

Abstract
Loss of nuclear TDP-43 is a hallmark of neurodegeneration in TDP-43 proteinopathies including ALS and frontotemporal dementia (FTD). TDP-43 mislocalization results in cryptic splicing and polyadenylation of pre-mRNAs (pre-mRNAs) encoding stathmin-2 (also known as SCG10), a protein required for axonal regeneration. Here we found that TDP-43 binding to a GU-rich region sterically blocked recognition of the cryptic 3’ splice site in STMN2 pre-mRNA. Targeting dCasRx or antisense oligonucleotides (ASOs) suppressed cryptic splicing, which restored axonal regeneration and stathmin-2 dependent lysosome trafficking in TDP-43 deficient human motor neurons. In mice gene-edited to contain human STMN2 cryptic splice/polyadenylation sequences, ASO injection into cerebral spinal fluid successfully corrected Stmn2 pre-mRNA misprocessing and restored stathmin-2 levels independent of TDP-43 binding.
One-Sentence Summary:
Rescue of stathmin-2 in human iPS-derived motor neurons and mouse brain by blocking cryptic splicing.
In the human nervous system, the ability to maintain proper RNA metabolism is thought to decline during aging (1). Disruption of RNA metabolism is a common feature of many human neurodegenerative disorders, including the fatal paralytic disease amyotrophic lateral sclerosis (ALS) and the two most common dementias - Alzheimer’s disease (AD) and frontotemporal dementia (FTD) (1–5). While the exact gene expression profiles and affected neuronal populations vary among the dementias and other neurodegenerative disorders, there is growing evidence supporting common molecular mechanisms (6, 7).
TDP-43 proteinopathy describes a set of neurological disorders that are characterized by mislocalization of the RNA-binding protein TDP-43 (encoded by the TARDBP gene). TDP-43 relocates from its typically nuclear location and accumulates in the cytoplasm of affected neurons in the form of aggregates. TDP-43 mislocalization and aggregation is found in 97% of ALS patients, about half of FTD patients, and 30–50% of AD patients (8–11). TDP-43 pathology has also been reported in a growing number of brain disorders (2), including Huntington’s disease (12), Perry syndrome (13), and chronic traumatic encephalopathy (CTE) (14). Recently, a subset of aged Alzheimer’s patients has been re-classified as limbic-predominant age-related TDP-43 encephalopathy (LATE disease) (15) because their post-mortem brain samples show aberrant TDP-43 instead of the expected amyloid beta (Aβ). While cytoplasmic accumulation of TDP-43 has been reported in ALS and FTD, nuclear clearance of TDP-43 is often observed even without apparent aggregation(s) (16, 17). Loss of nuclear TDP-43 affects expression and processing of multiple mRNA targets across different cell types and tissues (18–24).
TDP-43 closely regulates maturation of the pre-mRNA encoding stathmin-2 (also known as SCG10, encoded by the STMN2 gene) (25, 26). Stathmin-2 is a neuronally enriched protein that plays a crucial role in axonal outgrowth during development (27) and regeneration (25, 26, 28). Developmental deletion of mouse Stmn2 causes motor deficits with denervation of neuromuscular junctions (29, 30). Among the four members of the stathmin gene family, stathmin-2 has the highest expression in mouse and human motor neurons (25). Indeed, STMN2 is among the top twenty most enriched mRNAs in the ALS-vulnerable motor neurons of the anterior grey column of the spinal cord (25, 31). In postmortem FTD and ALS patient brain and spinal cord tissues, loss of nuclear TDP-43 results in the utilization of cryptic splice (25, 26, 32) and polyadenylation sites (25) within the first intron of the STMN2 pre-mRNA (25, 26). This leads to inclusion of a cryptic exon 2a and the production of an mRNA encoding only a truncated open reading frame (17 codons) (25), and suppression of the full-length 179 amino acid stathmin-2 protein (25, 26, 32).
Stathmin-2 is a tubulin-binding protein that is thought to affect microtubule dynamic instability (33, 34), although its mechanism of action in axons and axonal growth cones is unclear (35–38). Lowering stathmin-2 in induced pluripotent stem cell (iPSC) derived human motor neurons inhibits the regenerative capacity of injured axons (25, 26). Diminished regeneration capacity of injured motor neurons when TDP-43 function is reduced can be rescued by restoring stathmin-2 levels (25), which suggests that restoration of stathmin-2 expression in TDP-43 proteinopathies may provide a therapeutic strategy.
Here, we determined the regulatory elements through which TDP-43 regulates STMN2 pre-mRNA processing and identified steric binding antisense oligonucleotides (ASOs) capable of restoring normal stathmin-2 protein and RNA levels when administered within a mammalian nervous system.
A GU-motif confers TDP-43 binding-dependent maturation of stathmin-2 pre-mRNA
A previous analysis of datasets for RNAs bound by TDP-43 (using UV individual nucleotide resolution crosslinking, immunoprecipitation, and sequencing [iCLIP]) identified TDP-43 binding within the human (18), but not murine (19), STMN2 pre-mRNA, approximately 5.6kb from the start (5’ end) of the first STMN2 intron (25). The iCLIP-defined TDP-43 binding site is positioned between the cryptic splice and polyadenylation sites of the recently identified exon 2a (25), in a region containing a 24 base GU-rich segment comprised of three closely spaced GUGUGU hexamers, which are the consensus motif for TDP-43 binding (19, 39). We used CRISPR-Cas9 genome engineering to test whether TDP-43 binding at this locus could prevent misprocessing by blocking recognition of the cryptic RNA elements. We replaced the 24 base domain encoding the GU-motif sequence with a 19 base segment encoding the bacteriophage MS2 aptamer sequence, an RNA stem-loop structure which can be bound with high affinity [Kd = 2 ×10−10M] (40) by the MS2 coat protein (MCP) (Fig. 1A–C). In human neuronal SH-SY5Y cells carrying the MS2-binding site replacement in one STMN2 allele (Fig. S1A), steady-state full-length STMN2 mRNAs were reduced by 50%. This was accompanied by appearance of an abundant, truncated STMN2 RNA produced by use of the cryptic splice and polyadenylation sites, despite sustained TDP-43 levels (Fig. 1B). Thus, the GU motif bound by TDP-43 was required to suppress utilization of cryptic sites within the STMN2 pre-mRNA.
Figure 1. Human GU-motif removal and MS2-directed tethering demonstrate TDP-43 binding locus, while cryptic site mutations identify TDP-43 dependent misprocessing requiring cryptic splice acceptor.
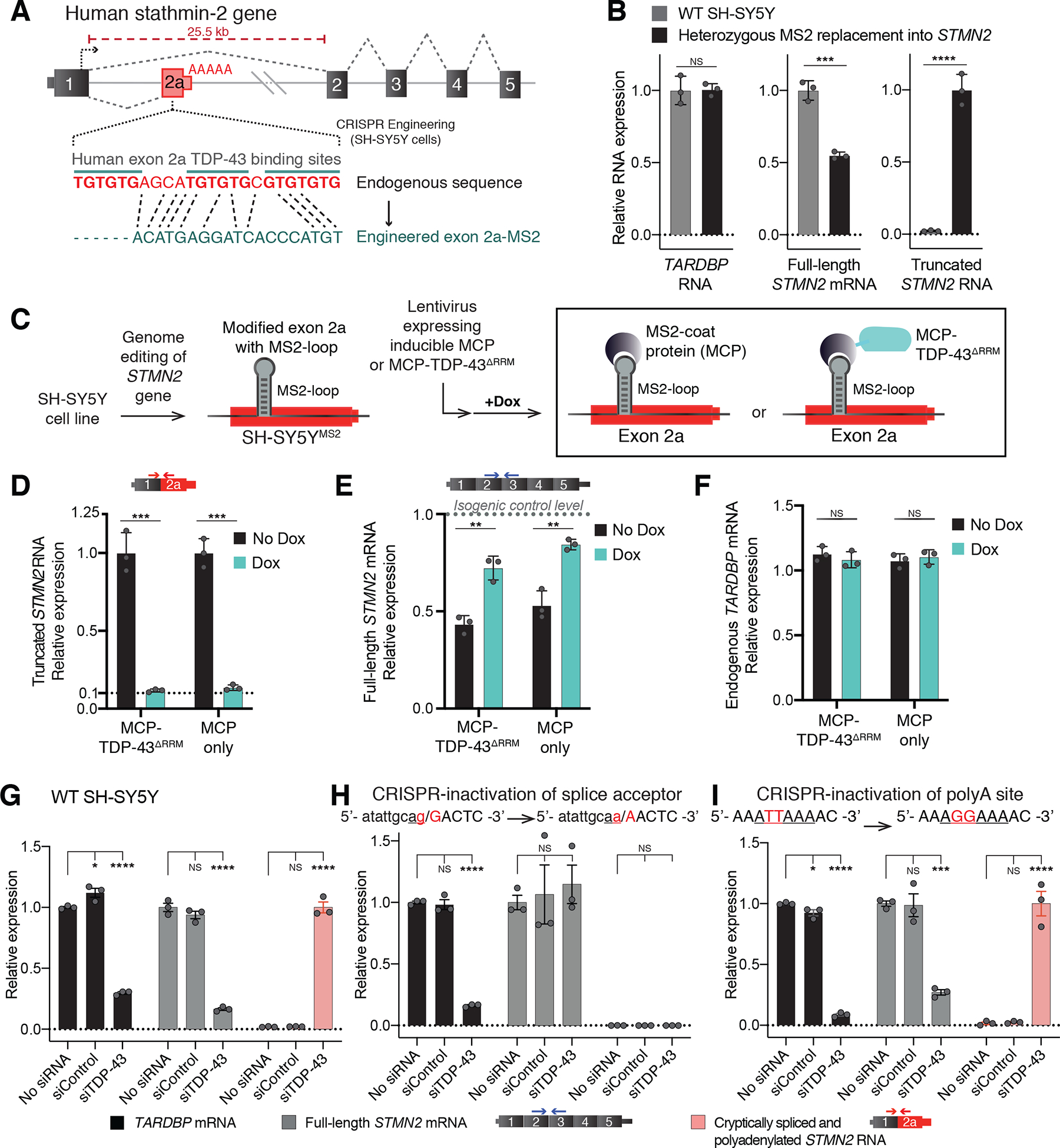
(A) Schematic of CRISPR-engineering strategy for conversion of the GU binding motif in exon 2a into an MS2 aptamer sequence in one STMN2 allele of diploid SH-SY5Y neuroblastoma cells. (B) qRT-PCR demonstrating that SH-SY5Y cells carrying heterozygous GU to MS2 edit misprocess STMN2 RNA, leading to 50% loss of stathmin-2 encoding mRNAs compared with wildtype cells and accumulation of truncated RNA. (C) Schematic depicting MS2:MCP directed strategy to direct MCP-tethered proteins to the normal TDP-43 binding locus. (D-F) qRT-PCR measurement of (D) truncated STMN2, (E) full-length STMN2 mRNA, or (F) endogenous TARDBP mRNA levels with and without induction of MCP-fusion protein expression in SH-SY5Y cells carrying heterozygous MS2 aptamer insertion. (G-I) qRT-PCR measured expression of TARDBP, full-length STMN2 mRNA and truncated STMN2 RNAs 96 hours after siRNA treatment with a control siRNA pool or a pool targeting TARDBP in (G) wildtype SH-SY5Y cells, (H) SH-SY5Y cells harboring homozygous mutation of the human exon 2a 3’ splice acceptor site and (I) SH-SY5Y cells harboring a homozygous mutation of the human exon 2a premature polyadenylation signal to the murine sequence AGGAAA. For all qPCR analysis individual data points are independently treated wells of cells. Error bars are SEM. Statistical significance was determined by 2-tailed Student’s T-test (B-F), or 1-way ANOVA with Dunnett correction (G-I). ****, p <0.0001; ***, p < 0.001; **, p < 0.01; *, p <0.05.
TDP-43 binding sterically blocks cryptic splicing and polyadenylation of STMN2
TDP-43 binding to the GU motifs could act directly to prevent use of cryptic processing sites in the STMN2 pre-mRNA by sterically blocking accessibility to those sites by additional splicing (41–44) or polyadenylation (45–48) factors. Alternatively TDP-43 binding to the GU motifs could act indirectly by promoting recruitment and assembly of additional RNA processing protein complexes whose presence occludes access of 3’ splice site or transcription termination and polyadenylation complexes. To distinguish between direct and indirect models for suppressing cryptic site utilization in the STMN2 pre-mRNA, we transduced our SH-SY5Y cells, in which the MS2 aptamer was inserted into one STMN2 allele, with a lentivirus carrying a doxycycline-inducible gene encoding a 29 kDa high-affinity MS2 coat protein dimer fused to an RNA-binding-incompetent human TDP-43 variant (MCP-TDP-43ΔRRM) missing both RNA-recognition motifs (ΔRRM). This allowed us to direct single-molecule binding of the fusion protein to the STMN2-embedded MS2 RNA aptamer, but not to TDP-43 binding sites on other endogenous pre-mRNAs (Fig. 1C). Within 24h of MCP-TDP-43ΔRRM induction, cryptically spliced and polyadenylated stathmin-2 mRNAs were reduced by 90% (Fig. 1D, left). Next, to determine the effect of simple steric binding at the replaced GU locus, we induced expression of MCP alone (without TDP-43 fusion) (Fig. 1C). Targeting exon 2a by MCP binding at the MS2 sequence similarly reduced accumulation of truncated STMN2 RNA by 90% (Fig. 1D, right). Correspondingly, full-length stathmin-2 mRNA levels were largely restored by binding of either MCP variant (Fig. 1E), while endogenous TARDBP mRNA levels remained unaltered (Fig. 1F). Thus, TDP-43 binding to the GU-domain of the STMN2 pre-mRNA blocked cryptic splicing and polyadenylation through a simple steric inhibition mechanism.
Genomic disruption of exon 2a cryptic splice site protects against misprocessing
We next tested whether recognition of either the cryptic splice or polyadenylation sites (which lie 203 bases from each other within the STMN2 pre-mRNA) were responsible for initiation of STMN2 pre-mRNA misprocessing when TDP-43 levels fall. Genome editing in SH-SY5Y cells was used to eliminate either the cryptic polyadenylation motif or the 3’ splice acceptor site (Fig. S1 B,C). As previously reported (25), siRNA mediated reduction of TDP-43 to 30% of its initial level in the parental cells resulted in an even larger (85%) reduction in stathmin-2-encoding mRNAs (Fig. 1G). In cells homozygously CRISPR-edited to convert the cryptic 3’ splice acceptor sequence from a functional AG/GA to a non-functional AA/AA sequence, siRNA reduction of TDP-43 resulted in no significant change in full-length STMN2 mRNA and complete abrogation of both cryptic splicing and polyadenylation (Fig. 1H).
By contrast, elimination of the identified cryptic polyadenylation signal was not protective against STMN2 pre-mRNA misprocessing. In cells homozygously edited to carry a disrupted cryptic polyadenylation signal (after converting the initial ATTAAA sequence to a non-functional AGGAAA (49)) (Fig. 1I), reduction in TDP-43 sharply reduced full-length STMN2 RNAs (Fig 1I, grey bars) and generated RNAs with exon 1 ligated onto exon 2a (Fig 1I, pink bars). Moreover, despite inactivation of the most proximal site of exon 2a polyadenylation, cryptically spliced STMN2 RNAs remained polyadenylated. A search of putative cryptic polyadenylation sites downstream within the intron 1 sequence identified 39 occurrences of the canonical AATAAA signal for polyadenylation and 22 additional instances of the ATTAAA variant, one or more of which must become new cryptic polyadenylation sites when the first ATTAAA was inactivated.
Cryptic human STMN2 splice and pA sites induce misprocessing of murine Stmn2
Despite 93% conservation in mRNA coding sequence and 100% conservation at the amino acid level, the murine Stmn2 gene has little conservation of sequence within the region of its first intron corresponding to human exon 2a, including absence of predicted or experimentally validated TDP-43 binding site(s) (19). Correspondingly, cryptic Stmn2 splicing and polyadenylation were not present in the mouse neuron-like N2A cell line after TDP-43 level was reduced by siRNA and full-length Stmn2 mRNA level remained unchanged (Fig. 2A).
Figure 2. Humanization of murine Stmn2 gene in N2A cells demonstrates human-specific inhibition of altered STMN2 pre-mRNA processing via non-conserved TDP-43 binding sites.
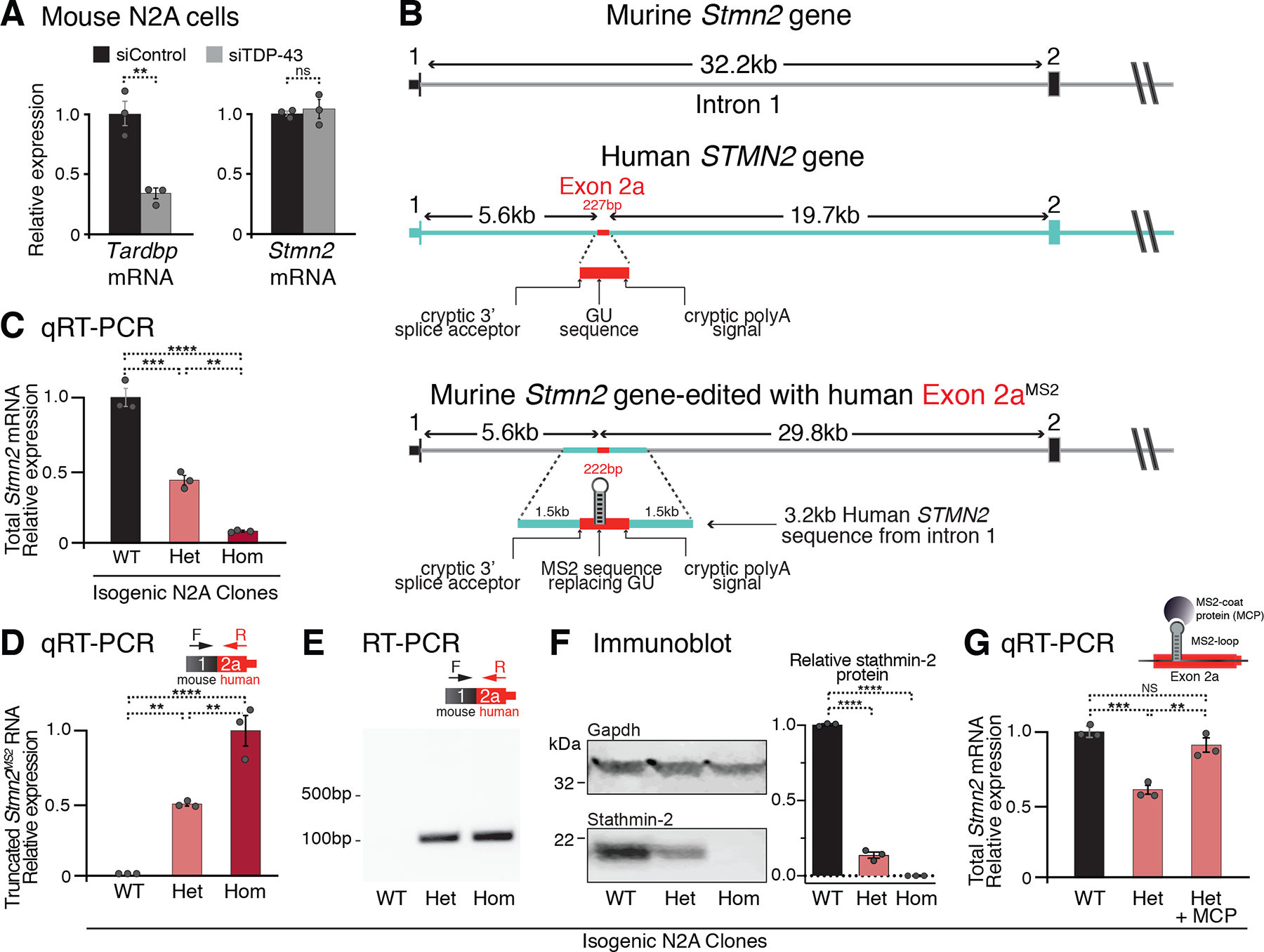
(A) qRT-PCR demonstrating that depletion of TDP-43 in wildtype murine N2A cells does not affect Stmn2 expression levels. (B) Schematics of the human and mouse Stmn2 genomic regions before and after genome-editing to insert a 3kb human fragment of STMN2 intron 1 into murine N2A cells. (C) qRT-PCR demonstrating dose-dependent reduction of N2A Stmn2 mRNA level that correlates with the number of alleles carrying human STMN2 gene fragment. (D) qRT-PCR and (E) RT-PCR confirming expression of chimeric Stmn2 truncated RNA with murine exon 1 spliced to human exon 2a, in N2A clones carrying the human STMN2 gene fragment. (F) Immunoblotting confirming reduced expression of full-length stathmin-2 protein levels in N2A clones that carry humanized Stmn2 gene fragment. (G) qRT-PCR showing restoration of normal Stmn2 pre-mRNA processing in N2A cells upon doxycycline induction of MCP expression. For all qPCR analyses, each data point represents an independently treated well of N2A cells. Error bars are SEM. Statistical significance was determined by 2-tailed Student’s T-test (A), or 1-way ANOVA with Dunnett (F) or Tukey (C, D, G) correction. ****, p <0.0001; ***, p < 0.001; **, p < 0.01; *, p <0.05.
We genome engineered one or both Stmn2 alleles in these mouse cells by insertion of a 3.2kb segment containing the exon 2a sequence of human STMN2 intron 1 after a 12 nucleotide modification to convert the GU-rich TDP-43 binding motif into an MS2 aptamer (Fig. 2B). The resultant edited murine Stmn2 gene contained human exon 2a sequence positioned as in the human gene, 5.6kb downstream of the 5’ splice site (5’ss) of intron 1. Heterozygous or homozygous insertions of the cryptic splicing and polyadenylation sites from human STMN2 resulted, respectively, in reduction or nearly complete elimination of mature murine Stmn2 mRNA (Fig. 2C), driven by constitutive use of the human cryptic 3’ splice and polyadenylation sites, leading to production of a chimeric murine-human truncated Stmn2 RNA (Fig. 2D,E). Murine stathmin-2 protein was accordingly reduced or eliminated in heterozygous and homozygous clones, respectively (Fig. 2F). Lentiviral-mediated expression of MCP restored levels of full-length murine Stmn2 mRNA (Fig. 2G). Thus, the cryptic human STMN2 3’ splice acceptor and polyadenylation elements could drive misprocessing of murine Stmn2 pre-mRNAs when TDP-43 was not bound.
Targeting of dCasRx blocks misprocessing of STMN2 pre-mRNA
The CRISPR effector RfxCas13d (CasRx) can be targeted to a specific RNA sequence with an appropriate ≥21 base guide RNA (50). The “nuclease-dead” variant of which (dCasRx) retains RNA-binding but not enzymatic cleavage activity and can be directed to pre-mRNA molecules to affect alternative splicing (50). We applied an initial test in human neuronal cells of the therapeutic potential in TDP-43 proteinopathies of dCasRx-directed targeting to sterically block use of cryptic splice and polyadenylation sites within the STMN2 pre-mRNA. This possibility is especially attractive for dCasRx given its small size and reported versatility in RNA binding that is independent of a protospacer adjacent motif (PAM) targeting requirement (50). Eleven guide RNAs were designed with sequences that tiled across a 353 base region spanning the cryptic splice and polyadenylation sites in the human STMN2 pre-mRNA (Fig. S2A). SH-SY5Y cells were genome edited to carry an ALS/FTD-linked TDP-43N352S/N352S mutation within both endogenous TARDBP alleles (25), resulting in a partial loss of TDP-43 function, production of a truncated STMN2 RNA (Fig. S2B,C), and a corresponding 50% reduction of the full-length stathmin-2 encoding mRNAs (Fig. S2B,D).
These cells were then transduced with a lentivirus encoding dCasRx and each of the 11 gRNAs from our tiling array. Four guides with binding sites covering a) the cryptic 3’ splice acceptor site (gRNA 3), b) overlapping or c) directly adjacent to the endogenous TDP-43 binding site (gRNA 6 and 7), and d) covering the cryptic polyadenylation signal (gRNA 10) (Fig. S2A) generated marked reduction of up to 70% in the levels of cryptically spliced, polyadenylated STMN2 RNA relative to a control TDP43N352S/N352S line expressing dCasRx without a gRNA (Fig. S2B,C). gRNA-mediated targeting of dCasRx to partly overlap the authentic TDP-43 binding site (gRNA 6) showed up to 40% increase in levels of full-length stathmin-2 encoding mRNAs, as did direct targeting of cryptic splice acceptor or polyadenylation signals (Fig. S2B,D). Guides directing binding to an intermediate locus upstream to the alternative polyadenylation signal produced further depletion of normal STMN2 levels in these cells (Fig. S2B, gRNA 8 and 9), perhaps displacing an additional RNA-binding factor with a role in pre-mRNA maturation. Stathmin-2 protein expression was restored proportionally to the mRNA levels, with gRNA 6 mediating 30% protein increase relative to no guide control (Fig. S2E,F). Thus, suppression of cryptic splicing and polyadenylation and partial restoration of STMN2 expression in cells with diminished TDP-43 activity was achieved by dCasRx-mediated steric binding on the exon 2a sequence.
Antisense oligonucleotide suppression of STMN2 pre-mRNA misprocessing
Antisense oligonucleotides (ASOs) that have been chemically modified to correct RNA processing defects without recruiting RNase H to catalyze RNA degradation have become an attractive therapeutic approach for neurodegenerative diseases (51, 52). Their usage to alter pre-mRNA splicing of the survival of motor neuron 2 (SMN2) gene (51) is an approved standard of care for spinal muscular atrophy (SMA) in the US and Europe. Correspondingly, 250 potential rescue ASOs (rASOs) were synthesized to target across the region spanning cryptic splice and polyadenylation sites in the STMN2 pre-mRNA (Fig. S3A) and evaluated for their ability to suppress cryptic splicing/polyadenylation of STMN2 pre-mRNAs. Using SH-SY5Y cells harboring a homozygous TDP-43N352S/N352S mutation, several rASOs were identified that within 24 hours produced dose-dependent restoration of full-length stathmin-2-encoding mRNAs to a level comparable to that in wildtype cells (Fig. S3B) and an accompanying reduction in levels of the truncated stathmin-2 RNA variant (Fig. S3C). Correspondingly, rASOs-mediated blockage of cryptic splicing and polyadenylation resulted in stathmin-2 protein restoration as early as 48 hours post-transfection (Fig. S3D,E).
rASOs restore axonal regeneration capacity in TDP-43 deficient motor neurons
To determine the therapeutic potential of rASOs in the neuronal population most affected in ALS, we generated motor neurons from induced pluripotent stem cells, reduced TDP-43 levels by addition of an ASO targeting TARDBP RNAs (25), and assayed for restoration of STMN2 mRNA and protein levels after addition to cell culture media of stathmin-2 rASOs (Fig. 3A). Reduction in TDP-43 resulted in accumulation of truncated STMN2 RNAs and suppression of full-length STMN2 mRNAs to 30% of their initial level (Fig. 3B,C). In the absence of a rASO, stathmin-2 protein levels dropped to an undetectable level in TDP-43 depleted neurons (Fig. 3D–F). rASO treatment increased STMN2 mRNAs in a dose-dependent manner in TDP-43-depleted motor neurons, with rASO-5 elevating STMN2 mRNA from 30% to an average of 86% of its normal level (Fig. 3B), while the truncated STMN2 RNA accumulation was almost completely reversed (Fig. 3C). Encouragingly, stathmin-2 protein level, which was below the detection limit in motor neurons with ASO-depletion of TDP-43, was rescued by rASOs (Fig. 3D), with rASO-5 returning stathmin-2 protein to a level nearly indistinguishable from that in control neurons with normal TDP-43 levels (Fig. 3E,F).
Figure 3. Dose dependent suppression of STMN2 cryptic splicing and polyadenylation by rASOs in iPSC-derived motor neurons with TDP-43 depletion.
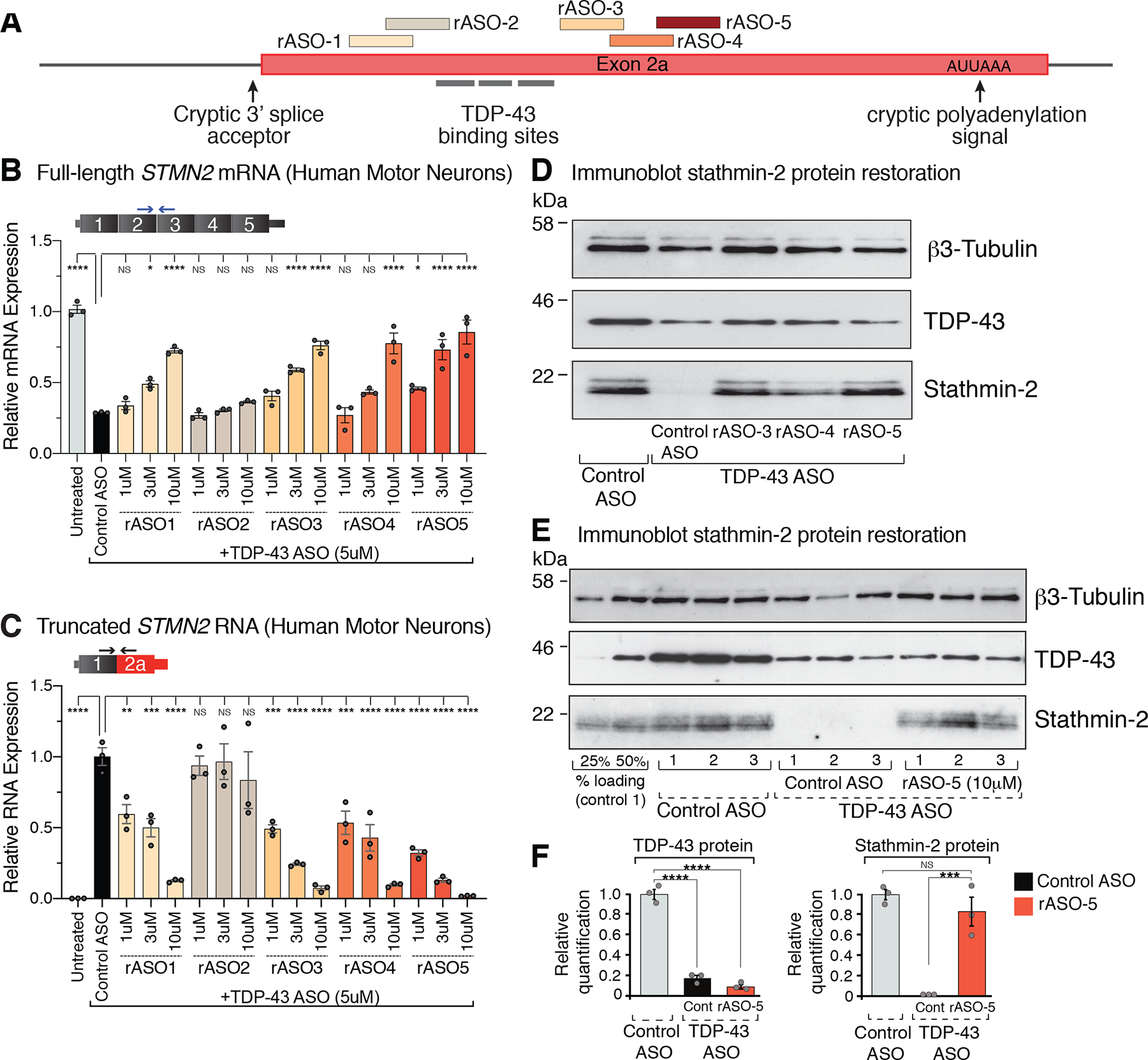
(A) Schematic representation of the exon 2a region of human STMN2 gene with TDP-43 binding sites and selected rASOs that show splice-modifying activity. (B) STMN2 mRNA restoration analyzed by qRT-PCR after treatment with 5 representative rASOs in iPSC-derived motor neurons depleted of TDP-43. Expression of TFRC mRNA was used as endogenous control. (C) qRT-PCR analysis of truncated STMN2 RNA levels after treatment with 5 representative rASOs in iPSC-derived motor neurons depleted of TDP-43. Expression of TFRC mRNA was used as endogenous control. (D) Immunoblot showing TDP-43 and stathmin-2 protein levels in iPS motor neurons treated with control or TDP-43 suppressing ASOs, subsequently treated with control or splice-rescuing rASOs 3, 4, or 5 to restore stathmin-2. Beta3 Tubulin used as an endogenous control. (E) Immunoblot showing TDP-43 and stathmin-2 levels in motor neurons depleted of TDP-43 with a targeted ASO and subsequently treated with control or rescue ASO-5. Linearity of antibody detection with 25% and 50% control treated motor neuron lysate loading controls included at the far-left side of the blot. Immunoblot quantified in (F). Each lane and data point represents an independently differentiated and ASO treated neuronal culture. Error bars are SEM. Statistical significance was determined by 1-way ANOVA with Dunnett correction. ****, p <0.0001; ***, p < 0.001; **, p < 0.01; *, p <0.05.
To determine the functional consequences of rASO-mediated stathmin-2 restoration on the regenerative capacity of motor axons, we utilized an iPSC-motor neuron axotomy and regrowth assay in microfluidic chambers (Fig. 4A) (25). Addition into the somatic compartment of an ASO targeting TDP-43-encoding mRNAs for RNase H-dependent degradation for 20 days led to loss of detectable stathmin-2 protein (Fig. 4B, green, middle panel) accompanied by inhibition of axonal regeneration capacity after mechanically induced axotomy (Fig. 4B,C, middle panels), in contrast to motor neurons treated with non-targeting control ASOs (Fig. 4B,C, left and top panels, respectively). Importance of stathmin-2 in axonal regeneration is established by direct reduction of stathmin-2 (using a STMN2 targeting ASO): loss of stathmin-2 alone is sufficient to almost eliminate axonal regeneration after axotomy (25).
Figure 4. Restoration of axonal regeneration capacity using rASOs that rescue stathmin-2 levels in iPSC-derived motor neurons with TDP-43 depletion.
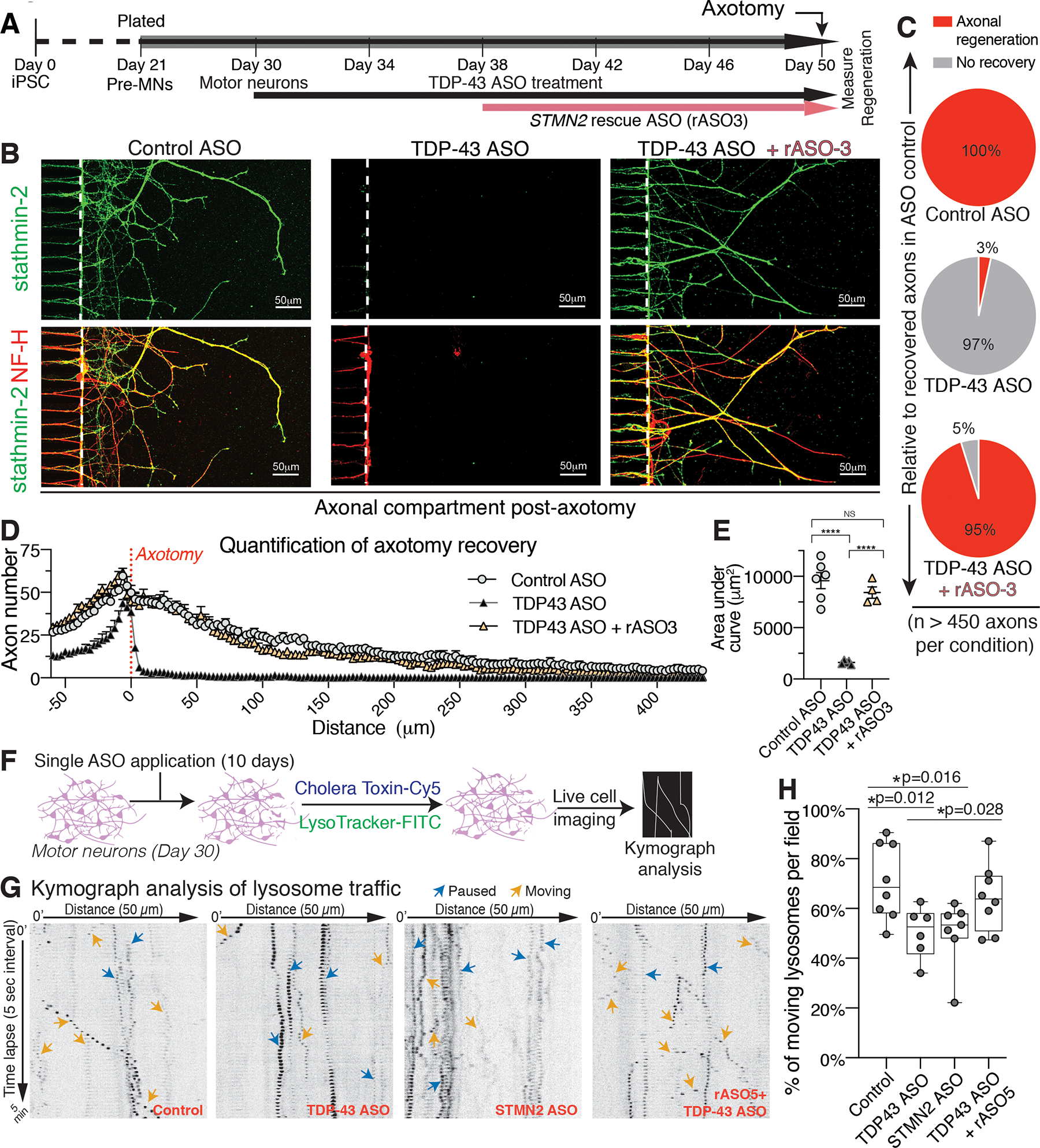
(A) Timeline of iPSC-derived motor neuron maturation, ASO/rASO treatment and axotomy. (B) Immunofluorescence images of microgrooves (left of dotted line) and distal compartments (right), 36 hours post-axotomy. Axonal regeneration and growth cones are observed by immunofluorescence detection of stathmin-2 (green) and NF-H (red) in the terminals of motor neurons. (C-E) Quantification of axonal recovery for at least 450 axons per condition represented as the percentage of recovered axons relative to control ASO treated motor neurons in (C), the overall number of axons per micrometer from the axotomy site plotted in (D), and corresponding area-under the curve in (E). Statistical significance determined by one-way ANOVA with Tukey’s multiple comparison correction. (F) Schematic of ASO treatment and live motor neuron lysosomal tracking and analysis. (G) Representative kymographs of lysosomal transport in axons of ASO-treated motor neurons. (H) Quantification of moving tracked axonal lysosomes after ASO treatment, statistical significance determined by one-way ANOVA indicated, three independently differentiated chambers quantified per condition. Error bars are SEM. ****, p <0.0001; ***, p < 0.001; **, p < 0.01; *, p <0.05.
Free uptake of an rASO added to the somatic compartment 8 days after initiation of ASO-dependent reduction in TDP-43 restored accumulation of axonal stathmin-2 protein levels (Fig. 4B, green, right panel). The ability of axotomized motor axons to regenerate into the distal compartment was also rescued, with nearly identical recovered axon number and density into the distal compartment when compared to control axons (Fig. 4B, right panels, quantified in Fig. 4C–E), despite apparent heterogeneity in stathmin-2 accumulation in individual axons (Fig. 4B). Indeed, analysis of regenerating axons after ASO-mediated reduction in stathmin-2 revealed that a residual level of stathmin-2 protein 25% of the normal level was sufficient to support axonal regeneration after injury in human motor neurons (Fig. S4A–C).
Recognizing that impaired axonal transport has been demonstrated in several ALS models (53–56), we next applied live-cell imaging to track organelle movement within cultured iPSC-derived motor neurons (Fig. 4F). Movement of lysosomes in iPSC-derived motor neurons treated with ASOs to degrade STMN2 or TARDBP mRNAs showed a decrease in actively moving lysosomes (reduction from 71% in axons exposed to control ASOs to 50% after stathmin-2 or TDP-43 suppression - Fig. 4G,H). rASO treatment to restore stathmin-2 levels significantly reversed the impaired lysosome trafficking phenotype despite sustained TDP-43 suppression, as evident by an increased proportion (to 64%, p=0.028) of lysosomes that were moving (Fig. 4H).
We then utilized electron microcopy imaging to provide ultrastructural analysis of excitatory synapses under conditions of stathmin-2 or TDP-43 suppression. Overall synaptic morphology did not show abnormalities (Fig. S5 A–E), including active zone and post synaptic density (PSD) length, and total amount of synaptic vesicles. However, in TDP-43 and/or stathmin-2 deficient motor neurons, multiple synapses showed an increase of electron dense material in the active zone, in comparison to motor neurons treated with a control ASO (Fig. S5 A–F). rASO treatment to specifically restore stathmin-2 levels in motor neurons, despite continuous TDP-43 suppression, fully restored the proportion of synapses showing normal pre-synaptic electron density to reflect that of control neurons (Fig. S5F, top and bottom bars).
rASO-mediated restoration of Stmn2 pre-mRNA processing in the mammalian CNS
Recognizing that murine Stmn2 pre-mRNA processing is independent of TDP-43 function (Fig. 2A) owing to absence of the regulatory sequences that define human exon 2a, we tested whether humanizing the murine Stmn2 pre-mRNA by introduction of human exon 2a and flanking intronic sequences was sufficient to confer TDP-43 dependency. For this, we generated humanized Stmn2 mice from mouse embryonic stem cells that had been CRISPR/Cas9 genome-edited to replace a 479bp segment of murine Stmn2 intron 1 with a 394bp segment of human STMN2 intron 1 containing exon 2a (227bp) and its flanking regions (75bp upstream and 92bp downstream). Exon 2a was positioned 5.5kb downstream of the 5’ splice site of murine exon 1, a spacing equivalent to its positioning in human STMN2 (Fig. S6A). Mice with heterozygous or homozygous humanized Stmn2 gene alleles were viable and developed normally.
Using primary cortical neurons from heterozygous Stmn2HumExon2a/+ embryos, we confirmed that the presence of human exon 2a and flanking sequences were sufficient to drive altered processing of the humanized Stmn2 pre-mRNA when TDP-43 was depleted (Fig. S6B,C). An 85% ASO-mediated reduction in TDP-43 level (Fig. S6C) triggered nearly complete suppression of full-length murine Stmn2 mRNAs from the humanized allele, as indicated by 50% reduction in total Stmn2 mRNA level in heterozygous cortical neuron cultures (Fig. S6D). Usage of cryptic splice/polyadenylation sites was confirmed by abundant, polyadenylated Stmn2 RNAs containing murine exon 1 spliced to human exon 2a and whose level was inversely proportional to the level of TDP-43 (Fig. S6C,E). Homozygous humanization of Stmn2 in a mouse line developing age-dependent motor neuron disease from expression of a disease-linked mutant TDP-43Q331K without loss of nuclear TDP-43 (57) neither exacerbated development of disease phenotype (Fig. S7A–D) nor induced cryptic Stmn2 pre-mRNA misprocessing (Fig. S7E) and loss of full-length Stmn2 mRNA (Fig. S7F) in brain or spinal cord by 12 months of age. These findings are consistent with misprocessing of the humanized Stmn2 pre-mRNA requiring loss of nuclear TDP-43 function.
ALS/FTD model mice developing cytoplasmic aggregation of TDP-43 from increased expression of wildtype TDP-43 die only days after weaning (58, 59) and do not provide an adequate therapeutic window for ASO-mediated restoration of stathmin-2 levels. To enable in vivo testing of rASOs for their efficacy in restoring stathmin-2 levels within the mammalian nervous system, we generated additional humanized mouse models. Two humanized Stmn2HumΔGU founder lines were generated by CRISPR/Cas9-mediated genome engineering of mouse ES cells to produce mice carrying a 3.2kb portion of human STMN2 intron 1 containing exon 2a (modified to replace the 24 base GU domain that is the TDP-43 binding site with the sequence for a 19-base MS2 aptamer) and inserted into the corresponding locus of the first intron of murine Stmn2 (Fig. 5A).
Figure 5. In vivo ASO-mediated restoration of Stmn2 pre-mRNA processing in a humanized mouse, engineered to constitutively misprocess Stmn2 RNAs from a partly humanized allele.
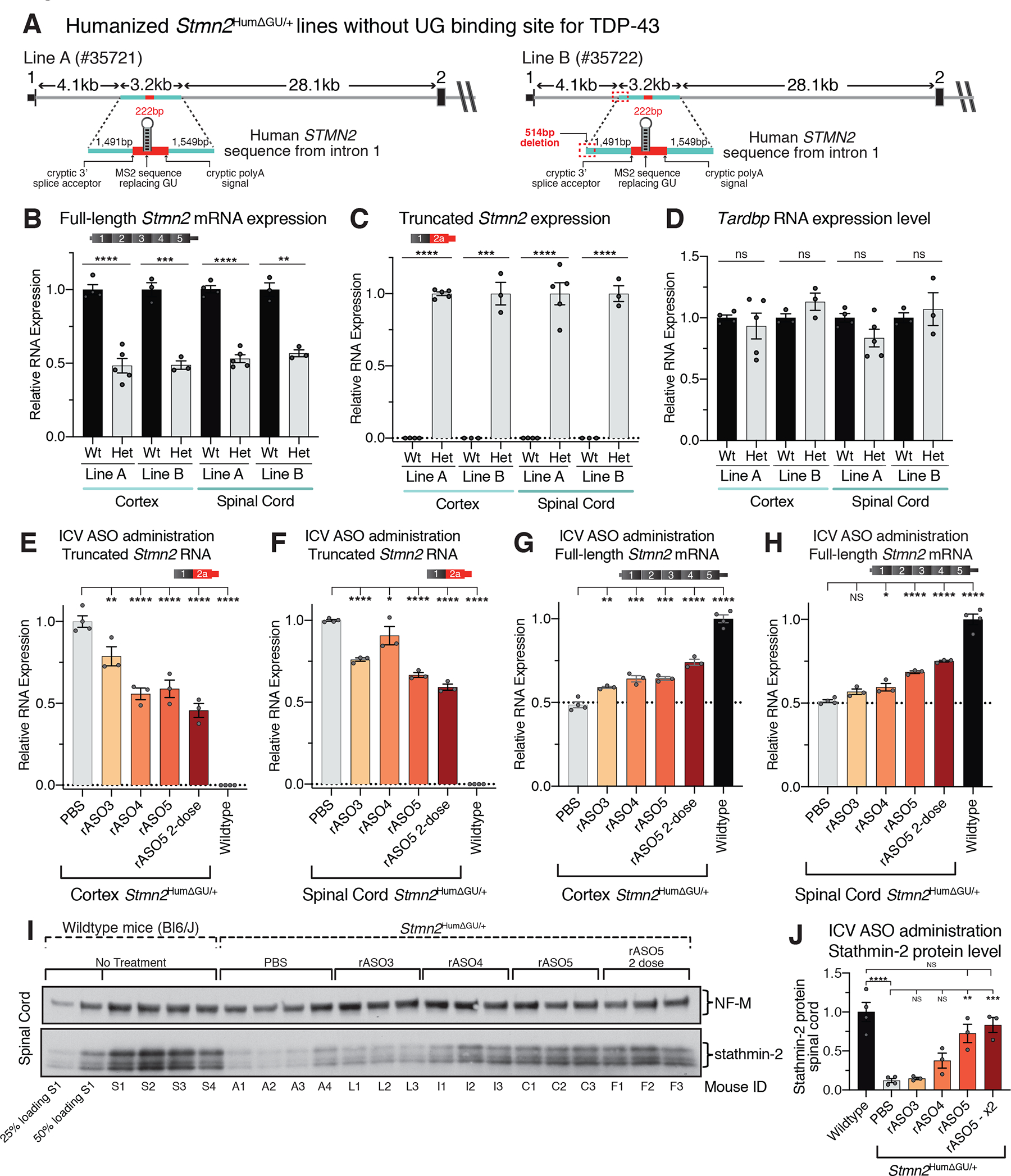
(A) Schematic showing the strategy to produce a Stmn2 human exon 2A knock-in mouse without a TDP-43 GU binding site to drive constitutive misprocessing of the humanized allele, and resulting mouse lines obtained after CRISPR editing. (B) qRT-PCR showing full-length murine Stmn2 mRNA levels were reduced by half in animals heterozygous for the humanized Stmn2HumΔGU/+ allele compared with wildtype littermate controls. (C) qRT-PCR showing that truncated chimeric RNAs consisting of a murine exon 1 fused to a modified human exon 2a were abundantly and specifically expressed in mice heterozygously carrying the modified humanized Stmn2HumΔGU/+ allele. (D) qRT-PCR showing normal murine Tardbp mRNA levels in both heterozygous Stmn2HumΔGU/+ mice and wildtype littermate controls. (E-F) qRT-PCR showing suppression of truncated chimeric RNA accumulation in both brain (E), and spinal cord (F), of mice dosed by ICV injection with rASOs. (G-H) qRT-PCR Sshowing rASO-mediated restoration of full-length murine Stmn2 mRNAs in both brain (G), and spinal cord (H), of mice after ICV injection of rASOs. (I) Immunoblot showing restoration of stathmin-2 protein in the spinal cords of heterozygous Stmn2HumΔGU/+ mice dosed by ICV injection with rASOs, compared with PBS injected animals or wildtype C57 BL/6J mice. 25% and 50% loading of lysates from wildtype mouse S1at the far left, NF-M shown as an endogenous loading control protein. (J) Quantification of relative stathmin-2 protein restoration. Each lane and data point represents an individual mouse. Error bars are SEM. Statistical significance was determined by 2-tailed Student’s T-test (B-D), or 1-way ANOVA with Dunnett (E-H) or Tukey (J) corrections. ****, p <0.0001; ***, p < 0.001; **, p < 0.01; *, p <0.05.
In heterozygotes of both lines, the inability of TDP-43 to bind exon 2aΔGU resulted in constitutive suppression of full-length murine Stmn2 mRNAs to 50% of wildtype levels (Fig. 5B) accompanied by chronic utilization of cryptic splicing and polyadenylation elements encoded by Stmn2HumΔGU pre-mRNAs (Fig. 5C) despite normal TDP-43 levels (Fig. 5D). While Stmn2HumΔGU heterozygous mice developed normally and matured without overt neurological symptoms, there was 80% reduced survival to weaning of Stmn2HumΔGU/HumΔGU homozygotes identified in heterozygous matings, evidence of developmental lethality driven by Stmn2 suppression.
Usage of Stmn2 cryptic splice and polyadenylation sites encoded by the modified human exon 2a was inhibited in cortex and spinal cord after intracerebral ventricular (ICV) injection of any of three rASOs into cerebral spinal fluid of 2-month-old Stmn2HumΔUG/+ mice (Fig. 5E,F). Two weeks after injection, rASO-5 suppressed inclusion of exon 2a by 50% and 35% in cortex and spinal cord, respectively (Fig. 5E,F). The suppression of cryptic processing was accompanied by significant restoration of full-length, stathmin-2 encoding mRNAs after injection of two of the three rASOs (rASO-4 and rASO-5) and was maximized by a second administration of rASO-5, yielding 53% and 45% increases of full-length Stmn2 mRNA production in the cortex and spinal cord and 55% and 40% decreases in truncated Stmn2 RNAs, respectively (Fig. 5E–H). The highest stathmin-2 protein restoration in the spinal cord was achieved in mice ICV-injected twice with rASO-5, resulting in almost 80% of its normal expression level in WT mice (Fig. 5I,J). Moreover, there was apparent, dose-dependent stabilization of stathmin-2 protein (Fig. 5I,J), with the reduced mRNA level in the Stmn2HumΔUG/+ mice yielding an even larger reduction in stathmin-2 protein, evidence supporting a therapeutic threshold for ASO-mediated STMN2 mRNA restoration inducing larger changes in stathmin-2 protein accumulation.
Discussion
Cytoplasmic accumulation coupled with nuclear clearance of the RNA binding protein TDP-43 is found in affected neurons of ~97% of ALS patients (10, 60, 61), which suggests that nearly all ALS-causing mechanisms converge on TDP-43 dysfunction. Nuclear loss of TDP-43 is also a common hallmark in approximately half of patients with FTD and Alzheimer’s disease (8–11). Although splicing and 3’ cleavage/polyadenylation of pre-mRNAs are co-transcriptionally coupled (62, 63), by introducing targeted gene editing, we have now determined that TDP-43 binding to the human STMN2 pre-mRNA sterically blocks recognition of a cryptic 3’ splice site in intron 1, thereby enabling correct pre-mRNA processing and production of a functional, stathmin-2 encoding mRNA. While therapeutic efforts to target several genetic forms of ALS and FTD are underway (64–66), direct approaches to restore normal TDP-43 protein localization and function are challenged by many factors, including as yet unidentified mechanisms leading to the initial TDP-43 dysfunction, apparently exquisite sensitivity of healthy neurons to reduction in TDP-43, and a tight autoregulatory mechanism that is proposed to gate TDP-43’s own pre-mRNA maturation and translation (19, 67, 68).
Here we have identified two approaches to enable restoration of endogenous stathmin-2 accumulation in neurons affected by TDP-43 pathology: targeting dCasRx or cryptic splice blocking ASOs to the STMN2 pre-mRNA. For the latter approach, we have engineered mice to carry a Stmn2 gene partially humanized by insertion of the human STMN2 cryptic splice and polyadenylation sequences but without TDP-43 binding. We then demonstrated in vivo proof-of-concept molecular efficacy of ASOs administered into the CSF in an adult mammalian nervous system to rescue stathmin-2 accumulation to a level that is sufficient to restore axonal regrowth and transport in affected human motor neurons. This approach follows from an initial success in the therapeutic use of a splice-modifying ASO whose action in spinal muscular atrophy restores SMN to motor neurons by correcting missplicing of the pre-mRNA of SMN2 (51, 69, 70). Important next steps for the development of stathmin-2 restoration as a potential therapy include: development of suitable TDP-43 loss-of-function mouse models with which to determine the phenotypic contribution of Stmn2 humanization, and direct experimental determination of the functional consequences of stathmin-2 loss from an aging adult mammalian nervous system. While non-splicing dependent mechanisms likely play a role in pathophysiology of disease and TDP-43-dependent cryptic splicing affects processing of many mRNA targets (e.g., UNC13A (71, 72)) whose contribution to disease phenotypes remain to be determined, our results provide direct support for ASO-mediated restoration of stathmin-2 as a potential therapeutic approach for ALS and other TDP-43 proteinopathies.
Materials and Methods:
RNA extraction, qRT-PCR quantification and RT-PCR
Cultured SH-SY5Y and N2A cells and neurons were directly lysed in Trizol reagent (Thermo). Tissue sections snap-frozen at time of collection were thoroughly mechanically homogenized in 1ml Trizol with a rotor stator homogenizer (Omni International), followed by chloroform addition (200μl) and RNA extraction according to the manufacturer instructions. Total RNA was quantified on a Nanodrop spectrophotometer (Thermo) and 1μg was taken forward to first-strand cDNA synthesis with SuperScript-III reverse transcriptase kit (Thermo) and oligo dT priming per manufacturer instructions. For qRT-PCR, cDNA was diluted to 1ng/μl, and 4ng were loaded into a 10μl reaction (BioRad). Three technical replicates were assayed per biological sample and analyzed on a C1000 thermocycler with a 384-well qPCR reaction module (BioRad) using primers/probes detailed in Supplementary Table 1. Murine Gapdh and Rps9 genes, and human GAPDH and TFRC genes were used as endogenous controls with equivalent results. Relative expression for each gene was calculated from delta-delta-Cq data, with graphing and statistical analysis performed in Prism8 software (GraphPad). All RT-PCR reactions were performed using Q5 High-Fidelity DNA polymerase (NEB), with products analyzed by electrophoresis on 2% agarose gels incubated with ethidium bromide for imaging.
Immunoblotting
Cell and tissue extracts were collected in radioimmunoprecipitation (RIPA) lysis buffer and protein concentrations were measured by Bradford assay (Bio-Rad) to equalize loading. Samples were boiled for 5 minutes in SDS buffer before being analyzed on polyacrylamide gels, transferred to nitrocellulose membrane (0.2 micron) and blocked for one hour in 5% milk 0.1% tween-20 tris buffered saline (TBST). All membranes were incubated overnight with primary antibody (GAPDH – Abcam # Ab8245; Stathmin-2 – Novus # NBP1-49461) immunoblots were then washed three times with TBST and probed with horseradish peroxidase conjugated secondary antibodies diluted 1:5,000–1:10,000 in 5% milk for 1 hour at room temperature before being exposed to films or imaged on a LICOR Fc imager.
SH-SY5Y and N2A cell culture
ATCC-derived human SH-SY5Y (ATCC# CRL-2266) neuroblastoma and murine N2A (ATCC# CCL-131) cells were cultured in DMEM/F12 (Gibco), and DMEM (Gibco), respectively, supplemented with 10% v/v fetal bovine serum (Omega) and 25mM HEPES (Gibco). Cells were grown in a 37°C incubator supplemented with 5% carbon dioxide.
siRNA Transfections
siRNA “smart pools” (Dharmacon) targeting either human TARDBP (Cat# L-012394-00-0020) or control sequences (Cat# D-001810-10-20) were reverse transfected into each well of a 50% confluent 12-well plate at a final concentration of 50nM using RNAiMax (Thermo) following manufacturer’s instructions. After 24h, cells were forward-transfected following manufacturer’s instructions with a second 50nM dose. Cells were collected for analysis 96-hours after the initial transfection.
Cell line genome editing
For SH-SY5Y cells, a plasmid expressing Cas9-T2A-GFP (a gift from the Zhang laboratory, Addgene #48138) was modified to insert the desired single-guide RNA protospacer sequence, the guide sequence GGCTTGTGGCACAGTTGACA was employed for editing the STMN2 cryptic polyadenylation sequence and TDP-43 binding site, and the guide sequence TATATTCATATTGCAGGACT was used for editing of the STMN2 cryptic 3’ splice acceptor site. Activity of each guide was confirmed by transfection into 293T cells and surveyor assay kit (IDT). For editing of the cryptic polyadenylation sequence and TDP-43 binding site, a 1630bp double-stranded homology directed repair donor template was prepared from DNA fragment spanning the human STMN2 exon 2a region (IDT), and desired mutations were made using site-directed mutagenesis. 30μg of DNA repair template was linearized and gel-purified. The cryptic 3’ splice acceptor repair template was prepared as a commercial 151 base single-stranded oligonucleotide (IDT) of sequence: 5’-CGAACTCATATACCTGGGGATTTTATTACTCTGGGAATTATGTGTTCTGCCCCATCACTCTCTCTTAATTGGATTTTTAAAATTATATTCATATTGAAAAACTCGGCAGAAGACCTTCGAGAGAAAGGTAGAAAATAAGAATTTGGCTCTC - 3’.
For each gene-editing assay, a 70% confluent 100mm dish of SH-SY5Y cells was trypsinized to single-cell suspension, pelleted, and resuspended in 100μl electroporation solution (Lonza Kit V) that contains 1μg of Cas9-sgRNA plasmid mixed with 10μg of double-stranded linearized HDR templates or 80 picomoles ssDNA template. Electroporation was performed in a 2mm cuvette using a Nucleofector 2B (Lonza), program A-023. Cells were immediately collected and resuspended in fresh media, pelleted by centrifugation to remove electroporation reagents, and replated. 72 hours post-electroporation, live GFP-positive cells were single-cell FACS sorted into six 96-well plates (SH800, Sony). Cells were maintained in 50% v/v sterile filtered conditioned media, refreshed every two days. gDNA extraction from surviving clones was carried out with a Quick-DNA 96 kit (Zymo Research), followed by PCR (for primers see Supplementary Table 1) and restriction digest. Identified positive gene-edited clones were then expanded into larger culture dishes. TOPO cloning of genomic DNA PCR products was performed with a Zero Blunt kit (Thermo), and resulting clones were Sanger-sequenced to determine diploid haplotypes.
For humanizing N2A cells at the Stmn2 locus, transfection of 1 μg plasmid for Cas9-T2A-GFP expression and an HDR template plasmid containing 3267bp human fragment flanked by 800bp murine homology “arms”, was carried out using XFect reagent (Takara) according to the manufacturer’s guideline. Cells were transfected at 50–70% confluency, maintained for 48h and then FACS-sorted for GFP-positive single-cells.
Mouse genome editing
Stmn2HumΔGU/+ mouse lines Stock # JR35721 and JR35722 were generated by CRISPR-Cas9 induced cleavage and homology repair (HDR) using an sgRNA encoded protospacer sequence ATCCGAAGCAGCTTCCCACC. A plasmid containing a 3262bp human STMN2 Intron1 fragment (in which the GU binding sequence of TDP-43 was replaced with an MS2 aptamer sequence for homology repair), flanked on both ends by ~800bp of corresponding mouse intronic sequence, was used a template for HDR. Strains were backcrossed twice to C57BL/6J inbred mice (#JR00664) to generate the N2 generation heterozygous mice, and homozygous mice derived from the matings of heterozygous mice were used for validation and characterization. Line JR35721 was used for in vivo rASO testing. Oligo dT-primed mRNA expression profiling in heterozygous mice revealed a corresponding suppression of Stmn2 expression from the humanized allele and accumulation of a truncated chimeric RNA derived from mouse exon 1 fused to human exon 2A.
Mouse phenotypic assessment
Grip strength was assessed using a grid attachment to the instrument, recording the front paws and all four paws strength, the average strength in grams of three consecutive trials was recorded. For Rotarod analysis, all the animals are subjected to two consecutive sessions, a “training session” on day one and a “test” session on day 2. On each session the mice were acclimated to the testing room for 60min. An Ugo-Basile accelerating rotarod model 47600 for mice was used. Mice were placed on a rotating rod at 4rpm which accelerates up to 40rpm over the course of 300 secs. Each mouse is subjected to four consecutive trials with a 45-sec resting interval. The time in seconds when the mouse falls from the rod is recorded. The average latency to fall of the last three by trials is reported. Compound action potential (CMAP) analysis was conducted in anesthetized animals using isoflurane (1.5–2%) and placed on a thermostatically regulated heating pad to maintain normal body temperature, CMAP responses were recorded from the proximal hindlimb using two recording electrodes. The active electrode was positioned over the tibialis anterior (TA) muscle. The reference electrode was positioned at the metatarsal region of the foot on the same limb. Supramaximal stimulation of the sciatic nerve was elicited via two needle electrodes placed subcutaneously. over the sacrum (anode) and the sciatic notch (cathode). Stimulations were delivered through a Stimulus Isolator (FE180, AD Instruments). CMAP amplitude was recorded and the peak-to-peak amplitude of the CMAPs was measured.
Primary cortical neuron cultures
Primary cortical neurons were dissected from embryonic day 16–17 (E16-E17) Stmn2+/HumanExon2a (#JR34060) mouse embryos. Cortical tissues were treated with 0.25% of Trysin+EDTA for 15 min at 37 C. Trypsin activity was inhibited using DMEM:F12 media supplemented with 10% of FBS and 1% of Penicillin/Streptomycin and tissues were then mechanically disaggregated using 1000 μL tips. A 4% BSA (in PBS1x) solution was further slowly added to the cell suspension to generate a bilayer solution (with the BSA at the bottom) and centrifuged for 10 min at 1000rpm. The cell pellet was resuspended in Neurobasal media supplemented with 2% of B27, 0.5mM of L-glutamine and 100U/mL of penicillin-streptomycin. Cells were seeded in culture plates previously coated with poly-D-lysine at the concentration of 700,000 cells/mL.
ASO treatments
For ASOs that rescue STMN2 altered pre-mRNA processing (rASOs), each base is 2’-O-methoxyethyl-modified and every internucleotide linkage is a phosphorothioate, all cytosine residues are 5-methylcytosines. For iPS-derived neurons, ASOs were added to the culture media for free uptake at the indicated concentration. For SH-SY5Y cells, ASOs were either electroporated as previously described or lipid transfected with Xfect transfection reagent (Takara) using the manufacturer instructions at an optimized ratio of 1.05μl Xfect polymer per 50μl of total buffer-diluted ASO, in 12-well plates. Stmn2HumΔGU/+ heterozygous mice were dosed by intracerebroventricular (ICV) injection with a 1000μg single dose, or alternatively in the case of the “rASO5 – 2 dose” condition two 500μg doses separated by two weeks of recovery, with tissues harvested at 14- and 28-days after initial injection, respectively. PBS ICV injection into heterozygous mice and naïve non-transgenic littermates were used as controls for truncated and full-length Stmn2 RNA accumulation.
Stable lentiviral vector transduction and selection
Constructs encoding a truncated variant encoding dCasRx, its guide RNA sequences, MCP- and MCP-TDP-43 mutant were all designed and codon optimized using SnapGene software and ordered as gBlock gene fragments (IDT). Fragments were assembled into their respective lentiviral transfer vectors by Gibson assembly (73). Virus particles were packaged by standard techniques using three-plasmid transfection (3:2:1 ratio of transfer vector : psPAX2 : pMD2G) into proliferating 293T cells with VirusGen transfection reagent (Mirus). Viral supernatants were collected at 48 and 72 hours and concentrated by sucrose centrifugation as previously described (74) then stored at −80°C. Cells were then spin-transduced with media containing the resulting virus in the presence of 8μg/ml polybrene at 1000 RCF for 2 hours at 32°C, then placed in normal media. 72 hours post-transduction cells were placed in antibiotic to select for successfully transduced clones. For inducible expression constructs, cells were subsequently transduced using a lentivirus encoding a constitutive Tet3G transactivator protein and a Neomycin-resistant gene. 72h later, cells were treated with Geneticin (Gibco) for 5 days, and selected cells were expanded. dCasRx guides were manually designed, ordered as complementary oligonucleotides (IDT) and integrated into a lentiviral transfer vector using BbsI restriction integration sites. Stable lentiviral integration of guide sequences was verified in the puromycin-selected cell lines by nested PCR and sanger sequencing. The dCasRx no guide control vector deleted the U6 promotor and guide direct-repeat elements by restriction digestion and re-ligation but was otherwise identical.
Generation and differentiation of iPSC-motor neuron
Previously generated J.Craig Venter iPSC line was used as described in (75). Briefly, iPSCs were grown in Matrigel-coated plates until 70–90% confluency in mTeSR-Plus medium. Day 1 of neural induction was marked by media change to N2B27 medium composed by (DMEM/F12 + GlutaMAX (ThermoFisher #10829018) supplemented with 1% PenStrep, 1:200 N2 supplement (ThermoFisher # 17502–048), 1:100 B27 Supplement (ThermoFisher # 17504–044), 150μM Ascorbic Acid (A4544-100g) supplemented with 10μM SB431542 (Tocris #1614), 1μM Dorsomorphin (Tocris #3093) and 3μM CHIR99021 (Tocris #4423). Daily media changes were done until day 7, when cells were passaged at 1:6 and medium was changed to N2B27 supplemented with 10μM SB431542, 1μM Dorsomorphin, 200nM Smoothened Agonist (SAG, EMD Millipore #566660) and 1.5μM Retinoic Acid (RA, Sigma #R2625-50mg). Media was changed every day with doubling volumes to adjust for cell density. Cells were passaged at day 18 to 10cm dishes coated with Matrigel using Accutase to detach the cells and plated at density of 7.5×106 cells per plate in media containing N2B27 medium supplemented with 200nM Smoothened Agonist (SAG) and 1.5μM Retinoic Acid (RA). Cells were passaged on day 22 to microfluidic chambers coated with 10μg/ml poly-D-lysine, 10μg/ml poly-L-ornithine, and subsequently 20μg/ml laminin. N2B27 medium supplemented with growth factors (2ng/ml BDNF (R&D Syst #248-BD), CTNF (R&D Syst #257-NT/CF) and GDNF (R&D syst #212-GD)), and 2μM DAPT (Tocris #2634). From day 25 onward, cells were fed every 2–3 days with N2B27 medium containing growth factors. During all passaging or thawing steps, the ROCK inhibitor Y27632 (Tocris) was used at a concentration of 10μM for 24 hours.
Compartmented microfluidic devices for axotomy
Microfluidic devices were fabricated and seeded with iPS-derived precursor motor neurons (pre-MNs) as previously described (25). Pre-MNs were matured for at least nine days before ASOs suppressing TDP-43 were added into the somatic compartment by free uptake. Eight days later, rASOs or control ASO were added to the media for 12 days. Half of media was changed every four days where ASOs targeting TDP-43 were replenished every 12 days and rASOs or control ASO were added to the media eight days after the first dosing. After 20 days of treatment, aspiration-induced axotomy was performed ensuring no axons remained in the distal compartment, and axonal regeneration capacity was evaluated for 36 hours. Chambers were prepared for immunofluorescence after 4% PFA fixation, blocking, and permeabilization with PBS, 0.5% Tween-20, 1.5% BSA for 1 hour. Staining was performed using primary antibodies stathmin-2 rabbit polyclonal Novus Biologicals NBP1-49461 (1:1000), and Neurofilament Heavy mouse monoclonal Novus Biologicals NBP2-29435 (1:500) diluted in PBS with 0.3% TritonX-100 overnight at room temperature. Primary antibodies were washed with PBS. (1:500) donkey anti-rabbit Alexa 488-, and donkey anti-mouse Alexa Cy3-coupled secondary antibodies (Jackson ImmunoResearch Cat 711-545-152 and 715-165-151) were incubated for 2h at room temperature before washing with PBS and images were acquired using a confocal microscope Leica SP8.
Modified Sholl Analysis for Quantification of Axotomy Data
Images from Control (n = 6), TDP-43 ASO (n = 5), and TDP-43 ASO rescued (n = 4) were processed using a modified sholl analysis to measure axonal complexity using ImageJ 1.53u. The neurofilament-heavy (NfH) channel was first filtered using a median filter and the site of the axotomy for each image was selected to align all images. Profile lines of NfH intensity were drawn in 3.125 μm (10 pixel) intervals perpendicular to the microgrooves across the entire image. Axon intersections were counted by identifying the number of peaks in each NfH profile plot. To assess axonal complexity, total axon intersections were plotted against distance from the site of axotomy and the area under this curve (AUC) was calculated for each image. A one-way ANOVA was performed to test for significant differences in axonal complexity (AUC) between groups. All statistical analysis was performed in GraphPad Prism 8.
Axonal transport live imaging
ASO treated iPSC-derived motor neurons were grown and treated in glass-bottom 24-well plates with 3 biological replicates per treatment group. Fresh medium was supplemented with 100nM Lysotracker Green and incubated with cells for 60 minutes at 37°C, followed by three washes with warm normal medium and live-cell imaging on a Yokogawa CQ1 system. 61 time-lapse images series were captured from seven fields per group of axonal transport at 5 second intervals in a controlled chamber at 37°C and 5% carbon dioxide. Analysis of particle transport distribution and density using kymograph analysis was performed with FIJI and KymoToolBox plugins. At least 220 particles from 22 axons were measured for each treatment.
Electron microscopy
For ultrastructural characterization of excitatory synaptic terminals, ASO treated human motor neurons were fixed for 1 hour with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, and spun down to a cell pellet. Subsequently, the pellet was blocked with 1% osmium tetroxide in 0.15 M sodium cacodylate buffer (LADD Research), stained with 2% uranyl acetate in double distilled water (LADD Research), and dehydrated with ethanol (LADD Research). The cells were then embedded in Durcupan resin (Sigma) and sectioned on an Ultracut UC6 Ultramicrotome (Leica) by using a diamond knife (Diatome). The sections were next transferred onto copper mesh grids (LADD Research) and post-stained with uranyl acetate and lead citrate (Electron Microscopy Sciences). Images were acquired using a JEOL1400 (JEOL, Peabody, MA) transmission electron microscope (supported by NIH equipment grant 1S10OD023527-01) at 80kV using a Oneview 4KGatan digital camera (Gatan, Pleasanton, CA).
Statistical tests and materials availability
Statistics were calculated using Prism 8 software by GraphPad. Individual data points are independent biological replicates (individual mice, individual neuron differentiations, or individual wells of independently treated cells, respectively). Statistical significance for 2 group comparisons was determined using 2-tailed Student’s t-tests; or for 3 or more comparisons one-way ANOVA with Tukey correction or Chi-squared with Yates correction as indicated in figure legends. ****, p <0.0001; ***, p < 0.001; **, p < 0.01; *, p <0.05; ns, p > 0.05; all error bars are SEM unless otherwise indicated. Observers and experimenters were blinded to genotype information where possible. Stem cells were used under UCSD IRB project #161575ZX, and animals were used in accordance with UCSD IACUC protocol S0022 or Jackson Laboratory AUS protocol 20029.
Supplementary Material
Acknowledgments:
The generation and development of the CRISPR/Cas9 mutant mouse models was supported by the Jackson Laboratory Genome Engineering Technology Core*.
Funding:
NINDS/NIH R01NS112503 to (DWC, CL-T)
NINDS/NIH RF1NS124203 (DWC, CL-T, CL)
ALS Finding a Cure (CL-T)
The Massachusetts Center for Alzheimer Therapeutic Science (CL-T)
The Sean M. Healey & AMG Center for ALS at Mass General (CL-T)
U42 Mutant Mouse Resource Research Center OD010921 (CL)
Ruth Kirschstein Institutional National Research Service Award T32 GM008666. (MWB, MSB), and T32 AG 66596-2 (MSB)
The Packard Center for ALS Research (DWC, MWB)
The ALS association (DWC, MWB, SVS)
MDA development grants (ZM and JL-E)
The BrightFocus Foundation (ARAAQ)
*Cancer Center Support Grant CA034196 to The Jackson Laboratory.
Footnotes
Competing interests:
A patent application has been filed describing composition and methods for restoring stathmin-2 expression; publication number US 2021-0252039. D.W.C. and Z.M. are inventors on the patent.
CFB, KL, PJN, FR, are employees of Ionis Pharmaceuticals.
DWC is a consultant for Ionis Pharmaceuticals.
Data and materials availability:
SH-SY5Y and N2A cells were obtained for research use via a material transfer agreement with ATCC. Plasmids were obtained by material transfer agreement with Addgene. All mouse strains have been made publicly available via The Jackson Laboratory. Other materials are available under MTA for non-commercial replication or extension of this work, upon request to corresponding authors. All other information is available in the manuscript or the supplementary material.
References and Notes
- 1.Nussbacher JK, Tabet R, Yeo GW, Lagier-Tourenne C, Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 102, 294–320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lagier-Tourenne C, Polymenidou M, Cleveland DW, TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19, R46–64 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicolas A et al. , Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 97, 1268–1283 e1266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weskamp K, Barmada SJ, RNA Degradation in Neurodegenerative Disease. Adv Neurobiol 20, 103–142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gitler AD, Shorter J, RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 5, 179–187 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ling SC, Polymenidou M, Cleveland DW, Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor JP, Brown RH Jr., Cleveland DW, Decoding ALS: from genes to mechanism. Nature 539, 197–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Josephs KA et al. , TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127, 811–824 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson BS et al. , TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284, 20329–20339 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arai T et al. , TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351, 602–611 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Amador-Ortiz C et al. , TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61, 435–445 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davidson Y et al. , TDP-43 in ubiquitinated inclusions in the inferior olives in frontotemporal lobar degeneration and in other neurodegenerative diseases: a degenerative process distinct from normal ageing. Acta Neuropathol 118, 359–369 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Mishima T et al. , Perry Syndrome: A Distinctive Type of TDP-43 Proteinopathy. J Neuropathol Exp Neurol 76, 676–682 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKee AC et al. , TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69, 918–929 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson PT et al. , Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142, 1503–1527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nana AL et al. , Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol 137, 27–46 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun M et al. , Cryptic exon incorporation occurs in Alzheimer’s brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol 133, 923–931 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tollervey JR et al. , Characterising the RNA targets and position-dependent splicing regulation by TDP-43; implications for neurodegenerative diseases. Nat Neurosci, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polymenidou M et al. , Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14, 459–468 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lagier-Tourenne C et al. , Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15, 1488–1497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown A-L et al. , TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma XR et al. , TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 603, 124–130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ling JP, Pletnikova O, Troncoso JC, Wong PC, TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeong YH et al. , Tdp-43 cryptic exons are highly variable between cell types. Mol Neurodegener 12, 13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melamed Z et al. , Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration Nat. Neurosci. 22, 180–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klim JR et al. , ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci 22, 167–179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chauvin S, Sobel A, Neuronal stathmins: a family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Prog Neurobiol 126, 1–18 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Shin JE, Geisler S, DiAntonio A, Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol 252, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guerra San Juan I et al. , Loss of mouse Stmn2 function causes motor neuropathy. Neuron, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krus KL et al. , Loss of Stathmin-2, a hallmark of TDP-43-associated ALS, causes motor neuropathy. bioRxiv, 2022.2003.2013.484188 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krach F et al. , Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol 136, 405–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prudencio M et al. , Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest 130, 6080–6092 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belmont LD, Mitchison TJ, Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell 84, 623–631 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Stein R, Mori N, Matthews K, Lo LC, Anderson DJ, The NGF-inducible SCG10 mRNA encodes a novel membrane-bound protein present in growth cones and abundant in developing neurons. Neuron 1, 463–476 (1988). [DOI] [PubMed] [Google Scholar]
- 35.Morii H, Shiraishi-Yamaguchi Y, Mori N, SCG10, a microtubule destabilizing factor, stimulates the neurite outgrowth by modulating microtubule dynamics in rat hippocampal primary cultured neurons. J Neurobiol 66, 1101–1114 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Riederer BM et al. , Regulation of microtubule dynamics by the neuronal growth-associated protein SCG10. Proc Natl Acad Sci U S A 94, 741–745 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duncan JE, Lytle NK, Zuniga A, Goldstein LS, The Microtubule Regulatory Protein Stathmin Is Required to Maintain the Integrity of Axonal Microtubules in Drosophila. PLoS One 8, e68324 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graf ER, Heerssen HM, Wright CM, Davis GW, DiAntonio A, Stathmin is required for stability of the Drosophila neuromuscular junction. J Neurosci 31, 15026–15034 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ayala YM et al. , Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 348, 575–588 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Lim F, Spingola M, Peabody DS, Altering the RNA binding specificity of a translational repressor. J Biol Chem 269, 9006–9010 (1994). [PubMed] [Google Scholar]
- 41.Rot G et al. , High-Resolution RNA Maps Suggest Common Principles of Splicing and Polyadenylation Regulation by TDP-43. Cell Rep 19, 1056–1067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batra R et al. , Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell 56, 311–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu H, Tang L, Mechano-regulation of alternative splicing. Curr Genomics 14, 49–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masuda A et al. , Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev 29, 1045–1057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin G, Gruber AR, Keller W, Zavolan M, Genome-wide analysis of pre-mRNA 3’ end processing reveals a decisive role of human cleavage factor I in the regulation of 3’ UTR length. Cell Rep 1, 753–763 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Di Giammartino DC, Nishida K, Manley JL, Mechanisms and consequences of alternative polyadenylation. Mol Cell 43, 853–866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.So BR et al. , A Complex of U1 snRNP with Cleavage and Polyadenylation Factors Controls Telescripting, Regulating mRNA Transcription in Human Cells. Molecular Cell 76, 590–599.e594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elkon R, Ugalde AP, Agami R, Alternative cleavage and polyadenylation: extent, regulation and function. Nature reviews 14, 496–506 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Tian B, Graber JH, Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip Rev RNA 3, 385–396 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konermann S et al. , Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676 e614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finkel RS et al. , Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Bennett CF, Kordasiewicz HB, Cleveland DW, Antisense Drugs Make Sense for Neurological Diseases. Annual Review of Pharmacology and Toxicology 61, 831–852 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alami NH et al. , Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guedes-Dias P, Holzbaur ELF, Axonal transport: Driving synaptic function. Science 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perlson E, Maday S, Fu MM, Moughamian AJ, Holzbaur EL, Retrograde axonal transport: pathways to cell death? Trends Neurosci 33, 335–344 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sleigh JN et al. , Mice Carrying ALS Mutant TDP-43, but Not Mutant FUS, Display In Vivo Defects in Axonal Transport of Signaling Endosomes. Cell Reports 30, 3655–3662.e3652 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arnold ES et al. , ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A 110, E736–745 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wils H et al. , TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Becker LA et al. , Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mackenzie IR et al. , Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61, 427–434 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Neumann M et al. , Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 314, 130–133 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Rigo F, Martinson HG, Functional coupling of last-intron splicing and 3’-end processing to transcription in vitro: the poly(A) signal couples to splicing before committing to cleavage. Mol Cell Biol 28, 849–862 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kyburz A, Friedlein A, Langen H, Keller W, Direct interactions between subunits of CPSF and the U2 snRNP contribute to the coupling of pre-mRNA 3’ end processing and splicing. Mol Cell 23, 195–205 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Miller T et al. , Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 383, 109–119 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, Shneider NA, Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nature Medicine 28, 104–116 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boros BD, Schoch KM, Kreple CJ, Miller TM, Antisense Oligonucleotides for the Study and Treatment of ALS. Neurotherapeutics 19, 1145–1158 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ayala YM et al. , TDP-43 regulates its mRNA levels through a negative feedback loop. Embo J, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Avendano-Vazquez SE et al. , Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev 26, 1679–1684 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR, Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 82, 834–848 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bennett CF, Krainer AR, Cleveland DW, Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu Rev Neurosci 42, 385–406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown AL et al. , TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ma XR et al. , TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 603, 124–130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gibson DG et al. , Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 74.Jiang W et al. , An optimized method for high-titer lentivirus preparations without ultracentrifugation. Sci Rep 5, 13875 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martinez FJ et al. , Protein-RNA Networks Regulated by Normal and ALS-Associated Mutant HNRNPA2B1 in the Nervous System. Neuron 92, 780–795 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SH-SY5Y and N2A cells were obtained for research use via a material transfer agreement with ATCC. Plasmids were obtained by material transfer agreement with Addgene. All mouse strains have been made publicly available via The Jackson Laboratory. Other materials are available under MTA for non-commercial replication or extension of this work, upon request to corresponding authors. All other information is available in the manuscript or the supplementary material.