

Abstract
Background:
Cardiac fibroblasts are the main non-myocyte population responsible for extracellular matrix (ECM) production. During perinatal development, fibroblast expansion coincides with the transition from hyperplastic to hypertrophic myocardial growth. Therefore, we investigated the consequences of fibroblast loss at the time of cardiomyocyte maturation by depleting fibroblasts in the perinatal mouse.
Methods and results:
We evaluated the microenvironment of the perinatal heart in the absence of fibroblasts and the potential functional impact of fibroblast loss in regulation of cardiomyocyte cell cycle arrest and binucleation. Cre-mediated expression of diphtheria toxin A in PDGFRα expressing cells immediately after birth eliminated 70–80% of the cardiac fibroblasts. At postnatal day 5, hearts lacking fibroblasts appeared similar to controls with normal morphology and comparable numbers of endothelial and smooth muscle cells, despite a pronounced reduction in fibrillar collagen. Immunoblotting and proteomic analysis of control and fibroblast-deficient hearts identified differential abundance of several ECM proteins. In addition, fibroblast loss decreased tissue stiffness and resulted in increased cardiomyocyte mitotic index, DNA synthesis, and cytokinesis. Moreover, decellularized matrix from fibroblast-deficient hearts promoted cardiomyocyte DNA replication. While cardiac architecture was not overtly affected by fibroblast reduction, few pups survived past postnatal day 11, suggesting an overall requirement for PDGFRα expressing fibroblasts.
Conclusions:
These studies demonstrate the key role of fibroblasts in matrix production and cardiomyocyte cross-talk during mouse perinatal heart maturation and revealed that fibroblast-derived ECM may modulate cardiomyocyte maturation in vivo. Neonatal depletion of fibroblasts demonstrated that although hearts can tolerate reduced ECM composition, fibroblast loss eventually leads to perinatal death as the approach simultaneously reduced fibroblast populations in other organs.
Keywords: Cardiac fibroblast, Extracellular matrix, Cardiomyocyte binucleation
1. Introduction
Although fibroblasts are present throughout the body and are considered the predominant producers of extracellular matrix (ECM), especially collagen, their contributions to organ maturation are poorly understood. In cardiac tissue, fibroblasts begin to produce and modify the ECM scaffolding shortly after their appearance [1], and ECM production continues after birth [2]. Although fibroblasts are likely to play a significant role in shaping heart organization and function, our knowledge of their essential roles in governing the extracellular environment during the dynamic process of heart growth is limited.
Rodent hearts undergo dynamic changes within the first several weeks after birth, including increased aerobic respiration, increased cardiac workload, and transition to a fatty acid metabolism [3]. Immediately after birth, cardiac growth is driven by proliferation. After postnatal day (P)4, fibroblast and cardiomyocyte proliferation subsides [4,5], and cardiomyocyte hypertrophy follows. As the heart transitions from hyperplastic to hypertrophic growth phases, the cardiac environment undergoes increased mechanical stiffness and ECM compositional changes [6,7].
One of the characteristics of this early adaptive time period is the ability of the heart to regenerate by increased cardiomyocyte proliferation [8]. As ECM compositional changes are concurrent with the decreased proliferation rate of both fibroblasts and cardiomyocytes, there is a potential role for fibroblasts and fibroblast-secreted ECM in regulation of cardiomyocyte proliferation. In support of this idea, in vitro experiments have identified that fibroblasts and fibroblast-derived ECM components including: fibronectin, collagen, periostin, heparin-binding epidermal growth factor [9], agrin [10] and nephronectin [11] can induce cardiomyocyte proliferation/cytokinesis [12,13]. The role of fibroblasts in modulating myocyte proliferation may not be a general fibroblast attribute as recent studies have suggested that a specific subtype of cardiac fibroblasts promotes cardiomyocyte maturation [2,14].
To investigate the contribution of fibroblasts to perinatal heart maturation in vivo, we generated mice with fewer fibroblasts by driving diphtheria toxin A (DTA) expression in PDGFRα expressing cells; PDGFRα being a common fibroblast protein [15–18]. Our data demonstrate that neonatal fibroblast ablation led to disrupted ECM composition, reduced collagen deposition, and decreased tissue stiffness in the heart. We also observed decreased cardiomyocyte binucleation at P5. Interestingly, neonatal cardiomyocytes exhibited decreased binucleation on ECM derived from fibroblast ablated hearts, demonstrating that the ECM produced by fibroblasts can contribute to the process of cardiomyocyte maturation. Our findings reveal an essential role for fibroblasts during perinatal heart development.
2. Methods
2.1. Mice
All animal protocols and experiments were approved by the University of Hawaii at Manoa Institutional Animal Care and Use Committee. Mice on a mixed C57Bl/6 background were used for these studies. R26RtdT [19] (Jackson labs, 007914) and R26RDTA [20] (Jackson labs, 010527) mice were purchased from Jackson Laboratory. Col1a1-GFP transgenic mice express cytoplasmic GFP under the control of a Col1a1 promoter/enhancer and were generated by Dr. David Brenner [21]. PDGFRα-CreERT2/+ mice were kindly provided by Dr. Brigid Hogan (Duke University Medical Center) [22]. DTA expression was induced between P1-P5 using a single intragastric injection of tamoxifen at 62.5 μg/g of body weight (AdipoGen, CDX-T0200). The fibroblast ablated genotype was PDGFRα-CreERT2/+; R26RtdT/DTA. All control mice used in these experiments were tamoxifen induced and age matched. Control genotypes were PDGFRα-CreERT2/+; R26RtdT/+ or PDGFRα+/+; R26RtdT/DTA (littermate controls). Littermate (Cre−) and non-littermate (Cre+) controls were used in all experiments, and no significant phenotypes were observed in these mice.
2.2. Screening for PDGFRα deletion
Deletion efficiency was tested by multiple approaches (Supplemental Table 1). For determining PDGFRα deletion, kidneys were collected, stored in RNAlater Stabilization Solution (Invitrogen, AM7021) and processed for RNA extraction as described below. Mice with greater than 50% of control PDGFRα expression were excluded from studies, as this indicated insufficient fibroblast ablation.
2.3. Western blot
Atria and valves were discarded from isolated hearts. Whole ventricle tissue was homogenized in RIPA buffer with protease inhibitor cocktail (Bimake, B14001) using a Dounce homogenizer. Samples were centrifuged at 16,000 xg for 20 min at 4 °C, and supernatant collected. Blots were probed with primary antibody overnight at 4 °C, and then incubated with the corresponding Li-Cor IRDye secondary antibody for 1 h at room temperature. Primary antibodies used for western blot are listed in Supplemental Table 2. GAPDH or α-tubulin was used as a loading control. An Odyssey CLx imaging system was used for detection and images were analyzed using Image Studio version 5.2.5 software (Li-Cor Biosciences).
2.4. Mass spectrometry
Whole ventricle tissue from P5 pups and adult hearts was minced and washed in distilled H2O with protease inhibitor cocktail (Bimake, B14001) for 30 min at room temperature with light agitation. P5 tissue was placed in 0.05% SDS in DPBS with protease inhibitor at room temperature with light agitation, and adult tissue in 1% SDS in DPBS with protease inhibitor until tissue was clear. Decellularized cardiac tissue was washed in distilled H2O with protease inhibitor 3 times for 5 min at room temperature with light agitation, and then washed overnight to completely remove detergent. Decellularized tissue was processed with Protein Extraction Reagent Type 4 (Sigma, C0356) with protease inhibitor and sonicated (20 kHz) 3–4 times for 5 s for P5 tissue and 10 times for 10 s for adult tissue.
Lysates were reduced, alkylated, and trypsin-digested into peptides. The peptides were cleaned using Sep-Pak Vac C18 cartridge (Waters Corporation) and label-free analysis was done by liquid chromatography-tandem mass spectrometry using a Q Exactive mass spectrometer (ThermoFisher). A 15 cm Å ~ 75 μm C18 column (5 μm particles with 100 Å pore size) was used and the nano-UPLC ran at 300 nL/min with a 150 min linear acetonitrile gradient (from 5 to 35% B over 150 min; A = 0.2% formic acid in water; B = 0.2% formic acid in 90% acetonitrile). Tandem mass spectrometry (MS/MS) was set with an exclusion of 25 s, and the samples were run with high-energy collisional dissociation fragmentation at normalized collision energy of 30% and an isolation width of 2 m/z. The resolution setting was 70,000 for target values of the MS at 1e6 ions and in MS2 at resolution setting of 17,500 for 1e5 ions. Mass spectrometry experiments were performed at the University of Mississippi Medical Center. Mass spectrometry analysis was performed blinded to the identity of the samples.
RAW files were analyzed using Proteome Discoverer 2.2 (Thermo Fisher). Precursor mass tolerance was set at 10 ppm and fragment mass tolerance was set at 0.6 Da. Dynamic modification was set to oxidation (+15.995 Da (M)) and static modification was set to carbamidomethyl (+57.021 Da (C)). Samples were searched against the reviewed mouse database downloaded from Uniprot (November 2018, with 16,977 sequences). A strict false discovery rate (FDR) of 1% was applied. Label-free quantification was done based on precursor ion intensity and normalization was done using the total peptide amount (from all peptides identified). Proteins were included only if they were identified by at least two high-confidence peptides. Identification of secreted proteins was based on annotation in UniProt (www.uniprot.org), i.e., presence of a signal peptide. The mass spectrometry proteomics data have been deposited to the Proteome Xchange Consortium via the PRIDE partner repository with the dataset identifier PXD021741.
2.5. Immunostaining and microscopy
Cardiac tissue was excised and fixed with freshly prepared 4% paraformaldehyde (PFA) in DPBS for 90 min at room temperature or 10% neutral buffered formalin (NBF) for 24 h at 4 °C. Immunostaining was performed as previously described [16]. Primary antibodies used for immunostaining are listed in Supplemental Table 2. A Zeiss Axiovert 200 microscope equipped with an Olympus DP71 camera or Leica SP8 DLS microscope was used for imaging. Figures were created in Adobe Photoshop CC (version 19).
5-ethynyl-2′-deoxyuridine (EdU; Lumiprobe, 20,540) was administered by intraperitoneal (IP) injection (50 mg/kg body weight) 1 h before isolation. Hearts were paraffin embedded and stained using a Click-iT EdU Alexa Fluor Imaging Kit (Invitrogen, C10337). For identification of cells undergoing cytokinesis, EdU labelling was performed as above and stained for Aurora B kinase and cardiac troponin T. Heat-induced epitope retrieval was performed on 5 μm tissue sections by heating slides to 98 °C in 10 mM sodium citrate buffer (pH 6.0) with 0.05% Tween-20 for 20 min. Aurora B kinase+ cardiomyocytes were classified as midline or asymmetric (Supplemental Fig. 8). A minimum of 200 Aurora B kinase+/EdU+ cardiomyocytes were counted per biological replicate.
2.6. qRT-PCR
RNA isolation was performed on whole ventricles using IBI Isolate DNA/RNA reagent (IBI Scientific, IB47602) and PureLink RNA mini kit (Thermo Fisher, 12,183,025) according to the manufacturer’s instructions. RNA quality and concentration were determined by spectrophotometry using a NanoDrop 2000 instrument (Thermo Fisher). cDNA was synthesized using M-MLV Reverse Transcriptase (Sigma, M1302) and random hexamer primer (Thermo Fisher, SO142). qPCR analysis was performed using PowerUp SYBR Green Master Mix (Thermo Fisher, A25742) and a LightCycler 480 instrument (Roche). Samples were run in triplicate and normalized to 18S expression. The 2−ΔΔCt method was used for determining relative gene expression levels. Primer sequences used for qRT-PCR are listed in Supplemental Table 3.
2.7. Hydroxyproline assay
Hydroxyproline assay (Cell Biolabs, STA-675) was performed on whole ventricle tissue isolated from P5 pups. Samples were homogenized in distilled H2O and hydrolyzed in 6 N HCl for 20 h at 95 °C. Hydrolyzed contents were processed according to manufacturer’s instructions. Absorbance of the supernatant was read using Molecular Devices SpectraMax M3 microplate reader at 540 nm wavelength.
2.8. Second harmonic generation
Cardiac tissue was isolated from P4 pups and fresh frozen embedded in OCT. Second harmonic generation (SHG) was performed on 6 μm coronal sections. SHG signals were acquired using Zeiss 780 laser-scanning confocal/multiphoton-excitation fluorescence microscope with a 34-channel GaAsP QUASAR detection unit and non-descanned detectors for two-photon fluorescence (Zeiss). The imaging settings were initially defined empirically to maximize the signal-to-noise ratio and to avoid saturation; and they were kept constant for all measurements for comparative imaging and results. Two-photon Chameleon laser tuned to 800 nm was used for excitation, and emission signals corresponding to the autofluorescence and SHG signals were detected simultaneously through non-de-scanned detectors. A total of 20 images were taken per heart and at least 4 hearts were analyzed per group. Matlab was used to analyze the percentage of collagen. SHG experiments were performed blinded to the identity of the samples.
2.9. Picrosirius red staining
Following deparaffinization and hydration of FFPE sections, Weigert’s hematoxylin was applied to sections for 8 min. After washing in running water for 10 min, sections were incubated in 1 g of Direct Red 80 (Sigma-Aldrich, 365548) in 100 mL saturated picric acid solution (Sigma-Aldrich, P6744) for 1 h at room temperature. The slides were rinsed in 0.5% acetic acid 2 times. After dehydration, the sections were mounted in Epredia Mounting Medium (Thermo Fisher, 22–11–610). Bright field and polarized images were acquired using a Leica Thunder Live Cell 3D microscope under the same exposure settings.
2.10. Gelatin zymography
Whole ventricle tissue isolated from P5 pups was homogenized in Triton X-100 lysis buffer (125 mM NaCl, 25 mM Tris-HCl, 1% Triton X-100, pH 8.5) with protease inhibitor cocktail (Bimake, B14001) using a Dounce homogenizer. Samples were centrifuged at 16,000 xg for 20 min at 4 °C, and the supernatant was collected. 5 μg of protein were loaded/lane. Novex 10% Zymogram Plus Gels (Thermo Fisher, ZY00105BOX) were used and gelatin zymography was performed according to the manufacturer’s instructions.
2.11. Atomic force microscopy
Cardiac tissue was isolated from P4 pups and fresh frozen embedded in OCT. Atomic force microscopy (AFM) was performed on 6 μm coronal sections. The nanomechanical and topographical properties of frozen, non-fixed coronal sections placed on glass slides were characterized using a NanoWizard® 4a (JPK Instruments). Briefly, the samples were immersed in PBS for 15 min to remove OCT and allowed to thaw. The sections were then covered with 1× protease inhibitor (Thermo Fisher, 78,443) and tissue stiffness was determined using a qp-BioAC-1 (NanoandMore) cantilever with a force constant in the range of 0.15 to 0.55 N/m. The cantilever was calibrated using the thermal oscillation method before each experiment. Tissue mapping was performed in quantitative imaging (QITM) mode. Coronal sections were scanned in a 400 μm2 area using a set point of 5 nN, a Z-length of 2 mm, and a pixel time of 50 ms with a resolution of 256 × 256 pixels. At least four scans were performed on the left ventricular (LV) free wall of each section, and from at least 3 different animals. The Hertz model was used to determine the tissue mechanical properties using the JPK software. AFM experiments were performed blinded to the identity of the samples.
2.12. Cardiomyocyte isolation
For EdU chase experiments EdU was administered 48 h before cell isolation. Cardiomyocytes from P5 and P9 hearts were isolated using a modified collagenase digestion protocol [23]. Myocyte cell suspensions were fixed with freshly made 4% PFA/DPBS for 15 min at room temperature. Fixed myocytes were washed twice with HBSS by centrifugation (50 ×g for 10 min). Cells were spun onto glass slides using a cytospin (Shandon Cytospin 4) at 1000 rpm for 3 min. EdU detection, primary antibody, and secondary antibody incubations were performed as described above. Nuclei were stained with DAPI. Microscope slides were mounted and imaged using a Zeiss Axiovert 200. 400–500 cardiomyocytes were counted per biological replicate.
2.13. Decellularization of tissue sections for culture
Hearts were isolated from P4 pups and fresh frozen embedded in OCT. 25 μm transverse sections from control and ablated hearts were adhered to an 8-well chamber slide (ibidi, 80,826). Sections were treated with 10 mM Tris and 1% wt/vol EDTA for 30 min and decellularized with 0.05% SDS in DPBS for 30 min at room temperature. Timing of decellularization was determined by DNA quantification using DAPI staining to confirm lack of cells in control and ablated tissue. After decellularization, sections were treated with 50 U/mL DNase and 1 U/mL RNase for 15 min at 37 °C. Decellularized sections were washed with DPBS and used for cardiomyocyte cell culture experiments as described above.
2.14. Cardiomyocyte/matrix co-culture
Cardiomyocytes from whole ventricles were isolated from P2 pups using the Pierce Primary Cardiomyocyte Isolation Kit (Thermo Fisher, 88,281). The cell suspension was filtered through a 70 μm nylon mesh strainer and pre-plated for 2 h to remove non-myocytes. Non-adherent cardiomyocytes were removed and centrifuged at 50 xg for 5 min. Cardiomyocytes were resuspended in medium 199 with 1× Pen/Strep, 5% horse serum, and 10 μM EdU, and seeded at 5.0 × 104 cells per 1.0 cm2 for 48 h on decellularized matrix or attachment factor (Invitrogen, S006100). Cultures were fixed with 4% PFA/DPBS for 15 min at room temperature, washed, permeabilized, and staining performed. >200 cardiomyocytes were counted per biological replicate. Quantifications were performed blinded to the identity of the culture substratum.
2.15. Statistical analyses
All statistical analyses were conducted using Prism 8 (Graphpad Software). To determine whether to use a parametric t-test, Shapiro-Wilk and Levene’s test was performed. If normal distribution and homogeneous variance was not found, statistical analyses between two groups were analyzed using Mann-Whitney U test. Comparisons between multiple groups were analyzed using two-way ANOVA with Tukey test. Statistical variability is expressed as mean ± SD; ns: not significant, P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. N values for experiments are indicated in figure legends.
3. Results
3.1. Postnatal cardiac ECM
Fibroblasts are first observed in the mouse heart around embryonic age (E)14.5 [24] and their numbers dramatically increase after birth [4]. Although differences between the embryonic/perinatal ECM and the adult ECM have been noted, the contribution of cardiac fibroblasts to these ECM differences has not been evaluated [2,10–12,25]. We compared ECM proteins from decellularized control P5 and adult ventricles using liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based analysis (Fig. 1A). Levels of some ECM proteins remained constant between perinatal and adult hearts (Supplemental Table 4). However, we found significant differences in several essential ECM components, including fibrillins, nidogens, laminins, and collagens (Fig. 1A and Supplemental Table 5).
Fig. 1.
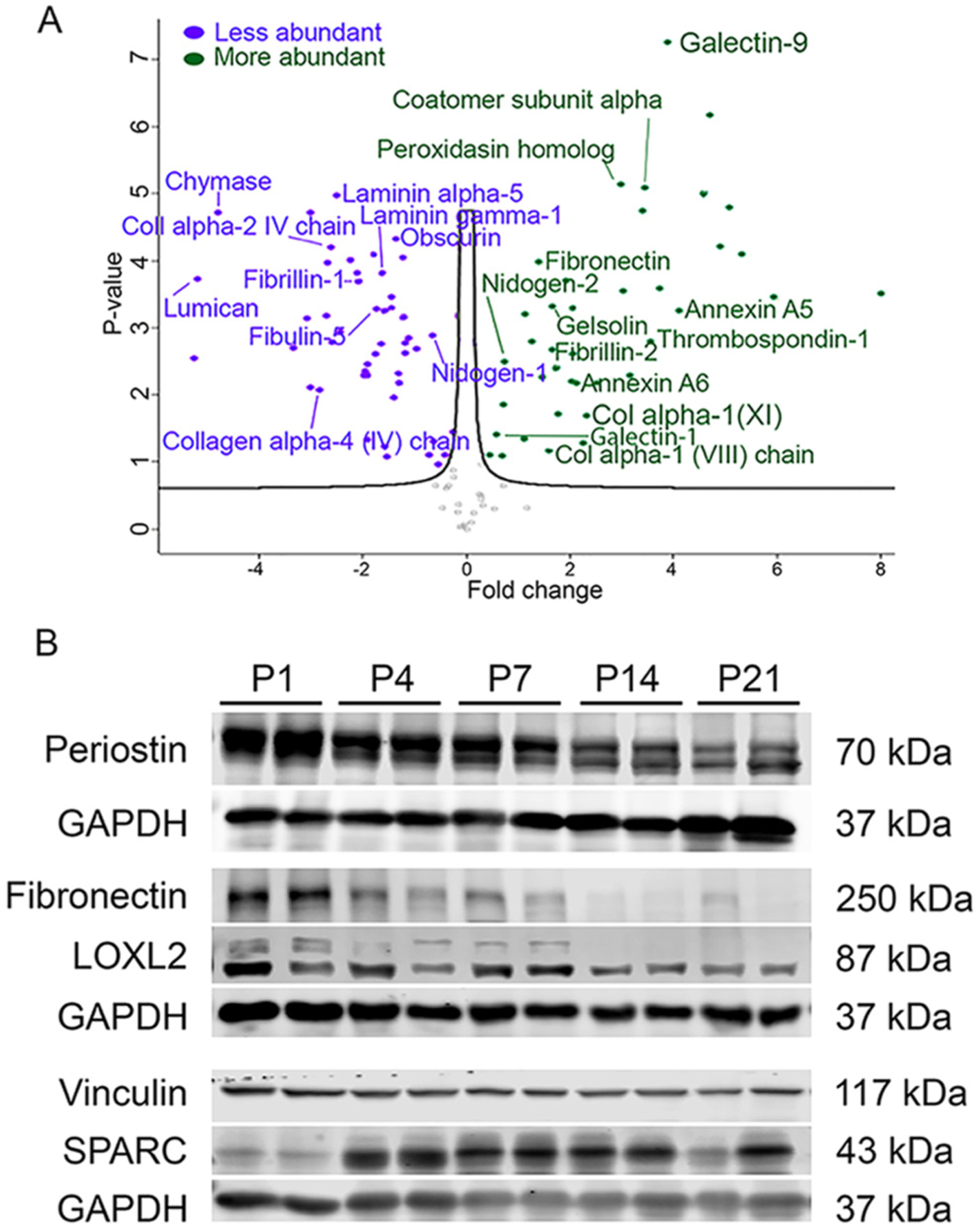
Fibroblast-related protein profiles in perinatal hearts. (A) Volcano plot with distribution of quantified secreted proteins. Fold change comparison is decellularized control perinatal day (P)5 relative to decellularized control adult (>3 months old) ventricle tissue using shotgun proteomics. All points are FDR < 0.05. p-values are calculated from the data of three biological replicates. (B) Western blots of proteins extracted from whole ventricle at indicated postnatal ages. Representative of a minimum of 4 pups at each time point.
We then determined the levels of common cardiac fibroblast-associated proteins [26] in total ventricle lysate by western blotting (Fig. 1B and Supplemental Fig. 1). To assess the relative abundance of fibroblast proteins, we used the level of PDGFRα, a protein expressed by resident cardiac fibroblasts as a surrogate [4,15,27]. Relative to P1 levels, PDGFRα protein increases by P4 (Supplemental Fig. 1), following the predicted increase in fibroblast numbers in the wild-type perinatal heart [4,5]. Examination of fibroblast-secreted ECM proteins revealed a dynamic and diverse profile of protein accumulation. Periostin, fibronectin, and LOXL2 protein levels demonstrated a similar profile with levels highest immediately after birth that gradually decreased by P21 (Fig. 1B). Other ECM proteins, such as active MMP8, tenascin X, and collagen I increased over time (Supplemental Fig. 1), whereas vinculin, a cytoskeletal protein, remained unchanged (Fig. 1B). SPARC protein content increased between P1 and P4. Our findings are consistent with previous observations of the composition of immature ECM in the fetal heart compared to mature ECM in the adult heart [2,12,28–30]. Results from both proteomics and western blot analyses suggest that changes in fibroblast ECM production in the perinatal heart coincide with heart maturation.
3.2. Perinatal fibroblast ablation
The perinatal heart experiences a dynamic change in ECM composition during maturation, therefore we chose to explore the importance of ECM by generating animals which lack the primary source of ECM, the cardiac fibroblast. We expressed diphtheria toxin A (DTA) in PDGFRα expressing cells, resulting in a dramatic reduction of cardiac fibroblasts. These animals are termed fibroblast “ablated.” Fibroblast proliferation peaks around 4 days after birth [4], therefore we initially induced DTA expression at P3 before this peak and examined hearts 7 days later (Fig. 2A). Using a tdTomato (tdT) Cre reporter to follow Cre recombinase activity, a marked decrease in lineage-tagged cells was found in ablated hearts when compared to controls (Fig. 2B, F). To determine if the observed decrease in PDGFRα lineage cells was due to an overall loss of cardiac fibroblasts, we used an unbiased reporter, collagen1a1-GFP (Col1a1-GFP) [21] for labeling cells with active collagen promoter activity [1,15,27,31]. Ablated hearts also demonstrated a decrease in GFP+ cells when compared to control hearts (Fig. 2C, G), indicating an overall fibroblast loss up to 70–80%. We examined additional fibroblast-generated proteins by immunostaining for vimentin and periostin. Ablated hearts contained reduced levels of these fibroblast-derived proteins as well (Fig. 2D, E). Western blot analysis of whole ventricle tissue verified a consistent and significant decrease in PDGFRα protein in ablated hearts (Fig. 2H–I). Examination of smooth muscle cell (SM22) and cardiomyocyte (sarcomeric α-actinin) protein levels showed that ablated hearts contained similar levels of SM22 and increased levels of sarcomeric α-actinin compared to controls (Fig. 2H–I). These data suggest that perinatal ablation of PDGFRα expressing cells is an effective means for reducing cardiac fibroblast numbers by 70–80% in the heart and that reduced fibroblast numbers do not grossly alter cardiac cellular composition.
Fig. 2.
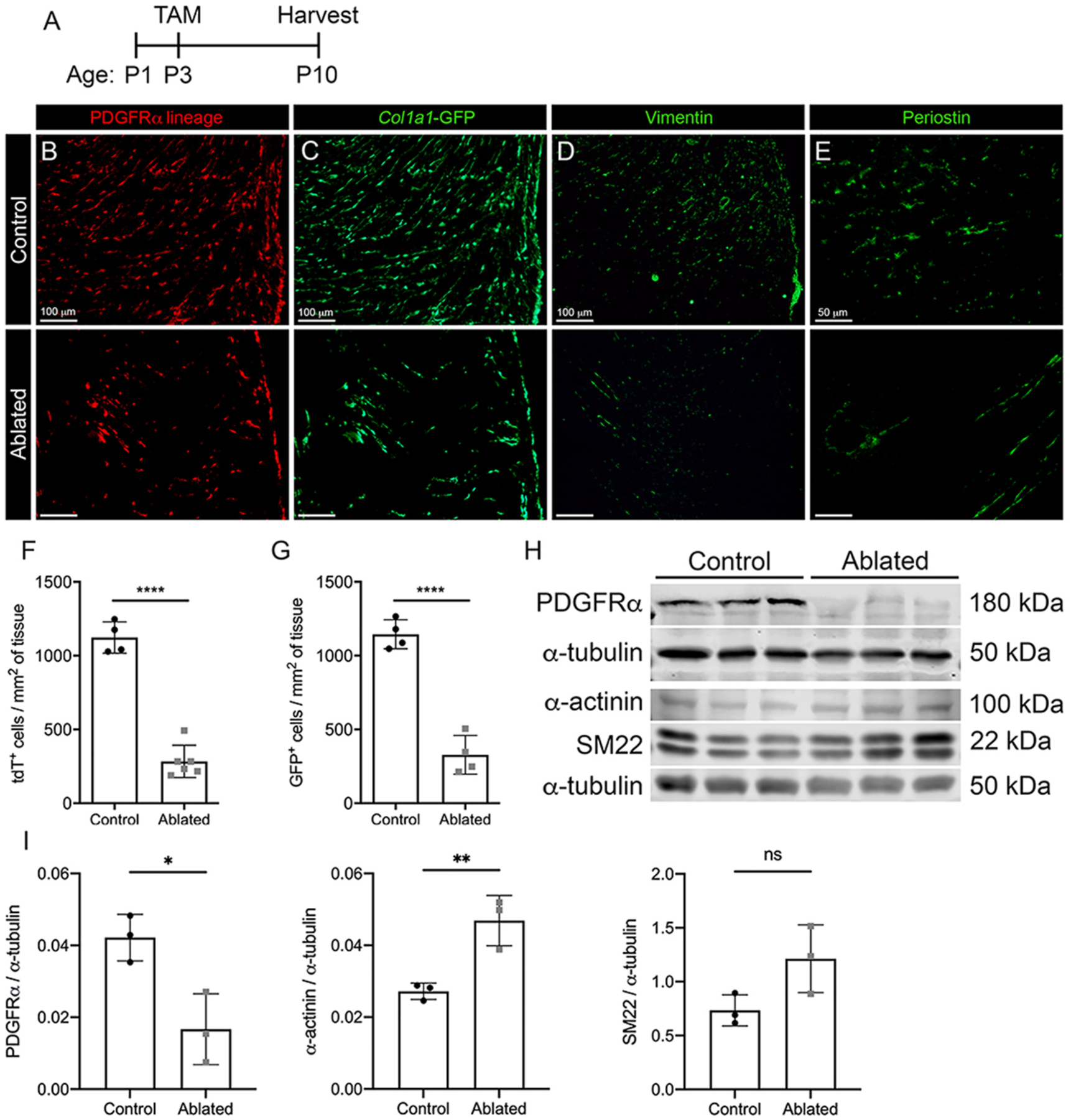
Perinatal fibroblast ablation. (A) Experimental scheme. (B-E) Representative images of left ventricular (LV) myocardium from PDGFRα-CreERT2/+; R26RtdT/+ (control) and PDGFRα-CreERT2/+; R26RtdT/DTA (ablated) hearts shown with endogenous labeling: (B) tdTomato and (C) Col1a1-GFP and immunostaining: (D) vimentin and (E) periostin. Representative from a minimum of 3 biological replicates for each staining. Quantification of (F) tdTomato and (G) Col1a1-GFP from control and ablated hearts. 3 fields of view (FOV) at 20× from 2 non-consecutive sections per biological replicate were quantified. Control: n = 4; ablated: n = 5–6. (H-I) Western blot analysis of whole ventricle lysate. n = 3 per group. Results are mean ± SD. Statistical significance was determined by unpaired t-test. ns: not significant, P > 0.05; *P ≤ 0.05 **P ≤ 0.01; ****P ≤ 0.0001.
Embryonic fibroblasts may have deposited a significant amount of ECM by P3, resulting in a milder ECM phenotype. Therefore, we induced fibroblast ablation at birth (P1). Pups with fibroblast ablation at P1 live up to 11 days (Supplemental Fig. 2A), thus, we were unable to extend our analyses of perinatal fibroblast ablation into adult stages. At P5, fibroblast ablated pups were slightly smaller than controls, but heart weight to body weight ratios were normal (Supplemental Fig. 2B–C). Lung histology revealed that ablated pups had defects in septation suggesting that the cause of death was likely due to lung abnormalities, although we can’t rule out other potential causes of demise (Supplemental Fig. 2D). Organ weights were proportionate to the animal weights (Supplemental Fig. 2E–F). Ablated hearts were indistinguishable from controls by hematoxylin staining of sections (Supplemental Fig. 3A–B). However, collagen was less mature in the mitral valve of ablated pups as indicated by picrosirius red staining (Supplemental Fig. 3C–D). Staining of sarcomeres and intercalated discs showed no obvious differences in organization (Supplemental Fig. 3E). Further examination of the hearts detected no significant apoptosis, autophagy, or increased residual fibroblast proliferation (Supplemental Fig. 4A–C, 6B). A metabolic panel revealed that there were significant differences in values related to kidney function and hematocrit (Supplemental Table 8). To determine how loss of fibroblasts affected postnatal heart development, we focused on P4 and P5 when heart to body weight ratios were similar (Fig. 3A, Supplemental Fig. 2B–C). Fibroblast ablation at P1 (Fig. 3A) resulted in decreased tdTomato+ and Col1a1-GFP+ cells by P5 (Fig. 3B–D). Substantial reduction in fibroblasts was observed in both ventricles and the interventricular septum (Fig. 3B–C). PDGFRα expression was examined by immunostaining, qPCR, and western blot analysis to verify ablation of PDGFRα expressing cells in the heart. Reduction in PDGFRα transcript and protein levels were observed 4 days after ablation (Fig. 3E, H, and Supplemental Fig. 5A–B).
Fig. 3.
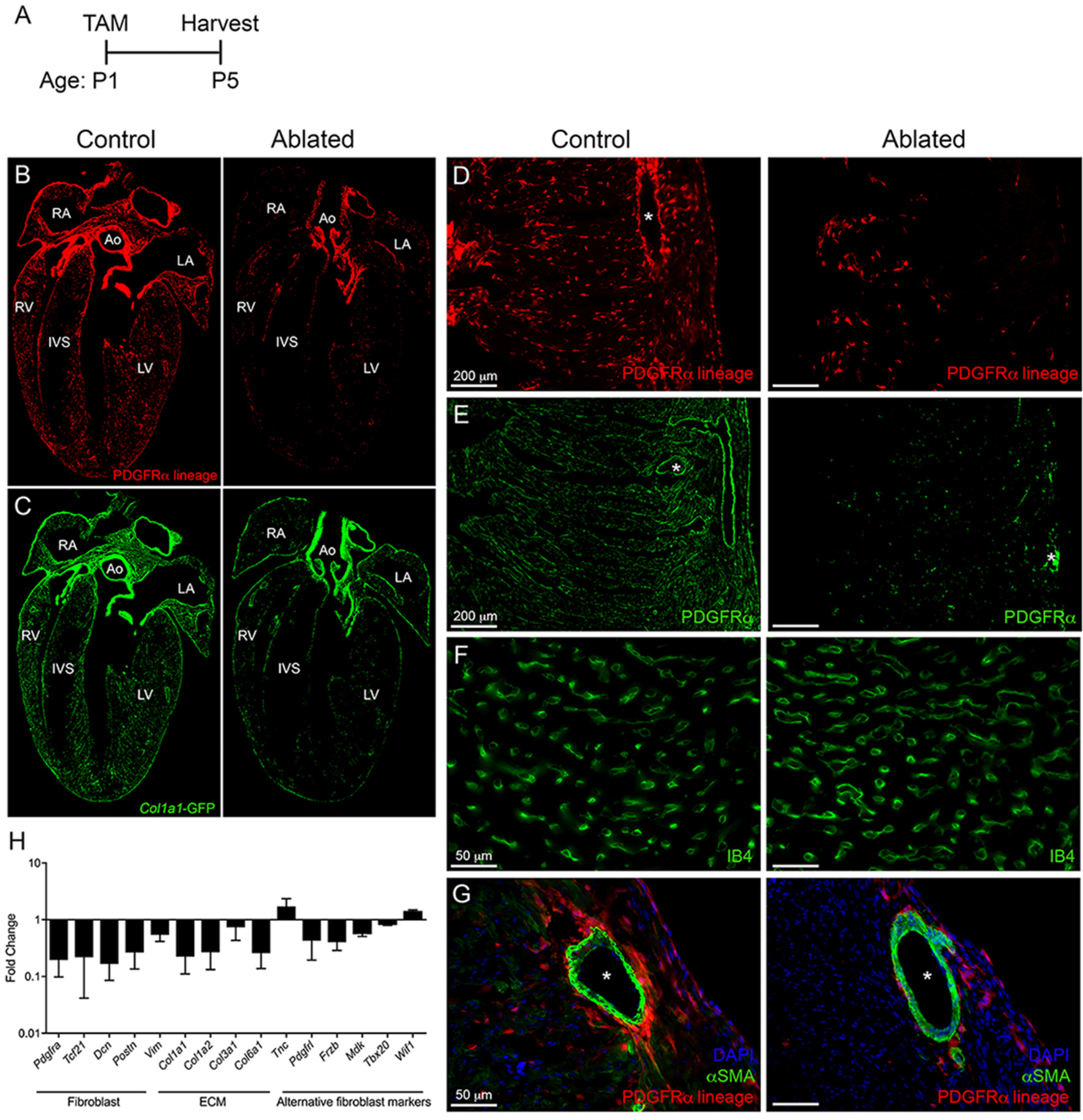
Ablation of fibroblasts in the perinatal proliferative window. (A) Experimental scheme. (B-C) 4 chamber view of (B) tdTomato and (C) Col1a1-GFP expression in control and ablated hearts. Ao: aorta, LA: left atria, RA: right atria, LV: left ventricle, RV: right ventricle, IVS: interventricular septum. (D-G) Representative images of (D) tdTomato, (E) PDGFRα, (F) isolectin B4 (IB4), and (G) αSMA immunostaining in the LV myocardium. Asterisks indicate blood vessels. Representative from a minimum of 3 biological replicates for each staining. (H) qPCR analysis of selected fibroblast, ECM, and alternative fibroblast genes in whole ventricle tissue from ablated hearts compared to controls. 18 s was used as a housekeeping gene. Control: n = 3; ablated: n = 6. Results are mean ± SD.
Despite the loss of fibroblasts, both small and large vessels appeared morphologically normal (Fig. 3F–G). Flow cytometry confirmed the reduction in fibroblast numbers with no change in endothelial and immune cell populations (Supplemental Fig. 6A). In ablated hearts, several known fibroblast-related genes were trending towards being decreased, such as Tcf21, Dcn, Postn, and Vim. Alternative cardiac fibroblast-specific genes identified in recent single cell RNAseq studies, including Pdgfrl, Frzb, and Mdk [32,33], were also trending towards reduced in ablated hearts, suggesting that another subpopulation of fibroblasts does not compensate for the loss of PDGFRα expressing fibroblasts (Fig. 3H). Moreover, examination of fibroblast proliferation index in control and ablated hearts indicated that no compensatory proliferation occurs in the remaining fibroblast population (Supplemental Fig. 6B). We also examined gene expression of several collagens expressed by fibroblasts, including Col1a1, Col1a2, Col3a1, and Col6a1, all of which were decreased in ablated hearts compared to controls (Fig. 3H). These data demonstrate that our model of fibroblast depletion is precise regarding timing and that targeting fibroblast ablation at P1 results in a severe reduction in fibroblast numbers in the heart within only 4 days of deletion time.
3.3. Fibroblast loss results in aberrant ECM deposition
Cardiac fibroblasts are responsible for most type I collagen production in the heart [34,35]. Unsurprisingly, we observed reduced collagen I levels by immunostaining (Fig. 4A) and reduced total collagen by hydroxyproline content in ablated hearts after P1 ablation (Fig. 4B). SHG microscopy and picrosirius red staining demonstrated a reduced concentration of fibrillar collagen and type I collagen, respectively, surrounding blood vessels in ablated hearts despite normal morphology (Fig. 4C–F). A collagen hybridizing peptide was used to identify areas of non-triple helical collagen, and reduced staining was observed both around blood vessels and in the interstitium of ablated hearts (Fig. 4G–H). While fibroblasts have not been shown to specifically secrete basement membrane proteins, fibroblast loss resulted in an irregular pattern of laminin surrounding cardiomyocytes (Fig. 4I).
Fig. 4.
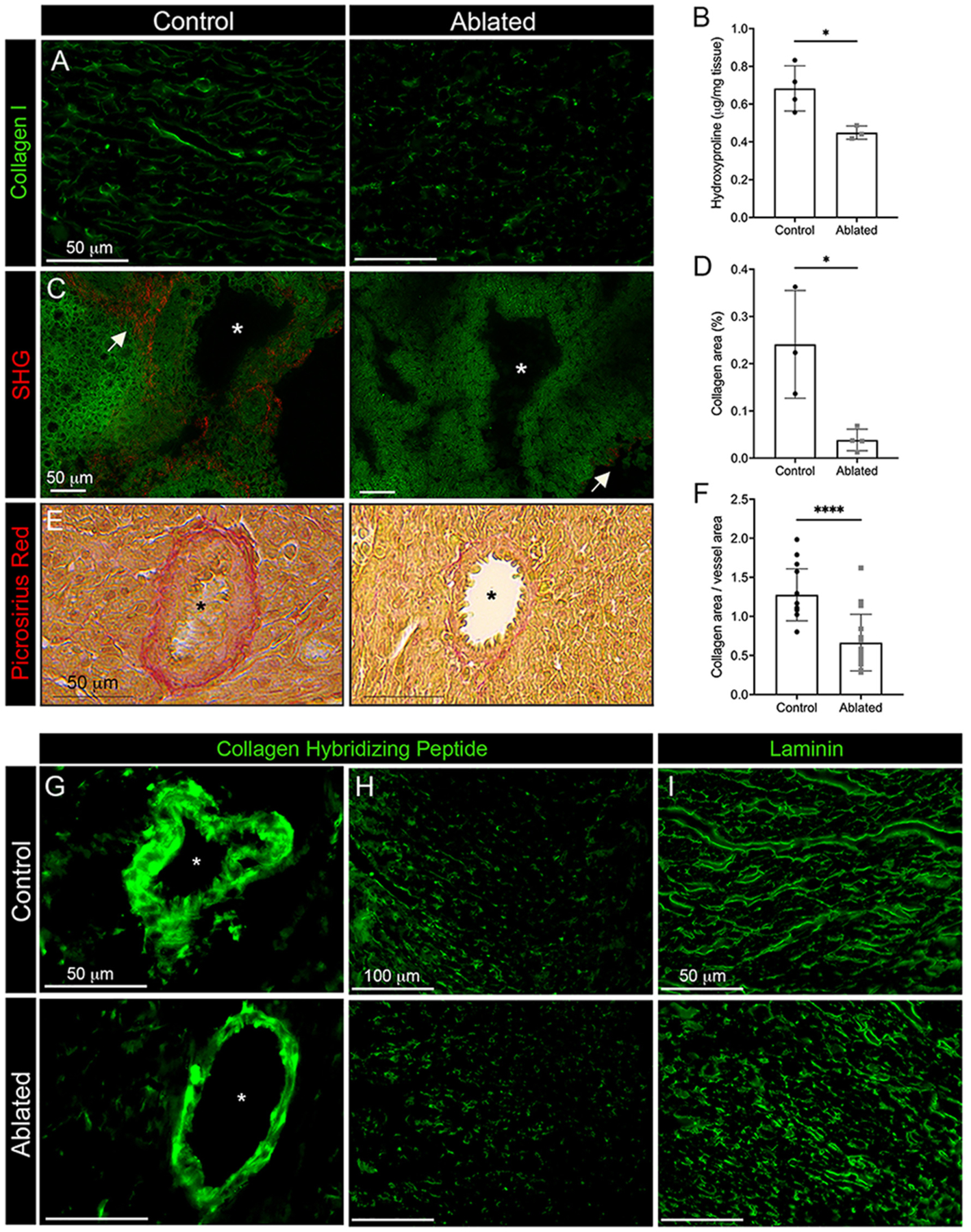
Reduced collagen deposition in fibroblast ablated hearts. Representative images of (A) collagen I immunostaining and (B) hydroxyproline content from whole ventricular tissue. Induction at P1 and isolation at P5. Control: n = 4; ablated: n = 3. (C-D) Quantification and representative images of fibrillar collagen analyzed by second harmonic generation (SHG). Induction at P1 and isolation at P4. 20 FOV per biological replicate were quantified. Control: n = 3; ablated: n = 4. (E-F) Picrosirius red staining and collagen area normalized to vessel area. Induction at P1 and isolation at P5. Control: 28 vessels across 3 biological replicates; ablated: 31 vessels across 4 biological replicates. (G-H) Representative images of collagen hybridizing peptide distribution surrounding blood vessels and within the ventricle wall, respectively, and (I) laminin immunostaining. Induction at P1 and isolation at P5. Representative from a minimum of 3 biological replicates for each staining. Asterisks indicate large blood vessels. Arrows indicate area of fibrillar collagen. Results are mean ± SD. Statistical significance was determined by unpaired t-test. *P ≤ 0.05; ****P ≤ 0.0001.
To identify potential ECM proteins secreted and/or maintained predominantly by fibroblasts, we examined ventricular proteins by western blot and proteomics. Periostin, SPARC, LOXL2, and type VI collagen protein levels were decreased on western blots in the absence of fibroblasts (Fig. 5A–B). Moreover, active MMP8 protein levels and MMP2/9 activity were reduced in ablated hearts (Fig. 5A–C). However, several proteins commonly associated with fibroblasts remained unchanged, such as fibronectin, vinculin, and agrin. These data suggest that PDGFRα expressing fibroblasts are not the main cell type responsible for secretion of these protein (Supplemental Fig. 5A–B). An unbiased comparison of decellularized ventricles by mass spectrometry revealed not only a reduction of some ECM proteins but also increases in some matrix-associated proteins (Fig. 5D and Supplemental Table 7). These results indicate that neonatal fibroblast ablation alters ECM composition.
Fig. 5.
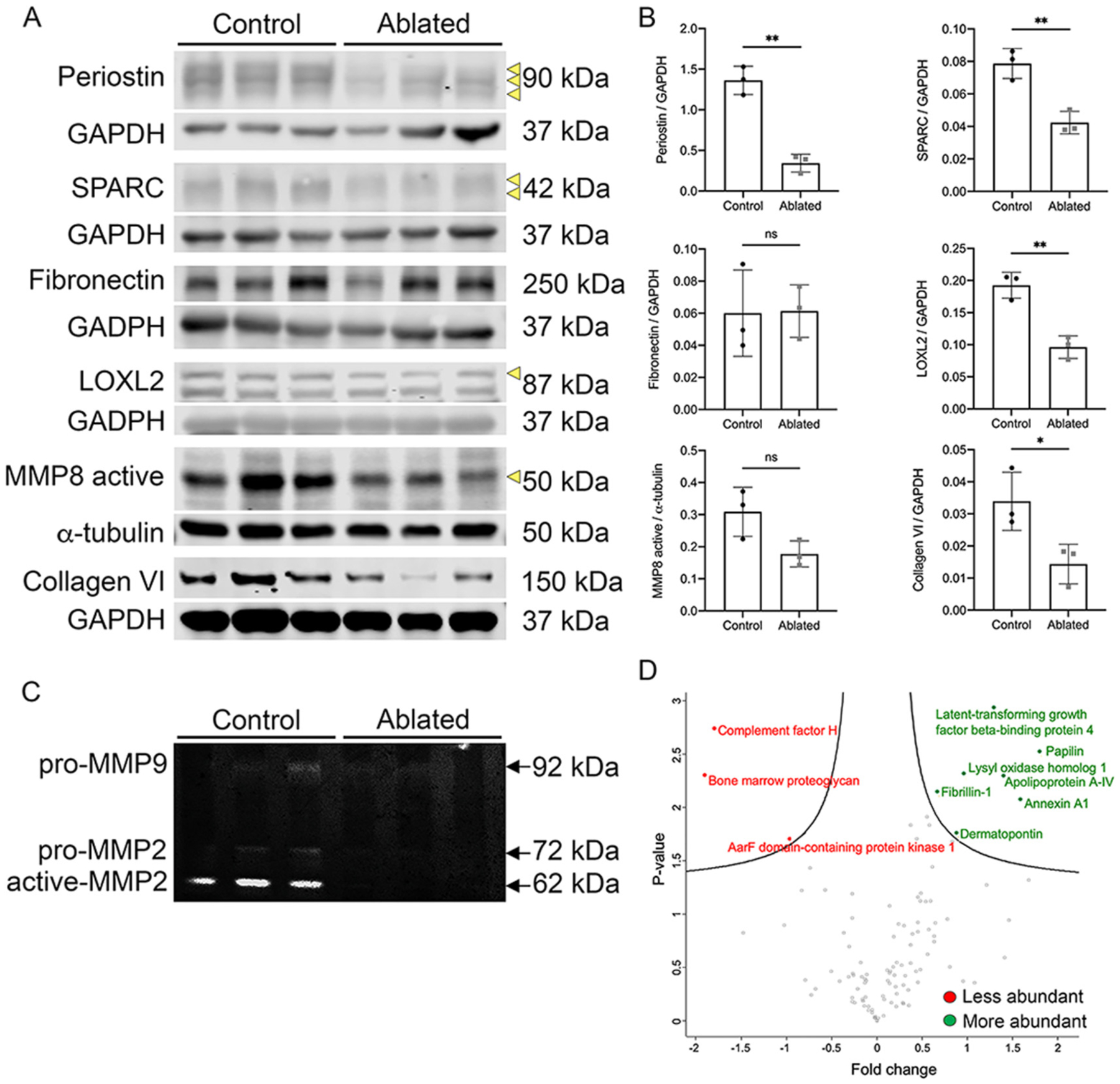
ECM changes observed with fibroblast loss. (A-B) Western blot analysis and quantification of whole ventricle lysates. Loading controls are shown below each blot. Arrows indicate protein band used for densitometry analysis. n = 3 per group. (C) Gelatin zymography of whole ventricle tissues. n = 3 per group. (D) Volcano plot of quantified proteins from decellularized control and ablated hearts. Fold change comparison is perinatal control relative to perinatal ablated. All points are FDR < 0.05. p-values are calculated from the data of three replicates. Induction at P1 and isolation at P5. Results are mean ± SD. Statistical significance was determined by unpaired t-test. ns: not significant, P > 0.05; *P ≤ 0.05; **P ≤ 0.01.
3.4. Fibroblast loss results in increasedd cardiac cell cycle activity
Substrate stiffness or compliance may be one of the factors that contribute to neonatal cardiomyocyte proliferation [13,36]. Recent studies demonstrated that cardiomyocytes cultured on a compliant matrix [13] or fetal heart-derived ECM [12] displayed an increase in proliferative markers and were able to reenter the cell cycle. Therefore, we determined if reduced collagen levels after fibroblast ablation resulted in cardiac tissue with a lower tensile strength and the potential for retention of cardiomyocyte proliferation ability. We measured the stiffness of the heart using AFM. As anticipated, ablated hearts were significantly less stiff than littermate controls at P4 (Fig. 6A–B). However, when fibroblasts were ablated at P5 and examined at P9, after the reported peak in myocyte proliferation [5], there was no significant difference in stiffness between control and ablated hearts (Fig. 6A–B). Interestingly, substrate stiffness was similar in control P1 and P9 hearts (Fig. 6B).
Fig. 6.
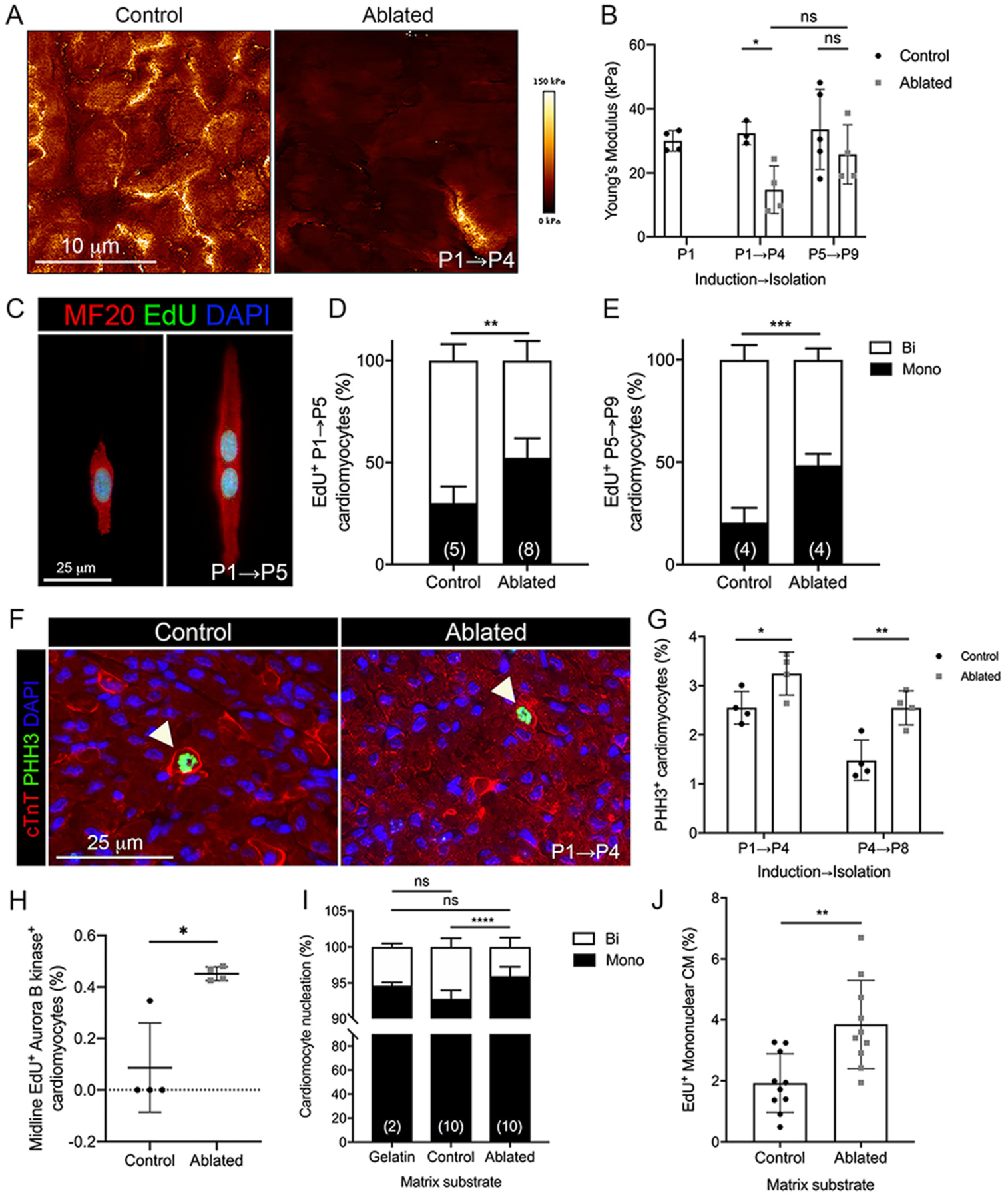
Cardiomyocyte cell cycle activity with fibroblast ablation. (A-B) Representative images of stiffness maps and the average Young’s modulus at indicated induction and isolation times. 4 scans of LV free wall per biological replicate were quantified. Control: n = 3–5; ablated: n = 4. (C) Representative images of EdU staining in isolated P5 cardiomyocytes. (D, E) Quantification of EdU+ mono and binucleated cardiomyocytes in (D) P5 and (E) P9 hearts induced at P1 and P5, respectively. Numbers within bar graphs represent biological replicate n values. 400–500 cardiomyocytes per biological replicate. (F) Representative images of immunostaining of cardiac troponin T (cTnT) and phosphohistone H3 (PHH3) in P4 heart. Arrows indicate PHH3+ cardiomyocytes. (G) Quantification of PHH3+ cardiomyocytes. n = 4 per group. 400–500 cardiomyocytes per biological replicate. (H) Quantification of midline EdU+/Aurora B kinase+ cardiomyocytes in P5 hearts. 200 cardiomyocytes per biological replicate. n = 4 per group. Quantification of (I) total mono and binucleated P2 cardiomyocytes and (J) EdU+ mononucleated P2 cardiomyocytes cultured on decellularized matrix from control and ablated P4 hearts for 48 h. Numbers within bar graphs represent biological replicate n values (control and ablated) or number of independent experiments (gelatin). 200 cardiomyocytes per biological replicate. Results are mean ± SD. Statistical significance was determined by unpaired t-test or Mann-Whitney U test (H). ns: not significant, P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
One feature of cardiomyocyte maturation is a switch from cytokinesis to binucleation via endomitosis, which correlates with a loss of cardiomyocyte proliferation and regenerative capacity [5]. Therefore, we compared the ratio of mono and binucleated cardiomyocytes in control and ablated hearts. To determine if cardiomyocytes in ablated hearts retain the ability to undergo cytokinesis, we injected control and ablated animals with EdU at P3 to label cells in S-phase. 48 h later at P5, we isolated primary neonatal cardiomyocytes and determined the number of EdU+ mono and binucleated cardiomyocytes (Fig. 6C–D). Fibroblast loss was associated with an increased number of EdU+ mononucleated cardiomyocytes compared to controls. To further investigate DNA synthesis in myocytes after the reported peak of proliferation [5], we examined the number of EdU+ mono- and binucleated cardiomyocytes from P9 hearts induced with tamoxifen at P5 (Fig. 6E). 48 h of EdU labeling revealed more mononucleated EdU+ cardiomyocytes in the absence of fibroblasts even at this later developmental time point. However, the total percentage of mono and binucleated cardiomyocytes remains the same between control and ablated hearts, suggesting that the mononucleated cells progress to binucleated cells eventually (Supplemental Fig. 7A). These data indicate that reduced fibroblast numbers may lead to a higher rate of cardiomyocyte cytokinesis, at a time when cardiomyocytes typically switch to binucleation. The in vivo cardiomyocyte mitotic index was visualized by phospho-histone H3 (PHH3) immunostaining in cardiomyocytes at P4 and P8 (Fig. 6F–G). We observed an increased mitotic index at both time points in ablated hearts, supporting the idea that loss of fibroblasts correlates with less mature cardiomyocytes that have an increased proliferative capacity. As midbody positioning with Aurora B kinase staining indicates cardiomyocyte cell division [37], we quantified the number of EdU+ cells with midline Aurora B kinase and found a significant difference between ablated and control hearts (Fig. 6H and Supplemental Fig. 8). Cardiomyocyte area measured in vivo showed that cardiomyocytes were smaller in ablated hearts (Supplemental Fig. 7B).
To determine if the observed increase in cardiomyocyte cell cycle activity was due to the extracellular environment generated by fibroblasts, we examined the percentage of mono and binucleated neonatal cardiomyocytes cultured on decellularized matrix from control and ablated P4 hearts after 48 h. We observed an increase in mononucleated cardiomyocytes on matrix derived from ablated hearts, as well as increased EdU incorporation in these mononucleated myocytes (Fig. 6I–J). These data suggest that the composition and architecture of the ECM may be, in part, responsible for a switch to myocyte binucleation. Taken together, these data demonstrate that changes in ECM composition due to fibroblast loss result in persistence of mononucleated cardiomyocytes.
4. Discussion
Resident cardiac fibroblasts are believed to be the predominant source of ECM in the heart. They are initially observed in the myocardium around E14.5 [24] and their numbers continually increase in the first week after birth [4]. In this study, we depleted fibroblasts at birth and investigated how a 70–80% reduction of fibroblasts affects the heart. Cardiac tissue appeared histologically normal despite a significant reduction in ECM components. While overall heart morphology was relatively normal, most pups did not survive past P11. A limitation of these studies is that systemic effects promoting cardiomyocyte proliferation cannot be discounted. Reduced lung septation may contribute to the cardiomyocyte phenotype, as systemic hypoxia can induce myocyte proliferation as reviewed in Kimura et al. [38]. An additional limitation is that we did not perform analyses that would permit a functional assessment of the heart.
Because fibroblasts are thought to be the main source of certain ECM components, especially type I collagen, we expected a reduction in these proteins with fibroblast loss. Interestingly, many ECM components were reduced in ablated hearts but not completely absent. There may be several explanations for this observation. First, we are initiating fibroblast loss after birth. Although fibroblasts are present in the embryonic heart prior to ablation, it is thought that collagen production and crosslinking increase substantially after birth [6]. Nonetheless, some fibroblast matrix production may occur in the late-gestational embryonic heart, and this embryonic ECM may be sufficient to maintain cardiac architecture in the first week of life even after removal of fibroblasts. Second, fibroblast depletion in the hearts we examined was incomplete. The remaining 20–30% fibroblasts may be sufficient to generate the required ECM, even though most fibroblast gene expression remains reduced. These observations suggest that the remaining fibroblasts do not substantially upregulate collagen gene expression. Third, recent single cell RNAseq studies have suggested that endothelial cells and cardiomyocytes can express ECM proteins, illustrating that fibroblasts are not the only cells that contribute ECM components such as type I collagen and fibronectin [39–45]. Therefore, these other cell types may partially compensate for the absence of fibroblasts. It would be interesting to examine ECM gene expression or production at the single cell level to determine if there is an increase in ECM expression by individual cells within any of the other cardiac cell populations.
Recent studies have indicated that fibroblasts within the heart exhibit heterogenous and dynamic gene expression and suggest that PDGFRα may not be uniformly expressed among fibroblasts [33], allowing the possibility that fibroblast removal may be incomplete in our studies. One subpopulation that has been described expresses a number of genes involved in the Wnt pathway (F-Wnt X) [32,33]. These unique fibroblasts have low PDGFRα expression, but our data suggest that expression of fibroblast genes is not upregulated in the absence of PDGFRα expressing cells. Instead, our data suggest that at this stage of heart development, PDGFRα may be expressed in many of the collagen-expressing cells including those derived from the endocardium and other subpopulations, as we see uniform depletion of fibroblasts in all four chambers as well as the septum of the heart. Another study identified heterogenous populations of fibroblasts that exist in the in vivo perinatal heart and demonstrated that a subset of periostin+ fibroblasts were highly proliferative after birth [2]. Ablation of these cells reduced mitotic activity and hypertrophic growth in cardiomyocytes. However, our results demonstrated that ablation of PDGFRα+ fibroblasts increased cell cycle activity in cardiomyocytes. A potential explanation for these contrasting outcomes could be that PDGFRα+ fibroblasts encompass a larger population of fibroblasts that possess the ability to alter collagen deposition, whereas periostin+ fibroblasts constitute a smaller subset of fibroblasts that do not overtly alter collagen deposition. Therefore, these populations of fibroblasts may have differing effects on cardiomyocyte maturation and binucleation.
A recent study demonstrated that cardiomyocytes cultured on matrix derived from P1 hearts were more proliferative compared to those cultured on matrix from P7 hearts [10]. Similarly, others have shown that fetal cardiac ECM promoted increased cardiomyocyte mitosis compared to adult cardiac ECM [12]. Additionally, mechanical properties of the matrix, such as stiffness, have been shown to regulate cardiomyocyte cell cycle arrest in vitro [13]. Manipulation of ECM crosslinking in vivo validated these results by showing that increased matrix plasticity rescued the ability of P3 hearts to regenerate after apical resection [28]. Our work supports the idea that fibroblasts contribute to heart maturation by dynamic secretion of ECM that potentially regulates cardiomyocyte binucleation rates. However, it is unlikely that this phenomenon is exclusively due to fibroblast abundance. These results suggest that the ECM secreted by fibroblasts is, in part, responsible for cardiomyocyte binucleation and cell cycle arrest.
The heart progressively loses the ability to generate new cardiomyocytes shortly after birth [8,46], and thus the mature heart has a limited capacity to repair itself after disease and damage. Therefore, identifying therapeutics to stimulate proliferation in post-mitotic cardiomyocytes is of great interest. While agrin supplementation can increase proliferation of adult cardiomyocytes and improve heart function after myocardial infarction [10], we found that agrin protein levels were relatively normal in fibroblast ablated hearts suggesting that fibroblasts are not required for agrin expression. While multiple studies have utilized injury models, such as apical resection and myocardial infarction in neonatal hearts to demonstrate regenerative capacity, we observed extended cell cycle activity past the normal proliferative window in the absence of injury, although the overall ratio of mononucleated cells to binucleated cells did not differ between control and fibroblast ablated hearts.
It is likely that multiple ECM components contribute to heart maturation. Nephronectin and SLIT2 were identified as two proteins preferentially expressed by embryonic fibroblasts that could enhance cardiomyocyte cell division [11]. We identified additional ECM proteins expressed by perinatal fibroblasts, including tenascin-X, fibrillin-1, and emilin-1. While we determined that increased cardiomyocyte DNA synthesis was in part due to changes in ECM generated by fibroblasts, other fibroblast activities, including paracrine signaling, may also regulate cardiomyocyte proliferation. We found that when fibroblasts were ablated after ECM maturation, tissue stiffness was not significantly reduced but increased DNA synthesis in mononucleated cardiomyocytes was sustained, suggesting that changes in ECM components and compliance are not solely responsible for increased cardiomyocyte cell cycle activity by fibroblasts. Tissue compliance remained relatively constant during the transition from hyperplastic to hypertrophic cardiomyocyte growth, further suggesting that compliance may not be a major determinant in cardiomyocyte regenerative capacity. Additionally, ECM and paracrine factors secreted by embryonic cardiac fibroblasts were shown to induce proliferation of cardiomyocytes through β1 integrin signaling in co-culture, highlighting the importance of fibroblast functions on cardiomyocyte biology [9]. Further investigation of fibroblast-specific ECM components responsible for cardiomyocyte cell arrest and binucleation will provide insight into the molecular mechanisms governing cardiac regeneration.
5. Conclusions
In summary, we observed that fibroblast ablation changed the extracellular environment and increased tissue compliance. Proteomic analysis of decellularized hearts revealed changes in ECM proteins identifying some ECM proteins that may be fibroblast-dependent. Modulation of ECM composition and complexity by fibroblasts resulted in an increased number of cardiomyocytes undergoing DNA replication and presented a higher ratio of cycling mononucleated to binucleated cells. Matrix from fibroblast ablated hearts sustained cardiomyocyte DNA synthesis demonstrating that fibroblast generated ECM proteins may participate in regulating cardiomyocyte cell cycle activity in the perinatal mouse heart. Our study highlights a role for PDGFRα fibroblasts in ECM production in the heart.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2022.05.003.
Supplementary Material
Acknowledgements
We thank Sharmaine Sebastian for her excellent technical support and all Tallquist lab members for critically reviewing this manuscript. We also thank Miyoko Bellinger and Kristen Ewell from the JABSOM Histology and Imaging Core.
Sources of funding
This work was supported by NIH HL156112 (MDT), HL144067 (MDT), HL147558 (TAM), HL116848 (TAM), HL127240 (TAM), DK119594 (TAM), NHLBI Institutional Cardiology Training Grant T32 HL115505 (JTK), NHLBI K25HL148386 (BP), American Heart Association Grants PRE29630019 (JTK) and GRNT33660474 (MDT), and FRN31400013 (TAM and BP) and by the Allen Distinguished Investigator Program, through support made by The Paul G. Allen Frontiers Group and the American Heart Association award 17DIA33820024 (SSA). The University of Hawaii Health Sciences Microscopy and Imaging Core was supported by RCMI-BRIDGES G12 MD007601. The Microscopy, Imaging, and Flow Cytometry Core at UHCC was supported by SIG: NIH S10OD028515. The Genomics and Bioinformatics Shared Resource (GBSR, RRID:SCR_019085) at UHCC is supported by NCI 5P30CA071789.
Non-standard abbreviations and acronyms:
- AFM
atomic force microscopy
- αSMA
alpha smooth muscle actin
- Cre
P1 bacteriophage Cre recombinase
- CreERT2
Cre recombinase fused to a mutant estrogen ligand-binding domain (ERT2)
- cTnT
cardiac troponin T
- DTA
diphtheria toxin A
- ECM
extracellular matrix
- EdU
5-ethynyl-2′-deoxyuridine
- GFP
green fluorescent protein
- LOXL2
lysyl oxidase-like 2
- MF20
myosin heavy chain
- MMP
matrix metalloproteinase
- PDGFRα
platelet derived growth factor receptor alpha
- PHH3
phosphohistone H3
- SHG
second harmonic generation
- Tcf21
transcription factor 21
- tdT
tandem dimer tomato
Footnotes
Disclosures
None.
References
- [1].Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, et al. , The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors, Development. 139 (12) (2012) 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hortells L, Valiente-Alandi I, Thomas ZM, Agnew EJ, Schnell DJ, York AJ, et al. , A specialized population of Periostin-expressing cardiac fibroblasts contributes to postnatal cardiomyocyte maturation and innervation, Proc. Natl. Acad. Sci. U. S. A 117 (32) (2020) 21469–21479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Maroli G, Braun T, The long and winding road of cardiomyocyte maturation, Cardiovasc. Res 117 (3) (2021) 712–726. [DOI] [PubMed] [Google Scholar]
- [4].Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z, Tallquist MD, Resident fibroblast expansion during cardiac growth and remodeling, J. Mol. Cell. Cardiol 114 (2018) 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ, Cardiomyocyte DNA synthesis and binucleation during murine development, Am. J. Phys 271 (5 Pt 2) (1996) H2183–H2189. [DOI] [PubMed] [Google Scholar]
- [6].Gershlak JR, Resnikoff JI, Sullivan KE, Williams C, Wang RM, Black LD 3rd., Mesenchymal stem cells ability to generate traction stress in response to substrate stiffness is modulated by the changing extracellular matrix composition of the heart during development, Biochem. Biophys. Res. Commun 439 (2) (2013) 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rozario T, DeSimone DW, The extracellular matrix in development and morphogenesis: a dynamic view, Dev. Biol 341 (1) (2010) 126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. , Transient regenerative potential of the neonatal mouse heart, Science. 331 (6020) (2011) 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, et al. , Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling, Dev. Cell 16 (2) (2009) 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O, et al. , The extracellular matrix protein agrin promotes heart regeneration in mice, Nature. 547 (7662) (2017) 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wu CC, Jeratsch S, Graumann J, Stainier D, Modulation of mammalian cardiomyocyte cytokinesis by the extracellular matrix, Circ. Res 127 (7) (2020) 896–907. [DOI] [PubMed] [Google Scholar]
- [12].Williams C, Quinn KP, Georgakoudi I, Black LD 3rd., Young developmental age cardiac extracellular matrix promotes the expansion of neonatal cardiomyocytes in vitro, Acta Biomater. 10 (1) (2014) 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yahalom-Ronen Y, Rajchman D, Sarig R, Geiger B, Tzahor E, Reduced matrix rigidity promotes neonatal cardiomyocyte dedifferentiation, proliferation and clonal expansion, Elife. 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang Y, Yao F, Wang L, Li Z, Ren Z, Li D, et al. , Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation, Nat. Commun 11 (1) (2020) 2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, et al. , Revisiting cardiac cellular composition, Circ. Res 118 (3) (2016) 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ivey MJ, Kuwabara JT, Riggsbee KL, Tallquist MD, Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival, Am. J. Physiol. Heart Circ. Physiol 317 (2) (2019) H330–h44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ivey MJ, Tallquist MD, Defining the cardiac fibroblast, Circ. J 80 (11) (2016) 2269–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moore-Morris T, Guimaraes-Camboa N, Yutzey KE, Puceat M, Evans SM, Cardiac fibroblasts: from development to heart failure, J. Mol. Med. (Berl) 93 (8) (2015) 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. , A robust and high-throughput Cre reporting and characterization system for the whole mouse brain, Nat. Neurosci 13 (1) (2010) 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wu S, Wu Y, Capecchi MR, Motoneurons and oligodendrocytes are sequentially generated from neural stem cells but do not appear to share common lineage-restricted progenitors in vivo, Development. 133 (4) (2006) 581–590. [DOI] [PubMed] [Google Scholar]
- [21].Yata Y, Scanga A, Gillan A, Yang L, Reif S, Breindl M, et al. , DNase I-hypersensitive sites enhance alpha1(I) collagen gene expression in hepatic stellate cells, Hepatology. 37 (2) (2003) 267–276. [DOI] [PubMed] [Google Scholar]
- [22].Chung MI, Bujnis M, Barkauskas CE, Kobayashi Y, Hogan BLM, Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation, Development. 145 (9) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Weldrick JJ, Abdul-Ghani M, Megeney LA, Burgon PG, A rapid and efficient method for the isolation of postnatal murine cardiac myocyte and fibroblast cells, Can. J. Physiol. Pharmacol 96 (5) (2018) 535–539. [DOI] [PubMed] [Google Scholar]
- [24].Dettman RW, Denetclaw W Jr., Ordahl CP, Bristow J, Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart, Dev. Biol 193 (2) (1998) 169–181. [DOI] [PubMed] [Google Scholar]
- [25].de Castro Bras LE, Toba H, Baicu CF, Zile MR, Weintraub ST, Lindsey ML, et al. , Age and SPARC change the extracellular matrix composition of the left ventricle, Biomed. Res. Int 2014 (2014), 810562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Abonnenc M, Nabeebaccus AA, Mayr U, Barallobre-Barreiro J, Dong X, Cuello F, et al. , Extracellular matrix secretion by cardiac fibroblasts: role of microRNA-29b and microRNA-30c, Circ. Res 113 (10) (2013) 1138–1147. [DOI] [PubMed] [Google Scholar]
- [27].Smith CL, Baek ST, Sung CY, Tallquist MD, Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling, Circ. Res 108 (12) (2011) e15–e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Notari M, Ventura-Rubio A, Bedford-Guaus SJ, Jorba I, Mulero L, Navajas D, et al. , The local microenvironment limits the regenerative potential of the mouse neonatal heart, Sci. Adv 4 (5) (2018) eaao5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Farhadian F, Contard F, Corbier A, Barrieux A, Rappaport L, Samuel JL, Fibronectin expression during physiological and pathological cardiac growth, J. Mol. Cell. Cardiol 27 (4) (1995) 981–990. [DOI] [PubMed] [Google Scholar]
- [30].Mays PK, McAnulty RJ, Campa JS, Laurent GJ, Age-related changes in collagen synthesis and degradation in rat tissues. Importance of degradation of newly synthesized collagen in regulating collagen production, Biochem. J 276 (Pt 2) (1991) 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, et al. , Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis, J. Clin. Invest 124 (7) (2014) 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, et al. , Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart, Cell Rep. 22 (3) (2018) 600–610. [DOI] [PubMed] [Google Scholar]
- [33].Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, et al. , Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury, Elife. 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Eghbali M, Czaja MJ, Zeydel M, Weiner FR, Zern MA, Seifter S, et al. , Collagen chain mRNAs in isolated heart cells from young and adult rats, J. Mol. Cell. Cardiol 20 (3) (1988) 267–276. [DOI] [PubMed] [Google Scholar]
- [35].Eghbali M, Blumenfeld OO, Seifter S, Buttrick PM, Leinwand LA, Robinson TF, et al. , Localization of types I, III and IV collagen mRNAs in rat heart cells by in situ hybridization, J. Mol. Cell. Cardiol 21 (1) (1989) 103–113. [DOI] [PubMed] [Google Scholar]
- [36].Young JL, Kretchmer K, Ondeck MG, Zambon AC, Engler AJ, Mechanosensitive kinases regulate stiffness-induced cardiomyocyte maturation, Sci. Rep 4 (2014) 6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hesse M, Doengi M, Becker A, Kimura K, Voeltz N, Stein V, et al. , Midbody positioning and distance between daughter nuclei enable unequivocal identification of cardiomyocyte cell division in mice, Circ. Res 123 (9) (2018) 1039–1052. [DOI] [PubMed] [Google Scholar]
- [38].Kimura W, Nakada Y, Sadek HA, Hypoxia-induced myocardial regeneration, J. Appl. Physiol 123 (6) (2017) 1676–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hu P, Liu J, Zhao J, Wilkins BJ, Lupino K, Wu H, et al. , Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts, Genes Dev. 32 (19–20) (2018) 1344–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, et al. , Single-cell resolution of temporal gene expression during heart development, Dev. Cell 39 (4) (2016) 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, et al. , Single-cell transcriptome analysis maps the developmental track of the human heart, Cell Rep. 26 (7) (2019) (1934–50.e5). [DOI] [PubMed] [Google Scholar]
- [42].Howard BV, Macarak EJ, Gunson D, Kefalides NA, Characterization of the collagen synthesized by endothelial cells in culture, Proc. Natl. Acad. Sci. U. S. A 73 (7) (1976) 2361–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kusuma S, Zhao S, Gerecht S, The extracellular matrix is a novel attribute of endothelial progenitors and of hypoxic mature endothelial cells, FASEB J. 26 (12) (2012) 4925–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Myers PR, Tanner MA, Vascular endothelial cell regulation of extracellular matrix collagen: role of nitric oxide, Arterioscler. Thromb. Vasc. Biol 18 (5) (1998) 717–722. [DOI] [PubMed] [Google Scholar]
- [45].Lundgren E, Gullberg D, Rubin K, Borg TK, Terracio MJ, Terracio L, In vitro studies on adult cardiac myocytes: attachment and biosynthesis of collagen type IV and laminin, J. Cell. Physiol 136 (1) (1988) 43–53. [DOI] [PubMed] [Google Scholar]
- [46].Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, et al. , Cardiomyocyte proliferation contributes to heart growth in young humans, Proc. Natl. Acad. Sci. U. S. A 110 (4) (2013) 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.