

Abstract
Type Ib glycogen storage disease (GSD-Ib) is caused by a deficiency in the glucose-6-phosphate (G6P) transporter (G6PT) that translocates G6P from the cytoplasm into the endoplasmic reticulum lumen, where the intraluminal G6P is hydrolyzed to glucose by glucose-6-phosphatase-α (G6Pase-α). Clinically, GSD-Ib patients manifest a metabolic phenotype of impaired blood glucose homeostasis and a long-term risk of hepatocellular adenoma/carcinoma (HCA/HCC). Studies have shown that autophagy deficiency contributes to hepatocarcinogenesis. In this study, we show that G6PT deficiency leads to impaired hepatic autophagy evident from attenuated expression of many components of the autophagy network, decreased autophagosome formation and reduced autophagy flux. The G6PT-deficient liver displayed impaired sirtuin 1 (SIRT1) and AMP-activated protein kinase (AMPK) signaling, along with reduced expression of SIRT1, forkhead boxO3a (FoxO3a), liver kinase B-1 (LKB1) and the active p-AMPK. Importantly, we show that overexpression of either SIRT1 or LKB1 in G6PT-deficient liver restored autophagy and SIRT1/FoxO3a and LKB1/AMPK signaling. The hepatosteatosis in G6PT-deficient liver decreased SIRT1 expression. LKB1 overexpression reduced hepatic triglyceride levels, providing a potential link between LKB1/AMPK signaling upregulation and the increase in SIRT1 expression. In conclusion, downregulation of SIRT1/FoxO3a and LKB1/AMPK signaling underlies impaired hepatic autophagy which may contribute to HCA/HCC development in GSD-Ib. Understanding this mechanism may guide future therapies.
Graphical Abstract
Graphical Abstract.

Introduction
Glycogen storage disease type Ib (GSD-Ib, MIM232220) is caused by pathogenic mutations in the SLC37A4 gene that encodes the glucose-6-phosphate (G6P) transporter (G6PT) (1,2). The primary function of the G6PT is to translocate G6P from the cytoplasm into the endoplasmic reticulum (ER) lumen, where it is hydrolyzed to glucose and phosphate by glucose-6-phosphatase-α (G6Pase-α; G6PC) in the terminal step of gluconeogenesis and glycogenolysis (1,2). G6PT and G6Pase-α are functionally co-dependent, and the G6PT/G6Pase-α complex maintains interprandial euglycemia (1,2). Clinically, GSD-Ib and GSD-Ia patients manifest a metabolic phenotype of impaired glucose homeostasis and long-term risks of hepatocellular adenoma/carcinoma (HCA/HCC) that develops in 75% of GSD-I patients over 25 years old (1–4). Unlike GSD-Ia, GSD-Ib patients also manifest neutropenia and myeloid dysfunction (1–4). There is no known cure for GSD-Ib. The current dietary therapies have enabled GSD-Ib patients to maintain a normalized metabolic phenotype, if adhered to strictly. However, the underlying pathological processes remain uncorrected, and HCA/HCC still occurs in metabolically compensated GSD-Ib patients (1–4).
Autophagy is an evolutionary conserved, degradative process that produces energy and building blocks through lysosomal degradation of intracellular proteins and organelles during nutrient deprivation and environmental stresses (5). We and others have shown that G6Pase-α deficiency leads to hepatic autophagy impairment (6–8) that can contribute to hepatocarcinogenesis (9). In this study, we focus specifically on the pathways contributing to hepatocarcinogenesis in GSD-Ib. We show that G6PT deficiency impairs hepatic autophagy in GSD-Ib and delineate the signaling pathways involved.
Autophagy can be regulated positively by sirtuin 1 (SIRT1) (10,11), AMP-activated protein kinase (AMPK) (12,13), the Forkhead box O (FoxO) transcription factor family members (14) and negatively by mammalian target of rapamycin (mTOR) (15). SIRT1 is an NAD+-dependent deacetylase that can regulate autophagy directly via deacetylation of key components of the autophagy network and indirectly via deacetylation and activation of FoxO factors that control the transcription of autophagy-related (ATG) genes (10). AMPK is a key enzyme that regulates cellular energy state, growth, inflammation and mitochondrial function (12,13). In the liver, AMPK is activated via phosphorylation of the AMPK α-subunit at residue T172 by the liver kinase B-1 (LKB1), a serine/threonine kinase which is also a tumor suppressor (16). The concerted action of LKB1–AMPK acts as a cellular energy sensor. In contrast, mTORC1 inhibits catabolic pathways, such as autophagy and promotes anabolic metabolism, such as synthesis of protein, lipid and nucleotides (15). Initiation of autophagy is mediated by signaling via the Unc-51-like kinase 1 (ULK1) complex which is reciprocally regulated by AMPK and mTORC1 (13,15). In G6Pase-α deficiency, underlying the disease GSD-Ia, downregulation of SIRT1 signaling was shown to underlie hepatic autophagy impairment (7,8). However, the studies failed to establish the role of AMPK in autophagy deficiency in GSD-Ia.
To study the biology and pathogenesis of GSD-Ib, we have generated two animal models. The global G6pt-deficient (G6pt−/−) mouse line fully recapitulates the phenotype of human GSD-Ib (17). However, the G6pt−/− mice suffer from fasting hypoglycemia, the hallmark of GSD-Ib, making mechanistic studies requiring fasting difficult. To address fasting studies, we also developed the liver-specific G6pt-deficient (L-G6pt−/−) mouse line (18). This line sustains fasting, reaches adulthood and manifests a liver phenotype mimicking that of human GSD-Ib, although it lacks the kidney and intestinal contributions to GSD-Ib.
In this study, we show that G6PT deficiency leads to impaired hepatic autophagy, characterized by attenuated expression of many components of the autophagy network, decreased autophagosome formation and reduced autophagy flux. The G6PT-deficient liver displayed reduced expression of SIRT1, FoxO3a, LKB1, the active p-AMPK along with impaired SIRT1 and AMPK signaling. Importantly, hepatic overexpression of either SIRT1 or LKB1 corrected defective autophagy, improved SIRT1 and AMPK signaling and increased hepatic levels of SIRT1, FoxO3a, LKB1 and p-AMPK, demonstrating that downregulation of SIRT1/FoxO3a and LKB1/AMPK signaling underlies hepatic autophagy deficiency in GSD-Ib.
Results
G6pt−/− and L-G6pt−/− mice display impaired hepatic autophagy
Autophagy progresses through vesicle initiation, nucleation and elongation, to fusion of the autophagosome–lysosome leading to degradation of abnormal protein aggregates and damaged organelles (19). We examined the expression of components of hepatic autophagy network in 3-week-old G6pt−/− mice (Fig. 1A) and 10-week-old L-G6pt−/−mice at 4-week post G6pt gene deletion (Fig. 1B). Compared with the controls, both groups of GSD-Ib mice displayed a similar reduction in protein expression of many components of the autophagy networks, consistent with impaired autophagy. These included: ATG101, involved in initiation; Beclin1 and Vps34 (class III phosphatidylinositol 3-kinase), involved in vesicle nucleation; ATG3, ATG12-ATG5 and LC3B-II (microtubule-associated protein 1 light chain 3B-II), involved in vesicle elongation and BNIP3 (BCL2/adenovirus E1B 19 kDa protein-interacting protein 3), involved in mitophagy. The conversion of the non-lipidated LC3-I to phosphatidylethanolamine-conjugated LC3-II is a critical step in autophagosome formation (19). The decrease in hepatic levels of LC3B-II in G6pt−/− (Fig. 1A) and L-G6pt−/− (Fig. 1B) mice is consistent with decreased autophagosome formation. Autophagy impairment is usually accompanied by the accumulation of p62, a selective substrate for autophagy (5). Compared with the controls, hepatic levels of p62 were markedly elevated in both G6pt−/− (Fig. 1A) and L-G6pt−/− mice (Fig. 1B), also supporting an impaired hepatic autophagy in GSD-Ib.
Figure 1.
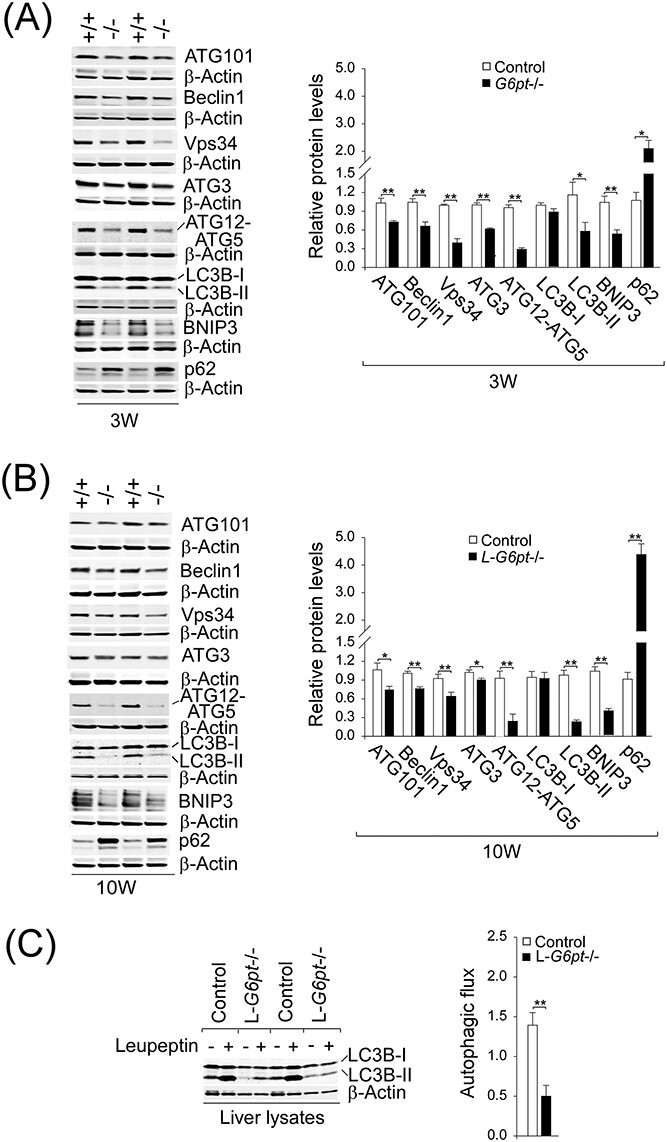
G6PT-deficient livers display impaired autophagy. (A) Western-blot analyses of hepatic ATG101, Beclin1, Vps34, ATG3, ATG12-ATG5, LC3B, BNIP3, p62 and β-actin in 3-week-old (3 W) control (n = 6–9) and G6pt−/− (n = 6–9) mice. Two sets of representative immune-stained lanes for controls (+/+) and G6pt−/− (−/−) mice are shown for each protein. Densitometric quantification was performed and normalized against β-actin. (B) Identical western blot analyses for 10-week-old (10 W) control (+/+; n = 6) and L-G6pt−/− (−/−; n = 6) mice. (C) Autophagic flux determination. Western blot analysis of hepatic LC3B and β-actin in 10-week-old control and L-G6pt−/− mice that were fasted for 20 h, then treated with either saline (−) or leupeptin (+). Autophagic flux (n = 6 for each group) was determined by the difference in LC3B-II protein levels, when normalized to β-actin, between mice treated with saline and mice treated with leupeptin. Values represent the mean ± SEM. *P < 0.05, **P < 0.01.
To confirm that the impaired autophagy seen in G6PT-deficient liver was resulting from reduced autophagosome formation and not from an increased lysosomal clearance of autophagosomes, we examined autophagic flux (20) in L-G6pt−/− and control mice that were fasted for 20 h to induce autophagy. The studies could not be performed accurately on G6pt−/− mice because of the frequent, severe hypoglycemic seizures manifested by the global G6pt−/− mice (17). Autophagic flux was determined by the difference in LC3B-II protein levels between 10-week-old mice treated with saline and mice treated with leupeptin, a lysosomal inhibitor. Compared with the controls, autophagic flux was significantly attenuated in the L-G6pt−/− livers (Fig. 1C), demonstrating the presence of hepatic autophagy impairment in GSD-Ib.
Impaired hepatic SIRT1/FoxO3a signaling in G6pt−/− mice
SIRT1 (10,11) and FoxO3a (14) are positive regulators of autophagy. Compared with the controls, hepatic protein levels of SIRT1 and FoxO3a were markedly decreased in the G6pt−/− mice (Fig. 2A). SIRT1 can increase the expression of FoxO3a (7,8) and deacetylate and activate transcriptional activity of FoxO factors (10). Consistent with the attenuated expression of SIRT1, the ratios of acetylated FoxO3a (inactive form) to total FoxO3a in nuclear extracts of G6pt−/− livers were higher than those of control livers (Fig. 2B), demonstrating that hepatic levels of active FoxO3a were also reduced in the G6pt−/− mice.
Figure 2.

Hepatic SIRT1-FoxO3a signaling in G6pt−/− mice and phenotype analysis. Experiments were conducted using 3-week-old (3 W) control (+/+; n = 6–8) and G6pt−/− (−/−; n = 6–8) mice or 10-week-old (10 W) control (n = 8) and L-G6pt−/− (n = 8) mice. (A) Western-blot analyses of hepatic SIRT1, FoxO3a, PPAR-α, PPAR-γ and β-actin. Three sets of representative immune-stained lanes for control and G6pt−/− mice are shown for each protein. Densitometric quantification was performed and normalized against β-actin. (B) Quantification of the ratios of acetylated FoxO3a to total FoxO3a. Liver nuclear extracts from 3 W control and G6pt−/− mice (n = 6 for each group) were subjected to immunoprecipitation with an antibody against FoxO3a and the resulting precipitates analyzed by western blot using an antibody against either the acetylated lysine or FoxO3a. (C) Quantification of the ratios of acetylated ATG5 or ATG7 to total ATG5 or ATG7. Liver extracts from 3 W control and G6pt−/− mice (n = 6 for each group) were subjected to immunoprecipitation with an antibody against acetylated lysine and the resulting precipitates analyzed by western blot using an antibody against either ATG5 or ATG7. (D) Hepatic triglyceride, glycogen, lactate and G6P levels in 3 W control and G6pt−/− mice. (E) Hepatic triglyceride levels in 10 W control and L-G6pt−/− mice. (F) H&E, Oil Red O and PAS staining of the livers in 3 W control and G6pt−/− mice or 10 W control and L-G6pt−/− mice. Scale bar, 20 μm. Values represent the mean ± SEM. *P < 0.05, **P < 0.01.
Studies have shown that SIRT1-mediated deacetylation of autophagy network proteins, including ATG5 and ATG7, positively regulates autophagy (10,19,21,22). During autophagic vesicle elongation, ATG7 and ATG10 facilitate the covalent conjugation of ATG12 to ATG5. The ATG12–ATG5 conjugate interacts with ATG16-like 1 (ATG16L1) and the resulting ATG12–ATG5–ATG16L1 complex promotes the conversion of LC3-I to LC3-II, a critical step in autophagosome formation (19,22). Consistent with the attenuated expression of the SIRT1 deacetylase, the acetylated forms of ATG5 and ATG7 were increased in the livers of G6pt−/− mice, compared with controls (Fig. 2C). Supporting this, hepatic levels of ATG12–ATG5 and LC3B-II were decreased in the G6pt−/− mice, compared with the controls (Fig. 1A).
Studies have shown that hepatosteatosis negatively regulates autophagy (23) and reduces SIRT1 expression (7). Hepatomegaly, a hallmark of GSD-Ib, is caused by increased accumulation of hepatic glycogen and neutral fat (1–4). Consistent with this, 3-week-old G6pt−/− (Fig. 2D) and 10-week-old L-G6pt−/− (Fig. 2E) mice had significantly higher hepatic levels of triglyceride than their age-matched controls, which was confirmed histologically by Oil Red O staining (Fig. 2F), which detects neutral triglycerides and lipid. These findings confirmed the presence of hepatosteatosis in both G6pt−/− and L-G6pt−/− mice, which is a characteristic of GSD-Ib patients (1–4). The marked glycogen storage in liver hepatocytes of both G6pt−/− and L-G6pt−/− mice was confirmed by H&E and periodic acid Schiff (PAS) staining (Fig. 2F). The liver of the G6pt−/− mice also displayed significantly higher levels of glycogen, G6P and lactate, compared with the controls (Fig. 2D).
Hepatic levels of peroxisome proliferator-activated receptor-α (PPAR-α), a transcription factor that stimulates fatty acid oxidation and increases SIRT1 expression (24) was also decreased in the G6pt−/− mice (Fig. 2A). In contrast, hepatic levels of PPAR-γ, a lipogenic nuclear receptor and a transcriptional inhibitor of SIRT1 (25) were markedly increased in G6pt−/− mice, compared with the controls (Fig. 2A). Collectively, the marked hepatosteatosis along with decreased PPAR-α and increased PPAR-γ contribute to downregulation of SIRT1 signaling in the G6pt−/− mice.
Impaired hepatic LKB1–AMPK but unaltered mTOR signaling in G6pt−/− mice
The positive autophagy regulator AMPK is activated by LKB1-mediated phosphorylation of the AMPK α-subunit at T172 (16). In G6pt−/− mice, hepatic levels of total AMPK were increased 1.7-fold over the controls, but levels of the active p-AMPK-T172 were 6.7-fold lower than the controls (Fig. 3A). This correlated with a 2.5-fold reduction of hepatic LKB1 protein in the G6pt−/− mice compared with their controls (Fig. 3A). On the other hand, hepatic levels of total mTOR, p-mTOR-S2448 and p-mTOR-S2481 were similar between G6pt−/− and control mice (Fig. 3A). The mTORC1 regulates protein synthesis and cell growth through downstream targets, the ribosomal protein S6 kinase 1 (S6K1) and 4E-binding protein 1 (4E-BP1) (26). The mTORC1 activates both proteins by phosphorylation of S6K1 at T389 and 4E-BP1 at T37/T46. While hepatic levels of total S6K1 were not statistically different between control and G6pt−/− mice, hepatic levels of p-S6K1-T389 and the ratios of p-S6K1 to S6K1 were decreased significantly in the G6pt−/− mice, compared with the controls (Fig. 3B). In G6pt−/− mice, hepatic levels of both total 4E-BP1 and p-4E-BP1-T37/T46 were increased significantly, compared with the controls, but the ratios of p-4EBP1 to 4E-BP1 in control and G6pt−/− mice were not statistically different (Fig. 3B).
Figure 3.
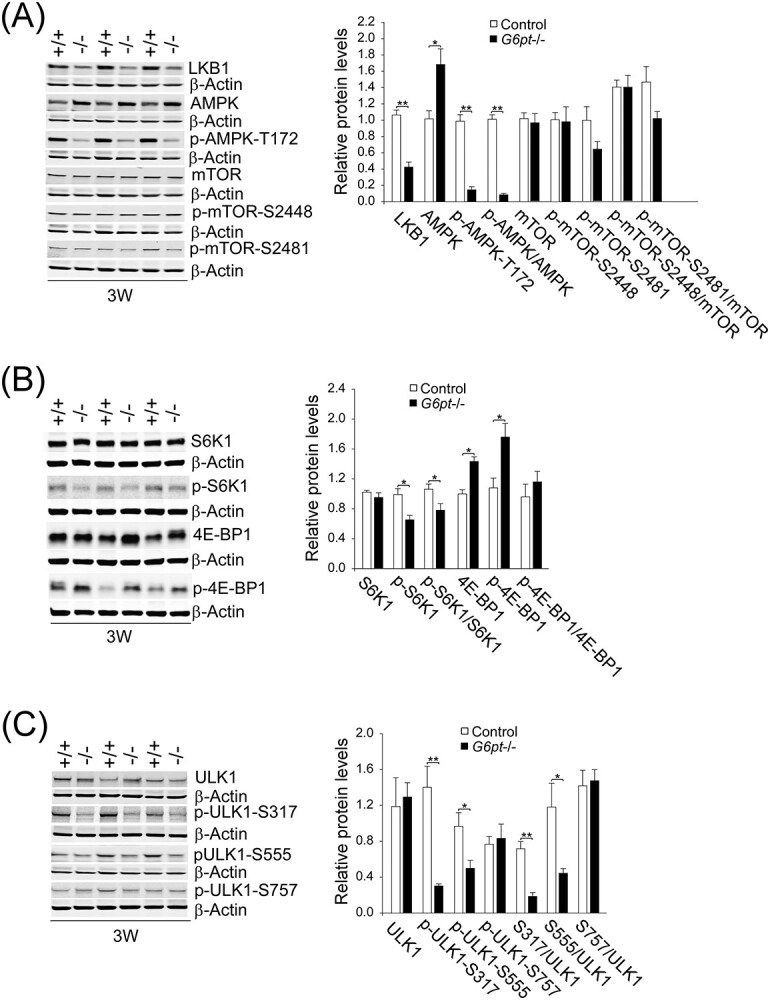
Hepatic LKB1-AMPK and mTOR signaling in G6pt−/− mice. Experiments were conducted using 3-week-old (3 W) control (+/+; n = 6) and G6pt−/− (−/−; n = 6) mice. (A) Western-blot analyses of hepatic LKB1, AMPK, p-AMPK, mTOR, p-mTOR and β-actin. (B) Western-blot analyses of hepatic S6K1, p-S6K1, 4E-BP1 and p-4E-BP1. (C) Western-blot analyses of hepatic ULK1, p-ULK1 and β-actin. Three sets of representative immune-stained lanes for control and G6pt−/− mice are shown for each protein. Densitometric quantification was performed and normalized against β-actin. Values represent the mean ± SEM. *P < 0.05, **P < 0.01.
Phosphorylation of ULK1 kinase plays a key role in the initial stages of autophagy (12,13,15). AMPK phosphorylates ULK1 at residues S317 and S555, leading to ULK1 activation and autophagy induction. In contrast, mTOR inhibits autophagy by phosphorylating ULK1 at residue S757, preventing phosphorylation and activation of ULK1 by AMPK (12,15). While hepatic levels of total ULK1 were not statistically different between control and G6pt−/− mice, hepatic levels of p-ULK1-S317 and p-ULK1-S555 were decreased significantly in the G6pt−/− mice compared with the controls (Fig. 3C). In contrast, the hepatic levels of p-ULK1-S757 were statistically similar between G6pt−/− and control mice (Fig. 3C). These data indicate that downregulation of LKB1/AMPK signaling contributes to autophagy deficiency in GSD-Ib independent of mTOR.
SIRT1 overexpression corrects impaired hepatic autophagy
To show that SIRT1 signaling plays a key role in autophagy impairment in GSD-Ib, we examined the effects of Ad-mediated SIRT1 overexpression on hepatic autophagy in G6pt−/− mice. Interestingly, SIRT1 overexpression substantially increased hepatic protein levels of FoxO3a, LKB1 and p-AMPK-T172 in G6pt−/− mice (Fig. 4A). Importantly, SIRT1 overexpression in G6pt−/− liver corrected defective hepatic autophagy when assessed by almost complete normalization of the expression of ATG101, Beclin1, Vps34, ATG3, ATG12-ATG5, LC3B-II, BNIP3 and p62 in Ad-SIRT1-treated G6pt−/− mice, as compared with the controls (Fig. 4B).
Figure 4.

SIRT1 overexpression corrects impaired hepatic autophagy in G6pt−/− mice. One-week-old control (+/+) and G6pt−/− (−/−) mice were treated with 2.5 × 1011 pfu/kg of Ad-SIRT1 and analyzed at age 3 weeks. Western blot analyses were performed in untreated and Ad-SIRT1-treated control and G6pt−/− mice (n = 6 for each group). (A) Western-blot analyses of hepatic SIRT1, FoxO3a, LKB1, AMPK, p-AMPK and β-actin. (B) Western-blot analyses of hepatic ATG101, Beclin1, Vps34, ATG3, ATG12-ATG5, LC3B, BNIP3, p62 and β-actin. Two sets of representative immune-stained lanes for untreated and Ad-SIRT1-treated control and G6pt−/− mice are shown for each protein. Densitometric quantification was performed and normalized against β-actin. Statistical analyses were performed between control and untreated or Ad-SIRT1-treated G6pt−/− mice, and between untreated and Ad-SIRT1-treated G6pt−/− mice. Values represent the mean ± SEM. *P < 0.05, **P < 0.01.
SIRT1 can deacetylate LKB1 at residue K48, leading to cytoplasmic translocation of LKB1 and activation of AMPK (27). In G6pt−/− mice, the ratios of acetylated-LKB1 to total LKB1 in liver extracts were markedly higher than those of the control mice (Fig. 5A). Compared with the controls, SIRT1 overexpression completely normalized the ratio of acetylated-LKB1 to total LKB1 in the G6pt−/− livers (Fig. 5A), suggesting activation of LKB1 signaling. Consistent with this, SIRT1 overexpression increased hepatic levels of the AMPK targets, p-ULK1-S317 and p-ULK1-S555 (Fig. 5B), confirming activation of hepatic LKB1-AMPK signaling in G6pt−/− mice. In summary, downregulation of SIRT1 signaling plays a key role in hepatic autophagy impairment in GSD-Ib.
Figure 5.
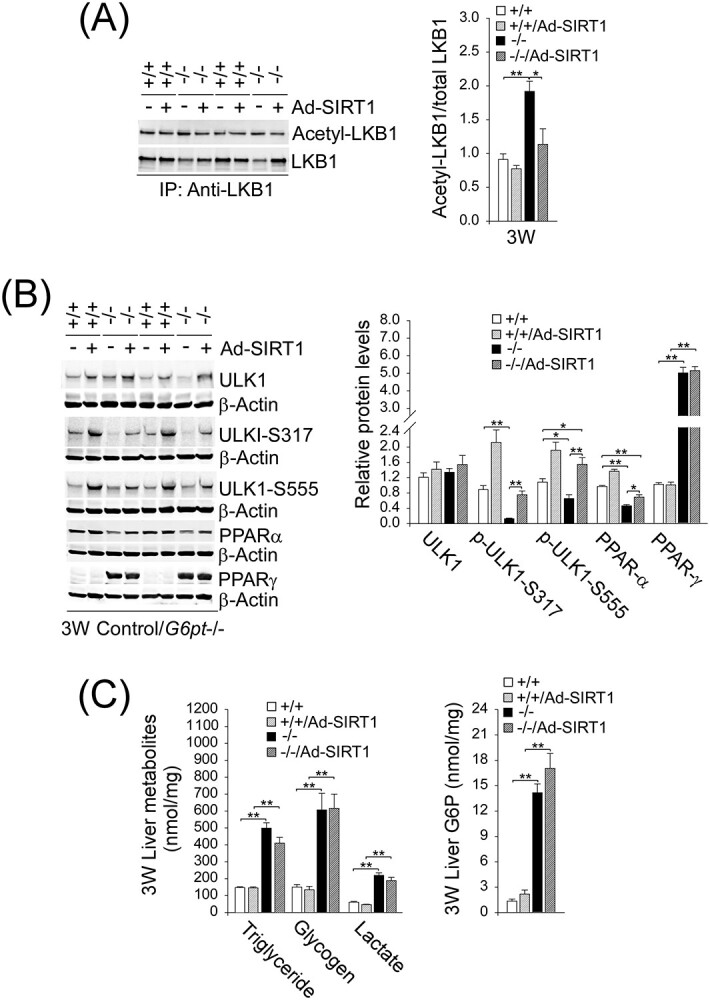
SIRT1 overexpression activates hepatic LKB1-AMPK signaling in G6pt−/− mice. One-week-old control (+/+) and G6pt−/− (−/−) mice were treated with Ad-SIRT1 at 2.5 × 1011 pfu/kg and analyzed at age 3 weeks. (A) Quantification of the ratios of acetylated-LKB1 to total LKB1. Liver extracts from untreated and Ad-SIRT1-treated control and G6pt−/− mice (n = 6 for each group) were subjected to immunoprecipitation with an antibody against LKB1 and the resulting precipitates analyzed by western blot using an antibody against either the acetylated lysine or LKB1. (B) Western-blot analyses of hepatic ULK1, p-ULK1, PPAR-α, PPAR-γ and β-actin in untreated and Ad-SIRT1-treated control and G6pt−/− mice (n = 6 for each group). Two sets of representative immune-stained lanes for untreated and Ad-SIRT1-treated control and G6pt−/− mice are shown for each protein. Densitometric quantification was performed and normalized against β-actin. (C) Hepatic levels of triglyceride, glycogen, lactate and G6P in untreated and Ad-SIRT1-treated control and G6pt−/− mice. For triglyceride, n = 14 for each group and for glycogen, lactate and G6P, n = 8 for each group. Statistical analyses were performed between control and untreated or Ad-SIRT1-treated G6pt−/− mice, and between untreated and Ad-SIRT1-treated G6pt−/− mice. Values represent the mean ± SEM. *P < 0.05, **P < 0.01.
Interestingly, SIRT1 overexpression also increased hepatic levels of PPAR-α although it failed to alter hepatic levels of PPAR-γ (Fig. 5B). Notably, while the Ad-SIRT1-treat G6pt−/− mice showed significant improvement in the markers for hepatic autophagy, SIRT1 overexpression did not improve metabolic abnormalities characteristic of GSD-Ib, namely elevated hepatic levels of triglyceride, glycogen, lactate and G6P, compared with the controls (Fig. 5C).
LKB1 overexpression corrects impaired hepatic autophagy
To demonstrate that LKB1/AMPK signaling also plays a key role in autophagy impairment in GSD-Ib, we examined the effects of Ad-mediated LKB1 overexpression on hepatic autophagy in G6pt−/− mice. An increase in hepatic LKB1 expression in G6pt−/− mice substantially increased hepatic levels of p-AMPK-T172 (Fig. 6A). Hepatic levels of total ULK1 were not statistically different between the untreated and Ad-LKB1-treated control and G6pt−/− mice (Fig. 6A). Notedly, hepatic levels of p-ULK1-S317 and p-ULK1-S555 were similar between the Ad-LKB1-treated G6pt−/− mice and their wild-type (WT) controls, demonstrating LKB1-activated AMPK signaling (Fig. 6A). Importantly, hepatic LKB1 overexpression in G6pt−/− mice corrected defective autophagy evidenced by complete normalization of hepatic levels of genes in the autophagy network, including ATG101, Beclin1, Vps34, ATG3, ATG12-ATG5, LC3B-II, BNIP3 and p62 in Ad-LKB1-treated G6pt−/− mice, as compared with the controls (Fig. 6B).
Figure 6.

LKB1 overexpression corrects impaired hepatic autophagy in G6pt−/− mice. One-week-old control (+/+) and G6pt−/− (−/−) mice were treated with 2.5 × 1011 pfu/kg of Ad-LKB1 and analyzed at age 3 weeks. Western-blot analyses were performed in untreated and Ad-LKB1-treated control and G6pt−/− mice (n = 6 for each group). (A) Western-blot analyses of hepatic LKB1, AMPK, p-AMPK, ULK1, p-ULK1 and β-actin. (B) Western-blot analyses of hepatic ATG101, Beclin1, Vps34, ATG3, ATG12-ATG5, LC3B, BNIP3, p62 and β-actin. Two sets of representative immune-stained lanes for untreated and Ad-SIRT1-treated control and G6pt−/− mice are shown for each protein. Densitometric quantification was performed and normalized against β-actin. Statistical analyses were performed between control and untreated or Ad-LKB1-treated G6pt−/− mice, and between untreated and Ad-LKB1-treated G6pt−/− mice. Values represent the mean ± SEM. *P < 0.05, **P < 0.01.
An increase in hepatic LKB1 expression also increased hepatic levels of SIRT1 and FoxO3a in the G6pt−/− mice (Fig. 7A). AMPK inhibits de novo synthesis of triglycerides and activates fatty acid β-oxidation (12), suggesting that the LKB-mediated activation of AMPK in G6pt−/− mice can reduce hepatosteatosis. Compared with untreated G6pt−/− mice, hepatic levels of triglyceride were significantly reduced in Ad-LKB1-treated G6pt−/− mice (Fig. 7B). Our study provides a potential link between LKB1/AMPK signaling upregulation and the increase in SIRT1 expression. However, LKB1 overexpression was unable to normalize hepatic levels of PPAR-α and PPAR-γ (Fig. 7A), nor to reduce hepatic levels of glycogen, lactate and G6P (Fig. 7B) in G6pt−/− mice.
Figure 7.
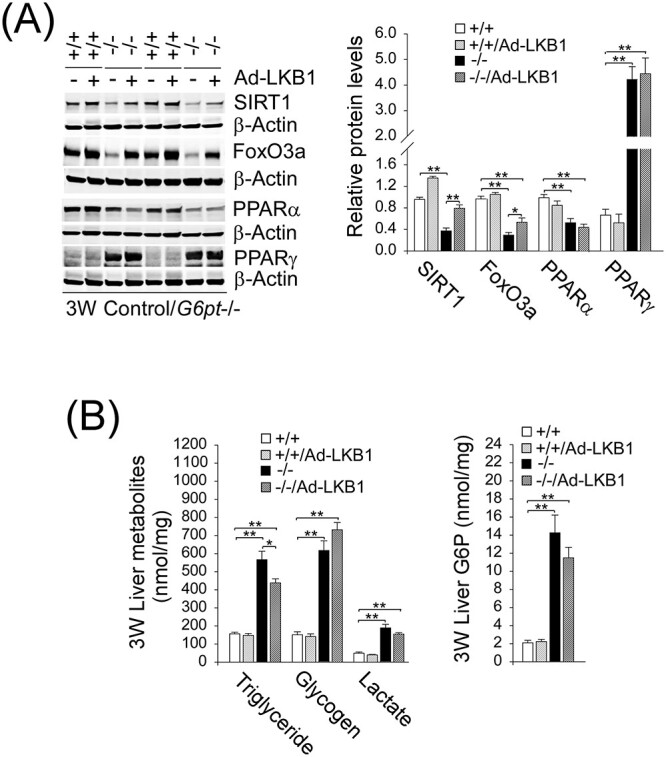
LKB1 overexpression increases hepatic levels of SIRT1 and FoxO3a in G6pt−/− mice. One-week-old control (+/+) and G6pt−/− (−/−) mice were treated with 2.5 × 1011 pfu/kg of Ad-LKB1 and analyzed at age 3 weeks. (A) Western-blot analyses of hepatic SIRT1, FoxO3a, PPAR-α, PPAR-γ and β-actin in untreated and Ad-LKB1-treated control and G6pt−/− mice (n = 6 for each group). Two sets of representative immune-stained lanes for untreated and Ad-SIRT1-treated control and G6pt−/− mice are shown for each protein. Densitometric quantification was performed and normalized against β-actin. (B) Hepatic levels of triglyceride, glycogen, lactate and G6P in untreated and Ad-LKB1-treated control and G6pt−/− mice (n = 8 for each group). Statistical analyses were performed between control and untreated or Ad-LKB1-treated G6pt−/− mice, and between untreated and Ad-LKB1-treated G6pt−/− mice. Values represent the mean ± SEM. *P < 0.05, **P < 0.01.
Discussion
Interprandial blood glucose homeostasis is primarily maintained by endogenous glucose production from the liver through hydrolysis of G6P in the ER. A rate limiting step in this pathway is the transport of cytoplasmic G6P, by G6PT, into the ER lumen where G6P is hydrolyzed to glucose by the compartmentalized hydrolase, G6Pase-α, for release into the blood (1,2). Consequently, GSD-Ib patients lacking a functional G6PT accumulate high levels of G6P in the liver, leading to reprogramming of G6P metabolism that results in marked increases in the accumulation of glycogen and neutral fat. Clinically, GSD-Ib patients manifest a metabolic phenotype of impaired glucose homeostasis and a long-term complication of HCA/HCC that develops in 75% of GSD-I patients over age 25 years old (1–4). We have previously shown that a deficiency of G6Pase-α in GSD-Ia leads to impaired hepatic autophagy (7,8) that can contribute to hepatocarcinogenesis (9). To understand the pathways contributing to hepatocarcinogenesis in GSD-Ib, we hypothesized that impaired hepatic autophagy is a significant contributor. In this study, we present evidence to support this hypothesis and show that hepatic G6PT deficiency reduced the expression of many components of the autophagy networks, decreased autophagosome formation, increased p62 accumulation, reduced autophagy flux, along with reduced SIRT1/FoxO3 and LKB1/AMPK signaling (Fig. 8A). Finally, we show that an increase in hepatic levels of either SIRT1 or LKB1 in G6PT-deficient livers almost completely ameliorates defective autophagy and activates SIRT1/FoxO3 and LKB1/AMPK signaling, demonstrating that downregulation of SIRT1/FoxO3a and LKB/AMPK signaling underlies hepatic autophagy deficiency in GSD-Ib (Fig. 8B).
Figure 8.
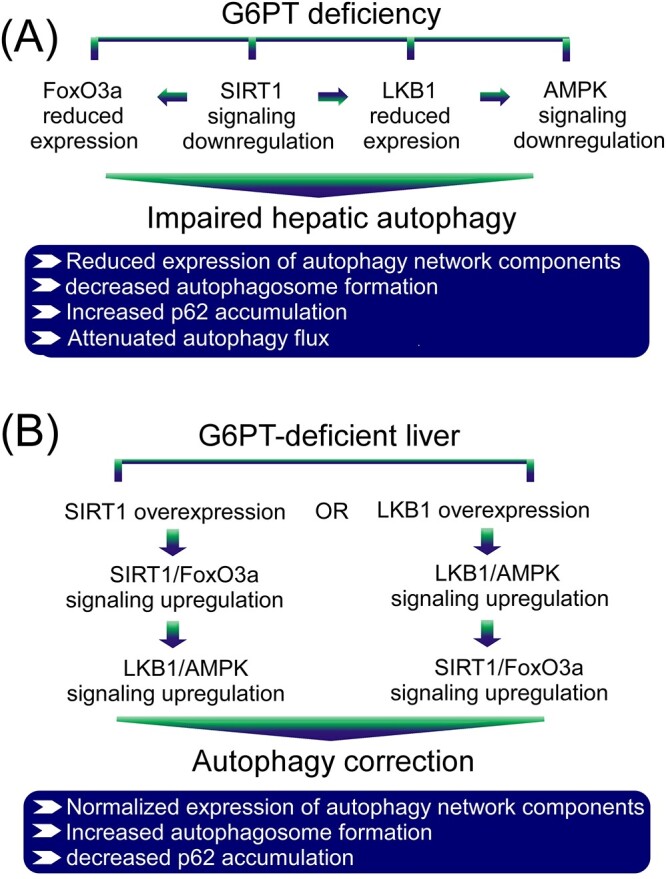
Molecular mechanism underlying impaired hepatic autophagy in G6PT deficiency. (A) G6PT deficiency leads to impaired hepatic autophagy that is mediated by downregulation of multiple autophagy-activating signaling pathways, including SIRT1/FoxO3a and LKB1/AMPK that lead to decreased expression of many components of the autophagy network, reduced autophagosome formation, increased p62 accumulation and attenuated autophagic flux. (B) Overexpression of either SIRT1 or LKB1 in G6PT-deficient liver upregulates both SIRT1/FoxO3a and LKB1/AMPK signaling and corrects impaired autophagy characterized by normalized expression of many components of the autophagy network, increased autophagosome formation and decreased p62 accumulation.
We demonstrated that the G6PT-deficient livers exhibited marked hepatosteatosis along with attenuated expression of PPAR-α and increased expression of PPAR-γ. PPAR-α is a nutrient sensing nuclear receptor that promotes fatty acid oxidation, serves as the master regulator of lipid metabolism, promotes autophagy (28) and increases SIRT1 expression (24). PPAR-γ is a ligand-regulated nuclear receptor that stimulates lipogenesis and a transcriptional inhibitor of SIRT1 (25). The marked hepatosteatosis along with altered expression of PPAR-α and PPAR-γ contribute to downregulation of SIRT1 signaling that underlies, at least in part, impaired hepatic autophagy in GSD-Ib. We have previously shown that hepatic autophagy impairment in GSD-Ia is mediated, at least in part, by SIRT1 signaling downregulation (7,8). However, it is unclear whether LKB1/AMPK signaling also plays a role in autophagy impairment in GSD-Ia. In this study, we showed, for the first time, that hepatic autophagy deficiency in GSD-Ib is mediated by downregulation of both SIRT1/FoxO3a and LKB/AMPK signaling.
SIRT1 can upregulate autophagy directly via deacetylation of autophagy network proteins and indirectly via deacetylation and activation of FoxO3a that transactivates ATG genes (10,14,19,21,22). Moreover, it is known that SIRT1 can increase the expression of LKB1 (8) as well as deacetylate and activate LKB1 (27). It is also established that liver specific deletion of SIRT1 impairs PPAR-α signaling, while an increase in hepatic SIRT1 levels stimulates the activity of PPAR-α, a positive autophagy regulator (29). In this study, we show that SIRT1 overexpression in G6PT-deficient livers corrected autophagy impairment, demonstrating that SIRT1 signaling downregulation plays a key role in hepatic autophagy deficiency in GSD-Ib. Interestingly, SIRT1 overexpression also increased hepatic levels of FoxO3a, LKB1, p-AMPK and PPAR-α, all positive autophagy regulators. AMPK stimulates autophagy via activating phosphorylation of ULK1 at residues S317 and S555 (12,13,15). Compared with the control littermates, hepatic levels of p-ULK1-S317 and p-ULK1-S555 decreased significantly in the G6pt−/− mice. On the other hand, the Ad-SIRT1-treated G6pt−/− mice expressed similar hepatic levels of p-ULK1-S317 and p-ULK1-S555 as the control mice, demonstrating, for the first time, that SIRT1 overexpression activates AMPK signaling in G6pt−/− mice. However, hepatic SIRT1 overexpression failed to normalize the expression of PPAR-γ, suggesting that in G6pt−/− mice, PPAR-γ is a transcription factor that works upstream of SIRT1.
Importantly, we showed that LKB/AMPK signaling also plays a key role in regulating hepatic autophagy impairment in GSD-Ib. First, the G6PT-deficient livers displayed decreased hepatic levels of LKB1 and p-AMPK. Second, LKB1 overexpression in G6PT-deficient livers corrected defective autophagy, increased levels of p-AMPK and completely normalized hepatic levels of the AMPK targets, p-ULK1-S317 and p-ULK1-S555 that play a key role in autophagy initiation (13,15). Third, hepatic LKB1 overexpression in G6pt−/− mice increased the expression of SIRT1 and FoxO3a that promote autophagy. The mechanism responsible for the increase in SIRT1 and FoxO3a in Ad-LKB1-treated mice is unclear. AMPK inhibits de novo synthesis of triglycerides and activates fatty acid β-oxidation (12) and the antidiabetic drug metformin lowers hepatic lipids via activating LKB1/AMPK signaling (30), indicating that LKB1-mediated increase in p-AMPK can reduce hepatosteatosis. Indeed, compared with the untreated G6pt−/− mice, hepatic levels of triglyceride were significantly decreased in Ad-LKB1-treated G6pt−/− mice. Our data provide a potential link between LKB1/AMPK signaling upregulation and the increase in SIRT1 expression. Taken together, our data indicate that downregulation of LKB1/AMPK signaling is an additional pathway that underlies autophagy deficiency in GSD-Ib. Since hepatic LKB1 overexpression failed to normalize the expression of PPAR-α and PPAR-γ, it suggests that these two transcription factors work upstream of LKB1.
The protein mTOR negatively regulates autophagy via phosphorylation of ULK1 at residue S757, preventing phosphorylation and activation of ULK1 by AMPK (15). The mTORC1 also promotes protein synthesis through the phosphorylation of S6K1 and 4EBP1 (26). We show that hepatic levels of total mTOR and active p-mTOR were not statistically different between control and G6pt−/− mice. The hepatic expression of p-ULK1-S757 and the ratio of p-ULK1-S757 to the total ULK1 were similar between the control and G6pt−/− mice. Both the hepatic level of p-S6K1 and the ratio of p-S6K1 to the total S6K1 were decreased in G6pt−/− mice, compared with the controls. While the hepatic levels of total 4E-BP1 and p-4E-BP1 were increased in the G6pt−/− mice, the ratio of p-4E-BP1 to total 4E-BP1 was unchanged between control and G6pt−/− mice. Our data demonstrate that mTOR plays a minimal role in regulating hepatic autophagy in GSD-Ib.
The G6PT is essential for maintaining liver homeostasis, and G6PT-deficient livers show downregulation of many stress-related energy sensors, including SIRT1, FoxO3a, LKB1 and p-AMPK. SIRT1 and AMPK regulate each other and share many common targets, resulting in many of the convergent biological effects on energy metabolism (31). AMPK can enhance SIRT1 activity by increasing cellular NAD+ levels, resulting in the deacetylation and modulation of the activity of many downstream SIRT1 targets (32). SIRT1 can deacetylate and activate FoxO3a that controls the transcription of many ATG genes (10) and deacetylate LKB1, leading to LKB1 cytoplasmic localization and activation (27). LKB1 activates AMPK via phosphorylation of the AMPK-α subunit at residue T-172 (16). In a previous study, we have shown that SIRT1 increases the expression of FoxO3a and LKB1 (7,8). We now show that hepatic SIRT1 overexpression increased hepatic levels of FoxO3a, LKB1 and p-AMPK, indicating that SIRT1 stimulates the expression of FoxO3a and LKB1 and activates both via deacetylation.
While LKB1 or SIRT1 overexpression have been used in this study to dissect the mechanisms that may contribute to impaired autophagy and the development of HCA/HCC in GSD-Ib, these approaches are not envisioned as likely therapeutic options for GSD-Ib. The most pressing need for GSD-Ib patients is the stabilization of glucose homeostasis, which may be resolved by a future G6PT gene augmentation strategy (33). Such a therapy should also hold the promise of removing the long-term risks of HCA/HCC (33). Current treatment for GSD-Ib patients depends on strict compliance with dietary therapies that improve hepatic glycogen load and metabolic controls and may reduce but not eliminate the risk of HCA/HCC development. Our work suggests there may be value in investigating ways to manipulate SIRT1 and LKB1/AMPK levels, perhaps using small molecule therapeutic approaches that stimulate SIRT1 (34) and/or AMPK (35) to minimize the long-term risk of GSD-Ib.
In summary, we have demonstrated that a deficiency in G6PT, that underlies GSD-Ib, leads to defective hepatic autophagy that can contribute to hepatocarcinogenesis, a long-term complication of GSD-I. We have further shown that downregulation of SIRT1/FoxO3a and LKB1/AMPK signaling underlies hepatic autophagy deficiency in GSD-Ib.
Materials and Methods
Animals
All animal studies were conducted under an animal protocol approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Animal Care and Use Committee. The G6pt−/− mice with a GSD-Ib phenotype and the age-matched G6pt+/+ and G6pt+/− mice, with indistinguishable, WT phenotype, have been described (17). In studies using the G6pt−/− mice, liver samples were collected from mice at age 3 weeks without fasting.
To generate the liver-specific G6pt−/− deficient (L-G6pt−/−) mice, we first crossed the G6pt fx/fx mice (18) harboring a conditional null allele for G6pt with the SAcreERT2/w mice (36) expressing a tamoxifen-dependent Cre-recombinase under the control of the serum albumin promoter. The L-G6pt−/− and L-G6pt+/− mice were then generated by tamoxifen-mediated excision of the G6pt exons in 6-week-old G6ptfx/fx.SAcreERT2/w and G6pcfx/w.SAcreERT2/w mice, respectively. L-G6pt−/− mice at 4 weeks post G6pc gene deletion were used in this study and liver samples were collected from mice following a 6-h fast, except for the autophagy flux study, described below, where mice were first subjected to a 20-h fast to induce autophagy. The L-G6pt+/+ and L-G6pt+/− were used as controls. For majority of our studies, we employed the G6pt−/− mice that fully recapitulate the phenotype of human GSD-Ib (17). However, for studies of autophagy flux that requires extended fasting of the animals, we employed the L-G6pt−/− mice. We have conducted parallel studies on impaired hepatic autophagy using both G6pt−/− and L-G6pt−/− mice and showed that both models manifest impaired hepatic autophagy.
To increased SIRT1 or LKB1 expression, 1-week-old G6pt−/− mice were treated with 1 × 109 plaque-forming unit (pfu)/mice (or 2.5 × 1011 pfu/kg) of a recombinant adenovirus (Ad) vector expressing either human SIRT1 (Ad-SIRT1) or human LKB1 (Ad-LKB1) (Vigene Biosciences, Rockville, MD, USA) via jugular vein injection and liver samples were collected from mice at age 3 weeks without fasting.
Hepatic metabolite analysis, hematoxylin & eosin, PAS and Oil Red O staining
Liver tissues were homogenized in 5% NP-40, incubated at 99°C for 5 min, and centrifuged to remove insoluble materials. The resulting supernatants were used to measure levels of G6P, lactate, glycogen and triglycerides using the respective assay kit from BioVision (Milpitas, CA, USA).
Hematoxylin and eosin (H&E) and PAS staining were performed on liver sections preserved in 10% neutral buffered formalin and Oil Red O staining was performed on cryopreserved OCT embedded liver sections following the standard procedures. The stained sections were visualized using the Imager A2m microscope with Axiocam 506 camera and the ZEN 2.6 software (Carl Zeiss, White Plains, NY, USA).
Western-blot analysis
Western-blot images were detected with the use of the LI-COR Odyssey scanner and the Image studio 3.1 software (Li-Cor Biosciences, Lincoln, NE, USA). The antibodies obtained from Cell Signaling Technology (Danvers, MA, USA) were acetylated-lysine (#9814), AMPK (#2532), ATG101 (#13492), ATG3 (#3415), ATG7 (#8558), BNIP3 (#12396), ATG12 (#4180), VPS34 (#4263), mouse β-actin (#3700), rabbit β-actin (#4970), Beclin1 (#3738), FoxO3a (#12829), LKB1 (#3047), mTOR (#2983), p-mTOR-S2448 (#5536), p-mTOR-S2481 (#2974), 4E-BP1 (#9644), p-4E-BP1-T37/46 (#2855), p70 S6 Kinase (S6K1) (#2708), Phospho-p70 S6 Kinase (Thr389) (p-S6K1-T389) (#9234), PARP (#9542), ULK1 (# 8054S), p-ULK1-S757 (#14202S) and p-ULK1-S555 (#5869S); the antibodies obtained from Abcam (Cambridge, MA, USA) were p-AMPK (ab133448), LC3B (ab51520) and PPAR-α (ab24509); the antibodies obtained from Santa Cruz Biotechnology (Dallas, TX, USA) were ATG5 (sc-515 347), LKB1 (sc32245) and PPAR-γ (sc7196); the antibody against SIRT1 (#07-131) was from Millipore (Burlington, MA, USA); the antibody against p62 (NBP1-49956) was from Novus Biologicals (Centennial, CO) and the antibody against p-ULK1-S317 (#LS-C800719) was from Lifespan Bioscience Inc. (Seattle, WA, USA).
Immunoprecipitation
To detect the acetylated LKB1, ATG5 or ATG7, liver tissues were homogenized in a cell lysis buffer (Cell Signaling # 9803) containing 1X Halt Protease and Phosphatase Inhibitor Cocktails (Thermo Scientific, Waltham, MA, USA), 1 mm okadaic acid (Sigma-Aldrich, St. Louis, MO, USA), and 1 mm PMSF (Cell Signaling # 8553). After centrifugation, the resulting supernatants were subjected to immunoprecipitation. To detect the acetylated LKB1, the liver tissue supernatants were immunoprecipitated with an antibody against LKB1 (Santa Cruz Biotechnology: sc32245) and the resulting precipitates examined by western-blot analysis using an antibody against either the acetylated lysine (Cell Signaling # 9814) or an antibody against LKB1 (Cell Signaling #3047). To detect the acetylated ATG5 or ATG7, the liver tissue supernatants were immunoprecipitated with an antibody against acetylated lysine and the resulting precipitates examined by western-blot analysis using an antibody against either ATG5 or ATG7.
To detect the acetylated FoxO3a, liver nuclear extracts were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific) containing 1X Halt Protease and Phosphatase Inhibitor Cocktails, 1 mm okadaic acid and 1 mm PMSF. After centrifugation, the resulting supernatants were subjected to immunoprecipitation with an antibody against FoxO3a (Cell Signaling #12829) and the resulting precipitates were examined by western-blot analysis using an antibody against either the acetylated lysine (Cell Signaling # 9814) or FoxO3a.
Autophagy flux determination
Autophagic flux was determined on L-G6pt−/− mice at 4 weeks post G6pt gene deletion as previously reported (7). Briefly, control and L-G6pt−/− mice were fasted for 20 h to induce autophagy. Then, mice were injected intraperitoneally with either saline or leupeptin (Sigma-Aldrich, 60 mg/kg body weight) and were sacrificed 4 h later. LC3B-II and β-actin in liver lysates were analyzed by western blots and quantified by densitometry. Autophagic flux was determined by the difference in LC3B-II protein levels, when normalized to β-actin, between mice treated with saline and mice treated with leupeptin.
Statistical analysis
The unpaired t-test was performed using the GraphPad Prism Program, version 8 (GraphPad Software, San Diego, CA, USA). Values were considered statistically significant at P < 0.05.
Funding
Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (NICHD—HD000912-38; https://intramural.nih.gov/search/searchview.taf?ipid=100503&ts=1513174914).
Conflict of Interest statement. None declared.
Authors’ Contributions
S.G. performed the experiments, analyzed the data and co-wrote the paper; L.Z., C.L., I.A., H.D.C. and R.R. performed the experiments and edited the manuscript; A.E. and B.C.M. analyzed the data and edited the manuscript. J.Y.C. designed the research, acquired the funding, analyzed the data and wrote the paper.
Contributor Information
Sudeep Gautam, Section on Cellular Differentiation, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20802, USA.
Lisa Zhang, Section on Cellular Differentiation, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20802, USA.
Cheol Lee, Section on Cellular Differentiation, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20802, USA.
Irina Arnaoutova, Section on Cellular Differentiation, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20802, USA.
Hung Dar Chen, Section on Cellular Differentiation, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20802, USA.
Roberta Resaz, Istituto Giannina Gaslini, Largo Gaslini 5, 16147, Genoa, Italy.
Alessandra Eva, Istituto Giannina Gaslini, Largo Gaslini 5, 16147, Genoa, Italy.
Brian C Mansfield, Section on Cellular Differentiation, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20802, USA.
Janice Y Chou, Section on Cellular Differentiation, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20802, USA.
References
- 1. Chou, J.Y., Jun, H.S. and Mansfield, B.C. (2010) Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat. Rev. Endocrinol., 6, 676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chou, J.Y., Jun, H.S. and Mansfield, B.C. (2015) Type I glycogen storage diseases: disorders of the glucose-6-phosphatase/glucose-6-phosphate transporter complexes. J. Inherit. Metab. Dis., 38, 511–519. [DOI] [PubMed] [Google Scholar]
- 3. Rake, J.P., Visser, G., Labrune, P., Leonard, J.V., Ullrich, K. and Smit, G.P. (2002) Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur. J. Pediatr., 161, S20–S34. [DOI] [PubMed] [Google Scholar]
- 4. Kishnani, P.S., Austin, S.L., Abdenur, J.E., Arn, P., Bali, D.S., Boney, A., Chung, W.K., Dagli, A.I., Dale, D., Koeberl, D. et al. (2014) Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet. Med., 16, e1–e29. [DOI] [PubMed] [Google Scholar]
- 5. Mizushima, N. and Komatsu, M. (2011) Autophagy: renovation of cells and tissues. Cell, 147, 728–741. [DOI] [PubMed] [Google Scholar]
- 6. Farah, B.L., Landau, D.J., Sinha, R.A., Brooks, E.D., Wu, Y., Fung, S.Y., Tanaka, T., Hirayama, M., Bay, B.H., Koeberl, D.D. et al. (2016) Induction of autophagy improves hepatic lipid metabolism in glucose-6-phosphatase deficiency. J. Hepatol., 64, 370–379. [DOI] [PubMed] [Google Scholar]
- 7. Cho, J.H., Kim, G.Y., Pan, C.J., Anduaga, J., Choi, E.-J., Mansfield, B.C. and Chou, J.Y. (2017) Downregulation of SIRT1 signaling underlies hepatic autophagy impairment in glycogen storage disease type Ia. PLoS Genet., 13, e1006819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gautam, S., Zhang, L., Arnaoutova, I., Lee, C., Mansfield, B.C. and Chou, J.Y. (2020) The signaling pathways implicated in impairment of hepatic autophagy in glycogen storage disease type Ia. Hum. Mol. Genet., 29, 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Madrigal-Matute, J. and Cuervo, A.M. (2016) Regulation of liver metabolism by autophagy. Gastroenterology, 150, 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ng, F. and Tang, B.L. (2013) Sirtuins' modulation of autophagy. J. Cell. Physiol., 228, 2262–2270. [DOI] [PubMed] [Google Scholar]
- 11. Galluzzi, L., Pietrocola, F., Levine, B. and Kroemer, G. (2014) Metabolic control of autophagy. Cell, 159, 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jeon, S.M. (2016) Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med., 48, e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tamargo-Gómez, I. and Mariño, G. (2018) AMPK: regulation of metabolic dynamics in the context of autophagy. Int. J. Mol. Sci., 19, 3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sengupta, A., Molkentin, J.D. and Yutzey, K.E. (2009) FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem., 284, 28319–28331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim, Y.C. and Guan, K.L. (2015) mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest., 125, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shaw, R.J., Lamia, K.A., Vasquez, D., Koo, S.H., Bardeesy, N., Depinho, R.A., Montminy, M. and Cantley, L.C. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science, 310, 642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen, L.-Y., Shieh, J.-J., Lin, B., Pan, C.-J., Gao, J.-L., Murphy, P.M., Roe, T.F., Moses, S., Ward, J.M., Westphal, H. et al. (2003) Impaired glucose homeostasis, neutrophil trafficking and function in mice lacking the glucose-6-phosphate transporter. Hum. Mol. Genet., 12, 2547–2558. [DOI] [PubMed] [Google Scholar]
- 18. Raggi, F., Pissavino, A.L., Resaz, R., Segalerba, D., Puglisi, A., Vanni, C., Antonini, F., Del Zotto, G., Gamberucci, A., Marcolongo, P. et al. (2018) Developmental and characterization of an inducible mouse model for glycogen storage disease type Ib. J. Inherit. Metab. Dis., 41, 1015–1025. [DOI] [PubMed] [Google Scholar]
- 19. Fullgrabe, J., Klionsky, D.J. and Joseph, B. (2014) The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol., 15, 65–74. [DOI] [PubMed] [Google Scholar]
- 20. Yoshii, S.R. and Mizushima, N. (2017) Monitoring and measuring autophagy. Int. J. Mol. Sci., 18, 1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee, I.H., Cao, L., Mostoslavsky, R., Lombard, D.B., Liu, J., Bruns, N.E., Tsokos, M., Alt, F.W. and Finkel, T. (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U. S. A., 105, 3374–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Narita, T., Weinert, B.T. and Choudhary, C. (2019) Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol., 20, 156–174. [DOI] [PubMed] [Google Scholar]
- 23. Yang, L., Li, P., Fu, S., Calay, E.S. and Hotamisligil, G.S. (2010) Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab., 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hayashida, S., Arimoto, A., Kuramoto, Y., Kozako, T., Honda, S., Shimeno, H. and Soeda, S. (2010) Fasting promotes the expression of SIRT1, an NAD+ −dependent protein deacetylase, via activation of PPARalpha in mice. Mol. Cell. Biochem., 339, 285–292. [DOI] [PubMed] [Google Scholar]
- 25. Han, L., Zhou, R., Niu, J., McNutt, M.A., Wang, P. and Tong, T. (2010) SIRT1 is regulated by a PPAR{γ}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res., 38, 7458–7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma, X.M. and Blenis, J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol., 10, 307–318. [DOI] [PubMed] [Google Scholar]
- 27. Lan, F., Cacicedo, J.M., Ruderman, N. and Ido, Y. (2008) SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem., 283, 27628–27635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim, K.H. and Moore, D.D. (2017) Nutrient-sensing nuclear receptors PPARα and FXR control liver energy balance. J. Clin. Invest., 127, 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Purushotham, A., Schug, T.T., Xu, Q., Surapureddi, S., Guo, X. and Li, X. (2009) Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab., 9, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zang, M., Zuccollo, A., Hou, X., Nagata, D., Walsh, K., Herscovitz, H., Brecher, P., Ruderman, N.B. and Cohen, R.A. (2004) AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem., 279, 47898–47905. [DOI] [PubMed] [Google Scholar]
- 31. Ruderman, N.B., Xu, X.J., Nelson, L., Cacicedo, J.M., Saha, A.K., Lan, F. and Ido, Y. (2010) AMPK and SIRT1: a long-standing partnership? Am. J. Physiol. Endocrinol. Metab., 298, E751–E760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cantó, C., Gerhart-Hines, Z., Feige, J.N., Lagouge, M., Noriega, L., Milne, J.C., Elliott, P.J., Puigserver, P. and Auwerx, J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature, 458, 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kwon, J.H., Lee, Y.M., Cho, J.-H., Kim, G.Y., Anduaga, J., Starost, M.F., Mansfield, B.C. and Chou, J.Y. (2017) Liver-directed gene therapy for murine glycogen storage disease type Ib. Hum. Mol. Genet., 26, 4395–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farghali, H., Kemelo, M.K. and Canová, N.K. (2019) SIRT1 modulators in experimentally induced liver injury. Oxidative Med. Cell. Longev., 2019, 8765954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim, J., Yang, G., Kim, Y., Kim, J. and Ha, J. (2016) AMPK activators: mechanisms of action and physiological activities. Exp. Mol. Med., 48, e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schuler, M., Dierich, A., Chambon, P. and Metzger, D. (2004) Efficient temporally controlled targeted somatic mutagenesis in hepatocytes of the mouse. Genesis, 39, 167–172. [DOI] [PubMed] [Google Scholar]