

Abstract
Lipids have been found to modulate tumor biology, including proliferation, survival, and metastasis. With the new understanding of tumor immune escape that has developed in recent years, the influence of lipids on the cancer–immunity cycle has also been gradually discovered. First, regarding antigen presentation, cholesterol prevents tumor antigens from being identified by antigen presenting cells. Fatty acids reduce the expression of major histocompatibility complex class I and costimulatory factors in dendritic cells, impairing antigen presentation to T cells. Prostaglandin E2 (PGE2) reduce the accumulation of tumor-infiltrating dendritic cells. Regarding T-cell priming and activation, cholesterol destroys the structure of the T-cell receptor and reduces immunodetection. In contrast, cholesterol also promotes T-cell receptor clustering and relative signal transduction. PGE2 represses T-cell proliferation. Finally, regarding T-cell killing of cancer cells, PGE2 and cholesterol weaken granule-dependent cytotoxicity. Moreover, fatty acids, cholesterol, and PGE2 can improve the activity of immunosuppressive cells, increase the expression of immune checkpoints and promote the secretion of immunosuppressive cytokines. Given the regulatory role of lipids in the cancer–immunity cycle, drugs that modulate fatty acids, cholesterol and PGE2 have been envisioned as effective way in restoring antitumor immunity and synergizing with immunotherapy. These strategies have been studied in both preclinical and clinical studies.
KEY WORDS: Lipids, Fatty acids, Cholesterol, Prostaglandin E2, Tumor immune escape, Cancer–immunity cycle, Immunotherapy, Combination therapy
Graphical abstract
Lipids regulate cancer immunity cycle by affecting antigen presentation, T-cell priming and activation and T-cell killing of tumor, which in turn promotes immune surveillance escape.
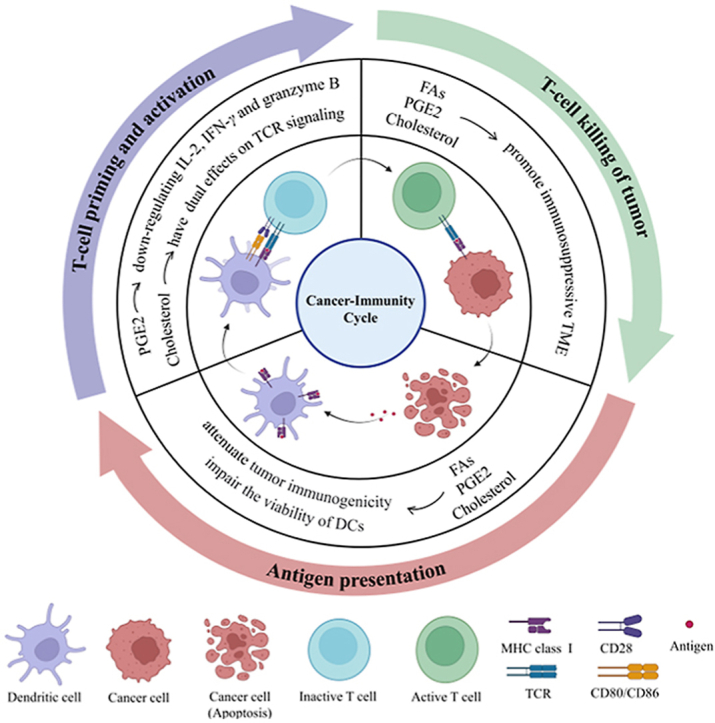
1. Introduction
Lipids comprise a complex group of biomolecules that differ in structure and function, and they have been classified into eight categories as follows: fatty acids (FAs), glycerolipids, glycerophospholipids, sphingolipids, sterols, prenols, saccharolipids, and polyketides1,2. Lipids execute important physiological roles in energy storage and the modulation of membrane fluidity, as well as various in trafficking and signaling events3, 4, 5, 6, 7. Among the various categories of lipids, FAs, cholesterol, and prostaglandins, which are arachidonic acid derivatives, have been shown to be critical in regulating tumor proliferation, survival, and metastasis8, 9, 10, 11. Increased fatty acid oxidation (FAO) and de novo lipid synthesis often occur in tumor and provide extra energy for tumor development and progression12. Prostaglandin E2 (PGE2) enhances intestinal adenoma growth via activation of the Ras-mitogen-activated protein kinase cascade13. PGE2 can cause decreased expression of programmed cell death in human colonic cancer cells by increasing NF-κB expression14. Cholesterol content within specific membrane regions can affect extrinsic (death receptor pathway) and intrinsic (mitochondrial) apoptotic pathways15. Moreover, cancer cells promote migration and invasion by balancing FA saturation levels16. With increased levels of cyclooxygenase-2 (COX-2)/PGE2, cancer cells become more migratory during epithelial–mesenchymal transition17,18.
In addition to its role in mediating biological characteristics of tumor cells, a large amount of evidence indicates that lipids alter the function and status of immune cells in the tumor microenvironment (TME)19, 20, 21, 22. Lipids, especially cholesterol, FAs and prostaglandins, can affect any part of the cancer immune cycle, thereby disrupting normal antitumor immunity (Fig. 1). Regarding antigen presentation, cholesterol changes the formation of tumor-associated antigens (TAAs), which enables tumor cells to escape immune surveillance23. FAs, especially saturated palmitic acid and short chain fatty acids (SCFAs), inhibit antigen presentation. They decrease the expression of major histocompatibility complex class I (MHC I) and costimulatory factor24,25. During T-cell priming and activation, cholesterol keeps T-cell receptors (TCR) in a resting conformation by binding to the TCRβ transmembrane region26. In contrast, cholesterol facilitates the formation of a larger TCR signalosome and promote T-cell signal transduction as part of membrane rafts27. In addition, PGE2 inhibits T-cell activation and cytotoxicity by downregulating interleukin 2 (IL-2), interferon-γ (IFN-γ) and granzyme B28, 29, 30. Regarding T-cell killing of cancer cells, FAs enhance the immunosuppressive function of myeloid-derived suppressor cells (MDSCs) and PGE2 promotes the infiltration of regulatory T cells (Tregs)31,32. Furthermore, FAs and PGE2 upregulate extrinsic immunosuppressive cytokines, such as interleukin 10 (IL-10) and transforming growth factor-β (TGF-β)33,34. FAs and cholesterol can also promote the expression of immune checkpoints34,35. Taken together, these data suggest that the abnormal accumulation of lipids hinders the establishment of a complete immune cycle and helps to establish an immunosuppressive environment. Strategies that decrease the level of lipids can improve antigen presentation, promote T-cell priming and activation and disrupt the immunosuppressive state.
Figure 1.

Lipids mediate the cancer–immunity cycle. The complete adaptive immune cycle is crucial for tumor immunotherapy, and mainly includes the following three components: (1) antigen presentation, (2) T-cell priming and activation, and (3) T-cell killing of tumors. Lipids, especially fatty acids, cholesterol, and prostaglandins, mainly have a negative effect on the cancer–immunity cycle. Combination drugs of modulating lipids with immunotherapy methods can potentially further improve the antitumor immune response in patients.
FAs, cholesterol, and PGE2 exhibit negative effects in the adaptive immune cycle. Therefore, recent preclinical experiments and clinical trials have tested the combination of lipid-based therapy and immunotherapy as a promising cancer treatment strategy36, 37, 38, 39, 40. The aim is to block FA synthesis and absorption through methods including the inhibition of the activities of fatty-acid synthase (FASN), fatty acid transport protein 2 (FATP2), and peroxisome proliferator-activated receptors (PPARs). PPARs are molecular sensors of FA and play a crucial role in regulating FAO and FA storage41. Statins are the most common pharmacological intervention option for cholesterol regulation. They are inhibitors of hydroxyl methylglutaryl-coenzyme-A (HMG-CoA), the key catalytic enzymes in the rate-limiting step of cholesterol biosynthesis42. PGE2 synthesis is governed by COX-2, and PGE2 interacts with the E prostanoid receptor (EPR) to activate downstream signals. Inhibitors of COX-2 and EPR can modulate the content and function of PGE2.
Given the importance of the complete immune cycle in the suppression of tumors and the profound impact of lipids on the immune system, here, we present a review describing how lipids affect the cancer adaptive immunity cycle and how lipid-based therapy regulates immune responses in cancer patients treated with immunotherapy. Moreover, we discuss potential innovative strategies for targeting immune cells with the modulation of lipids to develop potential combination immunotherapy targets.
2. Lipid-mediated cancer–immunity cycle: Cancer antigen presentation
Antigen presentation mainly includes two distinct events: tumor antigens are displayed on the cancer cell surface, and antigen presenting cells (APCs) take up antigens and cross-present to T cells. Lipids can act as TAAs directly to affect the immune response during initial step. Lipids can also modulate antigen presentation through APCs (Fig. 2).
Figure 2.

Lipids reshape tumor microenvironment. Lipids mainly regulate cancer cells, immune cells, and cytokines to form an immunosuppressive tumor microenvironment, which inhibits the immune response and promotes tumor immune evasion. In cancer cell plasma membranes, cholesterol binds to glycosphingolipids, a kind of TAA, to change their conformation so that they cannot be recognized. Moreover, FAO-derived acetyl-CoA upregulates CD47 transcription in glioblastoma multiforme to weaken macrophage phagocytosis. FASN stabilizes the PD-L1 protein by promoting PD-L1 palmitoylation. In DCs, saturated palmitic acid significantly reduces MHC I expression and lowers the conjugation of APCs and T cells and SCFA reduces the expression of costimulatory molecules CD80 and CD86 on DCs and ICOS on T cells during anti-CTLA4 treatment. Increased FA uptake reduces the ability of DCs to process and present antigens and increases the expression of PD-L1. PGE2 reduces the accumulation of TIDCs, which inhibits antigen uptake. In MDSCs, FATP2 promotes the production of PGE2, which causes the expansion of polymorphonuclear MDSCs and further suppression of the antigen-specific T-cell response. Moreover, FATP2-induced lipid accumulation generates ROS and promotes the immunosuppressive effects of MDSCs on T cells. FAO promotes the immunosuppressive function of tumor-infiltrating MDSCs. In T cells, cholesterol increases immune checkpoint expression (e.g., PD-1, 2B4, TIM-3, and LAG-3) and regulates TCR signaling. PGE2 inhibits T-cell proliferation and decreases IL-2 receptor expression in T cells to inhibit the early step of T-cell activation. In regulatory T cells, de novo fatty-acid synthesis mediated by FASN contributes to the functional maturation of Tregs, and COX-2 expression levels are positively correlated with the levels of tumor-infiltrating Foxp3+ Tregs. In macrophages, cholesterol and PGE2 induce macrophages to polarize to M2 TAM. For cytokines secreted by immune cells, lipids not only promote the secretion of immunosuppressive cytokines, such as IL-10 and TGF-β, but also inhibit the secretion of cytotoxic cytokines, such as TNF-α, IFN-γ, and granzyme B. Abbreviations: TAA, tumor-associated antigen; FAO, fatty acid oxidation; PD-L1, programmed cell death ligand 1; DC, dendritic cell; TIDC, tumor-infiltrating dendritic cell; Treg, regulatory T cell; MHC I, major histocompatibility complex class I; APC, antigen presenting cell; SCFA, short-chain fatty acid; ICOS, inducible co-stimulator; CTLA4, cytotoxic T lymphocyte antigen 4; FA, fatty acid; PGE2, prostaglandin E2; MDSCs, myeloid-derived suppressor cells; FATP2, fatty acid transport protein 2; PD-1, programmed cell death 1; TIM-3, T-cell immunoglobulin and mucin-domain containing-3; LAG-3, lymphocyte activation gene-3; IL-2, interleukin-2; IL-10, interleukin-10; TNF-α, tumor necrosis factor α; TGF-β, transforming growth factor-β; FASN, fatty-acid synthase; TCR, T-cell receptor; M1 TAM, M1 tumor-associated macrophage; M2 TAM, M2 tumor-associated macrophage; COX-2, cyclooxygenase-2; IFN-γ, interferon-γ; TME, tumor microenvironment; CoA, coenzyme-A.
Glycosphingolipids (GSLs) are widely present on tumor cells and cancer stem cells as a kind of TAAs that elicit an immune response in the host43,44. In cancer cell plasma membranes, cholesterol can bind to GSLs to reorient GSL carbohydrate to a membrane parallel, rather than perpendicular conformation44. Therefore, the GSL–cholesterol complex restricts tumor-associated GSL immunoreactivity and leads to ineffective immune surveillance and passive immunity in various tumors, such as breast carcinoma, prostate cancer, and colon carcinoma44.
DCs are the most potent APCs and initiate the majority of adaptive immune responses, but DCs in tumors usually exert immunosuppressive phenotypes. Saturated palmitic acid treatment was found to significantly reduce MHC I expression and lower APC–T-cell conjugation, which in turn inhibited the ability of APCs to active T cells. However, continued monounsaturated oleic acid treatment can normalize antigen presentation by sequestering palmitic acid into triglyceride-rich lipid droplets24. The study showed that saturated and unsaturated FAs exhibit differential regulation of antigen presentation and costimulatory molecule expression, so selective inhibition of FAs is needed. In addition, SCFAs, as butyrate and propionate, reduce the anti-cytotoxic T lymphocyte antigen 4 (CTLA4)-induced increase in the levels of the costimulatory molecules CD80 and CD86 on DCs and the inducible co-stimulator (ICOS) on T cells, which attenuates the therapeutic effect of anti-CTLA4 in metastatic melanoma25. Due to the increased FA uptake mediated by macrophage scavenger receptor 1, triglyceride levels are elevated in peripheral blood DCs in non-small lung cancer and renal cell carcinoma patients. DCs with increased levels of triglycerides exhibit impaired capacity to process and present TAAs, and a significantly lower ability to stimulate T cells, but the mechanism is not clear45. Other studies revealed that DCs containing high lipid levels not only exhibited lower MHC expression, but also lower costimulatory expression than DCs containing relatively low lipid level46. In cancer stem cells, Arf1 ablation causes the inhibition of lipolysis, which facilitates the engulfment of tumor-associated antigens on dendritic cells (DCs) and promotes DC antigen presentation, thereby enhancing T-cell infiltration and activation. Moreover, Arf1 ablation can efficiently synergize with programmed cell death 1 (PD-1) blockade to inhibit tumor growth47. PGE2, which is derived from arachidonic acid, both reduced the accumulation of tumor-infiltrating dendritic cells (TIDCs), thus reducing antigen uptake, and inhibit TIDC maturation within TME. Such a TME induced CD8+ T-cell tolerance upon migration to the tumor draining lymph nodes in murine renal cell carcinoma28.
Based on the above studies, it is speculated that modifying lipid metabolism can restore the vitality of DCs, which may be a promising anticancer immunotherapy. Blocking lipid synthesis with an acetyl-CoA carboxylase inhibitor restored the capacity of DCs and substantially enhanced the effects of cancer vaccines45. Moreover, the activity of FASN, a key enzyme involved in de novo lipogenesis, can be blocked by cerulenin, which can restore TIDC capacity to extend control of ovarian cancer46.
3. Lipid-mediated cancer–immunity cycle: T-cell priming and activation
TCR signaling and specific cytokines are responsible for T-cell growth, proliferation, and differentiation. Prolonged proliferation of T cells and their differentiation into effector T cells are critical for antitumor immunity48,49. In the TME, T-cell proliferation is impaired, and effector T-cell differentiation and function are altered or prevented48. Here, we discuss how lipids affect T-cell priming and activation in the TME (Fig. 2). By understanding these regulatory mechanisms of lipids, we can reorientate T-cell priming and activation by modulating lipid metabolism.
Previous studies have shown that cholesterol has a contradictory effect on TCR signaling depending on different biological functions. The direct binding motif of cholesterol appears in a variety of membrane proteins so that it can directly bind to membrane proteins, such as metabotropic glutamate receptor, adenosine A1 receptor, and γ-aminobutyric acid receptor50. On the surface of the T-cell membrane, cholesterol also directly modifies the TCR through allosteric signals to alter it into an inactive conformation that cannot be phosphorylated by active kinases51. Cholesterol sulfate, which has a structure similar to cholesterol, can replace the originally bound cholesterol, inhibit the phosphorylation of CD3, destroy the TCR polymer, and effectively inhibit signal transduction26. In contrast, cholesterol plays a positive role in regulating TCR signaling as part of membrane rafts. Yang et al.27 found that by inhibiting cholesterol esterification, plasma membrane cholesterol levels in CD8+ T cells can be increased, resulting in the formation of larger TCR microclusters. This feature is beneficial for improving the avidity of TCRs to tumor antigens and promoting the formation of a larger TCR signalosome. Therefore, acetyl-CoA acetyltransferase one is a key cholesterol esterification enzyme, and its pharmacological inhibitor (avasimibe) was used to treat melanoma in mice with significant antitumor effects. Combination therapy of avasimibe plus anti-PD-1 is superior to monotherapy in repressing melanoma and Lewis lung carcinoma (LLC) progression in mice27. Moreover, blocking cholesterol synthesis with lovastatin, a reversible competitive inhibitor of HMG-CoA, can dose-dependently inhibit CD3-induced T-cell proliferation52.
Prostaglandins inhibit T-cell proliferation and attenuate T-cell cytotoxicity. In the presence of PGE2, when T cells interacted with renal cell carcinoma cells, T-cell proliferation was inhibited, and intracellular IFN-γ expression and cell surface CD28 expression were reduced. The COX-2 inhibitor NS-398 increases cytotoxic T lymphocyte responses to renal cell carcinoma cells28. Moreover, PGE2 results in decreased IL-2 production, IL-2 receptor expression, and responsiveness to exogenous IL-2, which inhibit the early step of T-cell activation30. In addition, T cells primed by PGE2-matured DCs exhibit less granzyme B expression and poor tumor cytotoxicity. This result was also demonstrated in vivo29.
Cholesterol and PGE2 metabolic reprogramming are significantly associated with T-cell priming and activation. Therefore, this strategy is another effective means to increase the activation of T cells to improve the efficacy of existing immunotherapies.
4. Lipid-mediated cancer–immunity cycle: T-cell killing of tumor
The majority of T cells in the tumor microenvironment are depleted with an increase in inhibitory receptors, a decrease in effector cytokines, and impairment of cytotoxicity, potentially leading to tumor immune evasion. The regulatory mechanisms of T-cell exhaustion in the TME can be divided into the extrinsic pathway and intrinsic pathway53. Regarding extrinsic regulatory mechanisms, cancer cells and stromal cells (tumor-associated DCs, Tregs, tumor-associated macrophages (TAMs), and MDSCs) are the main exogenous cells that regulate T-cell exhaustion54, and both IL-10 and TGF-β are important extrinsic cytokines involved in the exhausted process of T cells55, 56, 57, 58. Regarding intrinsic regulatory mechanisms, the binding of immune checkpoint receptors and cognate ligands can exhaust T cells and inhibit their ability to kill tumor cells normally, so tumor cells can escape the immune surveillance of the host. On T cells, the immune checkpoint receptors include PD-1, CTLA-4, T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), B- and T-lymphocyte attenuator (BTLA), lymphocyte activation gene-3 (LAG-3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT). Their corresponding ligands, such as programmed cell death ligand 1 (PD-L1), galectin-9 and B7 homolog 3 (B7-H3) protein are expressed on tumor cells or MDSCs59, 60, 61. Much evidence suggests that lipids lead to the functional exhaustion of T cells and suppress adaptive immune responses through the extrinsic pathway and intrinsic pathway (Fig. 2).
4.1. Lipids regulate T-cell exhaustion by extrinsic mechanisms
Lipid metabolism is a key factor in regulating the function of MDSCs. Tumor-infiltrating MDSCs show increased FA absorption and thus increased levels of FAO as their energy sources. Such MDSCs represent a stronger immunosuppressive effect on CD8+ T cells than MDSCs with normal lipid content31,62. Pharmacological inhibition of FAO both reduced the immunosuppressive functions of tumor-infiltrating MDSCs and blocked their production of inhibitory cytokines. Monotherapy with the FAO inhibitor etomoxir or ranolazine significantly repressed the tumor growth of LLC and MCA-38 colon adenocarcinoma in a T-cell-dependent manner and synergized the antitumor effect of adoptive T-cell therapy62. In mice bearing LLC tumor, CT26 colon carcinoma, and B16F10 tumor, polymorphonuclear-MDSCs showed increased FATP2 expression, which suppressed the antigen-specific T-cell response in the TME. This phenomenon occurred because FATP2 promoted arachidonic acid uptake, and overload of arachidonic acid resulted in higher production of PGE2. This process promoted the expansion of polymorphonuclear-MDSCs and further suppressed the antigen-specific T-cell response63. In another study, FATP2-induced lipid accumulation in MDSCs generated ROS and promoted the immunosuppressive function of MDSCs on T cells by reducing T-cell proliferation and suppressing the ability of T cells to produce IFN-γ and tumor necrosis factor α (TNF-α)64. Therefore, reducing lipid accumulation by lipofermata, an inhibitor of FATP2, can block immunosuppressive activity in MDSCs and relieve T-cell exhaustion. More importantly, lipofermata can also enhance the antitumor effect of immunotherapy with anti-PD-L1 in B16F10 and LLC tumor-bearing mice64. PGE2, the derivative of FAs, was conducive to the formation of an immunosuppressive microenvironment in melanoma. Microsomal PGE synthase-1 was positively correlated with CD14 and integrin alpha M in MDSCs. Therefore, PGE2 depletion by PGE synthase knockout may reduce MDSC immunosuppressive activity on activated T cells65. Modulating PGE2 production with a COX-2 inhibitor has been demonstrated to efficiently improve the antitumor effect of immune checkpoint inhibitors (ICIs). Persistent tumor regression was observed in mice carrying prostaglandin E synthase KO tumors or in patients with metastatic melanoma and non-small cell lung cancer treated with a combination strategy of COX inhibitors and ICIs65,66. For pancreatic cancer, immunosuppression in TME is considered a major obstacle to the effectiveness of immunotherapeutic approaches to induce antitumor immune responses67. Pharmacological inhibition of COX-2 reduces the accumulation of MDSCs, thereby transforming the immunosuppressive microenvironment of pancreatic cancer into a T-cell-permissive environment68. This strategy increases cancer responses to the combination of checkpoint blockers and CD40 agonists68.
Tregs maintain immune tolerance but also lead to immunosuppression in the tumor microenvironment69,70. High blood levels of butyrate and propionate, which are the SCFAs, were associated with a higher proportion of Tregs in tumor-draining lymph nodes in tumor bearing mice (CT26 models)25. De novo fatty-acid synthesis mediated by FASN contributes to the functional maturation of Tregs, and FASN deletion from Tregs inhibits colon tumor growth, but the addition of palmitate, a product of FASN, restores the suppressive function of Tregs71. A retrospective analysis of 100 patients with non-small cell lung cancer showed that COX-2 expression is positively correlated with tumor-infiltrating foxp3+ Tregs, and patients with elevated COX-2 expression have shorter recurrence-free survival than patients without COX-2 expression32.
Macrophages can be polarized into classically activated (M1) and alternatively activated (M2) macrophages; the former foster an inflammatory response against tumor cells, whereas the latter tend to exert an immunosuppressive phenotype72. The inhibition of cholesterol efflux mediated by the ATP-binding cassette transporter G1 induces M2 macrophages to transform into the M1 type and enhances their cytotoxicity to kill cancer cells73. In glioblastoma multiforme, FAO-derived acetyl-CoA upregulated CD47 transcription to weaken macrophage phagocytosis74. Inhibition of FAO by etomoxir, a carnitine palmitoyltransferase 1 (CPT1) inhibitor, synergized with an anti-CD47 antibody to control regrown tumors with elevated macrophage phagocytosis74. In addition, PGE2 has been found to induce M2 macrophage polarization and recruit MDSCs to suppress host immunity, which in turn increases lung tumor stemness and epithelial–mesenchymal transition-like features75. In an immune-competent mouse model of C57 with inflammatory lung, using lower doses of prostaglandin E synthase inhibitors, Cay10526 can significantly inhibit lung metastasis75. Moreover, conditional depletion of COX-2 and PGE2 synthase in myeloid cells disrupt M2-like TAM function, which enhanced cytotoxic T-cell function in a mouse model of breast cancer through76.
DCs engulf FA-carrying tumor-derived exosomes, which can increase the secretion of TGF-β34. Moreover, in bone marrow-derived DCs, increasing PGE2 levels can promote the secretion of IL-10 through stimulation of EP2R and EP4Rs, which inhibits T-cell proliferation33. MF-766 blocks EP4R, synergistically improving the efficacy of anti-PD-1 therapy in CT26 and EMT6 tumor-bearing mice77.
These results indicate that therapeutic strategies blocking lipid synthesis can restore T-cell activity; enhance IFN-γ, IL-2, and TNF-α production; decrease the infiltration and function of immunosuppressive cells; and promote the infiltration of CD8+ T cells, natural killer cells and conventional DCs68,77. Therefore, these results suggest that drugs that regulate lipid metabolism can not only restore lipid homeostasis in tumors but also exert antitumor effects by increasing tumor immunity. In addition, these drugs can be used as immune sensitizers in combination with other drugs.
4.2. Lipids regulate T-cell exhaustion by intrinsic mechanisms
It is worth noting that increased lipids levels can increase immune checkpoint expression, suggesting that drugs blocking lipid synthesis can be used as novel immune checkpoint inhibitors to coordinate immunotherapy. A study revealed that FA-carrying tumor-derived exosomes significantly increase the expression of inhibitory checkpoint proteins on DCs, such as PD-L1 and signal regulatory protein α (SIRPα)34. FASN activity stabilizes the PD-L1 protein via PD-L1 palmitoylation to cause cisplatin resistance in bladder cancer78. In breast cancer, disruption of PD-L1 palmitoylation sensitizes breast cancer cells to T-cell killing and thus represses tumor growth79. These studies suggest that inhibition of lipid metabolism by targeting PD-L1 palmitoylation with small molecules may provide new avenues for improving the therapeutic efficacy of anti-PD-L1/PD-1 treatment.
Tumor tissues are rich in cholesterol, which leads to high cholesterol content in tumor-infiltrating CD8+ T cells. Cholesterol increases endoplasmic reticulum stress and then activates the endoplasmic reticulum stress sensor X-box binding protein 1 (XBP1), which regulates PD-1 and 2B4 transcription in CD8+ T cells35. Consequently, tumor-infiltrating CD8+ T cells can positively and progressively increase the expression of PD-1, 2B4, T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), and lymphocyte activation gene-3 (LAG-3)35. Reducing the level of cholesterol in CD8+ T cells can restore the less-exhausted phenotype of tumor-infiltrating CD8+ T cells and effectively enhance antitumor activity in a mouse melanoma model35. Simvastatin targets cholesterol biosynthesis and inhibits long noncoding RNA snhg29-mediated YAP activation, which in turn decreases PD-L1 transcription. Moreover, simvastatin inhibits colon cancer proliferation and synergizes with anti-PD-L1 against tumors80.
PGE2 in lung cancer tissue homogenate positively regulates the level of PD-1 in infiltrating CD8+ T cells by activating the EP2 and EP4 pathways81. In summary, exploring the mechanisms through which lipids regulate these extrinsic and intrinsic immunosuppression pathways may benefit our understanding of how immunosuppression is propagated in the tumor and which lipids are potential targets in lipid metabolism for immunotherapy.
5. Combination therapy
With the continuous development of research on the impact of lipids on the TME, the findings suggest the feasibility of combining drugs that modulating lipids and tumor immunotherapy in cancer treatment. Investigations of the combination of ICIs and drugs used to modulate FAs are currently mainly in preclinical research. PPARγ agonists (rosiglitazone and bezafibrate), that reduce FA storage, showed better inhibition of tumor growth and prolongation of survival time mainly in melanoma and lung cancer, when used in combination with immunotherapy, such as anti-PD-1 and cancer cell vaccines40,82,83. Preclinical studies also suggested that statins could potentiate immunotherapy in the treatment of lung cancer, melanoma and breast cancer80,84, 85, 86. Some retrospective studies have shown that the combined use of statins and PD-1 inhibitors was associated with longer tumor treatment fields and improved objective response rates (ORR) and progression-free survival (PFS). NSCLC patients treated with nivolumab and statins exhibited a statistically significant response (P = 0.02) and better ORR (40% versus 22%, P = 0.04) and PFS (median 7.8 versus 3.6 months, P = 0.03) than the non-statin group87,88. In malignant pleural mesothelioma, patients who were treated with statins showed an increased ORR (22% versus 6%, P = 0.05) and PFS (median 6.7 versus 2.4 months, P < 0.01)88. Moreover, the levels of cholesterol and SCFAs may be a biomarker of the efficacy of tumor immunotherapy89,90. Combination strategies using immunotherapy and COX inhibitors were mainly carried out, and a small number of drugs modulating FAs and cholesterol were also implemented in clinical trials, which are summarized in Table 1.
Table 1.
Clinical trials involving the combination of immunotherapy and lipid-mediated therapies.
| Adaptive immune cycle | Clinical trial identifier | Status | Phase | Cancer type | Drug (target) | Immunotherapy |
|---|---|---|---|---|---|---|
| Antigen presentation | NCT04093323 | Recruiting | 2 | HLA–A2+ refractory melanoma | Celecoxib (COX-2) | α-Type-1 polarized dendritic cells, recombinant interferon α-2b, PD-1 ligand inhibitor, PD-1 inhibitor |
| NCT02151448 | Completed | 1/2 | Peritoneal surface malignancies | Celecoxib (COX-2) | DC vaccine, interferon α-2b | |
| NCT01158534 | Completed | 2 | Renal cell cancer | Celecoxib (COX-2) | Interferon α-2b | |
| NCT03710876 | Active, not recruiting | 3 | Malignant pleural mesothelioma | Celecoxib (COX-2) | rAd-interferon | |
| NCT03403634 | Completed | 2 | Metastatic carcinoma in the liver, colorectal carcinoma | Celecoxib (COX-2) | Interferon α-2b | |
| NCT00081848 | Completed | 1 | Liver neoplasms | Celecoxib (COX-2) | Recombinant Fowlpox-GM-CSF | |
| T-cell priming and activation | NCT02922764 | Recruiting | 1 | Advanced solid malignancies and lymphoma | RGX-104 (LXR) | Ipilimumab (CTLA4) |
| NCT03396952 | Active, not recruiting | 2 | Cutaneous melanoma | Aspirin (COX) | Ipilimumab (CTLA-4) | |
| T-cell killing of tumor | NCT00193648 | Completed | 1 | Focal glomerulosclerosis | Rosiglitazone (PPARγ) | Atezolizumab (PD-L1) |
| NCT04114136 | Recruiting | 2 | Melanoma renal cell, carcinoma, NSCLC, hepatocellular carcinoma, urothelial cancer, gastric adenocarcinoma, HNSCC, esophageal adenocarcinoma, microsatellite instability-high solid malignant tumor | Rosiglitazone (PPARγ) | Nivolumab (PD-1)/pembrolizumab (PD-1) | |
| NCT02922764 | Recruiting | 1 | Advanced solid malignancies, lymphoma | RGX-104 (LXR) | Nivolumab (PD-1)/pembrolizumab (PD-1) | |
| NCT03638297 | Recruiting | 2 | Colorectal cancer | Aspirin (COX) | BAT1306 (PD-1) | |
| NCT03926338 | Recruiting | 1/2 | Colorectal cancer | Celecoxib (COX-2) | Toripalimab (PD-1) | |
| NCT03396952 | Active, not recruiting | 2 | Melanoma | Aspirin (COX) | Ipilimumab (PD-1) | |
| NCT03245489 | Recruiting | 1 | Head and neck cancer | Acetylsalicylic acid (COX) | Pembrolizumab (PD-1) | |
| NCT02659384 | Active, not recruiting | 2 | Ovarian neoplasms | Acetylsalicylic acid (COX) | Atezolizumab (PD-L1) |
6. Conclusions
Dysregulated cellular metabolism is the hallmark of cancer, in which lipid metabolism is now an undisputedly thought to be factor in supporting cancer growth and progression. Combined with recent findings in the field of tumor immune escape and cancer immunotherapy, we review the role of aberrant lipids, especially, FAs, cholesterol, and prostaglandins, an arachidonic acid derivative, in regulating tumor immunity and immunotherapy by acting on the cancer–immunity cycle.
De novo lipid synthesis, increased FA uptake and FAO can impair the capacity of DCs to process TAAs, promote the infiltration of Tregs and secretion of TGF-β and increase the expression of PD-L1 and CD47 on tumor or immune cells. Long- or short-chain saturated FAs can reduce the expression of MHC I or costimulatory factors respectively, thereby inhibiting the ability of APCs to activate T cells. Lipid synthesis, FA uptake, and FAO can be blocked by inhibiting acetyl-CoA, PPARγ, FATP2, and CPT1, respectively. These inhibitors synergize with immunotherapy in tumor-bearing mice (CT26, MC38, MCA-38 and LLC mice models). In clinical trials, only the PPARγ inhibitor rosiglitazone in combination with PD-1/PD-L1 monoclonal antibodies to treat solid tumors has entered phase 1/2 (Table 1). Moreover, ASC40, a novel potent oral inhibitor of FASN, has entered phase three clinical trials in combination with bevacizumab for the treatment of recurrent glioblastoma (NCT05118776) and exhibits promise as a drug candidate for synergistic immunotherapy. In addition, etomoxir, the pharmacologic inhibitor of CPT1, has been shown to synergize with the antitumor effect of adoptive T-cell therapy in preclinical research, but its clinical translation is less likely due to its severe hepatotoxicity91.
Cholesterol hindered DCs from recognizing GSL, a TAA, by changing its conformation, regulated TCR signaling, increased immune checkpoint expression in T cells and affected macrophage polarization to M2 macrophages. In preclinical research, simvastatin synergizes with anti-PD-L1 against colon cancer. However, the clinical trial investigating the combination of lovastatin and IFN α-2b was in a withdrawn state, but the reasons for the withdrawal were not specified (NCT00963664). RGX-104, a LXR agonist, protects cells from cholesterol overload92. It has been studied in combination therapy with ipilimumab, nivolumab, or pembrolizumab to treat advanced solid malignancies and lymphoma in phase 1, but no results have been published yet (Table 1).
PGE2 inhibited all three steps of the immune cycle. PGE2 reduces the accumulation of tumor infiltrating dendritic cell to inhibit antigen uptake. In addition, PGE2 inhibits T-cell proliferation and suppresses the secretion of IFN-γ, TNF-α, and granzyme B. Moreover, it increases the infiltration of immunosuppressive cells, such as Tregs and MDSCs, and the secretion of immunosuppressive cytokines, such as IL-10. It is worth noting that there are many clinical trials on the combination of COX inhibitors and immunotherapy against tumors (Table 1). This finding indicates the great potential of COX inhibitors in improving the efficacy of immunotherapy. COX-2-specific inhibitors may not affect the protective effect of COX-1 on the gastrointestinal tract, platelets, etc.93. This finding indicates that the COX-2 inhibitor celecoxib can be safer when used in combination with immunotherapy.
In summary, understanding how lipids affect antigen presentation, T-cell priming and activation, and T-cell killing of tumors and how they alter the immune context within the TME may provide new therapeutic targets against tumors. Whether new drug candidates can achieve safety, efficacy and tolerability in preclinical and clinical trials when used alone or in combination with other cancer treatments remains to be verified.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81930102 to Bo Yang), the National Natural Science Foundation of China (No. 82273949 to Ling Ding), the National Natural Science Foundation of China (No. 82104196 to Xi Chen).
Author contributions
Mingming Zheng designed and wrote the paper. Wenxin Zhang, Xi Chen, Hongjie Guo, Honghai Wu, Yanjun Xu, and Qiaojun He revised the manuscript. Ling Ding and Bo Yang were responsible for the conception and design of the review.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Ling Ding, Email: ld362@zju.edu.cn.
Bo Yang, Email: yang924@zju.edu.cn.
References
- 1.Fahy E., Subramaniam S., Murphy R.C., Nishijima M., Raetz C.R., Shimizu T., et al. Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res. 2009;50(Suppl):S9–S14. doi: 10.1194/jlr.R800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fahy E., Subramaniam S., Brown H.A., Glass C.K., Merrill A.H., Jr., Murphy R.C., et al. A comprehensive classification system for lipids. J Lipid Res. 2005;46:839–861. doi: 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.van Meer G. Cellular lipidomics. EMBO J. 2005;24:3159–3165. doi: 10.1038/sj.emboj.7600798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wymann M.P., Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- 5.Holthuis J.C., Menon A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature. 2014;510:48–57. doi: 10.1038/nature13474. [DOI] [PubMed] [Google Scholar]
- 6.Welte M.A., Gould A.P. Lipid droplet functions beyond energy storage. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:1260–1272. doi: 10.1016/j.bbalip.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pisani D., Ailhaud G. Involvement of polyunsaturated fatty acids in the control of energy storage and expenditure. OCL. 2019;26:37. [Google Scholar]
- 8.Rohrig F., Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16:732–749. doi: 10.1038/nrc.2016.89. [DOI] [PubMed] [Google Scholar]
- 9.Carracedo A., Cantley L.C., Pandolfi P.P. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuzu O.F., Noory M.A., Robertson G.P. The role of cholesterol in cancer. Cancer Res. 2016;76:2063–2070. doi: 10.1158/0008-5472.CAN-15-2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang D., Dubois R.N. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corn K.C., Windham M.A., Rafat M. Lipids in the tumor microenvironment: from cancer progression to treatment. Prog Lipid Res. 2020;80 doi: 10.1016/j.plipres.2020.101055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D., Buchanan F.G., Wang H., Dey S.K., DuBois R.N. Prostaglandin E2 enhances intestinal adenoma growth via activation of the Ras-mitogen-activated protein kinase cascade. Cancer Res. 2005;65:1822–1829. doi: 10.1158/0008-5472.CAN-04-3671. [DOI] [PubMed] [Google Scholar]
- 14.Poligone B., Baldwin A.S. Positive and negative regulation of NF-kappaB by COX-2: roles of different prostaglandins. J Biol Chem. 2001;276:38658–38664. doi: 10.1074/jbc.M106599200. [DOI] [PubMed] [Google Scholar]
- 15.Li Y.C., Park M.J., Ye S.K., Kim C.W., Kim Y.N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168:1107–1118. doi: 10.2353/ajpath.2006.050959. quiz 404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lingrand M., Lalonde S., Jutras-Carignan A., Bergeron K.F., Rassart E., Mounier C. SCD1 activity promotes cell migration via a PLD-mTOR pathway in the MDA-MB-231 triple-negative breast cancer cell line. Breast Cancer. 2020;27:594–606. doi: 10.1007/s12282-020-01053-8. [DOI] [PubMed] [Google Scholar]
- 17.Brouxhon S., Kyrkanides S., O'Banion M.K., Johnson R., Pearce D.A., Centola G.M., et al. Sequential down-regulation of E-Cadherin with squamous cell carcinoma progression: loss of E-Cadherin via a prostaglandin E2-EP2-dependent posttranslational mechanism. Cancer Res. 2007;67:7654–7664. doi: 10.1158/0008-5472.CAN-06-4415. [DOI] [PubMed] [Google Scholar]
- 18.Jang T.J., Cha W.H., Lee K.S. Reciprocal correlation between the expression of cyclooxygenase-2 and E-cadherin in human bladder transitional cell carcinomas. Virchows Arch. 2010;457:319–328. doi: 10.1007/s00428-010-0943-3. [DOI] [PubMed] [Google Scholar]
- 19.Yu Y., Gao L., Wang Y., Xu B., Maswikiti E.P., Li H., et al. A forgotten corner in cancer immunotherapy: the role of lipids. Front Oncol. 2021;11 doi: 10.3389/fonc.2021.751086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howie D., Ten Bokum A., Necula A.S., Cobbold S.P., Waldmann H. The role of lipid metabolism in T lymphocyte differentiation and survival. Front Immunol. 2017;8:1949. doi: 10.3389/fimmu.2017.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bleve A., Durante B., Sica A., Consonni F.M. Lipid metabolism and cancer immunotherapy: immunosuppressive myeloid cells at the crossroad. Int J Mol Sci. 2020;21:5845. doi: 10.3390/ijms21165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y., Wang Y., Ren Y., Zhang Q., Yi P., Cheng C. Metabolic modulation of immune checkpoints and novel therapeutic strategies in cancer. Semin Cancer Biol. 2022;86(Pt 3):542–565. doi: 10.1016/j.semcancer.2022.02.010. [DOI] [PubMed] [Google Scholar]
- 23.Janelle V., Rulleau C., Del Testa S., Carli C., Delisle J.S. T-cell immunotherapies targeting histocompatibility and tumor antigens in hematological malignancies. Front Immunol. 2020;11:276. doi: 10.3389/fimmu.2020.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaikh S.R., Mitchell D., Carroll E., Li M., Schneck J., Edidin M. Differential effects of a saturated and a monounsaturated fatty acid on MHC class I antigen presentation. Scand J Immunol. 2008;68:30–42. doi: 10.1111/j.1365-3083.2008.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coutzac C., Jouniaux J.M., Paci A., Schmidt J., Mallardo D., Seck A., et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat Commun. 2020;11:2168. doi: 10.1038/s41467-020-16079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang F., Beck-García K., Zorzin C., Schamel W.W.A., Davis M.M. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat Immunol. 2016;17:844–850. doi: 10.1038/ni.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang W., Bai Y., Xiong Y., Zhang J., Chen S., Zheng X., et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature. 2016;531:651–655. doi: 10.1038/nature17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmadi M., Emery D.C., Morgan D.J. Prevention of both direct and cross-priming of antitumor CD8+ T-cell responses following overproduction of prostaglandin E2 by tumor cells in vivo. Cancer Res. 2008;68:7520–7529. doi: 10.1158/0008-5472.CAN-08-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watchmaker P.B., Berk E., Muthuswamy R., Mailliard R.B., Urban J.A., Kirkwood J.M., et al. Independent regulation of chemokine responsiveness and cytolytic function versus CD8+ T cell expansion by dendritic cells. J Immunol. 2010;184:591–597. doi: 10.4049/jimmunol.0902062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vercammen C., Ceuppens J.L. Prostaglandin E2 inhibits human T-cell proliferation after crosslinking of the CD3-Ti complex by directly affecting T cells at an early step of the activation process. Cell Immunol. 1987;104:24–36. doi: 10.1016/0008-8749(87)90003-7. [DOI] [PubMed] [Google Scholar]
- 31.Yan D., Adeshakin A.O., Xu M., Afolabi L.O., Zhang G., Chen Y.H., et al. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front Immunol. 2019;10:1399. doi: 10.3389/fimmu.2019.01399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimizu K., Nakata M., Hirami Y., Yukawa T., Maeda A., Tanemoto K. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thorac Oncol. 2010;5:585–590. doi: 10.1097/JTO.0b013e3181d60fd7. [DOI] [PubMed] [Google Scholar]
- 33.Harizi H., Grosset C., Gualde N. Prostaglandin E2 modulates dendritic cell function via EP2 and EP4 receptor subtypes. J Leukoc Biol. 2003;73:756–763. doi: 10.1189/jlb.1002483. [DOI] [PubMed] [Google Scholar]
- 34.Yin X., Zeng W., Wu B., Wang L., Wang Z., Tian H., et al. PPARalpha inhibition overcomes tumor-derived exosomal lipid-induced dendritic cell dysfunction. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma X., Bi E., Lu Y., Su P., Huang C., Liu L., et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metabol. 2019;30:143–156.e5. doi: 10.1016/j.cmet.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rossi A., Filetti M., Taurelli Salimbeni B., Piras M., Rizzo F., Giusti R., et al. Statins and immunotherapy: togetherness makes strength the potential effect of statins on immunotherapy for NSCLC. Cancer Rep. 2021;4:e1368. doi: 10.1002/cnr2.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chae Y.K., Yousaf M., Malecek M.K., Carneiro B., Chandra S., Kaplan J., et al. Statins as anti-cancer therapy; can we translate preclinical and epidemiologic data into clinical benefit?. Discov Med. 2015;20:413–427. [PubMed] [Google Scholar]
- 38.Mukherjee P., Basu G.D., Tinder T.L., Subramani D.B., Bradley J.M., Arefayene M., et al. Progression of pancreatic adenocarcinoma is significantly impeded with a combination of vaccine and COX-2 inhibition. J Immunol. 2009;182:216–224. [PMC free article] [PubMed] [Google Scholar]
- 39.Markosyan N., Li J., Sun Y.H., Richman L.P., Lin J.H., Yan F., et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2) J Clin Invest. 2019;129:3594–3609. doi: 10.1172/JCI127755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chowdhury P.S., Chamoto K., Kumar A., Honjo T. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+ T cells and facilitates anti-PD-1 therapy. Cancer Immunol Res. 2018;6:1375–1387. doi: 10.1158/2326-6066.CIR-18-0095. [DOI] [PubMed] [Google Scholar]
- 41.Poulsen L., Siersbaek M., Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol. 2012;23:631–639. doi: 10.1016/j.semcdb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Langsjoen P.H., Langsjoen A.M. The clinical use of HMG CoA-reductase inhibitors and the associated depletion of coenzyme Q10. A review of animal and human publications. Biofactors. 2003;18:101–111. doi: 10.1002/biof.5520180212. [DOI] [PubMed] [Google Scholar]
- 43.Hakomori S., Zhang Y. Glycosphingolipid antigens and cancer therapy. Chem Biol. 1997;4:97–104. doi: 10.1016/s1074-5521(97)90253-2. [DOI] [PubMed] [Google Scholar]
- 44.Novak A., Binnington B., Ngan B., Chadwick K., Fleshner N., Lingwood C.A. Cholesterol masks membrane glycosphingolipid tumor-associated antigens to reduce their immunodetection in human cancer biopsies. Glycobiology. 2013;23:1230–1239. doi: 10.1093/glycob/cwt059. [DOI] [PubMed] [Google Scholar]
- 45.Herber D.L., Cao W., Nefedova Y., Novitskiy S.V., Nagaraj S., Tyurin V.A., et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16:880–886. doi: 10.1038/nm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang L., Fang X., Wang H., Li D., Wang X. Ovarian cancer-intrinsic fatty acid synthase prevents anti-tumor immunity by disrupting tumor-infiltrating dendritic cells. Front Immunol. 2018;9:2927. doi: 10.3389/fimmu.2018.02927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang G., Xu J., Zhao J., Yin W., Liu D., Chen W., et al. Arf1-mediated lipid metabolism sustains cancer cells and its ablation induces anti-tumor immune responses in mice. Nat Commun. 2020;11:220. doi: 10.1038/s41467-019-14046-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia A., Zhang Y., Xu J., Yin T., Lu X.J. T cell dysfunction in cancer immunity and immunotherapy. Front Immunol. 2019;10:1719. doi: 10.3389/fimmu.2019.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klebanoff C.A., Gattinoni L., Restifo N.P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fantini J., Di Scala C., Evans L.S., Williamson P.T.F., Barrantes F.J. A mirror code for protein–cholesterol interactions in the two leaflets of biological membranes. Sci Rep. 2016;6 doi: 10.1038/srep21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swamy M., Beck-Garcia K., Beck-Garcia E., Hartl F.A., Morath A., Yousefi O.S., et al. A cholesterol-based allostery model of T cell receptor phosphorylation. Immunity. 2016;44:1091–1101. doi: 10.1016/j.immuni.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 52.Chakrabarti R., Engleman E.G. Interrelationships between mevalonate metabolism and the mitogenic signaling pathway in T lymphocyte proliferation. J Biol Chem. 1991;266:12216–12222. [PubMed] [Google Scholar]
- 53.Jiang Y., Li Y., Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015;6 doi: 10.1038/cddis.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ostrand-Rosenberg S., Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith L.K., Boukhaled G.M., Condotta S.A., Mazouz S., Guthmiller J.J., Vijay R., et al. Interleukin-10 directly inhibits CD8+ T cell function by enhancing N-glycan branching to decrease antigen sensitivity. Immunity. 2018;48:299–312 e5. doi: 10.1016/j.immuni.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomas D.A., Massagué J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 58.Annacker O., Asseman C., Read S., Powrie F. Interleukin-10 in the regulation of T cell-induced colitis. J Autoimmun. 2003;20:277–279. doi: 10.1016/s0896-8411(03)00045-3. [DOI] [PubMed] [Google Scholar]
- 59.Waldman A.D., Fritz J.M., Lenardo M.J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jayaraman P., Jacques M.K., Zhu C., Steblenko K.M., Stowell B.L., Madi A., et al. TIM3 mediates T cell exhaustion during Mycobacterium tuberculosis infection. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurachi M. CD8+ T cell exhaustion. Semin Immunopathol. 2019;41:327–337. doi: 10.1007/s00281-019-00744-5. [DOI] [PubMed] [Google Scholar]
- 62.Hossain F., Al-Khami A.A., Wyczechowska D., Hernandez C., Zheng L., Reiss K., et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3:1236–1247. doi: 10.1158/2326-6066.CIR-15-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Veglia F., Tyurin V.A., Blasi M., De Leo A., Kossenkov A.V., Donthireddy L., et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569:73–78. doi: 10.1038/s41586-019-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adeshakin A.O., Liu W., Adeshakin F.O., Afolabi L.O., Zhang M., Zhang G., et al. Regulation of ROS in myeloid-derived suppressor cells through targeting fatty acid transport protein 2 enhanced anti-PD-L1 tumor immunotherapy. Cell Immunol. 2021;362 doi: 10.1016/j.cellimm.2021.104286. [DOI] [PubMed] [Google Scholar]
- 65.Kim S.H., Roszik J., Cho S.N., Ogata D., Milton D.R., Peng W., et al. The COX2 effector microsomal PGE2 synthase 1 is a regulator of immunosuppression in cutaneous melanoma. Clin Cancer Res. 2019;25:1650–1663. doi: 10.1158/1078-0432.CCR-18-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang S.J., Khullar K., Kim S., Yegya-Raman N., Malhotra J., Groisberg R., et al. Effect of cyclo-oxygenase inhibitor use during checkpoint blockade immunotherapy in patients with metastatic melanoma and non-small cell lung cancer. J Immunother Cancer. 2020;8 doi: 10.1136/jitc-2020-000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kiaie S.H., Sanaei M.J., Heshmati M., Asadzadeh Z., Azimi I., Hadidi S., et al. Immune checkpoints in targeted-immunotherapy of pancreatic cancer: new hope for clinical development. Acta Pharm Sin B. 2021;11:1083–1097. doi: 10.1016/j.apsb.2020.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conejo-Garcia J.R. Breaking barriers for T cells by targeting the EPHA2/TGF-beta/COX-2 axis in pancreatic cancer. J Clin Invest. 2019;129:3521–3523. doi: 10.1172/JCI130316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Savage P.A., Klawon D.E.J., Miller C.H. Regulatory T cell development. Annu Rev Immunol. 2020;38:421–453. doi: 10.1146/annurev-immunol-100219-020937. [DOI] [PubMed] [Google Scholar]
- 70.Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lim S.A., Wei J., Nguyen T.M., Shi H., Su W., Palacios G., et al. Lipid signalling enforces functional specialization of Treg cells in tumours. Nature. 2021;591:306–311. doi: 10.1038/s41586-021-03235-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin Y., Xu J., Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol. 2019;12:76. doi: 10.1186/s13045-019-0760-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sag D., Cekic C., Wu R., Linden J., Hedrick C.C. The cholesterol transporter ABCG1 links cholesterol homeostasis and tumour immunity. Nat Commun. 2015;6:6354. doi: 10.1038/ncomms7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang N., Xie B., Xiao W., Fan M., Xu S., Duan Y., et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat Commun. 2022;13:1511. doi: 10.1038/s41467-022-29137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang T., Jing B., Xu D., Liao Y., Song H., Sun B., et al. PTGES/PGE2 signaling links immunosuppression and lung metastasis in Gprc5a-knockout mouse model. Oncogene. 2020;39:3179–3194. doi: 10.1038/s41388-020-1207-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen E.P., Markosyan N., Connolly E., Lawson J.A., Li X., Grant G.R., et al. Myeloid cell COX-2 deletion reduces mammary tumor growth through enhanced cytotoxic T-lymphocyte function. Carcinogenesis. 2014;35:1788–1797. doi: 10.1093/carcin/bgu053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Y., Cui L., Georgiev P., Singh L., Zheng Y., Yu Y., et al. Combination of EP4 antagonist MF-766 and anti-PD-1 promotes anti-tumor efficacy by modulating both lymphocytes and myeloid cells. OncoImmunology. 2021;10 doi: 10.1080/2162402X.2021.1896643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shahid M., Kim M., Jin P., Zhou B., Wang Y., Yang W., et al. S-Palmitoylation as a functional regulator of proteins associated with cisplatin resistance in bladder cancer. Int J Biol Sci. 2020;16:2490. doi: 10.7150/ijbs.45640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang Y., Hsu J.M., Sun L., Chan L.C., Li C.W., Hsu J.L., et al. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019;29:83–86. doi: 10.1038/s41422-018-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ni W., Mo H., Liu Y., Xu Y., Qin C., Zhou Y., et al. Targeting cholesterol biosynthesis promotes anti-tumor immunity by inhibiting long noncoding RNA SNHG29-mediated YAP activation. Mol Ther. 2021;29:2995–3010. doi: 10.1016/j.ymthe.2021.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang J., Zhang L., Kang D., Yang D., Tang Y. Activation of PGE2/EP2 and PGE2/EP4 signaling pathways positively regulate the level of PD-1 in infiltrating CD8+ T cells in patients with lung cancer. Oncol Lett. 2018;15:552–558. doi: 10.3892/ol.2017.7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goyal G., Wong K., Nirschl C.J., Souders N., Neuberg D., Anandasabapathy N., et al. PPARgamma contributes to immunity induced by cancer cell vaccines that secrete GM-CSF. Cancer Immunol Res. 2018;6:723–732. doi: 10.1158/2326-6066.CIR-17-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Luu M., Riester Z., Baldrich A., Reichardt N., Yuille S., Busetti A., et al. Microbial short-chain fatty acids modulate CD8+ T cell responses and improve adoptive immunotherapy for cancer. Nat Commun. 2021;12:4077. doi: 10.1038/s41467-021-24331-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oechsle C.M., Showalter L.E., Novak C.M., Czerniecki B.J., Koski G.K. Statin drugs plus Th1 cytokines potentiate apoptosis and Ras delocalization in human breast cancer lines and combine with dendritic cell-based immunotherapy to suppress tumor growth in a mouse model of HER-2pos disease. Vaccines. 2020;8:72. doi: 10.3390/vaccines8010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tenesaca S., Vasquez M., Alvarez M., Otano I., Fernandez-Sendin M., Di Trani C.A., et al. Statins act as transient type I interferon inhibitors to enable the antitumor activity of modified vaccinia Ankara viral vectors. J Immunother Cancer. 2021;9 doi: 10.1136/jitc-2020-001587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu X., Bao X., Hu M., Chang H., Jiao M., Cheng J., et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588:693–698. doi: 10.1038/s41586-020-2911-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Omori M., Okuma Y., Hakozaki T., Hosomi Y. Statins improve survival in patients previously treated with nivolumab for advanced non-small cell lung cancer: an observational study. Mol Clin Oncol. 2019;10:137–143. doi: 10.3892/mco.2018.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cantini L., Pecci F., Hurkmans D.P., Belderbos R.A., Lanese A., Copparoni C., et al. High-intensity statins are associated with improved clinical activity of PD-1 inhibitors in malignant pleural mesothelioma and advanced non-small cell lung cancer patients. Eur J Cancer. 2021;144:41–48. doi: 10.1016/j.ejca.2020.10.031. [DOI] [PubMed] [Google Scholar]
- 89.Karayama M., Inui N., Inoue Y., Yoshimura K., Mori K., Hozumi H., et al. Increased serum cholesterol and long-chain fatty acid levels are associated with the efficacy of nivolumab in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2021;71:203–217. doi: 10.1007/s00262-021-02979-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nomura M., Nagatomo R., Doi K., Shimizu J., Baba K., Saito T., et al. Association of short-chain fatty acids in the gut microbiome with clinical response to treatment with nivolumab or pembrolizumab in patients with solid cancer tumors. JAMA Netw Open. 2020;3 doi: 10.1001/jamanetworkopen.2020.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Galluzzi L., Kepp O., Vander Heiden M.G., Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- 92.Baranowski M. Biological role of liver X receptors. J Physiol Pharmacol. 2008;59 Suppl 7:31–55. [PubMed] [Google Scholar]
- 93.Crofford L.J. COX-1 and COX-2 tissue expression: implications and predictions. J Rheumatol Suppl. 1997;49:15–19. [PubMed] [Google Scholar]