

Abstract
Background:
Genome-wide association studies (GWAS) have identified hundreds of loci associated with common vascular diseases such as coronary artery disease (CAD), myocardial infarction (MI), and hypertension. However, the lack of mechanistic insights for many GWAS loci limits their translation into the clinic. Among these loci with unknown functions is UFL1-FHL5 (chr6q16.1), which reached genome-wide significance in a recent CAD/MI GWAS meta-analysis. UFL1-FHL5 is also associated with several vascular diseases, consistent with the widespread pleiotropy observed for GWAS loci.
Methods:
We apply a multimodal approach leveraging statistical fine-mapping, epigenomic profiling, and ex vivo analysis of human coronary artery tissues to implicate Four-and-a-half LIM domain 5 (FHL5) as the top candidate causal gene. We unravel the molecular mechanisms of the cross-phenotype genetic associations through in vitro functional analyses and epigenomic profiling experiments in coronary artery smooth muscle cells.
Results:
We prioritized FHL5 as the top candidate causal gene at the UFL1-FHL5 locus through eQTL colocalization methods. FHL5 gene expression was enriched in the SMC and pericyte population in human artery tissues with coexpression network analyses supporting a functional role in regulating SMC contraction. Unexpectedly, under procalcifying conditions, FHL5 overexpression promoted vascular calcification and dysregulated processes related to extracellular matrix organization and calcium handling. Lastly, by mapping FHL5 binding sites and inferring FHL5 target gene function using artery tissue gene regulatory network analyses, we highlight regulatory interactions between FHL5 and downstream CAD/MI loci, such as FOXL1 and FN1 that have roles in vascular remodeling.
Conclusion:
Taken together, these studies provide mechanistic insights into the pleiotropic genetic associations of UFL1-FHL5. We show that FHL5 mediates vascular disease risk through transcriptional regulation of downstream vascular remodeling gene programs. These trans-acting mechanisms may explain a portion of the heritable risk for complex vascular diseases.
Keywords: coronary artery disease, smooth muscle cells, vascular calcification, cofactor, genome-wide association studies, Computational Biology, Basic Science Research, Vascular biology, Smooth Muscle Proliferation and Differentiation
Graphical Abstract
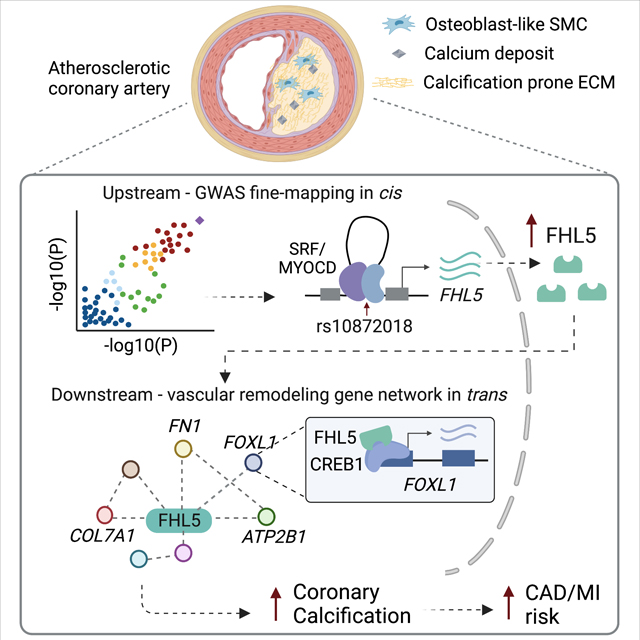
Introduction
Vascular diseases, such as atherosclerosis and aneurysm, encompass a wide range of disorders that affect blood vessels and perturb blood flow to critical tissues. Despite the heritability of these common pathologies, many of the associated genetic risk factors are unknown. Genome-wide association studies (GWAS) have uncovered the genetic basis for these complex traits which have implicated SMC dysfunction contributing to pathogenesis of these vascular diseases 1. Following injury or during disease, SMCs undergo phenotypic modulation to more closely resemble its precursor cell with reduced expression of SMC contractile markers 2. In addition to this progenitor-like cell state, SMCs can adopt a wide spectrum of phenotypic states resembling other cell types such as inflammatory macrophages, ECM depositing fibroblasts, and pro-calcifying osteoblasts 3.
In the case of coronary artery disease (CAD), which is among the leading causes of death worldwide, GWAS have identified over 200 loci 4,5,6. Many of these loci harbor genes that function independently of lipid metabolism and other classic risk factors. These studies highlight additional pathways, such as vascular remodeling and inflammation that directly contribute to CAD pathogenesis and prioritize novel therapeutic targets7,8. As demonstrated by lineage tracing and single-cell RNA sequencing (RNA-seq) studies in mice and humans, smooth muscle cells (SMCs) play a key role in both the development and progression of atherosclerosis9,10. SMCs undergo phenotypic transitions to give rise to diverse cell populations that drive atherosclerosis progression in early stages and contribute to fibrous cap stability in advanced lesions11. Intimal SMCs that acquire mesenchymal and ultimately osteoblast-like phenotypes deposit calcium-mineral in the collagenous matrix12. These remodeling events coupled with arterial calcification serve as strong predictors of adverse cardiovascular events13. Untangling the complex contribution of SMCs to CAD using human genetics may inform novel drug candidates targeting these primary disease processes in the vessel wall.
The most recent CAD and myocardial infarction (MI) meta-analysis of the combined UK Biobank and CARDIoGRAMplusC4D cohorts identified a genome-wide significant association of the UFL1-FHL5 locus (chr6q16.1)14. In addition to CAD/MI, UFL1-FHL5 is also associated with multiple vascular pathologies, including hypertension15, intracranial aneurysm16, and migraines17, similar to another pleiotropic locus PHACTR1-EDN118. The lead CAD/MI single nucleotide polymorphism (SNP), rs9486719 is located in the first intron of FHL5. FHL5 is a member of the four-and-a-half LIM (FHL) domain family of cofactors, which also includes structurally similar proteins, FHL1, FHL2, and FHL319. Among the FHL family, FHL5 is the most understudied, which may be attributed to its low expression in vitro and limited availability of suitable animal models. Early functional studies have focused on its expression in germ cells20,21, however FHL5 was recently linked to intimal hyperplasia in aortic SMCs by activating CREB target genes22,23.
Here, we present a comprehensive study that investigates the molecular underpinnings of the genetic association of the UFL1-FHL5 locus with CAD/MI and other vascular pathologies. We identify an FHL5-regulated transcriptional network that contributes to the maladaptive extracellular matrix (ECM) remodeling and vascular tone defects associated with clinical disease risk.
Methods
Data Availability
All raw and processed CUT&RUN and RNA-seq datasets are made available on the Gene Expression Omnibus (GEO) database (accession: GSE201572). A more detailed list of publicly available datasets used in the study is provided in the Online Supplemental Material.
Code Availability
All custom scripts used are available at https://github.com/MillerLab-CPHG/FHL5_Manuscript. All software tools used in this study are publicly available and full names and versions are provided in the reporting summary.
Statistical analysis
Data in bar graphs are presented with mean+/− standard error of mean (SEM) with each point represented as an individual replicate. Data in box plots are presented with lines denoting the 25th, median and 75th percentile with each point representing an individual donor. Pairwise comparisons were made using the student’s t test or Wilcoxon rank test as appropriate. Comparisons between more than two groups were assessed using a two-way ANOVA test or Kruskal-Wallis test for smaller sample sizes (n<6). The normality of the data for larger sample sizes (n>6) was assessed using the Shapiro-Wilk test, with P > 0.05, supporting a normal distribution. For each of these analyses, we considered P < 0.05 as significant. P-values < 0.01 are presented in scientific notation.
To identify differentially expressed genes, we used the false discovery rate (FDR) adjusted P< 0.05 threshold and log2FoldChange > 0.6. Heatmaps were created using the pheatmap package and represent normalized expression (Z-score) for genes, scaled across each row. Gene ontology enrichment analyses were performed relative to all expressed genes using Fisher’s Exact Test, with a significant threshold of 5% FDR.
Ethics statement
All research described herein complies with ethical guidelines for human subjects research under approved Institutional Review Board (IRB) protocols at Stanford University (#4237) and the University of Virginia (#20008), for the procurement and use of human tissues and information, respectively.
A detailed and expanded methods section can be found in the Online Supplemental Material.
Results
FHL5 is the top candidate causal gene associated with CAD/MI risk
The lead variant, rs9486719, tagging the UFL1-FHL5 locus (chr6q16.1) is associated with CAD (P = 1.1E-8) and MI (P = 6.8E-10) risk (Figure 1A and Figure S1A), as reported in the combined genome-wide association study (GWAS) meta-analysis of CARDIoGRAMplusC4D and UK Biobank (UKBB) data4,14. The lead MI SNP, rs9486719 is also associated with intermediate traits predictive of cardiovascular adverse events, such as blood pressure24,25 and hypertension15 (Figure S1B). We did not observe genetic associations of rs9486719 with lipid metabolism or other traditional CAD risk factors (e.g. type 2 diabetes or obesity) in the PhenoScanner database26,27 (Figure S1C and Table S1), implicating heritable CAD risk at this locus acting primarily in the vessel wall.
Figure 1. FHL5 is the top candidate causal gene at the UFL1-FHL5 locus associated with increased CAD/MI risk.
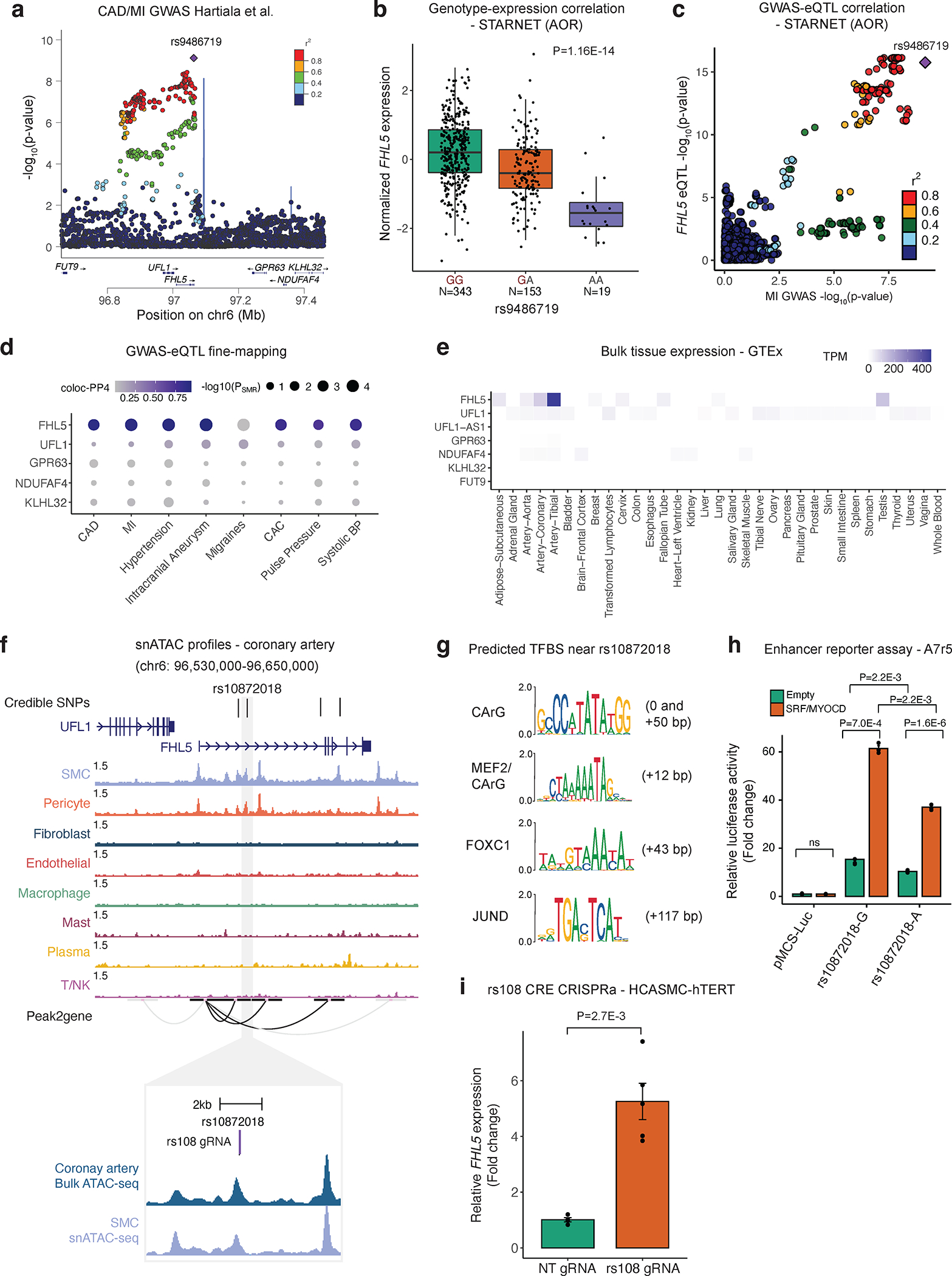
(a) LocusZoom plot showing the association of UFL1-FHL5 locus with myocardial infarction (MI) in the combined UKBB and CARDIoGRAMplusC4D meta-analysis. (b) Association of rs9486719 CAD/MI risk alleles with FHL5 gene expression in STARNET aortic tissue (AOR). CAD/MI risk allele at rs9486719 highlighted in red. Box plots represent the median with the box spanning the first and third quartiles and whiskers as 1.5 times IQR. (c) Locuscompare plot showing the associations of top FHL5 STARNET aortic tissue cis-eQTLs with MI. (d) Summary of SMR and coloc based fine-mapping analyses to prioritize candidate causal gene(s) at UFL1-FHL5 locus associated with vascular diseases and cardiovascular risk factors. The size of the dot reflects the -log10(pSMR) and the intensity of purple color represents the posterior probability of colocalization. (e) Normalized gene expression as transcripts per million (TPM) for all genes at the UFL1-FHL5 locus across GTEx tissues. (f) Genome browser view of human coronary artery snATAC-seq peaks showing overlap between CAD 95% credible set of SNPs (highlighting top candidate rs10872018) with putative enhancers correlated with the FHL5 promoter through peak2Gene analyses. Inset, genomic location of guide RNA targeting the cis-regulatory element (CRE) containing rs10872018 (rs108 gRNA) in purple. Bulk coronary and smooth muscle cell (SMC) snATAC tracks are shown below. (g) Predicted transcription factor binding sites (TFBS) at or around rs10872018 determined from JASPAR 2022. Distance (bp) of motif sequence is also shown relative to the rs10872018 SNP. (h) Luciferase reporter assay in A7r5 SMCs comparing rs10872018 allele-specific enhancer activity co-transfected with empty vector (control) or SRF and MYOCD expression constructs. Results are presented as fold change of empty vector control (pMCS-Luc) and values are mean ± SEM of triplicates. P-values determined using the Kruskal-Wallis test. (i) FHL5 upregulation upon CRISPR activation of rs10872018 CRE by expressing dCas9-p300 and rs108 gRNA in immortalized human coronary artery smooth muscle cells (HCASMC). Results are presented as fold change of non-targeting control (NT gRNA) and values are mean ± SEM of n=5 replicates.
rs9486719 is located in the fifth intron of FHL5, and similar to the majority of GWAS variants likely modulates gene expression to influence disease risk28,29 (Figure 1A). To prioritize the most likely target gene(s) at this locus, which includes 7 genes, we leveraged cis-expression quantitative trait loci (cis-eQTLs) in cardiometabolic tissues from Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task (STARNET)30. This SNP was strongly associated with FHL5 gene expression in both mammary arteries (MAM) with subclinical atherosclerotic disease and aortic root tissues (AOR) with atherosclerosis (Figure 1B). These results were consistent with GTEx eQTLs, in which rs9486719 was significantly associated with FHL5 expression in aorta (P=2.3E-7) (Figure S2A). Importantly, FHL5 eQTLs colocalized with CAD SNPs in aortic tissues, compared to eQTLs for neighboring genes (e.g., UFL1) (Figure 1C). In order to systematically prioritize the GWAS effector genes at the UFL1-FHL5 locus, we employed two complementary and independent fine-mapping methods, coloc31,32 and Summary-level based Mendelian Randomization (SMR)33,34. By integrating GTEx and STARNET artery tissue eQTLs for all the genes at the UFL1-FHL5 locus with GWAS summary statistics, we consistently identified FHL5 as the top candidate causal gene for CAD/MI and other common vascular traits (Figure 1D and Table S2). This was consistent with the tissue distribution of FHL5 gene expression, which was highly enriched in artery tissues in GTEx (Figure 1E) and STARNET (Figure S2B) compared to other genes at the locus. Together, these analyses suggest that the FHL5 risk allele (rs9486719-G) increases CAD/MI risk by increasing FHL5 gene expression in artery tissue (Figure S2C and S2D).
Epigenomic based fine-mapping of UFL1-FHL5 locus in human artery tissue
Next, we leveraged human coronary artery epigenomic profiles to resolve candidate causal variants at the UFL1-FHL5 locus. The MI 95% credible set determined from Probabilistic Annotation Integrator v3 (PAINTOR)35 using human coronary artery SMC and pericyte open chromatin profiles (snATAC-seq) as prior functional annotation weights consisted of 4 SNPs (Figure S3A and Table S3). By overlapping variants in this credible set with SMC Peak2Gene linkages36, we prioritized rs10872018, a SNP located in an active FHL5 regulatory element in vivo. (Figure 1F). These interactions were supported by the Activity-by-Contact Model (ABC)37 generated using ENCODE human coronary artery tissue ATAC-seq, H3K27ac, and HiChIP data, which further prioritized rs10872018 as the most likely causal variant (Table S4). This variant was identified as one of the top eQTLs in STARNET human vascular tissues (Figure S3B).
Since trait-associated SNPs are predicted to alter regulatory elements38, we scanned the genomic sequence +/− 100bp of rs10872018 for putative transcription factor binding sites and identified several motifs such as CArG, MEF2/CArG, FOXC1 and JUND (Figure 1G). Strikingly, multiple putative CArG boxes39 were identified proximal to rs10872018 (Figure S3C). This region containing the CArG boxes also shows a strong inter-species conservation (Figure S3D). The alternate allele (rs10872018-A) disrupts a non-canonical CArG box motif (Figure S3C), which is predicted to perturb binding of the SRF-Myocardin (MYOCD) transcriptional complex, a well-characterized regulator of SMC differentiation40,41. We validated this putative upstream regulatory mechanism using allele-specific enhancer luciferase reporter assays in A7r5 aortic SMCs. The risk allele (rs10872018-G) increased luciferase activity relative to the non-risk allele (rs10872018-A) (Figure 1H), consistent with the FHL5 eQTL direction in human artery tissues (Figure 1B and Figure S3B). SRF and MYOCD overexpression further potentiated the luciferase activity in an allele-specific manner (Figure 1H). To further confirm the function of this element in its endogenous chromatin environment, we leveraged the CRISPR-dCas9-p300 system. Targeting the rs10872018 harboring cis regulatory element (CRE), upregulated FHL5 gene expression ~5 fold, relative to the non-targeting (NT) gRNA control (Figure 1I). Together, these results demonstrate that altered SRF-MYOCD binding due to rs10872018 may partially explain the observed eQTL effects in human artery tissues during disease.
FHL5 is highly enriched in contractile mural cells in coronary arteries
To confirm expression of FHL5 protein in human artery tissues and identify its endogenous localization, we performed immunofluorescence on sections of human coronary arteries with subclinical atherosclerosis. FHL5 protein was enriched in the SMC-containing medial layer as well as the intima, colocalizing with F-actin positive cells. (Figure 2A, Figure S4A–B). Consistent with previous reports as a transcriptional regulator42, we observed nuclear localization of FHL5 in both the medial and intimal layers, but also perinuclear/cytoplasmic localization. To identify the cell-type specific expression profile, we queried a human coronary artery scRNA-seq dataset from 4 donor tissues with subclinical atherosclerosis11,43, which revealed FHL5 gene expression enriched in the mural cells (SMCs and pericytes) (Figure 2B). In fact, FHL5 was one of the more specific markers identified in SMC and pericyte clusters, compared to well-established contractile SMC markers, e.g., LMOD1 and MYOCD (Figure S4C). Similar results were observed in an advanced atherosclerotic carotid artery single-cell RNA-seq dataset44 (Figure S4D). Lastly, we corroborated these results in a single-nucleus ATAC-seq (snATAC-seq) dataset of 41 human coronary arteries with mostly subclinical atherosclerosis45, in which FHL5 had the highest gene score in the SMC and pericyte clusters, similar to LMOD1 (Figure 2C). Notably, a similar pattern of FHL5 expression was observed after integration of coronary snATAC-seq and scRNA-seq datasets (Figure S4C).
Figure 2. FHL5 expression is enriched in SMCs and pericytes in human coronary arteries.
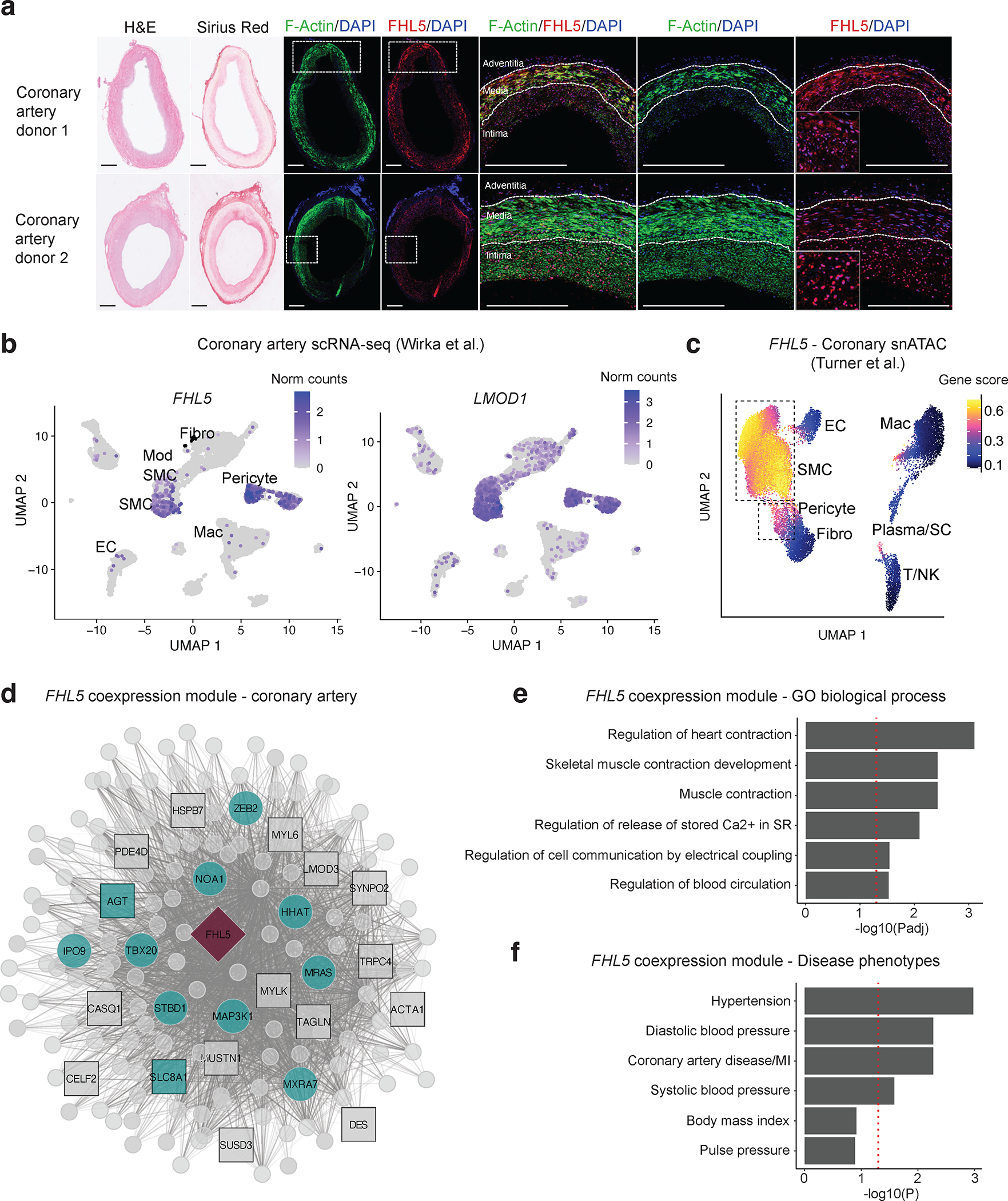
(a) Histological staining (H&E and Sirius Red) and immunofluorescence of F-actin (green) and FHL5 protein (red), and region of interest for the overlapping proteins in the medial and intimal layers of two representative human subclinical atherosclerotic coronary arteries (n=8 unique donors). Inset in the far-right image depicts a higher magnification of the nuclear FHL5 localization. Scale bars = 0.5 mm. (b) UMAP visualization of FHL5 and LMOD1 gene expression in different human coronary artery cell types from Wirka et al 11. (c) UMAP visualization of human coronary artery snATAC-seq cell clusters colored according to FHL5 gene score calculated in ArchR. (d) Undirected FHL5 coexpression network identified in human coronary arteries using weighted gene co-expression network analysis (WGCNA). Each node represents a gene and the lines connecting 2 nodes are weighted according to degree of correlation. The teal color highlights module genes associated with CAD/MI or blood pressure. The square nodes represent module genes that regulate SMC contraction. FHL5 is placed at the center and depicted as a red diamond. (e) Enrichment of gene ontology biological processes (GO-BP) and (f) cardiometabolic disease phenotypes in the FHL5 module protein-coding genes. The highlighted terms are a subset of the full ranked list found in Supp Table 6.
To gain insights into the functional role of FHL5 using a systems biology approach, we performed iterative Weighted Gene Co-expression Network Analysis (WGCNA)46,47 on transcriptomic data from 148 human coronary artery donors. After removing outliers and lowly expressed genes, we identified FHL5 in the light-green module, which included key SMC contractile mediators, such as MYLK, ACTA2, and TAGLN (Figure 2D and Table S5A). This module was enriched in SMC processes, such as muscle contraction (GO:0006936) and regulation of cell communication by electrical coupling (GO:0010649) as well as cardiometabolic GWAS candidate genes annotated in the Cardiovascular Disease Knowledge Portal (Figure 2E and Table S6). In further support of this link to vascular disease risk through regulation of SMC functions, FHL5 was identified as a key driver in module 152, a cross-tissue gene regulatory network (GRN) enriched in ECM and CAD candidate genes from the STAGE cohort (Figure S5A)48. The distinct enrichment in pathways among these two vascular tissues may reflect differences in the atherosclerotic lesion stage.
To further characterize FHL5 regulatory interactions in vivo, we constructed a Bayesian GRN incorporating STARNET aortic tissue eQTL data as priors (Figure S5B). A key driver analysis (KDA) of this network supported the regulatory potential of FHL5 which was predicted to function upstream of 2 pulse pressure genes, ACTG2 and MUSTN124 that have established roles in SMC contraction (Figure S5B and Table S5B). Notably, given its link to MI in a Japanese population49, ITIH3 was the key driver gene in this subnetwork. ACTG2 was also identified in the FHL5 network in STARNET (Figure S5A). Both ACTG2 and MUSTN1 are modestly expressed in coronary SMC relative to bulk coronary tissue (Figure S6), suggesting non-cell autonomous effects mediated by FHL5 in vivo. These integrative analyses suggest that this FHL5 GRN related to SMC contractility may contribute to pleiotropic association of FHL5 with common vascular diseases.
FHL5 expression increases SMC contraction and calcification
To overcome the limitations in replicative senescence in primary HCASMCs, we generated an immortalized coronary artery SMC line via overexpression of hTERT50. This immortalized cell line (HCASMC-hTERT) maintained protein expression of differentiated SMC markers and closely resembled primary HCASMCs at the transcriptomic level. (Figure S7A–C). Despite robust expression in intact arteries, FHL5 is potently downregulated in vitro, similar to the downregulation of other SMC contractile markers (Figure S8A). Therefore, to investigate its role in SMCs, we overexpressed either wildtype FHL5 or FHL5-NLS (FHL5 with a C-terminal nuclear localization signal) and confirmed protein expression (Figure 3A). The expression level of FHL5 was physiological and comparable to endogenous levels in human coronary arteries (Figure 3B). Motivated by our coexpression network analyses, we functionally validated the role of FHL5 in regulating SMC contractile pathways. Since calcium is a critical initiator of contraction, we also quantitated intracellular calcium levels. We observed increased SMC contraction and elevated calcium levels in the FHL5-NLS cells relative to HA control cells. While not statistically significant, FHL5 overexpressing cells trended in the same direction (Figure 3C and 3D). To further substantiate these findings and elucidate a potential mechanism underlying SMC contraction, we evaluated ser19 phosphorylation of myosin light chain (pMLC20) and myosin light chain kinase (MLCK) protein levels by western blot in HCASMC-hTert expressing FHL5 and FHL5-NLS (Figure S8B). Both FHL5 and FHL5-NLS expression resulted in increased pMLC20 and MLCK levels under basal and PE stimulated conditions. These findings suggest that FHL5 alters critical effectors of SMC contraction, contributing to the observed downstream contractile phenotype.
Figure 3. FHL5 regulates SMC contraction and calcification.
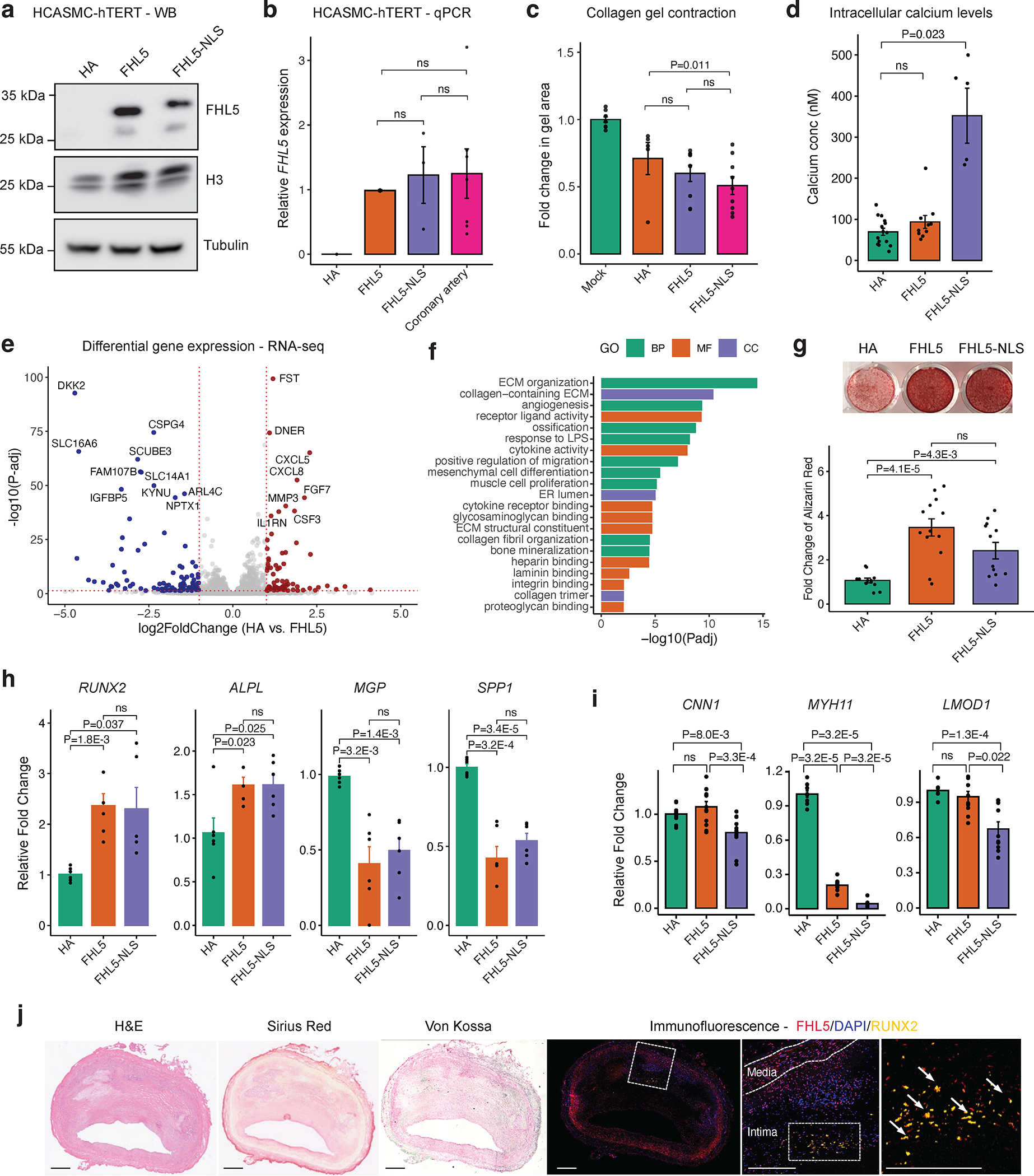
(a) Western blot of FHL5 showing expression of FHL5 and FHL5-NLS in HCASMC-hTERT cell lines with H3 and Tubulin as loading controls. (b) Relative expression of FHL5 in HCASMC-hTERT (HA, FHL5, FHL5-NLS) compared to endogenous levels of FHL5 in human coronary arteries (n=3 donors) determined using qPCR. (c) Change in collagen gel contraction (mm2) in HCASMC-hTERT (HA, FHL5, FHL5-NLS) relative to gel with no cells (n=4 independent replicates). (d) Quantification of intracellular calcium concentrations following stimulation with 10 μm phenylephrine (n=4 biological replicates). (e) Volcano plot showing differentially expressed genes (DEGs) following FHL5 overexpression in HCASMC-hTERT relative to HA, with upregulated genes in red and downregulated genes in blue. For clarity, genes with log2FoldChange > 5 and log2Foldchange <−5 are not represented. The full DEG list is provided in Table S7. (f) GO enrichment analysis of FHL5 DEGs showing top over-represented terms for biological process (BP), molecular function (MF), and cell compartment (CC). These highlighted terms were selected from the full ranked list found in Table S8. (g) Representative image of HCASMC-hTERT (HA, FHL5, FHL5-NLS) stained with Alizarin Red after 21 days under osteogenic conditions, and fold change (relative to HA) in calcium deposition quantified by measuring Alizarin Red staining in HCASMC-hTERT after 21 days in osteogenic media. (h) Relative expression of vascular calcification activators (RUNX2, ALPL) and inhibitors (MGP, SPP1) 14 days post treatment in osteogenic media measured by qPCR. (i) Relative expression of SMC markers 14 days post treatment in osteogenic media measured by qPCR. (j) Representative immunofluorescence staining of human coronary arteries (n=4 independent donors) showing FHL5 (red) colocalization with RUNX2 (yellow) in the intima layer near regions of calcium deposition; inset shows higher magnification of the regions of interest with overlapping FHL5 and RUNX2 protein. Adjacent sections subjected to histology staining for hematoxylin & eosin (H&E), collagen deposition (Sirius Red), and calcification (Von Kossa). Scale bars = 0.5 mm. All error bars represent mean+/−SEM. P-values determined from paired Student’s t-test. Individual points reflect replicates from at least n=3 independent experiments. ns: non-significant.
Next, to investigate the molecular mechanisms contributing to FHL5 regulation of SMC phenotypes, we performed bulk RNA-seq on HCASMC-hTERT expressing HA, FHL5 or FHL5-NLS constructs. While FHL5 and FHL5-NLS had distinct transcriptome signatures (Figure S8C and Table S7), the top differentially expressed genes had mainly concordant direction of effects between FHL5 and FHL5-NLS samples (Figure S8D–E and Table S7). When comparing FHL5 versus HA cells, we identified 377 differentially expressed genes (log2FoldChange > 0.6 and FDR < 0.05), of which 191 and 186 genes were upregulated and downregulated respectively (Figure 3E and Table S7). Top upregulated genes included various metalloproteinases (MMP1, MMP3, MMP10) and vessel wall matrisome proteins (DCN, COL5A3, ANGPTL4). We noted that FHL5-mediated differentially expressed genes (DEGs) were overrepresented in vascular remodeling pathways, such as cytokine activity/inflammation, ECM organization and ossification (Figure 3F and Table S8). Interestingly, FHL5 DEGs were also enriched with vascular and inflammatory disease candidate genes (Figure S8F).
Based on the perturbation of vascular remodeling and ossification pathways supported by RNA-seq, we next hypothesized that FHL5 may regulate vascular calcification to mediate CAD/MI risk. To this end, we treated SMCs with an osteogenic cocktail as done previously51,52. We observed increased mineral deposition quantified by alizarin red staining upon FHL5 and FHL5-NLS overexpression (Figure 3G). This increased calcification correlated with increased expression of osteogenic activators, RUNX2 and ALPL and reduced expression of osteogenic inhibitors, MGP and SPP1 (Figure 3H). Consistent with promoting SMC phenotypic transitions toward an osteogenic state, FHL5 and FHL5-NLS overexpression also coincided with downregulated expression of SMC markers, LMOD1, MYH11, and CNN1 (Figure 3I). Lastly, to validate the role of FHL5 in increasing vascular calcification, we immunostained sections of human coronary artery plaques. In accordance with our in vitro findings, we observed colocalization between FHL5 and RUNX2, a transcriptional activator of osteogenic differentiation near intimal calcium deposits (Figure 3J). These results were consistent with the direction of effect of the FHL5 genetic association with coronary artery calcification (CAC), where the CAC risk allele was associated with increased FHL5 gene expression. Together, these analyses suggest that FHL5 promotes a shift towards the SMC osteogenic phenotypic state in atherosclerotic lesions to increase CAD/MI risk.
FHL5 serves as a SMC cofactor to regulate disease-associated ECM interactions
In order to further decipher the direct transcriptional network regulated by FHL5 in SMCs under basal conditions, we used the Cleavage Under Targets and Release Using Nuclease (CUT&RUN) method to map genome-wide binding sites53,54. In total, we identified 17,201 FHL5 binding sites (qvalue < 0.01) that map to 6,776 unique genes, with a majority of binding sites located in annotated promoters and intronic regions (Figure 4A and Figure S9A)55,56. The active enhancer H3K27ac and promoter mark, H3K4me3 signals were enriched around the center of FHL5 peaks (Figure 4B, 4C and Figure S9B–D), with about 75% of binding sites overlapping primary HCASMC ATAC-seq peaks57 (Figure S9E).
Figure 4. FHL5 serves as a cofactor for the transcription factor, CREB, to regulate ECM organization in SMCs.
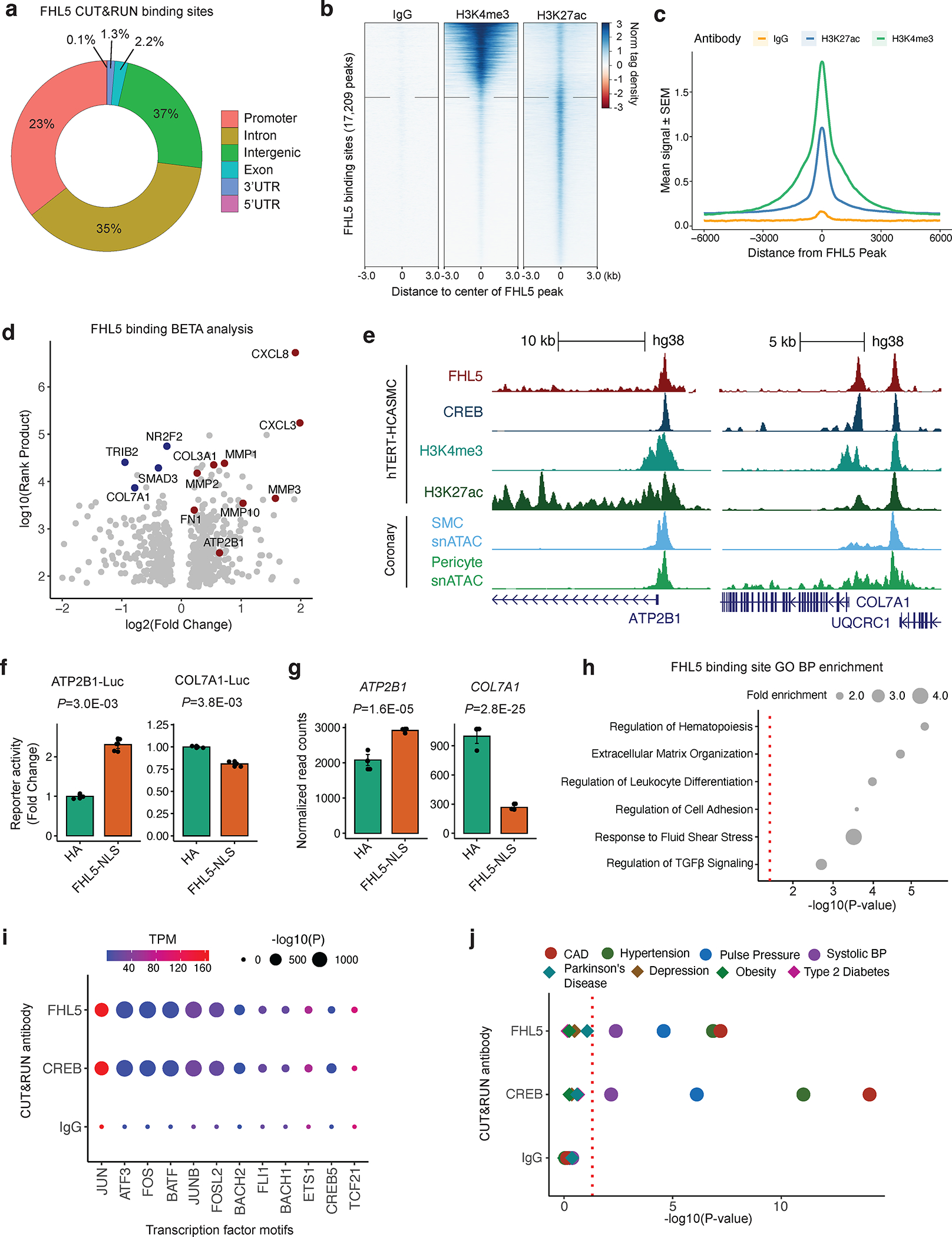
(a) Overlap of FHL5 peaks identified from CUT&RUN with genomic features. (b) Heatmap showing distribution of active chromatin histone marks (H3K4me3 and H3K27ac) +/−3kb from the center of FHL5 peaks, compared to IgG control. (c) Density plot showing genome-wide enrichment of active chromatin histone marks +/− 6kb from the center of FHL5 peaks. (d) Results of Binding and Expression Target Analysis (BETA) using FHL5 CUT&RUN peaks and FHL5 differentially expressed genes (DEGs) as input. Top candidate direct target genes highlighted in red, or blue based on upregulated and downregulated expression, respectively. Log normalized rank sum product score reflects the likelihood of direct transcriptional regulation for each gene. (e) UCSC genome browser tracks for FHL5, CREB, H3K27ac and H3K4me3 CUT&RUN performed in HCASMC-hTert, as well as snATAC-seq for SMCs and pericytes from coronary artery (Turner et al. 2022) at ATP2B1 and COL7A1 loci. (f) Relative change in luciferase reporter activity of ATP2B1 and COL7A1 enhancer regions in FHL5 overexpressing SMCs. (g) Relative change in RNA expression of ATP2B1 and COL7A1 in HCASMC-hTert overexpressing FHL5, shown as normalized read counts. (h) Top GO-BP overrepresented in FHL5 target genes identified from GREAT analysis of FHL5 peaks. These highlighted terms were selected from the full ranked list in Table S10. (i) Top transcription factor motifs enriched in FHL5, CREB, and IgG peaks identified from HOMER known motif analysis (dot size) as well as normalized (transcripts per million: TPM) expression level of the corresponding transcription factors in SMCs (color scale). (j) GREGOR analysis showing enrichment for vascular trait GWAS risk variants in FHL5, CREB, and IgG binding sites.
To examine the functional consequences of FHL5 binding on transcription, we integrated genome-wide FHL5 binding sites with differential expression analysis using the Binding and Expression Target Analysis (BETA) tool58. We observed substantial overlap between FHL5 DEGs and FHL5 peaks (P=6.3E-42, Fisher’s Exact Test), with a majority (51%) of these differentially expressed genes harboring at least 1 FHL5 binding site near its transcription start site. FHL5 directly upregulated key genes involved in vascular remodeling, including contractile genes, metalloproteinases, and ECM components (Figure 4D and Table S8). For example, we identified FHL5 binding at the 5’ regulatory elements near the ATP2B1, a CAD associated gene involved in calcium ion homeostasis, and at the gene promoter of COL7A1, a component of the basement membrane (Figure 4E). We validated the activity of these regulatory elements at the ATP2B1 and COL7A1 promoter in SMCs. FHL5 overexpression increased luciferase activity and correlated with increased ATP2B1 expression (Figure 4F and 4G). In contrast, FHL5 overexpression reduced the luciferase activity of the COL7A1 enhancer and COL7A1 gene expression, supporting the gene-specific activating or repressive role of FHL5 (Figure 4F and 4G). Gene ontology enrichment analysis of these high confidence target genes revealed FHL5 mediated direct regulation of pathways related to ECM organization and cell adhesion. We complemented this approach with Genomic Regions Enrichment of Annotations Tool (GREAT)59 analysis (Figure 4H and Table S10). FHL5 binding sites were proximal to genes related to inflammation, ECM organization and cell adhesion. These enrichments mirror the BETA results and further highlight the regulatory interactions between FHL5 and downstream vascular remodeling genes in mural cells.
Since FHL5 has not been reported to bind DNA directly, we performed motif enrichment analysis on genome-wide FHL5 peaks to identify candidate transcription factor binding partners, which revealed enrichment of AP1 and cAMP-response element (CRE) motifs within these peaks (Figure 4I). Regulatory elements proximal to upregulated genes after FHL5 overexpression were also enriched in AP1 motifs. Interestingly, consistent with previous studies, 48% of FHL5 binding sites overlapped CREB binding sites (Figure S9E), which were also strikingly enriched in AP1 binding motifs as well (Figure S9F and S9G). We further explored additional potential transcription factor binding partners using the Locus Overlap Analysis (LOLA) enrichment tool60 and assessed the enrichment of FHL5 binding sites in the ENCODE collection of transcription factor ChIP-seq datasets. Relative to the HCASMC ATAC-seq peak set, we observed significant enrichment of Pol2, AP1 members, (c-Fos and c-Jun), CREB and its common cofactor, p300 (Figure S9F). We validated the role of FHL5 as a cofactor for CREB through a cAMP response element (CRE) luciferase reporter assay, where FHL5 overexpression upregulated CRE activity (Figure S9H). This was further supported by co-immunoprecipitation of FHL5 and CREB proteins (Figure S9I), consistent with previous studies in germ cells61 and SMCs22.
FHL5 regulates a transcriptional network that contributes to CAD/MI risk by modulating SMC functions and vascular remodeling processes
As a transcriptional regulator of CAD-relevant pathways in the vessel wall, we next hypothesized that FHL5 regulation of downstream target genes contribute to the mechanistic link between FHL5 and vascular disease risk. To this end, we evaluated the enrichment of vascular disease GWAS SNPs in FHL5 binding sites using the Genomic Regulatory Elements and Gwas Overlap algoRithm (GREGOR) software package. Relative to a matched set of random SNPs62,63. FHL5 and CREB binding sites were highly enriched for CAD, MI, and blood pressure risk variants (Figure 4J), compared to non-vascular traits, further emphasizing the specific contribution of FHL5 to the heritability of common vascular diseases.
To further support this hypothesis and extend our findings to human artery tissues, we assessed the distal effects of FHL5 gene expression in STARNET cardiometabolic tissues. The top FHL5 cis-eQTL rs10872018 was associated with the expression of 743 and 568 eGenes in aortic and mammary artery tissues respectively at nominal significance (P < 0.05) (Table S11). These eGenes included key matrisome genes, such as FN1, COL4A4, and LUM. Integration of trans-eQTL target genes from STARNET artery tissues and FHL5 binding sites identified a network of downstream genes that harbor CAD risk variants (Figure 5A).
Figure 5. FHL5 regulates a network of CAD associated genes in human artery tissues.
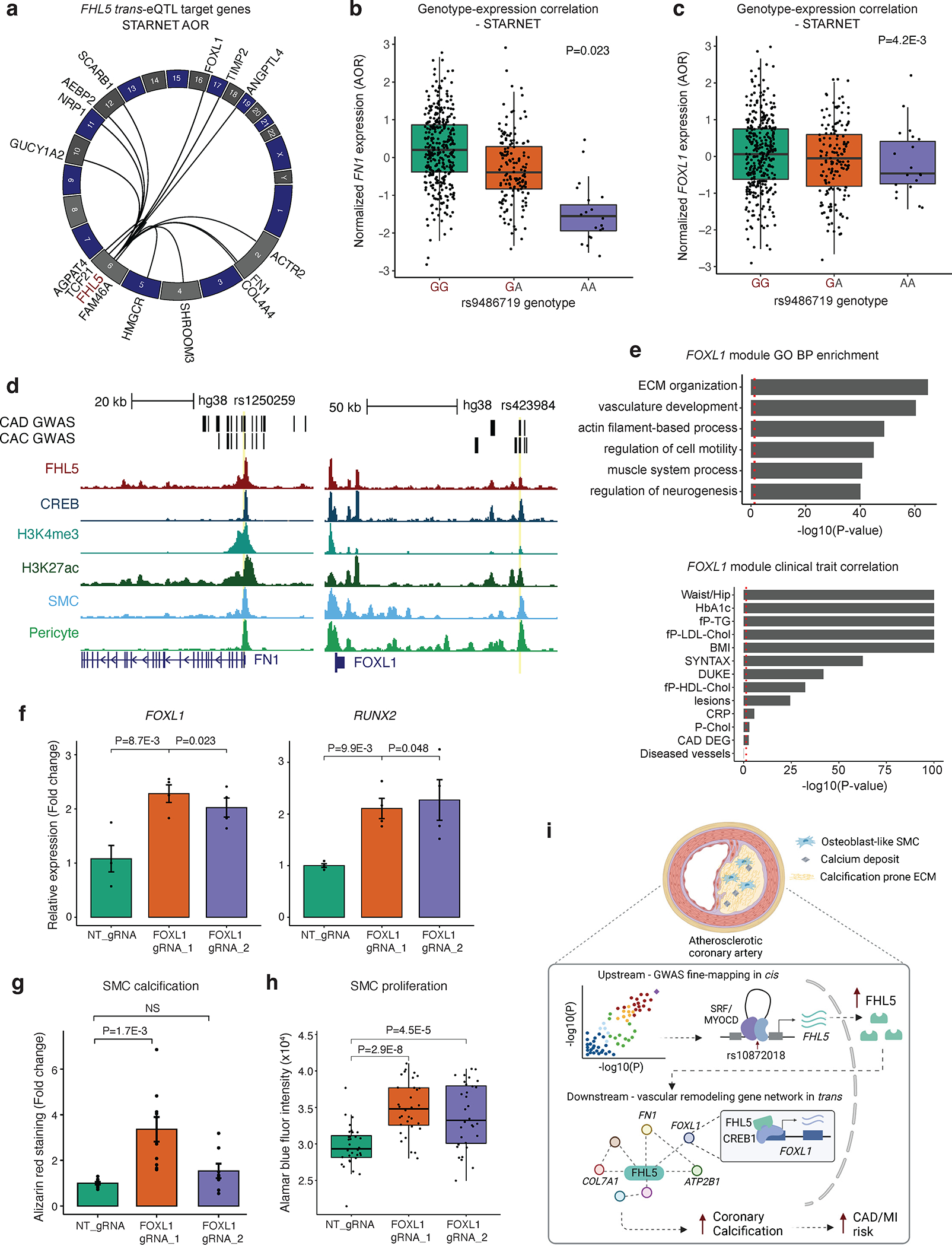
(a) Circos plot showing top trans-eQTL target genes for FHL5 lead SNP rs9486719 from STARNET artery tissues, which also harbor FHL5 binding sites in SMC. Genes are plotted according to genomic position with each black link representing significant trans-eQTL effects (P<0.05). (b) Association of rs9486719 with FOXL1 gene expression in atherosclerotic aortic artery (AOR) in STARNET. (c) Association of rs9486719 with FN1 gene expression in AOR tissue. The red allele color indicates the CAD risk allele (rs9486719-G). (d) UCSC genome browser tracks for FHL5, CREB, H3K27ac and H3K4me3 CUT&RUN performed in HCASMC-hTert, as well as snATAC-seq for SMCs and pericytes from human coronary artery (Turner et al. 2022) at the promoter of FN1 (left) and FOXL1 (right). (e) Top, enrichment of clinical traits in module 28 containing FOXL1 and FHL5 in STARNET cross-tissue networks. The red dotted line corresponds to P<0.05. Bottom, top GO-BP enriched in module 28. The red dotted line corresponds to a nominal threshold of FDR<0.05. These highlighted terms were subset from the full ranked list in Table S13a. (f) Relative expression of FOXL1 (left) and RUNX2 (right) in HCASMC-hTERT under osteogenic conditions where FOXL1 expression was activated using the CRISPR-SAM system and two different guide RNAs (FOXL1 gRNA_1 and FOXL1 gRNA_2), relative to non-targeting guide RNA (NT_gRNA). Values represent mean ± SEM of relative FOXL1 or RUNX2 expression normalized to GAPDH from n=4 replicates. (g) Alizarin red staining-based quantification of calcification in HCASMC expressing NT gRNA or FOXL1_gRNA_1, FOXL1_gRNA_2 under osteogenic conditions for 21 days. Values represent mean ± SEM from n=8 replicates. (h) Proliferation measured with Alamar blue fluorescence quantification after 4 days in FOXL1-CRISPRa HCASMC-hTERT. (i) Schematic depicting the proposed upstream and downstream mechanisms underlying the FHL5 genetic association with CAD/MI and other vascular traits. Top, risk alleles for candidate causal variants (e.g., rs10872018) are associated with increased FHL5 gene expression, which are predicted to function through SRF-MYOCD enhancers in cis. Bottom, this results in increased binding of FHL5 cofactor to CREB regulatory elements in vascular remodeling gene network in trans, ultimately leading to maladaptive ECM remodeling and vascular calcification in SMC and osteogenic-like SMC, thus increasing disease risk. Created with BioRender.com
We prioritized FN1 and FOXL1, two CAD/MI and CAC candidate genes, as likely downstream effectors of FHL5. The rs9486719-G CAD/MI risk allele associated with increased FHL5 gene expression was associated with both increased FN1 (Figure 5B) and FOXL1 (Figure 5C) expression in trans. Supporting this genetic regulation, we identified an FHL5 binding site overlapping active regulatory marks harboring CAD and CAC risk variants at both the FN1 promoter and upstream regulatory element of FOXL1 (Figure 5D). Interestingly, this candidate FOXL1 enhancer harbors rs423984, a variant in the MI 95% credible set that is also associated with CAC and FOXL1 gene expression in muscle tissue (Figure 5C and Figure S10A). As is common for intergenic variants at GWAS loci, this variant is likely correlated with expression of both FOXL1 and neighboring gene FOXC2, based on coronary artery snATAC peak2gene links (Figure S10A).
To provide insights into the function of FHL5 target genes in human arteries, we queried STARNET cross-tissue GRNs. FN1 is a member of module 39 in AOR tissue, which was enriched in genes predicted to regulate ECM organization and cell adhesion and correlated with clinical traits indicative of CAD severity (DUKE and SYNTAX score) (Table S12). Similarly, we identified FOXL1 and FHL5 in module 28, a cross-tissue GRN that also includes CAD-associated genes, SVEP1 and MFEG8. This module was functionally enriched in genes related to ECM organization, SMC contraction and calcium ion regulation (Figure 5E, Figure S10B, and Table S13), and correlated with atherosclerotic lesion presence and disease severity (Figure 5E). These enrichment analyses recapitulate the established role of FN1 in the vessel wall and its link to disease and highlight FOXL1 as a candidate effector gene during vascular remodeling.
To further explore the potential link between FHL5 and FOXL1, we observed FOXL1 was highly correlated with FHL5 gene expression in human coronary arteries11 (Figure S10C). Similar to FHL5, FOXL1 expression was highly enriched in SMC and pericytes (Figure S10D) and in bulk artery tissues in both STARNET and GTEx (Figure S10E and S10F). We next experimentally validated these putative contributions to SMC phenotypic modulation using the lentiviral dCas9 Synergistic Activator Mediator (SAM) system 64. This perturbation was associated with increased FOXL1 and RUNX2 gene expression, calcium deposition and SMC proliferation, mirroring the direction of effect of FHL5 overexpression (Figure 5F–H). Consistent with a role in promoting maladaptive SMC transitions, FOXL1 was elevated in atherosclerotic aortic tissues relative to healthy controls (P=1.4E-6) (Table S14). Taken together, FHL5 mediated regulation of FOXL1 and other ECM genes in trans may contribute in part to the mechanistic link between regulatory variants and vascular disease risk (Figure 5I).
Discussion
Despite the identification of over 200 loci associated with CAD, the mechanisms underlying these associations are largely unknown. Given at least half of these loci are predicted to function independently of traditional risk factors65, characterization of these novel loci may provide insights into some of the ‘hidden’ heritable risk factors. Our study addresses this gap by implicating FHL5 as the top candidate gene at the UFL1-FHL5 locus associated with vascular diseases. UFL1-FHL5 is also a well-established risk locus for migraine66, consistent with the genetic correlation and vascular etiology67,68. In this study, we elucidated the upstream regulatory mechanisms and downstream function of FHL5 in the human coronary artery. We showed that FHL5 promotes SMC calcification and contraction through dysregulation of vascular remodeling and calcium handling genes. Lastly, we characterized the FHL5 regulated SMC transcriptional network and highlighted trans-acting mechanisms mediated by FHL5 that contribute to the heritability of CAD and other common vascular diseases.
In atherosclerotic lesions, SMCs adopt phenotypes characteristic of other cell lineages, such as macrophages, fibroblasts, and osteoblasts2,11. SMCs that adopt this osteogenic phenotype participate in the deposition of intimal microcalcifications. Our findings support a mechanism where FHL5 mediated dysregulation of various metalloproteinases, collagens, and calcium signaling genes contributes to this pro-calcifying phenotype. Given the colocalization of RUNX2 and FHL5 in human coronary artery lesions, we postulate that FHL5 expression in this population of dedifferentiated SMCs alters the ECM composition to promote mineral deposition in the lesion. The significance of this calcification phenotype is underscored by the use of calcium scores in the clinic to identify patients at high risk for MI and other adverse cardiovascular events69,70. Despite observing increased contractility under basal conditions, which may reflect the homeostatic role of FHL5 in maintaining vascular tone, our studies under osteogenic conditions implicate FHL5 as a pro-calcifying factor as well. In support of a putative role in regulating both phenotypes, recent work by Karlöf et al71 reported that SMC contractile markers, which included FHL5, MYOCD and CNN1, were upregulated ~4-fold in highly calcified carotid artery plaques relative to lowly calcified plaques. This upregulation of SMC markers also correlated with increased expression of recently characterized SMC calcification markers (e.g. proteoglycan 4 (PRG4)72. This is further supported by gene ontology enrichment analysis highlighting calcium signaling, cytoskeletal rearrangements, and muscle contraction as overrepresented pathways among differentially expressed genes between highly and lowly calcified lesions71. To reconcile these phenotypes, we speculate that FHL5 regulation of intracellular calcium ion homeostasis contributes to both processes, given the critical role of calcium in the initiation of SMC contraction73,74, cell stiffness75,76, and vascular calcification41,77.
This study confirms and extends previous work identifying FHL5 as a cofactor for CREB 22 23. Our CUT&RUN data supports a similar mechanism where FHL5, through interactions with CREB, regulates target genes associated with vascular remodeling in SMCs. Previous studies have linked activation of the CREB/ATF3 signaling pathway to increased SMC proliferation, migration, and calcification78. In addition to the CRE motif, we also observed strong enrichment of the AP1 binding motif in FHL5 binding sites. This motif was also found to be enriched in accessible regulatory elements throughout the SMC transition to a fibroblast-like state79. Although we cannot rule out additional direct interactions with members of the AP1 family, this enrichment may also reflect epigenetic functions, similar to AP1 interactions at downstream CAD-associated loci, SMAD3 and CDKN2B-AS180. Lastly, we highlight that FHL5 expression is regulated by SRF-MYOCD in basal SMC, consistent with its positive effects on SMC contraction. However, unlike the expression of other classic SMC contractile genes (e.g., TAGLN, LMOD1, ACTA2), FHL5 expression is maintained in neointimal SMCs that contribute to vascular calcification. This may be attributed to the different SRF cofactors that influence cell proliferation and contractility). It would be interesting to map MYOCD binding sites in future studies to provide additional evidence for FHL5-mediated regulation in phenotypically modulated SMCs81.
Dissecting the regulatory network of disease associated transcription factors such as KLF4 and TCF21, have identified regulatory interactions with downstream CAD loci that govern SMC phenotypic modulation10,11. In support of the proposed omnigenic model, these studies highlight trans-acting mechanisms that may contribute to the majority of CAD heritability82,83. Our findings support the role of FHL5 as a “peripheral” transcriptional regulator of downstream “core” genes that have predicted functions in the vessel wall. By integrating STARNET vascular tissue trans-eQTL data, we identified a network of putative core genes that contribute to CAD pathogenesis, which include the well-characterized matrisome gene, FN1 and the transcription factor, FOXL1. FN1 has already been implicated as a regulator of SMC phenotypic modulation associated with CAD11,84,85,86. In contrast, the precise mechanism of how FOXL1 functions in artery tissues and atherosclerosis has not been fully characterized. Interestingly, FOXL1 and neighboring gene FOXC2 are both associated with CAD and bone mineral density87. As supported by our coronary artery SMC and STARNET cross-tissue GRNs, we propose that FOXL1 may regulate vascular remodeling pathways in vivo to impact disease risk. Functional dissection of other FHL5 target genes may uncover novel mechanisms and candidate genes contributing to heritable risk for CAD and other vascular diseases.
Although our study integrates multiple lines of human evidence to unravel the function of FHL5 in SMCs, we acknowledge known limitations. First, given that most participants in published GWAS and eQTL studies are of European descent, we estimated the LD pattern at the FHL5 locus using the 1000G European reference panel. However, due to the multi-ancestry cohorts included in many large-scale studies, these estimates do not perfectly reflect the entire population and represents a potential limitation of our fine mapping studies. Second, since we focus primarily on subclinical disease, this study does not address the potential roles for FHL5 in advanced disease stages. Future studies using more complex in vitro and human clinical samples will be needed to establish the causal link between FHL5 and indices of plaque stability and clinical outcomes. Given the lack of Fhl5 expression in murine artery tissues, investigations utilizing traditional models of atherosclerosis and vascular calcific disease may not be applicable. Third, this study focuses primarily on the transcriptional role of FHL5 in SMCs. Other FHL family members, FHL1 and FHL3, are known to function as scaffolds to regulate sarcomere formation in myoblasts19,88,89. We speculate that FHL5, due to the presence of similar conserved LIM domains, may mediate similar interactions with the actin cytoskeleton90,91 or focal adhesions92,93. While our study provides insights into the FHL5 mediated gene regulatory mechanisms, future studies will be needed to clarify its role in mediating mechano-transduction or other cell signaling events.
In summary, this work reveals a molecular mechanism by which FHL5, the top candidate causal gene at the pleiotropic UFL1-FHL5 locus, modulates vascular disease risk. We propose that FHL5 regulates a network of downstream genes in SMCs that participate in adverse vascular remodeling events driving disease progression. Similar to the effects of modulating other transcriptional regulators, modest increases in FHL5 gene expression may be propagated through its gene regulatory network in the vascular wall, ultimately increasing CAD/MI risk over time. Characterization of these trans effects in SMCs also highlights regulatory interactions at the molecular level governing atherogenic SMC phenotypic modulation. Future studies mapping genome-wide interactions of other disease associated transcriptional regulators may uncover new mechanisms and prioritize effector genes that impact primary disease processes, thereby accelerating the development of therapeutics to prevent MI and other vascular related pathologies.
Supplementary Material
Novelty and Significance:
What is known?
Common genetic variants at UFL1-FHL5 contribute to the genetic risk for several vascular diseases, including CAD/MI, hypertension, aneurysm, and migraines.
Expression quantitative trait locus analysis indicates that vascular disease risk variants modulate FHL5 gene expression in human artery tissues.
FHL5 was previously characterized as a potent coactivator during development and more recently was linked to vein graft intimal hyperplasia.
What new information does this article contribute?
FHL5 is the top candidate causal gene underlying the pleiotropic genetic associations with CAD/MI and other common vascular diseases.
FHL5 promotes the osteogenic SMC transition to increase vascular calcification in vitro under calcifying conditions and within human atherosclerotic lesions ex vivo.
FHL5 regulation of downstream gene programs harboring CAD/MI risk loci (e.g. FN1, FOXL1) linked to SMC phenotypic modulation and vascular remodeling, contributes to the heritable risk of common vascular diseases.
Although hundreds of loci are associated with common vascular diseases, the molecular mechanisms for many of these loci (e.g., UFL1-FHL5) are unresolved. FHL5, the top candidate causal gene implicated by eQTL analysis, encodes a cofactor that was first characterized as a transcriptional activator of sperm cell maturation. Despite recent reports of FHL5 exacerbating vein intimal hyperplasia, the molecular mechanism of its role in coronary artery SMC and contribution to common vascular diseases (e.g., CAD) is unknown.
Here, we dissect the upstream regulatory mechanisms at the UFL1-FHL5 locus and prioritize rs10872018 as a top candidate causal variant, with the CAD risk allele increasing FHL5 gene expression. We discover a previously unknown function of FHL5 in promoting the SMC osteogenic transition state, in which FHL5 overexpression increases mineral deposition and reduces levels of canonical SMC markers. We establish FHL5 transcriptionally regulated downstream processes, such as ECM remodeling and cell adhesion that contribute to the maladaptive vascular remodeling in the vessel wall. We also highlight regulatory interactions between FHL5 and downstream CAD/MI GWAS loci that may contribute to the pleiotropic associations of UFL1-FHL5 variants. Overall, this study illustrates how cis and trans-acting regulatory interactions contribute to complex disease risk.
Acknowledgements
We thank Dr. Pat Pramoonjago and Sheri VanHoose and members of the University of Virginia (UVA) histology core facilities for tissue sectioning and histological staining assistance and Dr. Katia Sol-Church and members of the UVA Genomics Core facility for assistance with library construction and sequencing. We also thank Dr. R. Kirk Riemer and Dr. Xiaoyuan Ma at Stanford University and Dr. Avril Somlyo at the University of Virginia for helpful discussions related to this study.
Sources of Funding
This work was supported by grants from: the National Institutes of Health (R01HL148239, R01HL164577, and R00HL125912 to C.L.M.; F31 HL156463 to D.W.; R01HL142809 to R.M.; K01HL164687 to C.L.L.C.) and the Fondation Leducq (‘PlaqOmics’ 18CVD02 to J.LM.B. and C.L.M.).
Non-standard Abbreviations and Acronyms:
- ABC
Activity-by-Contact
- AOR
Aortic root
- ATAC-seq
Assay for Transposase Accessible Chromatin sequencing
- CAC
Coronary Artery Calcification
- CAD
Coronary Artery Disease
- CRE
cis-regulatory element
- CREB
Cyclic AMP-Responsive Element-Binding Protein
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats
- CUT&RUN
Cleavage Under Targets and Release Using Nuclease
- dCas9
endonuclease deficient CRISPR associated protein 9
- ECM
Extracellular Matrix
- ENCODE
ENCyclopedia Of DNA Elements
- eQTL
expression Quantitative Trait Locus
- FHL1/2/3/5
Four-and-a-Half LIM domains 1/2/3/5
- FN1
Fibronectin 1
- FOXC1
Forkhead box L1
- GRN
Gene Regulatory Network
- GWAS
Genome-Wide Association Studies
- HCASMC
Human Coronary Artery Smooth Muscle Cells
- hTERT
human Telomerase Reverse Transcriptase
- KDA
Key Driver Analysis
- LMOD1
Leiomodin 1
- MAM
Mammary arteries
- MI
Myocardial Infarction
- MYOCD
Myocardin
- MYLK
Myosin Light chain Kinase
- NLS
Nuclear Localization Signal
- PAINTOR
Probabilistic Annotation Integrator
- PE
Phenylephrine
- pMLC20
phosphorylated Myosin Light Chain
- SMC
Smooth Muscle Cells
- SMR
Summary-level based Mendelian Randomization
- scRNA-seq
Single-Cell RNA sequencing
- snATAC-seq
Single-Nucleus Assay for Transposase Accessible Chromatin sequencing
- SNP
Single Nucleotide Polymorphism
- SRF
Serum Response Factor
- STAGE
STockholm Atherosclerosis Gene Expression
- STARNET
Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task
- UFL1
UFM1 Specific Ligase 1
Footnotes
Disclosures
Johan Björkegren is a shareholder in Clinical Gene Network AB and has an invested interest in STARNET. Jason Kovacic is the recipient of an Agilent Thought Leader Award (January 2022), which includes funding for research that is unrelated to the current manuscript. All other authors declare that they have no competing interests relevant to the contents of this paper to disclose.
Bibliography
- 1.Wong D, Turner AW, Miller CL. Genetic insights into smooth muscle cell contributions to coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2019;39:1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miano JM, Fisher EA, Majesky MW. Fate and state of vascular smooth muscle cells in atherosclerosis. Circulation. 2021;143:2110–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu M, Gomez D. Smooth muscle cell phenotypic diversity. Arterioscler. Thromb. Vasc. Biol. 2019;39:1715–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ. Res. 2018;122:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tcheandjieu C, Zhu X, Hilliard AT, Clarke SL, Napolioni V, Ma S, Lee KM, Fang H, Chen F, Lu Y, et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat. Med. 2022;28:1679–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishigaki K, Akiyama M, Kanai M, Takahashi A, Kawakami E, Sugishita H, Sakaue S, Matoba N, Low S-K, Okada Y, et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 2020;52:669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schunkert H, von Scheidt M, Kessler T, Stiller B, Zeng L, Vilne B. Genetics of coronary artery disease in the light of genome-wide association studies. Clin. Res. Cardiol. 2018;107:2–9. [DOI] [PubMed] [Google Scholar]
- 8.Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc. Res. 2018;114:1241–1257. [DOI] [PubMed] [Google Scholar]
- 9.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AAC, Greene ES, Straub AC, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015;21:628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alencar GF, Owsiany KM, Karnewar S, Sukhavasi K, Mocci G, Nguyen AT, Williams CM, Shamsuzzaman S, Mokry M, Henderson CA, et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation. 2020;142:2045–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, Kundu R, Nagao M, Coller J, Koyano TK, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019;25:1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Speer MY, Yang H-Y, Brabb T, Leaf E, Look A, Lin W-L, Frutkin A, Dichek D, Giachelli CM. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ. Res. 2009;104:733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mori H, Torii S, Kutyna M, Sakamoto A, Finn AV, Virmani R. Coronary Artery Calcification and its Progression: What Does it Really Mean? JACC Cardiovasc. Imaging. 2018;11:127–142. [DOI] [PubMed] [Google Scholar]
- 14.Hartiala JA, Han Y, Jia Q, Hilser JR, Huang P, Gukasyan J, Schwartzman WS, Cai Z, Biswas S, Trégouët D-A, et al. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur. Heart J. 2021;42:919–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canela-Xandri O, Rawlik K, Tenesa A. An atlas of genetic associations in UK Biobank. Nat. Genet. 2018;50:1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakker MK, van der Spek RAA, van Rheenen W, Morel S, Bourcier R, Hostettler IC, Alg VS, van Eijk KR, Koido M, Akiyama M, et al. Genome-wide association study of intracranial aneurysms identifies 17 risk loci and genetic overlap with clinical risk factors. Nat. Genet. 2020;52:1303–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gormley P, Anttila V, Winsvold BS, Palta P, Esko T, Pers TH, Farh K-H, Cuenca-Leon E, Muona M, Furlotte NA, et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 2016;48:856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, Emdin CA, Hilvering CRE, Bianchi V, Mueller C, et al. A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell. 2017;170:522–533.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shathasivam T, Kislinger T, Gramolini AO. Genes, proteins and complexes: the multifaceted nature of FHL family proteins in diverse tissues. J. Cell. Mol. Med. 2010;14:2702–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kotaja N, De Cesare D, Macho B, Monaco L, Brancorsini S, Goossens E, Tournaye H, Gansmuller A, Sassone-Corsi P. Abnormal sperm in mice with targeted deletion of the act (activator of cAMP-responsive element modulator in testis) gene. Proc Natl Acad Sci USA. 2004;101:10620–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macho B, Brancorsini S, Fimia GM, Setou M, Hirokawa N, Sassone-Corsi P. CREM-dependent transcription in male germ cells controlled by a kinesin. Science. 2002;298:2388–2390. [DOI] [PubMed] [Google Scholar]
- 22.Nakanishi K, Saito Y, Azuma N, Sasajima T. Cyclic adenosine monophosphate response-element binding protein activation by mitogen-activated protein kinase-activated protein kinase 3 and four-and-a-half LIM domains 5 plays a key role for vein graft intimal hyperplasia. J. Vasc. Surg. 2013;57:182–93, 193.e1. [DOI] [PubMed] [Google Scholar]
- 23.Uchida D, Saito Y, Kikuchi S, Yoshida Y, Hirata S, Sasajima T, Azuma N. Development of gene therapy with a cyclic adenosine monophosphate response element decoy oligodeoxynucleotide to prevent vascular intimal hyperplasia. J. Vasc. Surg. 2020;71:229–241. [DOI] [PubMed] [Google Scholar]
- 24.Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, Ntritsos G, Dimou N, Cabrera CP, Karaman I, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018;50:1412–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giri A, Hellwege JN, Keaton JM, Park J, Qiu C, Warren HR, Torstenson ES, Kovesdy CP, Sun YV, Wilson OD, et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat. Genet. 2019;51:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, Paul DS, Freitag D, Burgess S, Danesh J, et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. 2016;32:3207–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35:4851–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cano-Gamez E, Trynka G. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet. 2020;11:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hao K, Ermel R, Sukhavasi K, Cheng H, Ma L, Li L, Amadori L, Koplev S, Franzén O, d’Escamard V, et al. Integrative prioritization of causal genes for coronary artery disease. Circ. Genom. Precis. Med. 2022;15:e003365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franzén O, Ermel R, Cohain A, Akers NK, Di Narzo A, Talukdar HA, Foroughi-Asl H, Giambartolomei C, Fullard JF, Sukhavasi K, et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science. 2016;353:827–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallace C Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLoS Genet. 2020;16:e1008720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016;48:481–487. [DOI] [PubMed] [Google Scholar]
- 34.Wu Y, Zeng J, Zhang F, Zhu Z, Qi T, Zheng Z, Lloyd-Jones LR, Marioni RE, Martin NG, Montgomery GW, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 2018;9:918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kichaev G, Yang W-Y, Lindstrom S, Hormozdiari F, Eskin E, Price AL, Kraft P, Pasaniuc B. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 2014;10:e1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, Greenleaf WJ. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nasser J, Bergman DT, Fulco CP, Guckelberger P, Doughty BR, Patwardhan TA, Jones TR, Nguyen TH, Ulirsch JC, Lekschas F, et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593:238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benson CC, Zhou Q, Long X, Miano JM. Identifying functional single nucleotide polymorphisms in the human CArGome. Physiol. Genomics. 2011;43:1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long X, Bell RD, Gerthoffer WT, Zlokovic BV, Miano JM. Myocardin is sufficient for a smooth muscle-like contractile phenotype. Arterioscler. Thromb. Vasc. Biol. 2008;28:1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang H, Curinga G, Giachelli CM. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int. 2004;66:2293–2299. [DOI] [PubMed] [Google Scholar]
- 42.Fimia GM, Morlon A, Macho B, De Cesare D, Sassone-Corsi P. Transcriptional cascades during spermatogenesis: pivotal role of CREM and ACT. Mol. Cell. Endocrinol. 2001;179:17–23. [DOI] [PubMed] [Google Scholar]
- 43.Ma WF, Hodonsky CJ, Turner AW, Wong D, Song Y, Mosquera JV, Ligay AV, Slenders L, Gancayco C, Pan H, et al. Enhanced single-cell RNA-seq workflow reveals coronary artery disease cellular cross-talk and candidate drug targets. Atherosclerosis. 2022;340:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alsaigh T, Evans D, Frankel D, Torkamani A. Decoding the transcriptome of calcified atherosclerotic plaque at single-cell resolution. Commun. Biol. 2022;5:1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turner AW, Hu SS, Mosquera JV, Ma WF, Hodonsky CJ, Wong D, Auguste G, Sol-Church K, Farber E, Kundu S, et al. Single-nucleus chromatin accessibility profiling highlights regulatory mechanisms of coronary artery disease risk. Nat. Genet. 2022;54:804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greenfest-Allen E, Cartailler J-P, Magnuson MA, Stoeckert CJ. iterativeWGCNA: iterative refinement to improve module detection from WGCNA co-expression networks. BioRxiv. 2017; [Google Scholar]
- 48.Talukdar HA, Foroughi Asl H, Jain RK, Ermel R, Ruusalepp A, Franzén O, Kidd BA, Readhead B, Giannarelli C, Kovacic JC, et al. Cross-Tissue Regulatory Gene Networks in Coronary Artery Disease. Cell Syst. 2016;2:196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ebana Y, Ozaki K, Inoue K, Sato H, Iida A, Lwin H, Saito S, Mizuno H, Takahashi A, Nakamura T, et al. A functional SNP in ITIH3 is associated with susceptibility to myocardial infarction. J. Hum. Genet. 2007;52:220–229. [DOI] [PubMed] [Google Scholar]
- 50.Lee KM, Choi KH, Ouellette MM. Use of exogenous hTERT to immortalize primary human cells. Cytotechnology. 2004;45:33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pustlauk W, Westhoff TH, Claeys L, Roch T, Geißler S, Babel N. Induced osteogenic differentiation of human smooth muscle cells as a model of vascular calcification. Sci. Rep. 2020;10:5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malhotra R, Mauer AC, Lino Cardenas CL, Guo X, Yao J, Zhang X, Wunderer F, Smith AV, Wong Q, Pechlivanis S, et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat. Genet. 2019;51:1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skene PJ, Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skene PJ, Henikoff JG, Henikoff S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018;13:1006–1019. [DOI] [PubMed] [Google Scholar]
- 55.Zhu Q, Liu N, Orkin SH, Yuan G-C. CUT&RUNTools: a flexible pipeline for CUT&RUN processing and footprint analysis. Genome Biol. 2019;20:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu G, Wang L-G, He Q-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. [DOI] [PubMed] [Google Scholar]
- 57.Miller CL, Pjanic M, Wang T, Nguyen T, Cohain A, Lee JD, Perisic L, Hedin U, Kundu RK, Majmudar D, et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun. 2016;7:12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, Liu XS. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 2013;8:2502–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheffield NC, Bock C. LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 2016;32:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fimia GM, De Cesare D, Sassone-Corsi P. A family of LIM-only transcriptional coactivators: tissue-specific expression and selective activation of CREB and CREM. Mol. Cell. Biol. 2000;20:8613–8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmidt EM, Zhang J, Zhou W, Chen J, Mohlke KL, Chen YE, Willer CJ. GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics. 2015;31:2601–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khetan S, Kursawe R, Youn A, Lawlor N, Jillette A, Marquez EJ, Ucar D, Stitzel ML. Type 2 Diabetes-Associated Genetic Variants Regulate Chromatin Accessibility in Human Islets. Diabetes. 2018;67:2466–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NGD, Jansen H, Kanoni S, Nelson CP, Ferrario PG, König IR, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J. Am. Coll. Cardiol. 2017;69:823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hautakangas H, Winsvold BS, Ruotsalainen SE, Bjornsdottir G, Harder AVE, Kogelman LJA, Thomas LF, Noordam R, Benner C, Gormley P, et al. Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat. Genet. 2022;54:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winsvold BS, Nelson CP, Malik R, Gormley P, Anttila V, Vander Heiden J, Elliott KS, Jacobsen LM, Palta P, Amin N, et al. Genetic analysis for a shared biological basis between migraine and coronary artery disease. Neurol. Genet. 2015;1:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kurth T, Gaziano JM, Cook NR, Logroscino G, Diener H-C, Buring JE. Migraine and risk of cardiovascular disease in women. JAMA. 2006;296:283–291. [DOI] [PubMed] [Google Scholar]
- 69.Budoff MJ, Young R, Lopez VA, Kronmal RA, Nasir K, Blumenthal RS, Detrano RC, Bild DE, Guerci AD, Liu K, et al. Progression of coronary calcium and incident coronary heart disease events: MESA (Multi-Ethnic Study of Atherosclerosis). J. Am. Coll. Cardiol. 2013;61:1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Detrano R, Guerci AD, Carr JJ, Bild DE, Burke G, Folsom AR, Liu K, Shea S, Szklo M, Bluemke DA, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N. Engl. J. Med. 2008;358:1336–1345. [DOI] [PubMed] [Google Scholar]
- 71.Karlöf E, Seime T, Dias N, Lengquist M, Witasp A, Almqvist H, Kronqvist M, Gådin JR, Odeberg J, Maegdefessel L, et al. Correlation of computed tomography with carotid plaque transcriptomes associates calcification with lesion-stabilization. Atherosclerosis. 2019;288:175–185. [DOI] [PubMed] [Google Scholar]
- 72.Seime T, Akbulut AC, Liljeqvist ML, Siika A, Jin H, Winski G, van Gorp RH, Karlöf E, Lengquist M, Buckler AJ, et al. Proteoglycan 4 Modulates Osteogenic Smooth Muscle Cell Differentiation during Vascular Remodeling and Intimal Calcification. Cells. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adelstein RS, Sellers JR. Effects of calcium on vascular smooth muscle contraction. Am. J. Cardiol. 1987;59:4B–10B. [DOI] [PubMed] [Google Scholar]
- 74.Waugh WH. Role of calcium in contractile excitation of vascular smooth muscle by epinephrine and potassium. Circ. Res. 1962;11:927–940. [DOI] [PubMed] [Google Scholar]
- 75.Huang H, Sun Z, Hill MA, Meininger GA. A calcium mediated mechanism coordinating vascular smooth muscle cell adhesion during kcl activation. Front. Physiol. 2018;9:1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen X, Hu L, Li Z, Wang L, Pang X, Wen C-Y, Tang B. Extracellular calcium ion concentration regulates chondrocyte elastic modulus and adhesion behavior. Int. J. Mol. Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ. Res. 2011;109:e1–12. [DOI] [PubMed] [Google Scholar]
- 78.Choe N, Kwon D-H, Ryu J, Shin S, Cho HJ, Joung H, Eom GH, Ahn Y, Park WJ, Nam K-I, et al. miR-27a-3p Targets ATF3 to Reduce Calcium Deposition in Vascular Smooth Muscle Cells. Mol. Ther. Nucleic Acids. 2020;22:627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Örd T, Õunap K, Stolze LK, Aherrahrou R, Nurminen V, Toropainen A, Selvarajan I, Lönnberg T, Aavik E, Ylä-Herttuala S, et al. Single-Cell Epigenomics and Functional Fine-Mapping of Atherosclerosis GWAS Loci. Circ. Res. 2021;129:240–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao Q, Wirka R, Nguyen T, Nagao M, Cheng P, Miller CL, Kim JB, Pjanic M, Quertermous T. TCF21 and AP-1 interact through epigenetic modifications to regulate coronary artery disease gene expression. Genome Med. 2019;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nagao M, Lyu Q, Zhao Q, Wirka RC, Bagga J, Nguyen T, Cheng P, Kim JB, Pjanic M, Miano JM, et al. Coronary Disease-Associated Gene TCF21 Inhibits Smooth Muscle Cell Differentiation by Blocking the Myocardin-Serum Response Factor Pathway. Circ. Res. 2020;126:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu X, Li YI, Pritchard JK. Trans effects on gene expression can drive omnigenic inheritance. Cell. 2019;177:1022–1034.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell. 2017;169:1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, Yang DY, Trignano SB, Liu W, Shi J, et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation. 2020;142:2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kumra H, Sabatier L, Hassan A, Sakai T, Mosher DF, Brinckmann J, Reinhardt DP. Roles of fibronectin isoforms in neonatal vascular development and matrix integrity. PLoS Biol. 2018;16:e2004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shi F, Long X, Hendershot A, Miano JM, Sottile J. Fibronectin matrix polymerization regulates smooth muscle cell phenotype through a Rac1 dependent mechanism. PLoS ONE. 2014;9:e94988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Greenbaum J, Su K-J, Zhang X, Liu Y, Liu A, Zhao L-J, Luo Z, Tian Q, Shen H, Deng H-W. A multi-ethnic whole genome sequencing study to identify novel loci for bone mineral density. Hum. Mol. Genet. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cowling BS, McGrath MJ, Nguyen M-A, Cottle DL, Kee AJ, Brown S, Schessl J, Zou Y, Joya J, Bönnemann CG, et al. Identification of FHL1 as a regulator of skeletal muscle mass: implications for human myopathy. J. Cell Biol. 2008;183:1033–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van der Pijl RJ, Domenighetti AA, Sheikh F, Ehler E, Ottenheijm CAC, Lange S. The titin N2B and N2A regions: biomechanical and metabolic signaling hubs in cross-striated muscles. Biophys. Rev. 2021;13:653–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun X, Phua DYZ, Axiotakis L, Smith MA, Blankman E, Gong R, Cail RC, Espinosa de Los Reyes S, Beckerle MC, Waterman CM, et al. Mechanosensing through Direct Binding of Tensed F-Actin by LIM Domains. Dev. Cell. 2020;55:468–482.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Coghill ID, Brown S, Cottle DL, McGrath MJ, Robinson PA, Nandurkar HH, Dyson JM, Mitchell CA. FHL3 is an actin-binding protein that regulates alpha-actinin-mediated actin bundling: FHL3 localizes to actin stress fibers and enhances cell spreading and stress fiber disassembly. J. Biol. Chem. 2003;278:24139–24152. [DOI] [PubMed] [Google Scholar]
- 92.Samson T, Smyth N, Janetzky S, Wendler O, Müller JM, Schüle R, von der Mark H, von der Mark K, Wixler V. The LIM-only proteins FHL2 and FHL3 interact with alpha- and beta-subunits of the muscle alpha7beta1 integrin receptor. J. Biol. Chem. 2004;279:28641–28652. [DOI] [PubMed] [Google Scholar]
- 93.Nakazawa N, Sathe AR, Shivashankar GV, Sheetz MP. Matrix mechanics controls FHL2 movement to the nucleus to activate p21 expression. Proc Natl Acad Sci USA. 2016;113:E6813–E6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu B, Pjanic M, Wang T, Nguyen T, Gloudemans M, Rao A, Castano VG, Nurnberg S, Rader DJ, Elwyn S, et al. Genetic regulatory mechanisms of smooth muscle cells map to coronary artery disease risk loci. Am. J. Hum. Genet. 2018;103:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ottolini M, Daneva Z, Chen Y-L, Cope EL, Kasetti RB, Zode GS, Sonkusare SK. Mechanisms underlying selective coupling of endothelial Ca2+ signals with eNOS vs. IK/SK channels in systemic and pulmonary arteries. J Physiol (Lond). 2020;598:3577–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Artamonov MV, Sonkusare SK, Good ME, Momotani K, Eto M, Isakson BE, Le TH, Cope EL, Derewenda ZS, Derewenda U, et al. RSK2 contributes to myogenic vasoconstriction of resistance arteries by activating smooth muscle myosin and the Na+/H+ exchanger. Sci. Signal. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys. J. 2000;78:2655–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Daneva Z, Ottolini M, Chen YL, Klimentova E, Kuppusamy M, Shah SA, Minshall RD, Seye CI, Laubach VE, Isakson BE, et al. Endothelial pannexin 1-TRPV4 channel signaling lowers pulmonary arterial pressure in mice. eLife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci. STKE. 2004;2004:pl5. [DOI] [PubMed] [Google Scholar]
- 100.Woodruff ML, Sampath AP, Matthews HR, Krasnoperova NV, Lem J, Fain GL. Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J Physiol (Lond). 2002;542:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021;1:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). 2021;2:100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Janssens D CUT&RUN: Targeted in situ genome-wide profiling with high efficiency for low cell numbers. 2019; [DOI] [PubMed]
- 108.Yu F, Sankaran VG, Yuan G-C. CUT&RUNTools 2.0: a pipeline for single-cell and bulk-level CUT&RUN and CUT&Tag data analysis. Bioinformatics. 2021;38:252–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Picard Tools - By Broad Institute [Internet]. [cited 2022 Feb 4];Available from: https://broadinstitute.github.io/picard/
- 112.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Koplev S, Seldin M, Sukhavasi K, Ermel R, Pang S, Zeng L, Bankier S, Di Narzo A, Cheng H, Meda V, et al. A mechanistic framework for cardiometabolic and coronary artery diseases. Nat. Cardiovasc. Res. 2022;1:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, Bumgarner RE, Schadt EE. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008;40:854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed CUT&RUN and RNA-seq datasets are made available on the Gene Expression Omnibus (GEO) database (accession: GSE201572). A more detailed list of publicly available datasets used in the study is provided in the Online Supplemental Material.