

Abstract
Ariadne is a non-hallucinogenic analog in the phenylalkylamine chemical class of psychedelics that is closely related to an established synthetic hallucinogen, DOM, differing only by one methylene group in the α-position to the amine. Ariadne has been tested in humans including clinical trials at the Bristol-Myers pharmaceutical company that indicate a lack of hallucinogenic effects and remarkable therapeutic effects, such as rapid remission of psychotic symptoms in schizophrenics, relaxation in catatonics, complete remission of symptoms in Parkinson’s disease, and improved pro-cognitive effects in geriatric subjects. Despite these provocative clinical results, the compound has been abandoned as a drug candidate and its molecular pharmacology remained unknown. Here, we report a detailed examination of the in vitro and in vivo pharmacology of Ariadne and its analogs and propose a molecular hypothesis for the lack of hallucinogenic effects and therapeutic potential of this compound class. We also provide a summary of previous clinical and preclinical results to contextualize the molecular signaling data. Our results show that Ariadne is a serotonin 5-HT2 receptor agonist, exhibits modest selectivity over 5-HT1E and 5-HT1F receptors, has no relevant activity at 5-HT5,6,7 and other aminergic receptors, and no substantial affinity at plasma membrane monoamine transporters. Compared to DOM, Ariadne shows lower signaling potency and efficacy in multiple signaling pathways examined (Gq, G11, and β-arrestin2) coupled to 5-HT2A receptors. We confirmed the shift in signaling for an alpha-propyl analog and provide a molecular docking rationale for the progressive decrease in signaling potency with growing length of the α-substituent. Ariadne versus DOM exhibits no apparent change in the relative preference between Gq/11 activation and β-arrestin2 recruitment, instead there is a small but consistent drop in efficacy in these signaling channels. Ariadne acts as a 5-HT2A agonist in vivo in mice and shows markedly attenuated head twitch response (HTR) in comparison to its hallucinogenic analogs, consistent with previous studies in rabbits, cats, and dogs. Hence, we propose the lower 5-HT2A receptor signaling efficacy of this compound class as an explanatory model for the lack of hallucinogenic effects of Ariadne in humans, and the dramatically attenuated hallucinosis-like effects in animals (5-HT2A signaling efficacy hypothesis). In terms of reverse translation of the noted clinical therapeutic effects, we used an auxilin knock-out model of Parkinson’s disease where Ariadne rescued severe motor deficits in this mouse line, on par to effects of L-DOPA, a notable finding considering Ariadne’s lack of activity at dopamine receptors and transporters. Ariadne emerges as a prototype of a new drug class, non-hallucinogenic 5HT2A agonists, with considerable therapeutic potential across psychiatric and neurological indications.
Graphical Abstract
INTRODUCTION
Natural substances with hallucinogenic properties have been used by humans for millennia and likely played important roles in shaping cultural traditions of community integration, healing, spirituality and religion. In more recent history, since the human bioassay-guided isolation of the first hallucinogen mescaline, the interest in psychedelics has followed an oscillatory pattern with periods of intense activity. Currently the public, scientific, and medicinal interest in psychedelics is on the rise. This is driven by numerous factors, including the growing mental health epidemic, the limited efficacy of current medications, promising clinical results with prototypical psychedelics, existential despair in diverse communities, and interest in advancing spiritual connectedness, wellness and consciousness understanding. The biological effects of classic psychedelics such as mescaline, DMT, psilocybin, and LSD are largely mediated by serotonin receptors. Specifically, induction of transient altered states of consciousness and hallucinosis (a mental state characterized by the presence of hallucinations) by these substances is mediated by activation of the serotonin 2A (5-HT2A) receptors.1 Currently and historically, there are two standing questions in the field of psychedelic research: 1) Which therapeutic effects and modalities are facilitated by psychedelic experiences, and 2) Can substantial therapeutic effects be achieved by psychedelic-related compounds that lack the hallucinogenic activity?
Identification of psychedelic substances via isolation from natural sources or creation by synthesis provided a plethora of structural analogs that exhibit a varying degree of psychoactivity in humans.2 A systematic search for non-hallucinogenic psychedelic analogs have begun shortly after the discovery of LSD at Sandoz. This effort was guided by the concept of identifying ergoline derivatives with serotonin blocking effects or serotonin receptor antagonists.3 However, some of the ergoline compounds, for example lisuride or ergotamine, were later shown to act as 5-HT2A receptor agonists in vitro, while showing no psychedelic activity in humans, introducing a distinct class of pharmacological agents: non-hallucinogenic 5-HT2A agonists. While lisuride is a well-established example, its main therapeutic effect is mediated by the potent dopamine receptor agonism. Contribution of the 5-HT2A receptor modulation in vivo to lisuride’s therapeutic efficacy is not known due to its complex polypharmacology.4
In this study we focus on a much lesser-known example of non-hallucinogenic psychedelic analog – Ariadne (Figure 1A). This compound, 2-amino-1-(2,5-dimethoxy-4-methylphenyl)-butane, created and named by Alexander Shulgin, belongs to the mescaline lineage of phenylalkylamine psychedelics. It is closely related to an established synthetic psychedelic, DOM, differing only by one methylene group in the alpha-position to the amine.5 Shulgin also named Ariadne as 4C-D or “four-carbon DOM”, for the number of carbons of the alkyl chain connected to the aromatic ring and amine. This naming system is convenient, and we here elect to use it as it has already been broadly adopted for his more famous 2C-D (“two-carbon DOM”) series of analogs, such as 2C-E, 2C-B, 2C-T-7 etc.6
Figure 1.

Ariadne is a non-hallucinogenic analog of phenylalkylamine psychedelics with remarkable efficacy signals in clinical studies. (a) The mescaline molecular lineage of Ariadne, which contains an ethyl group in the alpha-position to the amine. The units represent an approximate measure of psycho-activity in humans relative to mescaline based on subjective reports after per oral administration.7 In contrast to DOM, a potent psychedelic, Ariadne showed no hallucinogenic effects at doses that induced notable clinical effects. The present work introduces novel analogs of Ariadne. (b) Broad receptor screen (SafetyScreen44, Eurofins-Panlabs) identified the serotonin 5-HT2A and 2B receptors as the only hits above the set threshold (more than 50% displacement of standard radioligands at 10 uM concentration of (rac)-Ariadne). TMA = 3,4,5-trimethoxyamphetamine; TMA-2 = 2,4,5-trimethoxyamphetamine; DOM = 2,5-dimethoxy-4-methyl-amphetamine.
In contrast to DOM, which is approximately 100-times more potent than mescaline in humans (per oral administration), Ariadne was reported to lack the hallucinogenic effects.6 In addition to reports from the psychonautic sphere (a community of people who self-administer psychedelic substances outside of legally approved research protocols), Bristol-Myers Company, a respectable pharmaceutical house in the U.S., has conducted clinical trials with the (R)-enantiomer of Ariadne in the 1970’s (designated as BL-3912A). These studies confirmed the lack of hallucinogenic effects up to and beyond 100 mg per day; notably, including patients with psychosis.8 Further, Bristol-Myers reported remarkable therapeutic effects including rapid remission of psychotic symptoms in patients suffering from schizophrenia and bipolar disorder (50-100 mg per day), relaxation in catatonics and increased sociability in anxious subjects, as well as nearly complete remission of symptoms in subjects with Parkinson’s disease (100 mg per day). Improvement in alertness, mobility and sociability was also reported in geriatric senile subjects (50 mg per day). Pro-cognitive effects (increased learning capacity) were also observed in these clinical trials.8 Unfortunately, the actual clinical data were never disclosed. According to Alexander Shulgin, (R)-Ariadne completed phase II of clinical trials with many patients, but further development was halted allegedly due to strategic economic considerations.9
Bristol-Myers has reported preclinical characterization of Ariadne using a multifactorial scale in cats, to assess Ariadne’s hallucinogenic-like profile in comparison to (R)-DOM. These results agree with the clinical findings by showing a low combined score of DOM-like behavioral features elicited by Ariadne (e.g., hissing, clawing, catatonia, piloerection, miosis, salivation, arched posture, and muscle rigidity).10 The low hallucinogenic-like effect of Ariadne enantiomers was confirmed in a rabbit hyperthermia assay.11 Ariadne also showed lack of stimulant effects compared to (S)-amphetamine, exhibiting a distinct behavioral profile from that of classic hallucinogens and psychostimulants in rats and conscious beagle dogs.12, 13 In the same dose range (10 mg/kg, s.c. or i.p.), (R)-Ariadne improved avoidance behavior in the absence of stimulant-like effects in rats. In rat drug discrimination assays, Ariadne substituted responding in LSD trained animals in one study, in another showed full substitution for MDMA stimulus.14,15 However, the molecular pharmacology of Ariadne has remained unknown. A rat smooth muscle spasmogenicity assays suggested a weak serotonergic-like effect of Ariadne compared to serotonin and (R)-DOM.11
Considering the remarkable clinical observations by Bristol-Myers, unknown mechanism of action, and our long-standing interest in psychoactive substances, we set out to examine the pharmacological profile of Ariadne in vitro and in mice. Therefore, we investigated Ariadne at many targets, including the serotonin receptors, which are typically involved in the expression of psychedelic effects. We examined Ariadne and its analogs compared to DOM, its structural homolog, in serotonin receptor signaling assays, and head-twitch response and behavioral assays in mice. Guided by these investigations, we provide the first molecular mechanistic hypothesis for the clinical therapeutic signals and the lack of hallucinogenic effects of this compound.
RESULTS
Synthesis of Ariadne and its analogs.
Majority of Ariadne analogs were prepared in two steps from commercially available aldehydes or aldehydes synthesized via established methods. The Henry reaction is a well-established means of generating nitroalkenes. We found that sonication of the appropriate substituted benzaldehyde and nitroalkane partners in the presence of butylamine catalyst in acetic acid produced the corresponding nitrostyrenes that were readily purified by either trituration with cold methanol or chromatography over a small silica pad using toluene-based eluent.16 The reduction of these nitrostyrenes was performed using lithium aluminum hydride in most cases. In cases where the 4-substituent was trifluoromethyl, iodo, bromo or chloro; alane (aluminum hydride) formed in situ was instead used to prevent dehalogenation.
For the 4-trifluoromethyl analog, a silver-catalyzed C–H trifluoromethylation approach employing silver fluoride and trifluoromethyltrimethylsilane was first attempted with yields consistently near 15%.17 While this is a serviceable method for discovery chemistry efforts, a copper (I) iodide-catalyzed approach from the corresponding 4-substituted iodo aldehyde afforded the product in 87% yield.18
Suzuki coupling of the 4-bromo-2,5-dimethoxybenzaldehyde with potassium cyclopropyltrifluoroborate salts using Molander’s conditions successfully yielded 4-cyclopropyl aldehyde (Scheme 2).19 The synthesis of 4-methoxymethyl aldehyde was first attempted in a similar fashion but was not successful. Instead, 4-bromo-2,5-dimethoxy benzaldehyde was reduced with NaBH4 to the alcohol, which was methylated to give the methoxymethyl aryl bromide. This was successfully formylated using nBuLi and DMF in diethyl ether (Scheme 3).20 The Ariadne enantiomers were obtained by separation via chiral SFC (Figure S1).
Scheme 2: Synthesis of aldehydes for 4C-TFM and 4C-cycPr.
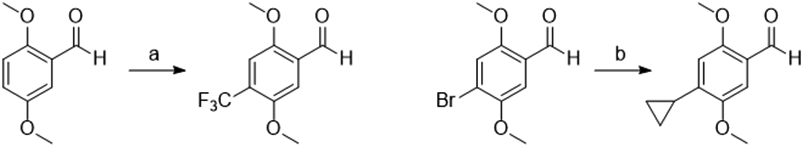
Reagents and Conditions: (a) TMS-CF3, AgF, PIDA, R.T., 20 h, 15% (b) potassium cyclopropyltrifluoroborate Pd(OAc)2, RuPhos, Cs2CO3, toluene:H2O (4:1), 95 °C, 40 h, 87%
Scheme 3: Synthesis of aldehyde for 4C-MOM.
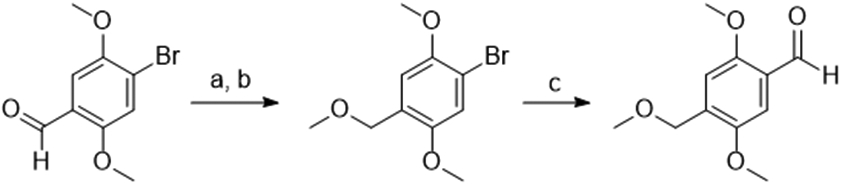
Reagents and Conditions: (a) NaBH4, MeOH, 0 °C to R.T. 1 h (b) MeI, NaH, DMF, 0 °C to R.T. 86% over two steps; (c) n-butyllithium, DMF, diethyl ether, 0 °C to 35 °C, 10 min, 66%
Ariadne is an agonist of 5-HT2 receptors.
We commenced the pharmacological exploration of Ariadne with a broad screen of 44 molecular targets (SafetyScreen44, Eurofins-Panlabs), which includes G protein-coupled receptors, steroid nuclear receptors, ion channels, neurotransmitter transporters, and metabolic and signaling enzymes. Only two molecular targets, 5-HT2A and 5-HT2B receptors, emerged above the positive hit threshold, defined as 50% displacement of standard radioligands by 10 uM concentration of Ariadne (Figure 1b, and Table S1). There was essentially no activity at the serotonin (SERT), dopamine (DAT) and norepinephrine (NET) transporters. The lack of SERT activity was confirmed in our laboratories in a SERT-mediated cell uptake assay, showing that Ariadne has very weak interaction with SERT (IC50 > 50 uM, Figure S4).
We next profiled racemic and (R)-Ariadne in a functional assay panel of 12 human 5-HT receptors using a G protein activation BRET assay (“5-HTome” screen, Figure 2b). Robust agonist activity at 5-HT2A/2B/2C receptors was confirmed (Emax>70%, EC50 < 1 uM) for racemic and (R)-Ariadne (Table S3; Figure 2). At 5-HT1 subtypes, (R-Ariande) possessed weak submicromolar potency activity at 5-HT1A and 5-HT1D/1e/1F receptors (EC50 range 300-600 nM for (R)-Ariadne) with weaker activity at 5-HT1B (EC50 = 4 uM).
Figure 2.

5-HT receptor binding and functional screening of Ariadne and its enantiomers. (a) Structures and binding affinities of Ariadne and enantiomers to 5-HT2A. (b) Heat map of (rac)-Ariande and (R)-Ariande in the 5-HTome G protein dissociation assays. Values represent Log (EMAX/EC50) from at least 3 independent experiments (see Supplemental Table S3) (c) Comparison of 5-HT2A/2B/2C receptor Gq dissociation and Ca2+ flux activities. Data represent average and S.E.M from three independent experiments performed in triplicate. All data were normalized to percent 5-HT response. (d) Summary of activity data in a tabular form. Data represent average and S.E.M from three independent experiments performed in triplicate.
To further examine the 5-HT2 receptor signaling induced by Ariadne, we performed both the Gq dissociation BRET and calcium flux assays (Fig 2c) on the racemate and isolated enantiomers. Ariadne showed more potent agonist activity at 5-HT2A (EC50 = 149 nM, (R)-Ariadne), versus 5-HT2B (EC50 = 739 nM, (R)-Ariadne), and 5-HT2C receptors (EC50 = 249 nM, (R)-Ariadne) in the Gq BRET assay. Racemic Ariadne showed similar dose-dependent curves with a small but consistent right-shift compared to (R)-Ariadne, serving as an internal control and indicating greater potency of the R- versus S-enantiomers (see below). Noteworthy is that racemate/enantiomers of Ariadne exhibited partial agonism versus 5-HT (Emax=83% relative to 5-HT, (R)-Ariadne). Therefore, an orthogonal assay measuring calcium flux was also performed for the 5-HT2 receptors.21 A similar range of potency, to that observed in Gq dissociation assay, was found at 5-HT2A receptor (EC50 = 30 nM, Emax=96% relative to 5-HT, (R)-Ariadne) where the (S)-enantiomer showed markedly lower signaling efficacy compared to the (R)- and (rac)-Ariadne (EC50=132 nM, Emax=63% for (S)-Ariadne, Figure 2c,d). A decrease in calcium signaling efficacy was measured at both the 5-HT2B receptor (Emax= 64% relative to 5-HT, Figure 2c), and 5-HT2C receptor (Emax= 60% relative to 5-HT). For the latter receptor, the low efficacy of calcium signaling is likely related to different kinetics of 5-HT2C activation relative to that of 5-HT2A/2B receptors, which calcium signaling assays are not adept at capturing at Gq-coupled GPCRs.22 The estimates of binding affinity at human 5-HT2A receptor were obtained via a radioligand displacement assay (standard radioligand, [125]I-DOI) for racemic Ariadne (Ki = 120 nM), (R)-Ariadne (Ki = 53 nM), and (S)-Ariadne (Ki = 220 nM) (Figures S2 & S3). The affinity values obtained from an agonist radioligand are consistent with the range of functional potencies determined in the 5-HT2A receptor signaling assays.
Comparison of Ariadne and hallucinogenic DOx analogs show consistent shift in signaling potency and efficacy at 5-HT2A receptors.
We next examined in more detail 5-HT2A receptor signaling induced by (rac)-Ariadne and its hallucinogenic counterpart (rac)-DOM in two primary effector assays using BRET: Gq dissociation and β-arrestin2 recruitment assays. In comparison to DOM, there is an approximate 4-to-6-fold weaker potency for Ariadne, depending on the G protein alpha subunit used in the assay (Gq versus G11), and a substantial decrease in signaling efficacy (Emax/Gq = 80% for Ariadne, versus Emax/Gq = 96% for DOM, 5-HT reference ligand). For β-arrestin2 recruitment, Ariadne also showed a similar shift to weaker potency (~4.5-fold) and efficacy (103% to 83%) (Figure 3A). To further examine the effect of changing the alpha-methyl to alpha-ethyl group, we compared the receptor signaling of n-propyl-Ariadne (4C-Pr) and DOPR, both compounds containing n-propyl group in the 4-position (Figure 3B), and measured a 20-30-fold decrease in potency and 15% loss in efficacy was measured in the Gq dissociation assays, and 10-fold decrease in potency and 15% drop in efficacy was seen in β-arrestin2 recruitment. Comparing iodo-Ariadne and DOI, a 10-fold decrease in potency and 10% loss of efficacy was found in the Gq dissociation assay, and a 5-fold decrease in potency and 20% decrease in efficacy in the β-arrestin2 recruitment assay (Figure 3C). Thus, comparison of non-hallucinogenic Ariadne and its analogs (alpha ethyl compounds) to the corresponding hallucinogenic compounds (alpha methyl compounds) shows that 1) there is no substantial effect in the bias or preference between the Gq and β-arrestin2 signaling pathways, 2) there is a modest but consistent loss of potency and efficacy in all examined signaling pathways (see below interpretation). Furthermore, the alpha-propyl analog (compound 5C-D) showed additional decrease in potency (~2-fold) and efficacy (~15 % drop) relative to Ariadne (Figure 3D).
Figure 3.

Comparison of hallucinogenic (DOx) and non-hallucinogenic (4C-x) analogs in Gq dissociation and βarrestin2 recruitment signaling assays. All data represent average and S.E.M from three independent experiments performed in triplicate. All data are normalized to % 5-HT response. (a-c) 4-methyl-, propyl- and iodo-substituted compounds show a shift toward lower potency and efficacy when alpha-methyl substituent is extended to ethyl. (d) Trend toward lower potency and efficacy continues as alpha-substituent is extended to the propyl group.
Receptor docking rationale for the alpha-substituent effect.
Comparative analysis of the structure of the active state 5-HT2A receptor (PDB ID: 6WHA) suggests that the bound agonist 25CN-NBOH is responsible for the induced fit at the bottom hydrophobic cleft of the orthosteric binding sub-pocket (OBP sub-pocket, Figure 4). Thus, compared to the closely related 5-HT2B (PDBID: 5TUD) and 5-HT2C (PDBID: 6BQH) structures with different ligands, the conserved W3366.48 and F3396.51 side chains are dramatically shifted to accommodate the phenoxy group of 25CN-NBOH. In 5-HT2B and 5-HT2C active state structures with ligands that avoid this OBP sub-pocket, it is partially collapsed. In such collapsed conformation of OBP sub-pocket, the long alkyl chain of the Adriane’s propyl analog would sterically clash with W3376.48 and F3406.51 side chains (Figure 4, the minimal distances from the propyl moiety are 3.15Å and 2.77Å respectively). In contrast, the smaller methyl group in (R)-DOM is well accommodated within the OBP sub-pocket, while the ethyl group in (R)-Adriane shows an intermediate fit (Figure S6).
Figure 4. Docking and comparative analysis of the (R)-Ariadne and alpha-propyl analog binding to human 5-HT2A receptor.

(a) Overview of the crystal structure of 5-HT2A (grey/cyan) receptor in active state with (R)-Ariadne predicted binding pose (pink spheres). (b) Close up on the orthosteric pocket with 25CN-NBOH (light blue) and docked (R)-Ariadne (pink sticks). (c) Superimposition of 5HT2A (cyan) and 5HT2B (orange) active state structures, showing a shift in W3376.48 and F3406.51 side chain conformations likely due to an induced fit around 25CN-NBOH (all other residues of the pocket conserved and have similar conformations). The propyl moiety of the (R)-Ariadne's propyl analog (“5C-D”, coral) is positioned to have moderate clashes with these two side chain conformations in the collapsed conformation (induced by agonists other than 25CN-NBOH) observed in 5HT2B and 5HT2C (not shown here).
The effect of 4-position substituent on 5-HT2A/2B/2C signaling.
Considering the known SAR of the 4-position in the 2C-x and DOx systems,23 we next explored the 4-position in the Ariadne series. We show that increasing the size and hydrophobicity of the 4-position led to an increase in the signaling potency at 5-HT2A receptor. For example, comparison of Ariadne (EC50/Gq = 185 nM) to iodo-Ariadne (EC50/Gq = 16 nM) leads to a >10-fold increase in potency (Figure 5). Similarly, there is a 20- to 30-fold increase in 5-HT2A Gq dissociation potency between DOM and DOPR or DOI (Figure 5), confirming the SAR trend in the 4-position across and Ariadne series.
Figure 5.

4-postion analogs of Ariadne compared at 5-HT2 receptors. (a) The methyl, trifluoromethyl, propyl, cyclopropyl and methoxymethyl analogs of Ariadne. (b) comparison of potency and selectivity of 4-substitued analogs. (c) Data summary in a tabular form. Data represent average and SEM from three independent experiments performed in triplicate. All data are normalized to percent 5-HT response.
We were also interested in examining the selectivity for 5-HT2A over 5-HT2B receptors, as the latter is a molecular target linked to adverse cardiac effects.24 We therefore synthesized a novel analog, TFM-Ariadne (4C-TFM), as a CF3 group is known to render a potent analog in the DOx series, DOTFM.25 Indeed, TFM-Ariadne was potent at 5-HT2A receptor and showed reduced signaling efficacy in the Gq dissociation assay (compared to 5-HT), in a similar range to iodo- and n-propyl(R)-Ariadne (Figure 5). Notably, this compound shows 8-fold selectivity for 5-HT2A over 5-HT2B as measured by Gq dissociation signaling potency. We also synthesized a novel analog with a methoxymethyl (MOM) group in the 4-position, MOM-Ariadne (4C-MOM), examining substituents with increased polarity relative to the n-Pr group.26 As expected, this compound was approximately 2-fold less potent at 5-HT2A compared to Ariadne, and twice as potent at 5-HT2B, with similar potency maintained at 5-HT2C. Similar signaling efficacies to Ariadne were found at 5-HT2A (Emax/Gq = 78%) and 5-HT2C (Emax/Gq = 82%) yet efficacy at 5-HT2B diminished by nearly 2-fold (Emax/Gq = 43%). The 4-cyclopropyl analog, cycP(R)-Ariadne (4C-cycPr), was also prepared and examined, showing similar signaling profile to Ariadne.
Ariadne and its analogs show attenuated head twitch behavior in mice, and effects in behavioral conflict tests.
The head twitch response (HTR) in mice and rats is becoming a widely used preclinical readout for assessing the hallucinogenic potential of 5-HT2A receptor agonists. It is characterized by intermittent twitches of rapid rotation of the head.27 There is a strong correlation between the potency of psychedelic effects in humans and HTR in mice for a broad spectrum of psychedelic substances.28 We used racemic DOPR as a reference phenylalkylamine psychedelic owing to its well-established hallucinogenic effects in humans, high potency agonism at 5-HT2A receptors in vitro and relatively simple pharmacological profile. DOPR showed a robust dose-dependent HTR response, peaking at 1 mg/kg (s.c. administration (Figure 6A). The time profile revealed a rapid increase in HTR frequency reaching the maximum in 10 minutes post-drug administration, followed by a slow decay (Figure 6B, the period used to determine dose-response is shown by the green band). Compared to racemic DOPR, racemic Ariadne showed a markedly attenuated HTR response (> 3-fold decrease in HTR count per 15 min) with the maximum at a one-log higher dose (10 mg/kg, s.c., Figure 6A). (rac)-Ariadne and the R- and S-enantiomers were numerically differentiated by the maximum number of head twitches in the order of their in vitro Gq protein signaling potency. Pharmacokinetic (PK) studies indicated high brain penetration by Ariadne (Figure 6C) and showed that the HTR collection period occurs in the ascending portion of the PK curve. The Ariadne analogs with varying 4-position substitutions were also examined, showing a comparable dose-dependent response to that of (R)-Ariadne, and consistent with in vitro mouse 5-HT2A potencies (Gq-BRET, Figure 6F, Table S8), except for 4-methoxymethyl-Ariadne (4C-MOM) which elicited only residual HTR (Figure 6D). Also, alpha-propyl-Ariadne (5C-D) induced HTR response that is indistinguishable from vehicle (Figure S13). The HTR of (R)-Ariadne and (rac)-4C-TFM was completely inhibited by MDL100,907 (volinanserin), a potent 5-HT2A receptor antagonist, demonstrating that the attenuated HTR of the Ariadne compound class is driven by 5-HT2A activation in vivo (Figures 6E and S12). General locomotion in response to a novel environment (open field test, OF) showed a minor sedative effect at the peak HTR dose of (R)-Ariadne (10 mg/kg, s.c.), and a marked sedation at 30 mg/kg, consistent with previous studies in rats (Figure 6G).13 DOPR showed minor to no effects on locomotion, trending toward an increased locomotion at its HTR peak dose (1 mg/kg), also consistent with minor stimulatory effects of phenylalkylamine psychedelics in mice (Figure 6H). These results indicate that there are no gross behavioral effects of Ariadne or DOPR in the ascending arm of the HTR dose range. Sedation becomes apparent in doses beyond the HTR maximum.
Figure 6. In vivo pharmacology of Ariadne and its analogs in comparison to hallucinogen DOPR.

(a) Mouse head twitch response (HTR) comparison of psychedelic (rac)-DOPR and non-psychedelic (rac)-Ariadne and its enantiomers (15 min period). (b) Time course of HTR events in vehicle, DOPR and (R)-Ariadne treated mice. Green band shows the 15-min. time period used for HTR scoring for dose-response studies. (c) Brain and plasma pharmacokinetic evaluation of Ariadne. Green band shows the time period used for HTR scoring for dose-response studies. N = 3 per time point. (d) HTR dose-response curves for DOPR, Ariadne and its analogs (15 min.). The same data for DOPR and Ariadne as in panel b, shown here for clarity of comparison. (e) Effect of 5-HT2A receptor antagonist MDL100907 (0.01 and 0.032 mg/kg, ip, 15 min prior) on (R)-Ariadne (10 mg/kg, sc) induced HTR (n = 5/group). The pre-treated mice exhibit comparable HTR counts to vehicle-treated mice. (f) Mouse 5-HT2A Gq-BRET dissociation assay for serotonin, DOPR, (R)-Ariadne and 4C-TFM. (g) Mouse open-field assay for evaluation of (R)-Ariadne’s effect on novelty induced locomotion, n=8/group, administered subcutaneously. (h) Mouse open-field assay for DOPR, n=8/group, administered subcutaneously. One-way ANOVA (E) with post-hoc Tukey’s test. All values represented as mean ± S.E.M, ****p < 0.0001. For a-d, n = 5/group, HTR behavior was assessed for 15 minutes (t = 5 to t = 20 minutes post subcutaneous injection).
Analysis of the OF data showed a trend for spending less time in the center of the arena for both (R)-Ariadne and DOPR (Figure S10), consistent with decreased entries and time spent in the open arms of the elevated plus maze (EPM), indicating acute anxiogenic-like effect of (R)-Ariadne (Figure 7C). Novelty suppressed feeding (NSF) performed in mice 7 days post-drug administration demonstrated a dose-dependent trend toward reducing latency to bite the presented food pellets at 10mg/kg for (R)-Ariadne, suggesting a lasting anxiolytic-like effect (Figure 7A, B).29
Figure 7. Effect of Ariadne in behavioral conflict paradigms.

(a) Survival plot of effect of (R)-Ariadne (10 mg/kg) on latency to feed in the novelty suppressed feeding (NSF) test 7 days post a single drug administration. (b) Effect of 6 mg/kg and 10 mg/kg (R)-Ariadne on latency to feed in the novelty suppressed feeding (NSF) test 7 days post a single drug administration. (c) Effect of (R)-Ariadne and (rac)-DOPR on the time spent in the open vs closed arms and (d) corresponding arm entries 30 minutes and 4 hours post a single subcutaneous injection. Kaplan–Meier survival analysis, Mantel–Cox log-rank test (a) and two-way ANOVA (c, d) with post-hoc Dunnett’s test. All values represented as mean ± S.E.M, n = 10 /group, ****p < 0.0001
(R)-Ariadne ameliorates motor abnormalities in a mouse model of Parkinson’s Disease.
In the 1978 patent from the Bristol Laboratories, the inventors noted complete remission of Parkinson’s symptoms in two patients.8 To address and examine these claims via reverse translation in mice, we used a novel model of PD based on auxilin knockout (KO) mice.30 Auxilin is a clathrin uncoating protein that participates in clathrin mediated endocytosis of synaptic vesicles (SV), enabling SV recycling and neurotransmission.30 Loss-of-function mutations in auxilin (DNAJC6/PARK19) cause PD. Auxilin KO mice display cardinal features of PD such as progressive motor deficits, nigral dopaminergic neuronal loss, α-synuclein pathology, and neuroinflammation. 9–12-month-old Aux-KO mice were previously shown to respond to L-DOPA treatment, which ameliorated PD-like motor abnormalities.30 Here, in a pilot study we treated 9-12 months old Aux-KO mice with (R)-Ariadne (10mg/kg body weight, intraperitoneal), followed by various motor behavior tests to evaluate if (R)-Ariadne can rescue the Parkinsonian phenotype akin to L-DOPA. Appropriate wildtype (WT) and vehicle controls were used. One week before the (R)-Ariadne treatment, all the mice were subjected to the balance beam, hind limb clasping and open field behavior tests (pre-treatment group). The balance beam test evaluates fine motor skills such as ability to walk on a raised narrow beam to reach a safety box, measured as a number of runs/minute and time taken to cross the beam (Figures 8A and B, Figure S5). Number of runs auxilin KOs could perform in a minute were significantly lesser when compared to WT mice before (R)-Ariadne treatment. Time taken to cross the beam was also significantly higher in Aux-KO mice (Figures 8C, Figure S5). Mice with neurological dysfunction typically show hind limb clasping reflex when picked up by the tail. Auxilin KOs show a significant increase in hind limb clasping score when compared to WTs before treatment (Figure S5). Though not significant, auxilin KOs also showed a trend toward an increase in clasping duration (Figure S5). In a sum, auxilin KO mice show a robust behavioral phenotype.
Figure 8. Effect of (R)-Ariadne in Auxilin-knockout mouse model of Parkinson’s Disease.

(a) Effect of (R)-Ariadne on number of balance beam runs performed over 60 seconds, grouped animal averages. (b) Effect of (R)-Ariadne on number of balance beam runs performed over 60 seconds, individual animal traces. (c) Effect of (R)-Ariadne on average time per run on balance beam. (d) Effect of (R)-Ariadne treatment on clasping duration. (e) Effect of (R)-Ariadne treatment on hindlimb clasping score. One-way ANOVA multiple comparisons with Tukey’s post hoc test (a,c), two-way ANOVA with Fisher’s LSD test (b), and Kruskal-Wallis multiple comparisons with Dunn’s post hoc tests (d, e). All values represented as mean ± S.E.M, ***p < 0.001, *p < 0.05
One week later, we repeated these behavior assays on the same set of mice 15 to 30 minutes after (R)-Ariadne treatment with appropriate vehicle controls (“Post-treatment”). After (R)-Ariadne treatment, Aux-KO mice showed a remarkable recovery on balance beam, where numbers of runs they could perform was significantly higher and comparable to WT mice (Figures 8A and B). A similar recovery was seen in duration of each run on the balance beam (Figure 8C). Surprisingly, even the WT mice showed an improvement in balance beam performance as measured by number of runs per minute after (R)-Ariadne treatment, when compared to how much they could run before treatment (Figure 8A and B). Auxilin KO mice also showed a significant recovery in hindlimb clasping behavior following (R)-Ariadne treatment. Both clasping score (Figure 8E) and clasping duration (Figure 8D) was down to zero in Aux-KO mice after (R)-Ariadne treatment, suggesting a remarkable recovery. Together, these results demonstrate that (R)-Ariadne treatment successfully and rapidly reversed selective motor behavior deficits seen in a PD mouse model, mirroring the effects of L-DOPA treatment, which is the current standard of care for PD patients. However, we note that this is a pilot study with mixed cohorts of males and females of different ages. A follow-up replication will be required in larger cohorts of both males and females.
DISCUSSION
Interpretation of HTR results, potency versus efficacy.
On their face value, the in vitro and in vivo pharmacological results in this study appear to be consistent; namely, the extension of the alpha-methyl group to the alpha-ethyl group leads to attenuated signaling potency and efficacy at 5-HT2A receptors in vitro, and decreased potency and efficacy of HTR in vivo. Thus, the HTR in vivo profile may be interpreted as a reverse translation of reduced psychedelic potential of Ariadne compounds. However, there are important caveats to be considered when interpreting HTR, especially the efficacy (i.e. the number of head twitches per selected time period). The strong correlation between the human psychedelic effects and mouse HTR of a wide range of hallucinogenic compounds is based on the potency of the effects.28 In terms of efficacy, the question of whether a relative magnitude of HTR, or HTR efficacy, can be used as a comparative proxy measure of a degree of psychedelic effects (from mild to strong psychedelic effects) has not been rigorously addressed. We note important confounds and caveats to this potential use of HTR data, related to the complexities of HTR as a pharmacodynamic readout; for example, as a function of off-target effects that modulate HTR response.27,31,32,33 To illustrate this point specifically, psilocin and psilocybin, the classic tryptamine psychedelics with well-established hallucinogenic effects in humans, show HTR counts in a similar range to that of Ariadne compounds (~ 15-25 HTR events per 15 min) as reported recently.21,34 Hence cautious interpretation is needed particularly for comparisons across different chemical scaffolds and pharmacophores, and for novel agents with no human data (during forward translation or de novo drug design). As with other animal readouts used in the past for this purpose, such as rabbit hyperthermia, HTR efficacy may still be useful for preclinical comparative examination of potential psychedelic-like efficacy, but within a series of close structural analogs with well-defined pharmacology.
In the present matter of Ariadne and its analogs, the mouse HTR was conducted in the context of reverse translation of human reports, as well as previous preclinical studies of hallucinosis-like effects in rabbits, cats, and dogs. The attenuated HTR potency and efficacy of Ariadne class in mice, as compared to closely related DOx hallucinogens, is consistent with no or dramatically attenuated hallucinogenic effects of Ariadne in humans.
Mechanistic hypothesis for lack of Ariadne’s hallucinogenic effects – signaling efficacy hypothesis.
The above results provide a molecular mechanistic picture for Ariadne and its analogs. Ariadne is a selective 5-HT2 receptor agonist, preferring the 5-HT2A/2C over 5-HT2B receptor subtypes, with modest selectivity over 5-HT1E and 5-HT1F receptors (0.5-1 log), no/low activity (EC50>10 uM) at 5-HT5,6,7, dopamine 1 and 2, and adrenergic receptors, and no relevant affinity at plasma membrane monoamine transporters. This pharmacological profile renders the interpretation of both the new results (acquired in the current study) and previous data relatively straightforward, in comparison to the widely known ergoline non-hallucinogens that in contrast have a very complex pharmacology.4 The HTR studies in mice described above indicate that Ariadne acts as a 5-HT2A agonist in vivo. However, in comparison to its hallucinogenic analogs, the HTR induced by Ariadne compounds shows markedly attenuated HTR efficacy in a lower potency range. Notwithstanding the caveats in the interpretation of HTR efficacy (see above), the results of mouse HTR are consistent with the previous studies in rabbits and cats, which demonstrated a behavioral profile distinct from the corresponding DOx amphetamine hallucinogens.10,11 This preclinical behavioral profile correlates with the in vitro pharmacology where Ariadne, in comparison to DOM, shows lower signaling potency and efficacy in the four signaling pathways examined (Gq, G11, and Ca2+, and β-arrestin2) coupled to 5-HT2A receptors. This shift in signaling was observed for two other pairs of alpha-ethyl and alpha-methyl analogs (4C-Pr vs DOPR, and 4C-I vs DOI). There is no apparent change in the relative bias of signaling between Gq/11 and β-arrestin2, instead there is a small but consistent drop in efficacy in all signaling channels. Hence, we propose the weaker 5-HT2A signaling efficacy as an explanatory model for the lack of hallucinogenic effects of Ariadne in humans, and the dramatically attenuated hallucinosis-like effects of Ariadne class in animals (the signaling efficacy hypothesis for the Ariadne compound class).
To examine this hypothesis further, we need to address an important question: is the lack of hallucinosis due to under-dosing of Ariadne in humans, or is there a true efficacy ceiling to psychedelic effects regardless of the dose (within reasonable and safe limits)? In other words, is Ariadne a true non-hallucinogenic 5-HT2A receptor agonist? The Bristol-Myers patents reported that doses up to 100 mg per day of (R)-Ariadne produced no hallucinations (routes of admin not specified).8 Shulgin reported 32 mg of racemate and 25 mg of (R)-Ariadne (p.o.) produced no hallucinogenic effect but a mild threshold psychoactive effect.6 An online Erowid experience report detailed an experiment with 50 mg (oral, male, 77 kg) with additional 25 mg (at t = 90 min post first dose) of Ariadne hydrochloride, leading to an MDMA-like effect but no psychedelic effects.35 It was not specified if the material was a racemate or the R-enantiomer. In another report, a subject (male, 24 years old, 77 kg) self-administered 75 mg of Ariadne orally, also leading to mild MDMA-like effects, but no obvious hallucinogenic effect.36 A report is available on the use of 125 mg of Ariadne orally (female, 28 years, racemate vs R not clarified), and the effect was described as a unique hybrid between MDMA-like and DOX/psychedelic-like activity.37 J. C. Winter confirms this, citing a personal communication with D. B. Vaupel regarding an unpublished clinical study carried out at the NIDA Addiction Research Center in Lexington, KY, where up to 270 mg of (R)-Ariadne was administered to healthy subjects (routes of administration not indicated). No hallucinations or perceptual alterations were observed but euphoric effects were noted.14 Finally, in a patent publication Shulgin refers to clinical studies in geriatric senile subjects where 100-300 mg of (R)-Ariadne was used with no psychedelics effects, but remarkable therapeutic effects.38
Although the recent human anecdotal evidence reported online (Bluelight, reddit Erowid) must be interpreted with caution, as there is no evidence provided about the identity and purity of the drug materials used, the reported effects are consistent with the reports of Shulgin and Bristol Laboratories. Thus, from the aggregate of human data we can conclude that > 50 mg of Ariadne leads to discernable psychoactive effects (in human subjects with average weight), while no hallucinogenic effects were observed up to 100-300 mg. For the corresponding hallucinogen DOM, 1-3 mg produce detectable psychoactive, sub-hallucinogenic effects, while > 3 mg produce classic hallucinogen effects.41
The preclinical profile of Ariadne - viewed as a reverse translation of the clinical observations - is largely consistent with its non-hallucinogen classification.42 Bristol-Myers’ data showed a 25-fold shift in potency between (R)-DOM and (R)-Ariadne in rabbit hyperthermia, a physiological readout that shows good correlation with human psychedelic effects when restricted to a well-defined, narrow structural class of hallucinogens.11 In cats, at 20-fold greater doses of (R)-Ariadne (10 mg/kg, s.c.) compared to (R)-DOM (0.5 mg/kg, s.c.), there were nearly no DOM-like effects noted. In the mouse HTR described in this study, there is at least 10-fold shift in potency and 3-fold drop in efficacy (head twitch counts per 15 min) between the alpha-ethyl and alpha-methyl analogs.
In the direction of forward translation, taking the upper limit of potency differences between DOM and Ariadne in animals, a factor of 25, and multiplying a decidedly psychedelic dose of DOM in humans, 5 mg, would yield a prediction that a dose range of > 125 mg of Ariadne could be psychedelic in humans. However, this is not the case according to the available data. Therefore, it is unlikely that the lack of hallucinosis for Ariadne is due to underdosing. These are approximate comparisons based on the sum of available data, which in the clinical realm is largely anecdotal and does not account for potential PK/bioavailability differences between the Ariadne and DOx compounds in humans. In summary, the available experimental and observational data supports the classification of Ariadne and its analogs as non-hallucinogenic 5-HT2A agonists.
Returning to the molecular hypothesis, assuming similar PK and brain penetration of the two compound classes, DOx and Ariadne (in humans), the 6-fold decrease in in vitro signaling potency between DOM and Ariadne observed in our studies does not explain the lack of psychedelic effects of Ariadne at > 100 mg. On the basis of our cell signaling data, we propose that the decrease in both signaling potency and signaling efficacy in Gq/11 (and potentially in β-arrestin pathways) results in the non-hallucinogenic behavioral phenotype of Ariadne in humans and animals. Although other aspects of signaling are likely to diverge between the DOx hallucinogens and Ariadne non-hallucinogens, the current experimental data points to signaling efficacy as the upstream signaling threshold (the signaling efficacy hypothesis).
To further support this hypothesis, we provide an example from the tryptamine class of psychedelics featuring closely related analogs diethyltryptamine (DET) and 6-fluoro-diethyltryptamine (6F-DET). These compounds were administered intramuscularly to alcoholic patients in a double-blinded fashion alongside with dipropyltryptamine (DPT) in a study by Szara39 Linton and Langs questionnaire with four subscales were used to assess a range of perceptual, cognitive, affective and somatic items in order to quantify the subject’s altered state. For all three drugs there were significant differences in the pre-drug vs drug subscale scores for somatic effects. However, in the subscales directed at loss of identity, loss of control or loss of contact with reality, 6F-DET subjects showed no significant differences in pre-drug vs drug scores, whereas significant differences were demonstrated between pre-drug vs drug scores in DET and DPT groups. Although limited by its small size (12 subjects in total), this clinical study suggests dramatic differences in psychedelic effects between DET (hallucinogen) and 6F-DET (non-hallucinogen); compounds of high structural similarity differing solely by one fluorine atom. In terms of in vitro pharmacology, as previously reported, the two compounds have nearly identical affinity for 5-HT2A receptors, but 6F-DET shows a 6-fold weaker signaling potency (EC50 values) and a 20% drop in efficacy in 5-HT2A-mediated phosphoinositide turnover (Emax = 82% for DET, Emax = 63% for 6F-DET).40 The modest loss of both the signaling potency and efficacy is strikingly similar between the DET and 6F-DET pair, and DOM and Ariadne pair of compounds, supporting the proposed signaling efficacy hypothesis. We note a clear caveat: this hypothesis is not generally applicable across different classes of 5-HT2A agonists, as illustrated by psilocin, which exhibits signaling efficacy of 50-70% (depending on the specific readout), that is signaling efficacy lower or on par to that of the above non-hallucinogens. However, psilocin in comparison to 6F-DET also has a 15-fold greater signaling potency at 5-HT2A receptors and 10-fold greater affinity at 5-HT2C receptors, among other pharmacological differences.40 Thus we re-iterate that the proposed signaling hypothesis is restricted to comparisons of structurally and pharmacologically closely related analogs. Also, it is the differential in signaling efficacy between close analogs - obtained by direct comparison in selected assays - that matters most, not the absolute values of signaling efficacy, which depend on the signaling assay (e.g. G-protein activation, IP3 signaling/IP1 formation, Ca2+ dynamics, and β-arrestin recruitment), assay conditions, cellular background, and lab-to-lab variability. The closely related pairs of compounds such as DOM and Ariadne, and DET and 6F-DET, will serve as important probes for addressing the signaling mechanism questions.
Mechanistic hypothesis for Ariadne’s clinical observations.
In Bristol-Myers’ patents, the inventors refer to highly provocative clinical effects in a range of indications including mood disorders, psychosis, and cognitive, behavioral and movement deficits in geriatric subjects. Based on our present work, we propose that these effects are predominantly driven by 5-HT2A/C agonism of Ariadne. This hypothesis in turn anticipates non-hallucinogenic 5-HT2A/C agonists as a novel class of medications with mood elevating and pro-cognitive effects, and perhaps a drug class with broad trans-diagnostic efficacy in psychiatric disorders with cognitive deficits. Citation of Ariadne-induced rapid remission of symptoms in subjects with Parkinson’s disease also suggests non-hallucinogenic 5-HT2A/C agonists as a novel experimental therapeutic class for PD and other neurological disorders.
We propose the following rationale for the rapid effects of Ariadne in the mouse PD model, as an initial guiding hypothesis for future studies. The in vitro profile suggests that Ariadne’s effect on dopamine neurotransmission is indirect, namely not via direct modulation of DAT or dopamine receptors. It has been demonstrated that 5-HT2A agonists increase dopamine release in nucleus accumbens and other regions of the mesolimbic system.43 It is therefore likely that 5-HT2A agonists also stimulate DA release in more dorsal areas of the striatum that are compromised by the PD pathology. The mechanism of this effect remains unclear; it is plausible that there are 5-HT2A receptors on dopamine neurons which directly stimulate dopamine release, and or there is a circuit effect where for example stimulation of PFC 5-HT2A receptors leads to an excitatory drive to the midbrain nuclei such as SNpc, VTA, DRN, and others,43 which in turn leads to compensatory stimulation of DA release in areas underpinning the motor deficits associated with PD. If this is the case, the present results indicate that non-hallucinogenic 5HT2A agonists of Ariadne class maintain this modulatory effect on the dopaminergic system. 5-HT2A receptor agonists have also been reported to increase biogenesis of mitochondria,44 which suggests an additional plausible mechanism for rescue effects, particularly in terms of longer lasting effects. Our future studies will examine Ariadne's long term therapeutic-like effects in this PD model.
Emerging class of non-hallucinogenic 5-HT2A agonists.
Ariadne is a prime example of a non-hallucinogenic 5-HT2A agonist with therapeutic potential based on human and clinical experience. Several other compounds of this category have been previously discovered, such as lisuride and ergotamine, which are close structural analogs of the potent classic hallucinogen LSD.4 Historically, non-hallucinogens have been largely overlooked as they lack the defining feature of psychedelics – the ability to induce transient altered brain-mind states. However, these compounds have the potential to become a new class of take-home psychiatric medications where different modalities of use can be envisioned. This paper presents another path for the use of these compounds in neurological disorders, such as PD, where accompanying psychiatric symptoms (such as hallucinations in a subset of PD patients) presents a critical barrier for the hallucinogens.
In more recent efforts, several 5-HT2A agonists with predicted non-hallucinogenic profiles have been introduced, such as tabernanthalog (an analog of azepinoindoles developed in the 1960’s by Upjohn company).45, 46 Preclinical data presented for these compounds is supportive of potential clinical use in treatment of depression and substance use disorders, however their psychoactive and therapeutic effects remain to be examined in humans.
There are a number of mechanistic hypotheses proposed to explain the distinct behavioral phenotypes induced by the non-hallucinogenic agonists, ranging from low receptor engagement (e.g. due to high metabolism/clearance or dose-limiting side effects), to modulation of other targets that may inhibit the expression of psychedelic responses (e.g. 5-HT1A receptor agonism), to divergent downstream 5-HT2A-linked signaling mechanisms (e.g. Gq/11versus β-arrestin or different G protein pathways).47, 48 In our present work, the direct comparison of the hallucinogen DOM and non-hallucinogen Ariadne demonstrates a consistent drop in the signaling efficacy across several 5-HT2A-signaling pathways. We therefore propose a signaling efficacy hypothesis for the lack of hallucinosis induced by Ariadne. For the retention of therapeutic effects, we speculate that different circuits may have different sizes of 5HT2A receptor reserve pools, and thus varying degrees of signal amplification of the receptor activation events. A partial agonist in hallucinosis/HTR-relevant circuits, where the receptor pool may be low, may act as a full agonist in other circuits, such as dopamine transmission-relevant circuits, where the reserve pool may be high. This is a theoretical framework that can be addressed experimentally in future studies.49
CONCLUSION
The case of Ariadne is a remarkable chapter in the history of neuropsychiatric drug development, one with acute importance and relevance to the current revival of psychedelic science. To date Ariadne provides the strongest support for the therapeutic potential of non-hallucinogenic 5-HT2A receptor agonists on the basis of the total available data summarized in this article. The rise and fall of this substance as a prototype of a novel medicinal class resembles the Greek myth of Ariadne, centered on a mythical princess after whom the compound was named. There are different versions of the myth, and we here add a new thread into the molecular dimension. Ariadne led Theseus out of the labyrinth, only to be abandoned by him. She was later found by Dionysus, deity of plentitude, joy and wine, to mother many children with him. The compound Ariadne, a substance of molecular simplicity, was leading Bristol-Myers out of innovation malaise to a new type of psychiatric medication, only to be abandoned by the company… Later to be found in the current revolution of psychiatric drug discovery, and we hope, guide us to many new medicines of this class.
MATERIALS AND METHODS
GPCR G protein-dissociation and β-arrestin2 recruitment BRET assays.
All BRET assays were conducted using BRET2 in HEK293T cells (ATCC CRL-11268; mycoplasma-free), which were subcultured in high-glucose DMEM (VWR) supplemented with 10% FBS (Life Technologies).
Constructs in G protein-dissociation BRET assays were derived from the codon-optimized Tango pcDNA3.1 library (Addgene) with V2tail/TEV/tTA encoding regions deleted to yield “de-Tango” constructs. 5-HT receptor constructs used in β-Arrestin2 recruitment BRET assays were also derived from the Tango library with V2tail/TEV/tTA encoding regions replaced with Renilla luciferase (Rluc8) using Gibson Assembly. All Gα-Rluc8, Beta, and GFP2-γ constructs were derived from the TRUPATH pcDNA5/FRT/TO library (Addgene). Geneblocks with N-terminal GFP2-fused human β-Arrestin2 constructs were synthesized by Integrated DNA Technologies (IDT) and subcloned into pcDNA3.1.
Approximately 48 hours before assays, cells were transfected using a reverse transfection method and plated in 1% dFBS at an approximate density of 15,000 cells per well into poly-L-lysine-coated 384-well white assay plates (Grenier Bio-One). For G protein-dissociation assays, cells were transfected in a 1:1:1:1 ratio of receptor: Gα-Rluc8: Beta: GFP2-γ constructs. For β-Arrestin2 recruitment assays, cells were transfected a 1:7.5 ratio of 5-HT-Rluc8: GFP2-fused human β-Arrestin2. All transfections were prepared in Opti-MEM (Invitrogen) and used a 3:1 ratio of TransIT-2020 (Mirus) uL:ug total DNA.
On the day of the assay, plates were decanted and 20 uL of drug buffer per well (1× HBSS, 20 mM HEPES, pH 7.4) was added using a Multidrop (ThermoFisher Scientific), and plates were allowed to equilibrate at 37°C in a humidified incubator before receiving drug stimulation. Drug dilutions of all compounds were performed in McCorvy buffer (1× HBSS, 20 mM HEPES, pH 7.4, supplemented with 0.3% BSA fatty acid free (GoldBio), and 0.03% ascorbic acid). Drugs were dispensed using a FLIPR Tetra (Molecular Devices). Next, plates were incubated at 37°C in a humidified incubator for 60 minutes or specified time point. Before reading, addition of coelenterazine 400a (5 uM final concentration; Nanolight Technology) was performed by the FLIPR Tetra. Immediately after, plates were read at 400 nm Rluc and fluorescent GFP2 emission at 510 nm for BRET2 at 0.8 second per well using a PheraStarFSX (BMB Lab Tech).
The BRET ratios of 510/400 luminescence were calculated per well and were plotted as a function of drug concentration using Graphpad Prism 5 or 9 (Graphpad Software Inc., San Diego, CA). Data were normalized to % positive control stimulation and analyzed using nonlinear regression “log(agonist) vs. response” to yield Emax and EC50 parameter estimates.
Calcium Flux Assays
Stably-expressing 5-HT2A/2B/2C receptor Flp-In 293 T-Rex Tetracycline inducible system (Invitrogen, mycoplasma-free) were used for calcium flux assays. Constructs used for these assays were derived from the codon-optimized Tango pcDNA3.1 library (Addgene) with V2tail/TEV/tTA encoding regions deleted to yield “de-Tango” constructs, and then shuttled into pcDNA5/FRT/TO using Gibson Assembly. Cell lines were maintained in high-glucose DMEM (VWR) containing 10% FBS (Life Technologies), 10 μg/mL Blasticidin (GoldBio), and 100 μg/mL Hygromycin B (GoldBio). Day before the assay, receptor expression was induced with tetracycline (2 ug/mL) and seeded into 384-well poly-L-lysine-coated black plates at a density of approximately 7,500 cells/well in DMEM containing 1% dialyzed FBS. On the day of the assay, plates were decanted and cells were incubated with Fluo-4 Direct dye (Invitrogen, 20 μl/well) for 1 h at 37°C, which was reconstituted in drug buffer (1× HBSS, 20 mM HEPES, pH 7.4) containing 2.5 mM probenecid. After dye load, cells were allowed to equilibrate to room temperature for 15 minutes, and then placed in a FLIPR Tetra fluorescence imaging plate reader (Molecular Devices). Drug dilutions were prepared at 5X final concentration in drug buffer (20 mM HEPES-buffered HBSS, pH 7.4) supplemented with 0.3% BSA fatty-acid free and 0.03% ascorbic acid. Drug dilutions were aliquoted into 384-well plastic plates and placed in the FLIPR Tetra for drug stimulation. Fluorescence for the FLIPR Tetra were programmed to read baseline fluorescence for 10 s (1 read/s), and afterward 5 μl of drug per well was added and read for a total of 5-10 min (1 read/s). Fluorescence in each well was normalized to the average of the first 10 reads for baseline fluorescence, and then either maximum-fold peak increase over basal or area under the curve (AUC) was calculated. Either peak or AUC was plotted as a function of drug concentration, and data were normalized to percent 5-HT stimulation. Data was plotted and non-linear regression was performed using “log(agonist) vs. response” in Graphpad Prism 8 to yield Emax and EC50 parameter estimates.
AuxKO Parkinson’s Model Mouse Behavior Assays.
WT and auxilin KO mice were either treated with vehicle or (R)-Ariadne. Accordingly, there were total of 4 groups: 1. WT treated with vehicle (n=5, age=9 months, sex balanced), 2. WT treated with (R)-Ariadne (n=5, age=9 months, sex balanced), 3. Auxilin KO treated with vehicle (n=6-7, age=9 months, sex balanced), and 4. Auxilin KO treated with (R)-Ariadne (n=7, age= 4 of 9 months, 3 of 12 months, sex balanced). Vehicle treatment groups received 0.85% saline, injected intraperitoneal, at a volume of 10ml/kg body weight. (R)-Ariadne treatment groups received 10 mg/kg body weight of (R)-Ariadne dissolved in 0.85% saline with the addition of 2 molar equivalents of glacial acetic acid, which was finally injected at a volume of 10ml/kg body weight. All groups were subjected to motor behavior assessments before treatment which were broadly grouped as “Pre-treatment”. One week later, 15-30 minutes after appropriate treatment with either vehicle or (R)-Ariadne as described above, same mice from all the groups were subjected to behavior tests again to assess the effect of treatment, together grouped as “Post-treatment”. Below is the brief description of behavior assays performed as per our established protocol 10.
Balance beam behavior
Balance beam test was used to use to assess fine motor skills by evaluating the ability of mice to walk on a raised narrow beam to reach a safety box (Supplementary vid. 1). Average time taken to cross the beam and the number of times a mouse can cross the beam in a minute were used as a measure of balance beam behavior.
Hind limb clasping
When mice are picked up by the base of the tail and lowered, they extend their hind limbs by reflex in anticipation of contact. A hind limb clasping instead of extension in such a scenario indicate neurological dysfunction. We performed this assay for 30 seconds and recorded the hind limb clasping. Hind limb clasping score (Supplementary vid. 2; 0: no clasp; 1: one hind limb clasp; 2: both the hind limbs clasp) and the total duration clasping were noted.
Open field
Mice tend to explore novel environment, which might be hindered if they have locomotory deficits. Open field test was used to assess overall locomotion, where movement of the mice in an open field arena was videotaped for 5 mins using Noldus Ethovision CT software. Total distance travelled and % time spent moving were used as measure of their overall locomotion.
5-HT Receptor Modeling
The receptor protein was extracted from the RCSB server for human 5-HT2A (PDBID: 6WHA) representing agonist-bound (25CN-NBOH) active state of the receptor. The mini-Galpha-q protein, beta/gamma subunits and an active-state stabilizing single-chain variable fragment (scFv16) objects were deleted from the 5-HT2A structure leaving the receptor protein subunit, and the crystallized ligand 25CN-NBOH, and the protein was processed via the addition and optimization of hydrogens and optimization of the side chain residues. Ligands were sketched and the formal charges were assigned. The ligand docking box for potential grid docking was defined as the whole extracellular half of the protein, and all atom docking was performed using Methyl- (R)-DOM, ethyl-based Adriane’s analog (S)-Adriane, (R)-Adriane and (R)-Adriane’s propyl analog ligands with a thoroughness value of 5. The docking was performed with D1553.32 residue set being flexible. The obtained conformations with the best-scored docking poses were further optimized by several rounds of minimization and Monte Carlo sampling of the ligand conformation, including the surrounding side-chain residues (within 5 Å of the ligand). All the above molecular modeling operations were performed in ICM-Pro v3.9-2b molecular modeling package.
For comparative modeling, the HTR2B was extracted from the RCSB server for human 5-HT2B (PDBID: 5TUD) representing agonist-bound (Ergotamine) active state of the receptor. The antibody object was deleted leaving the receptor protein subunit, and the crystallized ligand ergotamin, and the protein was prepared by addition and optimization of hydrogens and optimization of the side chain residues.
Supplementary Material
Scheme 1: General synthesis used for Ariadne analogs.
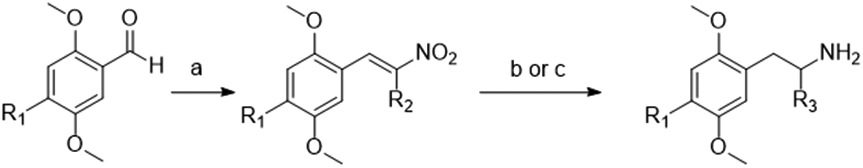
General synthesis scheme for Ariadne analogs: (a) nitropropane, butylamine, acetic acid, sonication, R.T. to 50 °C; (b) LiAlH4 and H2SO4 in THF, 0 °C to reflux when R1= I, Br, Cl, or CF3: (c) LiAlH4 in THF, reflux for all other R1 examples
Acknowledgements
We thank Zixing Huang in Richard U. Margolis lab in Molecular Pathobiology Department at NYU School of Dentistry and Phi Nguyen in René Hen’s lab at NYSPI/Columbia University for assistance with optimizing the NSF experimental conditions. Dr. Mu Yang and Dr. Peter Balsam for helpful discussion on mouse behavior assays and Dr. Yang’s consultation on the statistical tests. We thank Dr. David Sulzer for discussions about the potential mechanisms related to modulation of dopaminergic neurotransmission and the efficacy in the Parkinson’s mouse model. We also thank him for facilitating the collaboration with S.S.C. focused on the auxilin-KO model of Parkinson’s disease. We thank Risha Chakraborty for assisting in motor behavior evaluation of PD mouse model.
We thank Ksania Mariash for creating the Ariadne image incorporated in the graphical abstract and the cover art, inspired by violentcosmos (Tumblr) at: shorturl.at/krswJ.
Funding
D.S. thanks G.L. Freeman and Columbia University for support of this work.
Support for J.D.M. was provided by R35GM133421.
Support for S.S.C. was provided by Parkinson’s Foundation Research Center of Excellence (PF-RCE-1946), and Michael J. Fox Foundation Target Advancement Program grant (MJFF-020160) to S.S.C and V.D.J. S.S.C is funded by ASAP and are members of the ASAP CRN.
Footnotes
D.S. is a co-founder of Gilgamesh Pharmaceuticals and Kures, Inc.
M.J.C. is a co-founder of Gilgamesh Pharmaceuticals. M.J.C., D.S., and B.B. are inventors on a patent application related to the Ariadne compound class.
Supporting Information
The Supporting Information is available free of charge at acs.org
SI contains synthesis and characterization of compounds used in this study, additional in vitro and in vivo figures and experimental details.
Contributor Information
Michael J. Cunningham, Department of Chemistry, Columbia University, New York, NY 10027, United States of America
Hailey A. Bock, Department of Cell Biology, Neurobiology, and Anatomy, Medical College of Wisconsin, Milwaukee, WI 53226, United States of America
Inis C. Serrano, Department of Chemistry, Columbia University, New York, NY 10027, United States of America
Benjamin Bechand, Department of Chemistry, Columbia University, New York, NY 10027, United States of America.
D. J. Vidyadhara, Department of Neuroscience, Department of Neurology, Yale University, New Haven, CT 06510, United States of America
Emma M. Bonniwell, Department of Cell Biology, Neurobiology, and Anatomy, Medical College of Wisconsin, Milwaukee, WI 53226, United States of America
David Lankri, Department of Chemistry, Columbia University, New York, NY 10027, United States of America.
Priscilla Duggan, Department of Neuroscience, Barnard College, New York, NY 10027, United States of America.
Antonina L. Nazarova, Department of Quantitative and Computational Biology, Department of Chemistry, Dornsife Center for New Technologies in Drug Discovery and Development, Bridge Institute, Michelson Center for Convergent Biosciences, University of Southern California, Los Angeles, CA 90089, USA.
Andrew B. Cao, Department of Cell Biology, Neurobiology, and Anatomy, Medical College of Wisconsin, Milwaukee, WI 53226, United States of America
Maggie M. Calkins, Department of Cell Biology, Neurobiology, and Anatomy, Medical College of Wisconsin, Milwaukee, WI 53226, United States of America
Prashant Khirsariya, Department of Chemistry, Columbia University, New York, NY 10027, United States of America.
Christopher Hwu, Department of Chemistry, Columbia University, New York, NY 10027, United States of America.
Vsevolod Katritch, Department of Quantitative and Computational Biology, Department of Chemistry, Dornsife Center for New Technologies in Drug Discovery and Development, Bridge Institute, Michelson Center for Convergent Biosciences, University of Southern California, Los Angeles, CA 90089, USA..
Sreeganga S. Chandra, Department of Neuroscience, Department of Neurology, Yale University, New Haven, CT 06510, United States of America
John D. McCorvy, Department of Cell Biology, Neurobiology, and Anatomy, Medical College of Wisconsin, Milwaukee, WI 53226, United States of America
Dalibor Sames, Department of Chemistry, and Zuckerman Institute of Mind, Brain, Behavior, Columbia University, New York, NY 10027, United States of America.
REFERENCES
- 1).Nichols DE Psychedelics. Pharmacological Reviews 2016, 68 (2), 264–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Nichols DE Chemistry and Structure–Activity Relationships of Psychedelics. Behavioral Neurobiology of Psychedelic Drugs 2017, 1–43. [DOI] [PubMed] [Google Scholar]
- 3).Hofmann A LSD, my problem child: Reflections on sacred drugs, mysticism, and science; MAPS, Multidisciplinary Association for Psychedelic Studies: Santa Cruz, CA, 2009. [Google Scholar]
- 4).Marona-Lewicka D; Kurrasch-Orbaugh D; Selken J; Cumbay M; Lisnicchia J; Nichols D Re-Evaluation of Lisuride Pharmacology: 5-Hydroxytryptamine 1A Receptor-Mediated Behavioral Effects Overlap Its Other Properties in Rats. Psychopharmacology 2002, 164 (1), 93–107. [DOI] [PubMed] [Google Scholar]
- 5).Weingartner H; Snyder SH; Faillace LA DOM (STP), a New Hallucinogenic Drug: Specific Perceptual Changes. The Journal of Clinical Pharmacology and New Drugs 1971, 11 (2), 103–111. [DOI] [PubMed] [Google Scholar]
- 6).Shulgin AT; Shulgin A PIHKAL: A chemical love story; Transform Press: Berkeley, CA, 2020. [Google Scholar]
- 7).Shulgin AT; Shulgin LA; Jacob P A protocol for the evaluation of new psychoactive drugs in man. Methods & Findings in Experimental & Clinical Pharmacology. 1986, 8 (5), 313–20. [PubMed] [Google Scholar]
- 8).Partyka RA; Standridge RT; Howell HG; Shulgin AT 2-Amino-1-(2,5-dimethoxyphenyl)butanes, US 4,105,695, 1978. [Google Scholar]
- 9).Shulgin AT The ‘social-chemistry’ of pharmacological discovery. Social Pharmacology, 1987, 1 (3), 279–290. [Google Scholar]
- 10).Standridge RT; Howell HG; Tilson HA; Chamberlain JH; Holava HM; Gylys JA; Partyka RA; Shulgin AT Phenylalkylamines with Potential Psychotherapeutic Utility. 2. Nuclear Substituted 2-Amino-1-Phenylbutanes. Journal of Medicinal Chemistry 1980, 23 (2), 154–162. [DOI] [PubMed] [Google Scholar]
- 11).Standridge RT; Howell HG; Gylys JA; Partyka RA; Shulgin AT Phenylalkylamines with Potential Psychotherapeutic Utility. 1. 2-Amino-1-(2,5-Dimethoxy-4-Methylphenyl)Butane. Journal of Medicinal Chemistry 1976, 19 (12), 1400–1404. [DOI] [PubMed] [Google Scholar]
- 12).Buyniski JP, Smith ML, Bierwagen ME. Cardiovascular and gross behavioral effects of amphetamine, 2-amino-1-(2, 5-dimethoxy-4-methylphenyl) propane (DOM) and 2-amino-1-(2, 5-dimethoxy-4-methylphenyl) butane (BL-3912A) in the conscious dog. Research Communications in Chemical Pathology and Pharmacology 1974, 8 (2), 213–221. [PubMed] [Google Scholar]
- 13).Tilson HA; Chamberlain JH; Gylys JA Behavioral Comparisons of R-2-Amino-1-(2,5-Dimethoxy-4-Methylphenyl) Butane (BL-3912A) with R-DOM and S-Amphetamine. Psychopharmacology 1977, 51 (2), 169–173. [DOI] [PubMed] [Google Scholar]
- 14).Winter JC Effects of the Phenethylamine Derivatives, BL-3912, Fenfluramine, and SCH-12679, in Rats Trained with LSD as a Discriminative Stimulus. Psychopharmacology 1980, 68 (2), 159–162. [DOI] [PubMed] [Google Scholar]
- 15).Glennon RA MDMA-like Stimulus Effects of α-Ethyltryptamine and the α-Ethyl Homolog of Dom. Pharmacology Biochemistry and Behavior 1993, 46 (2), 459–462. [DOI] [PubMed] [Google Scholar]
- 16).Karlsen M; Liu HL; Berg T; Johansen JE; Hoff BH Synthesis of [13C6]-Labelled Phenethylamine Derivatives for Drug Quantification in Biological Samples. Journal of Labelled Compounds and Radiopharmaceuticals 2014, 57 (5), 378–387. [DOI] [PubMed] [Google Scholar]
- 17).Seo S; Taylor JB; Greaney MF Silver-Catalysed Trifluoromethylation of Arenes at Room Temperature. Chemical Communications 2013, 49 (57), 6385. [DOI] [PubMed] [Google Scholar]
- 18).Belser A; Canal CE; Greene BJ; Hartsel J; Nivorozhkin A; Palfreyman MG Therapeutic phenethylamine compositions and methods of use, February 24, 2022. [Google Scholar]
- 19).Molander GA; Ellis N Organotrifluoroborates: Protected Boronic Acids That Expand the Versatility of the Suzuki Coupling Reaction. Accounts of Chemical Research 2007, 40 (4), 275–286. [DOI] [PubMed] [Google Scholar]
- 20).Layton ME; Morales CA; Shair MD Biomimetic Synthesis of (−)-Longithorone a. Journal of the American Chemical Society 2002, 124 (5), 773–775. [DOI] [PubMed] [Google Scholar]
- 21).Klein AK; Chatha M; Laskowski LJ; Anderson EI; Brandt SD; Chapman SJ; McCorvy JD; Halberstadt AL Investigation of the Structure–Activity Relationships of Psilocybin Analogues. ACS Pharmacology & Translational Science 2020, 4 (2), 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Bdioui S; Verdi J; Pierre N; Trinquet E; Roux T; Kenakin T Equilibrium Assays Are Required to Accurately Characterize the Activity Profiles of Drugs Modulating GQ-Protein-Coupled Receptors. Molecular Pharmacology 2018, 94 (3), 992–1006. [DOI] [PubMed] [Google Scholar]
- 23).Nichols DE Chemistry and Structure–Activity Relationships of Psychedelics. Behavioral Neurobiology of Psychedelic Drugs 2017, 1–43. [DOI] [PubMed] [Google Scholar]
- 24 ).Rothman RB; Baumann MH Serotonergic Drugs and Valvular Heart Disease. Expert Opinion on Drug Safety 2009, 8 (3), 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Nichols DE; Frescas S; Marona-Lewicka D; Huang X; Roth BL; Gudelsky GA; Nash JF 1-(2,5-Dimethoxy-4-(Trifluoromethyl)Phenyl)-2-Aminopropane: A Potent Serotonin 5-HT2A/2C Agonist. Journal of Medicinal Chemistry 1994, 37 (25), 4346–4351. [DOI] [PubMed] [Google Scholar]
- 26).Kolaczynska KE; Luethi D; Trachsel D; Hoener MC; Liechti ME Receptor Interaction Profiles of 4-Alkoxy-Substituted 2,5-Dimethoxyphenethylamines and Related Amphetamines. Frontiers in Pharmacology 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Halberstadt AL; Geyer MA Effect of Hallucinogens on Unconditioned Behavior. Behavioral Neurobiology of Psychedelic Drugs 2016, 159–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Halberstadt AL; Chatha M; Klein AK; Wallach J; Brandt SD Correlation between the Potency of Hallucinogens in the Mouse Head-Twitch Response Assay and Their Behavioral and Subjective Effects in Other Species. Neuropharmacology 2020, 167, 107933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Samuels BA; Hen R Novelty-Suppressed Feeding in the Mouse. Mood and Anxiety Related Phenotypes in Mice 2011, 107–121. [Google Scholar]
- 30).Vidyadhara DJ; Somayaji M; Wade N; Yücel B; Zhao H; Shashaank N; Ribaudo J; Gupta J; Lam TKT; Sames D; Greene LE; Sulzer DL; Chandra SS Dopamine Transporter and Synaptic Vesicle Sorting Defects Initiate Auxilin-Linked Parkinson’s Disease. BioArxiv 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Collins GT; Gerak LR; France CP The Behavioral Pharmacology and Therapeutic Potential of Lorcaserin for Substance Use Disorders. Neuropharmacology 2018, 142, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Gewirtz JC; Marek GJ Behavioral Evidence for Interactions between a Hallucinogenic Drug and Group II Metabotropic Glutamate Receptors. Neuropsychopharmacology 2000, 23 (5), 569–576. [DOI] [PubMed] [Google Scholar]
- 33).Canal CE; Morgan D Head-Twitch Response in Rodents Induced by the Hallucinogen 2,5-Dimethoxy-4-Iodoamphetamine: A Comprehensive History, a Re-Evaluation of Mechanisms, and Its Utility as a Model. Drug Testing and Analysis 2012, 4 (7-8), 556–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Sherwood AM; Halberstadt AL; Klein AK; McCorvy JD; Kaylo KW; Kargbo RB; Meisenheimer P Synthesis and Biological Evaluation of Tryptamines Found in Hallucinogenic Mushrooms: Norbaeocystin, Baeocystin, Norpsilocin, and Aeruginascin. Journal of Natural Products 2020, 83 (2), 461–467. [DOI] [PubMed] [Google Scholar]
- 35).Xorkoth. Zen in a glittery powder. https://erowid.org/experiences/exp.php?ID=113083 (accessed Sep 14, 2022).
- 36).Psychestim. Mild, But Sweet. https://erowid.org/experiences/exp.php?ID=116183 (accessed Sep 14, 2022).
- 37).Kaleida. bridging the gap. https://erowid.org/experiences/exp.php?ID=113681 (accessed Sep 14, 2022).
- 38).Shulgin AT Treatment of senile geriatric patients to restore performance. U S. Patent 4,034,113, 5 Jul 1977.
- 39).Faillace L; Vourlekis A; Szara S Clinical Evaluation of Some Hallucinogenic Tryptamine Derivatives. The Journal of Nervous and Mental Disease 1967, 145 (4), 306–313. [DOI] [PubMed] [Google Scholar]
- 40).Blair JB; Kurrasch-Orbaugh D; Marona-Lewicka D; Cumbay MG; Watts VJ; Barker EL; Nichols DE Effect of Ring Fluorination on the Pharmacology of Hallucinogenic Tryptamines. Journal of Medicinal Chemistry 2000, 43 (24), 4701–4710. [DOI] [PubMed] [Google Scholar]
- 41).Snyder SH; Faillace L; Hollister L 2,5-Dimethoxy-4-Methyl-Amphetamine (STP): A New Hallucinogenic Drug. Science 1967, 158 (3801), 669–670. [DOI] [PubMed] [Google Scholar]
- 42).Shakhnovich V. It's Time to Reverse Our Thinking: The Reverse Translation Research Paradigm. Clinical and Translational Science 2018, 11 (2), 98–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Howell LL; Cunningham KA Serotonin 5-HT2 Receptor Interactions with Dopamine Function: Implications for Therapeutics in Cocaine Use Disorder. Pharmacological Reviews 2014, 67 (1), 176–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Fanibunda SE; Deb S; Maniyadath B; Tiwari P; Ghai U; Gupta S; Figueiredo D; Weisstaub N; Gingrich JA; Vaidya AD; Kolthur-Seetharam U; Vaidya VA Serotonin Regulates Mitochondrial Biogenesis and Function in Rodent Cortical Neurons via the 5-HT2A Receptor and SIRT1–Pgc-1α Axis. Proceedings of the National Academy of Sciences 2019, 116 (22), 11028–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Hester JB; Tang AH; Keasling HH; Veldkamp W Azepinoindoles. I. Hexahydroazepino[4,5-B] Indoles. Journal of Medicinal Chemistry 1968, 11 (1), 101–106. [DOI] [PubMed] [Google Scholar]
- 46).Cameron LP; Tombari RJ; Lu J; Pell AJ; Hurley ZQ; Ehinger Y; Vargas MV; McCarroll MN; Taylor JC; Myers-Turnbull D; Liu T; Yaghoobi B; Laskowski LJ; Anderson EI; Zhang G; Viswanathan J; Brown BM; Tjia M; Dunlap LE; Rabow ZT; Fiehn O; Wulff H; McCorvy JD; Lein PJ; Kokel D; Ron D; Peters J; Zuo Y; Olson DE A Non-Hallucinogenic Psychedelic Analogue with Therapeutic Potential. Nature 2020, 589 (7842), 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47).Rodriguiz RM; Nadkarni V; Means CR; Pogorelov VM; Chiu Y-T; Roth BL; Wetsel WC LSD-Stimulated Behaviors in Mice Require β-Arrestin 2 but not β-Arrestin 1. Scientific Reports 2021, 11 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).López-Giménez JF; González-Maeso J Hallucinogens and Serotonin 5-HT2A Receptor-Mediated Signaling Pathways. Behavioral Neurobiology of Psychedelic Drugs 2017, 45–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49).Bhowmik S; Galeta J; Havel V; Nelson M; Faouzi A; Bechand B; Ansonoff M; Fiala T; Hunkele A; Kruegel AC; Pintar JE; Majumdar S; Javitch JA; Sames D Site Selective C–H Functionalization of Mitragyna Alkaloids Reveals a Molecular Switch for Tuning Opioid Receptor Signaling Efficacy. Nature Communications 2021, 12 (1) 3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.