

Abstract
Background:
Protein-protein interactions (PPIs) are intriguing targets for designing novel small-molecule inhibitors. The role of PPIs in various infectious and neurodegenerative disorders makes them potential therapeutic targets . Despite being portrayed as undruggable targets, due to their flat surfaces, disorderedness, and lack of grooves. Recent progresses in computational biology have led researchers to reconsider PPIs in drug discovery.
Areas covered:
In this review, we introduce in-silico methods used to identify PPI interfaces and present an in-depth overview of various computational methodologies that are successfully applied to annotate the PPIs. We also discuss several successful case studies that use computational tools to understand PPIs modulation and their key roles in various physiological processes.
Expert opinion:
Computational methods face challenges due to the inherent flexibility of proteins, which makes them expensive, and result in the use of rigid models. This problem becomes more significant in PPIs due to their flexible and flat interfaces. Computational methods like molecular dynamics (MD) simulation and machine learning can integrate the chemical structure data into biochemical and can be used for target identification and modulation. These computational methodologies have been crucial in understanding the structure of PPIs, designing PPI modulators, discovering new drug targets, and predicting treatment outcomes.
Keywords: Protein-protein interactions, Computer-aided drug design (CADD), computational approaches, machine-based learning, molecular dynamics simulations, docking, screening
1. Introduction
PPIs are involved in almost all physiological functions, including cellular interaction, signal transduction, and metabolic pathway, so an in-depth understanding of PPIs is critical to explore their role in normal and diseased states. The PPI contacts, or interfaces as they are called, are highly specific as they are formed in defined regions of amino acids in the proteins, and they are meant to serve a specific function. The knowledge of PPIs can help us to explore not only the role of uncharacterized proteins but also their involvement in various pathophysiological states. A PPI is defined as ‘an interaction of two identical or non-identical proteins at their domain interfaces that regulates the function of the protein complex’ [1], and a modulator is a low-molecular-weight naturally derived agent with a complex structure that allows target specificity and strong binding affinity [1].
The human interactome [2,3], which is the sum of all PPIs in a cell, is expected to have about 130,000 to 650,000 binary PPIs. This complex network contributes significantly to the modification and accomplishment of an array of physiological functions [4]. But there has been an explosion of data related to the human interactome after the advancement in the high throughput screening techniques for PPIs, resulting in many unreliable and noisy data, limiting the true picture of all physiological interactions in the cells. On the other hand, given the physiological role of PPIs, they have been considered as potential drug targets. Their modulation with small molecules has resulted in drugs targeting over 50 PPIs, with >27 already in Phase I, II, and III clinical trials, primarily involving viral, autoimmune diseases, and cancer [5] So far, the FDA has approved ~5 PPI modulators to treat cancer, dry eye syndrome, autoimmune diseases, and disorders that do not respond to other treatments [5–7]. A rundown of some PPI modulators currently participating in clinical trials is listed in Table 1.
Table 1.
PPI modulators that are currently being studied in clinical studies. Data are acquired from https://clinicaltrials.gov.
| PPI | Disease | Drug | Status | NCT No | Refs |
|---|---|---|---|---|---|
| MDM2/p53 | Advanced stage cancer, lymphomas | ALRN-6924 | *P-I/II | 02264613 | [8] |
| CD40−/−L | Kidney transplantation | Bleselumab | *P-II | 02921789 | [9] |
| Relapsed diffuse large B-cell lymphoma | dacetuzumab | *P-II | 00435916 | [10] | |
| Lupus Nephritis | BI655064 | *P-II | 03385564 | [11] | |
| Advanced solid tumors | ABBV-428 | *P-I | 02955251 | [12] | |
| Ulcerative colitis | ABBV-323 | *P-II | 03695185 | [13] | |
| PD-1/L1 | Non-small lung cancer | Keytruda | **A | – | [14] |
| Merkel cell carcinoma | Bavencio | **A | – | [15] | |
| Unresectable or metastatic melanoma | JS001 | *P-III | 03430297 | [16] | |
| Advanced/metastatic solid malignancies | IBI308 | *P-I/II | 03568539 | [17] | |
| Locally advanced or metastatic urothelial bladder cancer | BGB-A317 | *P-II | 04004221 | [18] | |
| CD40/L | Kidney transplantation | Bleselumab | *P-II | 02921789 | [9] |
| MDM2/p53 | Acute myeloid leukemia | Idasanutlin | *P-III | 02545283 | [19] |
| Metastatic melanoma | AMG232 | *P-I/II | 02110355 | [20–22] | |
| Neoplasm malignant | SAR405838 | *P-I | 01636479 | [23] | |
| Bcl-2/Bax | Chronic lymphocytic leukemia | ABT-199 | **A | – | [24] |
| XIAP/caspase-9 | Relapsed or refractory multiple myeloma | LCL-161 | *P-II | 01955434 | [25] |
| Recurrent head and neck squamous | TL32711 | *P-I | 03803774 | [25] | |
| Solid cancers | GDC-0917 | *P-I | 01226277 | [26] | |
| Gp120/CCR5 | HIV | Maraviroc | **A | – | [27] |
| LFA-1/ICAM-1 | Dry eye | Lifitegrast | *P-IV | 03451396 | [28] |
| B-Catenin | Liver cirrhosis | RPI-724 | *P-I/II | 03620474 | [29] |
| Histone | Cardiovascular diseases | RVX-208 | *P-III | 02586155 | [30] |
| NUT midline carcinoma | GSK525762 | *P-I | 01587703 | [31] |
P stands for Phase,
A for Approved.
The traditional strategy for small molecule drug development focuses mostly on protein-ligand interactions, i.e. ion channels or receptors, enzymes, due to their distinct binding sites for better interaction [11]. Targeted modulation of PPIs using small molecules was considered challenging for a very long time due to their relatively flat and featureless surfaces and the absence of binding grooves, hence, the PPIs were thought to be ‘undruggable’ targets [12,13]. Other reasons that make this targeted modulation challenging include but are not limited to the following reasons.
(1) The interface area (1500–3000 Å2) between two proteins making a PPI is relatively large than a protein-ligand contact area (300–1000 Å2) [14,15].
(2) The residues involved in PPIs contribute strongly to binding affinities, making it difficult for small molecules to compete [16].
(3) PPI-targeting drugs have a relatively higher molecular weight (>400 Dalton) as compared to the typical ligands (200–500 Dalton), making it hard to meet Lipinski’s rule of 5 criteria [17,18].
(4) Furthermore, PPIs are incredibly flexible, making it difficult to detect the binding pocket in standard X-ray crystal structures [19].
Despite these challenges, significant advancement is being made in identifying and targeting PPIs, owing to progress in computational chemistry and structural biology. For classical targets, computational-guided modulator design is efficient, as described in our previous study [22,23]. These strategies increase their cost-effectiveness and throughput, allowing them to investigate dynamic PPI interfaces and shed light on PPI regulation, resulting in a number of successful examples [22,24]. PPIs have received much interest in the pharmaceutical industry in recent years. Because of the rapid advancements in this growing discipline, it is vital to review the most recent developments in the field of computational methodologies to guide future efforts.
In this review, a variety of in-silico strategies for designing PPI modulators are described. The review is focused on the current advances in in-silico techniques and presents a bird’s eye view of diverse methodologies. Following that, we give an overview of some published case studies to demonstrate the application of these methodologies to the design of targeted PPI modulators.
1.1. Aspects of the PPIs that are critical for modulation with a small molecule
Knowledge of common PPI features is essential for modulating PPIs and evaluating their biological effects. This includes a PPI’s overall structure, three-dimensional shape complementarity around the interaction area, and the physical/chemical components contributions to PPI stability. A recent review focusing on the protein features that generate favorable types of interactions provides a useful resource for building PPI modulators as well as biochemical and biophysical assays for discovering and evaluating them [25].
Based on this understanding, we can conclude that:
(1) PPIs are driven by a variety of chemical interations, including hydrophobic and electrostatic interactions and hydrogen bonding. These interactions affect the physical and chemical aspects needed to optimize binding complementarity.
(2) Most PPI interfaces couple and de-couple on a regular basis, resulting in intricate dynamic equilibria [26]. Furthermore, the difference in the Kd between μmol and pM determines how well a PPI can be modified. Knowing a target PPI’s Kd is crucial while working for its potential PPI modulators.
(3) The protein domain of the target PPI (or some part of it) might be intrinsically disordered or unfolded until it is stabilized by its partner protein [27]. Such a characteristic would make designing synthetic ligands on the protein surface highly challenging using computational techniques. Designing biochemical and biophysical experiments would be difficult too since unstructured proteins are unstable in solution without their protein partners.
(4) The protein domain of the target PPI might be same as the domains of partner proteins, with ~95%-80% homology [28].
In addition, some protein structures can change to utilize the same binding sites again, which makes them even less specific. ElonginB/ElonginC/VHL and ElonginB/ElonginC/SOCS2 are two examples that were found through X-ray crystallographic studies [29,30]. Therefore, it is important to know whether a protein can interact with multiple partners when trying to define and measure binding affinities of new inhibitors for its PPI modulation.
2. Approaches for hit identification of PPI modulators
Most ligands inhibit PPIs at hotspots and allosteric sites [25,31–34]. Below is a brief overview of these approaches that have been used to design efficient PPI modulators [35].
2.1. Hot-spots identification
As discussed earlier, the large interfacial area or contact area makes it hard to find a ‘shape complementary’ molecule, but the presence of certain residues that are mainly involved in binding makes the designing of drugs possible. Such residues are known as hot spots, which comprise key residues that are involved in PPIs and are usually present at the interfaces. Alanine scanning shows that tryptophan, arginine, and tyrosine are the residues that contribute mainly to PPIs (21%, 13.3%, and 12.3% respectively), while valine, lysine, or serine are the residues that are rarely involved [36]. The presence of hot spots in PPIs was first described in a study of a PPI complex of human growth hormone (hGH) and the extracellular domain of its receptor (gGHbd) [36]. Alanine substitution was conducted to explore the contribution of each residue involved in PPI. The results showed that only eight residues out of thirty-one contributed most (85%) to the total binding free energy. Today, the alanine-scanning technique is used in a combinatorial fashion to explore the binding contributions of the hot-spot residues to the total binding free energy of the complexes.
Hot-spot residues enable conformational changes for the ligand with little energy cost, which facilitates shape complementarity. This is a significant distinction between hot-spot residues and other residues that are located on the interfaces [37–39]. The chemical properties of hot-spot residues are responsible for their dynamical behaviors, which can be modeled by MD simulations. Indeed, MD simulations may provide an ensemble of probable conformations, revealing detailed structural and dynamical information for the binding pockets [40,41]. This is in contrast to traditional methodologies such as X-ray crystallography mainly used for visualizing the binding pockets.
The per-residue decomposition energy calculated from the MM/PB(GB)SA technique specifically shows the contribution of residues on the binding interface to the total binding free energy of the complex. The information can be used to infer hot-spot residues related to binding. MD can also help track dynamic movements, secondary structures, and transient pockets in IDPs (e.g. β amyloid, κ-casein, C-Myc, α-synuclein, and histone), that typically do not have specific conformations in PPIs [42,43]. However, 3D complex structures are required to conduct MD simulations. The large gap between the predicted PPIs and experimentally available PPI structures is thus a major challenge. When experimental structures are not available, efficient in-silico methods must be used, such as docking approaches. These are summarized in Table 2 [44].
Table 2.
Docking methods and the webservers with scoring functions and faster computing resources.
| Docking Methods | AutoDock | www.autodock.scripps.edu | [66] |
| AutoDock Vina | www.vina.scripps.edu | [67] | |
| Glide | www.schrodinger.com/products/glide | [68] | |
| GOLD | www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold | [69] | |
| Surflex | www.biopharmics.com | [70] | |
| FlexAID | www.biophys.umontreal.ca/nrg/resources.html | [71] | |
| DINC | www.dinc.kavrakilab.org | [72] | |
| rDock | www.rdock.sourceforge.net | [73] | |
| Faster Computing Sources | Rosetta software suite | www.rosettacommons.org/software | [74] |
| Rosetta Design Webserver | www.rosettadesign.med.unc.edu | [45] | |
| Modeller | www.salilab.org/modeller | [75] | |
| Robetta | www.robetta.bakerlab.org | [76] | |
| ROSIE | www.rosie.rosettacommons.org. | [52] | |
| FOLDX | www.foldxsuite.crg.eu | [77] | |
| ABS-Scan | www.proline.biochem.iisc.ernet.in/abscan | [53] | |
| DrugScorePPIs | www.cpclab.uni-duesseldorf.de/dsppi | [151] |
The consensus-binding site that is a subset of hot-spots is a druggable site that binds with numerous chemical probes [46]. Zerbe et al. have found that consensus sites of PPIs and hot spots have a close correlation and can interact with many small molecule inhibitors [47]. Therefore, identification of consensus-binding sites gives us another way to find hot-spots. Also, this strategy takes into account both the strong binding free energy and the fact that the topology is concave [47]. The probe-based MD simulation is a direct analog of the above strategy in silico, which has the added benefit of revealing the dynamical process of conformational changes [48,49]. In this method, the protein structure is solvated in a solution with a variety of solvents at different concentrations. This allows the solvent molecules to equilibrate and interact fully with the surface of the receptor. After MD, probes move around on their own and gather around the sites that bind well, revealing the consensus sites [49].
FTMap is a computational fragment mapping webserver that uses empirical energy functions to place different small-molecule probes in places where they work best [50]. The clusters of probes are ranked by how much energy they use on average. This means that the regions with multiple low-energy clusters will interact strongly with many low molecular weight probes, revealing the consensus sites.
2.2. Targeting allosteric sites
Enzymes modulate their function using allosteric regulation [51]. A small molecule binds at one site and stimulates a structural change at a remote region, modifying the active site’s conformation. Some PPIs may also use this mechanism. Thus, an inhibitor that binds to an allosteric site could in-principal disturb the major PPI, inhibiting its contact with the other protein (Figure 1). Allosteric modulation has many advantages [34]. It could offer better PPI modulation and improve specificity. It may be easier than hot-spot modulation, as accessible binding sites (e.g. grooves) may be present at several spots on a protein.
Figure 1.
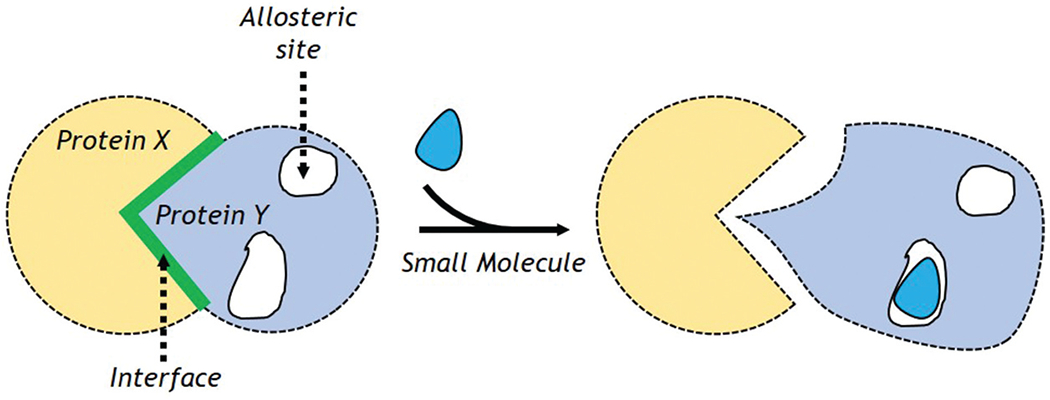
Mechanism for allosteric regulation.
High-throughput screening, ligand binding assays, fluorescence-based resonance energy transfer experiments, x-ray crystallography, phage display paired with crystallography are all employed to explore the allosteric mechanisms [52]. In addition, researchers have developed algorithms to predict allosteric sites [19]. These studies and developments have facilitated the success of many efforts to develop allosteric modulators of PPIs [1,53–56]. The CBF (core binding factor), a heterodimeric complex, is a modulator of normal hematopoiesis, and its gene is the drug-target in many human leukemias [1]. CBF is a PPI comprising CBF-SMMHC and Runx1. This PPI modulates CBF function and is necessary for leukemogenesis; thus, it could be a potential drug target [41].
Bushweller and co-workers resolved CBF’s structure by NMR [57] and employed alanine mutagenesis to study its binding interface with Runx1. Their interfacial contact area was explored to design PPI inhibitors [55]. The 35 putative ligands identified through virtual screening of 70,000 drug-like compounds were verified by physical screening. However, the NMR chemical shift data indicated that the most active compounds did not target the hot-spots but bind to an allosteric site. Finally, three small molecules that block the interaction of Runx1 and CBFβ were identified (Figure 2). It was observed that these molecules stopped the growth of ME-1 cells that caused leukemia. The IC50 value for these molecules was in the low μmol range. Their chemical properties, i.e. being low-molecular weight and water-soluble, present them as good candidates for further trials. Designing hot-spot ligands is difficult, due of CBFβ narrow hetero-dimerization interface. Therefore, this example shows how to identify PPI modulators targeting the allosteric mechanism.
Figure 2.
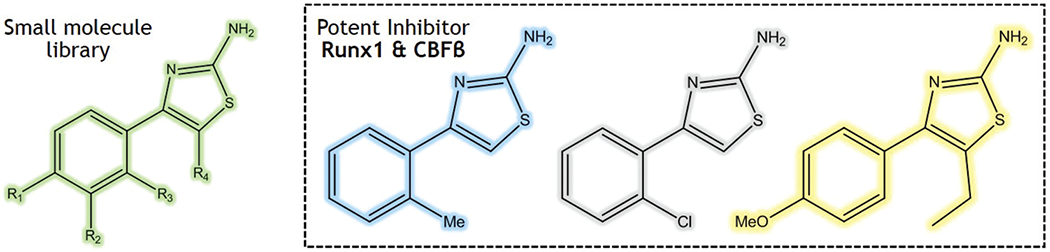
Well-known example of using allosteric modulation of PPIs to find new active molecules.
3. Strategies for the design of PPI modulators based on computational approaches
Investigating the human interactome vastness takes time, expense, and effort. PPI studies are extremely dependent on dynamical and physiological circumstances, causing difficulty differentiating real interactions from experimental artifacts and discrepancies in data, especially for transient interactions and IDPs. Computational methods have evolved as alternatives or complements to experimental procedures to fill in PPI gaps and give a basis for additional studies (Figure 3).
Figure 3.
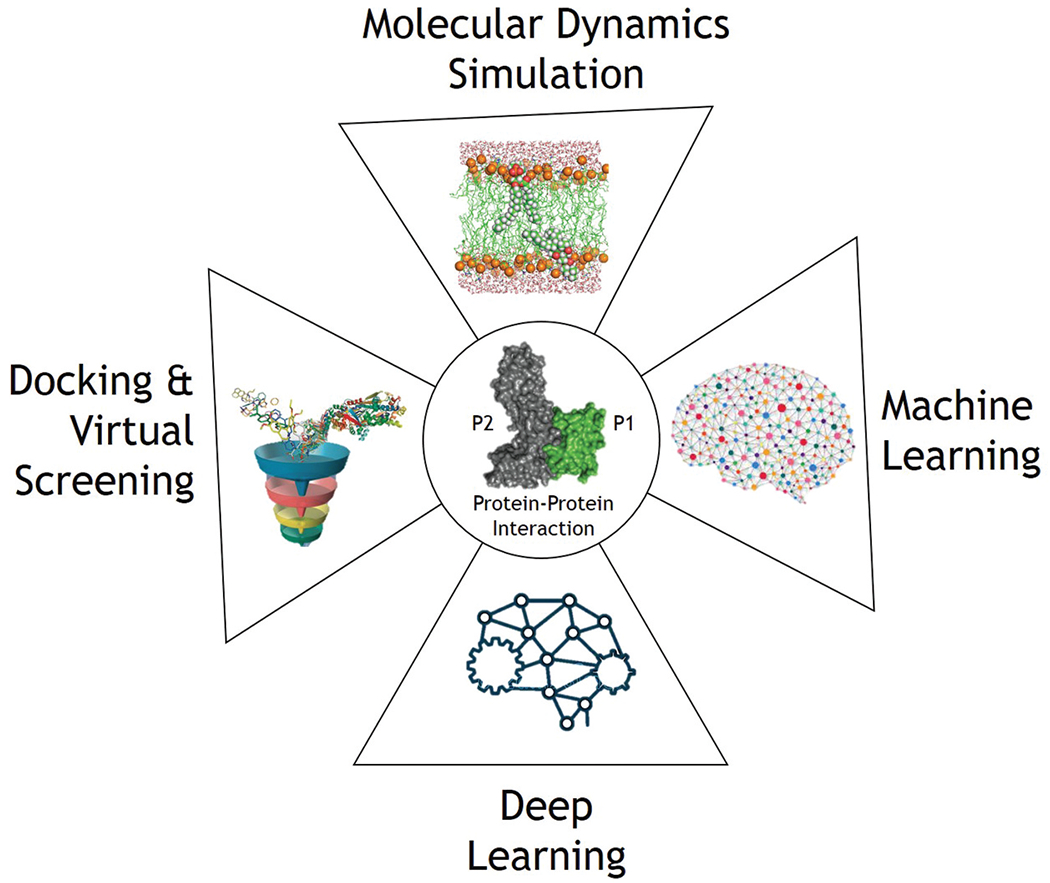
Computational strategies for PPI drug discovery. Diverse computational tools encompass various stages of PPI drug discovery.
3.1. Molecular dynamics simulations for PPI modulation
Even with the availability of large amounts of PPI data, there is still a lot to explore in terms of their structures and dynamics. PPI development is substantially slowed down by the lack of 3D structural data. One of the major drawbacks of crystallography is its inability to detect hot spots and grooves due to active protein-protein interactions. MD simulations provide a thorough evaluation of the structure and dynamics of PPI models. They shed light on PPI mechanisms, which can be leveraged to create PPI modulators. The dynamics of biological molecules is captured via MD simulations with starting structures provided by modeling tools (homology modeling or docking) or PPI databases. After the structural data is complete, the system is setup by defining the initial positions and velocities. Interaction forces among atoms are calculated using various force fields. Solution of the Newtonian equations of motion allows for tracking of time-dependent motions of all simulated atoms [58]. MD simulations can identify hot spots, structural and conformational changes, binding affinities, and molecular-level interactions, facilitating the PPI exploration (Figure 4). The following examples demonstrate how MD can help with PPI investigations.
Figure 4.
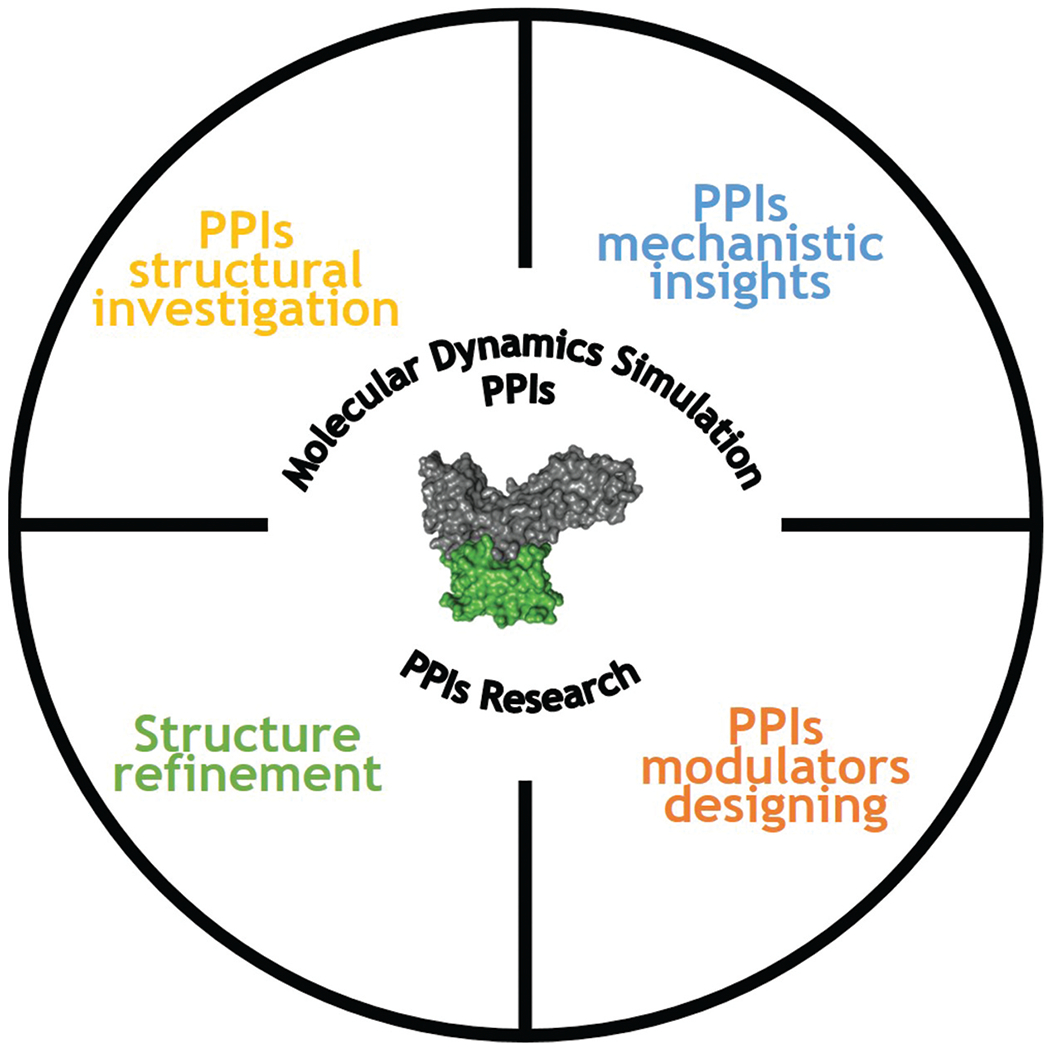
MD simulations have a wide range of applications in PPIs research. Several elements of PPIs can be explored using MD simulations.
Dixit et al. [59] used atomistic MD simulations and energy landscape analysis to study the function of the Hsp90 protein and identified significant binding regions and the hub of communication networks. In their study, MD simulations in combination with site-directed mutagenesis and Western blotting were used to explore the molecular-level interactions of Rho GTPases, Cdc42, and Rac1, with the scaffolding protein IQ motif-containing GTPase-activating protein 2 (IQGAP2) to figure out the binding mechanism of Rac1 and Cdc42 with GRD. It was discovered that Cdc42 and Rac1 govern actin aggregation during metastasis [60]. The heat shock protein Hsp90 is a promising target for the creation of novel anticancer treatments. Several attempts to create inhibitors against this polypeptide have focused on the C-terminal ATP-binding domain of the chaperone [61]. TRAP1 is a member of the HSP90 family that functions as a regulator of energy metabolism. It has significant effects on cancer, neurodegeneration, and ischemia. The Hsp90 levels and activities are higher in tumor cells. Selective inhibitors of TRAP1 could provide insights on its mechanisms of action and pave the way for the development of tailored drugs [62]. Several successful examples of TRAP1 inhibitor modification include the use of small molecules to target TRAP1 to reverse TRAP1-dependent succinate dehydrogenase inhibition, to inhibit the ATPase activity of Hsp90 to reduce tau aggregation in Alzheimer’s disease [63] to use allosteric TRAP1 inhibitors to inhibit tumorigenic growth of neoplastic cells [64], and the selective targeting of TRAP1 activity to provide new chaperone antagonists. These kinds of studies help understand the allosteric complexes and their dynamics and facilitate the development of inhibitors. It is well acknowledged that the majority of proteins act as oligomers. In another study, the oligomerization of peptide GNNQQNY from the yeast prion-like protein Sup35 into amyloid fibril was investigated. The results showed that during aggregation, antiparallel dimer forms predominate, followed by new peptides that can complement the parallel arrangement of assembly. This was exactly in accordance with the experimental crystal structure of the amyloid fibril [65]. A comprehensive MD simulation showed interactions between Aβ1–42 oligomers and full-length Amylin1–37 oligomers, revealing the association between type 2 diabetes and Alzheimer’s disease [66]. When used in conjunction with experimental screening techniques in PPI research, MD simulation is a critical supplement to those methods because of its high accuracy, thorough validation of interaction potentials, and wide availability of computational resources.
3.2. Free energy-based approaches
By sampling free energy landscapes, Monte Carlo (MC) [67,68] or Molecular Dynamics (MD) approaches capture kinetics, binding affinities, and mechanisms of action with a much higher accuracy. The difficulty in these approaches is limited timescales, as millisecond timescale is often required for binding events, which is not very common for current simulations. Despite advancements in specialized hardware [69], brute-force MD remains computationally infeasible for binding. Commonly, MD-based techniques are used as the final step in docking pipelines, resulting in refined models. Recent advancements in sophisticated sampling techniques and computer performance have made it possible to investigate peptide-protein interactions in novel ways.
Free energy perturbation (FEP) approaches calculate the free energy differences between the bound and unbound states and apply a path-dependent approach. The method is not reliable if the molecule scaffold, charge, or binding mode changes drastically [70–72]. The FEP success with small-sized systems is not yet applicable to larger systems such as protein-protein systems. Enhanced sampling methodologies allow researchers to examine peptide-peptide interactions and derive binding energies at a higher computational expense. To study peptide binding, generally there are two approaches, one that measures kinetics and mechanisms and the other that measures binding free energy.
Several peptide-protein systems [73,74] have been studied using frameworks like Markov State Models [75,76], weighted ensemble techniques [52,77], and milestoning [53], which use many classical MD trajectories to figure out kinetic and mechanistic details. To determine bound conformations [78–80], advanced sampling techniques pool together data from experiments and generalized ensemble techniques [81]. These methods provide answers to questions about the validation of simulations, data reproducibility, reliability, and interpretation.
3.3. Machine learning approaches
Machine learning (ML) is becoming increasingly common in biomedical research but needs a large number of training data. At many stages, such as merging diverse heterogeneous datasets, evaluating predictions, forecasting probable PPIs, and looking into extrapolated PPI networks, statistical and machine learning methods were applied [82–84].
Many ML techniques have been used to predict PPIs in the past, including k-nearest neighbor, gradient tree boosting (GTB) [85], DeepPPI [86], redundancy maximum relevance (mRMR) [87], naive Bayesian, L1-regularized logistic regression [85], neural networks, random forest, and many others [83,88,89]. To train interface predictors, ML algorithms leverage a collection of empirically validated PPI surfaces. The trained model is then used to identify hot spots at the PPI interface in query proteins [90]. The accuracy of the prediction model is highly susceptible to the quality of the input features utilized for training. As a result, figuring out the various protein properties required to train an ML system is critical. Models for predicting PPIs are built utilizing a variety of protein characteristics, either individually or in combination. PPIs cannot be predicted solely by one attribute.
Combining characteristics improves ML prediction. Model development makes use of amino acid types, protein expression data, solvent accessible surface area (SASA), physicochemical properties of amino acids, atomic and residue contacts, position-specific scoring matrices (PSSMs), residue energy, structural information, interface propensity, and evolutionary information [91,92]. Figure 5 depicts the five key PPI phases. In PPI-based ML, prediction models take sequence or structural features as input. Most ML interface predictions compare structure and sequence-based techniques. Several meta-based systems pool and re-compute the raw scores from prediction servers to enhance their performance. A number of ML predictors for the identification of PPIs were reviewed in Ref [41].
Figure 5.
ML-based PPI predictions follow a set of approved guidelines.
PPI prediction also used unsupervised ML techniques. Deep learning is a new ML discipline that utilizes neural networks (NNs) with multiple hidden layers. Deep learning is described in detail in Ref [93]. It can aid in decision-making, the comprehension of natural language, and the recognition of images and voice. The bioinformatics and pharmaceutical industries have also utilized this approach.
Sun et al. [94] recently predicted human PPIs based on their sequence using deep learning. Using stacked autoencoders (SAEs), they investigated PPIs in humans and other species and with the highest results on 10-fold cross validation and various external datasets ranging from 87.99 to 99.21% accuracy were developed. ML tools can also be used to build PPI libraries or to assess the drug-likeness or ADME characteristics of first PPI hits using various filtering methods. The details for the PPI modulators could be found in 2P2Idb, TIMBAL, and iPPI-DB [95,96]. A decision tree strategy, known as PPI-HitProfiler [97], is based on known PPI inhibitors, and it is implemented in the FAF (Free ADME-Tox Filtering tool)-Drugs webserver [98–100]. This method uses the structure of PPI inhibitors together with important descriptors such as radial distribution function and unsaturation index. Their approach correctly identified 70% of the validated active and 52% of the inactive using PPI complexes with ligand and bioassay data [97].
Another supported vector machine (SVM) tool (2P2IHunter) based on PPI modulator data from 2P2Idb [101] identified chemical features such as octanol-water partition coefficient, hydrophilicity, molecular weight, presence or absence of multiple bonds, aromaticity, H-bond donors and acceptors, and rotatable bonds as crucial features for PPI modulators. The SVM model was highly accurate with a high enrichment factor of 8, which is useful for removing non-PPI molecules from screening libraries, but unfortunately, this method had a low level of sensitivity.
In summary, machine learning is a promising general approach to predict PPIs, with the potential for better understanding the gigantic network of PPIs and their targeted modulation. However, there is still room for improvement in the prediction accuracy and computational efficiency in machine learning-based methods [102].
3.4. Screening approaches
In light of the labor-intensive nature of traditional experimental screening methods like high-throughput screening (HTS) and fragment screening, the virtual screening strategy has emerged as a useful alternative in the drug discovery process. There are two main types of virtual screening, i.e. structure-based virtual screening (SBVS) and ligand-based virtual screening (LBVS) [103,104]. The SBVS predicts the best interaction based on their binding affinity with the binding site. SBVS needs the 3D structure of the target to be known in order to predict the interaction energy. The LBVS technique compares the structure-activity data of potential compounds to that of known actives in cases where the 3D structure and/or homology model is not available. The reference structure for this method is a small molecule or a ligand. Compound databases are then searched for ligands with similar chemical or structural properties. The LBVS uses searches for similarity and substructure, quantitative structure-activity relationships (QSAR), and matching of pharmacophores and 3D shapes [105]. Virtual screening can help narrow down the number of candidates to a reasonable number.
Recently, Beekman et al. [106,107] suggested that in silico peptide-directed binding is a new and cost-effective way to find PPI modulators that are highly selective. This method used covalent docking to look through organic fragment libraries. A part of the lead peptide was removed to make the binding pocket easier to reach. Carlos et al. provided yet another successful example in which the use of a pharmacophore model derived from the conformational sampling of the active state of the receptor permitted large conformational sampling, widening the selectivity of the predicted ligands, and limiting the constraints commonly linked with the modulation of chemical scaffolds [108]. In addition, the great flexibility of PPI interfaces highlights the importance of multi-conformational virtual screening, which is in combination with MD can explore representative conformations effectively. On the other hand, when multiple conformations are used, it takes more computing power to look through a large ligand library. Kumar et al. have shown how cross-docking can be used to make a new virtual screening pipeline [109]. Using 3D shape similarities between the pockets and ligands, they found the best shape for each ligand in the collection. This method gets rid of the need for each chemical to dock with all structures. This makes multiple-receptor docking cheaper to compute. Less concentrated hot spots could imply a large number of binding pockets distributed across a large interface. When many binding pockets are physically close together, screening algorithms may only identify a subset of druggable sites, resulting in significantly decreased efficacy at lower concentrations than the peptide substrate.
The solution to this problem is the combination of virtual screening with rational design methodologies. The hits that target a certain hotspot region can begin rational drug design employing a structure-based drug design (SBDD) technique. The reactivity of hot spot residues could be tweaked by adding chemical groups to establish stronger interactions with the binding sites, hence increasing the compound’s activity. Sun et al., for example, utilized MD simulations and MM-GBSA free energy calculations to investigate the molecular determinants of binding between Keap1/Nrf2 PPI and revealed five sub-pockets, P1 and P2 being hot-spots [110].
3.5. Docking-based approaches
In the last few years, docking methods have changed in response to the growing interest in peptide-based medicines. There have been good reviews of these methods [111,112], and research that gives benchmarks for judging current and future methods. We explain the main ideas behind these techniques and simulations that try to predict how proteins and peptides will interact with each other. Multiple search modes based on system knowledge are used in docking approaches. The binding site and mechanism of the protein-peptide conformation must be restored. This is a problem of searching and scoring. Peptide-protein position, orientation, and internal structure are all factors in the search problem. Scoring finds correctly docked structures using a docking structure scoring function. Success is determined by top-scoring postures. Based on system knowledge, docking approaches reduce search space for computational efficiency. The peptide binds to all probable protein-receptor sites in a global search [113–117].
In the presence of previous data, local search narrows down the hot spot region resulting in a targeted search [118,119]. Template-based approaches circumvent this search by building flexibility on top of structural database models [120,121]. Methods can be further classified by protein receptor and peptide conformational freedom. Flexibility is a difficulty for protein-protein and protein-small molecule docking [122]. Modeling protein flexibility involves soft potentials [123,124], explore rotameric states [125], using various protein receptor structures [126,127] or refining with MD.
The difficulties in modeling peptides stem from the following: (1)→ Peptides can take on a variety of conformations depending on whether they are bound or unbound. (2)→ A single peptide can bind proteins in multiple conformations [128]. (3)→ Different amino acid sequences can interact with the same receptor in more than one way [129].
The majority of docking techniques employ a flexible conformational strategy for peptides. These strategies are either sequence-based or conformation-based. In sequence-based methods, the amino acid sequence is used to create or predict the PDB or the secondary structure [130]. The conformation-based methods employ multiple initial conformations often acquired by the peptides, i.e. helical or beta-sheet conformation [131]. The goal of scoring functions is to figure out which poses are biologically significant. The underlying binding affinities of various poses and substances should be reflected in these functions [132]. Empirical fits, knowledge-based, machine learning, and first principles are the four types of scoring functions [133].
The capacity to design good and bad poses is a hurdle in evaluation methods. As a result, decoy sets [134] have been developed, which are widely utilized as training sets for new functionalities. They directly impact the scoring algorithms that are developed along with them, which frequently lead to biases. Strategies to detect and counteract such biases have been proposed [134]. New scoring approaches are also being developed as scoring functions suitable for smaller proteins are not necessarily transferable to larger systems [135].
4. Case studies
Through a vast PPI network that includes a variety of caspases, the inhibitors of apoptosis (IAP) family proteins control cell death pathways. Apoptosis evasion is caused by a rise in the IAP family proteins in malignancies. Because of the high molecular variability of cancers, the IAP PPIs network is rewired differently in each individual. It is easier to choose pharmacodynamic and predictive biomarkers for IAP antagonists, when knowing the most recent status of the IAP PPIs network in a clinical sample. Advances in therapy have resulted in small gains in 5-year survival for women with ovarian cancer during the previous several decades. The significant incidence of recurrence with traditional chemotherapy highlights the need for novel chemo-resistant cell therapies. Apoptosis, or programmed cell death, is a tightly controlled process that is commonly interrupted in cancer.
Apoptosis signaling has both intrinsic (mitochondrial) and extrinsic pathways (death receptor-ligand). Intracellular damage stimulates the intrinsic pathway, increasing mitochondrial membrane permeability and releasing cytochrome c and the second mitochondria-derived caspase activator (SMAC). The release of cytochromeC and SMAC enhances apoptosome production and the degradation of IAPs respectively. Apoptosis pathways finally converge on a platform of cellular death driven by the activation of caspase-3 and 7.
4.1. The p53-mdm2/x interaction
p53 is known as the ‘guardian of the genome’ due to its tumor-suppressing properties. Through its various functional domains, it is predicted to be involved in over a thousand PPIs [136,137]. This paragraph will examine the association between the p53 transactivation domain and the MDM2 protein, which signals p53 for destruction. Inhibiting the link between p53 and MDM2/X is an important cancer target because it allows p53 to carry out its functions. Despite the similarity between MDM2 and MDMX, the development of dual inhibitor medications continues to be an important area of study, with several candidates in clinical trials. A short intrinsically disordered epitope from p53 terminal transactivation domain binds as a helix to MDM2 N-terminal domain in the p53-MDM2 interaction [138]. Three p53 residues are linked to a deep hydrophobic depression in MDM2, i.e. residue Phe19, Trp23, and Leu26 as shown in Figure 6.
Figure 6.
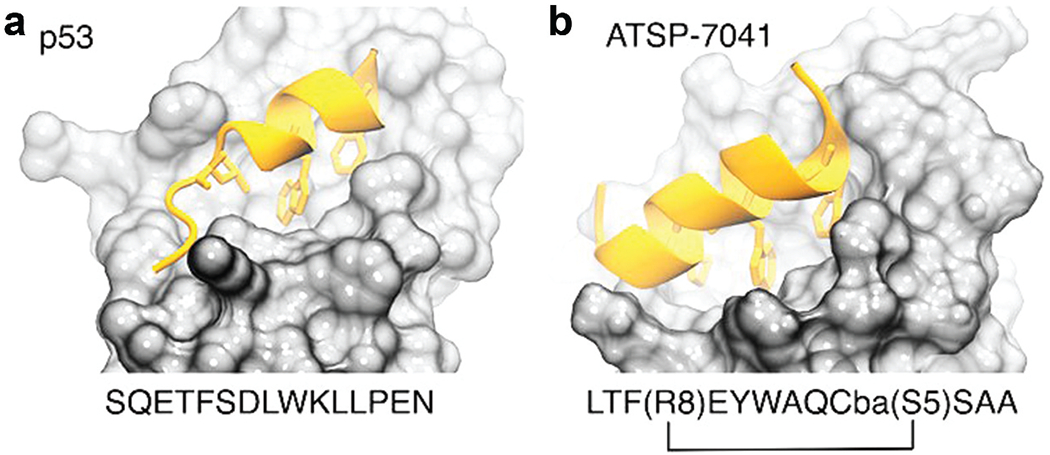
(a) p53 (PDB code 1ycr) and (b) a stapled peptide (MELD prediction) binding MDM2. Anchoring hydrophobic residues in the peptide represented as sticks.
MDMX and MDM2 binding sites are 80 percent identical, leading in p53 binding along the same mode. Despite their resemblance, MDMX has a shallower binding site, making binding inhibitors harder to develop. Computational approaches have been used in the rational design of small molecules and the synthesis of peptides as potential drug [139,140].
To explore the complexity of the p53-MDM2 interaction, MD simulations and docking approach have been used. On-rates for p53-MDM2 are currently close to experiment in studies employing MSM methods, while off-rates are difficult to quantify directly [74]. These investigations also reveal binding mechanisms, such as the helicity required a peptide to go from an induced-fit to a conformational selection binding paradigm [23]. Some research employs the longer MDM2 construct, which has a ‘lid’ piece that effectively lowers the amount of time the binding site is exposed to p53 binding. MD simulation approach gives precise information on the influence of the lid-disordered area on MDM2 binding energy surface when compared to p53 and other small molecule therapies [141].
The best scoring structure from the experimentally bound structure was 3.74, based on recent flexible docking simulations of the p53 peptide starting from unbound conformations and includes the disordered tails in MDM2 [142]. Nutlin (Figure 7) and its derivative and idasanutlin (Figure 7) are small molecule drugs that bind to and inhibit MDM2 and are used to treat refractory acute myeloid leukemia [143]. This class of compounds is ineffective against MDMX [144], and similar kind of results are obtained for other compounds such as AMG-232 (Figure 7), which binds to MDMX with a much smaller affinity as compared to MDM2. Small molecule MDMX inhibitors have also failed to work in cultured cells [145]. These molecules are designed to mimic the three hydrophobic residues found in the p53 binding epitope to reduce toxicity. Several of these designs are currently in clinical trials [146].
Figure 7.
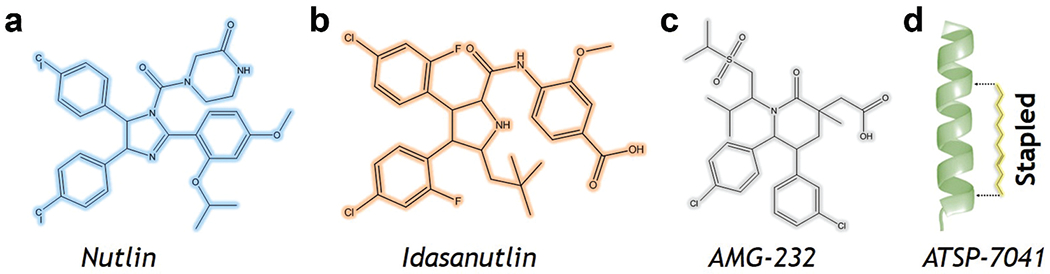
Chemical structures.
Using a known binding motif, a new technique for dual inhibition selects peptide sequences. As a result, several linear peptide designs with higher affinity than the original p53 peptide have been discovered [147]. In peptide designs, the three hydrophobic residues that interact in MDM2/X are retained, resulting in longer helices. The Brownian Dynamics method was used to compare the binding kinetics of various peptide sequences. Despite their increased affinity for MDM2 and MDMX, linear peptides have a poor ADME profile: they are easily degraded and have difficulty crossing barriers, limiting their application as pharmaceuticals. They are, nonetheless, excellent beginning points for peptidomimetic design. Another technique employs non-standard amino acid backbones to boost degradation resistance while retaining the side chains required for robust interactions with protein receptors [148]. Several of these peptides have advanced to clinical studies [149].
Stapled peptides are an attractive alternative to linear peptides because they may easily cross barriers are resistant to degradation and adopt stable helical conformations that enhance binding [150]. Tan and colleagues argued for rational design in order to incorporate chemical staples while preserving enthalpic interactions and minimizing entropic costs. In this area, they concentrated on finding bound conformations utilizing integrative modeling approaches based on MD simulations [151]. Hence, they were able to predict the binding of several linear and cyclic peptides, as well as the qualitative relative binding free energies. Moreover, they discover multiple peptide binding strategies [151,152]: ATSP-7041 (Figure 7) is a stapled peptide that binds in a disordered state as a helix, whereas p53 binds in a disordered state and then folds in the active site.
The latter condition needs partial unbinding and rebinding of the stapled inhibitor due to incorrect side chain orientation. Because simulations have long residence durations, even partial unbinding might be a slow step, resulting in slow convergence. A linear peptide with strong helical inclinations can be shown to rearrange its side chains by partially unfolding in the active site. Resulting, the time-consuming unbinding step is skipped by a linear peptide.
4.2. The BH3-Bcl-2 interaction
Apoptosis is a process of planned cell death that is essential for immune system function, tissue homeostasis, and embryonic development [153]. Overactive apoptosis might lead to an increased ischemia risk as a result of an increased burden of metabolic waste. The apoptotic pathway is modulated by complex PPI networks involving the B-cell lymphoma-2 (Bcl-2) family of proteins. Some members of this family are pro-apoptotic while others are pro-survival, and both of these modulate mitochondrial outer membrane permeability.
Over 20 Bcl-2 protein members have been identified. A sequence study shows that they share one or more Bcl-2 homology (BH) domains that are important for function, since their deletion by molecular cloning influences survival/apoptosis rates. The Pro-survival members, i.e. Bcl-2, Bcl-xL, etc., have four homology domains (BH1-4) and a transmembrane domain. The pro-apoptotic members include proteins with more than one BH domain, like the pro-apoptotic effectors Bax and Bak, which have four BH domains (BH1-4) linked to a transmembrane domain, and proteins with only one BH domain, like Bim, Bid, Puma, etc., whose sequences are very different [154]. Some BH3-only proteins such as Bim, Bid and to a minimal extent Puma activate the pro-apoptotic effector proteins directly. The remaining molecules act as sensitizers, binding to the pro-survival proteins and releasing the BH3-only activators [155] as shown in Figure 8.
Figure 8.
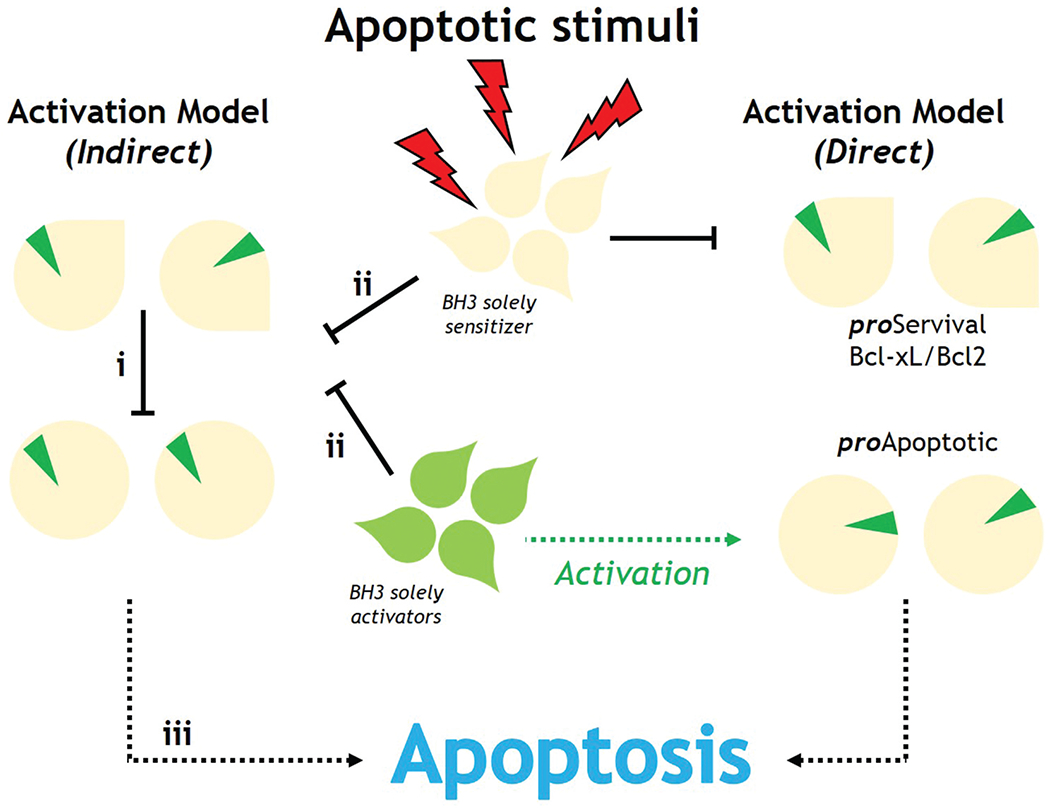
Direct and indirect Bak/Bax activation models in apoptosis.
Despite recent progress, the mechanism by which Bcl-2 regulates apoptosis remains unknown. Apoptosis susceptibility is determined by the amount of pro-apoptotic and prosurvival proteins in a cell, as well as their ability to form heterodimers. By attaching to pro-apoptotic proteins, pro-survival proteins prevent apoptosis. In cell-free systems and HeLa cells, pro-apoptotic proteins containing the BH3 domain-induced apoptosis, proving this notion.
The pro-survival Bcl-2 proteins sequester pro-apoptotic effectors and BH3-only proteins in healthy cells, inhibiting apoptosis. BH3-only proteins activate pro-apoptotic effectors either directly or indirectly by binding to pro-survival Bcl-2 proteins. By accumulating and activating pro-apoptotic effectors, this process enhances the permeability of the mitochondrial outer membrane [155]. Variable family affinities, as well as their modification by membrane implantation, are important factors.
Apoptotic downregulation is a vital phase in disease progression and maintenance, and BH3 domain peptide analogs have been identified as possible cancer therapeutics. The molecular pathways that regulate the intrinsic apoptotic pathway have been discovered by structural investigations on Bcl-2 family members. The first 3D structure of human Bcl-xL was solved using X-ray crystallography and NMR spectroscopy methodology [156]. Eight alpha helices are connected to BH domains. Helix 8, BH3 helix 2, and BH4 helix 1 are all followed by BH2. The linking loop is reached by extending BH1 along helices 4 and 5 (Figure 9A).
Figure 9.
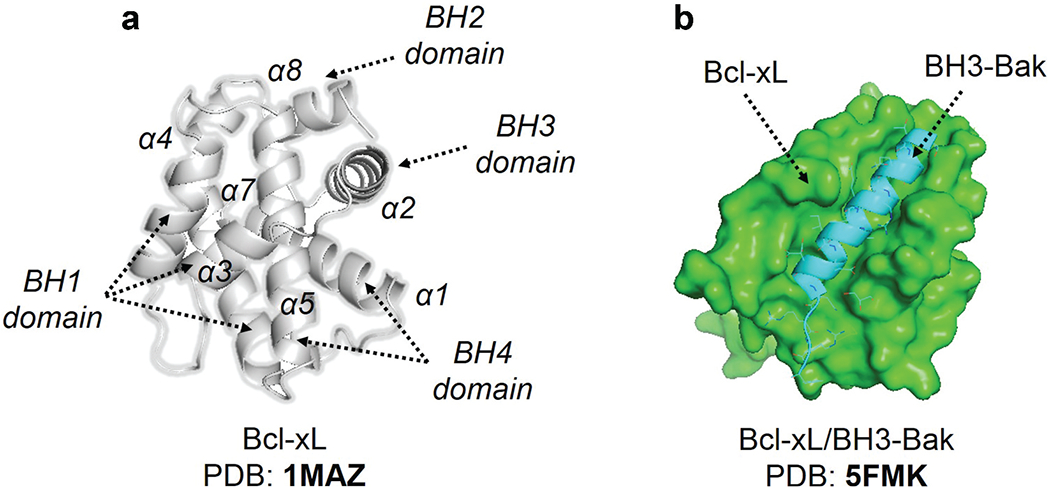
(a) The X-ray structure of Bcl-xL. (b) Illustrate the x-ray structure of the Bcl-xL and BH3-Bak complex. The diverse elements of secondary structure have been labelled.
The C-terminus of the protein acts as a membrane anchor and must be removed for structural analysis. In crystallographic structures, the broad hydrophobic groove created by BH1-BH3 domains corresponds to the interaction location of the BH3 domain. The apo structures of pro-survival Bcl-2 members have the same topology [157]. Despite their opposite responsibilities, Bak and Bax share the same structure. Most BH3-only proteins, except for Bid, are intrinsically disordered.
Currently, no heterodimer structures of the Bcl-2 family are available. Pro-survival Bcl-2 members interact with BH3-only (Bcl-2 homology 3 motif) proteins. The 3D structure of the Bcl-xL-Bak BH3 domain complex [158] is shown in Figure 9B. Four hydrophobic residues extend their side chains into the cleft, and the Asp83 residue makes an electrical contact with Arg139 in Bcl-xL, revealing a helical connection between the BH3 domain and a hydrophobic groove. Following that, many BH3 domains bound to various pro-survival Bcl-2 members were published in the literature, all of which shared the same general features as the Bcl-xL-Bak BH3 complex.
The consensus sequence is visible in the sequence alignment of the various BH3 domains; A1-B-XX-A2-XX-A3-B-C-A4-D, where An indicates the hydrophobic residue, B for residue with a short side chain, C is an acidic residue, D is a hydrophilic residue and X represents any residue [159]. All pro-apoptotic BH3-only members have a A2 leucine residue, whereas the rest of An have Val/Ile/Met/Leu or an aromatic residue. The pattern An display assurances their division on the same face of a helical structure. The hot spots in the hydrophobic binding cleft, which has four hydrophobic pockets (P1-4) and a conserved Arg residue are bound by these four residues and the conserved Asp residue. The pro-apoptotic BH3 domains have varying affinity for pro-survival proteins. Puma and Bim bind the pro-survival proteins in the same way. The Bcl-2/-xL/-w are bound by Bad and Bmf, while Mcl-1 and Bfl1/A1 are bound by Noxa.
The Bcl-xL/-1 and Bfl1/A1 are bound by Bid, Bik, and Hrk. Because of differences in their sequencing, they have varied binding preferences. Many structural and bioinformatics studies have been carried out to better understand the binding preferences of BH3 peptides and Bcl-2 family members, revealing more about the nature of these interactions [160]. According to computational and experimental investigations, BH3 peptides do not form helices in solution. Incorporating helix enhancer residues into the sequence, analogs with higher affinity can avoid negative configurational entropy effects. Analogs in solution had a limited helical shape as well. In early attempts to stabilize Bak BH3, lactam cross-links at locations I and i + 4 were employed. Despite their helical structure, none of these peptide analogs were able to connect to Bcl-2 due to steric hindrance. Hydrocarbon stapling worked out well. In this situation, α,α-di-substituted amino acids with olefin tethers provide the building blocks for macrocyclization between helix residues.
This method was utilized to successfully stabilize the Bid BH3 peptide [161], which was discovered to be helical, protease-resistant, and cell-permeable molecules with improved affinity for multidomain Bcl-2 member pockets. However, not all stapled BH3 helices boost bioactivity, therefore, a large number of modified peptides must be made and evaluated to identify potential candidates. Other kinds of fasteners have also been utilized. Recently, bisaryl cross-linkers have been used to reinforce peptide helices containing, for instance, two cysteines at positions i and i + 7. Using this approach, the Noxa BH3 peptide was stabilized, and it exhibited significant cell-killing efficacy against Mcl-1-overexpressing cancer cells. Such process results in a molecule with greater helicity than the native peptide leading to improved cell permeability and stability. Several BH3 mimetics have been discovered by a hit-to-lead structure-based optimization approach in combination with computational approaches.
Gossypol was used to identify potential Bcl-2 inhibitors such as sabutoclax [161] and TW-37 [162]. Obatoclax, a cytotoxic inhibitor of pro-survival BCL-2 family members that oppose Bax or Bak, was discovered using prodigiosin as a starting point. At low micromolar doses, WL-276 causes apoptosis in PC-3 cells. The chemical WEHI-539 was enhanced utilizing a structure-guided method after a hit was obtained by HTS. This drug binds to bcl-XL with a high affinity and specificity, efficiently killing cells by suppressing their pro/survival activity. It has the same inhibitory activity as BCl-2 but has a higher inhibitory activity than Bcl XL [163].
Combining NMR spectroscopy-based fragment screening with computational studies yielded additional small-molecule Bcl-2 inhibitors. ABT-737 was discovered to be a potent Bcl-2 inhibitor [164]. Although the medication is orally available, its pharmacokinetic profile when administered intraperitoneally is satisfactory. In April 2016, the US FDA authorized ABT-199 as a second-line treatment for chronic lymphocytic leukemia. The 4,5-diphenyl-pyrrole-3-carboxylic acid core structure of compound BM-957 inhibits cell proliferation in H1147 and H146 small-cell lung cancer cell lines [165].
4.3. Computational approaches in tissue engineering
The application of p53 and bcl2 notions to tissue engineering is as simple as finding naturally existing binding modalities and integrin-ECM interactions. This could lead to the discovery of new integrin-binding peptide motifs and chemical compounds. The RGD motif has been computationally and experimentally explored, because it binds a variety of integrin types. However, discrepancies in binding and recognition were caused by the motif internal structure and binding mechanism [166,167]. A scaffold with an RGD motif may adopt conformations that are recognized by a subset of integrins. A small molecule antagonist (i.e. RUC-1) was created using theoretical and experimental methods [168]. The RUC-1 antagonist binds to the IIb3 integrin without changing its shape [169]. Other computational algorithms aim to improve the peptide epitopes of membrane proteins like integrins [170]. The synthesis of material that self-assembles to act as a scaffold for cells is a second focus. To localize a drug to a specific physiological site, self-assembling peptides that can form hydrogels after injection into a patient are commonly employed [171].
Several peptides sequence-based experimental techniques for molecular self-assembly have already been disclosed [171]. To determine their activity, self-assembling peptides must contain a sequence capable of self-assembly as well as a sequence motif accessible and identifiable by integrins; see in ref [161]. This is an opportunity for computational approaches to investigate the best sequence design for achieving specified mechanical qualities (such mechanical stiffness), as well as rational optimization for active and accessible integrin-binding motif conformations. Smadbeck and colleagues used experimental approaches to describe small peptide motifs capable of self-assembly [172]. Another alternative is to use proteins that fold into stable structures and then self-assemble [173].
Despite these advances in computational design, the bulk of hybrid procedures still rely on experimental design followed by computational characterization, with molecular dynamics being the most common method [174,175]. Peptide secondary structure proclivities, protein structure and assembly prediction, and design principles are all relevant to this field of research [176].
4.4. 14-3-3 PPIs
The 14-3-3 protein family is a particularly intriguing topic for PPI modulation research because it has been discovered to have hundreds of protein-protein interactions. PPIs play a role in a variety of biological processes, including cell cycle regulation, signal transduction, protein trafficking, apoptosis, and cancer [177]. 14-3-3 proteins are also involved in phosphorylation-dependent PPIs, which regulate cell cycle progression, the initiation and maintenance of DNA damage checkpoints [178]. Besides this, 14-3-3 proteins are also involved in the progression of many neuropathological disorders [179,180], bound to tau-tangles and enhancing their aggregation as seen in Alzheimer’s patients [181]. Using small molecules to modify these PPIs is a crucial method for creating new drugs. The literature has reported a variety of natural, semi synthetic and synthetic compounds that perform their physiological functions by stabilizing complexes of their target proteins [182]. Fusicoccin-A (FC-A), a metabolite generated by the fungus Phomopsis amygdali, is an example of a natural stabilizer and was the first stabilizer to be reported for 14-3-3/client PPIs. The plasma membrane H+-ATPase (PMA2) and 14-3-3 complex was discovered to be stabilized by FC-A, with a 90 times increase in the affinity [183]. It was also found to stabilize 14-3-3/cystic fibrosis transmembrane conductance regulator (CFTR) complex that resulted in enhanced delivery to the plasma membrane. By looking at the examples above and many more [180,184], it can be deduced that FC-A might act as a potential chemical tool for investigating the role of 14-3-3 in various pathologies.
The semi-synthetic derivatives such as fusicoccin tetrahydrofuran (FC-THF) that bears an additional furan ring induces a 20 times increase in the stabilization of 14-3-3/potassium channel TASK-3 complex [185]. Fusicoccin-derivative (ISIR-005) is another semi-synthetic derivative that stabilizes the cancer-relevant interaction of the adaptor protein 14-3-3 and Gab2 [186]. Similar to natural and semi-synthetic stabilizers, there is another class of stabilizers of 14-3-3 PPIs that include synthetic products. These products include but are not limited to pyrrolidone 1 [187], Adenosine Monophosphate (AMP) [188], and The Molecular Tweezer CLR01 [189] etc.
5. Expert opinion
Due to their significance in cell signaling and regulation, PPIs are considered as potential therapeutic targets. But there are still many issues to explore about their interactions and modulation to fully define these massive networks and address PPI-based drug discovery challenges. No doubt, modern experimental techniques have expanded our knowledge of PPIs, but unfortunately, the size of the human interactome makes experimental methods insufficient, demanding more robust and efficient computational methods. Computational methods facilitate the characterization of PPIs by identifying their chemical structures, which in turn expedites and improves the design of PPI modulators. Advancement in computational resources and algorithms, coupled with a molecular-level understanding of proteins’ dynamics, has made in silico approaches successful in PPI drug discovery.
Recent advances in computational tools for evaluating and identifying PPI modulators have led many enterprises to invest in drug discovery, resulting in numerous peptide drugs on the market [164]. In the development of inhibitors that target PPIs, computational approaches can be used in a wide variety of ways. Low membrane permeability, a short half-life and low bioavailability of biomolecules are the significant issues that can be solved by combining computational techniques with experimental findings. Computational methods such as prediction and filtering tools are robust, especially for biomolecules with poor starting conformational data retrieved from experiments. In such a scenario, sequence-based methods can provide a useful insight into the starting structure and conformation of proteins that are crucial for ligand design later.
However, this information might not generate exact three-dimensional structures [190]. The majority of computational methods, such as free energy techniques, require the availability of structural data. Due to proteins’ inherent flexibility, approximations are often included in the algorithms when using these methods, but this creates biased systems; therefore, one must be extremely careful when choosing a computational technique for a specific problem [191]. Knowledge-based methods use the current structural data for proteins and ligands, meaning the results can be only as reliable as the data used to create them. Some PPI descriptors are more predictive than others [190]. A model built on one protein family or class of ligands might not accurately describe the properties of other protein families or ligand classes. Similarly, for screening methods, structural information is required, which makes this strategy unsuitable for unknown targets.
They provide no information about hot-spot regions or binding interactions. And like any other computation method, validation of the results is very important. However, validation for PPIs need more sensitive enhanced sampling methods and expertise owing to their disordered conformations [192]. Most of the biological phenomenon occur at a timescale that is inaccessible to most of the computational techniques. Achieving such timescale need a lot of GPU power which makes these methods computationally exhaustive. MD simulations can overcome such problems to some extent using enhanced sampling techniques such as meta-dynamics.
Growing interest in the use of MD for exploring PPIs has also been augmented by advancements in hardware [169,193]. The role of machine learning in predicting protein structures is exceptional, but when tested for PPI predictions, the results show some limitations [194], suggesting that these methodologies are still insufficient to capture all the details of the binding interactions at a molecular level. But we believe, with the availability of high atomic resolution structures, these discrepancies will be met. As of now, the use of computational tools to differentiate between functional and non-functional protein interactions based on their structure and dynamics is still controversial [195].
It’s important to remember that the reason for discussing the drawbacks of the above-mentioned computational methods is not to disregard them but to reinforce the need for additional research and to improve the current tools. In addition, the strengths of each method can be utilized to combine them with other methods. In CADD endeavors, advanced in-silico tools together with state-of-the-art experimental techniques can lead to a better understanding of PPI identification and modulation.
Article highlights.
PPIs were considered undruggable due to their flat surfaces, irregular conformations, and lack of grooves.
Rapid advances in computational biology have made PPIs a major research field in drug discovery.
Reviewed different in-silico approaches to find PPI interfaces and methods that are successfully applied to analyze PPI interfaces.
Reviewed examples that use computational methods to analyze PPIs and their impacts in diverse physiological processes.
This box summarizes key points contained in the article.
Funding
This paper was supported by Data Center of Management Science, National Natural Science Foundation of China - Peking University (81925034); Innovation Program of Shanghai Municipal Education Commission (2019-01-07-00-01-E00036); National Institute of Health/NIGMS (GM130367); National Key R&D Program of China (2021YFF0704000); National Natural Science Foundation of China (21977068-CHF & 32171242-CHF); The innovative research team of high-level local universities in Shanghai (SHSMU-ZDCX20212700); The Key Research and Development Program of Ningxia Hui Autonomous Region (022CMG01002 and 2022CMG01002).
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Footnotes
Reviewers disclosure
Peer reviewers in this manuscript have no relevant financial relationships or otherwise to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.••.Zinzalla G, Thurston DE. Targeting protein-protein interactions for therapeutic intervention: a challenge for the future. Future Med Chem. 2009. Apr 1;1:65–93. [DOI] [PubMed] [Google Scholar]; In the last 20 years, researchers have focused on modulating PPIs to develop new therapeutic approaches and target-selective drugs. This article presents several of these strategies along with a critical overview of the challenges and potential solutions for using PPIs as molecular targets
- 2.Venkatesan K, Rual JF, Vazquez A, et al. An empirical framework for binary interactome mapping. Nat Methods. 2009. Jan;6(1):83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koh GC, Porras P, Aranda B, et al. Analyzing protein-protein interaction networks. J Proteome Res. 2012. Apr 6;11(4):2014–2031. [DOI] [PubMed] [Google Scholar]
- 4.Gavin AC, Bösche M, Krause R, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002. Jan 10;415(6868):141–147. [DOI] [PubMed] [Google Scholar]
- 5.•.Cossar PJ, Lewis PJ, McCluskey A. Protein-protein interactions as antibiotic targets: a medicinal chemistry perspective. Med Res Rev. 2020. Mar;40(2):469–494. [DOI] [PubMed] [Google Scholar]; The authors of this article described 27 small molecule PPI modulators, a relatively new target for the development of antibiotic drugs that are currently undergoing Phase I–III clinical trials
- 6.Jin L, Wang W, Fang G. Targeting protein-protein interaction by small molecules. Annu Rev Pharmacol Toxicol. 2014;54:435–456. [DOI] [PubMed] [Google Scholar]
- 7.Nero TL, Morton CJ, Holien JK, et al. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer. 2014. Apr;14(4):248–262. [DOI] [PubMed] [Google Scholar]
- 8.Kimura K, Ikoma A, Shibakawa M, et al. Safety, tolerability, and preliminary efficacy of the anti-fibrotic small molecule PRI-724, a CBP/β-catenin inhibitor, in patients with hepatitis c virus-related cirrhosis: a single-center, open-label, dose escalation phase 1 trial. EBioMedicine. 2017. Sep;23:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey D, Jahagirdar R, Gordon A, et al. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J Am Coll Cardiol. 2010. Jun 8;55(23):2580–2589. [DOI] [PubMed] [Google Scholar]
- 10.Mirguet O, Gosmini R, Toum J, et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem. 2013. Oct 10;56(19):7501–7515. [DOI] [PubMed] [Google Scholar]
- 11.Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017. Jan;16(1):19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coyne AG, Scott DE, Abell C. Drugging challenging targets using fragment-based approaches. Curr Opin Chem Biol. 2010. Jun;14(3):299–307. [DOI] [PubMed] [Google Scholar]
- 13.Winter A, Higueruelo AP, Marsh M, et al. Biophysical and computational fragment-based approaches to targeting protein-protein interactions: applications in structure-guided drug discovery. Q Rev Biophys. 2012. Nov;45(4):383–426. [DOI] [PubMed] [Google Scholar]
- 14.Smith MC, Gestwicki JE. Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev Mol Med. 2012. Jul;26(14):e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng AC, Coleman RG, Smyth KT, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol. 2007. Jan;25(1):71–75. [DOI] [PubMed] [Google Scholar]
- 16.Ivanov AA, Khuri FR, Fu H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol Sci. 2013. Jul;34(7):393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001. Mar 1;46(1–3):3–26. [DOI] [PubMed] [Google Scholar]
- 18.Buchwald P. Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life. 2010. Oct;62(10):724–731. [DOI] [PubMed] [Google Scholar]
- 19.Eyrisch S, Helms V. What induces pocket openings on protein surface patches involved in protein-protein interactions? J Comput Aided Mol Des. 2009. Feb;23(2):73–86. [DOI] [PubMed] [Google Scholar]
- 20.Ni D, Wei J, He X, et al. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem Sci. 2020. Nov 2;12(1):464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ni D, Li Y, Qiu Y, et al. Combining allosteric and orthosteric drugs to overcome drug resistance. Trends Pharmacol Sci. 2020. May;41(5):336–348. [DOI] [PubMed] [Google Scholar]
- 22.Lu S, He X, Ni D, et al. Allosteric modulator discovery: from serendipity to structure-based design. J Med Chem. 2019. Jul 25;62(14):6405–6421. [DOI] [PubMed] [Google Scholar]
- 23.Wu J, Li D, Liu X, et al. IDDB: a comprehensive resource featuring genes, variants and characteristics associated with infertility. Nucleic Acids Res. 2021. Jan 8;49(D1):D1218–d1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geppert T, Bauer S, Hiss JA, et al. Immunosuppressive small molecule discovered by structure-based virtual screening for inhibitors of protein-protein interactions. Angew Chem Int Ed Engl. 2012. Jan 2;51(1):258–261. [DOI] [PubMed] [Google Scholar]
- 25.Keskin O, Gursoy A, Ma B, et al. Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chem Rev. 2008. Apr;108(4):1225–1244. [DOI] [PubMed] [Google Scholar]
- 26.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004. Jan 23;116(2):191–203. [DOI] [PubMed] [Google Scholar]
- 27.Gunasekaran K, Tsai CJ, Kumar S, et al. Extended disordered proteins: targeting function with less scaffold. Trends Biochem Sci. 2003. Feb;28(2):81–85. [DOI] [PubMed] [Google Scholar]
- 28.Keskin O, Nussinov R. Similar binding sites and different partners: implications to shared proteins in cellular pathways. Structure. 2007. Mar;15(3):341–354. [DOI] [PubMed] [Google Scholar]
- 29.Kamura T, Maenaka K, Kotoshiba S, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004. Dec 15;18(24):3055–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bullock AN, Debreczeni JE, Edwards AM, et al. Crystal structure of the SOCS2-elongin C-elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc Natl Acad Sci U S A. 2006. May 16;103(20):7637–7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.••.Burgoyne NJ, Jackson RM. Predicting protein interaction sites: binding hot-spots in protein-protein and protein-ligand interfaces. Bioinformatics. 2006Jun 1;22(11):1335–1342. [DOI] [PubMed] [Google Scholar]; Protein assemblies are poorly represented in structural databases, and elucidating their structure is a goal in biology. This article aims to analyze protein clefts that may be binding hotspots
- 32.Keskin O, Ma B, Nussinov R. Hot regions in protein–protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005. Feb 4;345(5):1281–1294. [DOI] [PubMed] [Google Scholar]
- 33.Tsai CJ, Del Sol A, Nussinov R. Allostery: absence of a change in shape does not imply that allostery is not at play. J Mol Biol. 2008. Apr 18;378(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.González-Ruiz D, Gohlke H. Targeting protein-protein interactions with small molecules: challenges and perspectives for computational binding epitope detection and ligand finding. Curr Med Chem. 2006;13(22):2607–2625. [DOI] [PubMed] [Google Scholar]
- 35.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007. Dec 13;450(7172):1001–1009. [DOI] [PubMed] [Google Scholar]
- 36.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995. Jan 20;267(5196):383–386. [DOI] [PubMed] [Google Scholar]
- 37.Jubb H, Blundell TL, Ascher DB. Flexibility and small pockets at protein-protein interfaces: new insights into druggability. Prog Biophys Mol Biol. 2015. Oct;119(1):2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arkin MR, Randal M, DeLano WL, et al. Binding of small molecules to an adaptive protein-protein interface. Proc Natl Acad Sci U S A. 2003. Feb 18;100(4):1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruncko M, Oost TK, Belli BA, et al. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem. 2007. Feb 22;50(4):641–662. [DOI] [PubMed] [Google Scholar]
- 40.Sheng C, Dong G, Miao Z, et al. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem Soc Rev. 2015. Nov 21;44(22):8238–8259. [DOI] [PubMed] [Google Scholar]
- 41.Macalino SJY, Basith S, Clavio NAB, et al. Evolution of in silico strategies for protein-protein interaction drug discovery. Molecules. 2018. 6; 23(8): Aug. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J, Kriwacki RW. Intrinsically disordered proteins: structure, function and therapeutics. J Mol Biol. 2018. Aug 3;430(16):2275–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wichapong K, Poelman H, Ercig B, et al. Rational modulator design by exploitation of protein-protein complex structures. Future Med Chem. 2019. May;11(9):1015–1033. [DOI] [PubMed] [Google Scholar]
- 44.Li F, Liu Z, Sun H, et al. PCC0208017, a novel small-molecule inhibitor of MARK3/MARK4, suppresses glioma progression in vitro and in vivo. Acta Pharm Sin B. 2020. Feb;10(2):289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zwier MC, Pratt AJ, Adelman JL, et al. Efficient atomistic simulation of pathways and calculation of rate constants for a protein-peptide binding process: application to the MDM2 protein and an intrinsically disordered P53 peptide. J Phys Chem Lett. 2016. Sep 1;7(17):3440–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kozakov D, Hall DR, Chuang GY, et al. Structural conservation of druggable hot spots in protein-protein interfaces. Proc Natl Acad Sci U S A. 2011. Aug 16;108(33):13528–13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zerbe BS, Hall DR, Vajda S, et al. Relationship between hot spot residues and ligand binding hot spots in protein-protein interfaces. J Chem Inf Model. 2012. Aug 27;52(8):2236–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sayyed-Ahmad A. Hotspot identification on protein surfaces using probe-based md simulations: successes and challenges. Curr Top Med Chem. 2018;18(27):2278–2283. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Lupala CS, Liu H, et al. Identification of drug binding sites and action mechanisms with molecular dynamics simulations. Curr Top Med Chem. 2018;18(27):2268–2277. [DOI] [PubMed] [Google Scholar]
- 50.Kozakov D, Grove LE, Hall DR, et al. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc. 2015. May;10(5):733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Liang J, Siu T, et al. Allosteric inhibitors of Akt1 and Akt2: discovery of [1,2,4]triazolo[3,4-f][1,6]naphthyridines with potent and balanced activity. Bioorg Med Chem Lett. 2009. Feb 1;19(3):834–836. [DOI] [PubMed] [Google Scholar]
- 52.Zhang BW, Jasnow D, Zuckerman DM. The “weighted ensemble” path sampling method is statistically exact for a broad class of stochastic processes and binning procedures. J Chem Phys. 2010. Feb 7;132(5):054107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Votapka LW, Amaro RE, Shehu A. Multiscale estimation of binding kinetics using brownian dynamics, molecular dynamics and milestoning. PLoS Comput Biol. 2015. Oct;11(10):e1004381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McMillan K, Adler M, Auld DS, et al. Allosteric inhibitors of inducible nitric oxide synthase dimerization discovered via combinatorial chemistry. Proc Natl Acad Sci U S A. 2000. Feb 15;97(4):1506–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gorczynski MJ, Grembecka J, Zhou Y, et al. Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins Runx1 and CBFbeta. Chem Biol. 2007. Oct;14(10):1186–1197. [DOI] [PubMed] [Google Scholar]
- 56.DeDecker BS. Allosteric drugs: thinking outside the active-site box. Chem Biol. 2000. May;7(5):R103–7. [DOI] [PubMed] [Google Scholar]
- 57.Huang X, Peng JW, Speck NA, et al. Solution structure of core binding factor beta and map of the CBF alpha binding site. Nat Struct Biol. 1999. Jul;6(7):624–627. [DOI] [PubMed] [Google Scholar]
- 58.Lee Y, Basith S, Choi S. Recent advances in structure-based drug design targeting class A G protein-coupled receptors utilizing crystal structures and computational simulations. J Med Chem. 2018. Jan 11;61(1):1–46. [DOI] [PubMed] [Google Scholar]
- 59.Dixit A, Verkhivker GM, Rutherford S. Probing molecular mechanisms of the Hsp90 chaperone: biophysical modeling identifies key regulators of functional dynamics. PLoS One. 2012;7(5):e37605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ozdemir ES, Jang H, Gursoy A, et al. Unraveling the molecular mechanism of interactions of the Rho GTPases Cdc42 and Rac1 with the scaffolding protein IQGAP2. J Biol Chem. 2018. Mar 9;293(10):3685–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar MV, Ebna Noor R, Davis RE, et al. Molecular insights into the interaction of Hsp90 with allosteric inhibitors targeting the C-terminal domain. Medchemcomm. 2018. Aug 1;9(8):1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paladino A, Woodford MR, Backe SJ, et al. Chemical perturbation of oncogenic protein folding: from the prediction of locally unstable structures to the design of disruptors of Hsp90-client interactions. Chemistry. 2020. Aug 3;26(43):9459–9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keegan BM, Koren J III, Blagg BS, et al. Disruptors of the Hsp90/ Aha1 complex that reduce tau aggregation: nonhuman/Lead optimization studies. Alzheimers Dement. 2020;16:e045193. [Google Scholar]
- 64.Triveri A, Sanchez-Martin C, Torielli L, et al. Protein allostery and ligand design: computational design meets experiments to discover novel chemical probes. J Mol Biol. 2022. Sep 15;434(17):167468. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Z, Chen H, Bai H, et al. Molecular dynamics simulations on the oligomer-formation process of the GNNQQNY peptide from yeast prion protein Sup35. Biophys J. 2007. Sep 1;93(5):1484–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baram M, Atsmon-Raz Y, Ma B, et al. Amylin-Aβ oligomers at atomic resolution using molecular dynamics simulations: a link between Type 2 diabetes and Alzheimer’s disease. Phys Chem Chem Phys. 2016. Jan 28;18(4):2330–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hansmann UH, Okamoto Y. New Monte Carlo algorithms for protein folding. Curr Opin Struct Biol. 1999. Apr;9(2):177–183. [DOI] [PubMed] [Google Scholar]
- 68.Metropolis N, Rosenbluth AW, Rosenbluth MN, et al. Equation of state calculations by fast computing machines. J Chem Phys. 1953;21(6):1087–1092. [Google Scholar]
- 69.Pan AC, Xu H, Palpant T, et al. Quantitative characterization of the binding and unbinding of millimolar drug fragments with molecular dynamics simulations. J Chem Theory Comput. 2017. Jul 11;13(7):3372–3377. [DOI] [PubMed] [Google Scholar]
- 70.Wallraven K, Holmelin FL, Glas A, et al. Adapting free energy perturbation simulations for large macrocyclic ligands: how to dissect contributions from direct binding and free ligand flexibility. Chem Sci. 2020. Feb 28;11(8):2269–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Ruiter A, Oostenbrink C. Advances in the calculation of binding free energies. Curr Opin Struct Biol. 2020. Apr;61:207–212. [DOI] [PubMed] [Google Scholar]
- 72.Gill SC, Lim NM, Grinaway PB, et al. Binding modes of ligands using enhanced sampling (blues): rapid decorrelation of ligand binding modes via nonequilibrium candidate monte carlo. J Phys Chem B. 2018. May 31;122(21):5579–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paul F, Wehmeyer C, Abualrous ET, et al. Protein-peptide association kinetics beyond the seconds timescale from atomistic simulations. Nat Commun. 2017. Oct 23;8(1):1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou G, Pantelopulos GA, Mukherjee S, et al. Bridging microscopic and macroscopic mechanisms of p53-MDM2 binding with kinetic network models. Biophys J. 2017. Aug 22;113(4):785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bowman GR, Ensign DL, Pande VS. Enhanced modeling via network theory: adaptive sampling of Markov state models. J Chem Theory Comput. 2010;6(3):787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Noé F, Schütte C, Vanden-Eijnden E, et al. Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations. Proc Natl Acad Sci U S A. 2009. Nov 10;106(45):19011–19016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huber GA, Kim S. Weighted-ensemble Brownian dynamics simulations for protein association reactions. Biophys J. 1996. Jan;70(1):97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lang L, Perez A. Binding ensembles of p53-MDM2 peptide inhibitors by combining bayesian inference and atomistic simulations. Molecules. 2021;26(1). DOI: 10.3390/molecules26010198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morrone JA, Perez A, Deng Q, et al. Molecular simulations identify binding poses and approximate affinities of stapled α-helical peptides to MDM2 and MDMX. J Chem Theory Comput. 2017. Feb 14;13(2):863–869. [DOI] [PubMed] [Google Scholar]
- 80.Morrone JA, Perez A, MacCallum J, et al. Computed binding of peptides to proteins with MELD-accelerated molecular dynamics. J Chem Theory Comput. 2017. Feb 14;13(2):870–876. [DOI] [PubMed] [Google Scholar]
- 81.Fukunishi H, Watanabe O, Takada S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: application to protein structure prediction. J Chem Phys. 2002;116(20):9058–9067. [Google Scholar]
- 82.Lu LJ, Xia Y, Paccanaro A, et al. Assessing the limits of genomic data integration for predicting protein networks. Genome Res. 2005. Jul;15(7):945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jansen R, Yu H, Greenbaum D, et al. A bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003. Oct 17;302(5644):449–453. [DOI] [PubMed] [Google Scholar]
- 84.Jansen R, Gerstein M. Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Curr Opin Microbiol. 2004. Oct;7(5):535–545. [DOI] [PubMed] [Google Scholar]
- 85.Yu B, Chen C, Zhou H, et al. GTB-PPI: predict protein-protein interactions based on L1-regularized logistic regression and gradient tree boosting. Genomics Proteomics Bioinformatics. 2020. Oct;18(5):582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Du X, Sun S, Hu C, et al. DeepPPI: boosting prediction of protein-protein interactions with deep neural networks. J Chem Inf Model. 2017. Jun 26;57(6):1499–1510. [DOI] [PubMed] [Google Scholar]
- 87.You ZH, Zhu L, Zheng CH, et al. Prediction of protein-protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set. BMC Bioinformatics. 2014;15 Suppl 15(Suppl15):S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patil A, Nakamura H. Filtering high-throughput protein-protein interaction data using a combination of genomic features. BMC Bioinformatics. 2005. Apr;18(6):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ofran Y, Rost B. ISIS: interaction sites identified from sequence. Bioinformatics. 2007. Jan 15;23(2):e13–6. [DOI] [PubMed] [Google Scholar]
- 90.Xue LC, Dobbs D, Bonvin AM, et al. Computational prediction of protein interfaces: a review of data driven methods. FEBS Lett. 2015. Nov 30;589(23):3516–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang B, Chen P, Huang DS, et al. Predicting protein interaction sites from residue spatial sequence profile and evolution rate. FEBS Lett. 2006. Jan 23;580(2):380–384. [DOI] [PubMed] [Google Scholar]
- 92.Sikić M, Tomić S, Vlahovicek K. Prediction of protein-protein interaction sites in sequences and 3D structures by random forests. PLoS Comput Biol. 2009. Jan;5(1):e1000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Du T, Liao L, Wu CH, et al. Prediction of residue-residue contact matrix for protein-protein interaction with Fisher score features and deep learning. Methods. 2016. Nov;1(110):97–105. [DOI] [PubMed] [Google Scholar]
- 94.Sun T, Zhou B, Lai L, et al. Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinformatics. 2017. May 25;18(1):277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Labbé CM, Kuenemann MA, Zarzycka B, et al. iPPI-DB: an online database of modulators of protein-protein interactions. Nucleic Acids Res. 2016. Jan 4;44(D1):D542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Labbé CM, Laconde G, Kuenemann MA, et al. iPPI-DB: a manually curated and interactive database of small non-peptide inhibitors of protein-protein interactions. Drug Discov Today. 2013. Oct;18(19–20):958–968. [DOI] [PubMed] [Google Scholar]
- 97.Reynès C, Host H, Camproux AC, et al. Designing focused chemical libraries enriched in protein-protein interaction inhibitors using machine-learning methods. PLoS Comput Biol. 2010. Mar 5;6(3):e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lagorce D, Sperandio O, Baell JB, et al. FAF-Drugs3: a web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015. Jul 1;43(W1):W200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lagorce D, Maupetit J, Baell J, et al. The FAF-Drugs2 server: a multistep engine to prepare electronic chemical compound collections. Bioinformatics. 2011. Jul 15;27(14):2018–2020. [DOI] [PubMed] [Google Scholar]
- 100.Miteva MA, Violas S, Montes M, et al. FAF-Drugs: free ADME/tox filtering of compound collections. Nucleic Acids Res. 2006. Jul 1;34(Web Server issue):W738–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.••.Hamon V, Bourgeas R, Ducrot P, et al. 2P2I HUNTER: a tool for filtering orthosteric protein-protein interaction modulators via a dedicated support vector machine. J R Soc Interface. 2014. Jan 6;11(90):20130860. [DOI] [PMC free article] [PubMed] [Google Scholar]; PPIs are the most screened target class in high-throughput screening (HTS). In this study they analyzed 40 2P2I small molecules and propose a protocol to filter general screening libraries using 11 Dragon descriptors. The resulting chemical space will support PPI inhibitor HTS campaigns
- 102.Bordner AJ, Abagyan R. Statistical analysis and prediction of protein-protein interfaces. Proteins. 2005. Aug 15;60(3):353–366. [DOI] [PubMed] [Google Scholar]
- 103.Cheng T, Li Q, Zhou Z, et al. Structure-based virtual screening for drug discovery: a problem-centric review. Aaps j. 2012. Mar;14(1):133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ripphausen P, Nisius B, Bajorath J. State-of-the-art in ligand-based virtual screening. Drug Discov Today. 2011. May;16(9–10):372–376. [DOI] [PubMed] [Google Scholar]
- 105.•.Wu KJ, Lei PM, Liu H, et al. Mimicking strategy for protein-protein interaction inhibitor discovery by virtual screening. Molecules. 2019. Dec 4;24(24):4428. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work outlined the process of employing virtual screening in order to identify mimicking protein partner inhibitors
- 106.Beekman AM, Cominetti MMD, Walpole SJ, et al. Identification of selective protein-protein interaction inhibitors using efficient in silico peptide-directed ligand design. Chem Sci. 2019. Apr 28;10 (16):4502–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Beekman AM, O’Connell MA, Howell LA. Peptide-directed binding for the discovery of modulators of α-helix-mediated protein-protein interactions: proof-of-concept studies with the apoptosis regulator Mcl-1. Angew Chem Int Ed Engl. 2017. Aug 21;56(35):10446–10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sanchez-Martin C, Moroni E, Ferraro M, et al. Rational design of allosteric and selective inhibitors of the molecular chaperone TRAP1. Cell Rep. 2020. Apr 21;31(3):107531. [DOI] [PubMed] [Google Scholar]
- 109.Kumar A, Zhang KYJ. A cross docking pipeline for improving pose prediction and virtual screening performance. J Comput Aided Mol Des. 2018. Jan;32(1):163–173. [DOI] [PubMed] [Google Scholar]
- 110.Jiang ZY, Lu MC, Xu LL, et al. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J Med Chem. 2014. Mar 27;57(6):2736–2745. [DOI] [PubMed] [Google Scholar]
- 111.Aderinwale T, Christoffer CW, Sarkar D, et al. Computational structure modeling for diverse categories of macromolecular interactions. Curr Opin Struct Biol. 2020. Oct;64:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.••.Porter KA, Desta I, Kozakov D, et al. What method to use for protein-protein docking? Curr Opin Struct Biol. 2019. Apr;55:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article present that what method to use for PP-docking. According to CAPRI-benchmark CASP’s set analysis, free docking of heterodimers produces acceptable or better predictions in the top 10 models for about 40% of structures. Template-based and free docking approaches may work better for targets with template structures
- 113.Zhou P, Jin B, Li H, et al. HPEPDOCK: a web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018. Jul 2;46(W1):W443–w450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu X, Yan C, Zou X. MDockPeP: an ab-initio protein-peptide docking server. J Comput Chem. 2018. Oct 30;39(28):2409–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Porter KA, Xia B, Beglov D, et al. ClusPro PeptiDock: efficient global docking of peptide recognition motifs using FFT. Bioinformatics. 2017. Oct 15;33(20):3299–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.de Vries SJ, Rey J, Schindler CEM, et al. The pepATTRACT web server for blind, large-scale peptide-protein docking. Nucleic Acids Res. 2017. Jul 3;45(W1):W361–w364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alam N, Goldstein O, Xia B, et al. High-resolution global peptide-protein docking using fragments-based PIPER-FlexPepDock. PLoS Comput Biol. 2017. Dec;13(12):e1005905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang Y, Sanner MF, Valencia A. AutoDock CrankPep: combining folding and docking to predict protein–peptide complexes. Bioinformatics. 2019. Dec 15;35(24):5121–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lamiable A, Thévenet P, Rey J, et al. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016. Jul 8;44(W1):W449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Obarska-Kosinska A, Iacoangeli A, Lepore R, et al. PepComposer: computational design of peptides binding to a given protein surface. Nucleic Acids Res. 2016. Jul 8;44(W1):W522–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee H, Heo L, Lee MS, et al. GalaxyPepDock: a protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015. Jul 1;43(W1):W431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.BR C, Subramanian J, Sharma SD. Managing protein flexibility in docking and its applications. Drug Discov Today. 2009. Apr;14(7–8):394–400. [DOI] [PubMed] [Google Scholar]
- 123.Fernández-Recio J, Totrov M, Abagyan R. Soft protein-protein docking in internal coordinates. Protein Sci. 2002. Feb;11(2):280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ferrari AM, Wei BQ, Costantino L, et al. Soft docking and multiple receptor conformations in virtual screening. J Med Chem. 2004. Oct 7;47(21):5076–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Leach AR. Ligand docking to proteins with discrete side-chain flexibility. J Mol Biol. 1994. Jan 7;235(1):345–356. [DOI] [PubMed] [Google Scholar]
- 126.Evangelista Falcon W, Ellingson SR, Smith JC, et al. Ensemble docking in drug discovery: how many protein configurations from molecular dynamics simulations are needed to reproduce known ligand binding? J Phys Chem B. 2019. Jun 27;123(25):5189–5195. [DOI] [PubMed] [Google Scholar]
- 127.Amaro RE, Baudry J, Chodera J, et al. Ensemble docking in drug discovery. Biophys J. 2018. May 22;114(10):2271–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huart A-S, Hupp TR. Evolution of conformational disorder & diversity of the P53 interactome. BioDiscovery. 2013;8:e8952. [Google Scholar]
- 129.Aiyer S, Swapna GVT, Ma LC, et al. A common binding motif in the ET domain of BRD3 forms polymorphic structural interfaces with host and viral proteins. Structure. 2021. Aug 5;29(8):886–898.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yan C, Xu X, Zou X. Fully blind docking at the atomic level for protein-peptide complex structure prediction. Structure. 2016. Oct 4;24(10):1842–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Weng G, Gao J, Wang Z, et al. Comprehensive evaluation of fourteen docking programs on protein-peptide complexes. J Chem Theory Comput. 2020. Jun 9;16(6):3959–3969. [DOI] [PubMed] [Google Scholar]
- 132.Ain QU, Aleksandrova A, Roessler FD, et al. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip Rev Comput Mol Sci. 2015. Nov-Dec;5(6):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li J, Fu A, Zhang L. An overview of scoring functions used for protein-ligand interactions in molecular docking. Interdiscip Sci. 2019. Jun;11(2):320–328. [DOI] [PubMed] [Google Scholar]
- 134.Morrone JA, Weber JK, Huynh T, et al. Combining docking pose rank and structure with deep learning improves protein-ligand binding mode prediction over a baseline docking approach. J Chem Inf Model. 2020. Sep 28;60(9):4170–4179. [DOI] [PubMed] [Google Scholar]
- 135.Tao H, Zhang Y, Huang SY. Improving protein-peptide docking results via pose-clustering and rescoring with a combined knowledge-based and MM-GBSA scoring function. J Chem Inf Model. 2020. Apr 27;60(4):2377–2387. [DOI] [PubMed] [Google Scholar]
- 136.Tan YS, Mhoumadi Y, Verma CS. Roles of computational modelling in understanding p53 structure, biology, and its therapeutic targeting. J Mol Cell Biol. 2019. Apr 1;11(4):306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene. 1999. Dec 13;18(53):7621–7636. [DOI] [PubMed] [Google Scholar]
- 138.Kussie PH, Gorina S, Marechal V, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996. Nov 8;274(5289):948–953. [DOI] [PubMed] [Google Scholar]
- 139.Tan YS, Lane DP, Verma CS. Stapled peptide design: principles and roles of computation. Drug Discov Today. 2016. Oct;21(10):1642–1653. [DOI] [PubMed] [Google Scholar]
- 140.Bowman AL, Nikolovska-Coleska Z, Zhong H, et al. Small molecule inhibitors of the MDM2-p53 interaction discovered by ensemble-based receptor models. J Am Chem Soc. 2007. Oct 24;129 (42):12809–12814. [DOI] [PubMed] [Google Scholar]
- 141.Bueren-Calabuig JA, Michel J. Elucidation of ligand-dependent modulation of disorder-order transitions in the oncoprotein MDM2. PLoS Comput Biol. 2015. Jun;11(6):e1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ciemny M, Kurcinski M, Kamel K, et al. Protein-peptide docking: opportunities and challenges. Drug Discov Today. 2018. Aug;23(8):1530–1537. [DOI] [PubMed] [Google Scholar]
- 143.Ding Q, Zhang Z, JJ L, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013. Jul 25;56(14):5979–5983. [DOI] [PubMed] [Google Scholar]
- 144.Joseph TL, Madhumalar A, Brown CJ, et al. Differential binding of p53 and nutlin to MDM2 and MDMX: computational studies. Cell Cycle. 2010. Mar 15;9(6):1167–1181. [DOI] [PubMed] [Google Scholar]
- 145.Reed D, Shen Y, Shelat AA, et al. Identification and characterization of the first small molecule inhibitor of MDMX. J Biol Chem. 2010. Apr 2;285(14):10786–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Burgess A, Chia KM, Haupt S, et al. Clinical overview of MDM2/X-targeted therapies. Front Oncol. 2016;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Phan J, Li Z, Kasprzak A, et al. Structure-based design of high affinity peptides inhibiting the interaction of p53 with MDM2 and MDMX. J Biol Chem. 2010. Jan 15;285(3):2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sang P, Shi Y, Lu J, et al. α-helix-mimicking sulfono-γ-AApeptide inhibitors for p53-MDM2/MDMX protein-protein interactions. J Med Chem. 2020. Feb 13;63(3):975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jiang L, Zawacka-Pankau J. The p53/MDM2/MDMX-targeted therapies-a clinical synopsis. Cell Death Dis. 2020. Apr 17;11(4):237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Meric-Bernstam F, Saleh MN, Infante JR, et al. Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDMX-and MDM2-mediated inhibition of WT p53 in patients with solid tumors and lymphomas. J Clin Oncol. 2017;35(15_suppl):2505. [Google Scholar]
- 151.Lang L, Perez A. Binding ensembles of p53-MDM2 peptide inhibitors by combining bayesian inference and atomistic simulations. Molecules. 2021;26(1):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee Y, Basith S, Choi S. Recent advances in structure-based drug design targeting class AG protein-coupled receptors utilizing crystal structures and computational simulations. J Med Chem. 2018;61(1):1–46. [DOI] [PubMed] [Google Scholar]
- 153.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. [DOI] [PubMed] [Google Scholar]
- 154.Chittenden T BH3 domains: intracellular death-ligands critical for initiating apoptosis. Cancer Cell. 2002. Sep;2(3):165–166. [DOI] [PubMed] [Google Scholar]
- 155.Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019. Mar;20(3):175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Muchmore SW, Sattler M, Liang H, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996. May 23;381(6580):335–341. [DOI] [PubMed] [Google Scholar]
- 157.Harvey EP, Seo HS, Guerra RM, et al. Crystal structures of anti-apoptotic BFL-1 and its complex with a covalent stapled peptide inhibitor. Structure. 2018. Jan 2;26(1):153–160.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sattler M, Liang H, Nettesheim D, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997. Feb 14;275(5302):983–986. [DOI] [PubMed] [Google Scholar]
- 159.Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene. 2008. Dec;27 Suppl 1 (Suppl1):S2–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vila-Julià G, Granadino-Roldán JM, Perez JJ, et al. Molecular determinants for the activation/inhibition of bak protein by BH3 peptides. J Chem Inf Model. 2020. Mar 23;60(3):1632–1643. [DOI] [PubMed] [Google Scholar]
- 161.Perez JJ, Perez RA, Perez A. Computational modeling as a tool to investigate PPI: from drug design to tissue engineering. Front Mol Biosci. 2021;8:681617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wei J, Stebbins JL, Kitada S, et al. BI-97C1, an optically pure Apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J Med Chem. 2010. May 27;53(10):4166–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shoemaker AR, Oleksijew A, Bauch J, et al. A small-molecule inhibitor of Bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res. 2006. Sep 1;66(17):8731–8739. [DOI] [PubMed] [Google Scholar]
- 164.Lee AC, Harris JL, Khanna KK, et al. A comprehensive review on current advances in peptide drug development and design. Int J Mol Sci. 2019. May 14;20:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pan R, Hogdal LJ, Benito JM, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014. Mar;4(3):362–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kapp TG, Rechenmacher F, Neubauer S, et al. A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins. Sci Rep. 2017. Jan;11(7):39805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dechantsreiter MA, Planker E, Mathä B, et al. N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J Med Chem. 1999. Aug 12;42(16):3033–3040. [DOI] [PubMed] [Google Scholar]
- 168.Zhu J, Zhu J, Negri A, et al. Closed headpiece of integrin αIIbβ3 and its complex with an αIIbβ3-specific antagonist that does not induce opening. Blood. 2010. Dec 2;116(23):5050–5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mittag T, Kay LE, Forman-Kay JD. Protein dynamics and conformational disorder in molecular recognition. J Mol Recognit. 2010. Mar-Apr;23(2):105–116. [DOI] [PubMed] [Google Scholar]
- 170.Shandler SJ, Korendovych IV, Moore DT, et al. Computational design of a β-peptide that targets transmembrane helices. J Am Chem Soc. 2011. Aug 17;133(32):12378–12381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Loo Y, Goktas M, Tekinay AB, et al. Self-assembled proteins and peptides as scaffolds for tissue regeneration. Adv Healthc Mater. 2015. Nov 18;4(16):2557–2586. [DOI] [PubMed] [Google Scholar]
- 172.Smadbeck J, Chan KH, Khoury GA, et al. De novo design and experimental characterization of ultrashort self-associating peptides. PLoS Comput Biol. 2014. Jul;10(7):e1003718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chen Z, Johnson MC, Chen J, et al. Self-assembling 2D arrays with de novo protein building blocks. J Am Chem Soc. 2019. Jun 5;141(22):8891–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cormier AR, Pang X, Zimmerman MI, et al. Molecular structure of RADA16-I designer self-assembling peptide nanofibers. ACS Nano. 2013. Sep 24;7(9):7562–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Leonard SR, Cormier AR, Pang X, et al. Solid-state NMR evidence for β-hairpin structure within MAX8 designer peptide nanofibers. Biophys J. 2013. Jul 2;105(1):222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Delgado J, Radusky LG, Cianferoni D, et al. FoldX 5.0: working with RNA, small molecules and a new graphical interface. Bioinformatics. 2019. Oct 15;35(20):4168–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Aghazadeh Y, Papadopoulos V. The role of the 14-3-3 protein family in health, disease, and drug development. Drug Discov Today. 2016. Feb;21(2):278–287. [DOI] [PubMed] [Google Scholar]
- 178.Wilker E, Yaffe MB. 14-3-3 Proteins–a focus on cancer and human disease. J Mol Cell Cardiol. 2004. Sep;37(3):633–642. [DOI] [PubMed] [Google Scholar]
- 179.Steinacker P, Aitken A, Otto M. 14-3-3 proteins in neurodegeneration. Semin Cell Dev Biol. 2011. Sep;22(7):696–704. [DOI] [PubMed] [Google Scholar]
- 180.•.Kaplan A, Ottmann C, Fournier AE. 14-3-3 adaptor protein-protein interactions as therapeutic targets for CNS diseases. Pharmacol Res. 2017. Nov;125(Pt B):114–121. [DOI] [PubMed] [Google Scholar]; This study clarifies the roles of 14-3-3 PPIs in neurons and the potential development of drugs that would specifically target 14-3-3 PPIs for the treatment of CNS diseases
- 181.Sluchanko NN, Gusev NB. Probable participation of 14-3-3 in tau protein oligomerization and aggregation. J Alzheimers Dis. 2011;27(3):467–476. [DOI] [PubMed] [Google Scholar]
- 182.Giordanetto F, Schäfer A, Ottmann C. Stabilization of protein-protein interactions by small molecules. Drug Discov Today. 2014. Nov;19(11):1812–1821. [DOI] [PubMed] [Google Scholar]
- 183.Oecking C, Eckerskorn C, Weiler EW. The fusicoccin receptor of plants is a member of the 14-3-3 superfamily of eukaryotic regulatory proteins. FEBS Lett. 1994. Sep 26;352(2):163–166. [DOI] [PubMed] [Google Scholar]
- 184.Molzan M, Kasper S, Röglin L, et al. Stabilization of physical RAF/14-3-3 interaction by cotylenin A as treatment strategy for RAS mutant cancers. ACS Chem Biol. 2013. Sep 20;8(9):1869–1875. [DOI] [PubMed] [Google Scholar]
- 185.Anders C, Higuchi Y, Koschinsky K, et al. A semisynthetic fusicoccane stabilizes a protein-protein interaction and enhances the expression of K+ channels at the cell surface. Chem Biol. 2013. Apr 18;20(4):583–593. [DOI] [PubMed] [Google Scholar]
- 186.••.Bier D, Bartel M, Sies K, et al. Small-molecule stabilization of the 14-3-3/Gab2 protein-protein interaction (PPI) interface. ChemMedChem. 2016. Apr 19;11(8):911–918. [DOI] [PubMed] [Google Scholar]; This study present an example of a semi-synthetic natural product derivative, ISIR-005, which stabilizes the Gab2-adap-tor PP’s interaction is associated for cancer
- 187.Richter A, Rose R, Hedberg C, et al. An optimised small-molecule stabiliser of the 14-3-3-PMA2 protein-protein interaction. Chemistry. 2012. May 21;18(21):6520–6527. [DOI] [PubMed] [Google Scholar]
- 188.Sato S, Jung H, Nakagawa T, et al. Metabolite regulation of nuclear localization of carbohydrate-response element-binding protein (ChREBP): role of amp as an allosteric inhibitor. J Biol Chem. 2016. May 13;291(20):10515–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bier D, Rose R, Bravo-Rodriguez K, et al. Molecular tweezers modulate 14-3-3 protein-protein interactions. Nat Chem. 2013. Mar;5(3):234–239. [DOI] [PubMed] [Google Scholar]
- 190.Aumentado-Armstrong TT, Istrate B, Murgita RA. Algorithmic approaches to protein-protein interaction site prediction. Algorithms Mol Biol. 2015;10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Baig MH, Ahmad K, Roy S, et al. Computer aided drug design: success and limitations. Curr Pharm Des. 2016;22(5):572–581. [DOI] [PubMed] [Google Scholar]
- 192.Murray CW, Rees DC. The rise of fragment-based drug discovery. Nat Chem. 2009. Jun;1(3):187–192. [DOI] [PubMed] [Google Scholar]
- 193.Dastidar SG, Lane DP, Verma CS. Multiple peptide conformations give rise to similar binding affinities: molecular simulations of p53-MDM2. J Am Chem Soc. 2008. Oct 15;130(41):13514–13515. [DOI] [PubMed] [Google Scholar]
- 194.Yuan Q, Chen J, Zhao H, et al. Structure-aware protein-protein interaction site prediction using deep graph convolutional network. Bioinformatics. 2021. Sep;22:8. [DOI] [PubMed] [Google Scholar]
- 195.Casadio R, Martelli PL, Savojardo C. Machine learning solutions for predicting protein–protein interactions. Wiley Interdiscip Rev Comput Mol Sci. 2022;12(6):e1618. [Google Scholar]