition in advanced melanoma. Front. Immunol 10:1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, et al. 2015. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520(7549):692–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Richters MM, Xia H, Campbell KM, Gillanders WE, Griffith OL, Griffith M. 2019. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 11:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang S, He Z, Wang X, Li H, Liu X-S. 2019. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. eLife 8:e49020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Şenbabaoğlu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, et al. 2016. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 17:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hausser J, Alon U. 2020. Tumour heterogeneity and the evolutionary trade-offs of cancer. Nat. Rev. Cancer 20(4):247–57 [DOI] [PubMed] [Google Scholar]
- 151.Creixell P, Schoof EM, Erler JT, Linding R. 2012. Navigating cancer network attractors for tumor-specific therapy. Nat. Biotechnol 30(9):842–48 [DOI] [PubMed] [Google Scholar]
- 152.Williams JB, Li S, Higgs EF, Cabanov A, Wang X, et al. 2020. Tumor heterogeneity and clonal cooperation influence the immune selection of IFN-γ-signaling mutant cancer cells. Nat. Commun 11:602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pieper R, Christian RE, Gonzales MI, Nishimura MI, Gupta G, et al. 1999. Biochemical identification of a mutated human melanoma antigen recognized by CD4+ T cells. J. Exp. Med 189(5):757–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang R-F, Wang X, Rosenberg SA. 1999. Identification of a novel major histocompatibility complex class II–restricted tumor antigen resulting from a chromosomal rearrangement recognized by CD4+ T cells. J. Exp. Med 189(10):1659–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang RF, Wang X, Atwood AC, Topalian SL, Rosenberg SA. 1999. Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science 284(5418):1351–54 [DOI] [PubMed] [Google Scholar]
- 156.Yotnda P, Firat H, Garcia-Pons F, Garcia Z, Gourru G, et al. 1998. Cytotoxic T cell response against the chimeric p210 BCR-ABL protein in patients with chronic myelogenous leukemia. J. Clin. Investig 101(10):2290–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yotnda P, Garcia F, Peuchmaur M, Grandchamp B, Duval M, et al. 1998. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J. Clin. Investig 102(2):455–62 [DOI] [PMC free article] [PubMed] [Google Scholar]