

Abstract
Neuroinflammation is now recognized to compound many central nervous system (CNS) pathologies, from stroke to dementia. As immune responses evolved to handle infections, studying CNS infections can offer unique insights into the CNS immune response and address questions such as: What defenses and strategies do CNS parenchymal cells deploy in response to a dangerous pathogen? How do CNS cells interact with each other and infiltrating immune cells to control microbes? What pathways are beneficial for the host or for the pathogen? Here, we review recent studies that use CNS-tropic infections in combination with cutting-edge techniques to delve into the complex relationships between microbes, immune cells, and cells of the CNS.
Introduction
The concept of the central nervous system (CNS) as an ‘immune-privileged’ organ arose in the 1960s with the recognition of the blood-brain barrier. Despite the relative intransigence of this concept, over multiple decades, many studies have challenged it [1]. In addition, the recognition that neuroinflammation potentiates many neurodegenerative diseases [2,3] makes understanding the nuances of neuroinflammation essential. One way to mechanistically define these nuances is by harnessing the processes inflammation evolved to handle: infections. Here, we will review how cutting-edge techniques have offered new insights into the complex interactions between microbes, cells of the CNS, and infiltrating immune cells. We will highlight how these studies challenge dogma about the capabilities of the CNS to generate a functional immune response and the consequences of neuroinflammation.
Microglia
Microglia arethe tissue resident macrophages of the brain and thus express many markers that are shared with peripheral macrophages/monocytes. In the noninflamed brain, the segregation of microglia from peripheral myeloid-lineage cells can be done by physical location, morphology, and expression or levels of expression of specific markers (e.g. microglia are CD11b+CD45lo-intermediate vs. peripheral myeloid cells CD11b+CD45hi). Unfortunately, in the setting of inflammation, these distinctions are less absolute and can vary by context. For example, CD11a-expression was recently shown to distinguish infiltrating myeloid-derived cells from microglia in the setting of Alzheimer’s disease mouse models and intracranial lipopolysaccharide injection, a component of bacterial cell walls that produces a strong proinflammatory response, but in the setting of chronic infection with Toxoplasma gondii, a eukaryotic parasite that naturally infects the CNS of humans and rodents, CD11a was now expressed on microglia [4]. This study highlights how CNS infections drive distinct neuroinflammatory responses and shows the difficulty of separating microglia from infiltrating myeloid cells during fulminant CNS inflammation. Until recently, this gap limited our ability to determine if microglia and infiltrating myeloid cells play overlapping or distinct roles during CNS infection.
Several groups have taken on the challenge of cleanly segregating the roles of microglia and infiltrating myeloid cells in infection. In a T. gondii infection model, Batista et al. leveraged the slow turnover of microglia compared to peripheral myeloid-derived cells in combination with a tamoxifen-dependent CX3CR1-Cre driver to generate mice in which only microglia express a green fluorescent protein (GFP). Isolation and profiling of microglia (GFP+) and infiltrating myeloid cells (GFP–) revealed that infiltrating myeloid cells expressed a strong NFкB signature and released inter-leukin (IL)-1β ex vivo. Conversely, microglia lacked this NFкB signature and exclusively released IL-1α ex vivo. In a series of follow-up studies, this work suggested that IL-1α signaling to the brain vasculature, which expresses the IL-1α receptor, helps to recruit infiltrating immune cells into the T. gondii-infected brain (Figure 1a). Collectively, this work is the first to strongly suggest that microglia and infiltrating myeloid cells control T. gondii in the CNS through non-overlapping functions [5].
Figure 1. Uninfected microglia recruit infiltrating immune cells and activate pathogen-specific T cells.
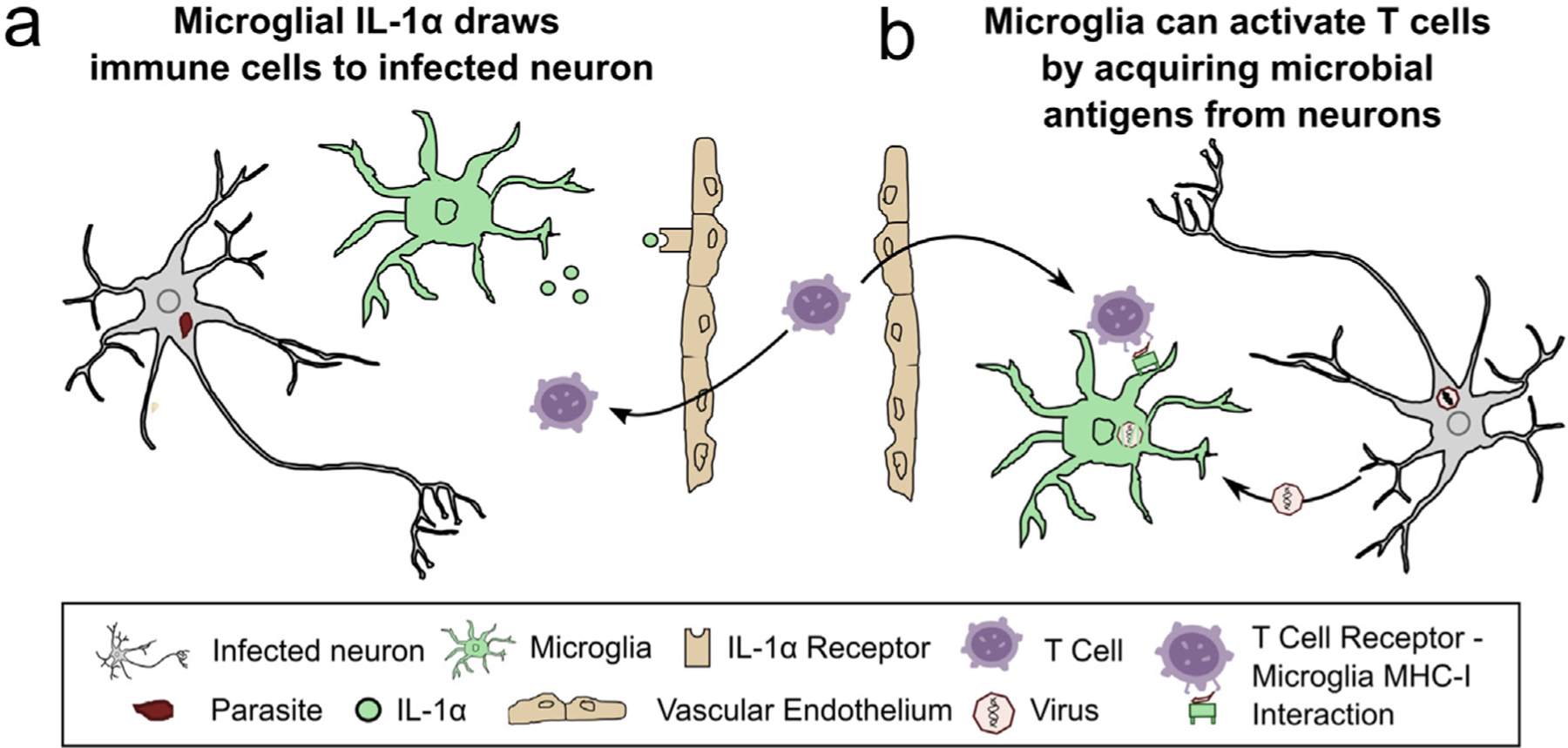
(a) In T. gondii-infected murine brains, microglia release IL-1α onto IL-1α receptors on the vascular endothelium, increasing T cell and myeloid-lineage cell recruitment into the CNS [5]. (b) Microglia acquire vesicular stomatitis virus (VSV) antigen from infected olfactory sensory neurons, which is then used to activate effector CD8+ T cells through T cell receptor– MHC-I interactions [6]. MHC, major histocompatibility complex.
Moseman et al., 2020 also used CX3CR1 tools to reveal a new way in which microglia help control vesicular stomatitis virus (VSV) in neurons. In this study, which used an intranasal inoculation model of VSV, olfactory sensory neurons (OSNs) were shown to be the primary infected CNS cell type and to clear the virus in a non-cytolytic, CD8+ T cell-dependent manner. Unexpectedly, the infected OSNs were not the cell type that was activating the virus-specific CD8+ T cells. Through a series of experiments in which CX3CR1 was used to mark or deplete microglia, the investigators found that uninfected microglia were acquiring VSV proteins/antigen from infected OSNs and then activating CD8+ T cells via microglial major histocompatibility complex (MHC)-I-T cell receptor (TCR) interactions [6] (Figure 1b). While microglial cross presentation has been suggested by a prior study that used whole body irradiation and model antigen injection into the CNS [7], Moseman et al. is the first to rigorously show microglial cross-presentation in a viral infection model.
Another recent approach to define the role of microglia in CNS infection has been systemic administration of PLX5622, a small molecule inhibitor of CSF1R that purportedly selectively depletes microglia, while not affecting peripheral myeloid cells, in non-infectious paradigms [8]. In the setting of viral infection (both peripheral and intracerebral inoculation), PLX5622 administration almost always correlates with higher CNS viral loads and transcriptional changes to infiltrating immune cells, suggesting that microglia potentially help control encephalitic viruses through effects on other immune cells [9–13]. Two recent studies have called these findings into question. One study showed that 14 days of PLX5622 significantly decreased the number of antigen-presenting cells in the blood in uninfected mice [11], while the other showed that prolonged PLX5622 administration caused significant decreases in the absolute numbers of immune cells in the spleen [13]. Thus, until more extensive evaluations of PLX5622’s effect on non-microglial immune cells are done, caution should be exercised when interpreting the reliability of PLX5622 to selectively affect only microglia.
Finally, not all microglial responses are beneficial to the host. A prior study of CNS toxoplasmosis found both a decrease in presynaptic clustering of GAD67 and an increase in seizure activity [14]. Follow-up work that included the use of serial block face scanning electron microscopy revealed that T. gondii-infected brains showed a loss of perisomatic inhibitory synapses in the hippocampus and layer V of the cortex. There was also an increase in infiltrating myeloid-derived cells and neurons ensheathed by microglia, including perisomatic inhibitory synapses. Using a variety of microglial hallmarks, including GFP-expression in T. gondii-infected Cx3cr1-GFP mice, these data suggested that in the infected brain, microglia remove perisomatic inhibitory synapses, leading to an increase seizure propensity [15]. The microglial-dependent elimination of synapses is consistent with prior work in West Nile Virus (WNV) encephalitis, in which CD8+ T cell-dependent inter-feron-γ production/release stimulates microglia to prune synapses, resulting in cognitive decline [16,17].
Astrocytes
Astrocytes have long been recognized as important participants in neuroinflammation [18]. A recent study highlighted the role astrocytes play in sensing an invading microbe by studying if the CNS used the alarmin IL-33 as a danger signal in the setting of T. gondii infection. Through a series of thoughtful experiments that included bone marrow chimeras and cell-specific deletions of Il1rl1—a receptor for IL-33—Still et al. showed that astrocytes both make and respond to IL-33. In turn, this astrocytic IL-33 signaling was important for recruiting T cells and myeloid-lineage cells into the CNS, likely through the downstream production of cytokines and chemokines such as the monocyte attractant ccl2 [19].
Recent work has also shown that astrocyte inflammatory responses cannot simply be categorized as harmful or helpful. Instead, these responses can vary by inciting stimuli or between astrocytes in different brain regions exposed to the same stimulus [18,20,21]. These regional differences in astrocytic response potentially underlie the clinical observation that the cerebellum is rarely infected with WNV compared to the cortex [22]. As T. gondii and several other CNS-tropic microbes also show predispositions for the cortex over the cerebellum [17,23,24], regional differences in astrocytic responses may have implications for other common neuro-infectious diseases.
This robust astrocytic inflammatory response is thought to prevent astrocytes from harboring latent viruses, but a recent paper suggests that, for certain viruses, astrocytes play a previously unrecognized role in latency. Poelart et al. used neurons that ectopically express the receptor for measles virus (MV) and astrocytes that do not express this receptor to show that neurons are acutely infected, followed by neuron-to-astrocyte transmission of viral ribonucleoproteins through excitatory amino acid transporters (Figure 2). This mode of transmission appears to avoid triggering a type I interferon response and enables MV spread to other astrocytes through membrane fusion, potentially making astrocytes the host cell for chronic MV infection [25]. Why astrocytes are permissive to chronic MV infection is unclear, but this study highlights how microbes exploit the crosstalk between CNS cell types.
Figure 2. Measles virus neurons transmit measles virus RNA to astrocytes through a non-canonical mechanism.
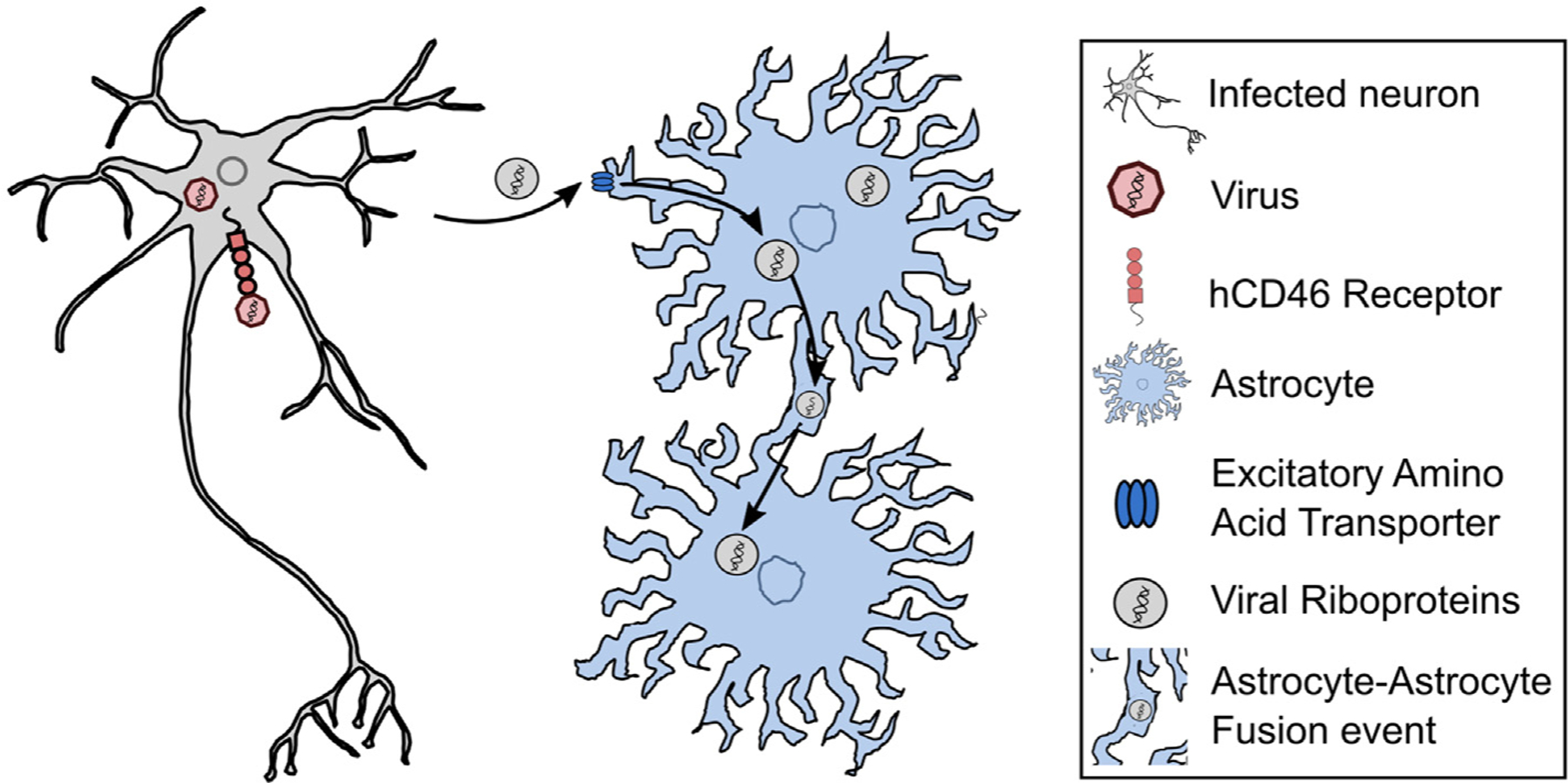
Measles virus (MV) infects murine neurons that ectopically express the human receptor hCD46. Viral riboproteins (RNP) can then spread to astrocytes that do not express hCD46. This spread requires direct contact with an infected neuron and glutamate/excitatory amino acid transporters. The MV genetic material can then spread from astrocyte-to-astrocyte through astrocyte-astrocyte membrane fusion events [25].
Oligodendrocytes
Oligodendrocytes (OLs) have typically not been considered major players in neuroinflammatory responses, though they have long been recognized as the target in immune responses that result in demyelination (e.g., multiple sclerosis and demyelination secondary to mouse hepatitis virus (MHV)). In intracranially inoculated MHV infections, OLs are the most commonly infected cell type but how these infected OLs contributed to the CNS immune response was unknown. Using a recombinant MHV strain that produces Cre recombinase to infect Cre reporter mice that express tdTomato only after Cre-mediated recombination, Pan et al. showed that by 30 days post infection (dpi) — a time point when actively replicating MHV cannot be isolated from the CNS — OLs made up the majority of the tdTomato+ cells in the brain and spinal cord. Interestingly, in the spinal cord, but not the rostral pons, tdTomato+ OLs were associated with demyelinating lesions and infiltrating immune cells, suggesting that demyelination was determined by regional differences in OLs and/or the infiltrating immune cell response to MHV. Transcriptional profiling of brain OLs showed that tdTomato+ OLs had persistent proinflammatory responses at 30 dpi compared to tdTomato– OLs and that all OLs from infected mice showed an upregulation of myelination genes compared to OLs from uninfected mice. These data suggest that OLs that clear MHV produce a chronic inflammatory state that leads to demyelination which, in turn, drives a compensatory upregulation of myelination genes in all OLs [26].
Neurons
Whether neurons mount traditional immune responses — including the ability to directly stimulate effector/ cytolytic CD8+ T cells — has been the subject of great interest and controversy, in part because neurons show low basal expression of classic immune response genes such as MHC-I or STAT1 [27]. While several microbial studies have shown neuron MHC-I-TCR engagement in vitro [28,29], no studies had shown this engagement in vivo. To probe neuron MHC-I-TCR engagement in vivo, Salvioni et al. generated a B6 transgenic mouse that ubiquitously expresses a floxed copy of an MHC-I allele (H2 Ld) associated with a low CNS T. gondii parasite burden. The control of T. gondii by this MHC-I allele is driven by the induction of a robust CD8+ Tcell response to a parasite peptide that is presented in a Ld- restricted manner [30–32]. The authors show that the removal of the Ld allele from neurons only resulted in a higher CNS parasite burden, indirectly suggesting that CD8+ TCR–neuron MHC-I interactions occur in vivo. In support of this possibility, transcriptional profiles from laser capture microdissected neurons that had been injected with T. gondii protein were enriched for CD8+ T cell transcripts, suggesting that CD8+ T cells lay in extreme proximity to these T. gondii-injected neurons [33]. These data are consistent with a recent paper showing that a subset of neurons expressing the influenza glycoprotein hemagglutinin had TCR-neuron MHC-I interactions [34]. How CD8+ T cell activation by neurons leads to the control of T. gondii remains unknown, but recent work suggests that cytokine stimulated neurons can clear intracellular parasites using cell-intrinsic, non-cytolytic mechanisms [38].
Recent work has also shed light on the downstream consequences of neuron–T cell interactions and/or cytokine stimulation. Di Liberto et al. used an innovative approach to determine the role of neuron STAT1 — a transcription factor activated by both type I and type II interferons — in CD8+ T cell-dependent neuronal damage. In this approach, neonatal mice are intracranially infected with an attenuated lymphocytic choriomeningitis virus (LCMV) strain that expresses Cre recombinase, leading to a long-term asymptomatic neuronal infection. In late adolescence, the mice are intravenously infected with wild-type LCMV, which induces Tcell-dependent encephalitis that results in neuronal synaptic pruning and motor symptoms. In this unusual paradigm, by infecting STAT1fl/fl mice, the Cre-expressing attenuated LCMV removes STAT1 only in neurons. Through a series of elegant experiments, the authors showed that the activation of neuronal STAT1 signaling leads to an increase in neuronal CCL2, which attracts phagocytic myeloid cells that prune the neuronal synapses [35]. In this paradigm, microglial synaptic stripping did not require C3/C4 complement, contrary to prior work in a WNV encephalitis model [16]. In a Zika virus model, T cell-dependent, interferon-γ mediated activation of microglia led to neuronal apoptosis that eventually resulted in post-infectious cognitive sequelae [17].
Neurons are not without their own internal defense systems. Two recent papers have shown that proteins primarily known for their role in the necroptotic death pathway (ZBP1, RIPK1, RIPK3) can also trigger nonnecroptotic anti-viral responses in neurons [36,37]. One paper demonstrated that WNV-infected or poly IC-stimulated neurons require RIPK1/RIPK3 signaling to generate the maximum production of cytokines such as CXCL10 and CCL2. Notably, this response seems neuron-specific as it was not seen with bone marrow-derived macrophages or microglia. In the setting of subcutaneous or intracranial WNV inoculation, a lack of RIPK3 led to increased mortality and WNV load in the CNS and decreased cytokine production and immune cell infiltration into the CNS [36]. In the second paper, Zika virus infection of neurons stimulated the RIP-activating nucleotide sensor ZBP1, initiating RIPK1/ RIKP3 signaling. With this pathogen, instead of inducing necroptosis, RIPK1/RIPK3 signaling upregulated an immunity related GTPase (IRG1), producing an anti-viral metabolic state via itaconate-induced blockade of succinate dehydrogenase [37]. Neuron-specific antimicrobial pathways appear to be an important line of defense and may become the topic of an increasing number of future studies.
What happens in vivo to neurons that clear intracellular pathogens or were manipulated but never infected? Such neurons could not be identified until the advent of Cre-expressing pathogens in combination with Cre reporter mice. Such systems have allowed researchers to identify and begin to profile these neurons. For example, in the study that employed VSV-Cre, virally marked but no longer infected OSNs were present at 50 dpi [6], showing that these neurons cleared the virus by non-cytolytic mechanisms. In a T. gondii-Cre model in which neurons injected with parasite protein express GFP regardless of infection status, single cell patch-clamping was used to assess the physiology of GFP+ neurons compared to nearby, uninjected ‘bystander’ (GFP–) neurons in ex vivo slices. The bystander neurons had relatively normal electrophysiology while the T. gondii-injected neurons were very depolarized and incapable of firing an action potential under normal physiologic conditions, indicating that these neurons were unhealthy and possibly dying. This possibility was confirmed by a 90% reduction in the number of T. gondii-injected neurons at 8 weeks post infection compared to 3 weeks post infection [24]. What dictates the survival or death of these neurons remains unclear.
Conclusions
While CNS-tropic microbes often cause high levels of morbidity and mortality, they can also be invaluable tools for teaching us how neuroinflammation works in the brain. These microbes can reveal intricate interactions and non-canonical means for self-defense. By studying CNS-tropic pathogens, which have evolved to handle the CNS immune responses with varying amounts of success, we will continue to learn how our brains handle inflammation.
Acknowledgments
The authors wish to thank the whole Koshy Lab for fruitful discussions. This work was supported by the National Institutes of Health (T32AG061897, R01NS095994, and R01AI157247) and University of Arizona, United States (BIO5 Institute).
Footnotes
Conflict of interest statement
Nothing declared.
References
- 1.Clough B, Frickel E-M: The Toxoplasma parasitophorous vacuole: an evolving host-parasite frontier. Trends Parasitol 2017, 33:473–488. [DOI] [PubMed] [Google Scholar]; Papers of particular interest, published within the period of review, have been highlighted as:* of special interest** of outstanding interest
- 2.Griciuc A, Tanzi RE: The role of innate immune genes in Alzheimer’s disease. Curr Opin Neurol 2021, 34:228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amor S, Puentes F, Baker D, van der Valk P: Inflammation in neurodegenerative diseases. Immunology 2010, 129: 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shukla AK, McIntyre LL, Marsh SE, Schneider CA, Hoover EM, Walsh CM, Lodoen MB, Blurton-Jones M, Inlay MA: CD11a expression distinguishes infiltrating myeloid cells from plaque-associated microglia in Alzheimer’s disease. Glia 2019, 67:844–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. * *.Batista SJ, Still KM, Johanson D, Thompson JA, OʼBrien CA, Lukens JR, Harris TH: Gasdermin-D-dependent IL-1α release from microglia promotes protective immunity during chronic Toxoplasma gondii infection. Nat Commun 2020, 11: 3687. [DOI] [PMC free article] [PubMed] [Google Scholar]; Isolation and profiling of microglia and infiltrating monocytes during T. gondii infection revealed unique inflammatory signatures between cells, suggesting non-overlapping functions.
- 6. * *.Moseman E Ashley, Blanchard Alexa C, Nayak Debasis, McGa- vern Dorian B: T cell engagement of cross-presenting microglia protects the brain from a nasal virus infection. Sci Immunol 2020, 5, eabb1817. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that uninfected microglia help control vesicular stomatitis virus (VSV) in neurons by acquiring VSV antigen from infected neuron debris, which allows the microglia to activate virus specific CD8+ T cells.
- 7.Jarry U, Jeannin P, Pineau L, Donnou S, Delneste Y, Couez D: Efficiently stimulated adult microglia cross-prime naive CD8+ T cells injected in the brain. Eur J Immunol 2013, 43: 1173–1184. [DOI] [PubMed] [Google Scholar]
- 8.Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, et al. : Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82:380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wheeler DL, Sariol A, Meyerholz DK, Perlman S: Microglia are required for protection against lethal coronavirus encephalitis in mice. J Clin Invest 2018, 128:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seitz S, Clarke P, Tyler KL: Pharmacologic depletion of microglia increases viral load in the brain and enhances mortality in murine models of flavivirus-induced encephalitis. J Virol 2018, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. *.Funk KE, Klein RS: CSF1R antagonism limits local restimulation of antiviral CD8+ T cells during viral encephalitis. J Neuroinflammation 2019, 16:22. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that 14 days of systemic PLX5622 treatment, a small molecule inhibitor of CSF1R popularly used to deplete microglia, significantly decreased the number of antigen-presenting cells in the blood in uninfected mice, suggesting that PLX5622 affects more than microglia.
- 12.Mangale V, Syage AR, Ekiz HA, Skinner DD, Cheng Y, Stone CL, Brown RM, O’Connell RM, Green KN, Lane TE: Microglia influence host defense, disease, and repair following murine coronavirus infection of the central nervous system. Glia 2020, 68:2345–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. *.Sanchez JMS, DePaula-Silva AB, Doty DJ, Hanak TJ, Truong A, Libbey JE, Fujinami RS: The CSF1R-microglia Axis has protective host-specific roles during neurotropic picornavirus infection. Front Immunol 2021, 12, 621090. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that prolonged PLX5622 administration (months) causes a significant decreases in the absolute numbers of immune cells in the spleen, highlighting that PLX5622 may affect more than just microglia.
- 14.Brooks JM, Carrillo GL, Su J, Lindsay DS, Fox MA, Blader IJ: Toxoplasma gondii infections alter GABAergic synapses and signaling in the central nervous system. mBio 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. *.Carrillo GL, Ballard VA, Glausen T, Boone Z, Teamer J, Hinkson CL, Wohlfert EA, Blader IJ, Fox MA: Toxoplasma infection induces microglia-neuron contact and the loss of perisomatic inhibitory synapses. Glia; 2020, 10.1002/glia.23816. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first paper to show that, in T. gondii-infected brains, microglia and infiltrating myeloid-lineage cells ensheath neurons, which is associated with the loss of perisomatic inhibitory synapses in the hippocampus and layer V of the cortex. This loss of inhibitory synapses likely contributes to an increased risk of seizures.
- 16.Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, et al. : A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534:538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. *.Garber C, Soung A, Vollmer LL, Kanmogne M, Last A, Brown J, Klein RS: T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat Neurosci 2019, 22: 1276–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]; In two encephalitic flaviviruses (West Nile Virus and Zika Virus) models, interferon-γ mediated activation of microglia by CD8+ T cells triggers synaptic elimination (WNV) and neuronal apoptosis (ZKV), eventually resulting in post-infectious cognitive sequelae.
- 18.Sofroniew MV, Vinters HV: Astrocytes: biology and pathology. Acta Neuropathol 2010, 119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. *.Still KM, Batista SJ, O’Brien CA, Oyesola OO, Früh SP, Webb LM, Smirnov I, Kovacs MA, Cowan MN, Hayes NW, et al. : Astrocytes promote a protective immune response to brain Toxoplasma gondii infection via IL-33-ST2 signaling. PLoS Pathog 2020, 16, e1009027. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that, in the setting of a T. gondii CNS infection, astrocytes produce and respond to the alarmin IL-33. This astrocytic IL-33 signaling recruits T cell and myeloid-lineage cells to the CNS, likely downstream cytokine and chemokine production.
- 20.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA: Genomic analysis of reactive astrogliosis. J Neurosci 2012, 32:6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, et al. : Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 2021, 24: 312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daniels BP, Jujjavarapu H, Durrant DM, Williams JL, Green RR, White JP, Lazear HM, Gale M, Diamond MS, Klein RS: Regional astrocyte interferon signaling restricts pathogenesis during neurotropic viral infection. J Clin Invest 2017, 127:843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boillat M, Hammoudi P-M, Dogga SK, Pagès S, Goubran M, Rodriguez I, Soldati-Favre D: Neuroinflammation-associated aspecific manipulation of mouse predator fear by Toxoplasma gondii. Cell Rep 2020, 30:320–334.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. * *.Mendez OA, Flores Machado E, Lu J, Koshy AA: Injection with Toxoplasma gondii protein affects neuron health and survival. Elife 2021, 10, e67681. [DOI] [PMC free article] [PubMed] [Google Scholar]; Patch clamp electrophysiology in ex vivo acute brain slices revealed that neurons injected with T. gondii proteins are depolarized and incapable of firing an action potential under normal physiologic conditions, indicating that these neurons are supremely unhealthy and possibly dying, while nearby neurons that were not injected with parasite proteins show little physiologic change compared to neurons in uninfected mice.
- 25. * *.Poelaert KCK, Williams RM, Matullo CM, Rall GF: Noncanonical transmission of a measles virus vaccine strain from neurons to astrocytes. mBio 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]; The investigators propose a new transmission modality for chronic measles virus (MV) infection. Using neurons that ectopically express the receptor for MV and astrocytes that do not express this receptor, this work shows that while neurons are acutely infected, neuron-to-astrocyte transmission of viral ribonucleoproteins occurs through excitatory amino acid transporters and enables MV spread to other astrocytes through membrane fusion.
- 26. * *.Pan R, Zhang Q, Anthony SM, Zhou Y, Zou X, Cassell M, Perlman S: Oligodendrocytes that survive acute coronavirus infection induce prolonged inflammatory responses in the CNS. Proc Natl Acad Sci USA 2020, 117:15902–15910. [DOI] [PMC free article] [PubMed] [Google Scholar]; In a mouse hepatitis virus (MHV) model of demyelination, the study shows that previously infected oligodendrocytes (OLs) persist even at 30 days post infection. In the spinal cord, but not the rostral pons, previously infected OLs show evidence of a prolonged inflammatory state which is associated with demyelinating lesions and infiltrating immune cells, suggesting regional differences in OL response.
- 27.Cullheim S, Thams S: Classic major histocompatibility complex class I molecules: new actors at the neuromuscular junction. Neuroscientist 2010, 16:600–607. [DOI] [PubMed] [Google Scholar]
- 28.Chevalier G, Suberbielle E, Monnet C, Duplan V, Martin-Blondel G, Farrugia F, Le Masson G, Liblau R, Gonzalez-Dunia D: Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PLoS Pathog 2011, 7, e1002393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. * *.Salvioni A, Belloy M, Lebourg A, Bassot E, Cantaloube-Ferrieu V, Vasseur V, Blanié S, Liblau RS, Suberbielle E, Robey EA, et al. : Robust control of a brain-persisting parasite through MHC I presentation by infected neurons. Cell Rep 2019, 27: 3254–3268.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that removing an MHC-I allelle associated with a low CNS parasite burden from neurons only resulted in an increase in CNS parasite burden, indirectly suggesting that CD8+ T cell receptor (TCR)– neuron MHC-I interactions occur in vivo.
- 30.Brown CR, McLeod R: Class I MHC genes and CD8+ T cells determine cyst number in Toxoplasma gondii infection. J Immunol 1990, 145:3438–3441. [PubMed] [Google Scholar]
- 31.Blanchard N, Gonzalez F, Schaeffer M, Joncker NT, Cheng T, Shastri AJ, Robey EA, Shastri N: Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat Immunol 2008, 9:937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frickel E-M, Sahoo N, Hopp J, Gubbels M-J, Craver MPJ, Knoll LJ, Ploegh HL, Grotenbreg GM: Parasite stage-specific recognition of endogenous Toxoplasma gondii-derived CD8+ T cell epitopes. J Infect Dis 2008, 198:1625–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merritt EF, Johnson HJ, Wong ZS, Buntzman AS, Conklin AC, Cabral CM, Romanoski CE, Boyle JP, Koshy AA: Transcriptional profiling suggests T cells cluster around neurons injected with Toxoplasma gondii proteins. mSphere 2020, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernard-Valnet R, Yshii L, Quériault C, Nguyen X-H, Arthaud S, Rodrigues M, Canivet A, Morel A-L, Matthys A, Bauer J, et al. : CD8 T cell-mediated killing of orexinergic neurons induces a narcolepsy-like phenotype in mice. Proc Natl Acad Sci USA 2016, 113:10956–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. * *.Di Liberto G, Pantelyushin S, Kreutzfeldt M, Page N, Musardo S, Coras R, Steinbach K, Vincenti I, Klimek B, Lingner T, et al. : Neurons under T Cell attack coordinate phagocyte-mediated synaptic stripping. Cell 2018, 175:458–471. e19. [DOI] [PubMed] [Google Scholar]; The authors used a LCMV neuron-specific, reactivation model which induced a T cell-dependent encephalitis, leading to neuronal synaptic pruning and motor symptoms. Using a Cre-expressing attenuated LCMV strain to infect STAT1fl/fl mice leads to removal of STAT1 only in neurons. Through a series of elegant experiments, the authors showed that activation of neuronal STAT1 signaling leads to an increase in neuronal CCL2, which attracts phagocytic myeloid cells that prune the neuronal synapses in a non-C3/C4 complement dependent fashion.
- 36.Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH, Tait SWG, Martinez J, Gale M, Loo Y-M, Oberst A: RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell 2017, 169:301–313.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daniels BP, Kofman SB, Smith JR, Norris GT, Snyder AG, Kolb JP, Gao X, Locasale JW, Martinez J, Gale M, et al. : The nucleotide sensor ZBP1 and kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immunity 2019, 50:64–76.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zika virus infection of neurons stimulated the RIP-activating nucleotide sensor ZBP1, initiating RIPK1/RIKP3 signaling. Instead of inducing necroptosis, RIPK1/RIPK3 signaling upregulated an immunity related GTPase (IRG1), producing an anti-viral metabolic state via itaconate-induced blockade of succinate dehydrogenase.
- 38.Chandrasekaran S, Kochanowsky JA, Merritt EF, Lagas JS, Swannigan A, Koshy AA: IFN-γ stimulated murine and human neurons mount anti-parasitic defenses against the intracellular parasite Toxoplasma gondii. Nature Communications 2022, 10.1038/s41467-022-32225-z. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]