

Abstract
This study examines the potential of Cannabis sativa L. plants to be repurposed as therapeutic agents for cancer treatment through designing of hybrid Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs). A set of 50 phytochemicals was taken from Cannabinoids and Terpenes and subjected for screening using Semi-flexible and Flexible Molecular Docking methods, MM-GBSA free binding energy computations, and pharmacokinetic/pharmacodynamic (ADME-Tox) predictions. Nine promising phytochemicals, Cannabidiolic acid (CBDA), Cannabidiol (CBD), Tetrahydrocannabivarin (THCV), Dronabinol (Δ-9-THC), Delta-8-Tetrahydrocannabinol (Δ-8-THC), Cannabicyclol (CBL), Delta9-tetrahydrocannabinolic acid (THCA), Beta-Caryophyllene (BCP), and Gamma-Elemene (γ-Ele) were identified as potential EGFR-TKIs natural product candidates for cancer therapy. To further validate these findings, a set of Molecular Dynamics simulations were conducted over a 200 ns trajectory. This hybrid early drug discovery screening strategy has the potential to yield a new generation of EGFR-TKIs based on natural cannabis products, suitable for cancer therapy. In addition, the application of this computational strategy in the virtual screening of both natural and synthetic chemical libraries could support the discovery of a wide range of lead drug agents to address numerous diseases.
Keywords: Cannabis sativa L., Virtual Screening, CADD, Semi-flexible & Flexible Molecular Docking, MM-GBSA free Binding energy, The inhibition constant (Ki), Molecular Dynamics, EGFR-TKIs, Breast & Lung Cancer, Cannabinoids & Terpenes
Graphical abstract

Highlights
-
•
Cannabinoids & Terpenes derived from Cannabis sativa L. screened in silico for targeting cancer via inhibiting the EGFR-TKD enzyme.
-
•
Hybrid approach of structure-based & biophysical simulations to identify EGFR-TKIs..
-
•
Semi-flexible & Flexible Molecular Docking methods, Drug-like, ADME-Tox, MM-GBSA and MD used to identify stable EGFR-TKIs.
-
•
The amino acid residues of Leucine, Phenylalanine, Valine, Lysine, Cysteine, & Threonine targeted for cancer cell line inhibition.
-
•
In silico drug discovery identified new generation of EGFR-TKIs as candidates for target therapy.
-
•
Natural substances from Cannabis sativa L. repurposed for therapeutics.
1. Introduction
Despite the fact that the Cannabis sativa L. plant has many uses, this plant is not without its utilitarian benefits, especially in fields of medicinal and pharmaceutical applications. It has therefore become a necessity to explore benefits of this plant by legalizing its use for the medical and industrial purposes [1]. The Cannabis sativa L. plant was originally popular in East Asia, but it has now become a popular plant all over the world due to the development of cultivation types [2]. Throughout a history, it has been cultivated and used as a source of artificial fiber, amusement, food, mood enhancement, and also in traditional medicine [3,4]. Two countries, Morocco and Afghanistan, are among the largest producers of hashish obtained from the cannabis plant, which is being distributed and consumed worldwide for its psychological effects [[5], [6], [7]]. Cannabis has already been found to have certain biological activities that could allow it to be used for medicinal purposes. Among the permitted therapeutic potentials, it mentions the ability to regulate sleep disorders [8], inflammation [9], neuro-degeneration [10], pain [11], nausea [12], anorexia [13], diseases of cancer and epilepsy [14].
Although the main psychoactive substance in cannabis is a Tetrahydrocannabinol (THC) [15], the cannabis plant contains several other active cannabinoids, including cannabis isolated (>120) such as Cannabidiol (CBD), Cannabinol (CNB), Terpenes (>120) [16,17], Flavonoids (∼34), and Phenolic compounds (∼42), etc. [1,[18], [19], [20]]. There are several categories of cannabis, the most important of which is the one based on the rate of THC and CBD and CNB [21]. The higher the THC level, the more the product belongs to the dope class, while the higher the CBD and CNB level, the more the product belongs to the cannabinoid class [22,23]. It should be noted that both Cannabidiol (CBD) and Cannabinol (CBN) do not have a psychoactive effect, but can block the effect of THC on the nervous system [23]. Although the THC (PubChem CID: 16078), and CBD (PubChem CID: 644019) and CNB (PubChem CID:2543) come from the same plant source, they have different biological effects on humans. The reason for these different therapeutic effects may be due to the structural diversities of these phytochemicals derived from cannabis as well as the specific cultivar of which they are derived [24]. This diversity is one of the rationales for studying differences between natural cannabis products and evaluating their effects on pharmacological properties in silico, in vitro and in vivo. In the context of the legalization of the use of cannabis products in the medical field, the biological activity of phytochemicals isolated from cannabis has been examined under different conditions, and here we highlight some effects of seven isolated cannabis include (Δ9-Tetrahydrocannabinol, Cannabidiol, Cannabigerol, Cannabichromene, Cannabidivarin, Tetrahydrocannabivirin) against Breast Cancer, Brain Cancer, Leukemia, Lung Cancer, Melanoma & Myeloma, Hepatocellular Carcinoma, Pancreatic, Prostate, Colon Cancer, Endometrial, Cervival, Oral Cancer, Glioma & Neuroblastoma, Cervical, Endometrial and Ovarian Cancer, etc. [25]. Among the seven cannabinoids reviewed, only Δ9-Tetrahydrocannabinol and Cannabidiol were able to reach clinical trials against brain cancer, leukemia, glioma cancer and neuroblastoma cancer [25]. While the rest of the isolated cannabis did not exceed the phase of in vivo testing as the highest estimate against the other types of cancer listed in the current review [25]. The reason for this could be due to the occurrence of undesirable side effects in living organisms submitted to the trial. Although the various functions of several bioactive compounds in cannabis have been reported to be beneficial against cancer, there is an almost complete lack of evaluation of the bioactivity of other natural products found in cannabis, such as the cannabinoids and terpenes. Due to the availability of wide varieties of natural products such as capsules, oils, extracts and tinctures based on terpenes, other medicinal applications of these agents are still unknown or have not yet been reported, especially their potential therapeutic against cancer. From this background, it will be the basis of our investigation to predict in silico the prospects of using terpenes and cannabinoids isolated for breast and lung cancer as a targeted model.
Due to the lack of efficient treatments accessible to all populations at the lowest cost and with less toxicity, efforts of researchers have turned towards medicinal plants such as Cannabis sativa L. and their therapeutic benefits against breast and lung cancer [[26], [27], [28]]. The World Health Organization (WHO) reported 10.44 million cancer deaths in 2020, including 2.26 million cases of breast cancer, 2.21 million cases of lung cancer, 1.93 million cases of colon and rectal cancer, 1.41 million cases of prostate cancer, 1.20 million cases of non-melanoma skin cancer, and 1.09 million cases of stomach cancer [[29], [30], [31]].
Despite tremendous progress in the cancer treatment methods around the world, the prospect of turning cancer into a disease that can be lived with or completely treated remains a long way off. However, this reality is regarded as a strong motivator for researchers in their various disciplines related to this context, with the horizon of achieving the desired goal. This is demonstrated by the wide range of available cancer therapies, including hormonal therapy, radiotherapy, surgery, targeted therapy with drugs/vaccines, gene therapy, and other chemotherapy treatments [32]. In this regard, targeted molecular therapies based on interference/interaction with specific molecular receptors such as proteins/enzymes have revolutionized the treatment of a variety of cancer diseases, including lung, breast, colorectal, and ovarian cancer, as a means of preventing cancer growth [33].
In this progression, The epidermal growth factor receptor (EGFR) tyrosine kinase domain (TKD) targeted therapies point outs a new era in precision oncology [34,35]. This is because the EGFR (or EGFR; ErbB-1; HER1) plays critical roles in the maintenance of epithelial tissue as well as the control of cell division and survival [36]. However, the receptor regulates many signaling pathways in the body, with the absence or low expression of EGFR and other receptor tyrosine kinases in humans leading to many diseases such as Alzheimer's disease [37,38], while overexpression of EGFR-TKD is linked to the development of many tumor diseases, including breast and lung cancers, which are currently the most lethal [35].
Focusing on ways to treat cancer tumors resulting from overexpression of the EGFR receptor tyrosine kinase, tyrosine kinase inhibitors (TKIs)have been and remain a successful therapeutic key in this regard [39,40]. Several EGFR therapies have been approved by the FDA for NSCLC and breast cancer other cancers, including Erlotinib and Gefitinib (first-generation TKIs); Afatinib, Dacomitinib, and Neratinib (second-generation TKIs); Osimertinib, Afatinib, and Lazertinib (third-generation TKIs); JBJ-0412502, BI, BI-4020 (fourth-generation TKIs) [35,41,42]. Combining chemotherapy with the targeted therapy through EGFR-targeted by the small molecule TKIs is emerging as a very important therapeutic model in the cancer treatment scenarios to achieve a longer life expectancy for patients. However, TKIs face many challenges, including a relatively limited range of targeted cancers, drug resistance after long-term treatment, and potential toxicity risks [43]. These reasons are sufficient to multiply studies and researches to develop and to screen new generations of EGFR-TKIs for an effective cancer treatment. In this regard, it appears that most FDA-approved EGFR-TKIs are based on small molecules, with the exception of some inhibitors such as Erlotinib (Tarceva) and Gefitinib (Iressa) which are based on phytochemicals Taxus brevifolia and Ancaria tomentosa, respectively [44,45]. In this context, four phytochemicals Curcumin [46], derived from Curcuma longa; Resveratrol [47], from Vitis vinifera; Quercetin [48], from Sophora japonica; and Pepperlongumene [49], from Piper longum, were studied and tested against cancer as a part of the EGFR-TKIs class. These compounds have demonstrated potential in their ability to inhibit/regulate EGFR-TKD activity and reduce resistance to several of its standard drug inhibitors. This highlights the need to expand the existing databases of prospective phytochemical agents for their use as EGFR-TKIs, through further research into different and diverse categories of natural products. With this in mind, the present study has focused on an untapped potential of Cannabis sativa L. based phytochemicals to develop a new generation of EGFR-TKIs with therapeutic applications for cancer and other diseases that are currently untreatable. With this insight, recent research has revealed that cannabis extracts may have potential for treating cancer [50]. Cannabinoids such as Cannabidiol (CBD), Cannabigerol (CBG), and Cannabinol (CNB) have been tested for their ability to bind to and inhibit the Epidermal Growth Factor Receptor (EGFR), which is associated with cancer progression [50]. This may be a turning point in the discovery of novel Cannabis sativa L-based drugs that specifically target cancers brought on by the overexpression of EGFR. Therefore, shifting research interest towards Cannabis sativa L-derived phytochemicals in the development of cancer therapies is a paradigm that merits further attention. With our study, we hope to promote this paradigm.
In light of these scenarios, consider the frightening statistics for breast and lung cancer deaths worldwide, which have been reported to account for nearly 50% of all deaths [51]. Because of the high deaths caused by these two cancers, our current research focuses on them as models for exploring potential therapeutic ways to reduce the rate of death caused by these cancers using phytochemicals isolated from Cannabis sativa L plant. To achieve this goal, we collected a database of 50 samples of cannabis extracts that were statistically categorized as the optimal extracts among the 279 samples statistically analyzed in a study conducted by Matan Birenboim et al. [1]. In the current study, we categorized the 50 phytochemical compounds into two sets. The first set consisted of 12 cannabinoids (C1–C12) and the second set consisted of 38 terpenes (T1-T38) (Table S1).
Based on molecular modeling techniques and their importance in computer-aided drug design (CADD) [52,53], we performed a comprehensive screening of 50 phytochemicals to determine their bioavailability, pharmacokinetics/pharmacodynamics, and their likely interactions with the Epidermal growth factor receptor tyrosine kinase domain (EGFR-TKD) involved in the proliferation of breast and lung cancer cell lines and several other types of cancer [[54], [55], [56]]. Based on predictions of drug-likeness and toxicity risks features, in silico ADME-Tox (Absorption, Distribution, Metabolism, Excretion and Toxicity) modeling, Molecular Docking Semi-Flexible& Flexible, theoretical values of inhibitory constants (Ki), MM-GBSA free binding energies, and Molecular Dynamics analysis, we screened top lead molecular scaffolds that would be best likely to inhibit breast and lung cancer cell growth by targeting the EGFR-TKD. To facilitate the identification of new candidate phytochemicals agents for their use as inhibitors of breast and lung cancer by targeting the Epidermal Growth Factor Receptor Tyrosine Kinase Domain (EGFR-TKD). In our present study, we use two drugs Tamoxifen (PubChem CID: 2733526 and Erlotinib (PubChem CID: 176870) as references to construct the hypothetical screening and make the rational comparisons. This is due to the broad clinical application of Erlotinib and Tamoxifen in patients with lung and breast cancer [57].
2. Material and methods
2.1. Identification of targeted receptor and standard drugs for drug design
The epithelial growth factor receptor (EGFR) mediates several tyrosine signaling pathways to transmit signals to the exterior/interior of the cell to perturb the cell function. The EGFR (Epidermal Growth Factor Receptor) is a member of the ErbB family of receptor tyrosine kinases [58]. The ErbB family consists of four receptor tyrosine kinases, including EGFR (ErbB1), ErbB2 (HER2), ErbB3 and ErbB4 [59]. The EGFR is the major receptor of the ErbB family and is responsible for transmitting signals from the cell surface ligands to an inside of the cell. It is involved in a variety of physiological processes, including cell growth, differentiation, migration, and survival [60]. In this regard, first and second generations of tyrosine kinase inhibitors (TKIs), the most important of which is Erlotinib, have often been a good and broad treatment alternative for several types of cancer including lung cancer through targeting EGFR-TKD. However, cases of resistance to these inhibitors leads to hyper-phosphorylation, enzyme overexpression, and the generation of new EGFR-TKD mutations [61]. Therefore, the patient resistance to this inhibitor limits its therapeutic efficacy, adversely affecting desired therapeutic endpoints. This emphasizes the need to discover stronger and more efficient alternatives to the conventional EGFR-TKIs, like erlotinib, which is used as a standard drug therapy for lung cancer in this study.
As for the treatment of breast cancer with EGFR-TKIs, some drugs such as lapatinib, Neratinib and Pyrotinib have already been shown significant clinical efficacy, but they are not unilateral and must be combined with other therapeutic agents [62]. Thus, the potential of EGFR-TKIs in the breast cancer treatment needs a further study and refinement until more suitable agents are found for the dual use, alone or in the combination with other therapies. Tamoxifen has been used as an effective anti-estrogen drug treatment for breast cancer for the last four decades. Tamoxifen inhibits estrogen (ER), which results in a significant downregulation of the ERBB2 protein, limiting the effects of estrogen in most areas of the body, including the breast [63,64]. Tamoxifen is still one of the most effective breast cancer treatments, reducing mortality by at least 30% [65]. However, resistance to this drug remains one of the major issues in the fight against breast cancer, as it has been observed that after the initial 5–10 year treatment period, breast cancer patients develop resistance to this drug [65,66]. In this context, a study conducted by Wang YK et al. [67] confirmed that ERBB2 and EGFR are membrane-bound tyrosine kinases with 95% similar structures, all of which, when overexpressed, can lead to cell transformation, cell proliferation and cancer. The high expression of ERBB2 has been linked to increased tumor invasiveness, metastasis, resistance to chemotherapy and poor prognosis. Another study, Tomoya T. et al. [68], reported that EGFR inhibition reduces tamoxifen resistance. As a result, the development of new potent EGFR inhibitors could provide a new therapeutic approach for breast cancer patients who have developed Tamoxifen resistance. These strong relationships between EGFR and ERBB2 in terms of Tamoxifen efficacy could make Tamoxifen an indirect pathway inhibitor of EGFR-TKD. More in-depth studies are needed to qualify this proposal. In this light, the repositioning of Tamoxifen as an ERBB2 inhibitor into a candidate EGFR-TKI could lead to further research and studies regarding the ability of this drug to inhibit the development of cancer cell lines resulting from EGFR overexpression, including breast cancer. This provision requires further and more extensive selective investigations. This is one of the main motivations for including this drug as a potential EGFR-TKI alongside Erlotinib in the current study. This insight may be useful in driving further research related to drug repositioning approaches.
2.2. Unlocking the potential of virtual screening: An exploration of methodology
• Selected Phytochemicals
This study used 50 phytochemicals derived from cannabinoids and Cannabis terpenes as inputs for early drug discovery using computational approaches to identify potential EGFR-TKIs candidate drugs for cancer therapy. For this, we adopted the Structure-Based Drug Design strategy as an input to implement this in silico computational study. In the light of this, our basic stage is to use a hybrid strategy based on the Molecular Docking approach to investigate the affinity and structural compatibility of the EGFR protein with the fifty cannabinoid/terpene phytochemicals. Table S1 in the supplementary information lists the identification details of these substances available in the PubChem library, including their chemical names, 2D structures, and ID codes.
-
•
Docking-Based Virtual Screening Strategy
Bioinformatics techniques have been successfully implemented in the drug design and discovery pipeline, resulting in many successes. These techniques allow for quick screening and prediction of compounds' biological activity, making them especially attractive to the pharmaceutical industry. Molecular docking is a particularly important application of these techniques in the virtual screening [69,70]. Automated docking tools such as HADDOCK, Z-Dock, Molecular Operating Environment (MOE), PyRx, and AutoDock have been developed for various applications. AutoDock is specifically designed to dock small molecules to a protein receptor, while Z-Dock and HADDOCK are tailored for predicting protein-protein docking poses [71]. In this study, we implemented the virtual screening platform by utilizing rigid and flexible Molecular Docking with Autodock Vina 1.1.2 and Autodock 4. These two tools rely upon genetic algorithms (GA) and empirical scoring function (SF) to accurately predict potential drug candidates from large sets of compounds [72,73]. By harnessing the computational power of modern computers, Autodock Vina and Autodock 4 can quickly identify novel molecules with high binding affinity which may have previously gone unnoticed. The combination of genetic algorithms and scoring functions make virtual screening (VS) using Autodock Vina and Autodock 4 powerful tools for drug discovery and development. On this basis, we applied this hybrid Molecular docking approach to construct a VS model, in order to increase the affinity between the ligand and target protein, as well as to identify the ideal orientation and conformation of the ligand in the target protein's active binding pocket. We leveraged Semi-flexible and Flexible hybrid molecular docking methodology to construct a virtual screening model that would improve the binding between the ligand and the target protein, and identify the orientation and optimal conformation of the ligand in the active binding pocket of the target protein (for more information, see the supplementary information document).
-
•
Assessing Crystal Structure Selection Using Criteria-Based Analysis
When selecting a crystal 3D structure from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) for a CADD study, resolution, ligand flexibility, size, symmetry, composition, existing data or literature, available tools, extraction method and organism type should be taken into consideration [74,75]. High-resolution structures can provide exact details, while highly flexible ligands may not be suitable for Structure-Based Drug Design (SBDD) modeling. Large complexes tend to be more challenging to model, while complexes with fewer components may be simpler. Symmetry can simplify the analysis, and existing data or literature can help in performing in silico drug design modeling. Necessary tools should be available for in silico modeling, and the extraction method and type of organism should also be taken into account.
•Structure of the targeted protein
The 3D crystal structure of EGFR complexed with Erlotinib (PDB code: 1M17) was obtained from the RCSB Protein Data Bank for this study (Fig. 1). The structure of EGFR in complex 1M17 consists of a single chain of 333 amino acids, with a molecular weight of 37.88 kDa, making it a suitable target for CADD-based studies. The small weight of the Erlotinib co-ligand (393.4 Da) is also beneficial for identifying an active site of interaction and the active amino acid residues in the EGFR-Erlotinib complex (1M17). Moreover, the experimental data such as Extraction Method (X-ray diffraction), Organism (Homo sapiens), Resolution (2.6 Å), R-Value Free (0.295), R-Value Work (0.251) and R-Value Observed (0.251), Expression System (Escherichia coli) further supports its suitability as an input for CADD-based studies [76]. In this study, we adopted the 3D crystal structure of the EGFR-Erlotinib complex (1M17) to implement a hypothetical assay based on molecular docking, MM-GBSA, and molecular dynamics simulations. The aim of this study was to assess the potential of phytochemical molecules to inhibit EGFR activity by using Erlotinib as a reference drug. This was in line with the subject of our current and previous studies [40,77], and it was also supported by the extensive use of this complex in the literature [76].
-
•
Identifying Potential Tyrosine Kinase Inhibitors
Fig. 1.

3D visualization of Erlotinib inhibitor in the active EGFR-TKD pocket.
The Erlotinib drug interactions in the EGFR active pocket of the 1M17 complex served as benchmarks in this study to identify candidate agents as novel EGFR-TKIs using molecular docking-based virtual screening [78,79]. We had selected Erlotinib as the standard drug in this study to identify and classify candidate molecules as EGFR-TKIs for lung cancer targeted therapy. However, given the critical importance of virtual screening based on molecular docking to reposition and target drugs for various therapeutic purposes, we included Tamoxifen as a standard drug to rank candidate molecules as EGFR-TKIs for targeted therapy against breast cancer. This ranking is established by comparing the binding energies of the examined phytochemical compounds with those of the standard drugs Tamoxifen and erlotinib in Protein-ligand complexes. Accordingly, the chance of a ligand binding to a protein increases with the amount of binding free energy produced by that protein's binding, lowering the complex's overall energy and making it more stable (more negative binding free energy). In light of this, we selected the ligands as prospective TKIs for dual-targeted therapy against lung and breast cancer that have more negative free binding energies than Erlotinib and Tamoxifen. Whereas, potential TKIs for targeted therapy against lung cancer includes ligands with more negative free binding energies compared to Erlotinib alone and less negative compared to Tamoxifen.
2.3. Preparation of the protein and the ligand
Using Molecular Operating Environment (MOE), Discovery Studio 2016, and AutoDockTools-1.5.6 tools, the EGFR-TKD protein structure was optimized and adapted for molecular docking simulation. During this step, missing side chains were added to the receptor backbone, polar hydrogens and a Gasteiger partial charge were added, water molecules and non-protein elements associated with the protein structure were also, removed. Using AUTOGRID based algorithms [80], the grid box coordinates of the ligand docking grid were determined in an accordance with the coordinates of the region occupied by the original Erlotinib ligand (4-anilinoquinazoline) inside the EGFR-TKD active pocket.
The depth of the XYZ input grid directions was fixed at 45 * 25 * 20 Å3 and the number of points in each direction were defined as x = 22.030 Å, y = 0.467 Å, z = 52.836 Å with a spacing of 0.375 Å. In this work, the seven rigid-flexible amino acid residues, Lysine (Lys721), Valine (Val702), Methionine (Met769), Alanine (Ala719), Leucine (Leu694 and Leu820) and Aspartic (Asp776) with which Erlotinib interacts were used as references in the analysis of ligand-protein interaction patterns contributing to the inhibition of EGFR-TKD enzymatic activity (Fig. 1). Cannabinoids (C1–C12) and Terpenes (T1-T38) ligands, as well as Tamoxifen and Erlotinib ligand structures, were obtained from the National Library of Medicine (https://pubchem.ncbi.nlm.nih.gov).
All collected 50 ligands were re-modeled and optimized for their use as inputs in molecular docking simulations using SYBYL-X 2.1.1, GaussView 6.0, Gaussian 09W, AutoDockTools-1.5.6, and Discovery Studio 2016 [81].
2.4. Receptor design validation and mapping of active site coordinates
Given the importance of molecular modeling based on molecular docking in a computer-aided drug design approach, this protocol of docking must be initially validated. Therefore, we validated the structure of the EGFR-TKD (PDB ID:1M17) protein and the performance of the adopted molecular docking algorithms before proceeding to the second stage of simulations. To do this, we re-docked the Erlotinib ligand in the EGFR-TKD pocket as defined in Fig. 1, then matched the original ligand with the re-docked and evaluated the level of superimposition between them by computing the DockRMSD (root-mean-square deviation) value (https://zhanggroup.org/DockRMSD/).
When the RMSD value is < 2 Å, the adopted molecular docking approach is suggested to be valid and reliable in predicting protein-ligand interactions [82,83].
2.5. Developing a virtual screening model
Following the validation of the molecular docking protocol, we docked 50 cannabinoid and terpene ligands into the active pocket of EGFR-TKD. This was done to evaluate the affinity energies of the ligands towards the active amino acid residues in the EFGR-TKD pocket. In the present screening study, we examined protein-ligand interactions through a molecular docking procedure using the AutoDockVina 1.1.2 and AutoDock 4.2.6 packages, respectively [72,84]. This selection was adopted due to the accuracy and quality of the simulation of protein-ligand interactions provided by algorithms of these tools compared to other programs, particularly with respect to the affinity energies of ligands towards protein receptors and the prediction of the non-covalent interaction patterns [85].
2.5.1. Autodock VINA virtual screening
During this screening, it should be noted that it is possible to perform a parallel set of semi-flexible molecular docking operations for multiple ligands to the same receptor and obtain separate data for each operation. Then, we inspected the output of the molecular docking by comparing the affinity energies of the ligand conformations to the active side chain residues of EGFR-TKD. In this systematic screening, we selected conformations (Flexible body ligands) whose affinity for the EGFR-TKD receptor (Rigid body protein) was more negative than or closer to that of the standard drugs, Tamoxifen and Erlotinib. In parallel, we eliminated EGFR-TKD incompatible phytochemicals that require very high binding energies to bind to them. This was done to ensure that we do not select structures that produce high affinities between the intermolecular and the receptor-ligand, which would likely cause an irreversible inhibition through the formation of covalent bonds (suicide inhibition). Whereas, we focused on ligands that produce low affinity (more negative binding energy) and lower force between intermolecular and receptor-ligand through the formation of non-covalent interactions (reversible inhibition).
2.5.2. Analysis of lead compounds for drug-like properties and ADME-Tox effects
After completing the screening of phytochemicals for their affinity energies with EGFR-TKD, we proceeded further to study their drug-like profiles, their pharmacokinetic/pharmacodynamic (ADME-Tox) profiles. This is a routine procedure in silico screening because of its great importance in the drug design and discovery process before moving to the experimental and drug trials phases. In addition, this screening procedure is very important in predicting the toxicity risks of molecules and their adverse effects on the efficacy of candidate drug molecules. For example, the properties of Erlotinib, which has shown in vitro, vivo and silico a set of undesirable deviations in safe drugs, as a result, clinical trials of this drug have shown unsatisfactory results [86]. It is therefore necessary to evaluate the pharmacokinetic properties of drug molecules in silico before making their use available.
2.5.3. Key criteria for lead compound selection
In this step, we identified candidate drug phytochemicals based on the detailed drug-like profiles of the desired bioavailability criteria of the drug molecules, based on the computational predictions provided via the SwissADME (http://www.swissadme.ch/index.php). Then, based on the pkCSM (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4434528/), we screened the ADME-Tox properties of the drug molecules that satisfied the bioavailability criteria proposed by Lipinski Rule of Five (RO5) and Viber rules [87]. In this regard, the most important parameters for which phytochemicals were evaluated for their lipophilicity (Log P), physicochemical properties (molecular weight, number of rotational bonds, number of H-bond acceptors and donors, Molar Refractivity), and medicinal chemistry (Bioavailability Score, synthetic accessibility) [87].
On the other hand, pharmacokinetic ADME-Tox parameters of phytochemicals were evaluated by predicting their absorption properties (skin permeability and human intestinal absorption) and distribution properties (human volume of distribution at steady state (VDss), unbound fraction, blood-brain barrier permeability (BBB), Central Nervous System permeability (CNS), metabolism properties (actions towards cytochrome P450 enzymes), excretion properties (Total Clearance index), toxicity parameters (AMES toxicity, Oral Rat Acute Toxicity (LD50), Hepatotoxicity, Maximum recommended tolerated dose). Drug-like and ADME-Tox predictions were supported by Osiris computations to evaluate the potential toxicity risks of selected phytochemicals (https://www.organic-chemistry.org/prog/peo/) [88].
Osiris computations provided molecular toxicity risk profiles, such as risks associated with mutagenicity, carcinogenicity, reproduction, and irritation that may be caused by undesirable radicals in the structure of SMILES-encoded molecules [89]. Once we had collected drug and pharmacodynamics property profiles of candidate molecules to inhibit EGFR-TKD enzymatic activity, we tested the binding interactions of these ligands to the active pocket of EGFR-TKD to confirm their safety. To achieve this goal, we followed the rational examination below.
2.6. Analysis of protein-ligand complex conformational stability
Despite the identification of drug-like and pharmacokinetics/pharmacodynamics properties of lead compounds, the pattern and requirements of the interaction of these compounds with target enzyme receptors remain a question that requires advanced biophysical modeling to solve. Therefore, in the present study, thus we decided to evaluate a set of helpful biophysical parameters to simplify the insight in this regard. For this purpose, we combined the following modelling techniques: Flexible Molecular Docking, Mechanized Generalized Surface Area (MM-GBSA) Calculations, and Molecular Dynamics Simulations (MDS) to provide biophysical profiles of the conformational pattern of Cannabis sativa L. as Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR TKIs). In this step of screening, we will consider the expected interaction patterns of the lead flexible ligands against the flexible active amino acid side chain in the EGFR-TKD active pocket, estimated binding energies (BE) of the ligands towards the receptor, estimate the micromolar (μM) values of the inhibitory concentrations (Ki), examine the structural and dynamic behavior of the proposed EGFR-TKIs.
-
•
Predict binding energies and inhibitory activity concentrations
For this purpose, using a Lamarckian genetic algorithm (LGA) and an empirical binding free energy function available in AutoDock 4.2.6, we can get: The top conformational binding mode profiles (lowest free binding energy with the minimum RMSD distance cutoff) in all selected EGFR-inhibitors complexes. Also, estimate the inhibitory concentration required to yield 50% of the maximum inhibition of the EGFR-TKD enzymatic activity based on the theoretical inhibition constants (, where R = 1.985 × 10−3 kcal K−1 × mol−1 and T = 298.15 K), the binding energy (BE) is the estimated free energy of binding, R is the ideal gas constant and T is the ambient temperature (298.15 K). (For details, see the supplementary information).
-
•
MM-GBSA Free Energies (ΔGbind) assessments
Using the Prime/MM-GBSA computations provided in Schrodinger Suite 2020-3, at pH 7 ± 2 the free binding energy ΔGbind for EGFR-TKIs systems were prepared according to the OPLS3e force field and the VSGB 2.1 solvent model [90,91]. This procedure was implemented as an aid protocol for molecular docking simulations towards the identification of potentially most stable systems for their validation by molecular dynamics simulation (for details, see the supplementary information). The examined EGFR-TKIs complexes were prepared and primed for Prime MM-GBSA scores using the protein prepwizard (Protein Preparation Wizard) on the Maestro 12.5 interface [92].
As part of the preparation, hydrogen was added to the 3D structure of the systems, disulfide bonds were formed, generated ionization/tautomeric states for all moieties (amino acid side chains and ligands), H-bond and system-based functional enhancement were optimized using OPLS3e (Optimized Potentials for Liquid Simulations) force field.
The Epik (Empirical pKa Prediction) function was incorporated during system preparation to provide appropriate resolution for protonation states of ligands during the drug discovery steps [93]. This was due to the pKa of the functional groups plays a major role in defining the pharmacokinetic profile and structural dynamics behavior of the functional groups that characterize the drug [93]. Epik module allowed us to reliably predict the protonated state of ligands, allowing us to understand the discrete interactions that cause ligands to bind to the target protein. However, in the virtual screening, we find a set of tools that provide known rigid structures containing all functional groups but do not take into account the protonation and tautomeric states of the ligands.
-
•
Molecular dynamics simulations
Using Desmond/GPU, multiple 200 ns molecular dynamics simulations were performed to analyze the dynamic and structural behavior of refined samples of EGFR-inhibitor systems under the OPLS3e force field and in a water-based SPC (Simple Point-Charge) model (for details, see the supplementary information). This simulation aided in the final stage of the overall learning process of the stability dynamics of free EGFR and EGFR-TKIs complexes in a laboratory-like virtual model. The most negative free binding energies of the ligands with EGFR, the estimated Ki concentrations, and the most stable conformations of the ligands in terms of functional groups used to select protein-drug samples for molecular dynamics tests.
3. Result and discussion
3.1. Validation of the structural model of EGFR-TKD protein (PDB ID:1M17)
Fig. 2 shows a 3D/2D overview of the conformation of the native (yellow) and re-docked (black) Erlotinib drug in the EGFR-TKD protein active pocket. Results of the molecular re-docking simulation showed that the binding affinity of Erlotinib towards EGFR-TKD was −8.2 kcal/mol and the ideal RMSD value of the conformational superimposition mode b/w the ligands (original/re-docked) was 0.155 Å. It was also observed that the re-docked Erlotinib interacted with the identical amino acid residues as the original Erlotinib in the 1M17 crystal complex. The RMSD value less than 2 Å and the close proximity between the interaction profiles of the original and re-docked Erlotinib validate the molecular docking protocol used in the present investigation. Therefore, the rest of the molecular docking simulations in this study can be completed with confidence.
Fig. 2.

The conformational positions of the original Erlotinib inhibitor (yellow)/re-docked (black) inside the active EGFR-TKD pocket. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.2. Screening the affinity of the ligands towards the active binding site
Table S2 reports the binding affinities of the ligands (Cannabinoids Isolated: C1–C12 and Cannabis Terpenes: T1-T38, Drugs: Tamoxifen, and Erlotinib) towards EGFR-TKD (PDB ID: 1M17). The results are presented in Table S2; which is compiled from the output of a semi-flexible molecular docking model performed by Autodock vina 1.1.2. From Table S2, we can notice that all values of binding affinities for docked ligands inside the active pocket of EGFR-TKD were significantly low (more negative). This indicates that Cannabis sativa L-based compounds are structurally consistent with EGFR-TKD, and that these compounds can establish a non-covalent binding with EGFR-TKD. Thus, these phytochemicals may be attractive reversible inhibitors of EGFR.
From Table S1, the binding affinities of ligands belonging to the isolated cannabinoids class recorded the highest value for C2 (−7.2 kcal/mol) and the lowest value for C11 (−11.3 kcal/mol). The ligands C1 (−9.9 kcal/mol), C4 (−9.8 kcal/mol), C5 (−9.9 kcal/mol), C6 (−10.8 kcal/mol), C7 (−10.7 kcal/mol), C8 (−10.1 kcal/mol), C9 (−10.0 kcal/mol), C10 (−8.4 kcal/mol) and C11 (−11.3 kcal/mol) recorded more negative binding affinities than Tamoxifen (−9.4 kcal/mol) and Erlotinib (−8.2 kcal/mol), which means that the structural properties of these molecules are more selective and compatible with EGFR- TKD compared to the reference drugs Tamoxifen and Erlotinib. Therefore, C1 (Cannabidiolic acid), C4 (Cannabidiol), C5 (Tetrahydrocannabivarin), C6 (Cannabinol), C7 (Dronabinol), C8 (Delta-8-Tetrahydrocannabinol), C9 (Cannabicyclol), andC11 (Delta9-tetrahydrocannabinolic acid) phytochemicals can be screened as potential anticancer reversible inhibitors against breast and lung cancers. Whereas the binding affinities of the ligands (C10 and C12) were less negative than those of Tamoxifen and more negative than those of Erlotinib, this means that both phytochemicals (C10: Cannabichromene and C12: Cannabichromenic Acid) have promising prospects against lung cancer.
On the other hand, from Table S1 we can notice that the binding affinities of ligands belonging to the Cannabis Terpenes class reached the highest value −5.5 kcal/mol for T2, T7 and T11, and the lowest value was −8.8 kcal/mol for T17. Apparently, all ligands (T1-T38) had a more limited negative binding affinities compared to Tamoxifen. This means that the interactions of the Cannabis Terpenes with EGFR-TKD are less likely to be compatible than those established by Tamoxifen and Cannabinoids isolated against breast cancer. However, the ligands T15 (−8.4 kcal/mol), T17 (−8.8 kcal/mol), T25 (−8.4 kcal/mol), and T28 (−8.5 kcal/mol) recorded more negative binding affinities than Erlotinib (−8.2 kcal/mol) toward the active EGFR-TKD pocket. This indicates that T15 (Beta-Caryophyllene), T17 (Nerolidol), T25 (Gamma-Elemene), and T28 (Beta-Eudesmene) are candidate reversible inhibitors agents for EGFR-TKD against lung cancer. Also, Table 1 shows that Tamoxifen has a higher negative binding energy towards EGFR (−9.4 kcal/mol) than Erlotinib (−8.2 kcal/mol), which may support its potential as a tyrosine kinase inhibitor that could specifically target EGFR. Therefore, repositioning Tamoxifen as a dual therapeutic strategy in lung and breast cancer targeted therapy may be effective, but further study and predictions are required. Table 1 summarizes filtered ligands based on their binding affinities and selectivity against breast and lung cancer.
Table 1.
Overview of the binding affinity of candidate EGFR-TKD inhibitors.
| Category of lead compound | Ligand name/2D Structure | binding affinity (kcal/mol) | Favorable affinity against |
|---|---|---|---|
| Isolated cannabinoids | C1 (CBDA) | −9.9 | breast and lung |
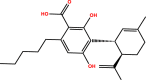 | |||
| C4 (CBD) | −9.8 | breast and lung | |
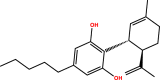 | |||
| C5 (THCV) | −9.9 | breast and lung | |
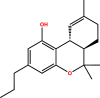 | |||
| C6 (CNB) | −10.8 | breast and lung | |
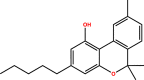 | |||
| C7 (Δ-9-THC) | −10.7 | breast and lung | |
 | |||
| C8 (Δ-8-THC) | −10.1 | breast and lung | |
 | |||
| C9 (CBL) | −10.0 | breast and lung | |
 | |||
| C10 (CBC) | −8.4 | lung | |
 | |||
| C11 (THCA) | −11.3 | breast and lung | |
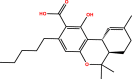 | |||
| C12 (CBCA) | −8.5 | lung | |
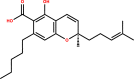 | |||
| Cannabis Terpenes | T15 (BCP) | −8.4 | breast and lung |
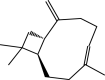 | |||
| T17 (NRD) | −8.8 | breast and lung | |
 | |||
| T25 (γ-Ele) | −8.4 | breast and lung | |
 | |||
| T28 (β-EUD) | −8.5 | lung | |
 | |||
| Standard drug | Tamoxifen | −9.4 | breast |
 | |||
| Erlotinib | −8.2 | lung | |
 |
3.3. Drug-like properties and toxicity risks
Table 2 provides a profile of the drug-like properties and potential toxicity risks of the Cannabis sativa L. phytochemicals proposed in this study. Concerning the bioavailability parameters estimated on the bases of Lipinski RO5 and Veber's Rule, we can notice that the identified Cannabis sativa L. compounds satisfy all conditions necessary for a good bioavailability. This can be confirmed by the absence of violations in Lipinski RO5 and Vebers' criterion. For standard drugs, Tamoxifen and Erlotinib, respectively, there were two violations in lipophilicity (LogP>5) and number of rotational bonds (nROB = 10 bonds). This means that the structural properties of these two drugs may be insufficient to ensure their proper absorption by the human intestine, which may cause a poor response of the drug as desired.
Table 2.
The Drug-like pattern and Toxicity risks of the screened molecules.
| Comp. | Bioavailability criteria |
DruglikenessVio. |
F |
SA |
Tox-R |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MW |
LogP |
nHBA |
nHBD |
nROB |
MR |
RO5 |
Viber |
>10% | 1–10 | +/±/- | |
| ≤500 D | <5 | <10 | <5 | <10 | 40–130 | 0 ≤ Vio≤2 | |||||
| C1 (CBDA) | 358.47 | 3.79 | 4 | 3 | 7 | 106.81 | 0 | 0 | 0.56 | 4.25 | - |
| C4 (CBD) | 314.46 | 4.31 | 2 | 2 | 6 | 99.85 | 0 | 0 | 0.55 | 4.05 | - |
| C5 (THCV) | 286.41 | 3.94 | 2 | 1 | 2 | 88.30 | 0 | 0 | 0.55 | 4.05 | - |
| C6 (CNB) | 310.43 | 4.23 | 2 | 1 | 4 | 97.10 | 0 | 0 | 0.55 | 3.39 | - |
| C7 (Δ-9-THC) | 314.46 | 4.39 | 2 | 1 | 4 | 97.91 | 0 | 0 | 0.55 | 4.27 | - |
| C8 (Δ-8-THC) | 314.46 | 4.39 | 2 | 1 | 4 | 97.91 | 0 | 0 | 0.55 | 4.21 | - |
| C9 (CBL) | 314.46 | 4.48 | 2 | 1 | 4 | 96.01 | 0 | 0 | 0.55 | 4.37 | - |
| C10 (CBC) | 314.46 | 4.31 | 2 | 1 | 7 | 100.34 | 0 | 0 | 0.55 | 4.26 | +RE, +MG |
| C11 (THCA) | 358.47 | 3.88 | 4 | 2 | 5 | 104.87 | 0 | 0 | 0.85 | 4.43 | - |
| C12 (CBCA) | 358.47 | 3.79 | 4 | 2 | 8 | 107.30 | 0 | 0 | 0.85 | 4.40 | +RE, +MG |
| T15 (BCP) | 204.35 | 4.63 | 0 | 0 | 0 | 68.78 | 0 | 0 | 0.55 | 4.51 | - |
| T17 (NRD) | 222.37 | 3.86 | 1 | 1 | 7 | 74.00 | 0 | 0 | 0.55 | 3.53 | +IRR |
| T25 (γ-Ele) | 204.35 | 4.53 | 0 | 0 | 2 | 70.42 | 0 | 0 | 0.55 | 3.81 | - |
| T28 (β-EUD) | 204.35 | 4.63 | 0 | 0 | 1 | 68.78 | 0 | 0 | 0.55 | 3.42 | - |
| Tam | 371.51 | 5.10 | 2 | 0 | 8 | 119.72 | 1 | 0 | 0.55 | 3.01 | +RE |
| Erlo | 393.44 | 1.89 | 6 | 1 | 10 | 111.40 | 1 | 0 | 0.55 | 3.19 | - |
MW: Molecular weight; MR: Molar Refractivity, LogP: Lipophilicity; nHBA: Num. H-bond acceptors; nHBD: Num. H-bond donors; nROB: Num. rotatable bonds; MR: Molar Refractivity, nVio: Num. volations, F: Bioavailability Score, SA: Synthetic accessibility.Tox-R: Potential toxicity risks: IRR: Irritant, RE:Reproductive effective, MG: Mutagenic, TUMO: Tumorigenic (+: highly toxic; -: not toxic; ±: slightly toxic. Tam: Tamoxifen, Erl: Erlotinib.
Regarding the bioavailability score (F), values of F> 50% indicates that more than 50% of the orally dosed drug can reach the systemic circulation and allow them to cross a variety of barriers in the process of drug metabolism. Regarding the synthetic accessibility index (SA), values of SA <10 for the examined compounds indicate the easy synthesis and evaluation of these compounds in vitro. Regarding the toxicity risk (Tox-R) parameters evaluated by Osiris calculations, we can notice that the structures of the molecules C10 (CBC), C12 (CBCA), C17 (NRD) and the reference drug Tamoxifen can cause toxicity risks represented by the influence on the reproduction, mutagenicity and irritation when these molecules are administered as a drug.
The overall drug-like predictions, combined with Osiris calculations, indicate that the phytochemical structures C1 (CBDA), C4 (CBD), C5 (THCV), C6 (CNB), C7 (Δ-9-THC), C8 (Δ-8-THC), C9 (CBL), C11 (THCA), T15 (BCP), T25 (γ-Ele), and T28 (β-EUD) screened from Cannabis sativa L. may be more compatible and suitable for use as a better anti-cancer drug compared to the reference drugs Tamoxifen and Erlotinib. Therefore, to avoid these risks, we exclude the phytochemicals C10 (CBC), C12 (CBCA) and T17 (NRD) from safe drug candidate selection. Consequently, we analyze in silico the ADME-Tox properties of the remaining phytochemicals C1 (CBDA), C4 (CBD), C5 (THCV), C6 (CNB), C7 (Δ-9-THC), C8 (Δ-8-THC), C9 (CBL), C11 (THCA), T15 (BCP), T25 (γ-Ele), and T28 (β-EUD).
3.4. In silico ADME-tox modeling
For our selected set of phytochemicals cannabinoids C1 (CBDA), C4 (CBD), C5 (THCV), C6 (CNB), C7 (Δ-9-THC), C8 (Δ-8-THC), C9 (CBL), C11 (THCA) and the cannabis terpenes T15 (BCP), T25 (γ-Ele), T28 (β-EUD), we have noticed that majority of compounds represented highest % human oral absorption (HOA) profiles (>90%), when calculated using ‘ADME-Tox’ (Table S3). Our best docked compound, C11 (>97.56% HOA) retained an absorption comparable to standard drugs, Tamoxifen (Tam) and Erlotinib (Erlo), i.e., Erlo (97.80% and 95.26% HOA, respectively). Furthermore, isolated cannabinoids (CBDA, CBD, THCV, CNB, Δ-9-THC, Δ-8-THC, CBL, THCA) demonstrated comparable skin permeability (log Kp) with respect to drugs, Tamoxifen and Erlotinib (log Kp: 2.73 each).
Among isolated cannabinoids, Δ-9-THC, Δ-8-THC, CBL are likely to exert lesser pharmacological actions, as denoted by little to no unbound fractions. Consequently, it is possible that these compounds have a structure appropriate for binding to the protein and not to the plasma. However, it is worthy to note that, cannabis terpenes, BCP and γ-Ele had noted with significant unbound fractions (0.26 and 0.14, respectively) comparable to drugs (Tam: 0.16 and Erl: 0.14). The steady state volume of distribution (VDss) and the blood-brain barrier (BBB) permeability properties were also found to be optimum for compounds in current study (Table S3). The total body clearance or total plasma (blood) clearance (CL) is commonly defined as the volume of plasma (blood) completely cleared of drug per unit time [94].
With this aspect, isolated cannabinoids and cannabis terpene showed to have high total clearance rates. Except compounds, CNB and β-EUD, all compounds were found to have no ‘AMES toxic’ profile. If a species has a high ORAT LD50 it means it has a high tolerance to the poison. A low ORAT LD50 means the species is highly susceptible to the poison. Considering this analogy, we noticed that except compounds, BCP, γ-Ele, and β-EUD, all others, isolated cannabinoids exhibited higher LD50 values than drug, Tamoxifen, but lesser than Erlotinib. Phytochemicals (CBDA, CBD, THCV, CNB, Δ-9-THC, Δ-8-THC, CBL, THCA) and (BCP, γ-Ele and β-EUD) depicted no hepatotoxicity. It is well known that Cytochrome P450 (CYP450) enzyme family plays crucial roles in metabolism of drug/xenobiosis. Substrate and inhibitory profiles for particular cytochrome enzyme, may lead alteration in drug metabolism and thus, generation of metabolites, which may alter pharmacological actions or excretion of drug. Phytochemicals (CBDA, CBD, THCV, CNB, Δ-9-THC, Δ-8-THC, CBL, THCA) and (BCP, γ-Ele and β-EUD) represented no substrate or inhibitory profiles against CYP-2D6 enzyme. Compounds, CBD, CNB, CBL, and β-EUD, including standards, found to be substrate for CYP-3A4. Further, except Δ-9-THC and Δ-8-THC, all others depicted non-inhibitory profile for CYP-3A4.
Phytochemicals, THCV, CNB, Δ-9-THC, Δ-8-THC, CBL exhibited inhibition against CYP-2C19. Among all phytochemicals, only C6 (CNB) would likely to show CYP-2C9 inhibition. All of these pharmacokinetics (PK) and pharmacodynamics (PD) properties are summarized in Table S3. Based on evaluation of the PK/PD properties (ADME-Tox) of the investigated phytochemicals as EGFR-TKIs, C1 (CBDA), C4 (CBD), C5 (THCV), C7 (Δ-9-THC), C8 (Δ-8-THC), and C9 (CBL), C11 (THCA), T15 (BCP), and T28 (γ-ELE) all have properties favorable for safe drug use, except for C6 (CNB) and T28 (β-EUD), which show potential toxicity. In this regard, an in vitro study conducted by Lamtha et al. [50] suggested that cannabinoid molecules CBD, CBG and CBN could act as EGFR-TKIs. However, their study did not assess the PK and PD properties of these molecules. In the present study, CNB was excluded from the drug candidates due to its potential toxicity, whereas CBD showed drug-like and ADME-Tox properties favorable for its use as a drug. On the other hand, CBG was not considered a suitable EGFR-TKI in this study due to its lower protein affinity (−7.2 kcal/mol) compared to the two standard drugs Erlotinib (−8.2 kcal/mol) and Tamoxifen (−9.4 kcal/mol).
3.5. Conformational stability analysis of EGFR- TKIs complexes
3.5.1. Characterization of screened EGFR-TKIs
Table S4 depicts the main results of the flexible molecular docking performed by Autodock 4.2.6 as well as the Prime MM-GBSA computations. The flexible molecular docking results generated the following parameters: estimated binding free energy (BE), estimated biological inhibitory activity (Ki), and interaction patterns between the ligands and the active site of EGFR-TKD.
Whereas, ΔGbind for EGFR-inhibitor complexes were generated from Prime/MM-GBSA calculations. The interactions of the proposed EGFR-TKIs (C1, C4, C5, C7, C8, C9, C11, T15, and T25) with the active EGFR-TKD pocket in the presence of Tamoxifen and Erlotinib are shown in Figs. S1 and S2, respectively, as predicted by flexible molecular docking and Prime MM-GBSA simulations.
The detailed findings described in Table S4 and Figs. S1 and S2 lead us to the following conclusions: The binding energies (BE) estimated by molecular docking ranged from −12.79 kcal/mol (EGFR-C11 complex) to −8.16 kcal/mol (EGFR-C4 complex) for the lead compounds belonging to isolated cannabinoids, and from −7.61 kcal/mol (EGFR-T15 complex) to −7.41 kcal/mol (EGFR-T25 complex) for the lead compounds belonging to cannabis terpenes. In comparison, the binding energy values of the two reference drugs ranged from −8.79 (EGFR-Tamoxifen complex) to −8.29 kcal/mol (EGFR-Erlotinib complex).
Inhibitory concentration (Ki) values were estimated by the flexible molecular docking ranged from 0.4115 E−3 μM (EGFR-C11 complex) to 1.0281 μM (EGFR-C4 complex) for lead compounds belonging to isolated cannabinoids, and from 2.603 μM (EGFR-T15 complex) to 3.650 μM (EGFR-T25 complex) for lead compounds belonging to cannabis terpenes. In comparison, the estimated inhibitory concentration values for the reference drugs were ranged from 0.4000 μM (EGFR-Tamoxifen complex) to 0.8253 μM (EGFR-Erlotinib complex).
The free binding energies (ΔGbind) were set to limits by Prime MM-GBSA module and ranged from −62.807 kcal/mol (EGFR-C11 complex) to −31.138 kcal/mol (EGFR-C1 complex) for isolated cannabinoids, and from −39.782 kcal/mol (EGFR-T15 complex) to −33.980 kcal/mol (EGFR-T25 complex) for cannabis terpenes. The binding energy values of the two standard drugs were ranged from −42.098 (EGFR-Tamoxifen complex) to −36.877 kcal/mol (EGFR-Erlotinib complex).
In all isolated cannabinoid complexes, the protein-ligand interactions were found to have hydrogen bond (conventional/carbon), hydrophobic, electrostatic (EGFR-C11 complex), and sulfur (EGFR-C5 and EGFR-C7 complexes), whereas in cannabis terpene complexes (EGFR-T15 and EGFR-T25), all interactions were of hydrophobic (Alkyl). Tamoxifen was interacted with the active amino acid residues in the EGFR pocket via hydrogen carbon bonds, hydrophobic interactions, and electrostatic (π-Cation) interactions, whereas Erlotinib interactions were hydrogen bond (conventional and carbon) and hydrophobic interactions.
Furthermore, in the inhibition of EGFR-TKD enzymatic activity, isolated cannabinoids and cannabis terpenes were able to generate multiple interactions in different modes with a set of reference amino acid residues. Visualizations of interacting candidate drug molecules with EGFR, are shown in Fig. S1. Following the MM-GBSA simulation, all of the examined ligands were not diverged from the active pocket of EGFR-TKD (Fig. S2), indicating that the proposed drug molecules' structure could achieve perfect conformance with the EGFR-protein TKD's structure.
Predictions of Epik (Empirical pKa Prediction) function under simulated physiological conditions were also indicated that the carboxylic acid moiety (COOH) in the C1 (CBDA) and C11 (THCA) structures was ionized to a carboxylate ion by loss of , whereas the other functional moieties of the ligands C4 (CBD), C5 (THCV), C7 (Δ-9-THC), C8 (Δ-8-THC), C9 (CBL), T15 (Δ-8-THC), T25 (γ-Ele) and Erlotinib were not protonated. Whereas Tamoxifen was ionized the ammonia () moiety by the receipt of the proton which formed the ammonium cation () on the Tamoxifen structure.
3.5.2. Validation of protein-ligand interactions' stability
In comparison to the two standard drugs (Tamoxifen and Erlotinib), overall results of the rational screening on Cannabis sativa L. showed that phytochemicals (CBDA, CBD, THCV, Δ-9-THC, Δ-8-THC, Δ-8-THC, THCA, Δ-8-THC, and γ-Ele) had flexible structural properties and favorable PK/PD properties to be used as safe cancer drugs. Because of their small molecular structures, these phytochemicals can bind to the active pocket of EGFR-TKD and perform several non-covalent interactions that are beneficial in inhibiting EGFR-TKD enzymatic activity. Therefore, there is great hope that these compounds will generate a new generation of EGFR-TKIs for targeted cancer therapy.
To validate this conclusion, we selected one sample from each category of Cannabinoids and Terpenes and closely examine their structural stabilities in situ inside the EGFR-TKD active pocket. Within each category, we have taken the sample with the best estimated inhibitory concentration (low Ki) and the lowest free binding energy estimated via Prime MM-GBSA computations as the samples for further examination. To this end, we had selected the phytochemical THCA (Ki = 0.4115 × 10−3 μM, ΔGbind = −62.807 kcal/mol) as a representative sample standard for the isolated cannabinoids category and the phytochemical BCP (Ki = 2.603 μM, ΔGbind = −33.980 kcal/mol) as a representative standard for the cannabis terpenes category and Tamoxifen (Ki = 0.400 μM), ΔGbind = −42.098 kcal/mol) and Erlotinib (Ki = 0.8253 μM, ΔGbind = −36.877 kcal/mol) as standarddrugs. Table 3 shows the most important parameters related to the interactions of the reference active amino acid residues (Leu694, Val702, Ala719, Lys721, Met769, Asp776, and Leu820) in the EGFR-TKD pocket with the samples C11 (THCA), T15 (BCP), Tamoxifen, and Erlotinib. Fig. 3 depicts a 2D visualization of the conformational profile of the selected samples' interactions in the EGFR-TKD active pocket (EGFR-C11, EGFR-T15, EGFR-Tamoxifen and EGFR-Erlotinib).
Table 3.
Interactions of sample TKIs with EGFR.
| Samples | EGFR-TKIs non-covalent Interactions |
Ki (μM) | ΔGbind (kcal/mol) | |||
|---|---|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic | Electrostatic | ||||
| THCA | EGFR-C11 | Conventional 7: THR830 (1.86, 1.85 Å), ASP831 (2.16, 2.99 Å), GLU738 (1.73 Å), LYS721 (2.21, 1.66 Å). Carbon 0. | Alkyl 16: ALA719 (4.99, 3.18 Å), MET769 (5.44, 4.60 Å), LEU820 (4.17, 4.60 Å), CYS773 (4.52 Å), VAL702 (4.94, 4.10, 5.15 Å), VAL768 (4.60 Å), LEU694 (5.46 Å), LEU723 (5.06 Å), ILE735 (4.88 Å), LYS721 (5.32 Å), LEU820 (4.70 Å). π-Alkyl 2: VAL702 (4.37 Å), LYS721 (4.40 Å). π-π T-shaped 0.π-Sigma 0 | π-Anion 1: ASP831 (4.70 Å) | 0.4115 × 10-3 | −62.807 |
| BCP | EGFR-T15 | ND | Alkyl 16: ALA719 (3.72, 3.99, 4.81 Å), VAL702 (4.77, 4.60, 3.75, 4.18 Å), LYS721 (5.32, 4.74, 4.07), MET769 (4.92, 4.45 Å), LEU820 (4.96, 4.92 Å), LEU694 (5.29 Å). | ND | 2.603 | −33.980 |
| Standard drugs | EGFR-Tamoxifen | Carbon 1: ASN818 (2.93 Å) | π-Sigma 1: ALA 719 (3.73 Å). Alkyl 5: ALA719 (4.11 Å), LEU694 (4.83 Å), VAL702 (5.22 Å), MET769 (4.30 Å), LEU820 (4.84 Å) π-Alkyl 6: VAL702 (4.98, 4.58, 5.23 Å), LYS721 (4.98, 4.02 Å), LEU820 (4.89 Å). π-π T-shaped 1: PHE699 (5.13 Å). | π-Cation 2: LYS721 (4.26, 4.96 Å) | 0.400 | −42.098 |
| EGFR-Erlotinib | Conventional 4: MET769 (2.65 Å), LYS721 (1.78, 2.22, 2.06 Å). Carbon 2: ASP831 (3.51 Å), GLU738 (2.83 Å). | π-Sigma 1: 1: VAL702 (3.91 Å). π-Alkyl 9: LEU694 (5.38, 4.17, 5.45 Å), ALA719 (5.24, 4.88 Å), LEU768 (5.41 Å), LEU769 (5.06 Å), MET769 Å (4.82 Å), LEU820 (5.14 Å) | ND | 0.825 | −36.877 | |
| Reference flexible amino acid residues | Leu694, Val702, Ala719, Lys721, Met769, Asp776 and Leu820 | |||||
- •EGFR-THCA interactions
Fig. 3.

EGFR-C11 interactions. (a) Flexible molecular docking predictions. (b) Prime MM-GBSA predictions. Amino acids surrounded by the red circle indicate reference sites, amino acids surrounded by the blue circle indicate novel active sites. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 3, Fig. 4, Fig. 5, Fig. 6 depict ligand interactions in the EGFR active pocket obtained through flexible molecular docking, while Fig. 3, Fig. 4, Fig. 5, Fig. 6 depict ligand interactions in the EGFR active pocket obtained through Prime MM-GBSA calculations in the VSGB 2.1 solvation model. For reference, Schrodinger algorithms change the names of standard residues such as ARG, ASP, GLU, LYS, and HIS to nonstandard residues RNA, ASH, GLH, LYN, HIE, and HIP as an informal convention for assigning different proton states to regular residues.
Fig. 4.

EGFR-T15 interactions. (a) Flexible molecular docking predictions. (b) Prime MM-GBSA predictions. Amino acids surrounded by the red circle indicate reference sites, amino acids surrounded by the blue circle indicate novel active sites. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 5.

EGFR-Erlotinib interactions. (a) Flexible molecular docking predictions. (b) Prime MM-GBSA predictions. Amino acids surrounded by the red circle indicate reference sites, amino acids surrounded by the blue circle indicate novel active sites. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 6.

EGFR-Tamoxifen interactions. (a) Flexible molecular docking predictions. (b) Prime MM-GBSA predictions. Amino acids surrounded by the red circle indicate reference sites, amino acids surrounded by the blue circle indicate novel active sites. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
For the EGFR-C11 complex, flexible molecular docking simulations were indicated that the binding of the C11 (THCA) to the EGFR active pocket was achieved through the formation of non-covalent interactions of conventional hydrogen bonds with residues THR830 (1.86, 1.85 Å), ASP831 (2.16, 2.99 Å), GLU738 (1.73 Å), LYS721 (2.21, 1.66 Å). In addition, it also formed hydrophobic alkyl interactions with residues ALA719 (4.99, 3.18 Å), MET769 (5.44, 4.60 Å), LEU820 (4.17, 4.60 Å), CYS773 (4.52 Å), VAL702 (4.94, 4.10, 5.15 Å), VAL768 (4.60 Å), LEU694 (5.46 Å), LEU723 (5.06 Å), ILE735 (4.88 Å), LYS721 (5.32 Å), LEU820 (4.70 Å). π-Alkyl 2: VAL702 (4.37 Å), LYS721 (4.40 Å) and π-Alkyl interactions with VAL702 (4.37 Å), LYS721 (4.40 Å), and interacted with residue ASP831 (4.70 Å) by the π-Anion electrostatic bonding. Among these interactions, the optimized THCA structure successfully formed multiple bonds with six side chain residues of the reference amino acid in the inhibition of EGFR enzyme activity, specifically LEU694, LEU820, ALA719, MET769, LYS721 and VAL702.
In addition, THCA interacted with seven novel active residues in the EGFR pocket, namely CYS773, LEU768, THR830, GLU738, ASP831, LEU723 and ILE735. On the other hand, the predictions of the Prime MM-GBSA energy calculations in VSGB solution indicated the stability of the THCA molecule structure within the EGFR active pocket and the maintenance of the same predicted interactions due to flexible molecular docking. This confirms that the THCA sample may be suitable for inhibiting EGFR enzymatic activity as well as achieving stability and compatibility with it in order to achieve the therapeutic goal. This means that the phytochemical compounds (CBDA, CBD, THCV, Δ-9-THC, Δ-8-THC, CBL and THCA) of the isolated cannabinoids may be potent new anticancer candidates based on targeting EGFR-TKD enzymatic activity.
-
•
EGFR-BCP interactions
For the EGFR-T15 complex, flexible molecular docking indicated that the interaction of BCP with the active EGFR pocket was done through the generation of non-covalent hydrophobic alkyl-like interactions with residues ALA719 (3.72, 3.99, 4.81 Å), VAL702 (4.77, 4.60, 3.75, 4.18 Å), LYS721 (5.32, 4.74, 4.07), MET769 (4.92, 4.45 Å), LEU820 (4.96, 4.92 Å), LEU694 (5.29 Å).
All amino acid residues with which T15 interacts in the EGFR active pocket are reference in the inhibition of EGFR-TKD enzymatic activity. Prime MM-GBSA simulations also indicated the stability of T15 within the active site in the EGFR pocket, confirming that the conformational structure of BCP is more suitable for inhibiting EGFR-TKD enzymatic activity. This suggests that cannabis terpene phytochemicals (BCP and γ-Ele) may be novel potent anticancer inhibitors of EGFR-TKD enzyme activity.
-
•
EGFR-Erlotinib
For the EGFR-Erlotinib complex, flexible molecular docking indicated that binding of Erlotinib to the EGFR active pocket was via the generation of non-covalent interactions via conventional hydrogen bonds with residues MET769 (2, 65 Å), LYS721 (1.78, 2.22, 2.06 Å) and carbon hydrogen bonds with residues ASP831 (3.51 Å), GLU738 (2.83 Å), as well as via the formation of hydrophobic π-Sigma interactions with VAL702 (3.91 Å) and with residues LEU694 (5.38, 4.17, 5.45 Å), ALA719 (5.24, 4.88) Å), LEU768 (5.41 Å), LEU769 (5.06 Å), MET769 Å (4.82 Å), LEU820 (5.14 Å) via π-Alkyl interactions. The interaction of Erlotinib in the EGFR active pocket was observed to preserve six reference interactions with residues LEU694, VAL702, ALA719, LYS721, MET769, LEU694.
The non-interaction with ASP776 is due to the optimization of the Erlotinib structure and also took into account the resulting torsional energies on the free energy emitted due to the rotational bonds found in the flexible ligand especially for the Dimethoxyethane moiety, as well as the flexibility of the amino acid residues used as references in the virtual flexible molecular docking screening. We can notice that following flexible molecular docking of the reference drug Erlotinib, the reference drug conformation generated new bonds with ASP831, GLU738, LEU768 and MET769. The conservation of all interactions in the EGFR-Erlotinib complex was confirmed by Prime MM-GBSA computations, thus validating the EGFR-Erlotinib model (PDB ID: 1M17) as a reference to simulate the mechanisms of Protein-drug interactions via its targeting. Thus, Erlotinib can be used as a reference in rational and theoretical comparisons for lung cancer drug design and discovery.
-
•
EGFR-Tamoxifen
For the EGFR-Tamoxifen complex, flexible molecular docking simulations indicated that Tamoxifen bound to the active EGFR pocket by generating non-covalent hydrogen-carbon bond interactions with ASN818 (2.93 Å), in addition it formed hydrophobic π-Sigma interactions with ALA719 (3.73 Å), alkyl interactions with residues ALA719 (4.11 Å), LEU694 (4.83 Å), VAL702 (5.22 Å), MET769 (4.30 Å), LEU820 (4.84 Å), π-Alkyl interactions with residues VAL702 (4.98, 4.58, 5.23 Å), LYS721 (4.98, 4.02 Å), LEU820 (4.89 Å), T-shaped π-π interactions with PHE699 (5.13 Å), and electrostatic π-Cation interactions with LYS721 (4.26, 4.96 Å). Prime MM-GBSA simulations showed that Tamoxifen retained most of its predicted interactions by molecular docking in the EGFR-TKD active pocket. Tamoxifen's interaction with the reference amino acid residues ALA719, VAL702, MET769, LEU820, and LYS721 further supports its potential as an EGFR inhibitor. Therefore, the selection of this drug as a reference in inhibiting EGFR enzymatic activity against breast cancer is valid for rational comparison and theoretical screening.
3.6. Molecular dynamics analysis (MDA)
The results of the obtained screenings indicated that the structures of the phytochemicals selected from the categories Cannabinoids and Terpenes are in high accordance with the structure of EGFR-TKD. This gives these phytochemicals a potential chance to form a new generation of EGFR-TKD inhibitors and to reach therapeutic targets against lung and breast cancer. To further verify, we simulated the molecular dynamics in the aqueous environment of C11 (THCA) and T15 (BCP) samples in the presence of the reference drugs Erlotinib and Tamoxifen. Molecular dynamics simulations of EGFR-TKD and its filtered complexes (EGFR-C11, EGFR-T15, EGFR-Erlotinib, and EGFR-Tamoxifen) were carried out for 200 ns using the same approach described in SI.
The dynamical and structural properties of the EGFR-TKIs sample systems were analyzed in terms of time scales of Root Mean Square Deviation/Fluctuation(RMSD/RMSF)/ of atomic positions, Protein-ligand interactions, Ligand structural properties (ligand RMSD, Radius of Gyration, Intra-molecular Hydrogen Bonds, Molecular Surface Area, Solvent-Accessible Surface Area, and Polar Surface Area), and thermodynamic properties of the (Total Energy, Potential Energy, Temperature, Volume, and Pressure).
3.6.1. RMSD and RMSF analysis
From Fig. 7a, we can deduce that the RMSD values of the EGFR backbone systems stabilized after ∼40 ns of the MD simulations path ranging up to ∼200 ns. The average values of the RMSD±SD evolutions of the average thermal structure derived between the reference frame (t = 0 ns) and the final frame (t = 200 ns) for the free EGFR, EGFR-Tamoxifen, EGFR-Erlotinib, EGFR-C11, EGFR-T15 backbone systems, were 2.10 ± 0.45 Å, 3.48 ± 0.34 Å, 2.14 ± 0.37 Å, 3.44 ± 0.81 Å and 3.35 ± 0.30 Å, respectively. SD (standard deviation) values less than 1 Å indicate that the structural conformation of the free and complexed EGFR protein did not show significant conformational changes during the MD simulations. This indicates that the structure of the EGFR protein is in perfect fit and equilibrium with the lead drag ligands THCA and BCP as well as with the reference drugs Tamoxifen and Erlotinib.
Fig. 7.

The MD simulations timelines for the EGFR systems uncomplexed, and complexed with the ligands C11, T15, Tamoxifen, and Erlotinib. (a) RMSD of C-alpha backbones of EGFR systems, (b) RMSF of side chains of EGFR systems, (c) RMSF of ligands complexed with EGFR.
The root means square fluctuations (RMSF) shown in Fig. 7b describe the local changes along the amino acid sequence of EGFR protein during the MD simulation run. The average values of RMSF±SD changes for the free EGFR and complexed with THCA, BCP, Tamoxifen and Erlotinib ligands were 2.02 ± 0.81 Å, 1.70 ± 0.95 Å, 1.72 ± 1.06 Å, 3.72 ± 1.22 Å, 1.58 ± 1.29 Å, respectively. It can be seen that the amino acid chain of the EGFR-Tamoxifen system underwent significant changes from the start to the end of the simulation, that can be explained by the significant average RMSF value of 3.72 Å. While the side chains of uncomplexed EGFR protein, EGFR-C11, EGFR-T15 and EGFR-Erlotinib, showed slight changes that can be explained by the average RMSF values ranging from 1.58 to 2.02 Å. It is also observed that the tails (N- and C-terminal) of the amino acid side chains fluctuate more than any other part of the protein, especially the amino acids numbered between 290 and 312 that are GLU961, ARG962, MET963, HIS964, LEU977, MET978, ASP9789, GLU978, GLU981, ASP982, MET983, ASP984, ASP985, VAL986, VAL987, ASP988, ALA989, ASP990, GLU991, TYR992, LEU993, ILE994, and PRO995.
Alignment of the protein-ligand complex to the uncomplexed protein backbone allows for the measurement of ligand-RMSF on the heavy atoms of the ligand. This provides insight into how the ligand fragments interact with the protein and bind in the active pocket of the protein. The ligand RMSF plot shown in Fig. 7c indicates the timeline of per-atom segmented ligand fluctuations, which corresponds to the two-dimensional structure of the C11 (THCA), T15 (BCP), Tamoxifen, and Erlotinib ligands.
The observed fluctuations of RMSF values for the ligands THCA, BCP, Tamoxifen, and Erlotinib reflect the fact that these ligands exhibit large internal atomic fluctuations during interaction with EGFR protein. This can be explained by the flexibility properties of this small molecule ligands. These properties allow the small molecules to provide various conformations and interaction patterns in the receptor protein cavity.
3.6.2. Protein-ligand interactions
Fig. 8 shows the profile of interactions that occur between the amino acid residues of EGFR with the ligands, C11 (THCA), T15 (BCP), Tamoxifen, and Erlotinib during the simulation time of the selected trajectory (0.00–200.00 ns). “Ligand-Protein Contacts” plots showing interactions that occur more than 10.0% of the simulation time between the atoms of the ligand and the amino acid residues of the protein. “Protein-ligand contacts” plots showing the time fractions of Protein-ligand interactions that maintained during the course of the simulation.
Fig. 8.

Detailed diagrams of local interaction dynamics of the ligands C11 (THCA), T15 (BCP), Tamoxifen and Erlotinib in the EGFR active pocket: (a) EGFR-C11 complex, (b) EGFR-T15 complex, (c) EGFR-Tamoxifen complex and (d) EGFR-Erlotinib.
“Timeline of interactions and contacts” diagram depicts timelines of interactions and contacts (H-bonds, hydrophobic, ionic, water bridges), the top panel shows the total number of specific contacts to the protein with which the ligand interacts during the simulation time course. The bottom panel of the diagram gives in detail the residues that interact with the ligand in each frame of the MD simulation course. Residues with more than one contact with the ligand are represented by darker shades of orange, as scaled on the right of the diagram.
From Fig. 8a, we can notice that the interactions of ligand THCA with the active amino acid residues in EGFR-C11 complex are stabilized via H-bond, hydrophobic, ionic, water-bridge like interactions. The amino acid residues ALA698, PHE698, PHE699, GLY700, VAL702, LYS721, ALA731, ILE735, GLU738, CYS751, LEU764, ASP813, LEU820, THR830, ASP831, PHE832, GLY833, and LEU834 have been identified as the main contacts with which the ligand C11 maintained their interactions during the MD simulation.
According to Fig. 8b (EGFR-T15 complex), the stability of the ligand BCP with the EGFR protein is due to hydrophobic interactions with the amino acid residues LEU694, PHE699, VAL702, ALA719, MET742, LEU768, MET769, CYS773, and LEU820.
According to Fig. 8c (EGFR-Tam complex), the majority of Tamoxifen-EGFR contacts were hydrophobic interactions with residues LEU694, PHE699, VAL702, ALA719, LYS721, LEU764, LEU768, LEU820, as well as H-bonding, water bridging, and ionic interactions with ASP831.
According to Fig. 8d (EGFR-Erlo complex), water bridges and hydrophobic interactions with residues LEU694, PHE699, VAL702, ALA719, LYS721, MET742, CYS751, LEU768, PRO770, ARG817, LEU820, THR830 as well as H-bond interactions with residues THR766 and MET769 are maintained between the Erlotinib drug and EGFR protein.
The residues LEU694, PHE699, VAL702, LYS721, CYS751, LEU820, THR830, LEU768, MET742 were identified as the major reference contacts with which the THCA and BCP ligands-maintained interactions during the MD simulation according to analysis of protein-ligand contact patterns using Tamoxifen and Erlotinib as references. As a result, the amino acid residues of Leucine, Phenylalanine, Valine, Lysine, Cysteine, and Threonine in the EGFR-TKD protein pocket can be considered as a key reference site favorable for threat modeling and achieving the desired biological function in order to inhibit the growth of breast and lung cancer cell lines by the phytochemical compounds of Cannabis sativa L (Cannabinoids and Terpenes).
3.6.3. Ligands properties
Fig. 9(a-d) shows the time courses of the most important properties of the ligands C11 (THCA), T15 (BCP), Tamoxifen and Erlotinib recorded during the MD simulation. The ligand RMSD plot shows the variations in the ligand's root mean square deviation (from 0.00 to 200.00 nsec) with respect to the first reference conformation (time t = 0). The plot of radius of gyration (rGyr) variations indicates the ligand's expansion, and the rGyr parameter reflects the ligand's main moment of inertia in this plot. The intramolecular hydrogen bonding (intraHB) plot represents the time tracking of the number of internal hydrogen bonds (HB) detected inside the ligand. The molecular surface area plot (MolSA) represents the molecular surface area calculated with a probe radius of 1.4 Å; the MolSA parameter measurement is equivalent to the van der Waals surface area. The surface area of a molecule that can be reached by one molecule of water is expressed by the solvent accessible surface area (SASA) plot. The polar surface area (PSA) plot is used to calculate the surface area of a molecule accessible to the solvent, to which only the oxygen and nitrogen atoms contribute.
Fig. 9.

Plots of ligand properties variations in the EGFR active pocket throughout the simulation trajectory (0.00–200.00 nsec): (a) C11 ligand (THCA), (b) T15 ligand (BCP), (c) Tamoxifen drug, (d) Erlotinib drug.
From Fig. 9(a-d), we can see that the RMSD property values for the ligands C11 (THCA), T15 (BCP), Tamoxifen, and Erlotinib stabilized within 0.5-1 Å (Fig. 9a), 0.3–0.9 Å (Fig. 9b), 1-1.5 Å (Fig. 9c), and 1-1.2 Å (Fig. 9d), respectively. For C11, T15, Tamoxifen, and Erlotinib ligands, respectively, the radius of gyration (rGyr) stability parameter ranged from 3.9-4 Å (Fig. 9a), 2.75–2.80 Å (Fig. 9b), 4.00–4.25 Å (Fig. 9c), and 4.80–4.95 Å (Fig. 9d). One intramolecular hydrogen bond (IntraHB) was found in the structure of the ligand THCA (Fig. 9a), whereas none were found in the structures of the ligands BCP (Fig. 9b), Tamoxifen (Fig. 9c), or Erlotinib (Fig. 9d). For the ligands THCA, BCP, Tamoxifen, and Erlotinib, respectively, the molecular surface area stability parameter (MolSA) ranged from about 350 to 360 Å2 (Fig. 9a), 232–236 Å2 (Fig. 9b), 375–390 Å2 (Fig. 9c), and 400–405 Å2 (Fig. 9d). For the ligands THCA, BCP, Tamoxifen, and Erlotinib, respectively, the stability of the solvent accessible surface area (SASA) parameter ranged from 50 to 55 Å2 (Fig. 9a), 0–100 Å2 (Fig. 9b), 140–160 Å2 (Fig. 9c), and 160–200 Å2 (Fig. 9d). For the ligands THCA, BCP, Tamoxifen, and Erlotinib, the polar surface area (PSA) stability parameters were 1120–120 Å2 (Fig. 9a), 0 Å2 (Fig. 9b), 5–10 Å2 (Fig. 9c), and 64–72 Å2 (Fig. 9d), respectively.
3.6.4. Thermodynamics properties
An overview of the quality of the MD simulation is given by the evaluation of thermodynamic parameters such as total energy (E), potential energy (EP), temperature (T), pressure (P), and volume (V) of protein-ligand systems. Table 4 and the graphical plots in Fig. 10(a-e) summarize the calculated mean values of thermodynamic properties and associated time scales that reflect the distribution of thermodynamic parameters E, EP, T, P, and V generated along a 200 ns trajectory from MD simulation runs for EGFR-C11 and EGFR-T15 samples, EGFR-Tamoxifen (Fig. 10c), EGFR-Erlotinib (Fig. 10d), and the free uncomplexed EGFR (Fig. 10e). Table 4 clearly demonstrates that the average values of E, EP, T, P, and V calculated for EGFR systems complexed with the ligands C11, T15, Tamoxifen, and Erlotinib were very close to the thermodynamic properties of the free EGFR protein. The stable distribution of the E, EP, T, P, and V parameter plots shown in Fig. 10a-e confirms these findings.
Table 4.
Computed average values of Total Energy (E), Potential Energy (EP), Temperature (T), Pressure (P), and Volume (V) properties for the EGFR protein systems examined.
| EGFR uncomplexed | EGFR-C11 | EGFR-T15 | EGFR-Tam | EGFR-Erlo | |
|---|---|---|---|---|---|
| Total energy (kcal/mol) | −141829.047 | −141737.555 | −141819.536 | −141574.711 | −141737.842 |
| Potential energy (kcal/mol) | −172395.959 | −172369.700 | −172428.177 | −172173.428 | −172326.123 |
| Temperature (K) | 298.690 | 298.687 | 298.695 | 298.679 | 298.693 |
| Pressure (bar) | 1.356 | 1.269 | 1.562 | 0.974 | 1.031 |
| Volume (Å3) | 505443.948 | 506482.028 | 506029.727 | 505868.647 | 505560.214 |
Fig. 10.

Thermodynamic properties timelines of Total Energy, Potential Energy, Temperature, Pressure and Volume for free EGFR (a) and the EGFR complexed w/C11 (b), T15 (c), Tamoxifen (d) and Erlotinib (e).
As a whole, the collected data generated by the post-docking validation procedures, including MM-GBSA computations and MD simulations conducted in this study, can support the structural match of EGFR-TKD with the nine phytochemical samples isolated from the Cannabinoids and Terpenes. Thus, these substances could become future TKIs against breast and lung cancer for use alone or in combination with other drugs such as Tamoxifen and Erlotinib.
By applying virtual screening models and combining them with computer-aided drug design approaches and biophysical simulations, this study identified nine cannabis compounds (CBDA, CBD, THCV, Δ-9-THC, Δ-8-THC, CBL, THCA, BCP, γ-Ele) as promising candidates for novel epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) in targeted cancer therapies. To further capitalize on these molecular scaffolds, further hit design and drug discovery efforts are warranted. To advance this concept and progress the lead compounds described in this work through the drug design pipeline, preliminary in vitro and in vivo assessments and analyses should be conducted. Finally, the advancements in scientific knowledge and technology have enabled the reintegration of natural products and their derivatives into drug design and development. This has provided an opportunity to overcome the technical barriers associated with detection, access, isolation, characterization, and valorization that had caused a decline in their use in the pharmaceutical industry beginning in the 1990s. Advances in analytical tools, engineering strategies applied to therapeutic research, and genome discovery in the agricultural sector have also facilitated this return. Consequently, the Cannabis sativa L. plant has been used as a model to challenge the stereotypes typically surrounding its recreational use in certain communities. Despite this, its production and consumption remain steady. In May 2021, the Moroccan state legalized the cultivation and export of Cannabis sativa L. for industrial, medical, and commercial purposes, excluding recreational purposes [95]. This could be a major shift in the region towards the legalization of the plant primarily for therapeutic use, considering Morocco's desirable climate, large territory, and convenient geographical location. Thus, this study aimed to identify and explore the potential opportunities in the field of Cannabis sativa L. to develop new medicinal agents, which could contribute to human safety and aid in the treatment of incurable diseases such as cancer.
4. Conclusion
Cancer is one of the leading causes of death worldwide, having accounted for more than 10 million deaths in 2020, equating to roughly one in every six deaths. Breast cancer, particularly in women, is the leading cause of cancer-related deaths, with an estimated 2.26 million lives lost to the disease in 2020. Lung cancer takes second place, responsible for 2.21 million deaths. In light of these alarming statistics, and the lack of effective treatment options, a study was conducted to explore the potential of phytochemicals derived from Cannabis sativa L to treat breast and lung cancers caused by abnormal enzymatic activity of EGFR-TKD (PDB ID:1M17). A total of fifty phytochemicals consisting of cannabinoids (C1–C12) and terpenes (T1-T38) were used to investigate their compatibility for binding interactions with the active pocket of EGFR-TKD (PDB ID:1M17). Tamoxifen and Erlotinib were used as references in the positive control in silico to identify the most promising lead candidate drugs for inhibiting the growth of breast and lung cancer cells.
Using Computer Aided Drug Design (CADD) methods and biophysical simulations, a multi-phase analysis of phytochemical compounds was conducted. Semi-flexible molecular docking simulations were then used to evaluate the ligands' affinity with the EGFR active site and rank them based on their most stable affinity energies in comparison to the reference drugs Tamoxifen and Erlotinib. Following this, an in silico predictive computational PK/PD models was developed combining Drug-like and ADME-Tox predictions with Osiris computations to select phytochemicals that met the criteria for oral bioavailability, pharmacokinetics, and pharmacodynamics. After screening the ligands, flexible molecular docking simulations were combined with MM-GBSA computations and molecular dynamics analysis to generate biophysical aspects related to Protein-ligand profiles interactions. This evaluation included parameters such as binding energies (BE), inhibition constants (Ki), free binding energies (ΔGbind), RMSD, RMSF, protein-ligand contacts, radius of gyration (rGyr), intramolecular hydrogen bonds (intraHB), molecular surface area (MolSA), solvent accessible surface area (SASA), polar surface area (PSA), total energy, potential energy, temperature. Results from the study indicated that the phytochemicals CBDA (Ki = 0.0175 μM, BE = −10.57 kcal/mol, ΔGbind = −31.138 kcal/mol), CBD (Ki = 1.0281 μM, BE = −8.16 kcal/mol, ΔGbind = −41.322 kcal/mol), THCV (Ki = 0.00967 μM, BE = −10.92 kcal/mol, ΔGbind = −46.578 kcal/mol), Δ-9-THC (Ki = 0.0350 μM, BE = −10.16 kcal/mol, ΔGbind = −40.446 kcal/mol), Δ-8-THC (Ki = 0.0665 μM, BE = −9.78 kcal/mol, ΔGbind = −41.353 kcal/mol), CBL (Ki = 0.1182 μM, BE = −9.44, ΔGbind = −35.748 kcal/mol), THCA (Ki = 0.0004115 μM, BE = −12.79 kcal/mol, ΔGbind = −62.807 kcal/mol), BCP (Ki = 2.603 μM, BE = −7.61 kcal/mol, ΔGbind = −33.980 kcal/mol), and γ-Ele (Ki = 3.650 μM, BE = −7.41 kcal/mol, ΔGbind = −39.782 kcal/mol) displayed a more suitable conformational and pharmacological pattern to inhibit EGFR enzymatic activity when compared to the reference drugs Tamoxifen (Ki = 0.4000 μM, BE = −8.72 kcal/mol, ΔGbind = −42.098 kcal/mol) and Erlotinib (Ki = 0.8253 μM, BE = −8.29 kcal/mol, ΔGbind = −36.877 kcal/mol).
The analysis of drug-like properties related to bioavailability, toxicity risk, and ADME-Tox indicated that the chosen candidate drug compounds had very favorable drug-use characteristics. In contrast, the reference drugs Tamoxifen and Erlotinib displayed several aberrations and toxicity risks. This suggests that the proposed drugs' phytochemicals structures are likely to be more stable in the EGFR protein pocket, thus enabling them to reach their therapeutic target against breast and lung cancer cell growth with greater efficacy than Tamoxifen and Erlotinib. Molecular dynamics simulations conducted over a 200 ns trajectory further confirmed the stability of the target EGFR protein structure, as well as the ideal fit between the EGFR protein structure and the isolated samples of cannabinoids and terpenes phytochemicals . Consequently, the phytochemicals CBDA, CBD, THCV, Δ-9-THC, Δ-8-THC, CBL, THCA, BCP, and γ-Ele discussed in this study could be employed as structural keys for the development of new drugs that act as EGFR-TKIs for breast and lung cancer.
Author contribution statement
Ossama Daoui: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Wrote the paper.
Suraj N. Mali, Kaouakeb Elkhattabi: Contributed reagents, materials, analysis tools or data.
Souad ElKhattabi, Samir Chtita: Conceived and designed the experiments; Contributed reagents, materials, analysis tools or data.
Data availability statement
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.
Consent for publication
All authors have agreed with the content of the manuscript.
Funding
No funding was received for this project.
Competing interests
The authors reported in the manuscript has no any conflict of interest.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2023.e15545.
Contributor Information
Ossama Daoui, Email: ossama.daoui@usmba.ac.ma.
Suraj N. Mali, Email: mali.suraj1695@gmail.com.
Kaouakeb Elkhattabi, Email: kaouakeb.elkhattabi@fmd.um5.ac.ma.
Souad Elkhattabi, Email: souad.elkhattabi@usmba.ac.ma.
Samir Chtita, Email: samirchtita@gmail.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Birenboim M., Chalupowicz D., Maurer D., Barel S., Chen Y., Fallik E., Paz-Kagan T., Rapaport T., Sadeh A., Kengisbuch D., Shimshoni J.A. Multivariate classification of cannabis chemovars based on their terpene and cannabinoid profiles. Phytochemistry. 2022;200 doi: 10.1016/j.phytochem.2022.113215. [DOI] [PubMed] [Google Scholar]
- 2.Chouvy P.-A. EchoGéo; 2019. Cannabis Cultivation in the World: Heritages, Trends and Challenges. [Google Scholar]
- 3.Aviram J., Lewitus G.M., Vysotski Y., Yellin B., Berman P., Shapira A., Meiri D. Prolonged medical cannabis treatment is associated with quality of life improvement and reduction of analgesic medication consumption in chronic pain patients. Front. Pharmacol. 2021:1199. doi: 10.3389/fphar.2021.613805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pagano C., Navarra G., Coppola L., Avilia G., Bifulco M., Laezza C. Cannabinoids: therapeutic use in clinical practice. Int. J. Mol. Sci. 2022;23:3344. doi: 10.3390/ijms23063344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tayyab M., Shahwar D. GCMS analysis of Cannabis sativa L. from four different areas of Pakistan. Egypt. J. Food Sci. 2015;5:114–125. [Google Scholar]
- 6.Bachir F., Eddouks M., Arahou M., Fekhaoui M. 2021. Origin, Early History, Cultivation, and Characteristics of the Traditional Varieties of Moroccan Cannabis sativa L., Cannabis and Cannabinoid Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Punja Z.K. Emerging diseases of Cannabis sativa and sustainable management. Pest Manag. Sci. 2021;77:3857–3870. doi: 10.1002/ps.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi S., Huang B.C., Gamaldo C.E. Therapeutic uses of cannabis on sleep disorders and related conditions. J. Clin. Neurophysiol. 2020;37:39–49. doi: 10.1097/WNP.0000000000000617. [DOI] [PubMed] [Google Scholar]
- 9.Kaushal N., Gupta M., Kulshreshtha E. Hempseed (Cannabis sativa) lipid fractions alleviate high-fat diet-induced fatty liver disease through regulation of inflammation and oxidative stress. Heliyon. 2020;6 doi: 10.1016/j.heliyon.2020.e04422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paes-Colli Y., Aguiar A.F., Isaac A.R., Ferreira B.K., Campos R.M.P., Trindade P.M.P., de Melo Reis R.A., Sampaio L.S. Phytocannabinoids and cannabis-based products as alternative pharmacotherapy in neurodegenerative diseases: from hypothesis to clinical practice. Front. Cell. Neurosci. 2022:273. doi: 10.3389/fncel.2022.917164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonagh M.S., Morasco B.J., Wagner J., Ahmed A.Y., Fu R., Kansagara D., Chou R. Cannabis-Based Products for Chronic Pain. Ann Intern Med. 2022;175:1143–1153. doi: 10.7326/M21-4520. [DOI] [PubMed] [Google Scholar]
- 12.Stith S.S., Li X., Orozco J., Lopez V., Brockelman F., Keeling K., Hall B., Vigil J.M. The effectiveness of common cannabis products for treatment of nausea. J. Clin. Gastroenterol. 2022;56:331–338. doi: 10.1097/MCG.0000000000001534. [DOI] [PubMed] [Google Scholar]
- 13.Doppen M., Kung S., Maijers I., John M., Dunphy H., Townsley H., Eathorne M.A., Semprini A., Braithwaite I. Cannabis in palliative care: a systematic review of current evidence. J. Pain Symptom Manag. 64. 2022:e260–e284. doi: 10.1016/j.jpainsymman.2022.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Elliott J., DeJean D., Clifford T., Coyle D., Potter B.K., Skidmore B., Alexander C., Repetski A.E., Shukla V., McCoy B. Cannabis-based products for pediatric epilepsy: a systematic review. Epilepsia. 2019;60:6–19. doi: 10.1111/epi.14608. [DOI] [PubMed] [Google Scholar]
- 15.Kopustinskiene D.M., Masteikova R., Lazauskas R., Bernatoniene J. Cannabis sativa L. Bioactive compounds and their protective role in oxidative stress and inflammation. Antioxidants. 2022;11:660. doi: 10.3390/antiox11040660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Namdar D., Voet H., Ajjampura V., Nadarajan S., Mayzlish-Gati E., Mazuz M., Shalev N., Koltai H. Terpenoids and phytocannabinoids co-produced in Cannabis sativa strains show specific interaction for cell cytotoxic activity. Molecules. 2019;24:3031. doi: 10.3390/molecules24173031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baram L., Peled E., Berman P., Yellin B., Besser E., Benami M., Louria-Hayon I., Lewitus G.M., Meiri D. The heterogeneity and complexity of Cannabis extracts as antitumor agents. Oncotarget. 2019;10:4091. doi: 10.18632/oncotarget.26983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shapira A., Berman P., Futoran K., Guberman O., Meiri D. Tandem mass spectrometric quantification of 93 terpenoids in cannabis using static headspace injections. Anal. Chem. 2019;91:11425–11432. doi: 10.1021/acs.analchem.9b02844. [DOI] [PubMed] [Google Scholar]
- 19.Radwan M.M., Chandra S., Gul S., ElSohly M.A. Cannabinoids, phenolics, terpenes and alkaloids of cannabis. Molecules. 2021;26:2774. doi: 10.3390/molecules26092774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berman P., Futoran K., Lewitus G.M., Mukha D., Benami M., Shlomi T., Meiri D. A new ESI-LC/MS approach for comprehensive metabolic profiling of phytocannabinoids in Cannabis. Sci. Rep. 2018;8:1–15. doi: 10.1038/s41598-018-32651-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramos-Guerrero L., Montalvo G., Cosmi M., García-Ruiz C., Ortega-Ojeda F.E. Classification of various marijuana varieties by Raman microscopy and chemometrics. Toxics. 2022;10:115. doi: 10.3390/toxics10030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andre C.M., Hausman J.-F., Guerriero G. Cannabis sativa: the plant of the thousand and one molecules. Front. Plant Sci. 2016;7:19. doi: 10.3389/fpls.2016.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong H., Cairns B.E. Cannabidiol, cannabinol and their combinations act as peripheral analgesics in a rat model of myofascial pain. Arch. Oral Biol. 2019;104:33–39. doi: 10.1016/j.archoralbio.2019.05.028. [DOI] [PubMed] [Google Scholar]
- 24.Aviz-Amador A., Contreras-Puentes N., Mercado-Camargo J. Virtual screening using docking and molecular dynamics of cannabinoid analogs against CB1 and CB2 receptors. Comput. Biol. Chem. 2021;95 doi: 10.1016/j.compbiolchem.2021.107590. [DOI] [PubMed] [Google Scholar]
- 25.Tomko A.M., Whynot E.G., Ellis L.D., Dupré D.J. Anti-cancer potential of cannabinoids, terpenes, and Flavonoids present in cannabis. Cancers. 1985;12 doi: 10.3390/cancers12071985. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar P., Mahato D.K., Kamle M., Borah R., Sharma B., Pandhi S., Tripathi V., Yadav H.S., Devi S., Patil U. Pharmacological properties, therapeutic potential, and legal statusof Cannabis sativa L.: an overview. Phytother Res. 2021;35:6010–6029. doi: 10.1002/ptr.7213. [DOI] [PubMed] [Google Scholar]
- 27.Pellati F., Borgonetti V., Brighenti V., Biagi M., Benvenuti S., Corsi L. Cannabis sativa L. and nonpsychoactive cannabinoids: their chemistry and role against oxidative stress, inflammation, and cancer. BioMed Res. Int. 2018:2018. doi: 10.1155/2018/1691428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nigro E., Formato M., Crescente G., Daniele A. Cancer initiation, progression and resistance: are phytocannabinoids from Cannabis sativa L. Promising compounds? Molecules. 2021;26:2668. doi: 10.3390/molecules26092668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferlay J., Colombet M., Soerjomataram I., Parkin D.M., Piñeros M., Znaor A., Bray F. Cancer statistics for the year 2020: an overview. Int. J. Cancer. 2021;149:778–789. doi: 10.1002/ijc.33588. [DOI] [PubMed] [Google Scholar]
- 30.Dyba T., Randi G., Bray F., Martos C., Giusti F., Nicholson N., Gavin A., Flego M., Neamtiu L., Dimitrova N. The European cancer burden in 2020: incidence and mortality estimates for 40 countries and 25 major cancers. Eur. J. Cancer. 2021;157:308–347. doi: 10.1016/j.ejca.2021.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bilal A., Naveed N., Haider M.S. A brief note on cancer and its treatment. Occup Med Health Aff. 2021;9:2. [Google Scholar]
- 32.Saha M. In: Cancer Diagnostics and Therapeutics : Current Trends, Challenges, and Future Perspectives. Basu S.K., Panda C.K., Goswami S., editors. Springer; Singapore: 2022. Medical oncology in cancer treatment; pp. 271–285. [DOI] [Google Scholar]
- 33.Lee Y.T., Tan Y.J., Oon C.E. Molecular targeted therapy: treating cancer with specificity. Eur. J. Pharmacol. 2018;834:188–196. doi: 10.1016/j.ejphar.2018.07.034. [DOI] [PubMed] [Google Scholar]
- 34.Duarte H.O., Reis C.A., Gomes J. Insights on ErbB glycosylation – contributions to precision oncology. Trends in Cancer. 2022;8:448–455. doi: 10.1016/j.trecan.2022.02.003. [DOI] [PubMed] [Google Scholar]
- 35.Levantini E., Maroni G., Del Re M., Tenen D.G. EGFR signaling pathway as therapeutic target in human cancers. Semin. Cancer Biol. 2022;85:253–275. doi: 10.1016/j.semcancer.2022.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Bains L., Chawla T. Gallbladder Cancer: Current Treatment Options. Springer; 2023. Anti-EGFR therapy in gallbladder cancer; pp. 331–349. [Google Scholar]
- 37.Jayaswamy P.K., Vijaykrishnaraj M., Patil P., Alexander L.M., Kellarai A., Shetty P. Implicative role of epidermal growth factor receptor and its associated signaling partners in the pathogenesis of Alzheimer's disease. Ageing Res. Rev. 2022 doi: 10.1016/j.arr.2022.101791. [DOI] [PubMed] [Google Scholar]
- 38.Ibrahim M.T., Uzairu A., Shallangwa G.A., Uba S. In-silico activity prediction and docking studies of some 2, 9-disubstituted 8-phenylthio/phenylsulfinyl-9h-purine derivatives as Anti-proliferative agents. Heliyon. 2020;6 doi: 10.1016/j.heliyon.2020.e03158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seshacharyulu P., Ponnusamy M.P., Haridas D., Jain M., Ganti A.K., Batra S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets. 2012;16:15–31. doi: 10.1517/14728222.2011.648617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daoui O., Elkhattabi S., Chtita S. Design and prediction of ADME/Tox properties of novel magnolol derivatives as anticancer agents for NSCLC using 3D-QSAR, molecular docking, MOLCAD and MM-GBSA studies. Lett. Drug Des. Discov. 2022;20:545–569. doi: 10.2174/1570180819666220510141710. [DOI] [Google Scholar]
- 41.Iancu G., Serban D., Badiu C.D., Tanasescu C., Tudosie M.S., Tudor C., Costea D.O., Zgura A., Iancu R., Vasile D. Tyrosine kinase inhibitors in breast cancer. Exp. Ther. Med. 2022;23:1–10. doi: 10.3892/etm.2021.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L. Two non-small cell lung cancer (NSCLC) patients with brain metastasis harboring epidermal growth factor receptor (EGFR) G719X and L861Q mutations benefited from aumolertinib: two cases report and review of the literature. Heliyon. 2022;8 doi: 10.1016/j.heliyon.2022.e10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan Q., Lu Y., Xie L., Wu D., Liu R., Gao W., Luo K., He B., Pu Y. Recent advances in boosting EGFR tyrosine kinase inhibitors-based cancer therapy. Mol. Pharm. 2023;20:829–852. doi: 10.1021/acs.molpharmaceut.2c00792. [DOI] [PubMed] [Google Scholar]
- 44.Naeem A., Hu P., Yang M., Zhang J., Liu Y., Zhu W., Zheng Q. Natural products as anticancer agents: current status and future perspectives. Molecules. 2022;27:8367. doi: 10.3390/molecules27238367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rahman Md.M., Bibi S., Rahaman Md.S., Rahman F., Islam F., Khan M.S., Hasan M.M., Parvez A., Hossain Md.A., Maeesa S.K., Islam Md.R., Najda A., Al-malky H.S., Mohamed H.R.H., AlGwaiz H.I.M., Awaji A.A., Germoush M.O., Kensara O.A., Abdel-Daim M.M., Saeed M., Kamal M.A. Natural therapeutics and nutraceuticals for lung diseases: traditional significance, phytochemistry, and pharmacology. Biomed. Pharmacother. 2022;150 doi: 10.1016/j.biopha.2022.113041. [DOI] [PubMed] [Google Scholar]
- 46.Chen P., Huang H.-P., Wang Y., Jin J., Long W.-G., Chen K., Zhao X.-H., Chen C.-G., Li J. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J. Exp. Clin. Cancer Res. 2019;38:254. doi: 10.1186/s13046-019-1234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xin R., Shen B., Huang Z.-Y., Liu J.-B., Li S., Jiang G.-X., Zhang J., Cao Y.-H., Zou D.-Z., Li W., Li C.-G., Ma Y.-S., Fu D. Research progress in elucidating the mechanisms underlying Resveratrol action on lung cancer. Curr. Pharmaceut. Biotechnol. 2023;24:427–437. doi: 10.2174/1389201023666220818085945. [DOI] [PubMed] [Google Scholar]
- 48.Huang K.-Y., Wang T.-H., Chen C.-C., Leu Y.-L., Li H.-J., Jhong C.-L., Chen C.-Y. Growth suppression in lung cancer cells harboring EGFR-C797S mutation by quercetin. Biomolecules. 2021;11:1271. doi: 10.3390/biom11091271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin H.-O., Park J.-A., Kim H.-A., Chang Y.H., Hong Y.J., Park I.-C., Lee J.K. Piperlongumine downregulates the expression of HER family in breast cancer cells. Biochem. Biophys. Res. Commun. 2017;486:1083–1089. doi: 10.1016/j.bbrc.2017.03.166. [DOI] [PubMed] [Google Scholar]
- 50.Lamtha T., Tabtimmai L., Songtawee N., Tansakul N., Choowongkomon K. Structural analysis of cannabinoids against EGFR-TK leads a novel target against EGFR-driven cell lines. Curr. Res. Pharmacol. Drug Discov. 2022;3 doi: 10.1016/j.crphar.2022.100132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnold M., Morgan E., Rumgay H., Mafra A., Singh D., Laversanne M., Vignat J., Gralow J.R., Cardoso F., Siesling S., Soerjomataram I. Current and future burden of breast cancer: global statistics for 2020 and 2040. Breast. 2022;66:15–23. doi: 10.1016/j.breast.2022.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daoui O., Elkhattabi S., Bakhouch M., Belaidi S., Bhandare R.R., Shaik A.B., Mali S.N., Chtita S. Cyclohexane-1,3-dione derivatives as future therapeutic agents for NSCLC: QSAR modeling, in silico ADME-Tox properties, and structure-based drug designing approach. ACS Omega. 2023 doi: 10.1021/acsomega.2c07585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khatabi K.E., El-mernissi R., Moukhliss Y., Hajji H., Rehman H.M., Yadav R., Lakhlifi T., Ajana M.A., Bouachrine M. Rational design of novel potential EGFR inhibitors by 3D-QSAR, molecular docking, molecular dynamics simulation, and pharmacokinetics studies. Chem. Data Collect. 2022;39 doi: 10.1016/j.cdc.2022.100851. [DOI] [Google Scholar]
- 54.Lo H.-W., Hsu S.-C., Hung M.-C. EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization. Breast Cancer Res. Treat. 2006;95:211–218. doi: 10.1007/s10549-005-9011-0. [DOI] [PubMed] [Google Scholar]
- 55.Thorat B.R., Mali S.N., Wagh R.R., Yamgar R.S. Synthesis, molecular docking, antioxidant, anti-TB, and potent MCF-7 anticancer studies of novel aryl-carbohydrazide analogues. Curr. Comput. Aided Drug Des. 2022;18:247–257. doi: 10.2174/1573409918666220610162158. [DOI] [PubMed] [Google Scholar]
- 56.Chakrabarti P., Chakravarty D. Intrinsically disordered proteins/regions and insight into their biomolecular interactions. Biophys. Chem. 2022;283 doi: 10.1016/j.bpc.2022.106769. [DOI] [PubMed] [Google Scholar]
- 57.Ko J.-C., Chiu H.-C., Syu J.-J., Jian Y.-J., Chen C.-Y., Jian Y.-T., Huang Y.-J., Wo T.-Y., Lin Y.-W. Tamoxifen enhances erlotinib-induced cytotoxicity through down-regulating AKT-mediated thymidine phosphorylase expression in human non-small-cell lung cancer cells. Biochem. Pharmacol. 2014;88:119–127. doi: 10.1016/j.bcp.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 58.Mishra R., Patel H., Alanazi S., Yuan L., Garrett J.T. HER3 signaling and targeted therapy in cancer. Onco Rev. 2018;12:355. doi: 10.4081/oncol.2018.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foley J., Nickerson N.K., Nam S., Allen K.T., Gilmore J.L., Nephew K.P., Riese D.J. EGFR signaling in breast cancer: bad to the bone. Semin. Cell Dev. Biol. 2010;21:951–960. doi: 10.1016/j.semcdb.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subramaniyan V., Fuloria S., Gupta G., Kumar D.H., Sekar M., Sathasivam K.V., Sudhakar K., Alharbi K.S., Al-Malki W.H., Afzal O., Kazmi I., Al-Abbasi F.A., Altamimi A.S.A., Fuloria N.K. A review on epidermal growth factor receptor's role in breast and non-small cell lung cancer. Chem. Biol. Interact. 2022;351 doi: 10.1016/j.cbi.2021.109735. [DOI] [PubMed] [Google Scholar]
- 61.Dong J.-K., Lei H.-M., Liang Q., Tang Y.-B., Zhou Y., Wang Y., Zhang S., Li W.-B., Tong Y., Zhuang G., Zhang L., Chen H.-Z., Zhu L., Shen Y. Overcoming erlotinib resistance in EGFR mutation-positive lung adenocarcinomas through repression of phosphoglycerate dehydrogenase. Theranostics. 2018;8:1808–1823. doi: 10.7150/thno.23177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yi M., He T., Wang K., Wei Y. Comparison of gefitinib plus chemotherapy versus gefitinib alone for advanced non-small-cell lung cancer: a meta analysis. Clinics. 2023;78 doi: 10.1016/j.clinsp.2022.100152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramadan W.S., Vazhappilly C.G., Saleh E.M., Menon V., AlAzawi A.M., El-Serafi A.T., Mansour W., El-Awady R. Interplay between epigenetics, expression of estrogen receptor- α, HER2/ERBB2 and sensitivity of triple negative breast cancer cells to hormonal therapy. Cancers. 2019;11:13. doi: 10.3390/cancers11010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Russnes H.G., Lingjærde O.C., Børresen-Dale A.-L., Caldas C. Breast cancer molecular stratification: from intrinsic subtypes to integrative clusters. Am. J. Pathol. 2017;187:2152–2162. doi: 10.1016/j.ajpath.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 65.Ali S., Rasool M., Chaoudhry H., Pushparaj P.N., Jha P., Hafiz A., Mahfooz M., Abdus Sami G., Azhar Kamal M., Bashir S., Ali A., Sarwar Jamal M. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation. 2016;12:135–139. doi: 10.6026/97320630012135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Droog M., Beelen K., Linn S., Zwart W. Tamoxifen resistance: from bench to bedside. Eur. J. Pharmacol. 2013;717:47–57. doi: 10.1016/j.ejphar.2012.11.071. [DOI] [PubMed] [Google Scholar]
- 67.Yk W., Cf G., Yun T., Chen Z., Xw Z., Xx L., Nl M., Wz Z. Assessment of ERBB2 and EGFR gene amplification and protein expression in gastric carcinoma by immunohistochemistry and fluorescence in situ hybridization. Mol. Cytogenet. 2011;4:14. doi: 10.1186/1755-8166-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takeda T., Tsubaki M., Matsuda T., Kimura A., Jinushi M., Obana T., Takegami M., Nishida S. EGFR inhibition reverses epithelial-mesenchymal transition, and decreases tamoxifen resistance via Snail and Twist downregulation in breast cancer cells. Oncol. Rep. 2022;47:109. doi: 10.3892/or.2022.8320. [DOI] [PubMed] [Google Scholar]
- 69.De B., Bhandari K., Singla R.K., Saha G., Goswami T.K. In silico molecular GRIP docking of some secondary metabolites combating diabesity. Bull. Natl. Res. Cent. 2020;44:66. doi: 10.1186/s42269-020-00327-7. [DOI] [Google Scholar]
- 70.Al-Nema M., Gaurav A., Lee V.S. Docking based screening and molecular dynamics simulations to identify potential selective PDE4B inhibitor. Heliyon. 2020;6 doi: 10.1016/j.heliyon.2020.e04856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iakovou G., Laycock S.D., Hayward S. Interactive flexible-receptor molecular docking in virtual reality using DockIT. J. Chem. Inf. Model. 2022;62:5855–5861. doi: 10.1021/acs.jcim.2c01274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Z., Sun H., Yao X., Li D., Xu L., Li Y., Tian S., Hou T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016;18:12964–12975. doi: 10.1039/c6cp01555g. [DOI] [PubMed] [Google Scholar]
- 74.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.R.P.D. Bank, RCSB PDB: Homepage, (n.d.). https://www.rcsb.org/(accessed December 14, 2022).
- 76.Stamos J., Sliwkowski M.X., Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 77.Mishra V.R., Ghanavatkar C.W., Mali S.N., Chaudhari H.K., Sekar N. Schiff base clubbed benzothiazole: synthesis, potent antimicrobial and MCF-7 anticancer activity, DNA cleavage and computational study. J. Biomol. Struct. Dyn. 2020;38:1772–1785. doi: 10.1080/07391102.2019.1621213. [DOI] [PubMed] [Google Scholar]
- 78.de Oliveira O.V., Rocha G.B., Paluch A.S., Costa L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2021;39:3924–3933. doi: 10.1080/07391102.2020.1772885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar S., Kumar S. In: In Silico Drug Design. Roy K., editor. Academic Press; 2019. Chapter 6 - molecular docking: a structure-based approach for drug repurposing; pp. 161–189. [DOI] [Google Scholar]
- 80.Mohammadi F., Moeeni M., Mahmudian A., Hassani L. Inhibition of amyloid fibrillation of lysozyme by bisdemethoxycurcumin and diacetylbisdemethoxycurcumin. Biophys. Chem. 2018;235:56–65. doi: 10.1016/j.bpc.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 81.Daoui O., Elkhattabi S., Chtita S. Rational identification of small molecules derived from 9, 10-dihydrophenanthrene as potential inhibitors of 3CLpro enzyme for COVID-19 therapy: a computer-aided drug design approach. Struct. Chem. 2022;33:1667–1690. doi: 10.1007/s11224-022-02004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Daoui O., Nour H., Abchir O., Elkhattabi S., Bakhouch M., Chtita S. A computer-aided drug design approach to explore novel type II inhibitors of c-Met receptor tyrosine kinase for cancer therapy: QSAR, molecular docking, ADMET and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2022;0:1–18. doi: 10.1080/07391102.2022.2124456. [DOI] [PubMed] [Google Scholar]
- 83.Poustforoosh A., Hashemipour H., Tüzün B., Pardakhty A., Mehrabani M., Nematollahi M.H. Evaluation of potential anti-RNA-dependent RNA polymerase (RdRP) drugs against the newly emerged model of COVID-19 RdRP using computational methods. Biophys. Chem. 2021;272 doi: 10.1016/j.bpc.2021.106564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kalbhor M.S., Bhowmick S., Alanazi A.M., Patil P.C., Islam M.A. Multi-step molecular docking and dynamics simulation-based screening of large antiviral specific chemical libraries for identification of Nipah virus glycoprotein inhibitors. Biophys. Chem. 2021;270 doi: 10.1016/j.bpc.2020.106537. [DOI] [PubMed] [Google Scholar]
- 86.Taiwade C., Fulfager A., Bhargave H., Soni G., Yadav K. Erlotinib hydrochloride novel drug delivery systems: a mini review unravelling the role of micro- and nanocarriers. Drug Deliv. Lett. 2021;11:295–306. doi: 10.2174/2210303111666210827094543. [DOI] [Google Scholar]
- 87.Islam M.A., Pillay T.S. β-secretase inhibitors for Alzheimer's disease: identification using pharmacoinformatics. J. Biomol. Struct. Dyn. 2019;37:503–522. doi: 10.1080/07391102.2018.1430619. [DOI] [PubMed] [Google Scholar]
- 88.Naßwetter L.C., Fischer M., Scheidt H.A., Heerklotz H. Membrane-water partitioning – tackling the challenges of poorly soluble drugs using chaotropic co-solvents. Biophys. Chem. 2021;277 doi: 10.1016/j.bpc.2021.106654. [DOI] [PubMed] [Google Scholar]
- 89.Akkoc S., Karatas H., Muhammed M.T., Kökbudak Z., Ceylan A., Almalki F., Laaroussi H., Ben Hadda T. Drug design of new therapeutic agents: molecular docking, molecular dynamics simulation, DFT and POM analyses of new Schiff base ligands and impact of substituents on bioactivity of their potential antifungal pharmacophore site. J. Biomol. Struct. Dyn. 2022;0:1–14. doi: 10.1080/07391102.2022.2111360. [DOI] [PubMed] [Google Scholar]
- 90.Daoui O., Elkhattabi S., Chtita S. Rational design of novel pyridine-based drugs candidates for lymphoma therapy. J. Mol. Struct. 2022;1270 doi: 10.1016/j.molstruc.2022.133964. [DOI] [Google Scholar]
- 91.Xu J., Zhang S., Wu T., Fang X., Zhao L. Discovery of TGFBR1 (ALK5) as a potential drug target of quercetin glycoside derivatives (QGDs) by reverse molecular docking and molecular dynamics simulation. Biophys. Chem. 2022;281 doi: 10.1016/j.bpc.2021.106731. [DOI] [PubMed] [Google Scholar]
- 92.Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 93.Greenwood J.R., Calkins D., Sullivan A.P., Shelley J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010;24:591–604. doi: 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- 94.Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kakudidi E., Tugume P., Asiimwe S., Anywar G. In: Cannabis/Marijuana for Healthcare. Agrawal D.C., Kumar R., Dhanasekaran M., editors. Springer Nature; Singapore: 2022. Traditional and modern Health uses of Cannabis sativa L. In africa and its phytochemical and pharmacological profile; pp. 189–210. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.