

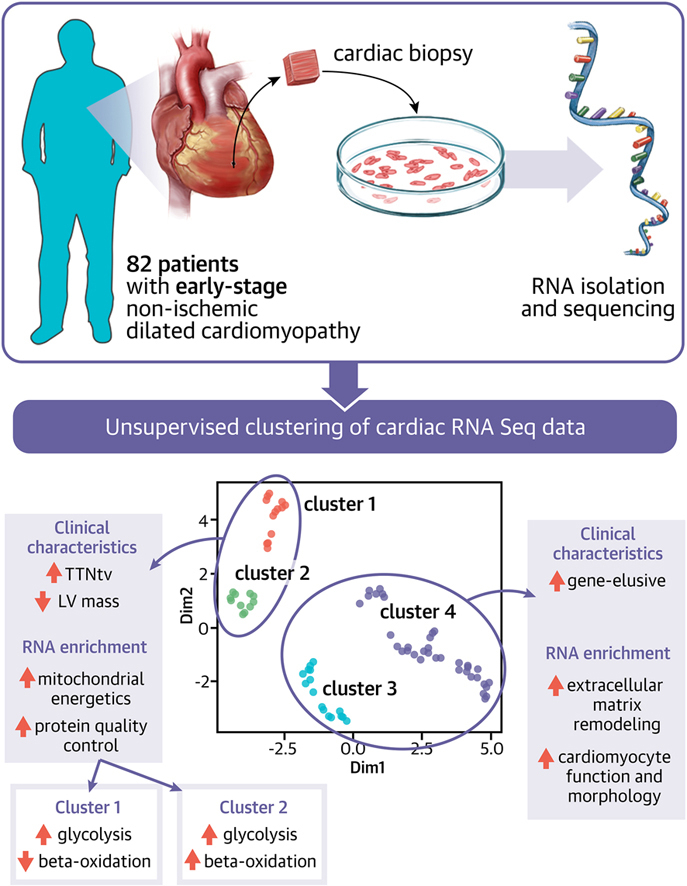
Visual Abstract
Key Words: clustering, dilated cardiomyopathy, genetics, transcriptomics
Highlights
-
•
Unsupervised clustering of the cardiac transcriptome reveals clinically relevant patient subgroups in early- and end-stage DCM cohorts.
-
•
A subgroup enriched by patients with a truncating titin variant had a strong cardiac upregulation of the OXPHOS system, dysregulation of the ubiquitin-proteasome pathway, and a shift in cardiac substrate utilization.
-
•
The cardiac transcriptome is partially influenced by the genotype and phenotype of the patient but also contains unique information. The addition of the cardiac transcriptome to the genotype and phenotype of the patient increases the possibility for individualized medicine.
Summary
Dilated cardiomyopathy is a heterogeneous disease characterized by multiple genetic and environmental etiologies. The majority of patients are treated the same despite these differences. The cardiac transcriptome provides information on the patient's pathophysiology, which allows targeted therapy. Using clustering techniques on data from the genotype, phenotype, and cardiac transcriptome of patients with early- and end-stage dilated cardiomyopathy, more homogeneous patient subgroups are identified based on shared underlying pathophysiology. Distinct patient subgroups are identified based on differences in protein quality control, cardiac metabolism, cardiomyocyte function, and inflammatory pathways. The identified pathways have the potential to guide future treatment and individualize patient care.
Dilated cardiomyopathy (DCM) is a nonischemic form of heart failure (HF) that is currently the leading global cause of heart transplantation.1 The disease is not seen as a single disease entity but rather as a nonspecific phenotype, which can be the result of a multitude of environmental or genetic triggers.2 Using unsupervised machine learning on phenotypical features, we previously identified clinical DCM subgroups with comparable disease course.3 The patients within the created clinical subgroups were clinically more homogeneous compared with dividing patients into subgroups based on etiology. Besides the clinical homogeneity, the patients also share a comparable transcriptomic profile in the heart, potentially indicating a shared disease mechanism. Distinct transcriptomic profiles are also present within the patient subgroup of titinopathies.4 Truncating variants in titin (TTNtv) are the most prevalent cause of monogenic DCM and are found in 10% of patients with DCM.5 Titin cardiomyopathy is thought to be a well-treatable form of HF, showing an increase in left ventricular ejection fraction (LVEF) after being treated with standard HF therapy.6 The identified subgroups within the group of titin cardiomyopathies are different in clinical presentation and disease course, showing that not every titin cardiomyopathy is, per definition, benign.7 This demonstrates the complexity of the influence of both genetic and clinical modifiers on the phenotype and underlying cardiac transcriptome. The cardiac transcriptome seems an accurate representation of disease activity and has the potential to guide treatment and discover novel treatment targets.
In the current study, the transcriptomic data are the primary input for unsupervised clustering, which is subsequently associated to the clinical and genetic data. This will provide further insight in the clinical profile of patients who share comparable disease processes in the heart.
Methods
Patient cohort and clinical data
The patient cohort was derived from the prospective Maastricht Cardiomyopathy Registry of the Maastricht University Medical Center (MUMC, Maastricht, the Netherlands), between 2012 and 2021. The DCM diagnosis was defined according to the World Health Organization criteria and the latest European Society of Cardiology (ESC) proposal.8,9 Enrolled patients presented with LVEF <50% at baseline echocardiographic evaluation in the absence of any of the following conditions: obstruction >50% of a major coronary artery branch (at coronary angiography [CAG]), pericardial diseases, congenital heart diseases, cor pulmonale, and active myocarditis. Patients, for whom it is not contraindicated, received guideline-directed medical therapy titrated to the maximal tolerated dose as well as device therapy (implantable cardioverter-defibrillator [ICD] and cardiac resynchronization therapy defibrillator [CRT-D] implantation) according to the latest ESC guidelines.10 The study was performed according to the declaration of Helsinki and approved by the local Institutional Review Board of the Maastricht University Medical Center. All patients gave written informed consent.
As part of the diagnostic protocol, all patients underwent physical examination, blood sampling, 12-lead electrocardiogram, 24-hour Holter monitoring, complete echocardiographic and Doppler evaluation, and CAG at baseline.3 Endomyocardial biopsies (EMBs) and cardiovascular magnetic resonance imaging were performed if patients consented and were able to undergo these procedures. Spare EMBs dedicated to research were used to isolate RNA from and to perform RNA sequencing. Clinical data and RNA-sequencing data gathered from previous studies were used as primary input for the current study.3,4,11,12
Validation cohort
To validate the application of our clustering methodology, we used publicly accessible RNA sequencing and clinical data from the MAGNet consortium (GSE141910).13 In contrast to our cohort, RNA was isolated from hearts of patients with end-stage DCM. As our cohort was Caucasian, we only retrieved the data from the Caucasian patients with DCM (n = 89). The clinical dataset was limited to 7 variables: age, sex, hypertension, diabetes, body mass index, atrial fibrillation, and LVEF. Genetic testing was not performed in these patients.
Genetic analysis
All patents received genetic counseling and DNA testing, using a panel comprising 47 DCM-associated genes. All detected variants were confirmed by Sanger sequencing. Detected variants were classified according to the latest American College of Medical Genetics (ACMG) and Association for Molecular Pathology (AMP) guidelines. Truncating variants in TTN were reported as pathogenic when they were detected in an exon with a percentage spliced in (PSI) above 99%. If no pathogenic or likely pathogenic was identified, a patient was classified as "gene-elusive."
RNA-sequencing
RNA was isolated from cardiac biopsies from patients with DCM. The mRNA-sequencing library was generated using TruSeq mRNA sample preparation kit (Illumina) and sequenced on the NextSeq 500 (Illumina) and checked for quality and integrity.
Analysis and clustering of RNA-sequencing data
We first filtered samples based on 3 quality metrics: the total final reads >5,000,000; the total present RNAs >40,000; and no lower-side outliers for heart myocardium biomarker genes, including MYL3, MYH6, MHRT, HSPB3, TNNI3, TECRL, and RYR2. The RNA-sequencing read counts were normalized using global geometric library size factor (GLSF) method with the iCellR package v 1.6.014 in the statistical programming language R (version 4.1.0). The top 2000 dispersed transcripts based on standard deviation were selected to run dimensionality reduction by principal component analysis (PCA), uniform manifold approximation and projection (UMAP), and K nearest-neighbor–based network graph drawing layout (KNetL). Clustering was performed using the dimensionality reduction combined with the graphic-based clustering method with iCellR package. The optimal number of clusters was determined inside the iclust function. To see the distances or similarities among resulted clusters, pseudotime abstract K (PAK) NetL map was calculated. In addition to the dimensionality reduction combined with the graphic-based clustering method, discrete data Poisson or negative-binomial model-based clustering methods were applied as validation. The optimal model and the number of clusters were determined based on connectivity, Dunn’s index, and silhouette width using optCluster package v1.3.0.
Differentially expressed genes and pathways
Differential expression analyses were performed on normalized read counts of samples by nonparametric Wilcoxon rank-sum test with the limma package v3.48.3.15 The fold change (FC) was calculated by comparing geometric means (log-average) of each part. The P values were adjusted for multiple comparisons by false discovery rate (FDR) with the Benjamini- Hochberg method. The transcripts with mean expression <10 counts were regarded as not expressed and excluded from the analysis. The marker transcripts of each cluster are defined as those significant differentially expressed with adjusted P value <0.05, and absolute FC >1.5, and mean expression >200 in the higher expressed part.
The marker transcripts were associated with biological function based on biological process GO annotation, using the topGO package v2.38.1.16 The analysis used all genes in RNAseq data (mapped to 27,053 unique gene IDs) as background, with Fisher’s exact test, and eliminating local similarities and dependencies between GO terms. The GO terms that were over-represented in the cluster analysis as top 3 most significant ones, with a P value <0.0005, and covering at least 2 genes, were selected as most prominent biological processes related to a cluster. The marker transcripts were plotted to a heatmap and grouped by hierarchical clustering, using ComplexHeatmap package v2.8.0.17 The pathways of interest were mapped with the results from differential expression analyses and visualized with the WikiPathways App, or in-house created map, in Cytoscape v 3.8.2.18
Genotype and clinical phenotype association analyses for the RNA-seq clusters
All statistical analyses were performed in the statistical programming language R (version 4.1.0). The genotype and clinical parameters of each cluster, including both categorical variables and quantitative variables, were analyzed in an over-representation analysis by the v-test with the FactoMineR package v2.4. The quantitative variables were log transformed before analyses. Clinical variables are presented using the mean ± SD or median with 25th to 75th percentiles (Q1-Q3) for continuous variables and count (percentage) for categorical variables. The overall associations were tested by chi-square or Kruskal-Wallis tests for categorical and continuous variables, respectively.
Survival analysis
The median follow-up time was 8.4 years (25th-75th percentiles: 5.9-12.2 years). Information about the occurrence of adverse events at follow-up was retrieved from the hospital medical records, the Dutch Personal Records Database, or telephone contact with the patient or their general practitioners. We collected information regarding 4 different adverse events: death caused by cardiovascular disease; heart transplantation or left ventricular assist device (LVAD) implantation; heart failure that required a nonelective hospitalization, despite optimal heart failure therapy, according to the ESC, American College of Cardiology (ACC), and American Heart Association (AHA) guidelines; and life-threatening arrhythmias (LTAs), defined as nonfatal ventricular fibrillation (with or without ICD-shock) or sustained ventricular tachycardia with appropriate ICD shock. The combined endpoint was defined as the occurrence of at least 1 of these adverse events. Patients who died of noncardiac causes were censored at the moment of death (n = 2); those patients did not reach the combined endpoint.
Kaplan-Meier survival curves were estimated and differences among groups were assessed by the log-rank test, using time of diagnosis as time zero. Calculations were done using R environment version 3.5 (R Foundation).
Results
Patient cohort
RNA was isolated from cardiac biopsies of 82 patients with DCM included in the Maastricht Cardiomyopathy Registry (mCMP-registry). The mean age of diagnosis was 55 years (SD: 12; range: 26-83). Thirty-two percent (26 of 82) of probands reported family histories of DCM. Seventy-three percent (60 of 82) of the patients were male. The average ejection fraction was 28% ± 9% ( range: 10%-47%), with a mean indexed ventricular end-diastolic diameter of 31 ± 4 mm/m2 (range: 23-47 mm/m2). Fifty percent of patients had genetic variants in DCM-associated genes (41 of 82). The most prevalent pathogenic gene variant was a truncating variant in TTN, which was detected in 31% (26 of 82), followed by LMNA (7 of 82), RBM20 (5 of 82), MYH7 (2 of 82), TNNT2 (1 of 82), and DES (1 of 82). Patients with pathogenic variants in genes other than TTN or LMNA were clustered together in a group referred to as "other genetic."
Clustering based on transcriptome
The top 2,000 transcripts of 708 unique genes were selected as input for the clustering analysis (Figure 1A). The dimensionality reduction combined with the graphic-based clustering based on UMAP identified 4 distinct clusters in the transcriptomic data, in which cluster 1-2 and 3-4 shared strong similarities (Figure 1B). Despite cluster 1-2 and 3-4 shared (mathematical) similarities, the individual clusters still reflect biological differences, which makes it valuable to proceed with 4 clusters. The final clustering was validated using OptCluster, in which the suggested superclusters showed a Jaccard similarity coefficient of 0.92 (Supplemental Figure 1). We did not observe a complete clustering based on genotype, confirmed by the Jaccard similarity coefficient of 0.22 (Figure 1D). Most patients with TTNtv were identified in cluster 1, although each cluster contained a number of patients with TTNtv. In contrast, cluster 4 was strongly enriched with patients who did not have proven genetic etiologies. Interestingly, most of the patients with genetic DCM with variants in a different gene than TTN were also grouped in cluster 3 and 4 based on their RNA profiles (Figure 1C).
Figure 1.

Clustering of Patients Based on Transcriptomic Profile
(A) Dispersion plot of RNA sequencing data. The top 2,000 transcripts were selected as input for the clustering analysis based on expression in the number of samples, base mean reads, and the dispersion of the reads among samples. (B) Uniform manifold approximation and projection (UMAP) plot identified 4 clusters. (C) The association between genotypes and RNA-seq clusters. (D) UMAP plot of samples labeled with genotype.
Gene expression profile of clusters and superclusters
In total, 3,513 unique genes were distinct markers for 1 of the 4 clusters, and 1,738 genes for superclusters (Supplemental Table 1, Supplemental Figure 2). The genotype had a strong influence on the distinct RNA-profile of cluster 1 and 2 (mainly TTNtv), although the genotype does not explain the complete RNA-profile (Figure 2, Supplemental Figure 3).
Figure 2.

Heatmap of Significant Differentially Expressed Genes per Cluster in Association With Genotype and Phenotype
The enriched biological process GO items in each gene group were summarized using biological blocks identified in the clusters. Samples were annotated to genotypes and important clinical parameters.
Biological function of RNA clusters
Pathway analysis was performed for both the 4 individual clusters and the 2 superclusters using biological process GO annotations, which were collectively grouped according to biological function (Figure 3). Cluster 1-2 is characterized by a strong upregulation of mitochondrial energetics and organization, and gene expression with decreased post-translational modifications (transcription and translation). Cluster 3-4 is characterized by active cardiomyocyte function and morphology, and extracellular matrix remodeling and signaling. The top 5 overexpressed genes in cluster 1-2 are all involved in mitochondrial function and response to oxidative stress (Supplemental Figure 2B). The top 5 overexpressed genes in cluster 3-4 are mainly related to the protein phosphorylation and signal transduction, cytoskeleton, and Golgi transport.
Figure 3.

Top GO Items of Each Cluster Grouped to Biological Functions
Results are displayed per cluster and the comparison between the 2 superclusters.
Important cardiac metabolic pathways related to mitochondrial function
Mitochondrial function appeared to be the most distinct upregulated feature of supercluster 1-2, including oxidative phosphorylation and biogenesis. The expression of the housekeeping gene involved in mitochondrial biogenesis (SSBP1) was significantly increased in cluster 1-2 (adjusted P = 5.2 × 10-10), showing an increase of more than 150% compared with cluster 3-4.
We further focused on the major energetic pathways in the heart: glucose uptake and glycolysis; lipid uptake, storage, and beta-oxidation;19,20 tricarboxylic acid (TCA) cycle; and the electron transport chain (Supplemental Figure 4). Both cluster 1 and cluster 2 showed a strong upregulation of the electron transport chain (Supplemental Figure 4D, Supplemental Figure 5D) but had distinct differences in the other metabolic pathways. In cluster 1, the glycolysis pathway was upregulated, but the malate-aspartate shuttle that is necessary in the heart to bring electrons produced in glycolysis to the mitochondria was downregulated (Supplemental Figure 4A). Also the step from pyruvate to acetyl-CoA and the complete TCA cycle was downregulated (Supplemental Figure 4C). Lipid uptake and beta-oxidation in both mitochondria and peroxisome were strongly downregulated (Supplemental Figure 4B). The uncoupling proteins UCP3 to 5, which are necessary to limit the concentration of reactive oxygen species (ROS), were strongly upregulated (adjusted P = 1.6 × 10-6) (Supplemental Figure 4). In contrast to cluster 1, the malate-aspartate shuttle, the beta-oxidation and TCA cycle were strongly upregulated in cluster 2 (Supplemental Figure 5). In addition, although genes in sarcomere organization were downregulated in patients from clusters 1 (Figure 3, Supplemental Figure 6), other structural sarcomere genes were strongly upregulated in cluster 1 and 2, such as troponin and tropomyosin (Supplemental Figure 6). The observed discrepancy in sarcomere genes and metabolic pathways between cluster 1 and 2 might imply a complex crosstalk between sarcomere function, mitochondria, and other metabolic pathways.
Association among clinical phenotype, genotype, and clusters
Next, we tested for differences in 53 clinical parameters among clusters. In this analysis, differences in LV mass, LV stroke volume, N-terminal pro b-type natriuretic peptide (NT-proBNP), New York Heart Association (NYHA) functional class and genotype characterized the generated clusters (Supplemental Table 2). All patients in cluster 1 had genetic etiologies, of which TTNtv was the most prevalent, and a significant lower LV mass and stroke volume compared with the other clusters (Figure 4, Supplemental Table 2). Patients from cluster 2 all were in NYHA class I-II, and patients in cluster 3 had the lowest NT-proBNP. Cluster 4 was characterized by an enrichment of patients with no genetic variant associated with DCM.
Figure 4.

Association Among Phenotype, Genotype, and Clusters Based on Cardiac Transcriptome
(A) Over-representation analysis showing the most distinct clinical variables per cluster (v-test). Only significant variables with P values <0.05 are listed. (B) Details of significant variables by grouped bar plot, or violin plot + box plot for categorical variables and quantitative variables, respectively.
There was no significant association between the clusters and the combined endpoint (cardiovascular death, heart transplantation, heart failure hospitalization, or a life-threatening arrhythmia; log-rank P = 0.20) (Figure 5). The 2 opposite clusters (1 and 4) were trending to have a worse outcome compared with the intermediate clusters (2 and 3). In total, 26 patients reached the combined endpoint; 2 additional patients died of noncardiac causes.
Figure 5.

Event-Free Survival Stratified by Transcriptomic Cluster
Kaplan-Meier curve for the combined outcome of life-threatening arrhythmias, cardiovascular death, heart transplantation, or heart-failure hospitalization stratified by transcriptomic cluster.
Validation of the clustering methodology in a cohort of patients with end-stage DCM
We validated the unsupervised clustering technique by applying it on RNA-sequencing data from hearts of patients with end-stage DCM. Compared with our cohort, these patients had lower LVEF and higher prevalence of atrial fibrillation (AF) (Supplemental Table 3). Three distinct clusters were identified in the transcriptomic dataset (Figure 6). The RNA expression profile of cluster 3 was the complete opposite from patients in cluster 1 and 2 (Figure 6C). Pathway analysis showed that the biological background of cluster 3 was very similar to the transcriptomic profile of cluster 1 in our early-stage DCM cohort: a strong upregulation of mitochondrial energetics and gene expression with decreased post-translational modification (transcription and translation) (Figure 6, Supplemental Figure 7). The top 5 overexpressed genes in cluster 3 are all mainly related to protein degradation and synthesis (Supplemental Figure 8). Cluster 2 formed an intermediate cluster, which was characterized by increased expression of genes associated with the inflammatory response and innate immune system. Only sex was strongly associated with the identified clusters; female patients were significantly over-represented in cluster 2 (Figure 6, Supplemental Figure 9). None of the other clinical variables was significantly associated with any of the clusters. Overall, we could successfully apply the clustering method in a cohort of patients with end-stage DCM to identify biologically relevant subgroups.
Figure 6.

Clustering of Patients With End-Stage Dilated Cardiomyopathy Based on Transcriptomic Profile
(A) UMAP plot identified 3 clusters. (B) UMAP plot of samples labeled by sex. (C) Heatmap of significant differentially expressed genes per cluster in association with phenotypic characteristics below. The enriched biological process GO items in each gene group were summarized using biological blocks identified in the clusters on the left. Abbreviation as in Figure 1.
Discussion
We explored the potential of the cardiac transcriptomic profile to recognize distinct clinical relevant patient subgroups. In a well-phenotyped cohort of patients with early-stage DCM, 4 molecular clusters were identified, which could be roughly divided into 2 superclusters, showing differences in underlying biological pathways and genotype—mainly TTNtv—with mild differences in phenotype. In a cohort of end-stage DCM, 3 molecular clusters were identified, showing differences in biological pathways, partly explained by sex differences.
Identified biological pathways in patients with dilated cardiomyopathy
The activity of molecular pathways in the heart is a multifactorial process, influenced by intrinsic and extrinsic stressors on the myocardium.2 The generated clusters in the early-stage DCM cohort are associated with the presence of a pathogenic variant in a DCM-associated gene, which partly explains the distinct transcriptomic profiles. Truncating variants in TTN are the most prevalent and also have an important contribution to the generated patient clusters, as shown in the over-representation analysis (Figure 4). A previous report described major differences in the transcriptomic profile between end-stage failing DCM hearts and nonfailing hearts.21 In contrast to our findings, the transcriptome could not differentiate between patients with DCM with and without TTNtv. The cardiac tissue from terminally failing hearts reflect the pathophysiology of advanced HF and masks the early differences in the pathogenicity of TTNtv, which we observed in our early-stage cardiac biopsies. Titin is a major structural protein of the sarcomere and also fulfills an important signaling function in the cardiomyocyte.22 A combined disease mechanism of both haploinsufficiency and a poison peptide-dominant negative concept is suggested in TTNtv-associated DCM.21,22 The presence of truncated titin proteins in the cardiomyocyte is a huge burden for the ubiquitin-proteasome system (UPS), which mediates the degradation of truncated titin.23 The continuous degradation will saturate the UPS mechanism of the cell, contributing to HF in patients with TTNtv DCM in time. We observed a dysregulation in protein degradation pathways (UPC, autophagy) in cluster 1, which is necessary for a high turnover of the large sarcomeric proteins in the heart muscle (Supplemental Figures 10 and 11). The importance of proteostasis in the development of DCM is further highlighted by loss-of-function variants in BAG3 and BAG5, both important chaperones in proteostasis, which lead to severe DCM.24,25 Disturbance or saturation of the proteostasis process seems to be a turning point toward development of DCM, which is especially observed in cluster 1. This might also explain the over-representation of patients with TTNtv in this cluster. Proteostasis is an energy- demanding process, which requires a large percentage of the cells' ATP, and the observed increase in mitochondrial pathways in cluster 1 could be a secondary consequence to meet the increased energy demand.
Cardiac energy metabolism as center of the disease
A continuous generation of energy is essential for the heart to maintain contractile function.26 The healthy heart is flexible and can use any substrate to generate energy. The loss of substrate flexibility is one of the first alterations toward HF and represents a common change in heart disease independent of etiology. As an adaptive response, the heart changes toward glycolysis, which is more efficient compared with lipid oxidation.27 The timing of metabolic adaptation can differ per etiology. For example, metabolic adaptation will occur in an earlier stage in patients with TTNtv.12,28,29 This does not exclude that additional factors can aggravate onset of disease and accelerate the metabolic remodeling.30,31 Although cluster 1 and 2 formed a supercluster, the main difference between the clusters was related to the metabolic substrate pathways: cluster 2 had an increased activity of the beta-oxidation, whereas cluster 1 had higher expression of mitochondrial uncoupling proteins, possibly linked to a higher lipotoxicity or stress conditions.19 The significant differences in cardiac metabolism could indicate 2 different stages of metabolic remodeling and thus different stages of disease in which cluster 2 has still more reserve to cope with the energy demand of the cardiomyocyte and showed milder clinical symptoms and a better survival compared with cluster 1. In addition, we observed differences in the genes involved in sarcomere function and organization between cluster 1 and 2, indicating a link between the energy metabolism and sarcomere function of the heart in different stages of the disease. However, based on our data, we cannot infer a direct causal relationship between sarcomere and metabolic dysfunction.
Clinical relevance
Our study underpins the heterogeneity of dilated cardiomyopathy, both in early- and end-stage disease. The cardiac transcriptome is influenced by genotype and phenotype, which are strongly correlated, as we know from described genotype-phenotype associations.32 We reveal 4 molecular clusters based on cardiac transcriptomic profile in early-stage DCM, in which cluster 1 and 4 reflect 2 opposites and cluster 2 and 3, 2 intermediates (Figure 2). The clusters were not associated with prognosis, although there was a trend that cluster 1 and 4 had worse prognoses compared with the intermediate clusters. In addition, we describe 3 identified clusters in the cohort of patients with end-stage DCM. These clusters illustrate that the clinical parameters to determine heart function reflect a small part of the pathophysiological processes driving progression of heart failure (eg, also patients with early-stage DCM with a mildly reduced ejection fraction were clustered in group 1, and only female sex was distinct for a cluster in the end-stage DCM cohort. Also, the genetic information partially influences the transcriptomic profile in addition to the clinical parameters. The cardiac transcriptome profile forms a unique entity, providing complementary information to the genotype and phenotype to recognize patient subgroups (Figure 7). A post hoc genetic analysis in the POSEIDON-DCM (Percutaneous Stem Cell Injection Delivery Effects on Neomyogenesis in Dilated Cardiomyopathy) trial showed that the genotype contributes to the response of treatment in the trial, probably caused by differences in underlying pathophysiological processes in the heart.33 As hypothesized earlier, proteostasis and metabolic adaptation are 2 key (coping) mechanisms in the development of DCM. As an example, the presence of a pathogenic TTN variant can drive the heart into metabolic adaptation irrespective of LVEF, mimicking metabolic changes as seen in patients with advanced HF. The presence of a truncated titin protein is a burden on the protein quality-control pathways. Proteasome inhibition has been suggested as potential therapy for patients with TTNtv.23 We show that the activity of the processes implicated in protein control vary per patient with TTNtv, indicating that not every patient will benefit equally from the treatment. It is important to note that the generated clusters are not static but reflect the dynamics of the disease processes. The RNA isolated from the cardiac tissue reflects a snapshot in the disease course of a patient. Although the transcriptome elegantly shows the activity of gene expression, additional omics (eg, proteomics or ribo-seq) are necessary to measure the actual protein turnover. Circulating biomarkers have the potential as a noninvasive measure of process activity (eg, proteostasis) in the heart, which could help to guide treatment of the individual patient (Figure 7).
Figure 7.

Integration of the Cardiac Transcriptomic Profile in Association With the Genotype and Phenotype
The cardiac transcriptome is unique per individual patient and is influenced by the genotype and phenotype of the patient. The transcriptome reflects the activity of (patho)physiological mechanisms, which possibly is reflected in biomarkers in the blood. Such biomarker profile could be used to help guide the individual patient's treatment in selecting the right medication and timing it at the best moment in the disease stage.
Study limitations
The patient cohort was biased by patients with genetic cardiomyopathies (61%) in which patients with TTNtv were over-represented (31%). We used publicly available RNA-sequencing data of hearts from patients with end-stage DCM. In contrast to our cohort, the clinical dataset was very limited. There was no genotype information available, which prevented us from making any correlations between the identified clusters and genotype in the patients with end-stage disease.
Conclusions
Four distinct molecular clusters were identified in a cohort of patients with early-stage DCM, based on differences in protein quality control, cardiac metabolism, and cardiomyocyte function. In a cohort of patients with end-stage DCM, 3 clusters were identified based on mitochondrial energetics and inflammatory pathways. The cardiac transcriptome profile forms a unique entity, which has the potential to determine disease stage and guide future treatment, complementary to the genotype and phenotype.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Patients with dilated cardiomyopathy and a comparable activity of disease processes in the heart, as reflected in the cardiac transcriptome, do not always have the same clinical phenotype, probably because of differences in metabolic adaptation capacity. This might explain why not every patient responds adequately to given therapy, based on clinical parameters.
TRANSLATIONAL OUTLOOK 1: The cardiac transcriptome is currently determined via RNA gathered from cardiac biopsies, which are obtained through an invasive procedure. Circulating biomarkers have the potential as a noninvasive measure of cardiac process activity. Future studies should combine the cardiac transcriptome with circulating biomarkers to associate serum markers with the cardiac process activity.
TRANSLATIONAL OUTLOOK 2: The addition of the cardiac transcriptome to the genotype and phenotype of the patient will create a more individualized characterization of a patient. Future studies should determine how a better patient characterization would lead to a better response to medication for heart failure.
Funding Support and Author Disclosures
The research leading to these results has received funding from the DCVA DOUBLE DOSE grant. We acknowledge the support from the Netherlands Cardiovascular Research Initiative, an initiative with support of the Dutch Heart Foundation and CVON Arena-PRIME, 2017-18. Drs Verdonschot and Nabben are supported by a Dutch Heart Foundation grant. Heymans receives personal fees for scientific advice to Astra-Zeneca, Pfizer, Novo Nordisk and CSL Behringer. All other authors have reported that they have no relationships relevant to the contents of this paper.
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
Appendix
For supplemental tables and figures, please see the online version of this paper.
Appendix
References
- 1.Japp A.G., Gulati A., Cook S.A., Cowie M.R., Prasad S.K. The diagnosis and evaluation of dilated cardiomyopathy. J Am Coll Cardiol. 2016;67:2996–3010. doi: 10.1016/j.jacc.2016.03.590. [DOI] [PubMed] [Google Scholar]
- 2.Verdonschot J.A.J., Hazebroek M.R., Ware J.S., Prasad S.K., Heymans S.R.B. Role of targeted therapy in dilated cardiomyopathy: the challenging road toward a personalized approach. J Am Heart Assoc. 2019;8 doi: 10.1161/JAHA.119.012514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verdonschot J.A.J., Merlo M., Dominguez F., et al. Phenotypic clustering of dilated cardiomyopathy patients highlights important pathophysiological differences. Eur Heart J. 2021;42:162–174. doi: 10.1093/eurheartj/ehaa841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verdonschot J.A.J., Derks K.W.J., Hazebroek M.R., et al. Distinct cardiac transcriptomic clustering in titin and lamin A/C-associated dilated cardiomyopathy patients. Circulation. 2020;142:1230–1232. doi: 10.1161/CIRCULATIONAHA.119.045118. [DOI] [PubMed] [Google Scholar]
- 5.Verdonschot J.A.J., Hazebroek M.R., Krapels I.P.C., et al. Implications of genetic testing in dilated cardiomyopathy. Circ Genom Precis Med. 2020;13:476–487. doi: 10.1161/CIRCGEN.120.003031. [DOI] [PubMed] [Google Scholar]
- 6.Jansweijer J.A., Nieuwhof K., Russo F., et al. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur J Heart Fail. 2016;19:512–521. doi: 10.1002/ejhf.673. [DOI] [PubMed] [Google Scholar]
- 7.Anderson J.L., Christensen G.B., Escobar H., et al. Discovery of TITIN gene truncating variant mutations and 5-year outcomes in patients with nonischemic dilated cardiomyopathy. Am J Cardiol. 2020;137:97–102. doi: 10.1016/j.amjcard.2020.09.026. [DOI] [PubMed] [Google Scholar]
- 8.Pinto Y.M., Elliott P.M., Arbustini E., et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37:1850–1858. doi: 10.1093/eurheartj/ehv727. [DOI] [PubMed] [Google Scholar]
- 9.Richardson P., McKenna W., Bristow M., et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 10.McDonagh T.A., Metra M., Adamo M., et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021;42:3599–3726. doi: 10.1093/eurheartj/ehab368. [DOI] [PubMed] [Google Scholar]
- 11.Verdonschot J.A.J., Merken J.J., van Stipdonk A.M.W., et al. Cardiac inflammation impedes response to cardiac resynchronization therapy in patients with idiopathic dilated cardiomyopathy. Circ Arrhythm Electrophysiol. 2020;13 doi: 10.1161/CIRCEP.120.008727. [DOI] [PubMed] [Google Scholar]
- 12.Verdonschot J.A.J., Hazebroek M.R., Derks K.W.J., et al. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J. 2018;39:864–873. doi: 10.1093/eurheartj/ehx808. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y., Morley M., Brandimarto J., et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 2015;105:83–89. doi: 10.1016/j.ygeno.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khodadi-Jamayran A., Tsirigos A. Graph drawing-based dimensionality reduction to identify hidden communities in single-cell sequencing spatial representation. bioRxiv. 2020 doi: 10.1101/2020.05.078550. [DOI] [Google Scholar]
- 15.Ritchie M.E., Phipson B., Wu D., et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexa A., Rahnenfuhrer J., Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics (Oxford, England) 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 17.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics (Oxford, England) 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 18.Kutmon M., Lotia S., Evelo C.T., Pico A.R. WikiPathways app for cytoscape: making biological pathways amenable to network analysis and visualization. F1000Research. 2014;3:152. doi: 10.12688/f1000research.4254.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopaschuk G.D., Ussher J.R., Folmes C.D., Jaswal J.S., Stanley W.C. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 20.Colasante C., Chen J., Ahlemeyer B., Baumgart-Vogt E. Peroxisomes in cardiomyocytes and the peroxisome/peroxisome proliferator-activated receptor-loop. Thromb Haemost. 2015;113:452–463. doi: 10.1160/TH14-06-0497. [DOI] [PubMed] [Google Scholar]
- 21.McAfee Q., Chen C.Y., Yang Y., et al. Truncated titin proteins in dilated cardiomyopathy. Sci Transl Med. 2021;13 doi: 10.1126/scitranslmed.abd7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabish A.M., Azzimato V., Alexiadis A., Buyandelger B., Knoll R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys Rev. 2017;9:207–223. doi: 10.1007/s12551-017-0265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fomin A., Gärtner A., Cyganek L., et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci Transl Med. 2021;13 doi: 10.1126/scitranslmed.abd3079. [DOI] [PubMed] [Google Scholar]
- 24.Hakui H., Kioka H., Miyashita Y., et al. Loss-of-function mutations in the co-chaperone protein BAG5 cause dilated cardiomyopathy requiring heart transplantation. Sci Transl Med. 2022;14 doi: 10.1126/scitranslmed.abf3274. [DOI] [PubMed] [Google Scholar]
- 25.Domínguez F., Cuenca S., Bilińska Z., et al. Dilated cardiomyopathy due to BLC2-associated athanogene 3 (BAG3) mutations. J Am Coll Cardiol. 2018;72:2471–2481. doi: 10.1016/j.jacc.2018.08.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ussher J.R., Elmariah S., Gerszten R.E., Dyck J.R. The emerging role of metabolomics in the diagnosis and prognosis of cardiovascular disease. J Am Coll Cardiol. 2016;68:2850–2870. doi: 10.1016/j.jacc.2016.09.972. [DOI] [PubMed] [Google Scholar]
- 27.Heggermont W.A., Papageorgiou A.P., Heymans S., van Bilsen M. Metabolic support for the heart: complementary therapy for heart failure? Eur J Heart Fail. 2016;18:1420–1429. doi: 10.1002/ejhf.678. [DOI] [PubMed] [Google Scholar]
- 28.Ware J.S., Cook S.A. Role of titin in cardiomyopathy: from DNA variants to patient stratification. Nat Rev Cardiol. 2018;15:241–252. doi: 10.1038/nrcardio.2017.190. [DOI] [PubMed] [Google Scholar]
- 29.Schafer S., de Marvao A., Adami E., et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49:46–53. doi: 10.1038/ng.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Pavia P., Kim Y., Restrepo-Cordoba M.A., et al. Genetic variants associated with cancer therapy-induced cardiomyopathy. Circulation. 2019;140:31–41. doi: 10.1161/CIRCULATIONAHA.118.037934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ware J.S., Amor-Salamanca A., Tayal U., et al. Genetic etiology for alcohol-induced cardiac toxicity. J Am Coll Cardiol. 2018;71:2293–2302. doi: 10.1016/j.jacc.2018.03.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kayvanpour E., Sedaghat-Hamedani F., Amr A., et al. Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol. 2016;106:127–139. doi: 10.1007/s00392-016-1033-6. [DOI] [PubMed] [Google Scholar]
- 33.Rieger A.C., Myerburg R.J., Florea V., et al. Genetic determinants of responsiveness to mesenchymal stem cell injections in non-ischemic dilated cardiomyopathy. EBioMedicine. 2019;48:377–385. doi: 10.1016/j.ebiom.2019.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.