

Background:
Macrophage-inducible C-type lectin (Mincle) is expressed on hepatic macrophages and senses ethanol (EtOH)-induced danger signals released from dying hepatocytes and promotes IL-1β production. However, it remains unclear what and how EtOH-induced Mincle ligands activate downstream signaling events to mediate IL-1β release and contribute to alcohol-associated liver disease (ALD). In this study, we investigated the association of circulating β-glucosylceramide (β-GluCer), an endogenous Mincle ligand, with severity of ALD and examined the mechanism by which β-GluCer engages Mincle on hepatic macrophages to release IL-1β in the absence of cell death and exacerbates ALD.
Method and Results:
Concentrations of β-GluCer were increased in serum of patients with severe AH and correlated with disease severity. Challenge of hepatic macrophages with lipopolysaccharide and β-GluCer induced formation of a Mincle and Gsdmd-dependent secretory complex containing chaperoned full-length gasdermin D (Hsp90-CDC37-NEDD4) with polyubiquitinated pro-IL-1β and components of the Caspase 8-NLRP3 inflammasome loaded as cargo in small extracellular vesicles (sEVs). Gao-binge EtOH exposure to wild-type, but not Mincle −/− and Gsdmd −/− , mice increased release of IL-1β-containing sEVs from liver explant cultures. Myeloid-specific deletion of Gsdmd similarly decreased the formation of sEVs by liver explant cultures and protected mice from EtOH-induced liver injury. sEVs collected from EtOH-fed wild-type, but not Gsdmd −/− , mice promoted injury of cultured hepatocytes and, when injected into wild-type mice, aggravated Gao-binge EtOH-induced liver injury.
Conclusion:
β-GluCer functions as a danger-associated molecular pattern activating Mincle-dependent gasdermin D-mediated formation and release of IL-1β-containing sEVs, which in turn exacerbate hepatocyte cell death and contribute to the pathogenesis of ALD.
INTRODUCTION
Alcohol-associated liver disease (ALD) ranges from steatosis to hepatitis, fibrosis, cirrhosis, and HCC.1 Severe alcohol-associated hepatitis (sAH) and chronic ALD are primary drivers of liver disease morbidity and mortality in the US, but effective treatment strategies are not available.2,3 Inflammatory responses are critical contributors to progression of ALD. Impaired intestinal barrier integrity and changes in the microbiome contribute to increased circulating concentrations of microbes and their metabolites in patients with ALD and in animal models of ALD.4 Recognition of lipopolysaccharide (LPS) by Toll-like receptor 4 on resident hepatic macrophages stimulates the expression of inflammatory cytokines, including TNFα and IL-1β. These inflammatory mediators in turn impact the functions of hepatocytes, liver sinusoidal endothelial cells, and stellate cells, linking inflammation to loss of hepatocellular function and death.5–7 Multiple programmed cell death pathways are associated with ALD, including apoptosis, necroptosis, and pyroptosis.8,9 Caspase (Casp) 3, commonly associated with apoptosis, a relatively noninflammatory form of cell death,10 is activated in livers of ethanol (EtOH)-fed mice and patients with AH.11,12 Recent evidence suggests that Casp11-mediated pyroptosis also plays a fundamental role in EtOH-induced liver injury.13
Abundant evidence indicates that the combination of increased circulating endotoxin and EtOH-induced hepatocellular death drives hepatic inflammation in ALD.1 However, the mechanisms by which the relatively low concentrations of endotoxin present in the context of alcohol consumption initiates chronic low-grade inflammation and how this is amplified in the progression of ALD are unknown. We reported that low concentrations of endotoxin, reflecting the relevant pathophysiological concentrations in both patients with ALD and EtOH-fed mice, induce expression of macrophage-inducible C-type lectin (Mincle/Clec4e), a sensor for cell death, via IRAKM-dependent Toll-like receptor 4 signaling in hepatic macrophages.5 Mincle detects molecules released by dead hepatocytes, including β-glucosylceramide (β-GluCer), spliceosome-associated protein 130 (SAP130), and cholesterol sulfate, and activates inflammasomes and IL-1β production.14–16 Therefore, we proposed that Mincle serves as a critical link between cell death and inflammation in ALD.5
IL-1β and IL-18 production by inflammasomes are critical drivers of hepatic inflammation and progression of ALD.1 The inflammasome is a Casp-containing multiprotein complex that processes pro-IL-1β and pro-IL-18 into their mature active forms.17–19 Mice deficient in inflammasome components, defective in IL-1β signaling, or provided with exogenous IL-1β receptor antagonist are protected from EtOH-induced liver injury.20,21 IL-1β and IL18 concentrations are increased in patients with AH and are associated with disease severity.22–26 Two multicenter double-blind, randomized, placebo-controlled trials are currently evaluating the efficacy of canakinumab (anti-IL-1β) (NCT037751090) or anakinra (IL1 receptor antagonist) (NCT04072822) in AH.
Canonical IL-1β secretion involves initial processing of the inactive precursor of IL-1β by inflammasomes, followed by release of mature IL-1β from lytic cells.17 However, multiple noncanonical forms of nonlytic/cell death-independent release of IL-1β have also been described and are particularly important for export of mature IL-1β from neutrophils and macrophages in response to challenge with microbial products.27,28 Human monocytes also release IL-1β in a Casp8-dependent alternative inflammasome activation pathway that is independent of pyroptosis.29 The contributions of noncanonical pathways of IL-1β release in the context of ALD are not well understood.
Gasdermin D (GSDMD) is classically associated with pyroptotic cell death, whereby GSDMD is cleaved by Casp1/11 and the N-terminal fragment of GSDMD then forms oligomeric pores in the plasma membrane, resulting in lytic cell death.30 Our recent study utilizing intestinal epithelial cells (IECs) revealed a novel form of IL-1β secretion mediated by a GSDMD-dependent nonpyroptotic release of small extracellular vesicles (sEVs). In IECs, we found that GSDMD is required for formation and release of sEVs containing polyubiquitinated pro-IL-1β on activation of a Casp8-NLRP3 inflammasome.31 Mechanistically, full-length GSDMD is chaperoned by an Hsp90-CDC37 complex in IECs. In response to stimulus 1 (LPS), the chaperoned full-length GSDMD engages NEDD4, an E3 ubiquitin ligase, and brings it into the proximity of pro-IL-1β. The pro-IL-1β is then captured in the Casp8-NLRP3 inflammasome on stimulus 2 (ATP). NEDD4 subsequently catalyzes the polyubiquitination of pro-IL-1β; polyubiquitination promotes the loading of the entire complex into vesicles destined for release into the extracellular space.31
As Mincle expressed on hepatic macrophages senses EtOH-induced danger signals, contributing to IL-1β release and inflammatory responses,5 here we hypothesized that Mincle-dependent IL-1β production in hepatic macrophages also relies on the GSDMD-dependent pathway we discovered in IECs. Although in previous studies we identified SAP130 as an important danger-associated molecular pattern–activating Mincle in response to EtOH,5 here we report accumulation of another Mincle ligand, β-GluCer, in the circulation of patients with severe alcohol-associated hepatitis (sAH) and in mice after chronic EtOH feeding. Both Mincle and Gsdmd were required for release of IL-1β-containing sEVs from hepatic macrophages, and myeloid Gsdmd-deficient mice were protected from EtOH-induced liver injury. Provision of sEVs isolated from wild-type, but not Gsdmd-deficient, mice exacerbated EtOH-induced liver injury in mice. Taken together, these data identify β-GluCer-Mincle-GSDMD signaling in the regulation of IL-1β secretion in sEVs, providing an important link between hepatocellular injury and inflammation to drive the pathogenesis of ALD.
EXPERIMENTAL PROCEDURES
Additional experimental details can be found in Supplemental Information (http://links.lww.com/HC9/A243).
Patient samples
Deidentified serum and plasma samples, along with basic clinical and demographic data, were obtained from the Northern Ohio Alcohol Center biorepository (NCT03224949). Patients with AH were stratified as moderate AH [model for end-stage liver disease (MELD) <20] or severe AH (MELD ≥20). Descriptive demographic and clinical data are provided in Supplemental Table 1 (http://links.lww.com/HC9/A243). For western blots, samples from 5 livers explanted from patients with severe AH during liver transplantation and 5 wedge biopsies from healthy donor livers were snap frozen in liquid nitrogen and stored at −80 °C. AH and healthy donor samples were provided by the NIAAA R24 Clinical Resource for Alcoholic Hepatitis Investigations at Johns Hopkins University. Clinical and demographic data on these subjects were previously reported.32 This study was approved by the Institutional Review Board at Cleveland Clinic (IRB 17-718), and all study participants consented before collection of data and blood samples.
Mouse model
All mice were on C57BL/6 background. Mincle-deficient, Gsdmd-deficient, and Gsdmd fl/fl(Cyagen Biosciences) mice were previously described.5,31,33 Wild-type C57BL/6J mice were purchased from Jackson Laboratories. All procedures involving animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Ten- to twelve-week-old female knockout and heterozygous littermate mice were exposed to the Gao-binge (acute on chronic) model of EtOH exposure.34
Cell and liver explant culture
Primary hepatocytes and primary hepatic macrophages from mice were isolated and cultured as previous described.5,34 The immortalized mouse KC line (imKC) was purchased from Sigma (Cat#SCC119). For liver explant culture, mouse livers were minced (~2 mm) and cultured overnight in serum-free culture medium (DMEM). Culture media were collected and used for isolation of EVs.
Exosome analysis
EVs were collected from culture medium of mice liver explant cultures or cultured cells. EVs were isolated using an exosome isolation kit (Invitrogen, 4478359) according to the manufacturer’s instructions and subjected to nanoparticle tracking ZetaView analysis for quantification and sizing or for measurement of IL-1β by ELISA. The polyethylene glycol-based density gradient method was used for enrichment of extracellular vesicles from human plasma.
Data analysis and statistics
Data are expressed as mean±SEM. Mouse feeding trials were as follows: n=4–6 for pair fed and n=6–8 for EtOH fed. For cell culture experiments, at least 3 independent experiments were conducted. GraphPad Prism 7 was used for data analysis when Student t test was required, as well as for data representation. SAS (Carey, IN) was used for ANOVA using the general linear model procedure, and follow-up comparisons made by least square means testing. Data were log-transformed as necessary to obtain a normal distribution. p < 0.05 was considered significant.
RESULTS
Serum β-GluCer was increased in patients with AH and mice exposed to Gao-binge EtOH feeding
We recently reported that challenging peripheral blood mononuclear cells from patients with sAH with low concentrations of LPS, equivalent to those detected in the circulation of patients with ALD, increases Mincle expression35; secondary challenge with LPS and/or the Mincle ligand, trehalose-6,6-dibehenate (TDB), induced higher IL-1β expression in peripheral blood mononuclear cells from patients with sAH compared with healthy controls.35 β-GluCer, an endogenous Mincle ligand released by damaged or dying cells, functions as an endogenous danger-associated molecular pattern to amplify inflammatory responses.15 Circulating concentrations of β-GluCer are elevated in patients with diverse chronic inflammatory diseases, implicating β-GluCer as a functional biomarker for tissue damage and inflammation.36 Here we find that serum concentrations of the abundant d18:1/16:0 species of β-GluCer were increased in patients with severe AH and alcohol-associated cirrhosis compared with healthy controls (Figure 1A). MELD score, an indicator of severity of AH, was positively correlated with β-GluCer (d18:1/16:0) (Figure 1B). The concentration of β-GluCer (d18:1/16:0) was also higher in plasma from mice after Gao-binge (acute on chronic) EtOH feeding compared with pair-fed controls (Figure 1C). Challenge of primary hepatocyte cultures with 150 mM EtOH for 24 hours also increased the accumulation of β-GluCer in the culture media (Figure 1D).
FIGURE 1.
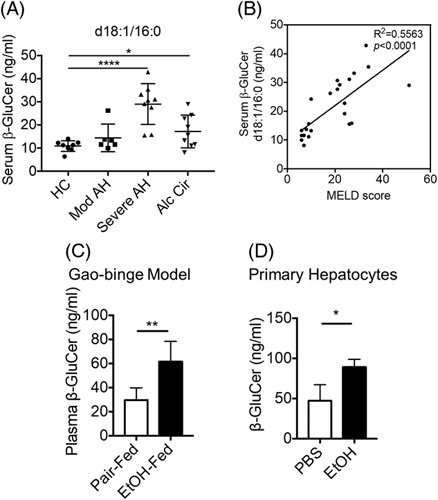
The macrophage-inducible C-type lectin ligand, β-GluCer, was increased in the circulation of patients with AH and mice after exposure to Gao-binge EtOH feeding. (A) Concentration of β-GluCer (d18:1/16:0) in serum of HCs (n=8) and patients with moderate AH (n=6), severe AH (n=9), or alcohol-associated cirrhosis (n=9). (B) Concentration of β-GluCer was correlated with MELD scores of patients with moderate and severe AH. (C) Concentration of β-GluCer in plasma of mice after Gao-binge EtOH feeding. (D) β-GluCer in culture medium of primary hepatocytes treated with 150 mM EtOH for 24hrs. β-GluCer concentrations were measured by mass spectroscopy. Data represent mean±SEM. ANOVA (A) or 2-tailed unpaired Student t test (C, D). *p < 0.05, **p < 0.01, ****p < 0.0001. Abbreviations: AH, alcohol-associated hepatitis; β-GluCer, β-glucosylceramide; EtOH, ethanol; MELD, model for end-stage liver disease.
β-GluCer promoted Mincle-dependent IL-1β production without triggering cell death in hepatic macrophages
We have previously reported that expression of Mincle on hepatic macrophages is increased in response to chronic EtOH exposure and amplifies inflammatory responses in the liver by sensing the hepatocyte-derived danger signal SAP130 to promote IL-1β secretion.5 As β-GluCer, another endogenous Mincle ligand with potent immunostimulatory activity,15 is elevated in patients with sAH and mice exposed to EtOH, we hypothesized that β-GluCer would also activate Mincle-expressing hepatic macrophages. To test this hypothesis, we knocked down Mincle (Mincle-KD) in an imKC using small hairpin RNA. Cells were then primed with low-concentration LPS (100 pg/mL for 12 h), followed by challenge with the Mincle ligands, β-GluCer (20 µg/mL for 2 h) or TDB (2 µg/mL for 2 h). β-GluCer, similar to TDB,5 increased expression of inflammatory cytokines IL1β, TNFα, and IL6 in a Mincle-dependent manner (Supplemental Figure 1, http://links.lww.com/HC9/A243). We next investigated the interaction of Mincle and β-GluCer on release of IL1β. Primary cultures of hepatic macrophages from Mincle +/− and Mincle-deficient mice were first primed with LPS (10 ng/mL for 12 h) and then stimulated with β-GluCer (20 µg/mL for 12 h) or ATP (1 h), as a positive control. Pro-IL-1β expression in cell lysates was not affected by genotype or treatments (Figure 2A). In contrast, β-GluCer stimulation increased IL-1β cleavage (Figure 2A) and secretion into the cell culture media (Figure 2B). The LPS/β-GluCer-stimulated secretion of IL-1β was modest compared with that in LPS/ATP-treated cells (Figure 2B). Importantly, LPS/β-GluCer-stimulated, but not LPS/ATP-stimulated, IL-1β expression was dependent on Mincle (Figure 2A/B). Intriguingly, although hepatic macrophages treated with LPS/ATP produced IL-1β that was coupled with cell death, challenge of hepatic macrophages with LPS/β-GluCer did not impact cell viability, assessed by lactate dehydrogenase release (Figure 2C). These data indicated that β-GluCer-activated Mincle signaling in hepatic macrophages led to IL-1β secretion in the absence of lytic cell death.
FIGURE 2.
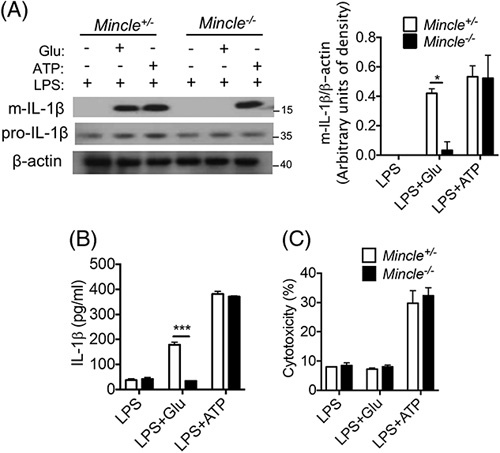
β-GluCer promoted Mincle-dependent IL-1β production without triggering cell death in hepatic macrophages. Primary hepatic macrophages isolated from Mincle +/- and Mincle −/− mice were primed overnight with 10 ng/mL LPS and then stimulated with 20 µg/mL GluCer for 12 hours or 2.5 mM ATP for 1 hour. (A) Cell lysates were collected for western blot analysis of pro- and mature- (m) IL-1β, with β-actin used as a loading control. (B/C) Cell culture medium was collected and used to (B) measure for IL-1β concentration by ELISA or (C) measure LDH release to assess cytotoxicity. Data represent mean±SEM. Two-tailed unpaired Student t test. *p < 0.05, ***p < 0.001. Abbreviations: Mincle, macrophage-inducible C-type lectin; LPS, lipopolysaccharide.
Mincle-dependent IL-1β secretion was mediated by GSDMD-guided formation and release of sEVs
We next investigated the mechanism for the nonlytic release of IL-1β in response to β-GluCer-activated Mincle signaling in hepatic macrophages. Emerging evidence supports an important function for cell death-independent release of IL-1β,27–29 including our recent discovery of a nonpyroptotic role of GSDMD in the formation and release of IL-1β-containing sEVs from IECs.31 Therefore, we asked whether Mincle-dependent IL-β release from hepatic macrophages is also achieved via activation of this GSDMD-mediated nonlytic pathway. To test this hypothesis, we knocked down Gsdmd (Gsdmd-KD) or Mincle (Mincle-KD) in imKCs using small hairpin RNA. We primed wild-type, Gsdmd-KD, and Mincle-KD imKCs with low-concentration LPS (100 pg/mL for 12 h), followed by challenge with the Mincle ligands, β-GluCer (20 ug/mL for 12 h) or TDB (2 ug/mL for 12 h). Both LPS/β-GluCer and LPS/TDB induced a robust release of sEVs in wild-type imKCs; the size of the sEVs is illustrated by electron microscopy (Figure 3A). Importantly, knockdown of Gsdmd or Mincle impaired the LPS/β-GluCer-induced and LPS/TDB-induced release of sEVs from imKCs (Figure 3A).
FIGURE 3.
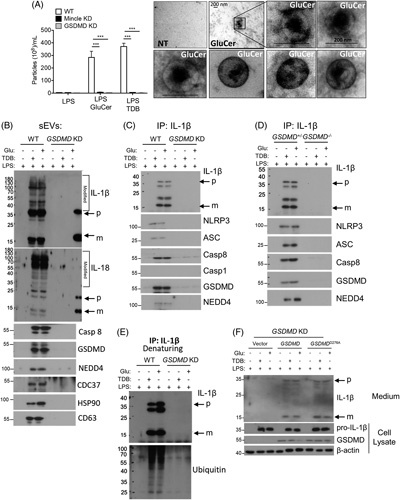
Mincle signal-dependent release of IL-1β-containing sEVs from hepatic macrophages requires GSDMD. (A–D) immortalized murine KCs (imKCs) were transfected with small hairpin RNA to knockdown Gsdmd or Mincle and then primed overnight with 100 pg/mL LPS followed by stimulation with 20 µg/mL GluCer or 2 µg/mL TDB for 12 hours. (A) sEVs released from imKC were quantified by ZetaView and visualized by electron microscopy. (B) Western analysis of sEVs from WT and Gsdmd KD imKCs using antibodies against the indicated proteins (Rec: recombinant protein as + control; p: precursor; m: mature IL-1β or IL-18). Note: For all western using sEVs, sEVs isolated from an equal volume of cell culture media were loaded in gels; sEVs were not normalized for particle number. (C) IL-1β was immunoprecipitated from WT and Gsdmd-KD imKC culture media followed by western analyses with antibodies against the indicated proteins. (D) IL-1β was immunoprecipitated from the cell culture media from WT and Gsdmd-KD imKCs and probed with antibodies to IL-1β or ubiquitin under denaturing conditions. (E) Primary KC isolated from heterozygous littermates, and Gsdmd −/− mice were primed overnight with 100 pg/mL LPS followed by stimulation with 20 µg/mL GluCer or 2 µg/mL TDB for 12 hours. IL-1β was immunoprecipitated from culture media and analyzed by western blot as in (C). (F) Expression vectors containing WT Gsdmd or D276A mutated Gsdmd were transfected into Gsdmd-KD imKCs, then primed with 100 pg/mL LPS overnight, and stimulated with 20 µg/mL GluCer or 2 µg/mL TDB for 12 hours. Cell lysates and culture medium were collected for western analyses for indicated proteins. N=3 independent experiments. Data were represent mean±SEM. One way ANOVA ***p < 0.001. Abbreviations: GluCer, glucosylceramide; GSDMD, gasdermin D; Gsdmd-KD knocked down Gsdmd; LPS, lipopolysaccharide; Mincle-KD, knocked down Mincle; sEVs, small extracellular vesicles; TDB, trehalose-6,6-dibehenate; WT, wild-type.
We have previously reported that when the Casp8 inflammasome is activated in IECs, full-length GSDMD is chaperoned by CDC37/HSP90 and recruits NEDD4 (an E3 ligase) to the complex.31 NEDD4 in turn ubiquitinates pro-IL-1β; this facilitates the loading of pro-IL-1β into the cargo of the sEVs.31 Importantly, sEVs released from LPS/β-GluCer-treated imKCs contained high–molecular-weight modified pro-IL-1β, pro-Il-18, Casp8, full-length GSDMD, NEDD4, HSP90, and CDC37 (Figure 3B). The IL-1β/IL-18-containing sEVs from LPS/β-GluCer-treated imKCs were also positive for the exosome marker CD63 (Figure 3B). Taken together, these data are consistent with the hypothesis that β-GluCer-activated Mincle signaling in hepatic macrophages uses the GSDMD-mediated nonlytic pathway to release IL-1β/IL-18-containing sEVs.
Although the sEVs contained both IL-1β and IL-18 inflammasome products, we focused our mechanistic studies on IL-1β. When IL-1β was immunoprecipitated from the culture media of LPS/β-GluCer-treated imKCs (Figure 3C) or primary hepatic macrophages (Figure 3D), these same GSDMD-interacting partners were present in a secretory complex along with pro-IL-1β and mature-IL-1β and Casp8-NLRP3 inflammasome components (Figure 3C, D). Importantly, Gsdmd was required for both the release of sEVs containing IL-1β, IL-18, GSDMD, NEDD4, and Casp8 (Figure 3B) and the release of the IL-1β secretory complex (Figure 3C, D). Lysates from the Gsdmd-KD imKCs and primary hepatic macrophages from Gsdmd −/− mice had similar expression of pro-IL-1β, Casp8, and NEDD4 (Supplemental Figure 2A, B, http://links.lww.com/HC9/A243). Finally, given the presence of the E3 ligase NEDD4 in both the sEVs and secretory complex from wild-type hepatic macrophages, we asked whether the high-molecular-weight/modified forms of IL-1β in the sEVs were polyubiquitinated. Immunoprecipitates of IL-1β from the media of wild-type, but not Gsdmd-KD, imKCs, performed under denaturing conditions, had more ubiquitin immunoreactivity in response to LPS/β-GluCer and LPS/TDB (Figure 3E). Taken together, our results indicate that signaling by Mincle ligands mediates IL-1β secretion via the release of GSDMD-dependent CD63+ sEVs from hepatic macrophages.
Pyroptotic activation of GSDMD requires cleavage of D276, in the linker region of GSDMD, in response to inflammasome assembly.37 As β-GluCer-activated Mincle signaling in hepatic macrophages led to IL-1β secretion in the absence of lytic cell death (Figure 2C), we hypothesized that expression of the inactive D276A form of GSDMD in Gsdmd-KD imKCs would restore their ability to release IL-1β-containing sEVs. Indeed, when Gsdmd-KD imKCs were transduced with either wild-type or GSDMDD276A, both LPS/β-GluCer and LPS/TDB treatment stimulated the release of IL-1β into the culture media (Figure 3F).
Mincle or Gsdmd deficiency impaired the release of IL-1β-containing sEVs from liver explants from mice after Gao-binge EtOH feeding
As both Mincle −/− and Gsdmd −/− were required for the release of IL-1β-containing sEVs from hepatic macrophages, we next explored their role in the release of sEVs from mouse liver. Mincle −/− , Gsdmd −/− , and their respective heterozygous littermates were subjected to Gao-binge EtOH feeding. Liver explants were cultured overnight, and exosomes were isolated from the culture media by standard methods. The size distribution of sEVs released by liver explants was not affected by genotype or EtOH feeding (Figure 4A/B). In contrast, Gao-binge EtOH increased the number of sEVs and their IL-1β cargo released from liver explants from heterozygous littermates but not Mincle −/− or Gsdmd −/− mice (Fig. 4C/D). To characterize the composition of the secretory complexes released from Gsdmd +/− and Gsdmd −/− mice, we normalized total sEV numbers released from liver explants between genotypes and immunoprecipitated IL-1β. Gao-binge EtOH feeding induced the formation of a secretory complex containing full-length GSDMD (Hsp90-CDC37-NEDD4) with high-molecular-weight modified pro-IL-1β and Casp8 in wild-type, but not Gsdmd −/− , mice (Figure 4E). We next asked if extracellular vesicles circulating in patients with moderate and sAH also contained modified high-molecular-weight IL-1β. Indeed, exosomes isolated from patients with AH contained pre-dominantly high-molecular-weight forms of IL-1β (Figure 4F). Taken together, these data indicate that Gao-binge EtOH triggers a Mincle-GSDMD-dependent release of CD63+ IL-1β-containing sEVs.
FIGURE 4.
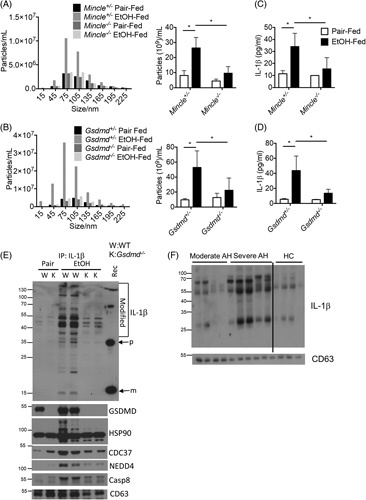
Mincle and GSDMD promoted IL-1β secretion by small extracellular vesicles (sEVs). (A–E) Gsdmd −/− or Mincle −/− and heterozygous littermate mice (n=6) were exposed to Gao-binge ethanol feeding. (A and B) In all, 100 mg of liver explants was cultured overnight. sEVs were isolated from the cell culture media and analyzed by nanoparticle tracking with ZetaView for size distribution and total EV numbers/mL media. (C and D) Concentration of IL-1β in the collected sEVs was measured by ELISA. (E) IL-1β was immunoprecipitated from culture medium for western blotting with the indicated antibodies. Recombinant IL1-β (Rec) was included as a positive control. (F) EVs isolated from plasma of HC and patients with AH were analyzed by western blotting with antibodies IL1-β and the additional indicated proteins. Data were represent mean±SEM. ANOVA *p < 0.05, Abbreviations: AH, alcohol-associated hepatitis; EtOH, ethanol; GSDMD, gasdermin D; HC, healthy controls; K, knock-out; m, mature IL-1β; Mincle, macrophage-inducible C-type lectin; p, precursor; W, wild type.
Myeloid Gsdmd deficiency protects mice from Gao-binge-induced liver injury
Consistent with the hypothesis that the Mincle-GSDMD-mediated release of IL-1β-containing sEVs in response to EtOH contributes to the progression of EtOH-induced liver injury, we have previously reported that Mincle −/− mice are protected from chronic EtOH-induced liver injury.5 Similarly, GSDMD has been implicated in progression of EtOH-induced liver injury.13,38 Hepatocyte overexpression of a constitutively active GSDMD exacerbated liver injury13, and global Gsdmd −/− mice are protected from Gao-binge-induced liver injury.38 We also confirmed that global Gsdmd −/− mice are protected from Gao-binge EtOH with reduced circulating ALT, hepatic triglycerides, and inflammatory cytokine gene expression (Supplemental Figure 3, http://links.lww.com/HC9/A243). Although these previous studies have focused on GSDMD activity in hepatocytes, our data on GSDMD activity in hepatic macrophages led us to hypothesize the myeloid Gsdmd deficiency would be critical for the formation of IL-1β sEVs and the development of EtOH-induced liver injury. We generated LysM-Cre Gsdmd fl/fl mice to test this hypothesis. As expected, Gsdmd fl/fl mice developed the typical profile of Gao-binge-induced liver injury, including increased circulating ALT, hepatic steatosis, and increased expression of inflammatory cytokines and serum amyloid A, an acute-phase protein (Figure 5A–D). However, myeloid-Gsdmd-deficient mice were protected from Gao-binge induced liver injury (Figure 5A–D). Importantly, while the number of sEVs released from liver explants was not affected by genotype (Figure 5E), the IL-1β content in sEVs was reduced in myeloid-Gsdmd-deficient mice (Figure 5F).
FIGURE 5.
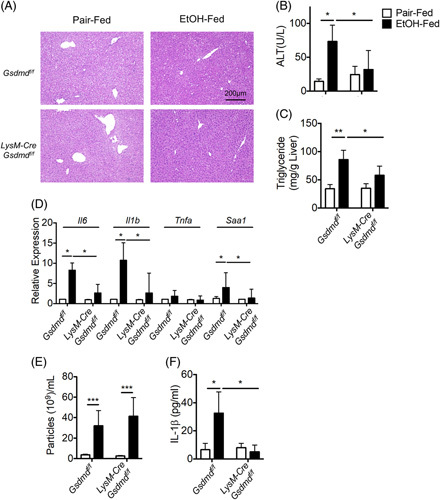
Myeloid GSDMD promoted release of IL-1β-containing small extracellular vesicles (sEVs). LysM-Cre Gsdmdf/f and Gsdmdf/f mice (n=6) were exposed to Gao-binge ethanol feeding. (A) H&E staining for liver histology. (B) ALT activity in plasma. (C) Hepatic triglyceride content. (D) IL-6, IL-1β, TNFα, and SAA1 mRNA expression. (E) sEVs from liver explants cultures were quantified by ZetaView, as described in Figure 4. (F) sEV numbers were equalized, and the concentration of IL-1β was measured by ELISA. Data represent mean±SEM. ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001. Abbreviations: ALT, alanine aminotransferase; EtOH, ethanol; GSDMD, gasdermin D.
IL-1β-containing sEVs promote hepatocyte death and liver injury
Hepatocytes are highly responsive to cytokines/chemokines produced by hepatic macrophages, such as IL-1β, contributing to the pathogenesis of ALD. We recently reported that IL-1β induced expression of the acute-phase protein SAA1; this response, in combination with EtOH, increased Casp3 cleavage and hepatocyte death.34 These data led us to hypothesize that IL-1β-containing sEVs may exert their pathogenic role, at least in part, via induction of the acute-phase protein SAA1. As expected, challenge of hepatocytes with EtOH or recombinant IL-1β induced expressionof SAA and cytotoxicity (Figure 6A, B).34 Importantly, IL-1β-containing sEVs isolated from liver explant cultures of wild-type mice after Gao-binge EtOH exposure also induced the expression of SAA1 and death of primary hepatocytes (Figure 6A, B). The mechanism by which the IL-1β cargo in sEVs becomes accessible to IL-1 receptors is currently unknown, but it will be the focus of future investigations.
FIGURE 6.
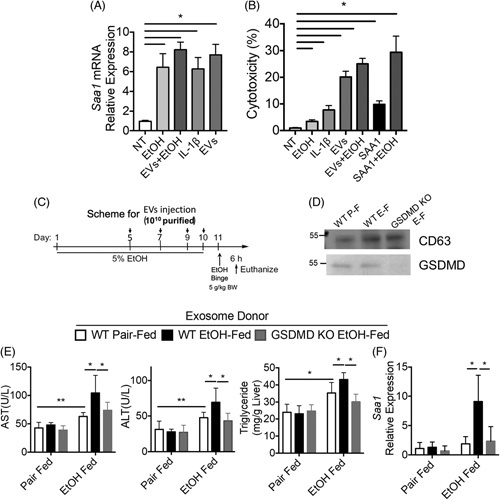
Exosomes from liver explant cultures of Gao-binge mice promote liver injury. (A and B) Primary hepatocytes were treated with recombinant IL-1β (10 ng/mL) or small extracellular vesicles (sEVs) isolated from liver explant cultures of Gao-binge EtOH-fed WT mice. (A) Expression of SAA1 mRNA. (B) Cytotoxicity was assessed by LDH assay. (C–F) (C) Schematic diagram illustrating the protocol for exosome donor experiment. sEVs were isolated from liver explant cultures from WT pair-fed, WT EtOH-fed, and Gsdmd −/− EtOH-fed mice. WT mice were injected with 1010 sEVs during Gao-binge acute on chronic ethanol feeding. (D) sEV numbers were normalized, and CD63 and GSDMD were assessed by western blot. (E) AST/ALT activity in plasma and hepatic triglyceride content. (F) SAA1 mRNA expression. Data represent mean±SEM. ANOVA. *p < 0.05, **p < 0.01. Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; EtOH, ethanol; GSDMD, gasdermin D; WT, wild-type.
These cell culture data suggest that IL-1β-containing sEVs might contribute to chronic EtOH-induced liver injury. Therefore, to determine the in vivo biological activity of sEVs, sEVs were collected from liver explants from wild-type and Gsdmd −/− mice and 1010 particles were injected intraperitoneally in wild-type mice during the course of Gao-binge EtOH feeding (Figure 6C). Expression of CD63 was equal across the purified sEVs, but GSDMD was absent from the sEVs isolated from Gsdmd-deficient mice (Figure 6D). Importantly, sEVs purified from liver explant cultures of wild-type EtOH-fed, but not pair-fed, mice increased hepatic injury, steatosis, and expression of inflammatory cytokines and SAA (Figure 6E, F). This exacerbation was not observed with sEVs purified from liver explant cultures of Gsdmd −/− EtOH-fed mice (Figure 6E, F). Taken together, these results suggest that GSDMD-mediated production of IL-1β-containing sEVs contributes to the pathogenesis of ALD.
DISCUSSION
In this study, we report Mincle-dependent IL-1β secretion via GSDMD-guided formation and release of sEVs from hepatic macrophages during EtOH-induced liver injury. This pathway is triggered by β-GluCer, an endogenous Mincle ligand released by dying hepatocytes after EtOH exposure. Importantly, serum β-GluCer is elevated in patients with AH and positively correlates with disease severity. β-GluCer is also elevated in the circulation of mice exposed to Gao-binge EtOH feeding. Gsdmd or Mincle deficiency impaired the release of IL-1β-containing sEVs, and IL-1β-containing sEVs exacerbated hepatocyte cell death. Intravenous injection of IL-1β-containing sEVs purified from liver explant cultures of EtOH-fed, but not pair-fed, mice markedly increased EtOH-induced hepatic injury and steatosis, indicating that IL-1β-containing sEVs contribute to the pathogenesis of ALD.
Previous studies have suggested that Mincle ligands induce activation of the ASC-NLRP3 inflammasome, which leads to Casp8-dependent IL-1β production.5,39 In this study, we showed that the Mincle ligand β-GluCer induces GSDMD and its interacting partners (including E3 ligase NEDD4) to form a secretory complex with the Casp8-inflammasome and polyubiquitinated pro-IL-β (Figure 7). This secretory complex is loaded into sEVs, which are marked by the exosome marker CD63. We recently reported the downstream signaling events of Casp8-inflammasome activation in IECs, where full-length GSDMD, chaperoned by CDC37/HSP90, recruits NEDD4 to mediate polyubiquitination of pro-IL-1β, followed by cargo loading into CD63+ sEVs.31 Pyroptotic activation of GSDMD is triggered via cleavage at D276, located in its linker region, by either Casp1 or Casp8 in response to inflammasome assembly. β-GluCer-activated Mincle signaling in hepatic macrophages led to the release of IL-1β-containing sEVs in the absence of cytotoxicity. Importantly, when Gsdmd-deficient imKCs (Figure 4F) or IECs31 are transduced with either wild-type or GSDMDD276A mutant, release of IL-1β-containing sEVs is restored, demonstrating a nonlytic function of GSDMD in the process of sEV release. Taken together, these results indicate that β-GluCer-activated Mincle signaling in hepatic macrophages uses a nonlytic GSDMD-mediated mechanism to release IL-1β-containing sEVs.
FIGURE 7.
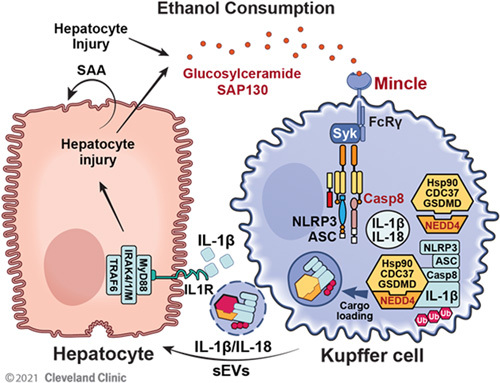
Model for the critical link between β-GluCer, β-glucosylceramide (β-GluCer) and the production of IL-1β-containing sEVs. Ethanol exposure results in the release of β-GlucCer from injury hepatocyte. Mincle, expressed by hepatic macrophages, senses β-GluCer and stimulates the release of IL-1β-containing sEVs in a GSDMD-dependent mechanism. The sEVs, in turn, interact with hepatocytes to stimulate the expression of the acute-phase protein SAA. The Mincle-GSDMD-IL-1β pathway provides a mechanism linking hepatocyte injury to inflammation to perpetuate chronic inflammation in alcohol-associated liver disease. Abbreviations: Mincle, macrophage-inducible C-type lectin; GSDMD, gasdermin D; sEVs, small extracellular vesicles.
The concentration of β-GluCer is tightly regulated and restricted to endoplasmic reticulum and Golgi apparatus in normal living cells. Elevated circulating β-GluCer concentrations are observed in various human diseases, including Gaucher disease, multiple sclerosis, and NAFLD. Gaucher disease is an inherited genetic disorder caused by mutation of a critical GluCer hydrolysis enzyme (glucocerebrosidase), leading to accumulation of GluCer in multiple organs, including the liver.40 The fact that liver disease is common in Gaucher disease41 implicates the critical pathogenic impact of GluCer on hepatic cellular function. In support of this, a recent study indicated that GluCer accumulation in sphingomyelin synthase 1-deficient mouse liver resulted in steatosis, steatohepatitis, and fibrosis.42 Consistently, pharmaceutic inhibition of glucosylceramide synthase alleviated the hepatic steatosis and fibrosis in obese mice.43 Collectively, the current study, in combination with previous work, identifies β-GluCer, a danger signal released by damaged cells, as a potent pathogenic mediator in driving the progression of liver disease. Out study, in particular, identifies a critical link between β-GluCer and the production of pathogenic IL-1β-containing sEVs, which could be an important target for the development of future therapeutic strategies for the prevention or treatment of ALD, as well as liver diseases of other etiologies.
Supplementary Material
AUTHOR CONTRIBUTIONS
Quanri Zhang performed experiments and wrote the manuscript. Weiwei Liu, Katarzyna Bulek, Han Wang, Megan R. McMullen, Renliang Zhang, and Xiaoqin Wu performed experiments. Nicole Welch, Jaividhya Dasarathy, and Srinivasan Dasarathy collected patient samples. Laura E. Nagy and Xiaoxia Li initiated the idea and wrote the manuscript.
ACKNOWLEDGMENTS
The authors sincerely thank Dr Zhaoli Sun at Johns Hopkins University for the human liver specimens.
FUNDING INFORMATION
This work was in part supported by R01AA023722 (Xiaoxia Li and Laura E. Nagy), P50AA024333 (Laura E. Nagy, Srinivasan Dasarathy, and Jaividhya Dasarathy), R01AA027456 and U01AA026938 (Laura E. Nagy), K08AA028794 (Nicole Welch), RO1 GM119174; RO1 DK113196; RO1 AA021890; 3U01AA026976; UO1 AA 026976; R56HL141744;UO1 DK061732; R21 AR 071046 (Srinivasan Dasarathy); K99AA029146 (Xiaoqin Wu). The acquisition of human liver specimens was supported by R24 AA025017 (Zhaoli Sun).
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: AH, alcohol-associated hepatitis; ALD, alcohol-associated liver disease; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Casp, caspase; CDC37 cell division cycle 37; EtOH, ethanol; GSDMD, gasdermin D; Gsdmd-KD, knocked down Gsdmd; HC, healthy control; HSP90, heat shock protein 90; imKC, immortalized KC; LDH, lactate dehydrogenase; LPS, lipopolysaccharide; MELD, model for end-stage liver disease; Mincle, macrophage inducible C-type lectin; Mincle-KD, knocked down Mincle; NEDD4, neural precursor cell expressed developmentally down-regulated protein 4; NLRP3, NLR family pyrin domain containing 3; SAA1, serum amyloid A1; sAH, severe alcohol-associated hepatitis; SAP130, spliceosome-associated protein 130; sEVs, small extracellular vesicles; TDB, trehalose-6,6-dibehenate; WT, wild-type; β-GluCer, β-glucosylceramide.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal’s website, www.hepcommjournal.com.
Contributor Information
Quanri Zhang, Email: zhangq2@ccf.org.
Weiwei Liu, Email: liuw@ccf.org.
Katarzyna Bulek, Email: bulekk@ccf.org.
Han Wang, Email: wangh3@ccf.org.
Megan R. McMullen, Email: mcmullm2@ccf.org.
Xiaoqin Wu, Email: wux@ccf.org.
Nicole Welch, Email: welchn@ccf.org.
Renliang Zhang, Email: zhangr1@ccf.org.
Jaividhya Dasarathy, Email: jdasarathy@metrohealth.org.
Srinivasan Dasarathy, Email: dasaras@ccf.org.
Laura E. Nagy, Email: nagyl3@ccf.org.
Xiaoxia Li, Email: xli73337@gmail.com.
REFERENCES
- 1. Nagy LE, Ding WX, Cresci G, Saikia P, Shah VH. Linking pathogenic mechanisms of alcoholic liver disease with clinical phenotypes. Gastroenterology. 2016;150:1756–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12:231–242. [DOI] [PubMed] [Google Scholar]
- 4. Jiang L, Schnabl B. Gut microbiota in liver disease: what do we know and what do we not know? Physiology (Bethesda). 2020;35:261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou H, Yu M, Zhao J, Martin BN, Roychowdhury S, McMullen MR, et al. IRAKM-Mincle axis links cell death to inflammation: pathophysiological implications for chronic alcoholic liver disease. Hepatology. 2016;64:1978–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Inzaugarat ME, Johnson CD, Holtmann TM, McGeough MD, Trautwein C, Papouchado BG, et al. NLR family pyrin domain-containing 3 inflammasome activation in hepatic stellate cells induces liver fibrosis in mice. Hepatology. 2019;69:845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilkinson AL, Qurashi M, Shetty S. The role of sinusoidal endothelial cells in the axis of inflammation and cancer within the liver. Front Physiol. 2020;11:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miyata T, Nagy LE. Programmed cell death in alcohol-associated liver disease. Clin Mol Hepatol. 2020;26:618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2015;7:a026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou Z, Sun X, Kang YJ. Ethanol-induced apoptosis in mouse liver: Fas- and cytochrome c-mediated caspase-3 activation pathway. Am J Pathol. 2001;159:329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Natori S, Rust C, Stadheim LM, Srinivasan A, Burgart LJ, Gores GJ. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol. 2001;34:248–253. [DOI] [PubMed] [Google Scholar]
- 13. Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology. 2018;67:1737–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. 2008;9:1179–1188. [DOI] [PubMed] [Google Scholar]
- 15. Nagata M, Izumi Y, Ishikawa E, Kiyotake R, Doi R, Iwai S, et al. Intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc Natl Acad Sci U S A. 2017;114:E3285–e3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kostarnoy AV, Gancheva PG, Lepenies B, Tukhvatulin AI, Dzharullaeva AS, Polyakov NB, et al. Receptor Mincle promotes skin allergies and is capable of recognizing cholesterol sulfate. Proc Natl Acad Sci U S A. 2017;114:E2758–e2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 2020;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol. 2016;16:7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cui K, Yan G, Xu C, Chen Y, Wang J, Zhou R, et al. Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1β in mice. J Hepatol. 2015;62:1311–1318. [DOI] [PubMed] [Google Scholar]
- 22. McClain CJ, Cohen DA, Dinarello CA, Cannon JG, Shedlofsky SI, Kaplan AM. Serum interleukin-1 (IL-1) activity in alcoholic hepatitis. Life Sci. 1986;39:1479–1485. [DOI] [PubMed] [Google Scholar]
- 23. Sekiyama KD, Yoshiba M, Thomson AW. Circulating proinflammatory cytokines (IL-1 beta, TNF-alpha, and IL-6) and IL-1 receptor antagonist (IL-1Ra) in fulminant hepatic failure and acute hepatitis. Clin Exp Immunol. 1994;98:71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ludwiczek O, Vannier E, Moschen A, Salazar-Montes A, Borggraefe I, Gabay C, et al. Impaired counter-regulation of interleukin-1 by the soluble IL-1 receptor type II in patients with chronic liver disease. Scand J Gastroenterol. 2008;43:1360–1365. [DOI] [PubMed] [Google Scholar]
- 25. Hanck C, Manigold T, Böcker U, Kurimoto M, Kölbel CB, Singer MV, et al. Gene expression of interleukin 18 in unstimulated peripheral blood mononuclear cells of patients with alcoholic cirrhosis. Gut. 2001;49:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spahr L, Garcia I, Bresson-Hadni S, Rubbia-Brandt L, Guler R, Olleros M, et al. Circulating concentrations of interleukin-18, interleukin-18 binding protein, and gamma interferon in patients with alcoholic hepatitis. Liver Int. 2004;24:582–587. [DOI] [PubMed] [Google Scholar]
- 27. Chen KW, Groß CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 2014;8:570–582. [DOI] [PubMed] [Google Scholar]
- 28. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48:35–44.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44:833–846. [DOI] [PubMed] [Google Scholar]
- 30. Gaidt MM, Hornung V. Pore formation by GSDMD is the effector mechanism of pyroptosis. Embo J. 2016;35:2167–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bulek K, Zhao J, Liao Y, Rana N, Corridoni D, Antanaviciute A, et al. Epithelial-derived gasdermin D mediates nonlytic IL-1β release during experimental colitis. J Clin Invest. 2020;130:4218–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miyata T, Wu X, Fan X, Huang E, Sanz-Garcia C, Ross CKC, et al. Differential role of MLKL in alcohol-associated and non-alcohol-associated fatty liver diseases in mice and humans. JCI Insight. 2021;6:e140180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li S, Wu Y, Yang D, Wu C, Ma C, Liu X, et al. Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J Exp Med. 2019;216:2562–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang H, Zhou H, Zhang Q, Poulsen KL, Taylor V, McMullen MR, et al. Inhibition of IRAK4 kinase activity improves ethanol-induced liver injury in mice. J Hepatol. 2020;73:1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim A, Bellar A, McMullen MR, Li X, Nagy LE. Functionally diverse inflammatory responses in peripheral and liver monocytes in alcohol-associated hepatitis. Hepatol Commun. 2020;4:1459–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurz J, Brunkhorst R, Foerch C, Blum L, Henke M, Gabriel L, et al. The relevance of ceramides and their synthesizing enzymes for multiple sclerosis. Clin Sci (Lond). 2018;132:1963–1976. [DOI] [PubMed] [Google Scholar]
- 37. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. [DOI] [PubMed] [Google Scholar]
- 38. Luan J, Chen W, Fan J, Wang S, Zhang X, Zai W, et al. GSDMD membrane pore is critical for IL-1β release and antagonizing IL-1β by hepatocyte-specific nanobiologics is a promising therapeutics for murine alcoholic steatohepatitis. Biomaterials. 2020;227:119570. [DOI] [PubMed] [Google Scholar]
- 39. Zhang Q, Liu W, Wang H, Zhou H, Bulek K, Chen X, et al. TH17 cells promote CNS inflammation by sensing danger signals via Mincle. Nat Commun. 2022;13:2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29:567–583. [DOI] [PubMed] [Google Scholar]
- 41. Adar T, Ilan Y, Elstein D, Zimran A. Liver involvement in Gaucher disease—review and clinical approach. Blood Cells Mol Dis. 2018;68:66–73. [DOI] [PubMed] [Google Scholar]
- 42. Li Z, Chiang YP, He M, Worgall TS, Zhou H, Jiang XC. Liver sphingomyelin synthase 1 deficiency causes steatosis, steatohepatitis, fibrosis, and tumorigenesis: an effect of glucosylceramide accumulation. iScience. 2021;24:103449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao H, Przybylska M, Wu IH, Zhang J, Maniatis P, Pacheco J, et al. Inhibiting glycosphingolipid synthesis ameliorates hepatic steatosis in obese mice. Hepatology. 2009;50:85–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.