

Abstract
Reprogramming of energy metabolism is one of the basic characteristics of cancer and has been proved to be an important cancer treatment strategy. Isocitrate dehydrogenases (IDHs) are a class of key proteins in energy metabolism, including IDH1, IDH2, and IDH3, which are involved in the oxidative decarboxylation of isocitrate to yield α-ketoglutarate (α-KG). Mutants of IDH1 or IDH2 can produce d-2-hydroxyglutarate (D-2HG) with α-KG as the substrate, and then mediate the occurrence and development of cancer. At present, no IDH3 mutation has been reported. The results of pan-cancer research showed that IDH1 has a higher mutation frequency and involves more cancer types than IDH2, implying IDH1 as a promising anti-cancer target. Therefore, in this review, we summarized the regulatory mechanisms of IDH1 on cancer from four aspects: metabolic reprogramming, epigenetics, immune microenvironment, and phenotypic changes, which will provide guidance for the understanding of IDH1 and exploring leading-edge targeted treatment strategies. In addition, we also reviewed available IDH1 inhibitors so far. The detailed clinical trial results and diverse structures of preclinical candidates illustrated here will provide a deep insight into the research for the treatment of IDH1-related cancers.
Key words: IDH1, Cancer, Regulatory mechanisms, IDH1 inhibitors, D-2HG, Metabolic reprogramming, Epigenetics, Immune microenvironment
Graphical abstract
Isocitrate dehydrogenase 1 (IDH1) is a potential target for cancer treatment. Mutant IDH1 promotes the occurrence and development of cancer by influencing metabolism, immune microenvironment, epigenetics, and phenotype.

1. Introduction
Cancer seriously threatens human life and health. On January 6, 2022, the International Agency for Research on Cancer (IARC) under the World Health Organization released the IARC Biennial Report 2020–2021. The latest assessment shows that in this century, cancer is expected to overtake cardiovascular disease and become the main cause of premature death in most countries. From 2000 to 2022, Professor Robert A. Weinberg (Massachusetts Institute of Technology, USA) and Professor Douglas Hanahan (Agora Translational Cancer Research Center, Switzerland) summarized and developed the hallmarks of cancer every ten years to explain the mechanisms of the occurrence, development, and treatment response characteristics of malignant tumors1, 2, 3. Characteristics of cancer summarized in Fig. 1 will provide a reasonable explanation for the multi-level process of human tumor pathology, and will also have an outstanding impact on the development of cancer treatment pathways.
Figure 1.

Hallmarks of cancer from 2000 to 2022. Original hallmarks are the initially identified cancer feature; emerging hallmarks are the features that have not been determined in the corresponding period and need further research and confirmation; enabling characteristics are the features that have been proposed in the corresponding period; colored circular arrows represent the time when the cancer hallmarks were presented; “…” represents the time when new cancer characteristics may be proposed in the future.
In normal tissues, glucose is converted to pyruvate by glycolysis, and then enters the mitochondria and is oxidized to supply energy for life activities through the tricarboxylic acid cycle, when the oxygen supply is sufficient. Only when the oxygen is deficient, pyruvate undergoes lactic acid fermentation for energy. However, tumor cells still preferentially obtain energy through glycolysis and produce lactic acid as the end product, even when the oxygen supply is sufficient. This phenomenon was first observed by Warburg in 1924, that is, the Warburg effect, which is an important feature of tumor energy metabolism4,5 (Fig. 2A). Metabolic changes are thought to play an important role in the development of cancers, but the mechanism of metabolic changes in cancer cells is controversial. A study in 2006 revealed the molecular mechanism of the Warburg effect for the first time, proposing that hypoxia-inducible factor 1 (HIF-1) can cause the reprogramming of energy metabolism, including increased glucose uptake, glycolysis, and lactate production, under the condition of hypoxia or loss of Von Hippel-Lindau gene function6. In 2008, c-Myc and p53 were also proposed as key energy metabolism regulators, and it was pointed out that many oncogenes and tumor suppressor genes played corresponding roles through the above three transcription factors7. Since 2011, with the recognition of energy metabolism reprogramming as one of the top ten characteristics of cancer, energy metabolism-related targets, such as IDH1/2, Hexokinase 2, and M2-type pyruvate kinase 2, have become research hotspots in the field of cancer treatment8. In addition, the research on the molecular mechanism of energy metabolism reprogramming is gradually deepened9,10. From 2016 to 2022, the research on cancer energy metabolism reprogramming gradually entered the range of fatty acid, amino acid, and cholesterol metabolism11,12. More interestingly, studies on immunity, inflammation, microenvironment, ubiquitination system, and intestinal microbiota have increased people's understanding of cancer energy metabolism13, 14, 15, 16, 17. To meet the needs of rapid proliferation, cancer cells acquire the ability to rearrange their energy metabolism, which is the most fundamental manifestation of cancer adaptation to the environment. Therefore, targeting cancer energy metabolism is undoubtedly a very important cancer treatment method with broad potential.
Figure 2.

Research timeline of cancer energy metabolism reprogramming and IDH1/2. (A) Milestone events of cancer energy metabolism reprogramming from 1924 to 2022. (B) Milestone events of IDH1/2 research from 2006 to 2021. The description of key events is on the opposite side of the time.
As an important metabolic enzyme and tumor biomarker of many cancers, IDHs are considered as a valuable target for cancer treatment. Significant advances in cancer genetics have shown that genes encoding IDHs are frequently mutated in a variety of human malignancies. A series of groundbreaking studies further elucidated the biological effects of IDH mutations, revealing the potential role of IDH mutations in tumorigenesis. In 2006, a sequencing study revealed for the first time that IDH1 mutation was associated with tumors18. With the development of sequencing technology, mutation sites and frequencies of IDH1/2 in different cancers have also been proposed one by one19,20 (Fig. 2B). Up to now, no mutation of IDH3 has been found. In a study of 5149 patients with solid tumors, 205 IDH mutations (3.78%) were found, including 145 IDH1 mutations (2.68%) and 63 IDH2 mutations (1.16%). Compared with IDH2, IDH1 has a higher mutation frequency and involves more types of cancer20. In addition, according to the information provided by Cortellis Drug Discovery Intelligence database, there are 320 IDH1-related clinical trials, 137 IDH2-related clinical trials, and zero IDH3-related clinical trials, which further indicates that targeting IDH1 has a greater prospect in the field of clinical application. Therefore, in this review, we summarized the impacts of IDH1 on four aspects of cancer, including metabolic reprogramming, epigenetics, cancer immune microenvironment, and cancer phenotype. In addition, the IDH1 inhibitors and drug combinations were summarized to provide ideas for IDH1-related cancer treatment and the development of drugs.
2. The cancer regulatory mechanisms of IDH1
2.1. Physiological function and structure of IDH1
IDHs are the rate-limiting enzyme in the tricarboxylic acid cycle involved in cellular energy metabolism, catalyzing the oxidative decarboxylation of isocitrate to α-KG and CO2, and converts NAD(P)+ into NAD(P)H. IDHs are divided into NADP+-dependent cytoplasm/peroxisome IDH1, mitochondrial IDH2, and NAD+-dependent mitochondrial IDH321,22. IDH1 and IDH2 are involved in the metabolism of reduced glutamine during changes in electron transfer receptors and during hypoxia23, 24, 25. These two isomers also play an important role in cell resistance to oxidative damage through their forward oxidative decarboxylation reaction26. Furthermore, their reverse reductive carboxylation reaction plays a key role in the regulation of adipogenesis and glycolysis27,28. IDH3 catalyzes the irreversible conversion of isocitrate to α-KG during the tricarboxylic acid cycle29 (Fig. 3).
Figure 3.

The selected function and structure of IDH1. The structure of IDH1WT (PDB ID: 4KZO). The up circular picture is a magnified view of the binding site of IDH1WT. The red arrow points to NADPH, and the blue arrow points to α-KG, green sphere is the divalent metal ion. The down circular picture is the enlarged view of the most common mutation site of IDH1WT. IDH1R132H is the most common mutation. IDH1 is composed of two subunits, heterozygous mutation (one subunit is mutated, and the other subunit is not mutated) of IDH1 can catalyze α-KG to yield D-2HG. Succinyl-coenzyme A (SucCoA); oxaloacetate (OAA); coenzyme A (CoA).
IDH1 gene locates in zone 3, band 4 (2Q34) of chromosome 2, with a total length of 18,917 nucleotides. IDH1 is an asymmetric homodimer composed of two subunits, each with 414 amino acid residues. Each protein subunit is composed of 3 domains: large functional domain (located at AA1–103 and 286–414), and has a typical Rossmann fold; small domain (located at AA104–136 and 186–285), forming α/β sandwich structure; Clasp domain (located at AA137–185), and folds into two anti-parallel β-sheets30. The large and small domains are connected by β-sheets, and there are two cracks on their sides31 (Fig. 4). The function was performed by forming two protein subunits into hydrophilic active sites. Deep fissures include NADP binding sites and isocitrate metal ion binding sites (Fig. 3), which can regulate the active and inactive state of IDH1 and the release of α-KG and NADPH30. After completion of catalysis, IDH1 can recombine NADP+ and isocitrate, changing its conformation back to its inactive form30. Shallow fissures are involved in the conformational changes of the homodimer IDH1.
Figure 4.

Amino acid sequence, mutation site, and mutation-related disease of IDH1. The human IDH1 is composed of 414 amino acids. In this figure, rectangles of different colors are used to show the structural characteristics of IDH1. The black rectangle marks the mutation site of IDH1, and the table below shows the disease corresponding to the mutation. Acute myeloid leukemia (AML); adenoid cystic carcinoma (AdCC); astrocytoma (A); breast neoplasm (BN); enchondromatosis (E); lymphoma (L); glioblastoma (GBM); glioblastoma multiforme (GM); glioma susceptibility 1 (GLM1); hepatocellular carcinoma (HCC); lung adenocarcinoma (LUAD); malignant melanoma of skin (CMM); medulloblastoma (MDB); metaphyseal chondromatosis (MC); multiple myeloma (MM); myelodysplastic syndrome (MDS); neoplasm of brain (NB); neoplasm of the large intestine (NLI); oligodendroglioma (ODGM); prostate adenocarcinoma (PAAD). The above information cited from the Uniport Database. Update [2020]. URL: https://www.uniprot.org/uniprotkb/O75874/feature-viewer.
2.2. IDH1 mutation is associated with cancer
In 2006, a sequencing study on human breast cancer and colon cancer revealed for the first time that IDH1 mutation was associated with cancer18. In 2008, the researchers set the research object as glioblastoma (GBM) and determined the point mutation of IDH1R132 by using the whole exome sequencing technology, suggesting that the mutation occurred mostly in young and secondary cancer patients19. Another group detected IDH1 mutation in acute myeloid leukemia (AML) for the first time32. And IDH2 mutation was found in some cancers33 (Fig. 2B). Since the significance of IDH1/2 mutation was clarified, there have been a lot of studies all over the world to analyze their mutation frequencies in different cancer species. The next-generation sequencing technology has greatly promoted research progress20.
In AML with IDH1/2 mutation, the mutation frequency is about 20%34. In gliomas, IDH1/2 mutations are mainly seen in secondary GBM, oligodendroglioma, astrocytoma, and other low-grade malignant gliomas35. In 2021, a next-generation sequencing study involving 20 common solid tumors (more than 28,000 patients in total) showed that the incidence of IDH1/2 mutation is only 1.3%, and the mutation frequency in lung cancer, colorectal cancer, liver cancer, stomach cancer, and other solid tumors is only 0.5%–3%20. The unique solid tumor with IDH1/2 mutation frequency exceeding 5% is cholangiocarcinoma (low incidence)36,37. In addition, IDH1/2 mutation frequency is also high in rare cancers such as chondrosarcoma (38%–86%), undifferentiated carcinoma of the paranasal sinuses (49%–82%), and angioimmunoblastic T-cell lymphoma (20%–30%)38. At present, IDH1 mutation mainly occurs in the arginine132 residue, including R132H (the most common type), R132C, R132L, R132S, R132G, and R132P (Fig. 4). R172K is the most common IDH2 mutation. However, IDH1 and IDH2 mutations are mutually exclusive and rarely occur simultaneously. Currently, the conclusions of various studies are consistent, that is, cancers with IDH1/2 mutations are not common. Even so, targeted IDH1 mutations have also brought precise treatment for AML, glioma, cholangiocarcinoma, and rare cancers39.
2.3. The regulation mechanism of IDH1 on cancer
2.3.1. IDH1-mediated metabolic reprogramming
2.3.1.1. IDH1 mutation leads to accumulation of D-2HG
Under physiological conditions, IDH1 catalyzes the oxidative decarboxylation of isocitrate to α-KG39 (Fig. 3). Once the gain of function mutation occurs, the affinity between mutant IDH1 and substrate decreases, and the activity of IDH1WT is inhibited by forming heterodimers40 (Fig. 3). Further, α-KG is catalyzed by mutant IDH1 to generate a large amount of cancer-promoting metabolite D-2HG41. It has been reported that an amazing 30 mmol/L D-2HG can be found in IDH1 mutated cancers42. D-2HG has a similar structure with α-KG. D-2HG can competitively suppress many α-KG-dependent enzymes, mainly including hydroxylation-related enzymes (such as prolyl-hydroxylase (PHD)), histone methylation-related enzymes, and DNA/RNA methylation-related enzymes, which will greatly disrupt normal physiological activities43.
Hypoxia inducible factors-1α (HIF-1α) is a key component of the transcription factor HIF-1, which plays an important role in hypoxia conditions. It can sense low oxygen levels in cells, regulate the expression of genes related to angiogenesis, glucose metabolism, and other signaling pathways critical for tumor growth. HIF-1α is also prevalent in solid tumors44. PHD can adjust the stability of HIF-1α. Under normal partial pressure of oxygen, proline residues of HIF-1α were hydroxylated by PHD, then ubiquitinated, and finally degraded by the proteasome. Under anoxic conditions, however, hydroxylation will not occur, which will eventually lead to HIF-1α accumulation43. The HIF-1α subunit is transferred to the nucleus to form a heterodimer with the HIF-1β ligand subunit (aryl hydrocarbon receptor nuclear translocator), and then specifically binds to the hypoxia-responsive elements, in turn, induces the transcription of hypoxia-related genes, and promotes tumorigenesis45,46 (Fig. 5). Therefore, mutant IDH1 inhibits the activity of PHD and improves the expression level of HIF-1α, and then, expression of target genes such as vascular endothelial growth factor, hexokinase, glucose transporter-1, phosphofructokinase, and phosphoglycerate kinase 1 increases, which may promote tumor cell growth, invasion, angiogenesis, and metastasis40,47.
Figure 5.

IDH1-mediated metabolic reprogramming. In general, IDH1 mutation increases the level of D-2HG and the expression of cancer-promoting proteins through the HIF-1α signal pathway. In addition, IDH1 mutation also inhibits the synthesis of ATP, results in a decline in antioxidant capacity, and causes erythroid cell maturation disorder via the inhibition of α-KG dehydrogenase. In the low-nutritional stated PDAC cells, the levels of IDH1 and NADPH increased, enhancing the mitochondrial function to maintain the growth of cancer cells. The black arrow means the promotion effects. The black T-shaped arrow means inhibition effects. The red arrow means the final effect caused by IDH1 mutations. The blue arrow means the change of IDH1 level in the low-nutritional stated PDAC cells and the resulting final effect. Vascular endothelial growth factor (VEGF), hexokinase (HK), glucose transporter-1 (GLUT1), phosphofructokinase (PFK), phosphoglycerate kinase 1 (PGK1), prolyl-hydroxylase (PHD), hypoxia inducible factors-1α (HIF-1α), hypoxia-responsive elements (HERs), heme oxygenase-1 (HO-1), reactive oxygen species (ROS), glutathione (GSH).
Electron transport chain is composed of five main complexes. Complexes I, III, and IV establish proton gradients by transferring electrons to oxygen molecules, and finally, complex V uses this proton gradient to promote ATP synthesis. It has been reported that D-2HG can inhibit complex IV48 and complex V49, which will greatly reduce the proportion of energy supply through oxidative phosphorylation. In order to better meet the energy demand, cancer cells will adjust the energy supply mode to aerobic glycolysis (Fig. 5).
Recently, it has been pointed out that mutant IDH1 can also cause heme synthesis disorder and reduce the level of heme catabolites (biliverdin and bilirubin). Mice with Idh1 mutation will suffer from erythrocytic dysplasia50. D-2HG produced by mutant IDH1 can directly inhibit the activity of α-KG dehydrogenase, which in turn, leads to the reduction of succinyl-coenzyme A, causes the disorder of heme synthesis, and finally blocks the differentiation of erythroblasts in the late stage. At the same time, heme synthesis disorder affects the expression of heme oxygenase-1 and reduces the level of heme catabolite. Finally, it will promote the excessive accumulation of reactive oxygen species in cells, induce the death of IDH1 mutant erythroid cells, lead to the imbalance of myeloid erythroid development of bone marrow precursor cells, and participate in the occurrence of myeloid tumors50 (Fig. 5).
2.3.1.2. IDH1 mutation leads to the decrease of NADPH level
NADPH is a necessary co-factor for cell functions such as lipid metabolism, glucose metabolism, and anti-oxidative stress28,51,52. NADPH is an important electron donor for glutathione, thioredoxin, and other transcription factors. Moreover, NADPH plays an important role in regulating the redox state of cells. IDH1 catalyzes the conversion of isocitrate to α-KG is accompanied by the production of NADPH, which has the function of maintaining the balance of cell redox reaction and regulating the level of reactive oxygen species (ROS). NADPH converts glutathione disulfide into glutathione (GSH), and GSH is the main antioxidant of ROS43 (Fig. 5). IDH1 mutation leads to the decrease of NADPH level, which makes cells more susceptible to damage by ROS, causing cell membrane damage and enzyme activity changes. At the same time, DNA damage leads to genome instability, which ultimately leads to carcinogenesis. Research by Shi et al.53 showed that in glioma cells overexpression of the IDH1 mutant gene, the intracellular NADPH level is reduced, which in turn leads to inhibition of the growth of glioma cells. Studies have reported that the treatment of glioblastoma can be enhanced by targeting IDH1-mediated NADPH biosynthesis54.
ROS is the main molecule produced by the body during oxidative stress and has long been considered as an important factor for the development and recurrence of cancers. ROS accumulation can induce cell apoptosis, but moderate ROS production is one of the important components of inflammatory characteristics of innate immune response55. Lipopolysaccharide (LPS) extracted from Gram-negative bacteria are typical stimuli that trigger inflammatory cascades in vitro and in vivo. IDH1 helps to reduce the ROS induced by LPS or H2O2 treatment56,57. In vitro studies have found that LPS can induce the expression of IDH1 and reduce the ROS induced by LPS or H2O2 in rat murine macrophages RAW 264.7, at the same time, the overexpression of IDH1 can reduce the level of intracellular peroxides, which may reduce ROS level in this way, thereby inhibiting tumor development57. In vivo studies have shown that IDH1 protects mouse liver cells from damage caused by endotoxin-induced oxidative stress by regulating the ratio of NADP+/NADPH in the cell58. This suggests that stimulating IDH1 activity in inflammatory responses, including in the early stages of septic shock, may be an effective therapeutic strategy to reduce oxidative stress. Under the induction of tumor promoter Tissue polypeptide antigen and UVC, IDH2 in JB6 P+ was not significantly changed. IDH1 knockout and overexpression enhances and inhibits Tissue polypeptide antigen-induced tumor-like transformation of cells, respectively59. Therefore, regulating the activity of IDH1 may be one of the effective ways to reduce inflammatory oxidative stress during tumorigenesis and development.
2.3.1.3. Metabolic remodeling induced by wild-type IDH1
Researchers found an RNA-binding protein, human antigen R (HuR), which can enhance mitochondrial function and antioxidant capacity when nutrition is deficient60. After HuR knockdown, the level of IDH1 is also significantly reduced. In addition, some studies have shown that HuR participates in the pre-translation modification of IDH1. Therefore, researchers believe that IDH1 and HuR are closely related and are also crucial to the survival of cells in a low-nutrient environment61. Researchers found that ROS in tumor cells accumulated in the early stage under low glucose culture conditions, but decreased on the third day, accompanied by a compensatory increase in NADPH over time. Only when IDH1WT is knocked down, the survival rate of cancer cells in low glucose culture will be reduced. In addition, the IDH1WT-knockout PDAC cell line lost its antioxidant capacity, suggesting that IDH1WT plays an important role in the survival of tumor cells in a low glucose environment62. After exogenous supplement of α-KG, the level of related metabolites in cells recovered, and mitochondria were able to continue to maintain the survival of cells, suggesting that the IDH1 metabolite α-KG influences the survival of cells under hypoxia by mediating mitochondrial function62 (Fig. 5). More importantly, this study pointed out that IDH1 mutant inhibitors can inhibit wild-type IDH1 in the cell environment with low magnesium ion concentration, which will provide new therapeutic ideas for the treatment of IDH1-related cancers. At the same time, it also puts forward higher requirements for clinical diagnosis and detection technology.
2.3.2. Effect of IDH1 on epigenetics
D-2HG can occupy the same binding pocket of α-KG and competitively inhibit many α-KG-dependent enzymes, such as DNA demethylase and histone demethylase, which can lead to DNA and histone hypermethylation, and then make the abnormal epigenetic regulation, block cell differentiation, cause abnormal expression of a series of oncogenes, suppressor oncogene, and signal transduction genes, resulting in the occurrence of cancers63, 64, 65, 66, 67, 68.
2.3.2.1. D-2HG inhibits DNA demethylase
TET oncogene family member 2 (TET2) plays an important role in stem cell differentiation, epigenetic regulation, and the occurrence of hematopoietic malignancies. TET2 achieves DNA demethylation by converting 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC)69. The loss of TET2 function can cause DNA hypermethylation of hematopoietic stem cells, which in turn leads to abnormal gene expression70. The TET2 gene is mutated in myeloid diseases including acute myeloid leukemia71. So far, more and more evidences show that TET2 mutations play an important role in AML. The study of Figueroa et al.72 showed that abnormal DNA methylation is a hallmark of AML. Clinical observations have demonstrated that IDH1 and IDH2 mutations lead to a hypermethylated phenotype, destroy the function of TET2, and impair hematopoietic differentiation72. In addition, IDH1/2 mutations in AML and TET2 mutations have similar DNA methylation phenotypes. Importantly, they are mutually exclusive72, implying that they have the same pathway of action. The research further supports the latest data on this cooperative mechanism73. The oncogenome map is used to detect 207 patients with glioblastoma multiforme and found that IDH1 mutation is closely related to glioma-CpG island methylator phenotype74. Among patients with DNA hypermethylation, 78% of patients have IDH1 gene mutations, while no IDH1 mutations are found in patients without DNA hypermethylation74. These studies show that the mutant IDH1R132H and its product D-2HG can inhibit TET2 from catalyzing the production of 5hmC, leading to DNA hypermethylation, which in turn leads to cancer formation (Fig. 6).
Figure 6.

Effect of IDH1 on Epigenetics. IDH1 mutation causes the level of D-2HG to increase, which in turn promotes the hypermethylation of DNA and histones, and eventually induces cancer. The black arrow means the promotion effects. The black T-shaped arrow means inhibition effects. The black dotted arrow means the time sequence. “Me” means methylation. Receptor-interacting protein 3 (RIP3), DNA methyltransferase 1 (DNMT1), TET oncogene family member 2 (TET2), 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), JmjC domain containing histone demethylase (JHDM), zinc finger E-box binding homeobox 1 (ZEB1), epithelial–mesenchymal transition (EMT).
In addition to inhibiting the DNA hypermethylation of IDH1/2 mutant cells by mediated TET2 activity, D-2HG can also regulate DNA methylation through DNA methyltransferase 1 (DNMT1). D-2HG binds to DNMT1 and promotes its separation from the receptor-interacting protein 3 (RIP3) promoter, induces hypermethylation, inhibits RIP3 protein, thereby inhibits RIP3-dependent cell necrosis, and promotes tumorigenesis75. The D-2HG produced by IDH1 mutant cells promotes the binding of DNMT1 to the Fibulin-5 promoter, leading to methylation, and ultimately enhancing the migration and proliferation of non-small cell lung cancer cells76 (Fig. 6).
2.3.2.2. D-2HG inhibits histone demethylase
Histone methylation is an important form of epigenetic modification. JmjC domain containing histone demethylase (JHDM) is an important histone demethylase, which mainly catalyzes the demethylation of histones H3K4, H3K9, H3K27, H3K36, and H4K2077, 78, 79, 80, 81, 82. Histone demethylases play an important role in human diseases such as neurological disorders and cancer83,84. In vitro studies have shown that D-2HG inhibits the activity of histone demethylase in cells65. Xu et al.65 further used in vivo studies to verify that D-2HG can inhibit the activity of a variety of histone demethylases. Lu et al.66 found that a variety of histone methylation markers increased in the cultured cell models expressing IDH1R132H mutation or treated with cell-permeable D-2HG. Interestingly, as the number of cell passages increased, DNA methylation also appeared in cells. It is worth noting that the appearance of histone methylation is always earlier than DNA methylation85. The study indicated that astrocytes expressing mutant IDH1R132H showed increased H3K9me2, H3K27me3 and H3K36me3 levels, as well as decreased 5hmC and hypermethylation, further proving that IDH1 mutation is the molecular basis of G-CIMP86. Research also showed that the expression of IDH1 and IDH2 mutants inhibited 5mC hydroxylation and histone demethylation65. In the azoxymethane mouse bowel cancer model, the level of D-2HG in the tumor is elevated87. Colvin et al.88 found that D-2HG can induce histone modifications, leading to increased gene expression in the promoter region of Zinc finger E-box binding homeobox 1 (ZEB1) gene, which is the main regulator of epithelial–mesenchymal transition (EMT). D-2HG can also directly induce EMT of colorectal cancer cells88. EMT promotes cancer cells to invade local tissues and enter the blood, leading to distant organ metastasis89. D-2HG levels are elevated in clinical samples of colorectal cancer, especially those related to distant metastasis, suggesting the role of D-2HG in tumor metastasis88 (Fig. 6).
2.3.3. Effect of IDH1 mutation on cancer immune microenvironment
At present, many studies have shown that abnormal accumulation of metabolites can lead to tumorigenesis. More and more studies show that IDH1 is involved in the regulation of the cancer microenvironment (acquired immunity and natural immunity), and the combination of IDH1 inhibitors and cancer immunotherapy drugs shows good therapeutic effects.
2.3.3.1. Regulation of IDH1 on acquired immune system
D-2HG produced by mutant IDH1 may cause the immune microenvironment of GBM and other cancers to be suppressed90. By inhibiting the expression and activation of signal transducer and activator of transcription 1, D-2HG reduces the secretion of chemokine CXC chemokineligand-10 in glioma cell lines, thereby inhibiting cytotoxic T lymphocyte infiltration at tumor sites91. The transporter solute carrier family 13 member 3 assists T lymphocytes in uptake of D-2HG. Excess D-2HG inhibits ATPase, reduces ATP production, and weakens phospholipase C gamma phosphorylation, both leading to the reduction of nuclear translocation of activated T cell nuclear factor, and eventually reducing the activation of T lymphocytes92. In high-grade glioma, D-2HG can enhance the tryptophan-2,3-dioxygenase activity in macrophages, thus promoting the metabolism of l-tryptophan to the aryl hydrocarbon receptor ligand kynurine. Kynurine induced aryl hydrocarbon receptor translocation to the nucleus, where it increased interleukin 10 production and decreased the expression of costimulatory molecules cluster of differentiation 86, cluster of differentiation 80, and major histocompatibility complex II. This leads to reduced antigen presentation and increased T cell inhibition, thus driving a more immunosuppressive tumor microenvironment93. In addition, D-2HG can also induce transient hypermethylation of programmed cell death-ligand 1 promoter, thereby reducing the expression of PD-L194. Some studies have shown that the combination of IDH1 inhibitor and anti-PD-L1 can significantly prolong the survival period of IDH1 mutant glioma mice95. In general, D-2HG inhibits anti-tumor T cell immunity. Inspired by this, targeted mutant IDH may have a synergistic effect with immunotherapy (Fig. 7).
Figure 7.

Effect of IDH1 mutation on cancer immune microenvironment. IDH1 mutation will cause the cancer microenvironment to be in a state of immunosuppression. The black arrow means the promotion effects. The black T-shaped arrow means inhibition effects. The red arrow means the final effects caused by IDH1 mutation. Signal transducer and activator of transcription 1 (STAT1), CXC chemokine ligand-10 (CXCL10), solute carrier family 13 member 3 (SLC13A3), phospholipase C gamma (PLC-γ), nuclear translocation of activated T cell nuclear factor (NFAT), tryptophan-2,3-dioxygenase (TDO), l-tryptophan (L-Trp), aryl hydrocarbon receptor (AhR), kynurine (Kyn), interleukin 10 (IL-10), cluster of differentiation 86 (CD86), cluster of differentiation 80 (CD80), major histocompatibility complex II (MHCII), programmed cell death-ligand 1 (PD-L1), natural killer cell group 2D (NKG2D), NKG2D ligand (NKG2DL), natural killer (NK).
2.3.3.2. Regulation of IDH1 on natural immune system
D-2HG also inhibits the natural immune function and inactivates complement in IDH1 mutant astrocytes. The mechanism is that D-2HG inhibits the assembly of C5 convertase in the classical pathway of complement activation and inactivates the assembled C3/C5 convertase. At the same time, it inhibits the assembly of C3/C5 convertase in the alternative pathway96. In these ways, glioma cells can resist complement mediated lysis and phagocytosis. The receptor natural killer cell group 2D (NKG2D) on natural killer cells activates NK cells when it connects with the NKG2D ligand (NKG2DL) on the surface of target cells (such as tumor cells), thereby killing tumor cells. The expression level of NKG2D on the surface of IDH1 mutant astrocytes is lower than that of wild-type IDH1, which may be related to the hypermethylation of NKG2D promoter97. The down-regulation of NKG2D helps tumor cells escape the cytolysis of NK cells (Fig. 7).
2.3.4. Effect of IDH1 on cancer phenotype
2.3.4.1. IDH1 induces apoptosis or autophagy depending on cell type
The study of Gilbert et al.98 showed that D-2HG can trigger the apoptosis of LN18 cells, while the apoptosis of U87MG cells did not change. This indicates that the apoptotic response to D-2HG is cell type specific. In addition, both cell lines did not show significant changes in the activity of caspase 8-dependent exogenous pathways98. D-2HG also increased the formation of autophagosomes in U87MG cells, which is a sign of autophagosome formation98. Their research showed that IDH1 mutations can induce apoptosis and autophagy, but these effects vary greatly with cell types.
2.3.4.2. IDH1 induces apoptosis through multiple signaling pathways
Prostate apoptosis response-4 (Par-4) is a tumor suppressor protein. Par-4 can promote the apoptosis of a variety of cancer cells. Par-4 can kill human cancer cells from pancreas99, cervix100, lung100, prostate101, kidney101, endometrium102, and colon103. D-2HG can inhibit the transcription of Par-4 in vitro by inhibiting promoter activity and enhancing mRNA degradation104. The apoptosis-inducing selectivity in the cancer cell domain within Par-4 is highly active on glioma cells. Among IDH1 wild-type high-grade gliomas, gliomas expressing more Par-4 have a significantly longer median survival104 (Fig. 8).
Figure 8.

The effect of IDH1 on cancer phenotype and its molecular mechanism. The black arrow means the promotion effects. The black T-shaped arrow means inhibition effects. “+” means synergistic effect. The colors of targets represent the influence on IDH1WT and IDH1 (upstream), or the influence on cancer cell phenotype (downstream). Prostate apoptosis response-4 (Par-4), B-cell lymphoma-2 (Bcl-2), B-cell lymphoma-XL (Bcl-XL), myeloid cell leukemia 1 (Mcl-1), adenosine 5′-monophosphate-activated protein kinase (AMPK), mechanistic target of rapamycin (mTOR), C/EBP homologous protein (CHOP), c-Jun N-terminal kinase (JNK), poly ADP-ribose polymerase (PARP), paired box 5 (PAX5), phosphoinositide 3-kinase (PI3K), protein kinase B (AKT).
Anti-apoptotic B-cell lymphoma-2 (Bcl-2) family members, such as B-cell lymphoma-XL (Bcl-XL) and myeloid cell leukemia 1 (Mcl-1), are highly expressed in human glioblastoma. Compared with wild-type IDH1 cells, the apoptosis induced by Bcl-XL inhibition was significantly more in IDH1 mutant cells105. In anaplastic astrocytoma, the level of Mcl-1 in IDH1 mutant cells is lower than that in IDH1 wild-type cells. The specific knockdown of Mcl-1 makes glioblastoma cells sensitive to apoptosis mediated by Bcl-XL inhibition105. The energy expenditure mediated by D-2HG activates adenosine 5′-monophosphate-activated protein kinase (AMPK), which leads to weakening of protein synthesis and mechanistic target of rapamycin (mTOR) signal, and ultimately to the decrease of Mcl-1105. These data indicate that IDH1 mutant gliomas are susceptible to Bcl-XL inhibition (Fig. 8).
The C/EBP homologous protein (CHOP) is a member of the C/EBP family, and its expression level is relatively low during the growth of normal cells, but it is significantly increased when the endoplasmic reticulum is stressed106. Since CHOP lacks a stable DNA binding domain, it needs to be heterodimerized with other members of the C/EBP family to transcriptionally regulate the expression of the response gene107. Research by Yang et al.106 found that in melanoma cells, endoplasmic reticulum stress increases the expression and activity of CHOP, which directly binds to the IDH1 promoter region after forming a heterodimer with C/EBPβ, transactivating IDH1 expression. The activated IDH1 promotes the degradation of HIF-1α and down-regulates it, which in turn makes melanoma cells apoptosis induced by hypoxia (Fig. 8).
Research by Li et al.108 showed that D-2HG competitively inhibits succinate dehydrogenase (SDH), preferentially inducing succinyl-CoA accumulation and excessive succinylation in mitochondria. IDH1 mutation or SDH inactivation can cause excessive succinylation, cause respiratory depression, and induce cancerous metabolism and mitochondrial depolarization108. These mitochondrial dysfunctions cause Bcl-2 to accumulate on the mitochondrial membrane and cause apoptosis resistance in hypersuccinylated cells108 (Fig. 8).
c-Jun N-terminal kinase (JNK), belonging to the family of mitogen-activated protein kinases, responds to stress stimuli such as serum starvation, and also plays an important role in the apoptosis pathway. Jiang et al.109 knocked IDH1R132Q into mutant mouse cells and found that D-2HG inhibited JNK activation induced by serum starvation and prevented cell apoptosis. During starvation, cell division cycle 42 (Cdc42) usually destroys the self-inhibition of mixed lineage kinase 3 (MLK3) and triggers the MLK3–MKK4/7 (mitogen-activated protein kinase kinase, MKK) –JNK–Bim (Bcl-2 interacting mediator of cell death) apoptosis cascade109. D-2HG binds to Cdc42 and eliminates its binding to MLK3, causing MLK3 inactivation and apoptosis109 (Fig. 8).
In the study of Rosiak et al.110, IDH1R132H-positive neural stem cells and their derivatives have a high percentage of apoptotic cells. By analyzing the activity of poly ADP-ribose polymerase (PARP) and caspase-3, it was confirmed that the expression of IDH1R132H increased the apoptosis sensitivity of neural stem cells and their derivatives110. Strong apoptosis causes insufficient differentiation of cells expressing IDH1R132H (Fig. 8).
According to reports, long non-coding RNAs (IncRNAs) are important regulators in tumorigenesis. IDH1 antisense RNA 1 (IDH1-AS1) is an IncRNA that can interact with genes to regulate the Warburg effect. The study by Zhang et al.111 found that paired box 5 is a transcriptional activator of IDH1-AS1, and the up-regulation of IDH1-AS1 induced by paired box 5 promotes the proliferation and apoptosis of prostate cancer by regulating autophagy related protein 5 (ATG5)-mediated autophagy. Wang et al.112 used primary glioblastoma cell lines U251 and U87-MG to study the effect of IDH1-AS1 on the growth of glioma cells, and found that IDH1-AS1 overexpression inhibited cell proliferation and blocked the cell cycle in the G1 phase, and the protein expression levels of cyclinD1, cyclin A, cyclin E, CDK2 and CDK4 (cyclin dependent kinase, CDK) decrease, and cell apoptosis increases (Fig. 8).
p53 is an important tumor suppressor protein. On one hand, it can activate various responses, including arresting the cell cycle and promoting apoptosis113. These all seem to help suppress cancers. On the other hand, p53 responds to stress caused by factors such as oncogene activation, hypoxia or starvation113. These can promote the development of cancers. In addition, p53 can also play a role in normal development. The activation of p53 may also have adverse effects and may cause disease. Parsons et al.19 found that the frequency of p53 mutations in human glioblastoma with IDH1 mutation is very high. The study by Hu et al.114 showed that patients with osteosarcoma with high IDH1 expression have very high p53 expression. Therefore, IDH1 may be related to p53. Subsequent studies have shown that IDHR132H mutation is significantly related to p53, but is negatively related to epidermal growth factor receptor (EGFR) mutation115. Next, we should investigate whether these three molecules participate in a common signal pathway (Fig. 8).
2.3.4.3. Effects of IDH1 on the proliferation, migration, and drug resistance of cancer
The Wnt/β-catenin signaling pathway plays a key role in normal embryonic development and promotes the metastasis of several cancers116, 117, 118, 119, 120. Research by Cui et al.121 showed that the R132H mutation in IDH1 negatively regulates Wnt/β-catenin signaling, thereby reducing the proliferation, cell survival and invasion of human gliomas. IDH1 mutation causes cell cycle arrest in G1 phase, reduces the ratio of G2/M phase, inhibits cell proliferation, down-regulates cell invasion, and improves the prognosis of patients with glioma122. Research by Wang et al.123 also showed that IDH1 mutations can cause cell cycle arrest in G1 phase, reduce cell proliferation and invasion, and increase sensitivity to chemotherapy (Fig. 8).
Shen et al.124 studied the function of IDH1 in cell migration. Studies have shown that IDH1 regulates the migration of primary GBM cells by changing the level of α-KG. The function of the IDH1/α-KG axis may depend on the regulation of phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mTOR pathway. Overexpression of IDH1 or α-KG treatment both promoted the PI3K/AKT/mTOR pathway124. Treatment with mTOR-specific inhibitor rapamycin can reverse the increase in cell migration caused by overexpression of IDH1 and α-KG treatment (Fig. 8).
Studies have found that overexpression of the wild-type IDH1 gene does not affect cell cycle, apoptosis, and invasion ability. However, it leads to resistance to high-dose temozolomide (TMZ) chemotherapy in vivo and in vitro. The mechanism is that overexpression of wild-type IDH1 reduces the Bcl-2-associated X (Bax)/Bcl-2 ratio and caspase-3 activity, and inhibits TMZ-induced apoptosis123. Calvert et al.125 demonstrated that wild-type IDH1 is overexpressed in primary GBM, and the genetic or pharmacological inhibition of IDH1 activity reduces the growth of tumor cells, part of the mechanism is to increase the drug's sensitivity to apoptosis. This finding indicates that the up-regulation of IDH1 represents a common metabolic adaptation of GBM for macromolecule synthesis and treatment of drug resistance. In addition, some researchers first proposed the mechanism of acquired drug resistance of IDH through clinical case studies. The research results show that when receiving the same type of inhibitor treatment, the conversion between IDH1 and IDH2 mutants will occur (IDH1 inhibitor will cause mutant IDH1 to change into mutant IDH2), and cancer cells can continue to produce D-2HG126. These findings confirm the roles of continuous D-2HG production in cancer progression and propose a therapeutic strategy to prevent or overcome drug resistance, that is, the combination of IDH1 and IDH2 inhibitors. Therefore, DNA sequencing and D-2HG content determination are necessary for the early stage of IDH-targeted therapy (Fig. 8).
3. IDH1 inhibitors
In Table 1, we listed IDH1 inhibitors in the clinical research stage and showed their clinical therapeutic potential. In Table 2, we summarized IDH1 inhibitors in the preclinical research stage based on the core skeleton. The structural diversity of these compounds laid the foundation for obtaining inhibitors with higher activity and better selectivity. In addition, in Table 3, we have collected the IDH1 combination drug cases currently in the clinical research stage, which may provide new treatment methods for the treatment of refractory and acquired drug-resistant cancers.
Table 1.
IDH1 inhibitors in the clinical research stage.
| Code name | NCT No. | Condition | Clinical efficacy |
|---|---|---|---|
| AG-120 (ivosidenib) Launched, 2018 |
NCT02989857 (Phase III) NCT03839771 (Phase III) NCT03173248 (Phase III) |
Cholangiocarcinoma; Gliomas; AML; Myelodysplastic syndrome |
Cholangiocarcinoma132: mPFS: 6.9 months, SAE: 30%; Advanced cholangiocarcinoma with IDH1 mutation134: mOS: 10.3 months, SAE: 2%; Gliomas135: ORR: 2.9%, mPFS: 13.6 months; AML128: ORR: 41.6%, CRR: 21.6%. |
| AG-221 (enasidenib) Launched, 2017 |
NCT03839771 (Phase III) NCT02577406 (Phase III) NCT04822766 (Phase III) |
Myeloid leukemia; AML; Myelodysplastic syndrome |
AML136: ORR: 40.3%, mOS: 19.7 months; Myelodysplastic syndromes137: ORR: 53%, mOS: 16.9 months. |
| AG-881 (vorasidenib) | NCT04164901 (Phase III) | Glioma | Glioma138: ORR: 18%, mPFS: 36.8 months. |
| DS-1001b |
NCT04458272 (Phase II) NCT05303519 (Phase II) |
Glioma | Glioma139: ORR: 17%, mPFS: 10.4 months. |
| FT-2102 (olutasidenib) |
NCT04013880 (Phase I/II) NCT03684811 (Phase I/II) NCT02719574 (Phase I/II) |
AML; Myelodysplasia; Glioma; Cholangiocarcinoma |
Glioma140: ORR: 48%. |
| BAY-1436032 |
NCT03127735 (Phase I) NCT02746081 (Phase I) |
AML; Metastatic cancer; Glioma |
AML149: ORR: 15%; Solid Tumors141: ORR: 11%. |
| IDH-305 |
NCT02977689 (Phase II) NCT02987010 (Phase II) |
Glioma; AML; myelodysplastic syndrome |
AML142: CR/CRi: 32%. |
| LSN-3410738 (LY-3410738) |
NCT04521686 (Phase I) NCT04603001 (Phase I) |
Metastatic cancer; AML; Chronic myelomonocytic leukemia; Myelodysplasia |
– |
| HMPL-306 |
NCT04272957 (Phase I) NCT04764474 (Phase I) NCT04762602 (Phase I) |
Leukemia; Myelodysplastic syndrome; Myeloid leukemia |
– |
| IDH1RpepvaccH (vaccine) | NCT02771301 (Phase II) | Neurologic cancer Glioma |
Glioma143: ORR: 84.4%, Three-year survival rate: 84%, TRAE: 90.6%, SAE: 3.1%. |
| PEPIDH1M (vaccine) | NCT02193347 (Phase I) | Glioma | – |
ORR, overall response rate; mOS, median overall survival; mPFS, median progression-free survival; SAE, serious adverse events; CRR, rate of complete remission; CR/Cri, complete remission/complete remission with incomplete recovery; TRAE, treatment related adverse events.
Table 2.
Inhibitors targeting IDH1 in the preclinical study stage.
| Compound | Structure | In vitro activity | In vivo activity/PK/ADME | Ref. |
|---|---|---|---|---|
| AGI-5198 |  |
IDH1R132H (enzyme): IC50 = 0.07 μmol/L; U87 (cell lines): IC50 = 0.07 μmol/L |
U87 R132H tumor xenograft mouse model: 2-HG inhibition: 89.4% (BID) and 69% (Single) |
158 |
| ML309 |  |
IDH1R132H (enzyme): IC50 = 96 nmol/L; U87 MG cells: 2-HG assay: EC50 = 509 nmol/L |
Male BALB/c nude mice: Tmax = 1.0 h; Cmax = 3625 ng/mL; t1/2 = 3.76 h |
160 |
| (R)-1-(4-Cyanopyridin-2-yl)-N-((S)-1-((3,3-difluorocyclobutyl)carbamoyl)-2,3-dihydro-1H-inden-1-yl)-N-(3,5-difluorophenyl)-5-oxopyrrolidine-2-carboxamide (6f) | 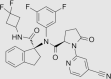 |
IDH1R132H (enzyme): IC50 = 45 nmol/L; IDH1R132C HT-1080 cells: 2-HG inhibition: IC50 < 5 nmol/L |
– | 161 |
| IDH-C227 | 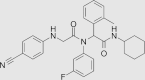 |
IC50 < 0.1 μmol/L against HT-1080 and 0.25 μmol/L against U87MG cells. | – | 187 |
| N-(2-(Cyclohexylamino)-1-(4-methoxy-2-methylphenyl)-2-oxoethyl)-N-(4-nitrophenyl)propiolamide (43) | 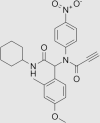 |
IDH1R132H (enzyme): IC50 = 961.5 nmol/L IDH1R132H U87 cells 2-HG inhibition: EC50 = 208.6 nmol/L |
– | 162 |
| IDH125 |  |
IDH1R132H (enzyme): IC50 = 0.22 μmol/L; IDH1R132H HCT116 cells 2-HG inhibition: IC50 = 0.66 μmol/L |
– | 163 |
| IDH662 | 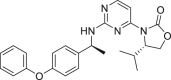 |
IDH1R132H (enzyme): IC50 = 0.01 μmol/L; IDH1R132H HCT116 cells 2-HG inhibition: IC50 = 0.022 μmol/L |
Plasma protein binding >99% |
163 |
| IDH889 |  |
IDH1R132H (enzyme): IC50 = 0.02 μmol/L; IDH1R132H HCT116 cells: 2-HG inhibition: IC50 = 0.014 μmol/L |
HCT116 R132H tumor xenograft mouse model (10 mg/kg, Oral) AUC: 3.6 μmol/(L·h), Cmax: 1.7 μmol/L Brain/plasma ratio: 1.4 |
163 |
| NI-1 |  |
IDH1R132H (enzyme): IC50 = 96 nmol/L |
– | 166 |
| (R)-4-(Fluoromethyl)-3-(2-(((S)-1-(1-(4-(trifluoromethyl)phenyl)-1H-imidazole-4-yl)ethyl)amino)pyrimidin-4-yl)oxazolidin-2-one (19) |  |
IDH1R132H HCT116 cells: 2-HG inhibition: IC50 = 0.039 μmol/L |
Rat liver microsomal (10 mg/kg, Oral) Clint: 7 μL/min/mg AUC: 180 μmol/(L‧h) |
164 |
| (R)-3-(2-((1-(1-(3-Chloro-4-fluorophenyl)-1H-imidazole-4-yl)cyclopropyl)amino)-5-fluoropyrimidin-4-yl)-4-((S)-1-fluoroethyl)oxazolidin-2-one (5t) | 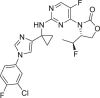 |
IDH1R132H (enzyme): IC50 = 35 nmol/L; IDH1R132H HT-1080 cells 2-HG inhibition: IC50 = 18 nmol/L |
Rat liver microsomal: Clint: 45 mL/min/g |
165 |
| (S)-4-Isopropyl-3-(6-(((S)-1-(2′-methoxy-[1,1′-biphenyl]-4-yl)ethyl)amino)pyrazin-2-yl)oxazolidin-2-one (3g) | 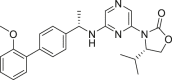 |
2-HG levels (%, IDH1 R132H): 31.9% (10 μmol/L) 2-HG levels (%, IDH1 R132C): 40.6% (10 μmol/L) |
PAMPA-BBB assaya: Permeability (×10−6 cm/s): 6.65 ± 0.42 |
166 |
| I-8 |  |
IDH1R132H (enzyme): IC50 = 135.6 ± 17.9 nmol/L IDH1R132C (enzyme): IC50 = 174.2 ± 22.1 μmol/L |
HT-1080 R132C tumor xenograft BALB/c mice 150 mg/kg could induce 30% inhibitory of 2-HG production |
167 |
| IDH1-IN-6 | 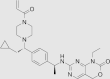 |
IDH1R132H (enzyme): IC50 = 6.27 nmol/L; IDH1R132C (enzyme): IC50 = 36.9 nmol/L; IDH1R132C HT-1080 cells: 2-HG inhibition: IC50 = 1.28 nmol/L |
– | 168 |
| SYC-435 (Compound 2) |  |
IDH1R132H (enzyme): Ki = 0.19 μmol/L; IDH1R132C (enzyme): Ki = 0.12 μmol/L |
– | 169 |
| 1-Hydroxy-6-(4-hydroxybenzyl)-4-methylpyridin-2(1H)-one (3) | 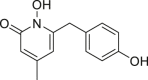 |
IDH1R132H (enzyme): Ki = 0.28 μmol/L; IDH1R132C (enzyme): Ki = 0.27 μmol/L |
– | 169 |
| Thiohydantoin16 (16) |  |
IDH1R132H (enzyme): Ki = 0.75 μmol/L; IDH1R132C (enzyme): Ki = 1.2 μmol/L |
– | 171 |
| (E)-5-((5-oxo-2-Thioxoimidazolidin-4-ylidene)methyl)pyridin-2(1H)-one (18) | 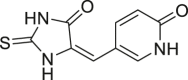 |
IDH1R132H (enzyme): Ki = 0.42 μmol/L; IDH1R132C (enzyme): Ki = 2.3 μmol/L |
– | 171 |
| GSK321 | 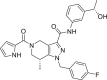 |
HT-1080 cells: 2-HG inhibition: EC50 = 85 nmol/L; IDH1R132H (enzyme): IC50 = 4.6 nmol/L; IDH1R132C (enzyme): IC50 = 3.9 nmol/L; IDH1R132G (enzyme): IC50 = 2.9 nmol/L |
2-HG inhibition: (IDH1R132C HT-1080 fibrosarcoma cells): EC50 = 320 nmol/L |
172 |
| GSK864 |  |
IDH1R132H (enzyme): IC50 = 15.2 nmol/L; IDH1R132C (enzyme): IC50 = 8.8 nmol/L; IDH1R132G (enzyme): IC50 = 16.6 nmol/L |
– | 172 |
| (6aS,7S,10aR)-7-Methyl-8-oxo-10a-phenyl-2-(phenylamino)-5,6,6a,7,8,10a-hexahydrobenzo[h]quinazoline-9-carbonitrile (1) |  |
IDH1WT (enzyme): IC50 = 410 nmol/L |
– | 172 |
| BRD2879 |  |
IDH1R132H (enzyme): IC50 = 50 nmol/L; IDH1R132C (enzyme): IC50 = 2.5 μmol/L |
Human plasma protein binding: 99.5% | 173 |
| (S)-2-((1-(6-Chloro-2-oxo-1,2-dihydroquinolin-3-yl)ethyl)amino)-4-methoxypyrimidine-5-carbonitrile (63) |  |
IDH1R132H (enzyme): IC50 = 18 nmol/L; IDH1R132C (enzyme): IC50 = 130 nmol/L; IDH1R132H HCT116 cells: 2-HG inhibition: IC50 = 45 nmol/L; IDH1R132C HCT116 cells: 2-HG inhibition: IC50 = 233 nmol/L |
HCT116 R132H/R132C xenograft bearing female BALB/c nude mice: 2-HG inhibition: IC50 = 49 nmol/L (R132H); IC50 = 46 nmol/L (R132C). |
174 |
| Bisimidazole 3 |  |
IDH1R132H (enzyme): IC50 = 13 ± 5 nmol/L; IDH1R132H HEK-293 cells: 2-HG inhibition: IC50 = 81.5 nmol/L |
– | 175 |
| 3-(1-(3-(1H-Imidazole-1-yl)propyl)-6-chloroindolin-3-yl)-4-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-pyrrole-2,5-dione (11e) |  |
IDH1R132H (enzyme): IC50 = 0.16 ± 0.04 μmol/L |
– | 176 |
| Licochalcone A | 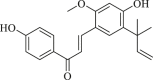 |
IDH1R132H (enzyme): IC50 = 76.87 μmol/L; IDH1R132C (enzyme): IC50 = 5.176 μmol/L |
– | 165 |
| CRUK-MI 20a (20a) |  |
IDH1WT (enzyme): IC50 = 0.27 ± 0.22 μmol/L |
– | 177 |
| HMS-101 | 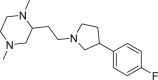 |
IDH1R132H (enzyme): IC50 = 5 μmol/L; IDH1R132C (enzyme): IC50 = 4 μmol/L |
– | 178 |
| Clomifene | 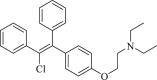 |
IDH1R132H (enzyme): IC50 = 50.20 ± 0.12 μmol/L; IDH1R132C (enzyme): IC50 = 42.33 ± 0.31 μmol/L; IDH1R132H HT-1080 cells: 2-HG inhibition: IC50 = 37.86 ± 0.32 μmol/L |
HT-1080 R132H tumor xenograft mouse model: 100 mg/kg Clomifene could induce 57.38% inhibitory of D-2HG production. |
179 |
| ZX06 | 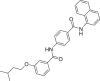 |
2-HG levels (%, IDH1 R132H): 54.9% (10 μmol/L); 2-HG levels (%, IDH1 R132C): 48.4% (10 μmol/L) |
PAMPA-BBB assay: Permeability (×10−6 cm/s): 8.15 ± 0.29 |
180 |
| DC_H31 | 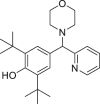 |
IDH1R132H (enzyme): IC50 = 0.41 μmol/L; IDH1R132C (enzyme): IC50 = 2.7 μmol/L |
– | 181 |
| KRC-09 | 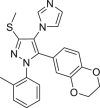 |
IDH1R132H (enzyme): IC50 = 76.87 μmol/L; 2-HG levels: (IDH1 R132H): 45% (50 μmol/L) |
– | 182 |
| α-Mangostin |  |
IDH1R132H (enzyme): Ki = 2.85 μmol/L |
– | 183 |
| (8R,10R,13R)-17-((2R,5R,E)-5,6-Dimethylhept-3-en-2-yl)-8-hydroxy-10,13-dimethyl-1,2,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one (3) |  |
IDH1R132H HT-1080 cells: 2-HG inhibition: IC50 = 35.97 μmol/L |
– | 184 |
| DOA | 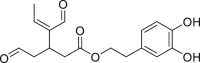 |
IDH1R132H (enzyme): IC50 = 12 μmol/L |
– | 185 |
PAMPA-BBB: blood–brain barrier specific parallel artificial membrane permeability assay.
Table 3.
The combination therapy of IDH1 inhibitors and other targeted drugs.
| IDH1 inhibitor | Combined drug | NCT No. | Condition | Conclusion/objective |
|---|---|---|---|---|
| Ivosidenib (AG-120) | Azacytidine (DNMT1 inhibitor) | NCT03173248 (Phase III) | AML | Recovery of blood counts; rates of febrile neutropenia and infections were reduced |
| Ivosidenib (AG-120) | Azacytidine (DNMT1 inhibitor) | NCT03503409 (Phase II) | AML; Myelodysplasia |
Evaluate the efficacy and safety in patient with IDH1 mutation-positive myelodysplastic syndrome |
| Ivosidenib (AG-120) | Nivolumab (anti-PD-1) | NCT04056910 (Phase II) | Metastatic cancer; Glioma |
Evaluate response to treatment, survival and safety events |
| Ivosidenib (AG-120) | Nivolumab (anti-PD-1) | NCT04044209 (Phase II) | AML; Myelodysplasia |
Evaluate safety and efficacy |
| Ivosidenib (AG-120) | Enasidenib (IDH2 inhibitor) | NCT02632708 (Phase I) | AML | Decrease in plasma and bone marrow 2-HG concentrations; CCR: 18.4% |
| Ivosidenib (AG-120) | Enasidenib (IDH2 inhibitor) | NCT03839771 (Phase III) | AML; Myelodysplasia |
Evaluate safety and efficacy |
| Ivosidenib (AG-120) | Enasidenib (IDH2 inhibitor) | NCT02677922 (Phase I/II) | AML | Treatment-emergent adverse events (TEAEs) including nausea, anemia and thrombocytopenia were reported |
| Ivosidenib (AG-120) | Vorasidenib (AG-881) | NCT03343197 (Phase I) | Glioma | Activation of IFN signaling and increased T-cell infiltration was observed |
| Ivosidenib (AG-120) | Venetoclax (Bcl-2 inhibitor) | NCT03471260 (Phase I/II) | AML; Hematologic cancer; Myelodysplasia; Myeloproliferative diseases |
Evaluate the safety and efficacy |
| Ivosidenib (AG-120) | Itraconazole (Hedgehog inhibitor) | NCT02831972 (Phase I) | Healthy volunteers | Ivosidenib alone or with itraconazole were well-tolerated with similar favorable safety profiles |
| Ivosidenib (AG-120) | Enasidenib (IDH2 inhibitor) Fedratinib (JAK2 inhibitor) |
NCT04955938 (Phase 1b) | Chronic myeloid leukemia; Myelodysplasia; Myelofibrosis; Polycythemia vera; Thrombocythemia |
Evaluate the safety and efficacy |
| Ivosidenib (AG-120) | Azacytidine (DNMT1 inhibitor) Venetoclax (Bcl-2 Inhibitor) |
NCI-2018-00921 (Phase I/II) | Myeloid leukemia | One-year overall survival were 75%, 50% and 100% in newly diagnosed AML, relapsed/refractory (R/R) AML and myelodysplatic syndrome, CRR: 67% |
| Ivosidenib (AG-120) | Enasidenib (IDH2 inhibitor) Azacytidine (DNMT1 inhibitor) |
NCT02677922 (Phase I/II) | AML; Myelodysplasia; Myeloproliferative diseases |
ORR: 78% |
| Enasidenib (AG-221) | Azacytidine (DNMT1 inhibitor) | NCT02677922 (Phase I/II) | AML | TRAE: 44% |
| Enasidenib (AG-221) | Azacytidine (DNMT1 inhibitor) | NCT03683433 (Phase II) | AML | TEAE: 85% |
| Enasidenib (AG-221) | Azacytidine (DNMT1 inhibitor) | NCT03383575 (Phase II) | AML | All patients reported leukocytosis. |
| Enasidenib (AG-221) | Azacytidine (DNMT1 inhibitor) | AG221-AML-005 (Phase I/II) | AML | mOS remained unchanged (22.0 months) |
| Enasidenib (AG-221) | Azacytidine (DNMT1 inhibitor) | NCT03013998 (Phase II) | AML | Low early death rate; High CR/CRi: 47%; yielded durable remissions |
| Enasidenib (AG-221) | Azacytidine (DNMT1 inhibitor) | NCT03683433 (Phase II) | AML; Chronic myelomonocytic leukemia; Myelodysplasia |
Evaluate the clinical activity of enasidenib in combination with azacitidine for patients with relapsed/refractory acute myeloid leukemia |
| Enasidenib (AG-221) | Cobimetinib (MEK1 inhibitor) | NCT05441514 (Phase Ib) | AML | Evaluate the efficacy and safety |
| Enasidenib (AG-221) | Venetoclax (Bcl-2 inhibitor) | 19-5939 (Phase I/II) | AML; Myelodysplasia; Myeloproliferative diseases |
Evaluate the safety, tolerability, efficacy, and best dose of venetoclax administered in combination with enasidenib in patients with blood cancers |
| Enasidenib (AG-221) | Daunorubicin (topoisomerase II) Cytarabine (DNA polymerase) |
NCT03825796 (Phase II) | AML | Evaluate the efficacy and safety |
| Enasidenib (AG-221) |
Azacytidine (DNMT1 inhibitor) Venetoclax (Bcl-2 inhibitor) |
NCT03683433 (Phase II) | AML | 6-month OS was 70%; CR/CRi: 100% (ND AML); CR/CRi: 58% (R/R AML) |
| Olutasidenib (FT-2102) | Cedazuridine (cytidine deaminase (CDA) inhibitor) Decitabine (deoxycytidine analog antimetabolite and a DNA methyltransferase inhibitor) |
NCT04013880 (Phase I/II) | AML; Myelodysplasia |
Evaluate the efficacy and safety |
| Olutasidenib (FT-2102) | Azacytidine (DNMT1 inhibitor) | NCT03684811 (Phase Ib/II) | Glioma | Dose-limiting toxicities ( ≥grade 3 transaminase elevations) were noted in combination group, meeting stopping criteria |
| Olutasidenib (FT-2102) | Azacytidine (DNMT1 inhibitor) | NCT02719574 (Phase I/II) | AML; Myelodysplasia |
mOS: 37.7 (monotherapy) versus 52.5 (combination therapy) weeks |
| Vorasidenib (AG-881) | Omeprazole (PPI) | NCT04128787 (Phase I) | Healthy volunteers | Evaluate the safety and tolerability |
| Vorasidenib (AG-881) | Lamotrigine (anticonvulsant agent) | NCT04015687 (Phase I) | Healthy volunteers | Evaluate the safety and pharmacokinetics |
| IDH1RpepvaccH (vaccine) | Avelumab (anti-PD-L1) | NCT03893903 (Phase I) | Glioma | Evaluate safety, tolerability and immunogenicity |
| IDH1RpepvaccH (vaccine) | Temozolomide (DNA alkylating) | NCT02454634 (Phase I) | Astrocytoma; Glioma; Oligodendroglioma |
TRAE: 90.6% |
| PEPIDH1M (vaccine) | Temozolomide (DNA alkylating) | NCT02193347 (Phase I) | Glioma | Evaluate the safety |
ND, new diagnosis; ORR, overall response rate; mOS, median overall survival; CRR, rate of complete remission; CR/Cri, complete remission/complete remission with incomplete recovery; TRAE, treatment related adverse events; TEAEs, treatment-emergent adverse events.
3.1. IDH1 inhibitors in the clinical trial
AG-120 (ivosidenib) is a potent inhibitor of mutated IDH1, which has been shown to significantly inhibit the production of D-2HG in tumor models127. In phase I clinical trials of solid and hematological malignancies, AG-120 showed promising clinical activity127. Ivosidenib is a promising new drug for the treatment of AML with IDH1 mutation. Ivosidenib can permanently relieve relapsed or refractory AML with IDH1 mutations128. From a phase I trial, Choe et al.129 investigated the molecular mechanism of resistance to ivosidenib in 174 patients with mIDH1 relapsed/refractory AML. Studies have found that mutations in the receptor tyrosine kinase pathway are associated with primary resistance to ivosidenib, and multiple mechanisms lead to acquired resistance, especially the disappearance of receptor tyrosine kinase pathway mutations and D-2HG recovery mutations129. Ivosidenib may also slow down the progression of IDH1 mutant gliomas130. In patients with advanced glioma, ivosidenib (500 mg) once a day is associated with prolonged disease control, good safety, and decreased growth of non-enhancing tumors131. The results of a phase III study of IDH1 mutations and chemotherapy-refractory cholangiocarcinoma showed that ivosidenib significantly improved progression-free survival, and it was well tolerated132. A phase I clinical study of ivosidenib in patients with advanced chondrosarcoma showed that ivosidenib has good safety and clinical activity133 (Table 1 and Fig. 9).
Figure 9.

Structures of IDH1 inhibitor in the clinical trial stage. Assay type and activity data are displayed on the right side of the compound's structure.
AG-221 is a dual inhibitor of IDH1 and IDH2, its conditions are leukemia, myelodysplastic syndrome, and solid tumor. The product was first marketed in the United States in 2017 for the treatment of adult patients with recurrent or refractory AML. In addition, phase III clinical trials are ongoing at celgene for the treatment of patients 60 years or older with AML refractory to or relapsed after second or third-line AML therapy. The drug is also in phase II clinical development at the company for the treatment of patients with high-risk IDH2-mutant myelodysplastic syndrome, as monotherapy or in combination with azacytidine144 (Table 1 and Fig. 9).
IDH-305 (13) is an oral inhibitor of IDH1 mutants that can effectively inhibit the production of D-2HG in a variety of xenograft models145. IDH305 (13) has entered human clinical trials for the treatment of cancers with IDH1 mutations, such as AML, chondrosarcoma, and cholangiocarcinoma. IDH305 (13) also showed good brain penetration145, indicating its potential in the treatment of IDH1 mutant brain cancer (Table 1 and Fig. 9).
DS-1001b is a selective, orally bioavailable, mutant IDH1 inhibitor that can destroy the proliferation of chondrosarcoma cells with IDH1 mutations in vitro and in vivo, and reduce the level of D-2HG146. DS-1001b also reduced the levels of H3K4me3 and H3K9me3, restored the abnormal histone modifications induced by D-2HG146. Inhibition of mutant IDH1 by DS-1001b is a promising treatment for chondrosarcoma (Table 1 and Fig. 9).
BAY-1436032 is a pan-inhibitor of IDH1 protein with different codon 132 mutations147. It works through an allosteric inhibition mechanism. Except for the inhibition of angiotensin 2 at an IC50 of 4.2 μmol/L, the researchers did not detect any relevant off-target effects. In other words, BAY-1436032 is a highly specific and effective inhibitor against IDH1 proteins with R132H or R132C mutations. BAY-1436032 can strongly reduce D-2HG levels in cells carrying IDH1R132H, IDH1R132C, IDH1R132G, IDH1R132S, and IDH1R132L mutations147. In vitro or in vivo, BAY-1436032 showed no toxicity147. BAY-1436032 can also be taken orally. BAY-1436032 also significantly prolonged the survival of mice transplanted with human astrocytomas carrying the IDH1R132H mutation147. Unfortunately, researchers have not yet obtained the co-crystallization of BAY-1436032 and mutant IDH1 protein. BAY-1436032 is very effective against all major types of IDH1 mutant AML148. In IDH1 mutant AML, the results of a phase I clinical study showed that BAY-1436032 is safe and moderately effective as a monotherapy149. But at the highest dose tested, BAY-1436032 still has a low overall remission rate and incomplete target inhibition149, so it is not suitable for further clinical development in AML. In another study, in a patient-derived IDH1 mutant AML xenograft model in vivo, BAY-1436032 combined with chemotherapy can delay the transplantation of leukemia cells150. BAY-1436032 can also play a strong synergistic effect with azacitidine by inhibiting mitogen-activated protein kinase/extracellular regulated protein kinase and Retinoblastoma gene/E2F transcription factor 2 signaling, significantly prolonging the survival period of AML patients with IDH1 mutations151 (Table 1 and Fig. 9).
Vorasidenib (AG881) is an effective, oral, blood–brain barrier permeable, dual inhibitor of mIDH1 and mIDH2, and is the first dual mIDH1/2 inhibitor reported so far152. Konteatis et al.152 determined the crystal structure of the complex formed by IDH1R132H and IDH2R140Q homodimers and Vorasinib in combination with NADPH, with a resolution of 2.1 and 1.99 Å, respectively. This increases our understanding of double mutant suppression. In 2018, Ma et al.153 resolved the crystal structures of IDH1R132H/NADPH/AG881 and IDH2R140Q/NADPH/AG881 complexes. In addition, in the orthotopic glioma mouse model, vorasinib can penetrate the brains of several preclinical species and greatly inhibit the production of D-2HG in glioma tissues153. Vorasidenib is currently in clinical development and has shown promising clinical activity in early clinical trials154 (Table 1 and Fig. 9).
FT-2102 is an effective, brain-permeable, orally selective mIDH1 inhibitor, which can effectively inhibit the production of D-2HG in a xenograft model in vivo155. Caravella et al.155 analyzed the crystal structure of FT-2102 coordinated with mIDH1R132H. FT-2102 has brain permeability, so it can be used to treat mIDH1-driven central nervous system cancers. Currently, FT-2102 is undergoing clinical research in hematological malignancies, solid tumors, and gliomas with mIDH1155. In summary, two IDH1 inhibitors (Idhifa and Tibsovo) have been approved for marketing worldwide. In addition to being effective for acute myeloid leukemia, it is also expected to become a new targeted therapy for patients with IDH1 mutations in cholangiocarcinoma. More drugs targeting at IDH1 remain to be discovered (Fig. 9 and Table 1).
HMPL-306 is a new small molecule dual inhibitor of IDH1 and IDH2. At present, three international phase I clinical studies of HMPL-306, led by MD Anderson Cancer Center, have been launched for the treatment of patients with advanced solid tumors and malignant blood tumors. All patients will receive drug treatment in March 2021. The HMPL-306 structure has not been disclosed at present. Hehuang Pharmaceutical (Chi-Med) holds global ownership. HMPL-306 is expected to become the first IDH1/IDH2 dual-targeting inhibitor in the world156 (Table 1).
PEPIDH1M is a peptide vaccine in phase I clinical trials at Duke University for the intradermal treatment of patients with IDH1 positive recurrent grade II glioma. PEPIDH1M vaccine is made up of a peptide that spans the mutated region of IDH1R132H. The peptide is administrated with granulocyte macrophage colony stimulating factor mixed with montanide ISA51 (vaccine adjuvant). According to the research results released in March 2021, in the clinical trial of 24 people (ClinicalTrials.gov Identifier: NCT02193347), the vaccine had a great impact on the cardiovascular system, causing hypertension (100%) and anemia (83.3%). The main adverse reactions of the gastrointestinal tract were constipation (45.83%) and nausea (41.67%). In addition, it caused the increase of alanine aminotransferase and aspartate aminotransferase, with a probability of about 30%157 (Table 1).
3.2. IDH1 inhibitors in the preclinical study stage
3.2.1. IDH1 inhibitors based on phenyl-glycine scaffold
AGI-5198 is the first reported IDH1R132H inhibitor, showing strong D-2HG inhibition in tumor xenograft models158. Since high levels of D-2HG have been shown to change the epigenetic state and biology of cells, the utility of this molecule is very important to evaluate the biological consequences of IDH mutations and the potential of IDH inhibitors to treat IDH mutant tumors. In addition, it can induce the demethylation of histone H3K9me3 and the re-expression of genes related to differentiation159. ML309 is an effective inhibitor targeting IDH1R132H, which can reduce the production of D-2HG in U87MG glioblastoma cells160. ML309 possesses a good in vitro ADME and in vivo PK profile160. But it has no obvious blood–brain barrier penetration ability in healthy mice160. AGI-5198 and ML309 are IDH1R132H inhibitors obtained by hit compound optimization. They have the same phenyl glycine scaffold, but they are synthesized by different routes. AGI-5198 adopted the Ugi reaction, which greatly reduced the difficulty of synthesis. A detailed structure–activity relationship analysis was carried out in the study of ML309. The above methods and conclusion will provide valuable information for the research of IDH1 inhibitors based on this core structure. AG-120 was designed and synthesized based on this scaffold. After that, a series of new IDH1 inhibitors were obtained by optimizing AG-120 through a reasonable structure-based design. Compound 6F is of excellent cellular potency (IC50 < 10 nmol/L) and also shows selectivity for wild-type IDH1 (79-fold) and mutant IDH2R140Q (>2000-fold). Pharmacokinetic studies showed low clearance and high bioavailability (>30%)161. Compound 43 also derives from the optimization of AG-120, but its activity still needs further optimization162 (Table 2).
3.2.2. IDH1 inhibitors based on 3-pyrimidin-4-yl-oxazolidin-2-one scaffold
IDH125 was identified as a potential IDH1R132H inhibitor by high-throughput screening and pharmacochemical methods (IDH1R132H enzyme inhibition assay: IC50 = 0.22 μmol/L). To obtain highly potent compounds, 20 compounds were synthesized. Among them, IDH662 and IDH889 are the compounds with the best in vitro activity. However, IDH662 has a high plasma protein binding rate of 99%, which limits its in vivo activity. In the HCT116 IDH1R132H tumor xenograft model, IDH889 can significantly reduce D-2HG163. It is noteworthy that, in addition to the possible treatment of AML, chondrosarcoma, cholangiocarcinoma, and other forms of mutant IDH1-driven cancers, IDH889 also shows brain penetration exposure163, suggesting its potential in the treatment of IDH1 mutant brain cancer patients. IDH305 was further optimized by IDH889. It is currently in phase II clinical trials for the treatment of glioma and AML. In addition, compound 19 was also obtained based on IDH889 optimization164. Through rational design based on structure, Zheng et al.165 discovered and optimized imidazole cyclopropyl amide analogs. The best compound 5t can effectively inhibit the activity of IDH1R132H, reduce the production of D-2HG, and has moderate liver microsome stability and PK characteristics165. Although the efficacy of compound 5t is twice that of IDH305, the oral exposure of compound 5t is not enough for an efficacy study. More efforts will be made to improve in vivo exposure to expand its further development. Ma et al.166 synthesized a series of mIDH1 inhibitors containing the backbone of 3-pyrazine-2-yl-oxazolidin-2-one. Further evaluation found that compound 3g and the positive drug NI-1 have similar inhibitory activity, and neither concentration of compound 3g shows significant toxicity166. Compound 3g has a strong inhibitory effect on IDH1132H and IDH1R132C, and higher selectivity for IDH1WT. In addition, compound 3g shows a good ability to penetrate the blood–brain barrier166. These findings indicate that compound 3g is worthy of further optimization, looking for inhibitors with lower toxicity for the treatment of IDH1 mutant brain cancer patients. Compound I-8 specifically inhibits the production of D-2HG in IDH1 mutant cells, reduces histone methylation levels, and induces differentiation167. In addition, I-8 can also be taken orally167. IDH1-IN-6 is a potent, selective, and orally active mutant isocitrate dehydrogenase (IDH) inhibitor with IC50 of 6.27 and 3.71 nmol/L for IDH1R132H and IDH1R132C, respectively. IN-6 is less active on inhibiting the IDH wild-type enzymes. IN-6 inhibits the production of D-2HG in HT-1080 cells with an IC50 of 1.28 nmol/L, indicating the inhibition of mutant IDHlR132C in cells168 (Table 2).
3.2.3. IDH1 inhibitors based on 1-hydroxypyridin-2-one scaffold
Zheng et al.169 discovered two 1-hydroxypyridine-2-one (compounds 2 and 3), which are effective inhibitors of IDH1R132H and IDH1R132C, with Ki values as low as 120 nmol/L. These compounds can inhibit the production of D-2HG in IDH1 mutant cells169. They are 60 times more selective than IDH1WT and are not cytotoxic to human cells. The researchers also determined the X-ray structure of IDH1R132H that forms a complex with compound 2 or 3, revealing the exact combination of these two compounds and the structural basis for high selectivity. The crystal structure shows that these inhibitors bind with IDH1R132H through hydrogen bonds, electrostatic and hydrophobic interactions169. Liu et al.170 reported the discovery, design, synthesis, and structure–activity relationship of a series of 1-hydroxypyridine-2-one type compounds against IDH1 mutants. 1-Hydroxypyridine-2-one type compounds 4 and 7 are low micromolar inhibitors of IDH1R132H. Under the guidance of structure–activity relationship and the X-ray structure of the IDH1R132H/IDH2 complex, Liu et al.170 designed and synthesized 61 derivatives, of which several effective inhibitors have Ki values of 140–270 nmol/L. 1-Hydroxypyridine-2-one type inhibitors have blood–brain barrier permeability170. It is necessary to further develop this type of inhibitors (Table 2).
3.2.4. IDH1 inhibitors based on 2-thiohydantoins scaffold
Wu et al.171 synthesized a series of novel 2-thiohydantoins and related compounds. These compounds can reduce the cell concentration of D-2HG in BT142 gliomas with IDH1R132H mutation, reduce histone methylation levels, and selectively inhibit the self-renewal ability of glioma stem-like cells with IDH1R132H mutation171. The author also analyzed the crystal structure of the complex formed by IDH1R132H and compound 16 or 22, showing the inhibitor–protein interaction, and laying the foundation for further structure-based inhibitor design171. In addition, the most effective inhibitor 18 is a competitive inhibitor relative to α-KG, which exerts a non-competitive mode of action on NADPH171. These compounds are new chemical probes for studying IDH1 mutations in cancer and provide new scaffolds for drug discovery against IDH1 mutation cancers (Table 2).
3.2.5. IDH1 inhibitors based on tetrahydropyrazolopyridine scaffold
Okoye-Okafor et al.172 analyzed the crystal structure of GSK321 bound to IDH1R132H homodimer in the presence of NADP+. The results of crystal and biochemical studies indicate that GSK321 binds to the allosteric site of IDH1, making the enzyme in a catalytically inactive conformation172. GSK321 stably reduced the production of D-2HG in several different IDH1 mutant AML cells172. It induces the differentiation of granulocytes and can reverse the methylation of DNA cytosine172. In addition, GSK864 also has nanomolar inhibitory activity against IDH1 mutants, showing that tetrahydropyrazolopyridine scaffold is of great research value. Compound 1 is a hit compound obtained by high-throughput screening together with GSK321. It shows inhibitory activity against IDH1WT with an IC50 value of 410 nmol/L. Three compounds 11, 13, and 15 with better activity were obtained through further structure optimization172. The above results provide a very potential core structure for the development of IDH1 inhibitors (Table 2).
3.2.6. IDH1 inhibitors based on 8-membered ring sulfonamides scaffold
BRD2879 inhibits the production of D-2HG in cells without significant toxicity173. However, the high molecular weight, lipophilicity, and low solubility of the specific inhibitor BRD2879 limit its use in vivo. BRD2879 represents a new structural class of mutant IDH1 inhibitors that, with optimization, may prove useful in the study of this enzyme and its role in cancer (Table 2).
3.2.7. IDH1 inhibitors based on quinolinone scaffold
Lin et al.174 discovered and optimized a series of quinolinones. Through rational design based on the structure, the researchers identified compound 63. It can effectively inhibit IDH1 mutants R132H, R132C, R132G, and R132L174 with good cell permeability, ADME/PK properties, and oral bioavailability. In the HCT116-IDH1R132H or HCT116-IDH1R132C xenograft BALB/c nude mice model, compound 63 can significantly reduce the level of D-2HG174. Preclinical studies have shown that compound 63 may have the potential to treat GBM, AML, or other forms of mIDH1-driven cancer (Table 2).
3.2.8. Other IDH1 inhibitors
3.2.8.1. IDH1 inhibitors with other core structures
Bisimidazole 3 (bisimidazoline scaffold) non-competitively inhibits IDH1R132H relative to NADPH and α-KG and the production of D-2HG in cells175. Studies have shown that bisimidazole 3 selectively inhibits IDH1 mutations by binding to allosteric sites, and the inhibition is competitive with Mg2+ 175. Hu et al.176 designed and synthesized a series of 3-(7-azaindolyl)-4-indolyl maleimides. Among them, 11a, 11c, 11e, 11g, and 11s showed a good inhibitory effect on IDH1R132H with high selectivity for wild-type IDH1176. Compounds 11a, 11c, 11e, 11g, and 11s can effectively inhibit the production of D-2HG in U87MG cells expressing IDH1R132H 176. Their research provides new information for the design of new IDH1R132H inhibitors.
Licochalcone A (imidazole cyclopropyl amine) is a selective inhibitor of IDH1R132C with IC50 value of 5.176 μmol/L186. Compared with the R132C mutation, the R132H mutation is not conducive to the binding of licochalcone A to the IDH1 protein186. Licochalcone A can induce apoptosis and cell cycle arrest in HT-1080 cells186. Jones et al.177 obtained a series of inhibitors targeting IDH1R132H through high-throughput screening. Among them, compound 20a promoted the differentiation of human IDH1R132H AML cells derived from patients, but did not promote differentiation in IDH1 wild-type AML cells177. In addition, the researchers also clarified the crystal structure of the complex formed by IDH1R132H and compound 20a, and the position of the previously unresolved protein loop can be observed177. More complete structure lays the foundation for the future development of IDH1R132H inhibitors. HMS-101 binds to the active site of mutant IDH1, inhibits cell proliferation, and induces differentiation of IDH1 mutant leukemia cells178. HMS-101 can also inhibit the production of D-2HG in syngeneic mutant IDH1 mouse models and human AML xenograft models in patients178. In addition, in cells treated with HMS-101, differentiation-related transcription factors CEBPA and PU.1 were significantly increased, while cell cycle regulator cyclin A2 decreased178. Besides, it also weakens the hypermethylation of histones178. This study provides clinical evidence for the further development of IDH1 mutant competitive inhibitors and the treatment of IDH1 mutant AML patients (Table 2).
3.2.8.2. IDH1 hit compounds found based on virtual screening
Virtual screening technology provides a fast and economical method for discovering new active substances by selecting compounds in a large database for screening. This method also provides many potential hit compounds for the discovery of IDH1 inhibitors. Through virtual screening, it was found that clomiphene can selectively inhibit the activity of IDH1R132H in a non-competitive manner and through an allosteric inhibition mechanism179. In vivo studies have shown that oral clomiphene can significantly inhibit tumor growth in HT-1080-bearing CB-17/Icr-scid mice179. An in-depth study of mechanism of clomiphene has great benefits for the treatment of many patients with glioma or AML. Zou et al.180 obtained 7 compounds through virtual screening based on cross-docking. They have a moderate mIDH1 inhibitory effect, of which ZX06 is the most effective and safest180. ZX06 can also penetrate the blood–brain barrier180. Therefore, ZX06 should be used as a lead compound to be further optimized for the treatment of IDH1 mutant brain cancer patients. In addition, this new virtual screening strategy should be further optimized to introduce more novel scaffold mIDH1 inhibitors. DC_H31 is a new type of IDH1R132H/C inhibitor, which acts through an allosteric mechanism181. At the cellular level, DC_H31 can inhibit cell proliferation in HT-1080 cells, promote cell differentiation, and reduce the production of D-2HG181. In vivo and in vitro, DC_H31 can promote the development of more effective pan-inhibitors against IDH1R132H/C through further structural optimization. KRC-09 can effectively inhibit the activity of IDH1R132H mutants and reduce the concentration of D-2HG in the U-87 MG cell line containing IDH1R132H 182. Although it is not as good as known inhibitors (such as AGI-5198), it has a novel scaffold that provides an idea for the future development of effective IDH1R132H inhibitors (Table 2).
3.2.8.3. Natural inhibitors of IDH1
α-Mangostin, a new selective inhibitor of IDH1R132H, competitively inhibits the binding of α-KG to IDH1R132H, but has no inhibitory effect on IDH1183. It can selectively promote the demethylation of 5-methylcytosine and histone H3 trimethylated lysine residues in IDH1R132H MCF10A cells183. It should be noted that there is no structural similarity between α-mangostin and the previous IDH1R132H selective inhibitors. Through structure-based virtual screening, it was found that compound 3 inhibited the mutant IDH1 in a non-competitive manner184. Compound 3 treatment can reduce the concentration of D-2HG in HT-1080 cells and reduce the level of histone H3K9me3 methylation184. Compound 3 may be the lead compound for anticancer drug candidates. The decarboxymethyl oleuropein aglycone (DOA) present in extra-virgin olive oil (EVOO) can specifically inhibit IDH1R132H and reduce the production of D-2HG185. DOA can restore the activity of histone demethylase inhibited by D-2HG, and also restore the expression of PD-L1 epigenetics185. This study shows that phenolic compounds in olive oil are inhibitors of IDH1 mutant and can be used as scaffolds for drug discovery (Table 2).
3.3. The combination therapy of IDH1 inhibitors and other targeted drugs
Cancer cells often upregulate different growth-promoting factors, which can act independently or interact in cells through signal networks. Cancer cells can easily acquire drug resistance by upregulating alternative factors or converting other signal pathways that promote proliferation. Therefore, there are lots of limitations in the treatment targeting only a single target. To overcome the shortcomings of single target drugs, it has become a recognized method to combine two (or more) different targets related to cancer development to achieve synergistic anticancer efficacy. At present, there are 31 IDH1 combined drug projects in the clinical research stage. AG-120 and AG-221 have the largest number of combined drug studies, accounting for 74.2% of all studies. Among them, AG-120 has the largest number of combined targets, and AG-221 and Azacitidine (DNMT1 inhibitor) are studied the most widely. In addition, there were 6 experiments (19.4%) in which the three drugs were combined for treating cancer (Fig. 10).
Figure 10.

Combined application of IDH1 inhibitors and other target drugs in the clinical research. Different IDH1 inhibitors are listed on the left side of the heatmap. Targets of drug combination are located below the heat map. The color depth represents the number of clinical experiments.
3.3.1. Combination of IDH1 inhibitors and DNMT1 inhibitors
The combination of IDH1 inhibitors and DNMT1 inhibitor (azacitidine) is the most common. The clinical trial results showed that AG-120 combined with azacitidine significantly improved the event-free survival (EFS, 33%) of patients compared with placebo and azacitidine group in the treatment of IDH1 mutant acute myeloid leukemia. The median overall survival (mOS) was 24.0 months with AG-120 and azacitidine, but only 7.9 months with placebo and azacitidine. More importantly, the rate of grade 3 and higher-level adverse events in this combined drug strategy was lower than that in the control group, 28% vs. 34%187. In addition, the combined application of FT-2102 and azacitidine also showed good clinical effects on the treatment of acute mycoid leukemia, mOS: 37.7 (monotherapy) vs. 52.5 (combination therapy) weeks, but this combined application needs to adjust the dosage to eliminate the toxicity of dose limitation. In the clinical trial of AG-221 combined with azacitidine, it is necessary to further consider how to reduce treatment-emergent adverse events to achieve higher safety (Table 3).
3.3.2. Combination of IDH1 inhibitors and anti-PD-1/anti-PD-L1
Since programmed cell death protein 1 (PD-1) antibody did not prolong the survival of glioblastoma patients in phase III clinical study, in order to further explore effective immunotherapy strategies, the researchers analyzed the immune regulatory targets. The results showed that adenosine a2a receptor/CD73/CD39 (cluster of differentiation, CD) pathway had the highest expression frequency in glioma patients, followed by the PD-1 pathway. Mechanism studies have shown that in IDH1 mutant glioma patients, D-2HG can upregulate CD73 expression on immune cells188. These results explain the phase III clinical results of PD-1 antibody to a certain extent, and also provide a basis for the combined application of IDH1 and PD-1/PD-L1 antibodies. At present, three studies on the combination of IDH1 inhibitors and PD-1 (including PD-L1) antibodies are conducting safety evaluation, which may provide a new and effective means for glioma immunotherap (Table 3).
3.3.3. Combination of IDH1 and IDH2 inhibitors
The mutual conversion of IDH1 and IDH2 is one of the mechanisms of acquired drug resistance caused by IDH1 or IDH2 inhibitor single drug application126, which makes the combination of IDH1 and IDH2 inhibitors have great therapeutic potential. The most common combination of IDH1 and IDH2 inhibitors is AG-120 and AG-221, as well as AG-120 and AG-881. In the three clinical studies of AG-120 and AG-221, the subjects were all patients with acute myeloid leukemia. The results showed that the combination of the two drugs could reduce the concentration of D-2HG in bone marrow and plasma, and the rate of complete remission was 18.4%. However, treatment-emergent adverse events (TEAEs) including nausea, anemia, and thrombocytopenia were reported. NCT03343197 is a phase I clinical study with glioma patients as the research object. After AG-120 and AG-881 were administrated in combination, activation of interferon signaling and increased T-cell infection were observed. Some studies have pointed out that differentiation syndrome is a serious adverse reaction of IDH inhibitors ivosidenib and enasidenib in patients with IDH1 and IDH2-mutated AML, respectively189. Therefore, during the combined application of IDH1 and IDH2 inhibitors, we need to pay high attention to this serious drug-related adverse reaction (Table 3).
3.3.4. Combination of IDH1 inhibitors and Bcl-2 inhibitors
AML is a highly heterogeneous disease, and multiple factors can affect the prognosis, including age, cytogenetic abnormalities, and molecular genetic abnormalities (IDH1/2, nucleophosmin 1, p53, and Fms-like tyrosine kinase 3). The over-expression of Bcl-2 is associated with the formation of drug resistance. Through large-scale RNAi screening, some researchers found that in acute myeloid leukemia, IDH1R132H mutation has a strong dependence on Bcl-2, an anti-apoptotic gene. Treatment with Venetoclax (ABT-199), a specific inhibitor of Bcl-2, will lead to more apoptosis of cells containing IDH1R132H mutation. This research result indicated that patients with acute myeloid leukemia carrying IDH1/2 mutations may respond to Bcl-2 inhibitors, which provides a clear molecular basis for the combined application of drugs that block the activity of mitochondrial electron transport chain in the treatment of AML48. At present, the clinical study of ABT-199, AG-120 and azacytidine show great therapeutic potential, one-year overall survival is 75%, 50%, and 100% in newly diagnosed AML, relapsed/refractory AML, and myelodysplastic syndrome, respectively, and rate of complete remission is 67% (Table 3).
3.3.5. Combination of IDH1 inhibitors and other target inhibitors
Itraconazole (R51211) is a triazole antifungal agent and an effective oral active hedgehog signal pathway antagonist with an IC50 about 800 nmol/L, which has anticancer and antiangiogenic effects190. At present, the combined application of AG-120 and itraconazole has shown good tolerance in healthy volunteers. The combined application of other target inhibitors such as cobimetinib (MEK1 inhibitor), omeprazole (proton pump inhibitor), lamotrigine (anticonvulsant agent), temozolomide (DNA alkylating), and corresponding IDH1 inhibitors is mostly at the stage of safety evaluation. These combinations will expand the application of IDH1 inhibitors in the field of cancer treatment (Table 3).
4. Conclusions
Cancer is the main cause of human death, and its morbidity and mortality are increasing year by year. The occurrence and development of cancer are closely watched by researchers, but the mechanism of cancer occurrence is quite complicated, and it is still a major problem that needs to be solved urgently in today's society. Abnormal energy and metabolism are important characteristics of malignant tumors, which play an important role in the progression of cancers. In 2011, Robert A. Weinberg proposed ten characteristics of cancer, including the abnormal energy metabolism. Isocitrate dehydrogenase (IDH) plays a very important role in the process of energy metabolism. In the tricarboxylic acid cycle, isocitrate dehydrogenase can catalyze the conversion of isocitrate to α-KG. After IDH is mutated, its enzymatic activity changes, which can convert α-KG into D-2HG, leading to high levels of D-2HG in the body. D-2HG is a recognized cancer metabolite and can promote cancer cell proliferation through a variety of mechanisms. Therefore, IDH gene mutations are closely related to the occurrence and development of cancers.
IDH1 plays an important role in the process of life activities including glutamine metabolism, cell active oxygen regulation, fat synthesis, phospholipid metabolism, insulin secretion, etc. Recent studies have found that the changes in IDH1 expression and the mutation of amino acid 132 are closely related to many cancers. IDH1 is one of the most widely mutated metabolic enzymes in human cancers discovered so far. The relationship between IDH1 (including its mutants) and cancer development was first discovered in gliomas. With the deepening of research, it is found that it has a certain correlation with many cancers. IDH1 mutations have been found in various cancers such as cholangiocarcinoma, acute myeloid leukemia, brain glioma, and chondrosarcoma. Mutant IDH1 acquires a new catalytic function, which can catalyze the production of D-2HG from α-KG, and then regulate the induction, growth, metastasis, metabolism, and drug resistance of cancer through various mechanisms. Inhibitors against IDH1 mutations can significantly reduce the level of D-2HG, which has become a hot spot in the development of some oncology drugs.
In this paper, we discussed the impacts of IDH1 on the occurrence and development of cancer from four perspectives. In the respect of metabolic reprogramming, IDH1 mutation causes a large amount of D-2HG accumulation, inhibits mitochondrial function, and promotes aerobic glycolysis to provide energy for cells. In addition, mutant IDH1 inhibits wild-type IDH1, causing depletion of NADPH, making cells unable to resist oxidative stress, and thus causing DNA damage. We also noticed that in some cancers, a low nutrition statement will increase the expression of IDH1WT, increase the level of NADPH, and enhance the function of mitochondria to ensure the normal growth of cancer cells. Cancer cells have a strong adaptive capacity. For cancer treatment based on energy metabolism, it is necessary to assess the nutritional status of patients, detect the expression of energy metabolism targets and their metabolite levels, formulate stricter drug indications, and achieve individualized treatment. D-2HG produced by IDH1 mutant can mediate hypermethylation of DNA and histones and promote the occurrence of cancer. Compared with genetic factors of cancer, reversible epigenetic variation can be regulated by chemical drugs, showing broad prospects in cancer treatment. At present, the drugs targeting epigenetics cannot be widely used in cancer treatment because of their low efficacy, low bioavailability, poor stability, high toxicity, poor drug compliance, etc. Therefore, the combination of IDH1/2 inhibitors and DNMT1 inhibitors may be a potential therapeutic strategy. Many clinical studies have been carried out on the combination of IDH1/2 inhibitors and DNMT1 inhibitors. With the deepening of research, epigenetic drugs will be able to meet the clinical needs better. Moreover, IDH1 mutation will cause the immune microenvironment to be in a state of inhibition. The analysis of this state will provide a basis for the combined application of IDH1 inhibitors and cancer immunotherapy methods. Cancer is a multi-dimensional and complex biological system. The impacts of IDH1 on cancer phenotype can further verify this point of view. IDH1 affects the proliferation, apoptosis, migration, and drug resistance of cancer cells through different signal pathways. Therefore, the interpretation of IDH1-related cancer signal pathway network will provide all-around guidance for cancer treatment based on IDH1.
Currently, the research and development of IDH1 inhibitors are mainly concentrated in the field of small molecules. In addition, vaccines targeting mutant IDH1 are in the clinical research stage. The clinical trials of combination drugs and dual-targeted drugs foreshadowed the trend of IDH1 inhibitors and further confirmed the limitations of single-target therapy. Inspired by the dual-targeted drugs and proteolysis-targeting chimeras (PROTAC) technology, we initiated the concept of “dual PROTAC”, which achieved the simultaneous degradation of EGFR and PARP by a single drug191, providing a new idea for the development of IDH1 inhibitors. Many studies have shown that PROTACs have excellent anti-cancer activity, which can solve the problem of drug resistance, and reduce the toxic side effects through selectively targeted degradation. The design of IDH1 mutant small molecules with better selectivity will greatly promote the research of IDH1 degraders and provide more treatment options for IDH1-related cancer.
Acknowledgments
This study was supported by National Natural Science Foundation of China (NSFC) (No. 81773637, 82141216, U1803122), Chunhui Program-Cooperative Research Project of the Ministry of Education, Shenyang Young and Middle-aged Innovative Talents Support Program (RC210446, China), and Liaoning Province Natural Science Foundation (Nos. 2022-MS-241, 2020-MZLH-31, China). We acknowledged the support from National-Local Joint Engineering Research Center for Molecular Biotechnology of Fujian & Taiwan TCM, Fujian Key Laboratory of Chinese Materia Medica, Fujian University Key Laboratory for Research and Development of TCM Resources, at Fujian University of Traditional Chinese Medicine.
Author contributions
Lixia Chen, Lidian Chen and Hua Li: Conceptualization, Writing-Reviewing and Editing. Yang Liu, Wei Xu: Writing-Original draft preparation and Visualization. Mingxue Li, Yueying Yang, and Dejuan Sun: Writing-Original draft preparation. All authors have approved the final version of the article.
Conflicts of interest
All the authors declared that they have no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Lidian Chen, Email: cld@fjtcm.edu.cn.
Hua Li, Email: 5402118@qq.com.
Lixia Chen, Email: syzyclx@163.com.
References
- 1.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46. doi: 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 4.Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBerardinis R.J., Chandel N.S. We need to talk about the Warburg effect. Nat Metab. 2020;2:127–129. doi: 10.1038/s42255-020-0172-2. [DOI] [PubMed] [Google Scholar]
- 6.Semenza G.L. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr. 2007;39:231–234. doi: 10.1007/s10863-007-9081-2. [DOI] [PubMed] [Google Scholar]
- 7.Yeung S.J., Pan J., Lee M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis – the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65:3981–3999. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jang M., Kim S.S., Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med. 2013;45:e45. doi: 10.1038/emm.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherwood V., Chaurasiya S.K., Ekström E.J., Guilmain W., Liu Q., Koeck T., et al. WNT5A-mediated β-catenin-independent signalling is a novel regulator of cancer cell metabolism. Carcinogenesis. 2014;35:784–794. doi: 10.1093/carcin/bgt390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masui K., Tanaka K., Akhavan D., Babic I., Gini B., Matsutani T., et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18:726–739. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z., Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci. 2016;73:377–392. doi: 10.1007/s00018-015-2070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang B., Song B.L., Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020;2:132–141. doi: 10.1038/s42255-020-0174-0. [DOI] [PubMed] [Google Scholar]
- 13.Cerezo M., Rocchi S. Cancer cell metabolic reprogramming: a keystone for the response to immunotherapy. Cell Death Dis. 2020;11:964. doi: 10.1038/s41419-020-03175-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qu D., Shen L., Liu S., Li H., Ma Y., Zhang R., et al. Chronic inflammation confers to the metabolic reprogramming associated with tumorigenesis of colorectal cancer. Cancer Biol Ther. 2017;18:237–244. doi: 10.1080/15384047.2017.1294292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang E., Wang X., Gong Z., Yu M., Wu H., Zhang D. Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct Target Ther. 2020;5:242. doi: 10.1038/s41392-020-00359-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang W., Shi B., Cong R., Hao M., Peng Y., Yang H., et al. RING-finger E3 ligases regulatory network in PI3K/AKT-mediated glucose metabolism. Cell Death Discov. 2022;8:372. doi: 10.1038/s41420-022-01162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan Y., Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19:55–71. doi: 10.1038/s41579-020-0433-9. [DOI] [PubMed] [Google Scholar]
- 18.Sjöblom T., Jones S., Wood L.D., Parsons D.W., Lin J., Barber T.D., et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 19.Parsons D.W., Jones S., Zhang X., Lin J.C., Leary R.J., Angenendt P., et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen D., Zhang J., Yuan K., Zhao J., Zhao Z., Cui L., et al. Landscape of IDH1/2 mutations in Chinese patients with solid tumors: a pan-cancer analysis. Mol Genet Genom Med. 2021;9:e1697. doi: 10.1002/mgg3.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vinekar R., Verma C., Ghosh I. Functional relevance of dynamic properties of dimeric NADP-dependent isocitrate dehydrogenases. BMC Bioinf. 2012;13:S2. doi: 10.1186/1471-2105-13-S17-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang L., Su S.M. Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem. 2017;86:305–331. doi: 10.1146/annurev-biochem-061516-044732. [DOI] [PubMed] [Google Scholar]
- 23.Mondesir J., Willekens C., Touat M., Botton S. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J Blood Med. 2016;7:171–180. doi: 10.2147/JBM.S70716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dimitrov L., Hong C.S., Yang C., Zhuang Z., Heiss J.D. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int J Med Sci. 2015;2:201–213. doi: 10.7150/ijms.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metallo C.M., Gameiro P.A., Bell E.L., Mattaini K.R., Yang J., Hiller K., et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S.M., Koh H.J., Park D.C., Song B.J., Huh T.L., Park J.W. Cytosolic NADP+-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic Biol Med. 2002;32:1185–1196. doi: 10.1016/s0891-5849(02)00815-8. [DOI] [PubMed] [Google Scholar]
- 27.Filipp F.V., Scott D.A., Ronai Z.A., Osterman A.L., Smith J.W. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012;25:375–383. doi: 10.1111/j.1755-148X.2012.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh H.J., Lee S.M., Son B.G., Lee S.H., Ryoo Z.Y., Chang K.T., et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J Biol Chem. 2004;279:39968–39974. doi: 10.1074/jbc.M402260200. [DOI] [PubMed] [Google Scholar]
- 29.Zhu S., Huang J., Xu R., Wang Y., Wan Y., McNeel R., et al. Isocitrate dehydrogenase 3b is required for spermiogenesis but dispensable for retinal viability. J Biol Chem. 2022;298 doi: 10.1016/j.jbc.2022.102387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu X., Zhao J., Xu Z., Peng B., Huang Q., Arnold E., et al. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem. 2004;279:33946–33957. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y., Tang S., Lai H., Jin R., Long X., Li N., et al. Discovery of novel IDH1 inhibitor through comparative structure-based virtual screening. Front Pharmacol. 2020;11 doi: 10.3389/fphar.2020.579768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mardis E.R., Ding L., Dooling D.J., Larson D.E., McLellan M.D., Chen K., et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan H., Parsons D.W., Jin G.L., McLendon R., Rasheed B.A., Yuan W.Y., et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duchmann M., Micol J.B., Duployez N., Raffoux E., Thomas X., Marolleau J.P., et al. Prognostic significance of concurrent gene mutations in intensively treated patients with IDH-mutated AML: an ALFA study. Blood. 2021;137:2827–2837. doi: 10.1182/blood.2020010165. [DOI] [PubMed] [Google Scholar]
- 35.Han S., Liu Y., Cai S.J., Qian M., Ding J., Larion M., et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer. 2020;122:1580–1589. doi: 10.1038/s41416-020-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kipp B.R., Voss J.S., Kerr S.E., Barr Fritcher E.G., Graham R.P., Zhang L., et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol. 2012;43:1552–1558. doi: 10.1016/j.humpath.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Kim N.I., Noh M.G., Kim J.H., Won E.J., Lee Y.J., Hur Y., et al. Frequency and prognostic value of IDH mutations in Korean patients with cholangiocarcinoma. Front Oncol. 2020;10:1514. doi: 10.3389/fonc.2020.01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pirozzi C.J., Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol. 2021;18:645–661. doi: 10.1038/s41571-021-00521-0. [DOI] [PubMed] [Google Scholar]
- 39.Philip B., Yu D.X., Silvis M.R., Shin C.H., Robinson J.P., Robinson G.L., et al. Mutant IDH1 promotes glioma formation in vivo. Cell Rep. 2018;23:1553–1564. doi: 10.1016/j.celrep.2018.03.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao S., Lin Y., Xu W., Jiang W., Zha Z., Wang P., et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dang L., White D.W., Gross S., Bennett B.D., Bittinger M.A., Driggers E.M., et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karpel-Massler G., Nguyen T.T.T., Shang E., Siegelin M.D. Novel IDH1-targeted glioma therapies. CNS Drugs. 2019;33:1155–1166. doi: 10.1007/s40263-019-00684-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang C., Moore L.M., Li X., Yung W.K., Zhang W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro Oncol. 2013;15:1114–1126. doi: 10.1093/neuonc/not087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu J., Zhang J., Gong Y., Testa C.L., Klein-Szanto A.J. Regulation of HIF-1 alpha by the proprotein convertases furin and PC7 in human squamous carcinoma cells. Mol Carcinog. 2015;54:698–706. doi: 10.1002/mc.22131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Partch C.L., Gardner K.H. Coactivators necessary for transcriptional output of the hypoxia inducible factor, HIF, are directly recruited by ARNT PAS-B. Proc Natl Acad Sci U S A. 2011;108:7739–7744. doi: 10.1073/pnas.1101357108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mimeault M., Batra S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J Cell Mol Med. 2013;17:30–54. doi: 10.1111/jcmm.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Armitage E.G., Kotze H.L., Allwood J.W., Dunn W.B., Goodacre R., Williams K.J. Metabolic profiling reveals potential metabolic markers associated with hypoxia inducible factor-mediated signalling in hypoxic cancer cells. Sci Rep. 2015;5:15649–15660. doi: 10.1038/srep15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan S.M., Thomas D., Corces-Zimmerman M.R., Xavy S., Rastogi S., Hong W.J., et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21:178–184. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu X., Chin R.M., Vergnes L., Hwang H., Deng G., Xing Y., et al. 2-Hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 2015;22:508–515. doi: 10.1016/j.cmet.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu Y., Yang R., Yang Y., Zhao Y., Wakeham A., Li W.Y., et al. IDH1 mutation contributes to myeloid dysplasia in mice by disturbing heme biosynthesis and erythropoiesis. Blood. 2021;137:945–958. doi: 10.1182/blood.2020007075. [DOI] [PubMed] [Google Scholar]
- 51.Ronnebaum S.M., Ilkayeva O., Burgess S.C., Joseph J.W., Lu D., Stevens R.D., et al. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J Biol Chem. 2006;281:30593–30602. doi: 10.1074/jbc.M511908200. [DOI] [PubMed] [Google Scholar]
- 52.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi J., Zuo H., Ni L., Xia L., Zhao L., Gong M., et al. An IDH1 mutation inhibits growth of glioma cells via GSH depletion and ROS generation. Neurol Sci. 2014;35:839–845. doi: 10.1007/s10072-013-1607-2. [DOI] [PubMed] [Google Scholar]
- 54.Wahl D.R., Dresser J., Wilder-Romans K., Parsels J.D., Zhao S.G., Davis M., et al. Glioblastoma therapy can be augmented by targeting IDH1-mediated NADPH biosynthesis. Cancer Res. 2017;77:960–970. doi: 10.1158/0008-5472.CAN-16-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lambeth J.D. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 56.Maeng O., Kim Y.C., Shin H.J., Lee J.O., Huh T.L., Kang K.I., et al. Cytosolic NADP+-dependent isocitrate dehydrogenase protects macrophages from LPS-induced nitric oxide and reactive oxygen species. Biochem Biophys Res Commun. 2004;317:558–564. doi: 10.1016/j.bbrc.2004.03.075. [DOI] [PubMed] [Google Scholar]
- 57.Itsumi M., Inoue S., Elia A.J., Murakami K., Sasaki M., Lind E.F., et al. Idh1 protects murine hepatocytes from endotoxin-induced oxidative stress by regulating the intracellular NADP+/NADPH ratio. Cell Death Differ. 2015;22:1837–1845. doi: 10.1038/cdd.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim J.Y., Shin J.Y., Kim M., Hann S.K., Oh S.H. Expression of cytosolic NADP+-dependent isocitrate dehydrogenase in melanocytes and its role as an antioxidant. J Dermatol Sci. 2012;65:118–125. doi: 10.1016/j.jdermsci.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 59.Reitman Z.J., Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102:932–941. doi: 10.1093/jnci/djq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zarei M., Lal S., Vaziri-Gohar A., O'Hayer K., Gunda V., Singh P.K., et al. RNA-binding protein HuR regulates both mutant and wild-type IDH1 in IDH1-mutated cancer. Mol Cancer Res. 2019;17:508–520. doi: 10.1158/1541-7786.MCR-18-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zarei M., Lal S., Parker S.J., Nevler A., Vaziri-Gohar A., Dukleska K., et al. Posttranscriptional upregulation of IDH1 by HuR establishes a powerful survival phenotype in pancreatic cancer cells. Cancer Res. 2017;77:4460–4471. doi: 10.1158/0008-5472.CAN-17-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vaziri-Gohar A., Cassel J., Mohammed F.S., Zarei M., Hue J.J., Hajihassani O., et al. Limited nutrient availability in the tumor microenvironment renders pancreatic tumors sensitive to allosteric IDH1 inhibitors. Nat Cancer. 2022;3:852–865. doi: 10.1038/s43018-022-00393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Semukunzi H., Roy D., Li H., Khan G.J., Lyu X., Yuan S., et al. IDH mutations associated impact on related cancer epidemiology and subsequent effect toward HIF-1α. Biomed Pharmacother. 2017;89:805–811. doi: 10.1016/j.biopha.2017.02.083. [DOI] [PubMed] [Google Scholar]
- 64.Molenaar R.J., Radivoyevitch T., Maciejewski J.P., Van Noorden C.J., Bleeker F.E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim Biophys Acta. 2014;1846:326–341. doi: 10.1016/j.bbcan.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 65.Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.H., et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu C., Ward P.S., Kapoor G.S., Rohle D., Turcan S., Abdel-Wahab O., et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chowdhury R., Yeoh K.K., Tian Y.M., Hillringhaus L., Bagg E.A., Rose N.R., et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koivunen P., Lee S., Duncan C.G., Lopez G., Lu G., Ramkissoon S., et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Losman J.A., Kaelin W.G. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feng Y., Li X., Cassady K., Zou Z., Zhang X. TET2 function in hematopoietic malignancies, immune regulation, and DNA repair. Front Oncol. 2019;9:210–218. doi: 10.3389/fonc.2019.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muto T., Sashida G., Oshima M., Iwama A. Mutations of epigenetic regulator genes and myeloid malignancies. Rinsho Ketsueki. 2015;56:2287–2294. doi: 10.11406/rinketsu.56.2287. [DOI] [PubMed] [Google Scholar]
- 72.Figueroa M.E., Lugthart S., Li Y., Erpelinck-Verschueren C., Deng X., Christos P.J., et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gaidzik V.I., Paschka P., Späth D., Habdank M., Köhne C.H., Germing U., et al. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Clin Oncol. 2012;30:1350–1357. doi: 10.1200/JCO.2011.39.2886. [DOI] [PubMed] [Google Scholar]
- 74.Noushmehr H., Weisenberger D.J., Diefes K., Phillips H.S., Pujara K., Berman B.P., et al. Cancer Genome Atlas Research Network, identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Z., Jiang B., Wang Y., Ni H., Zhang J., Xia J., et al. 2-HG inhibits necroptosis by stimulating DNMT1-dependent hypermethylation of the RIP3 promoter. Cell Rep. 2017;19:1846–1857. doi: 10.1016/j.celrep.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 76.Yan B., Hu Y., Ma T., Wang Y. IDH1 mutation promotes lung cancer cell proliferation through methylation of Fibulin-5. Open Biol. 2018;8:180086–180093. doi: 10.1098/rsob.180086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsukada Y., Fang J., Erdjument-Bromage H., Warren M.E., Borchers C.H., Tempst P., et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 78.Tahiliani M., Mei P., Fang R., Leonor T., Rutenberg M., Shimizu F., et al. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- 79.Fodor B.D., Kubicek S., Yonezawa M., O'Sullivan R.J., Sengupta R., Perez-Burgos L., et al. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20:1557–1562. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu W., Tanasa B., Tyurina O.V., Zhou T.Y., Gassmann R., Liu W.T., et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsukada Y., Fang J., Erdjument-Bromage H., Warren M.E., Borchers C.H., Tempst P., et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 82.Klose R.J., Kallin E.M., Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 83.Kooistra S.M., Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 84.Pedersen M.T., Helin K. Histone demethylases in development and disease. Trends Cell Biol. 2010;20:662–671. doi: 10.1016/j.tcb.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 85.Tamaru H., Selker E.U. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature. 2001;414:277–283. doi: 10.1038/35104508. [DOI] [PubMed] [Google Scholar]
- 86.Turcan S., Rohle D., Goenka A., Walsh L.A., Fang F., Yilmaz E., et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Montrose D.C., Zhou X.K., Kopelovich L., Yantiss R.K., Karoly E.D., Subbaramaiah K., et al. Metabolic profiling, a noninvasive approach for the detection of experimental colorectal neoplasia. Cancer Prev Res. 2012;5:1358–1367. doi: 10.1158/1940-6207.CAPR-12-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Colvin H., Nishida N., Konno M., Haraguchi N., Takahashi H., Nishimura J., et al. Oncometabolite d-2-hydroxyglurate directly induces epithelial–mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci Rep. 2016;6:36289–36299. doi: 10.1038/srep36289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kalluri R., Weinberg R.A. The basics of epithelial–mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Du X., Hu H. The roles of 2-hydroxyglutarate. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.651317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kohanbash G., Carrera D.A., Shrivastav S., Ahn B.J., Jahan N., Mazor T., et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127:1425–1437. doi: 10.1172/JCI90644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bunse L., Pusch S., Bunse T., Sahm F., Sanghvi K., Friedrich M., et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. 2018;24:1192–1203. doi: 10.1038/s41591-018-0095-6. [DOI] [PubMed] [Google Scholar]
- 93.Friedrich M., Sankowski R., Bunse L., Kilian M., Green E., Ramallo Guevara C., et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat Cancer. 2021;2:723–740. doi: 10.1038/s43018-021-00201-z. [DOI] [PubMed] [Google Scholar]
- 94.Röver L.K., Gevensleben H., Dietrich J., Bootz F., Landsberg J., Goltz D., et al. PD-1 (PDCD1) promoter methylation is a prognostic factor in patients with diffuse lower-grade gliomas harboring isocitrate dehydrogenase (IDH) mutations. EBioMedicine. 2018;28:97–104. doi: 10.1016/j.ebiom.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kadiyala P., Carney S.V., Gauss J.C., Garcia-Fabiani M.B., Haase S., Alghamri M.S., et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J Clin Invest. 2021;131 doi: 10.1172/JCI139542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang L., Sorensen M.D., Kristensen B.W., Reifenberger G., McIntyre T.M., Lin F. d-2-Hydroxyglutarate is an intercellular mediator in IDH-mutant gliomas inhibiting complement and T cells. Clin Cancer Res. 2018;24:5381–5391. doi: 10.1158/1078-0432.CCR-17-3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang X., Rao A., Sette P., Deibert C., Pomerantz A., et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. 2016;18:1402–1412. doi: 10.1093/neuonc/now061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gilbert M.R., Liu Y., Neltner J., Pu H., Morris A., Sunkara M., et al. Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol. 2014;127:221–233. doi: 10.1007/s00401-013-1194-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Azmi A.S., Philip P.A., Zafar S.F., Sarkar F.H., Mohammad R.M. PAR-4 as a possible new target for pancreatic cancer therapy. Expert Opin Ther Targets. 2010;14:611–620. doi: 10.1517/14728222.2010.487066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Burikhanov R., Zhao Y., Goswami A., Qiu S., Schwarze S.R., Rangnekar V.M. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009;138:377–388. doi: 10.1016/j.cell.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Santos R.V.C., de Sena W.L.B., Dos Santos F.A., da Silva Filho A.F., da Rocha Pitta M.G., da Rocha Pitta M.G., et al. Potential therapeutic agents against Par-4 target for cancer treatment: where are we going?. Curr Drug Targets. 2019;20:635–654. doi: 10.2174/1389450120666181126122440. [DOI] [PubMed] [Google Scholar]
- 102.Saegusa M., Hashimura M., Kuwata T., Okayasu I. Transcriptional regulation of pro-apoptotic Par-4 by NF-κB/p65 and its function in controlling cell kinetics during early events in endometrial tumourigenesis. J Pathol. 2010;221:26–36. doi: 10.1002/path.2680. [DOI] [PubMed] [Google Scholar]
- 103.Sharma A.K., Kline C.L., Berg A., Amin S., Irby R.B. The Akt inhibitor ISC-4 activates prostate apoptosis response protein-4 and reduces colon tumor growth in a nude mouse model. Clin Cancer Res. 2011;17:4474–4483. doi: 10.1158/1078-0432.CCR-10-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu Y., Gilbert M.R., Kyprianou N., Rangnekar V.M., Horbinski C. The tumor suppressor prostate apoptosis response-4 (Par-4) is regulated by mutant IDH1 and kills glioma stem cells. Acta Neuropathol. 2014;128:723–732. doi: 10.1007/s00401-014-1334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ubeda M., Wang X.Z., Zinszner H., Wu I., Habener J.F., Ron D. Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol Cell Biol. 1996;16:1479–1489. doi: 10.1128/mcb.16.4.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang X., Du T., Wang X., Zhang Y., Hu W., Du X., et al. IDH1, a CHOP and C/EBPβ-responsive gene under ER stress, sensitizes human melanoma cells to hypoxia-induced apoptosis. Cancer Lett. 2015;365:201–210. doi: 10.1016/j.canlet.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 107.Ron D., Habener J.F. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- 108.Li F., He X., Ye D., Lin Y., Yu H., Yao C., et al. NADP+-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol Cell. 2015;60:661–675. doi: 10.1016/j.molcel.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 109.Jiang B., Zhang J., Xia J., Zhao W., Wu Y., Shi M., et al. IDH1 mutation promotes tumorigenesis by inhibiting JNK activation and apoptosis induced by serum starvation. Cell Rep. 2017;19:389–400. doi: 10.1016/j.celrep.2017.03.053. [DOI] [PubMed] [Google Scholar]
- 110.Rosiak K., Smolarz M., Stec W.J., Peciak J., Grzela D., Winiecka-Klimek M., et al. IDH1R132H in neural stem cells: differentiation impaired by increased apoptosis. PLoS One. 2016;11 doi: 10.1371/journal.pone.0154726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang N., Li Z., Bai F., Zhang S. PAX5-induced upregulation of IDH1-AS1 promotes tumor growth in prostate cancer by regulating ATG5-mediated autophagy. Cell Death Dis. 2019;10:734. doi: 10.1038/s41419-019-1932-3. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang J., Quan Y., Lv J., Dong Q., Gong S. LncRNA IDH1-AS1 suppresses cell proliferation and tumor growth in glioma. Biochem Cell Biol. 2020;98:556–564. doi: 10.1139/bcb-2019-0465. [DOI] [PubMed] [Google Scholar]
- 113.Vousden K.H., Lane D.P. p53 in health and disease. Folia Neuropathol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 114.Hu X., Yu A.X., Qi B.W., Fu T., Wu G., Zhou M., et al. The expression and significance of IDH1 and p53 in osteosarcoma. J Exp Clin Cancer Res. 2010;29:43–52. doi: 10.1186/1756-9966-29-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Birner P., Toumangelova-Uzeir K., Natchev S., Guentchev M. Expression of mutated isocitrate dehydrogenase-1 in gliomas is associated with p53 and EGFR expression. Folia Neuropathol. 2011;49:88–93. [PubMed] [Google Scholar]
- 116.Cai J., Guan H., Fang L., Yang Y., Zhu X., Yuan J., et al. MicroRNA-374a activates Wnt/β-catenin signaling to promote breast cancer metastasis. J Clin Investig. 2013;123:566–579. doi: 10.1172/JCI65871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wu Y.Y., Ginther C., Kim J., Mosher N., Chung S.Y., Slamon D., et al. Expression of Wnt3 activates Wnt/β-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res. 2012;10:1597–1606. doi: 10.1158/1541-7786.MCR-12-0155-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nik N., Xi Z., Liu M., Li Y., Yao M., Liu T., et al. Advances in therapeutic agents targeting quiescent cancer cells. Acta Mater Medica. 2022;1:56–71. [Google Scholar]
- 119.Valenta T., Hausmann G., Basler K. The many faces and functions of β-catenin. EMBO J. 2012;31:2714–2736. doi: 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li T., Guo H., Song Y., Zhao X., Shi Y., Lu Y., et al. Loss of vinculin and membrane-bound β-catenin promotes metastasis and predicts poor prognosis in colorectal cancer. Mol Cancer. 2014;13:263–277. doi: 10.1186/1476-4598-13-263. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 121.Cui D., Ren J., Shi J., Feng L., Wang K., Zeng T., et al. R132H mutation in IDH1 gene reduces proliferation, cell survival and invasion of human glioma by downregulating Wnt/β-catenin signaling. Int J Biochem Cell Biol. 2016;73:72–81. doi: 10.1016/j.biocel.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 122.Wang J.B., Dong D.F., Gao K., Wang M.D. Mechanisms underlying the biological changes induced by isocitrate dehydrogenase-1 mutation in glioma cells. Oncol Lett. 2014;7:651–657. doi: 10.3892/ol.2014.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang J.B., Dong D.F., Wang M.D., Gao K. IDH1 overexpression induced chemotherapy resistance and IDH1 mutation enhanced chemotherapy sensitivity in Glioma cells in vitro and in vivo. Asian Pac J Cancer Prev. 2014;15:427–432. doi: 10.7314/apjcp.2014.15.1.427. [DOI] [PubMed] [Google Scholar]
- 124.Shen X., Wu S., Zhang J., Li M., Xu F., Wang A., et al. Wild-type IDH1 affects cell migration by modulating the PI3K/AKT/mTOR pathway in primary glioblastoma cells. Mol Med Rep. 2020;22:1949–1957. doi: 10.3892/mmr.2020.11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Calvert A.E., Chalastanis A., Wu Y., Hurley L.A., Kouri F.M., Bi Y., et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. 2017;19:1858–1873. doi: 10.1016/j.celrep.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Harding J.J., Lowery M.A., Shih A.H., Schvartzman J.M., Hou S., Famulare C., et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov. 2018;8:1540–1547. doi: 10.1158/2159-8290.CD-18-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Popovici-Muller J., Lemieux R.M., Artin E., Saunders J.O., Salituro F.G., Travins J., et al. Discovery of AG-120 (Ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett. 2018;9:300–305. doi: 10.1021/acsmedchemlett.7b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.DiNardo C.D., Stein E.M., De Botton S., Roboz G.J., Altman J.K., Mims A.S., et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386–2398. doi: 10.1056/NEJMoa1716984. [DOI] [PubMed] [Google Scholar]
- 129.Choe S., Wang H., DiNardo C.D., Stein E.M., De Botton S., Roboz G.L., et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020;4:1894–1905. doi: 10.1182/bloodadvances.2020001503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Anonymous The IDH1 inhibitor ivosidenib may slow IDH1-mutant glioma progression. Cancer Discov. 2020;10:OF6. [Google Scholar]
- 131.Mellinghoff I.K., Ellingson B.M., Touat M., Maher E., De La Fuente M.I., Holdhoff M., et al. Ivosidenib in isocitrate dehydrogenase 1 mutated advanced glioma. J Clin Oncol. 2020;38:JCO1903327. doi: 10.1200/JCO.19.03327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Abou-Alfa G.K., Macarulla T., Javle M.M., Kelley R.K., Lubner S.J., Adeva J., et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21:796–807. doi: 10.1016/S1470-2045(20)30157-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tap W.D., Villalobos V.M., Cote G.M., Burris H., Janku F., Mir O., et al. Phase I study of the mutant IDH1 inhibitor ivosidenib: safety and clinical activity in patients with advanced chondrosarcoma. J Clin Oncol. 2020;38:1693–1701. doi: 10.1200/JCO.19.02492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhu A.X., Macarulla T., Javle M.M., Kelley R.K., Lubner S.J., Adeva J., et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation: the Phase 3 randomized clinical ClarIDHy trial. JAMA Oncol. 2021;7:1669–1677. doi: 10.1001/jamaoncol.2021.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mellinghoff I.K., Ellingson B.M., Touat M., Maher E., De La Fuente M.I., Holdhoff M., et al. Ivosidenib in isocitrate dehydrogenase 1-mutated advanced glioma. J Clin Oncol. 2020;38:3398–3406. doi: 10.1200/JCO.19.03327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stein E.M., DiNardo C.D., Pollyea D.A., Fathi A.T., Roboz G.J., Altman J.K., et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–731. doi: 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stein E.M., Fathi A.T., DiNardo C.D., Pollyea D.A., Roboz G.J., Collins R., et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: a phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. 2020;7:e309–e319. doi: 10.1016/S2352-3026(19)30284-4. [DOI] [PubMed] [Google Scholar]
- 138.Mellinghoff I.K., Penas-Prado M., Peters K.B., Burris H.A., Maher E.A., Janku F., et al. Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial. Clin Cancer Res. 2021;27:4491–4499. doi: 10.1158/1078-0432.CCR-21-0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Natsume A., Arakawa Y., Narita Y., Sugiyama K., Hata N., Muragaki Y., et al. The first-in-human phase I study of a brain penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro Oncol. 2022;20:noac155. doi: 10.1093/neuonc/noac155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.De La Fuente M.I., Colman H., Rosenthal M., Van Tine B.A., Levacic D., Walbert T., et al. Olutasidenib (FT-2102) in patients with relapsed or refractory IDH1-mutant glioma: a multicenter, open-label, phase 1b/2 trial. Neuro Oncol. 2022;25:noac139. doi: 10.1093/neuonc/noac139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wick A., Bähr O., Schuler M., Rohrberg K., Chawla S.P., Janku F., et al. Phase I assessment of safety and therapeutic activity of BAY1436032 in patients with idh1-mutant solid tumors. Clin Cancer Res. 2021;27:2723–2733. doi: 10.1158/1078-0432.CCR-20-4256. [DOI] [PubMed] [Google Scholar]
- 142.DiNardo C.D., Schimmer A.D., Yee K.W.L., Hochhaus A., Kraemer A., Carvajal R.D., et al. A phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Blood. 2016;128:1073. [Google Scholar]
- 143.National Center for Tumor Diseases, Heidelberg. Phase I trial of IDH1 peptide vaccine in IDH1R132H-mutated grade III-IV gliomas (NOA-16). ClinicalTrials.gov. Updated [2018 November 7]. Available from: https://clinicaltrials.gov/ct2/show/NCT02454634?term=NCT02454634&draw=2&rank=1.
- 144.Clarivate. Drug discovery intelligence. Updated [2022]. Available from: https://www.cortellis.com/drugdiscovery/result/796c3f0c-3692-7d1d-de78-b0ca4b087fa0/drugs/productList.
- 145.Cho Y.S., Levell J.R., Liu G., Caferro T., Sutton J., Shafer C.M., et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med Chem Lett. 2017;8:1116–1121. doi: 10.1021/acsmedchemlett.7b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nakagawa M., Nakatani F., Matsunaga H., Seki T., Endo M., Ogawara Y., et al. Selective inhibition of mutant IDH1 by DS-1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene. 2019;38:6835–6849. doi: 10.1038/s41388-019-0929-9. [DOI] [PubMed] [Google Scholar]
- 147.Pusch S., Krausert S., Fischer V., Balss J., Ott M., Schrimpf D., et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017;133:629–644. doi: 10.1007/s00401-017-1677-y. [DOI] [PubMed] [Google Scholar]
- 148.Chaturvedi A., Herbst L., Pusch S., Klett L., Goparaju R., Stichel D., et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia. 2017;31:2020–2028. doi: 10.1038/leu.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Heuser M., Palmisiano N., Mantzaris I., Mims A., DiNardo C., Silverman L.R., et al. Safety and efficacy of BAY1436032 in IDH1-mutant AML: phase I study results. Leukemia. 2020;34:2903–2913. doi: 10.1038/s41375-020-0996-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gupta C., Kaulfuss S., Görlich K., Othman B., Chaturvedi A., Heuser M. Combination treatment of an IDH1 inhibitor with chemotherapy in IDH1 mutant acute myeloid leukemia. Ann Hematol. 2020;99:1415–1417. doi: 10.1007/s00277-020-04001-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chaturvedi A., Gupta C., Gabdoulline R., Borchert N.M., Goparaju R., Kaulfuss S., et al. Synergistic activity of IDH1 inhibitor BAY1436032 with azacitidine in IDH1 mutant acute myeloid leukemia. Haematologica. 2020;106:565–573. doi: 10.3324/haematol.2019.236992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Konteatis Z., Artin E., Nicolay B., Straley K., Padyana A.K., Jin L., et al. Vorasidenib (AG-881): a first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med Chem Lett. 2020;11:101–107. doi: 10.1021/acsmedchemlett.9b00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ma R., Yun C.H. Crystal structures of pan-IDH inhibitor AG-881 in complex with mutant human IDH1 and IDH2. Biochem Biophys Res Commun. 2018;503:2912–2917. doi: 10.1016/j.bbrc.2018.08.068. [DOI] [PubMed] [Google Scholar]
- 154.Mellinghoff I.K., Cloughesy T.F., Wen P.Y., Taylor J.W., Maher E.A., Arrillaga-Romany I., et al. ACTR-66. A phase 1, open-label, perioperative study of ivosidenib (AG-120) and vorasidenib (AG-881) in recurrent IDH1 mutant, low-grade glioma: updated results. Neuro Oncol. 2019;21:vi28–vi29. [Google Scholar]
- 155.Caravella J.A., Lin J., Diebold R.B., Campbell A.M., Ericsson A., Gustafson G., et al. Structure-based design and identification of FT-2102 (olutasidenib), a potent mutant-selective IDH1 inhibitor. J Med Chem. 2020;63:1612–1623. doi: 10.1021/acs.jmedchem.9b01423. [DOI] [PubMed] [Google Scholar]
- 156.HUTCHMED's worldwide business. Pipeline-and-products. Updated [2020]. Available from: https://www.hutch-med.com/sc/pipeline-and-products/our-pipeline/.
- 157.Archer Gary. 2021. IDH1 peptide vaccine for recurrent grade II glioma (RESIST)https://clinicaltrials.gov/ct2/show/results/NCT0219334 Available from: [Google Scholar]
- 158.Popovici-Muller J., Saunders J.O., Salituro F.G., Travins J.M., Yan S., Zhao F., et al. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med Chem Lett. 2012;3:850–855. doi: 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rohle D., Popovici-Muller J., Palaskas N., Turcan S., Grommes C., Campos C., et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Davis M., Pragani R., Popovici-Muller J., Gross S., Thorne N., Salituro F., et al. Probe Reports from the NIH molecular libraries program [Internet] National Center for Biotechnology Information (US); Bethesda (MD): 2010. ML309: a potent inhibitor of R132H mutant IDH1 capable of reducing 2-hydroxyglutarate production in U87 MG glioblastoma cells. updated [2013 May 8]. PMID: 23905201. [PubMed] [Google Scholar]
- 161.Zheng Q., Tang S., Fu X., Chen Z., Ye Y., Lan X., et al. Discovery and structure–activity-relationship study of novel conformationally restricted indane analogues for mutant isocitric dehydrogenase 1 (IDH1) inhibitors. Bioorg Med Chem Lett. 2017;27:5262–5266. doi: 10.1016/j.bmcl.2017.10.029. [DOI] [PubMed] [Google Scholar]
- 162.Zhou X., Zheng M., Zhao N., Hu Y., Yang K., Huo J., et al. Discovery of linear unnatural peptides as potent mutant isocitrate dehydrogenase 1 inhibitors by Ugi reaction. Bioorg Chem. 2022;119 doi: 10.1016/j.bioorg.2021.105569. [DOI] [PubMed] [Google Scholar]
- 163.Levell J.R., Caferro T., Chenail G., Dix I., Dooley J., Firestone B., et al. Optimization of 3-pyrimidin-4-yl-oxazolidin-2-ones as allosteric and mutant specific inhibitors of IDH1. ACS Med Chem Lett. 2017;8:151–156. doi: 10.1021/acsmedchemlett.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhao Q., Manning J.R., Sutton J., Costales A., Sendzik M., Shafer C.M., et al. Optimization of 3-pyrimidin-4-yl-oxazolidin-2-ones as orally bioavailable and brain penetrant mutant IDH1 inhibitors. ACS Med Chem Lett. 2018;9:746–751. doi: 10.1021/acsmedchemlett.8b00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zheng Q., Chen Z., Wan H., Tang S., Ye Y., Xu Y., et al. Discovery and structure–activity-relationship study of novel imidazole cyclopropyl amine analogues for mutant isocitric dehydrogenase 1 (IDH1) inhibitors. Bioorg Med Chem Lett. 2018;28:3808–3812. doi: 10.1016/j.bmcl.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 166.Ma T., Zou F., Pusch S., Yang L., Zhu Q., Xu Y., et al. Design, synthesis and biological activity of 3-pyrazine-2-yl-oxazolidin-2-ones as novel, potent and selective inhibitors of mutant isocitrate dehydrogenase 1. Bioorg Med Chem. 2017;25:6379–6387. doi: 10.1016/j.bmc.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 167.Jia P., Wu Y., Du H., Yang L., Zhang Z., Ma T., et al. I-8, a novel inhibitor of mutant IDH1, inhibits cancer progression in vitro and in vivo. Eur J Pharm Sci. 2019;140:105072–105082. doi: 10.1016/j.ejps.2019.105072. [DOI] [PubMed] [Google Scholar]
- 168.Bauer RA, Boulet SL, Burkholder TP, Gilmour R, Hahn PJ, Rankovic Z, inventors; Eli Lilly and Company, assignee. 7-Phenylethylamino-4H-pyrimido[4,5-d] [1,3] oxazin-2-one compounds as mutant IDH1 and IDH2 inhibitors. US paptent US16349873. December 8 2017.
- 169.Zheng B., Yao Y., Liu Z., Deng L., Anglin J.L., Jiang H., et al. Crystallographic investigation and selective inhibition of mutant isocitrate dehydrogenase. ACS Med Chem Lett. 2013;4:542–546. doi: 10.1021/ml400036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liu Z., Yao Y., Kogiso M., Zheng B., Deng L., Qiu J.J., et al. Inhibition of cancer-associated mutant isocitrate dehydrogenases: synthesis, structure–activity relationship, and selective antitumor activity. J Med Chem. 2014;57:8307–8318. doi: 10.1021/jm500660f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wu F., Jiang H., Zheng B., Kogiso M., Yao Y., Zhou C., et al. Inhibition of cancer-associated mutant isocitrate dehydrogenases by 2-thiohydantoin compounds. J Med Chem. 2015;58:6899–6908. doi: 10.1021/acs.jmedchem.5b00684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Okoye-Okafor U.C., Bartholdy B., Cartier J., Gao E.N., Pietrak B., Rendina A.R., et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. 2015;11:878–886. doi: 10.1038/nchembio.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Law J.M., Stark S.C., Liu K., Liang N.E., Hussain M.M., Leiendecker M., et al. Discovery of 8-membered ring sulfonamides as inhibitors of oncogenic mutant isocitrate dehydrogenase 1. ACS Med Chem Lett. 2016;7:944–949. doi: 10.1021/acsmedchemlett.6b00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lin J., Lu W., Caravella J.A., Campbell A.M., Diebold R.B., Ericsson A., et al. Discovery and optimization of quinolinone derivatives as potent, selective, and orally bioavailable mutant isocitrate dehydrogenase 1 (mIDH1) inhibitors. J Med Chem. 2019;62:6575–6596. doi: 10.1021/acs.jmedchem.9b00362. [DOI] [PubMed] [Google Scholar]
- 175.Deng G., Shen J., Yin M., McManus J., Mathieu M., Gee P., et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J Biol Chem. 2015;290:762–774. doi: 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hu Y., Gao A., Liao H., Zhang M., Xu G., Gao L., et al. 3-(7-Azaindolyl)-4-indolylmaleimides as a novel class of mutant isocitrate dehydrogenase-1 inhibitors: design, synthesis, and biological evaluation. Arch Pharm. 2018;351 doi: 10.1002/ardp.201800039. [DOI] [PubMed] [Google Scholar]
- 177.Jones S., Ahmet J., Ayton K., Ball M., Cockerill M., Fairweather E., et al. Discovery and optimization of allosteric inhibitors of mutant isocitrate dehydrogenase 1 (R132H IDH1) displaying activity in human acute myeloid leukemia cells. J Med Chem. 2016;59:11120–11137. doi: 10.1021/acs.jmedchem.6b01320. [DOI] [PubMed] [Google Scholar]
- 178.Chaturvedi A., Goparaju R., Gupta C., Weder J., Klünemann T., Araujo Cruz M.M., et al. In vivo efficacy of mutant IDH1 inhibitor HMS-101 and structural resolution of distinct binding site. Leukemia. 2020;34:416–426. doi: 10.1038/s41375-019-0582-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zheng M., Sun W., Gao S., Luan S., Li D., Chen R., et al. Structure based discovery of clomifene as a potent inhibitor of cancer-associated mutant IDH1. Oncotarget. 2017;8:44255–44265. doi: 10.18632/oncotarget.17464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zou F., Pusch S., Hua J., Ma T., Yang L., Zhu Q., et al. Identification of novel allosteric inhibitors of mutant isocitrate dehydrogenase 1 by cross docking-based virtual screening. Bioorg Med Chem Lett. 2018;28:388–393. doi: 10.1016/j.bmcl.2017.12.030. [DOI] [PubMed] [Google Scholar]
- 181.Duan Z., Liu J., Niu L., Wang J., Feng M., Chen H., et al. Discovery of DC_H31 as potential mutant IDH1 inhibitor through NADPH-based high throughput screening. Bioorg Med Chem. 2019;27:3229–3236. doi: 10.1016/j.bmc.2019.05.040. [DOI] [PubMed] [Google Scholar]
- 182.Kang C.H., Choi S.U., Son Y.H., Lee H.K., Jeong H.G., Yun C.S., et al. Discovery of a novel chemical scaffold against mutant isocitrate dehydrogenase 1 (IDH1) Anticancer Res. 2020;40:4929–4935. doi: 10.21873/anticanres.14496. [DOI] [PubMed] [Google Scholar]
- 183.Kim H.J., Fei X., Cho S.C., Choi B.Y., Ahn H.C., Lee K., et al. Discovery of α-mangostin as a novel competitive inhibitor against mutant isocitrate dehydrogenase-1. Bioorg Med Chem Lett. 2015;25:5625–5631. doi: 10.1016/j.bmcl.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 184.Zheng M., Tang R., Deng Y., Yang K., Chen L., Li H. Steroids from ganoderma sinense as new natural inhibitors of cancer-associated mutant IDH1. Bioorg Chem. 2018;79:89–97. doi: 10.1016/j.bioorg.2018.04.016. [DOI] [PubMed] [Google Scholar]
- 185.Verdura S., Cuyàs E., Lozano-Sánchez J., Bastidas-Velez C., Llorach-Parés L., Fernández-Arroyo S., et al. An olive oil phenolic is a new chemotype of mutant isocitrate dehydrogenase 1 (IDH1) inhibitors. Carcinogenesis. 2019;40:27–40. doi: 10.1093/carcin/bgy159. [DOI] [PubMed] [Google Scholar]
- 186.Hu C., Zuo Y., Liu J., Xu H., Liao W., Dang Y., et al. Licochalcone A suppresses the proliferation of sarcoma HT-1080 cells, as a selective R132C mutant IDH1 inhibitor. Bioorg Med Chem Lett. 2020;30:126825–126834. doi: 10.1016/j.bmcl.2019.126825. [DOI] [PubMed] [Google Scholar]
- 187.Montesinos P., Recher C., Vives S., Zarzycka E., Wang J., Bertani G., et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med. 2022;386:1519–1531. doi: 10.1056/NEJMoa2117344. [DOI] [PubMed] [Google Scholar]
- 188.Ott M., Tomaszowski K.H., Marisetty A., Kong L.Y., Wei J., Duna M., et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight. 2020;5 doi: 10.1172/jci.insight.134386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Norsworthy K.J., Mulkey F., Scott E.C., Ward A.F., Przepiorka D., Charlab R., et al. Differentiation syndrome with ivosidenib and enasidenib treatment in patients with relapsed or refractory IDH-mutated AML: a U.S. Food and Drug Administration systematic analysis. Clin Cancer Res. 2020;26:4280–4288. doi: 10.1158/1078-0432.CCR-20-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim J., Tang J.Y., Gong R., Kim J., Lee J.J., Clemons K.V., et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17:388–399. doi: 10.1016/j.ccr.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zheng M., Huo J., Gu X., Wang Y., Wu C., Zhang Q., et al. Rational design and synthesis of novel dual PROTACs for simultaneous degradation of EGFR and PARP. J Med Chem. 2021;64:7839–7852. doi: 10.1021/acs.jmedchem.1c00649. [DOI] [PubMed] [Google Scholar]