

Abstract
RAD52 protein is a coveted target for anticancer drug discovery. Similar to poly-ADP-ribose polymerase (PARP) inhibitors, pharmacological inhibition of RAD52 is synthetically lethal with defects in genome caretakers BRCA1 and BRCA2 (∼25% of breast and ovarian cancers). Emerging structure activity relationships for RAD52 are complex, making it challenging to transform previously identified disruptors of the RAD52–ssDNA interaction into drug-like leads using traditional medicinal chemistry approaches. Using pharmacophoric informatics on the RAD52 complexation by epigallocatechin (EGC), and the Enamine in silico REAL database, we identified six distinct chemical scaffolds that occupy the same physical space on RAD52 as EGC. All six were RAD52 inhibitors (IC50 ∼23–1200 μM) with two of the compounds (Z56 and Z99) selectively killing BRCA-mutant cells and inhibiting cellular activities of RAD52 at micromolar inhibitor concentrations. While Z56 had no effect on the ssDNA-binding protein RPA and was toxic to BRCA-mutant cells only, Z99 inhibited both proteins and displayed toxicity towards BRCA-complemented cells. Optimization of the Z99 scaffold resulted in a set of more powerful and selective inhibitors (IC50 ∼1.3–8 μM), which were only toxic to BRCA-mutant cells. RAD52 complexation by Z56, Z99 and its more specific derivatives provide a roadmap for next generation of cancer therapeutics.
Graphical Abstract
Graphical Abstract.
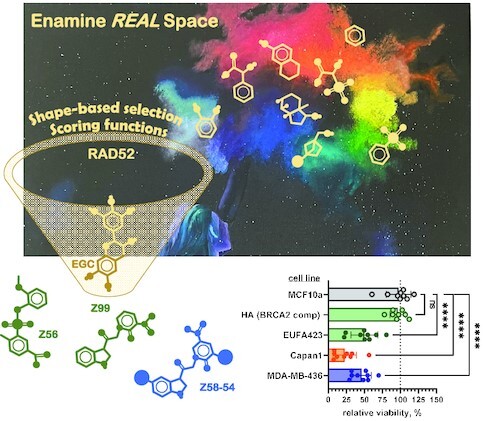
Exploration of the Enamine in silico REAL space of billions of potential compounds yielded new drug-like inhibitors of the DNA repair protein RAD52. The compounds were further optimized to improve efficacy and specificity in targeting RAD52 in vitro and killing BRCA1 and BRCA2 deficient cancer cells.
INTRODUCTION
Human DNA repair protein RAD52 (1,2), and more specifically the RAD52–ssDNA interaction, is an attractive target for the development of small molecule therapeutics specific to cancers displaying BRCAness phenotype, as well as ATM (ataxia–telangiectasia mutated) deficient cancers (3–10). While RAD52 pharmacological inhibition is selectively toxic to cells carrying biallelic mutations in BRCA1, BRCA2 and PALB2 (8,11–13), and to cells deficient in ATM serine/threonine kinase (10), its inactivation due to mutations is well tolerated ((14–16) and LOEUF score of 0.64 at gnomAD database). This situation is unique among DNA repair and genome maintenance proteins that share synthetic lethality with BRCA deficiency but whose mutations are much more rare (see (3) for review). RAD52, however, is a challenging target, as it does not possess any bona fide enzymatic activities, and the nature of its synthetic relationship with BRCAness and ATM deficiency is only beginning to emerge. Instead, many of RAD52′s cellular functions depend on its capacity to bind ssDNA, single-stranded features of complex DNA substrates, or ssDNA bound by Replication Protein A (RPA), the main ssDNA binding protein in eukaryotic cells (3,17,18).
Efficient pharmacological targeting of protein-DNA interactions with small molecules presents a formidable challenge due to the nature of DNA binding sites on proteins. This has been met in recent years by advancements in computer-aided drug discovery, which yielded drug candidates that directly disrupt protein-nucleic acid interactions of several transcription factors (19,20). In contrast to transcription factors, however, RAD52 binds ssDNA and displays no sequence specificity. The narrow continuous binding cavity of the RAD52–ssDNA binding site spans the circumference of the protein oligomeric ring (21–25). An effective and specific inhibitor of the RAD52–ssDNA interaction would require the ability to outcompete multivalent RAD52–ssDNA interactions, while at the same time displaying specificity for RAD52 over the multitude of cellular ssDNA binding proteins which include RPA, RAD51, and BRCA2 among many. The RAD52–ssDNA binding groove offers a repetitive pattern of potential small molecule binding pockets with reasonable druggability scores, though their nature is quite distinct from the typical enzyme active sites and small ligand binding pockets on receptors (8).
Previously, in Hengel et al. (8), we identified a variety of small molecule natural products in a small HTS screen of the MicroSource Spectrum library. In a parallel in silico, structure-based analysis we built a predictive pharmacophoric model of RAD52 inhibition. This structural model was highly successful in identifying novel natural products that bind to RAD52 and inhibit its interaction with ssDNA, which provided a valuable proof of concept. An important aspect of that study was that epigallochatechin (EGC) had a desirable selectivity, in that it competed with ssDNA binding to RAD52, but not to RPA. It was also selectively toxic to BRCA2-depleted cells confirming that pharmacological inhibition of RAD52–ssDNA interaction can perform the same function as RAD52 depletion. Selectivity for RAD52 over RPA was a unique property of EGC among compounds identified in our initial HTS screen and subsequent round of in silico natural products screening. Our structural models indicated that EGC binds to a distinct hot spot within the RAD52–DNA binding groove, suggesting that future scaffold hopping into drug-like space should primarily focus on this region. The approach taken in the current study is a multi-tiered workflow that uses Enamine's REAL database of theoretical drug-like compounds (described in more detail below; Figure 1A). The workflow first employs shape selectivity (based on the shape of the bound form of EGC, using the stress tested MD-based model from (8). Docking of the compounds to the EGC local hot spot is then followed by either the use of classic scoring functions or a Protein–Ligand Interaction Fingerprints (PLIFs) based on the RAD52–EGC complex from (8), as an alternative ligand scoring approach. Both the scoring function and PLIF approaches yielded synthetically tractable novel drug-like inhibitors of RAD52, some of which possessed the desired selectivity towards RAD52 over RPA in vitro, as well as selective toxicity towards BRCA1 and BRCA2-mutant cells. Additionally, we used one of the lead compounds, Z99, as the basis for a much larger search of REAL chemical space, consisting of 31 billion of novel compounds, in order to identify synthetically tractable heavier derivatives, which were synthesized and purified and found to have augmented affinity for RAD52. Importantly, although these Z99 derivatives had enhanced inhibition of RAD52 most of these scaffold expansion compounds had the same or reduced RPA inhibition compared to the parent Z99 compound. Because of this selectivity they were non-toxic to BRCA-proficient cells, while killing BRCA-deficient cells. These studies show a tremendous structural traction in the design of selective inhibitors that act via disruption of RAD52–ssDNA complexation, and highlight the feasibility of small drug-like molecules to selectively disrupt protein-nucleic acid interactions. Notably, our advancement in developing inhibitors of such a challenging target as RAD52 is rooted in a new workflow we have developed for using and narrowing down the ultra-large virtual chemical space of favourable chemotypes.
Figure 1.

Identification of RAD52 with new chemical scaffolds. (A) Workflow for scaffold hopping from EGC. Three million compound ENAMINE REAL database was narrowed to 7700 based on shape similarity to EGC and docked into the EGC pocket within the RAD52–ssDNA binding site (see text for details). Two methods were used to score the compounds: In the first approach (orange flow) CCG/MOE PLIF tool was used to create fingerprints for RAD52–ligand interactions, in order to find compounds mimicking the contacts of EGC and RAD52; in the second approach (green flow) the London dG scoring function was used to rank order compounds. Three top scoring compounds from each method were ordered. (B) Final poses for the EGC (green), Z99 (orange), and Z56 (gold) compounds bound to RAD52. (C) Top compounds selected from each computational approach, PLIF (orange box) and docking/scoring function (gold box).
MATERIALS AND METHODS
Docking and scoring by (i) molecular mechanics (MM) and generalized born (GB)/surface area (SA) (MMGB/SA) and (ii) PLIF (Protein–Ligand Interaction Fingerprints
Virtual screening of REAL (drug-like) chemical space to identify RAD52 inhibitors that have affinity to the EGC-hot spot: The first phase of this computational workflow to identify drug-like small molecules that bind to the EGC-hot spot of RAD52 involved a shape-similarity screening, using the bound EGC–RAD52 complex from Hengel et al. (8), against the Enamine's REAL chemical space; REadily AccessibLe through parallel synthesis using in-stock building blocks with pre-validated chemical reactivity (26,27). The concept is that these are drug-like compounds that are built using well-established chemical reactions, in order to achieve high retrosynthetic scores, and consists of ∼ 5.5 billion enumerated novel compounds that are constructed from 284 thousand building blocks. Our shape-based screening began with a representative 3 million REAL database, from which 500K compounds were selected for shape similarity with the bound form of EGC. This screening yielded ∼ 7.7 thousand compounds, which were next used in docking into the EGC hot spot. The hot spot was defined in a 5 Å boundary around the EGC from Hengel et al. (8).
Docking was performed using the docking utility of MOE 2016, as described in Hengel et al. (8). Briefly, classical docking, using the Triangle Matcher approach and scoring using either, the London dG scoring function (an empirical scoring function which attempts to approximate the binding energy of the docked ligand) in MOE 2016 (Chemical Computing Group, 2016), followed by force field (MMFF94x (28))-based ligand refinement and finally rescoring using an MM/GBSA-based approach (which is described in more detail below), or scoring by employing Protein–Ligand Interaction Fingerprints (PLIF) application in MOE 2016, based on the contacts and placement of EGC using the hybrid MD-docking approach described in (8,29). In the scoring function approach, the top 30 poses for each docked and scored (London dG scoring function) were subjected to energy minimization with a rigid RAD52 receptor using the MMFF94x force field, followed by rescoring (in order to estimate the ΔG of binding) of each distinct pose with the MM/GBSA methodology (30), which includes an implicit solvation energy calculation and captures changes in the solvent exposed surface area of the pose, which is a highly parameterized version of the popular MM/PBSA and MM/GBSA methodologies (31,32). In the PLIF approach, the scoring was entirely based on the similarity to the EGC–RAD52 complex. For both scoring methods, van der Waals surface overlap with the EGC complex was assessed in the top 500 hits as well. The top three compounds from each method were then submitted to Enamine for attempted synthesis, and were successfully synthesized to high purity (Supplementary Table S1).
Scaffold expansion of RAD52 inhibitor Z99 by a search of REAL chemical space using InfiniSee
The program InfiniSee (BiosolveIT, version 2.4.0) is designed to use a fragment buildup scheme to very rapidly search defined chemical spaces (e.g. REAL space) against a query compound. This approach allows a much more expanded search of REAL space ∼3.2 × 1010 virtual products; based on 165 Enamine reactions, 100 526 in-stock reagents, and 182 381 selected building blocks) than originally conceived in the REAL database, as described above, allowing one to assess billions of compounds for their fingerprint similarity to the query. We employed InfiniSee to identify slightly heavier (∼15%) and with >97% chemical similarity to Z99. Six compounds resulting from this search were submitted to Enamine for attempted synthesis, which was achieved at a high purity (see QC in Supplementary Information section). These compounds were also docked into the EGC pocket, using the protocol described above.
Evaluation of the compound-RPA complexes
In order to assess how various Z99 derivatives, as well as EGC and EGCG bind to RPA, the compounds were docked into the DNA binding pocket of PDB 1JMC (33). AutoDock VINA (34) was employed within the YASARA environment (35). Ligands and the receptor were prepared for docking as described in (8). Each minimized pose of each ligand was docked with 16 runs each to the receptor using AutoDock VINA.
Materials
The HPLC purified 30-nucleotide poly(dT) ssDNA, fluorescently labelled with Cy3 and Cy5 dies at the 3′ and 5′ termini, respectively (Cy3–dT30–Cy5), and all oligonucleotides were purchased from the Integrated DNA Technologies (Coralville, IA, USA). All chemicals were reagent grade (Sigma-Aldrich, St. Louis, MO). The sources of the antibodies are indicated with their respective experimental procedures below.
Compound nomenclature and properties
The compounds full identification numbers from the Enamine REAL database are shown in Table 1, Table 2, and Figures 1 and 6. For simplicity, the initial six compounds are referred to by their first two or three numbers yielding the unique identifiers within our set of compounds, while the compounds with the expanded Z99 scaffold are referred to as Z58 followed by the last two numbers in their respective designations. All compounds identified through in silico screening of the Enamine REAL Database (≥90% purity) were synthesized by Enamine Ltd (Kyiv, Ukraine). Compounds were used without further purification. Identity of these compounds was confirmed by liquid chromatography–mass spectrometry (LC–MS). High-resolution LC–MS was performed using a Q Exactive Orbitrap Mass Spectrometer (ThermoFisher). Ten microliter of diluted sample (1 ng/ul) was injected onto a Waters Acquity UPLC BEH C18 column (130 Å, 1.7 μm, 2.1 mm × 100 mm) using a mobile phase of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) at a flow rate of 300 μl/min with the following linear gradient A/B (v/v): 95/5 to 5/95 from 0–17 min. Mass detection was carried out in full scan mode with polarity switching over mass range of 70–1000 Da. Data acquisition and analysis were performed using Xcalibur software (ThermoFisher). Molecular ions (M−) or negative ions ([M−H]−) were detected for all compounds in negative ion mode. Positive ions ([M+H]+) were detected for compounds Z99, Z56, Z16, Z134, and Z58-54, 72, 83, 87, 62. Compound information and LC–MS results, including retention time (RT) and relative abundances (%) of ions, are included in the Supplementary Table S1.
Table 1.
Hits from the original in silico workflow
| Compound | Inhibition of the RAD52–ssDNA interaction, IC50 (μM) | Inhibition of the RPA–ssDNA interaction, IC50 (μM) | Inhibition of the RPA–ssDNA–RAD52 complex, IC50 (μM) | Toxicity towards BRCA-deficient cells (at 100 μM) | Toxicity towards BRCA-proficient cells (at 100 μM) |
|---|---|---|---|---|---|
| PLIF hits | |||||
| Z991573902 | 50.5 ± 5.8 | 14.0 ± 1.8 | 42.5 ± 6.7 | yes (EUFA423F, Capan1, MDA-MB-436) | Yes (EUFA423F HA, MCF10a) |
| Z1633286133 | 248.8 ± 9.5 | 255.4 ± 10.4 | 54.9 ± 3.1 | not tested | not tested |
| Z1347718341 | 1236 ± 75 | N/A | 96.9 ± 10.0 | not tested | not tested |
| Docking/scoring function hits | |||||
| Z45684214 | 23.1 ± 1.5 | N/A | 28.9 ± 5.5 | no | no |
| Z56785913 | 35.1 ± 2.6 | N/A | 8.28 ± 0.55 | yes (EUFA423F, Capan1, MDA-MB-436) | no |
| Z133609104 | 933 ± 46 | N/A | 46.7 ± 4.9 | not tested | not tested |
N/A, no activity.
Table 2.
Hits from Z99 scaffold expansion
| Compound | Inhibition of the RAD52–ssDNA interaction, IC50 (μM) | Inhibition of the RPA–ssDNA interaction, IC50 (μM) | Inhibition of the RPA–ssDNA–RAD52 complex, IC50 (μM) | Toxicity towards BRCA-deficient cells (at 10 μM) | Toxicity towards BRCA-proficient cells (at 10 μM) |
|---|---|---|---|---|---|
| Z5862216254 | 7.26 ± 0.61 | 18.0 ± 1.3 | N/A | yes (EUFA423F, Capan1, MDA-MB-436) | no |
| Z5862231762 | 2.84 ± 0.32 | 28.8 ± 4.0 | 4.55 ± 0.16 | yes/no (Capan1, MDA-MB-436) | no |
| Z5862148469 | 2.02 ± 0.12 | 1.12 ± 0.13 | 0.76 ± 0.05 | yes/no (Capan1, MDA-MB-436) | no |
| Z5862209772 | 7.97 ± 0.84 | 15.9 ± 1.4 | 7.00 ± 0.29 | yes (EUFA423F, Capan1, MDA-MB-436) | no/yes (MCF10a in colony formation assay) |
| Z5862135983 | 6.21 ± 0.42 | 54.0 ± 9.7 | 54.3 ± 16.2 | yes (EUFA423F, Capan1, MDA-MB-436) | no/yes (MCF10a in colony formation assay) |
| Z5862219887 | 3.98 ± 0.35 | 8.49 ± 1.43 | 6.43 ± 1.01 | yes (EUFA423F, Capan1, MDA-MB-436) | no |
N/A – no activity
Figure 6.

Compounds generated by expansion of the Z99 scaffold show improved efficacy in vitro and in cells. (A) Six new compounds were designed based on the Z99 scaffold. (B) Final poses of the scaffold expanded compounds docked into the EGC pocket of RAD52. Z99 is shown using surface representation, while the six new compounds are shown in stick representation. (C) IC50 values for the disruption of the RAD52–ssDNA interaction (blue bars), of the RPA–ssDNA interaction (green), and of the RPA–ssDNA-RAD52 complex (grey). Error bars represent fitting errors. The inhibition curves for all compounds are shown in Supplementary Figure S5. (D) Cell viability was measured using CellTiter-Glo assay for five cell lines after 72 h treatment with 10 μM of indicated compounds. Hundred percent live cells corresponds to DMSO only control. The data are shown as average ± standard deviation for nine independent wells. The detailed statistical analysis for each compound is shown in Supplementary Figure S7.
Proteins
We used a QuikChange Lightning site-directed mutagenesis kit (Agilent) to introduce K152A/R153A (for RAD52 IB mutant) and K102A/K133A/K169A/R173A (for RAD52 OB mutant) mutations in the RAD52 coding sequence of pET15b-6HIS-RAD52 plasmid. The following primer pairs were used; lower case, underlined letters represent mutation sites: GTTGACCTCAACAATGGCgcgTTCTACGTGGGAGTCTG and CAGACTCCCACGTAGAAcgcGCCATTGTTGAGGTCAAC (K102A), GTTAGTGAGGGCCTCgcgTCCAAGGCTTTATCTTTGG and CCAAAGATAAAGCCTTGGAcgcGAGGCCCTCACTAAC (K133A), GGAGGCGGTGACAGACGGGCTGaagcgaGCCCTCAGGAGTTTTGGGAATGC and GCATTCCCAAAACTCCTGAGGGCtgccgcCAGCCCGTCTGTCACCGCCTCC (K152A/R153A), and GCACTTGGAAACTGTATTCTGGACgcaGACTACCTGgcaTCACTAAATAAGCTTCCACGCCAG and CTGGCGTGGAAGCTTATTTAGTGAtgcCAGGTAGTCtgcGTCCAGAATACAGTTTCCAAGTGC (K169A/R173A). The presence of mutations was confirmed by Sanger sequencing (University of Iowa Institute of Human Genetics). The 6xHis-tagged human RAD52 proteins (wild type and mutants) and untagged human RPA were expressed and purified as previously described (8,21,36). RPA and RAD52 protein concentrations were determined by measuring absorbance at 280 nm using extinction coefficients of 88 830 M−1 cm−1 and 40 470 M−1 cm−1, respectively.
Cell lines
EUFA423F cells that are derived from a Fanconi anemia patient with complementation group D1 and have biallelic mutations (7691 insAT and 9900 insA) in BRCA2 that result in two different truncated forms of BRCA2 (37) and its counterpart complemented with expression vector for full-length BRCA2 (EUFA HA) were a generous gift from Dr Simon Powell (Molecular Biology Program, Radiation Oncology, Sloan-Kettering Cancer Center, New York, NY). Capan 1 (generous gift from Dr Simon Powell) cells are derived from a human pancreatic carcinoma and exhibit loss of heterozygosity with respect to the BRCA2 gene. One copy of the BRCA2 gene is lost and the remaining copy has a 6174delT mutation. This mutation disrupts BRC repeats 7 and 8 and causes a frameshift that introduces a stop codon which prematurely truncates the protein (38,39). This is a pathogenic mutation with several independent origins in both Jewish Ashkenazi and non-Jewish populations (40). MDA-MB 436 (generous gift from Dr. Carola Neumann, University of Pittsburgh) is a triple negative breast cancer cell line derived from a 51-year-old patient. It contains a BRCA1 5396 + 1G > A mutation in the splice donor site for exon 20 that results in a protein with truncated BRCT domain, accompanied by the loss of the second BRCA1 allele (41). EUFA423F, Capan-1 and MDA-MB 436 have been previously shown to depend on RAD52 for viability (6,11,13). MCF10a (generous gift from Dr Kris DeMali, University of Iowa) is a non-tumorigenic, breast epithelial cell line isolated from the mammary gland of a 36-year-old female with fibrocystic disease, and are spontaneously immortalized without defined factors (42).
MRC5SV40 cells were a gift from Dr. Patricia Kannouche (CNRS, Institute de Recherches sur le Cancer Gustave Roussy, Villejuif, France). The stable cell lines expressing the shRAD52 cassette were generated from MRC5SV40 and were described (43). Media for each cell line is summarized in Supplementary Table S2.
FRET-based DNA binding and inhibition assays
FRET-based assays that monitor the RAD52–ssDNA, RPA–ssDNA and RAD52–RPA–ssDNA binding were carried out as previously described (8,21,44) using Cary Eclipse Spectrofluorimeter (Agilent) at 25°C, in 5mm quartz cuvettes, and in buffer containing 30 mM Tris-acetate (pH 7.5) and 1mM DTT. Cy3 dye was excited at 530 nm and its emission was monitored at 565 nm. Emission of Cy5 acceptor fluorophore excited through the energy transfer from Cy3 donor is monitored at 660 nm simultaneously with emission of Cy3 dye. Both the excitation and the emission slits were set to 10 nm. All experiments were carried out using 1 nM (molecules) Cy3–dT30–Cy5 ssDNA. The optimal concentrations of the RAD52 (6 nM) and RPA (1 nM) proteins were selected by titrating the respective protein into 1 nM Cy3–dT30–Cy5 solution, and chosen based on the largest separation of the FRET values corresponding to the protein-bound and free DNA.
The inhibition of the RAD52–ssDNA, RPA–ssDNA and RAD52–RPA–ssDNA complexes was evaluated by titrating each compound to the respective complex, measuring Cy3 and Cy5 fluorescence, and calculating FRET as previously described (8).
Spectral properties of many of our compounds overlapped with the Cy3 and Cy5 fluorescence. To correct the calculated FRET values for the compound signal, the fluorogenic compounds were titrated into buffer and into solution of 1 nM Cy3–dT30–Cy5. For all tested compounds, the change in the signal in the Cy3 and Cy5 channels with and without Cy3/Cy5-labeled DNA present was the same. We therefore concluded that the compounds do not change the ssDNA conformation and are unlikely to interact with ssDNA. To account for the dye fluorescence, for each compound concentration, we have subtracted the apparent change in FRET of the ssDNA in the presence of the compound from the respective FRET signal of the RAD52–ssDNA, RPA–ssDNA or RAD52–ssDNA–RPA complex at the same inhibitor concentration. The IC50 values were calculated by fitting the corrected FRET data to the four parameter dose response model using GraphPad Prism. All data are shown as an average for at least three independent experiments ± standard deviation. The IC50 values are presented with their respective fitting errors.
Intrinsic protein fluorescence was used to evaluate direct interactions between compounds and RAD52. Fluorescence was measured using Cary Eclipse Spectrofluorimeter (Agilent) at 25°C, in 150 μl quartz cuvettes, and in buffer containing 30 mM Tris-Acetate pH 7.5, 100 mM KCl, 1 mM DTT. Tyrosine fluorescence in RAD52 was excited at 280 nm and emission spectra were recorded between 300 and 400 nm. All spectra were collected in triplicates after addition of the indicated concentration of the compound or equivalent amount of DMSO. The spectra were plotted using GraphPad Prism.
Mass photometry experiments were performed using the Refeyn TwoMP mass photometry instrument (Refeyn Ltd, Oxford, UK) in buffer containing 20 mM Tris (pH 7.4), 100mM KCl, and 1mM DTT. Cover slides were cleaned by sequential washing with miliQ water and 100% isopropanol twice and then with miliQ water and subsequently dried under an air stream. Silicon buffer gaskets were rinsed sequentially with miliQ water, isopropanol and miliQ water, dried at room temperature. Dried silicon gaskets were attached to the glass slide by applying a mild pressure and mounted on a Refeyn TwoMP mass photometer. Molecular weight calibrations were performed using two protein oligomer solutions, β-amylase (56, 112 and 224 kDa) and Thyroglobulin (670 kDa). In each experiment, 400nM of RAD52 was incubated with 200 μM of respective inhibitor for 45 min at room temperature. The protein-inhibitor solution was then diluted 4 times into the buffer-filled gasket yielding final concentration of 100 nM RAD52 and 50 μM inhibitor. Individual molecular weights collected from 3000 frames (59.9 s) were binned in 5 kDa bins and plotted as frequency histograms. GraphPad Prism was used to fit the molecular weight distributions to multiple Gaussians.
Cell proliferation assay
Selective toxicity of the compounds towards human BRCA1 and BRCA2 deficient cells was tested in MDA-MB 436 (BRCA1 deficient), EUFA423F (BRCA2 deficient), its complement EUFA423F HA (BRCA2 complemented), Capan1 (BRCA2 deficient), and MCF10a (BRCA positive) cells using CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI). The cell lines were maintained in media summarized in Supplementary Table S2. EUFA423F HA cells were routinely supplemented with G418 (IBI Science, Dubuque, IA) in order to maintain the BRCA2 expression plasmid. Cell lines were seeded in sterile tissue culture treated, white opaque 384-well plates (Perkin Elmer Inc., Waltham, MA; 2500–5000 cells/well) or 96-well plates (10 000 cells/well) and allowed to adhere for 24 h. Compounds dissolved in DMSO were added in the concentration range of 0–100 μM. Controls were treated with equivalent DMSO volumes. The cells were treated with compounds for 72 h (to ensure that all cells experienced at least two S-phases). Post treatment, the cells were washed, treated with the CellTiter-Glo® reagent, and the plate was analysed as using a Gen5 (ver2.1) luminosity plate reader. The luminescence that reports on ATP generation by the surviving cells was normalized for the DMSO treated control as 100%. The normalized luminescence (average ± standard deviation for at least three independent measurements obtained with different cell passages) was plotted in GraphPad Prism as a function of compound concentration.
Clonogenic survival
Cells were seeded in a 96-well plate. Compounds dissolved in DMSO were added to final concentrations of 0 and 100 μM for Z56 and Z99, and 0, 10 μM for Z58-54, Z58-62, Z58-69, Z58-72, Z58-83 and Z58-87. The cells were treated with compounds for 72 h. Posttreatment, cell cultures were harvested by collecting all non-adherent cells in the well, trypsinization of all adherent cells, combining, pelleting and then resuspending in fresh media. Cultures were subsequently counted using a hemocytometer. Cells (200 cells for EUFA423F, EUFA423F HA, 1000 cells for Capan1 and 500 cells for MCF10a) were plated in a 60 mm plate and colonies were allowed to grow until the colony sizes reached at least 50 cells. For each treatment and cell line, the cells were plated in triplicate. After expanding the colonies, cultures were fixed with a solution of 70% ethanol and crystal violet for clonogenic survival analysis. Pictures of individual wells were taken using GelCount (Version 1.3.0.4) by Oxford Optronix (Abingdon, UK). The number of surviving colonies following treatment are presented as a percentage of number of colonies in the experiment where the cells were treated with DMSO only.
In situ proximity ligation assay (PLA)
In situ PLA (DuoLink, Merck) was performed according to the manufacturer's instructions. To detect parental ssDNA-RAD52 interaction, cells were labelled with 50 μM IdU for 20 h, released in fresh DMEM for 2 h and then treated as indicated. After treatment, cells were permeabilized with 0.5% Triton X-100 for 10 min at 4°C, fixed with 3% formaldehyde/2% sucrose solution for 10 min, and then blocked in 3% BSA/PBS for 15 min. After washing with PBS, cells were incubated with the two relevant primary antibodies. The primary antibodies used were: rabbit polyclonal anti-RAD52 (Aviva 1:150), and an anti-IdU (mouse monoclonal anti-BrdU/IdU; clone b44 Becton Dickinson, 1:10). In control experiments, parallel samples were probed with each primary antibody alone. Samples were incubated with secondary antibodies conjugated with PLA probes MINUS and PLUS (DuoLink, Merck). Incubation with primary and secondary antibodies was accomplished in a humidified chamber for 1 h at 37°C. PLA probes MINUS and PLUS were ligated using two connecting oligonucleotides to produce a template for rolling-cycle and hybridisation with TRITC-labelled oligonucleotide. Samples were mounted in Prolong Gold anti-fade reagent with DAPI to counterstain nuclei. Images were acquired randomly using Eclipse 80i Nikon Fluorescence Microscope, equipped with a Virtual Confocal (ViCo) system.
Neutral comet assay
Slides were dipped into 1% agarose in PBS and left to dry. After treatments cells were resuspended in cold PBS and kept on ice to inhibit DSBs repair. Aliquots of cell suspensions were mixed with low melting point agarose solution (0.5% LMPA in PBS) at 37°C and pipetted onto agarose-covered surface of the slide. Agarose-embedded cells were lysed by submerging slides in lysis solution (30 mM EDTA, 0.1% sodium dodecyl sulphate (SDS)) and incubated at 4°C, 1 h in the dark. After lysis, slides were washed in Tris Borate EDTA (TBE) 1X running buffer (Tris 90 mM; boric acid 90 mM; EDTA 4 mM) for 1 min. Electrophoresis was performed for 20 min in TBE 1× buffer at 1 V/cm. Slides were subsequently washed in distilled water and finally dehydrated in ice-cold methanol. Nuclei were stained with GelRed (1:1000) and visualised using fluorescence.
Detection of ssDNA by native IdU assay
To detect nascent ssDNA, cells were labelled for 20 min with 100 μM IdU (Sigma-Aldrich), immediately prior to the indicated treatments. For immunofluorescence, cells were washed with PBS, permeabilized with 0.5% Triton X-100 for 10 min at 4°C and fixed in 3% PFA/2% sucrose. Fixed cells were then incubated with mouse anti-IdU antibody (Becton Dickinson) for 1 h at 37°C in 1% BSA/PBS, followed by species-specific fluorescein-conjugated secondary antibodies (Alexa Fluor 488 Goat Anti-Mouse IgG (H + L), highly cross-adsorbed—Life Technologies). Slides were analysed with Eclipse 80i Nikon Fluorescence Microscope, equipped with a Virtual Confocal (ViCo) system. For each sample, at least 100 nuclei were examined and pictures were taken at 40×. Quantification of fluorescence intensity was carried out using the ImageJ software.
RESULTS
In silico campaign targeting the EGC binding pocket of RAD52 oligomeric ring yielded six synthetic small molecule inhibitors disrupting the RAD52–ssDNA interaction
Our goal was to exploit our predictive models about RAD52–EGC complexation, based on (8,29), in order to identify drug-like compounds that have affinity for the EGC hot spot within the RAD52 primary DNA binding site. Figure 1A summarizes the total workflow, which starts with the 3 million drug-like compounds representing diversity of scaffolds found in the Enamine REAL Database. MOE shape screening application was used to select REAL compounds from a randomly selected 500K subset database with a 90% similarity to that of the EGC from the EGC–RAD52 complex (8). This shape screening yielded ∼7.7 thousand compounds, which were used as input for docking simulations into the hot spot occupied by EGC, using the MOE docking utility (see Materials and Methods for details). Due to the many false positives that can occur with classic scoring functions, we elected to also capitalize on our confidence in the RAD52–EGC complex to employ an alternative approach which uses PLIF in order to determine how similar a drug-like ligand's complexation is compared to that of EGC. For both methods the Van der Waals surface overlap was examined for the top 500 compounds (i.e. for both scoring function-based ranking and for PLIF-based ranking), and the top 3 compounds from each approach were selected for synthesis and purification. Figure 1B shows overlap between EGC (green) and representatives of both PLIF (Z99, orange) and scoring function (Z56, gold) bound to RAD52 (grey). Chemical structures of all six selected compounds are shown in Figure 1C; the QC data are shown in Supplementary Table S1, and ligand maps for representative compounds vis-à-vis EGC are shown in Figure 2.
Figure 2.

Ligand maps for compounds Z99, Z56 and EGC. The interaction key is shown on the right. Among notable contacts, all three compounds interact with R55, one of the key residues in the DNA binding site of RAD52. In contrast to EGC, however, whose interactions are dominated by van der Waals interaction and water-mediated contacts, Z99 and Z56 are involved in more direct interactions including hydrogen bonding.
Both in silico strategies, docking and PLIF, identified compounds that disrupt the RAD52–ssDNA and RAD52–RPA–ssDNA complexes in vitro
Six compounds with distinct chemical scaffolds were tested for their ability to disrupt the RAD52–ssDNA interaction using a Förster resonance energy transfer (FRET) based assay, which has been previously used to characterize the RAD52 inhibitors (8). Upon binding to ssDNA, RAD52 and RPA change the ssDNA geometry in a very specific manner. While in a stoichiometric complex with RAD52, the ssDNA is wrapped around the oligomeric protein ring (8,21,44). In contrast, RPA extends the ssDNA to nearly counter length (44,45). In our binding assays, FRET between the two fluorescent dyes (FRET donor Cy3 and FRET acceptor Cy5) conjugated at the ends of a synthetic DNA substrate Cy3–dT30–Cy5, reports on the ssDNA conformation and therefore on the state of the protein–ssDNA complexes (44,46). We have selected experimental conditions (see Materials and Methods for details) that allow for the best discrimination between free ssDNA (FRET ≈ 0.49), RAD52-bound ssDNA (FRET ≈ 0.82), RPA-bound ssDNA (FRET ≈ 0.35), and RAD52 bound to the RPA–ssDNA complex (FRET ≈ 0.62) (Figure 3A, B, and Supplementary Figure S1). All six compounds identified through the in silico workflow were able to disrupt the RAD52–ssDNA complexes with IC50 values in the mid to high micromolar range (Table 1, Supplementary Figure S1, blue circles and lines).
Figure 3.

Specificity of the new synthetic compounds towards RAD52 versus RPA is important for selective killing of BRCA2-deficient cells. The in vitro FRET-based assays follow inhibition of the RAD52–ssDNA interaction (blue circles and lines), RPA–ssDNA interactions (green circles and lines), and the interaction between RAD52 and RPA–ssDNA complex (black circles and lines) by Z56 (A) and Z99 (B) compounds. Complexes at the starting point of inhibitor titrations are depicted schematically on the left with respective FRET values indicated. The FRET values for these complexes are color cored on the graphs matching the respective inhibition curves. The free ssDNA is shown on the right with its respective FRET value and the light orange bar across the graphs that also signifies the endpoint of the inhibition reaction for displacing RAD52, RPA or RAD52 and RPA from ssDNA. The endpoint of the reactions where RAD52 is displaced from RPA-coated ssDNA is represented by the light green bar. The data shown as an average ± standard deviation for at least three independent measurements. Where invisible, the error bars are smaller than the respective symbols. Calculated IC50 values are shown above each graph. (C) Intrinsic tyrosine fluorescence-based analysis of the RAD52 complexation with Z56 and Z99 inhibitors. Tyrosine fluorescence, excited at 280 nm is presented as fluorescence spectra (each is an average ± standard deviation for three scans). Concentrations of each inhibitor are shown by their respective spectra. The change from the darker to lighter shades of blue correspond to increasing compound concentrations. Changes in fluorescence upon addition to equivalent amounts of DMSO are shown in the left panel. (D) Diagram of the BRCA2 truncations in two defective BRCA2 alleles of the EUFA423F cell line, and Capan1 cell line (left panel), and BRCA1 truncation in MDA-MB-436 cell line (right panel). In HA, the BRCA2 deficiency is complemented by expression of the full-length protein, while MCF10a cells have unaltered BRCA1 and BRCA2 genes. (E and F) Cell viability of BRCA-proficient (HA), BRCA1-mutated MDA-MB-436, and BRCA2-mutated EUFA423F cells as a function of Z56 (E) or Z99 – concentrations was evaluated using CellTiter-Glo assay. The data are plotted as average ± standard deviation for at least three independent measurements. Hundred percent live cells corresponds to DMSO only control. (G and H) Cell viability was measured for 5 cell lines after 72 h treatment with 100 μM of Z56 (G) or Z99 (H). The data are shown as individual measurements along with average ± standard deviation for 9 independent measurements. The response of each cell lines was compared to MCF10a (ns = not significant P> 0.05; *P< 0.05; **P< 0.01; ***P< 0.001; ****P < 0.0001; ordinary ANOVA).
While our rationale for targeting the EGC pocket within the RAD52–ssDNA binding groove, and not the pocket where a stronger, but promiscuous inhibitor epigallocatechin gallate (EGCG) binds, was to enhance selectivity towards RAD52 over other ssDNA binding proteins. Since RPA is one of the most important ssDNA binding proteins (18,47) and because the RPA–ssDNA complex is a physiological substrate for many cellular functions of RAD52, we tested the ability of the six compounds to inhibit the RPA–ssDNA interaction (Table 1, Figure 3A, B, Supplementary Figure S1 green circles and lines) and to displace RAD52 from the RPA-bound ssDNA without affecting the RPA–ssDNA complex (Table 1, Figure 3A, B, Supplementary Figure S1, black circles and lines). These reactions contained a stoichiometric complex of 1 nM RPA bound to 1 nM Cy3–dT30–Cy5. Among the tested compounds, Z99 and Z16 did not show specificity for RAD52 and were able to inhibit RPA–ssDNA with IC50 values comparable to (Z16) or better (Z99) than those for RAD52. Both of these compounds were PLIF hits.
All six compounds were able to remove RAD52 from the RPA–ssDNA complex. For the four compounds that did not inhibited RPA (Z45, Z56, Z133 and Z134), the final product at the saturating inhibitor concentrations was the RPA–ssDNA complex (FRET ≈ 0.35) further confirming specificity for RAD52 over RPA. The IC50 values for the disruption of the ternary RPA–ssDNA–RAD52 complex were several fold lower than the respective values for the RAD52–ssDNA complex for four compounds, Z16, Z56, Z133 and Z134. This difference was likely due to the different ssDNA contacts within the ternary complex.
Figure 3 shows the in vitro inhibition data for two compounds, Z56 (Figure 3A) which is specific to RAD52 and Z99 (Figure 3B) which is more promiscuous. Increasing concentrations of both Z56 and Z99 applied to the RAD52–ssDNA complex resulted in the FRET decrease from the values corresponding to the wrapped RAD52–ssDNA complex (0.82) to the values corresponding to free ssDNA (0.49). Addition of Z56 does not change the FRET values of the RPA–ssDNA complex (0.35) and decreases the FRET values of the RPA–ssDNA-RAD52 complex (0.62) to the values corresponding to the RPA–ssDNA complex (0.35). In contrast, titration of Z99 into both RPA–ssDNA and RPA–ssDNA-RAD52 complex yields FRET values corresponding to the free ssDNA (0.49). Because of their IC50 values and difference in specificity, these two compounds were selected for the cell-based analyses discussed below.
Z56 and Z99 inhibitors directly interact with RAD52 protein
To validate that selected compounds bind to RAD52 directly, we analysed the compound-RAD52 interaction by following RAD52 intrinsic fluorescence (Figure 3C). RAD52 has several tyrosine residues including Y65 and Y126 that are present in the EGC binding pocket. When excited at 280 nm, tyrosine residues of RAD52 generate a fluorescence emission spectrum with a peak at 338 nm (Figure 3C, dark blue lines). Changes in the environment and solvent exposure of these aromatic residues due to ligand complexation is expected affect their fluorescence. Since addition of DMSO (which is used to solubilize the compounds) may also affect the protein fluorescence, we first recorded RAD52 intrinsic fluorescence spectra in the presence of DMSO at equivalent concentrations to those added with the respective inhibitors (left panel). Addition of Z56 (middle panel) and Z99 (right panel) resulted in a much more significant quenching of RAD52 fluorescence compared to DMSO alone confirming the presence of a direct interaction between these compounds and RAD52.
Z56 and Z99 inhibitors bind within the inner binding groove of the RAD52 oligomeric ring
Biochemical and structural studies identified two distinct DNA binding sites in the RAD52 protein (24,48,49). The ‘inner’ or ‘primary’ binding site is a narrow groove that spans the circumference of the protein and accommodates ssDNA. A specific pocket within this groove was used in our computational workflow. Positively charged residues deep in the groove make electrostatic contacts with the DNA backbone. Among these key ssDNA contacts, R55 makes strong interactions with both Z56 and Z99. The ‘outer’ or ‘secondary’ DNA binding site of RAD52 binds both ssDNA and dsDNA and may provide a platform for DNA annealing (24,48). We have purified two mutants, inner binding defective mutant (IB, K152A/R153A, Supplementary Figure S2A, B, purple), and outer binding defective mutant (OB, K102A/K103A/K169A/R173A, Supplementary Figure S2 teal). Both mutants retained the capacity to bind ssDNA and RPA–ssDNA complex, but distorted the DNA to a lesser degree than the wild type RAD52 (Supplementary Figure S2C, D). Both Z56 and Z99 were able to inhibit ssDNA binding to IB and OB mutants. The IC50 value for Z56 inhibition of the wild type and IB mutant RAD52 were within the fitting error. In contrast, IC50 value for OB mutant, which can only engage the inner binding site for DNA binding was 6-fold lower. This correlates well with the computed binding position of this compound (Supplementary Figure S2E). Notably, while the IC50 values for Z56 inhibiting the RPA–ssDNA-RAD52(mutant) complex for both mutants were within fitting error of the corresponding values for RAD52(mutant)-ssDNA complex, for the wildtype protein the IC50 value for the RPA–ssDNA–RAD52 complex was 6-fold lower than for the RAD52–ssDNA complex, suggesting plasticity of the ssDNA binding within the RAD52 oligomeric ring (Table 1). Z99 had enhanced inhibition efficiency for both IB and OB mutant compared to the wild type RAD52, but similarly to Z56 had 6-fold increased efficiency against OB mutant compared to IB mutant.
The new RAD52 inhibitors compete with DNA binding, and not with the oligomerization of RAD52
RAD52 forms a ring shaped oligomer, with a range of monomers in the oligomeric ring identified in biochemical studies and an undecameric ring visualized by X-ray crystallography (23–25,50). Among the RAD52 inhibitors identified to date, one compound, 6-OH-dopa was proposed to act by interfering with RAD52 oligomerization and interaction between RAD52 protein rings (11). The oligomeric state of RAD52 in the presence and absence of our new inhibitors was evaluated using mass photometry, an interferometric light scattering technique for accurate, label-free mass measurement of single macromolecular complexes in solution, which is insensitive to the molecules’ shape (51). At 100 nM, RAD52 exists in solution as a mixture of oligomeric species (Supplementary Figure S3). The best fit to the binned mass frequency histograms was a sum of three Gaussians consistent with the majority of the protein existing as decameric and undecameric species, with a small fraction (5%) of monomers present. The mass distribution of RAD52 oligomers was not affected by any of the compounds. Notably, 200 μM is close to the solubility limit for some of the compounds where they start forming aggregates in aqueous solution. A small amount of such aggregates was observed as appearance and disappearance (negative mass) of low molecular weight species. The fact that the compounds, even at high concentration, do not affect the RAD52 oligomers or cause protein aggregation is an additional confirmation of their specific mode of action.
Z56 and Z99 selectively kill BRCA-mutant cells but promiscuity of Z99 causes toxicity
Among the identified compounds, three compounds Z45, Z56 and Z99 had sufficiently low IC50 values to be tested in cell-based experiments. Notably, these compounds represent both computational approaches, PLIFs and scoring function, and also represent compounds that are specific to RAD52 (e.g. Z56) or can promiscuously inhibit RPA (e.g. Z99). The inhibitors used in this experiment were Z45, Z56 and Z99. Z16, Z133 and Z134 were excluded as the volume of DMSO required to dissolve the inhibitor would have a significant negative impact on cell growth. Cell-based viability experiments were carried out using CellTiter-Glo® Luminescent Cell Viability Assay (see Materials and Methods for details) and an isogenic pair of cell lines: EUFA423F is a Fanconi anemia cell line that harbors biallelic mutations (7691 insAT and 9900 insA) in BRCA2 gene resulting in two different truncated forms of BRCA2 (37) and its counterpart complemented with expression vector for full-length BRCA2 (Figure 3C). The two BRCA2 truncations in EUFA423F are missing the C-terminal RAD51 binding region important for the BRCA2 function at stalled replication forks (52–55), and the OB-fold containing region important in BRCA2–ssDNA interaction (56–59) (Figure 3D). EUFA423F are sensitive to PARP inhibitor Velaparib (ABT-888) and are believed to represent a good BRCA-deficient cell model (60). The complemented cell line (HA) has a fully functional BRCA2, and is resistant to veliparib (60) and DNA cross-linking agent mitomycin C (37). MDA-MB-436 was used as a model for BRCA1-deficient cancer cell line. MDA-MB-436 is a triple negative breast cancer cell line with one BRCA1 allele lost and the second allele containing a pathogenic BRCA1 5396 + 1G > A mutation and expressing BRCA1 protein with truncated BRCT domain (schematically illustrated in Figure 3D) (41).
Among three tested compounds, only two compounds, Z56 (Figure 3E, green curves) and Z99 (Figure 3F, green curves) were capable of selectively targeting BRCA2-mutated cells. While both Z56 and Z99 were selectively toxic to BRCA2-mutated EUFA423F cells and BRCA1-mutated MDA-MB-436 cells, high concentrations of Z99 affected the viability of BRCA2-complementd HA cells. The reason for this toxicity is likely the capacity of Z99 to inhibit the ssDNA binding by RPA and/or other ssDNA binding proteins. In contrast, Z56, which had no effect on the RPA–ssDNA complex in FRET-based assays was also selective in cell viability experiments, leaving the growth of BRCA2-complemented HA cells unaffected, even at high (250μM) concentrations of the compound. Concentrations of Z56 at which the half amplitude of viability effect was achieved in the EUFA423F and MDA-MB-436 cells (∼30 μM) were very similar to the IC50 value for the disruption of the RAD52–ssDNA complex in FRET-based studies. The viability profiles of MDA-MB-436 for both Z56 (Figure 3E, blue curve) and Z99 (Figure 3F, blue curve) are similar to those of BRCA2-deficent EUFA423F cells suggesting that our new inhibitors work in both BRCA1 and BRCA2-deficent cells. The relative luminescence of the CellTiter-Glo® reagent saturated at about 40–50% of the initial signal. Similarly, previous reports indicated that RAD52 depletion by siRNA, PARP inhibitor Olaparib, and RAD52 inhibitor 6-OH-dopa reduce viability of MDA-MB-436 approximately two-fold (11).
Activity of Z56 and Z99 compounds was then analysed in two additional cell lines at 100 μM of respective inhibitor (Figure 3G and H). We used Capan1 as a model BRCA2-deficint cancer cell line. In Capan1, one copy of the BRCA2 gene is lost and the remaining copy has a pathogenic 6174delT mutation. This mutation disrupts BRC repeats 7 and 8 and causes a frameshift that introduces a stop codon which prematurely truncates the protein (schematically depicted in Figure 3D) (38,39). This is a pathogenic mutation with several independent origins in both Jewish Ashkenazi and non-Jewish populations (40). As a model for non-cancerous BRCA-proficient cell line we used MCF10a, a breast epithelial cell line spontaneously immortalized without defined factors (42). Similar to EUFA423F and MDA-MB-436, Capan1 cells were sensitive to both Z56 and Z99, while MCF10a cells were only sensitive to Z99 similar to BRCA2-complemented EUFA423F HA (Figure 3G and H).
The effect of the Z56 and Z99 compounds on cell viability was also confirmed in clonogenic survival assays. Supplementary Figure S4 shows quantification of colonies seeded by the cells pre-treated for 72 h with 100 μM of inhibitor or an equivalent amount of DMSO. Similar to viability experiments, Z56 reduced clonogenic potential of BRCA-deficient cells, while Z99 was toxic also to BRCA-proficient cells.
Z56 and Z99 specifically interfere with RAD52 cellular functions
RAD52 plays several genome stabilizing functions in human cells (see (3,7) for recent reviews). RAD52 participates in DSB repair by both homology-directed repair and via single-strand annealing (61,62), has a residual activity in supporting homologous recombination (6), functions to protect stalled and damaged DNA replication forks (43), cooperates with MUS81 nuclease in fork cleavage (63), antagonizes DNA polymerase θ (64), promotes early steps in mitotic DNA synthesis (65,66), and participates in RNA-templated DNA repair (67). To assess the specificity of our inhibitors in targeting RAD52 in cells, we focused on two specific function of RAD52, promoting formation of DSBs by MUS81 nuclease (63) and protection of stalled DNA replication forks (43). The latter was selected because of the recent observation that postreplicative ssDNA gaps may underlie the hypersensitivity of BRCA-deficient cancers to chemotherapy (68), and our previous discovery that RAD52 inhibition leads to replication fork restoration with ssDNA gaps (17,43).
Upon prolonged replication stress, RAD52 depletion or its inhibition with EGC leads to impaired MUS81-dependent replication fork cleavage (8,43,63). To assess the ability of Z56 and Z99 to interfere with the RAD52 function in promoting MUS81-dependent DSBs following replication stress, we performed neutral Comet assay in MRC5SV40 fibroblasts treated with HU for 24h in the presence of Z56, Z99 or EGC (Figure 4A, B). As expected, prolonged replication arrest induced by HU led to elevated levels of DSBs (8,63,69), which were significantly reduced by EGC (Figure 4B). Z56 and Z99 were also able to significantly reduce DSBs formation in HU-treated cells even at concentrations below their respective IC50s. To assess if the effect of Z56 and Z99 treatment was specific, we compared the formation of HU-induced DSBs in RAD52-expressing cells (MRC5SV40 fibroblasts) and cells stably depleted of RAD52 (MRC5SV40 shRAD52) (Figure 4C). As previously reported (63), depletion of RAD52 greatly reduced spontaneous and HU-induced DSBs. Most strikingly, while treatment with Z56 or Z99 reduced DSBs in RAD52-proficient cells it failed to further decrease their amount in the RAD52-depleted cells confirming that RAD52 is indeed the target of our inhibitors. RAD52 inhibition also affects DSBs formed at deprotected forks in the absence of BRCA2 (8). To further investigate the phenotypic effect of Z56 or Z99 in the cells, we evaluated formation of DSBs in a cell model in which an shRNA cassette targeting BRCA2 is under the control of doxycycline. As shown in Figure 4D, depletion of BRCA2 (+DOX) resulted in a significant increase in the number of DSBs. This increase was largely suppressed when cells were treated with Z56 or Z99. As also shown in Figure 4B, Z56 and Z99 outcompeted EGC leading to comparable DSBs suppression at a much lower dose (10 μM versus 50 μM).
Figure 4.

Z56 and Z99 interfere with RAD52-MUS81 function at stalled DNA replication forks. (A) Cartoon representation of fork cleavage by the RAD52-MUS81 axes. The presence of the DSBs can be detected using comet assay (B–D). (B) Evaluation of DSBs using Neutral Comet Assay. Cells were treated as indicated. Graph shows the mean of tail moment as mean ± SE (ns = not significant; ***P< 0.001; ****P < 0.0001; Mann–Whitney test). When indicated, cells were treated with inhibitors for 30 min before HU exposure. (C) Analysis of DSBs in MRC5 SV40 and MRC5 shRAD52 after HU exposure in presence or absence of RAD52 inhibitors (30 min before HU treatment). Graph shows the mean of tail moment ± SE (ns = not significant; *P< 0.1; ****P < 0.0001; Mann–Whitney test). (D) After 48 h of doxycycline induction cells were treated to perform neutral comet assay. Western blot shows BRCA2 level after doxycycline induction. LAMIN B1 was used as a loading control. Graph shows the mean of tail moment ± SE (****P < 0.0001; Mann–Whitney test).
RAD52 plays an important role in protecting DNA replication forks from reversal and subsequent degradation (43). Previously (43,69), we have established experimental conditions that are able to distinguish generation of nascent ssDNA that occurs at the reversed replication forks upon MRE11-dependent fork degradation and that occurring at processed of DSBs. These conditions include the timing of IdU pulse and the duration of HU treatment (43). Having demonstrated that Z56 and Z99 affect formation of DSBs upon prolonged replication stress, interfering specifically with RAD52 function, we next evaluated if they might similarly affect the role of RAD52 as antagonist of fork reversal and fork degradation (Figure 5A) (43). To assess fork degradation at reversed forks, we analysed exposure of nascent ssDNA by native anti-IdU IF (70) in HU-treated cells (Figure 5B). As shown in Figure 5B (blue circles) and 5C, a short treatment with HU did not lead to exposure of nascent ssDNA, implying that there was low, if any, fork degradation or DSBs processing. In sharp contrast, treatment with EGC significantly increased the level of ssDNA in HU-treated cells. Interestingly and consistent with the effect on DSBs formation, Z56 and Z99 were able to induce exposure of ssDNA at the nascent strand. To confirm that the effect of Z99 and Z56 on the stimulation of ssDNA exposure in the nascent strand of stalled forks is related to RAD52 inhibition, we performed the native anti-IdU assay in cells that have been stably-depleted of RAD52 using an shRNA cassette (43). Of note, all RAD52 inhibitors failed to induce any further accumulation of nascent ssDNA in cells stably-depleted of RAD52 (Figure 5C, compare blue and red circles).
Figure 5.

Z56 and Z99 interfere with RAD52-dependent protection of stalled DNA replication forks. (A) Cartoon representation of SMARCAL1-mediated fork reversal. RAD52 inhibition stimulates fork reversal and nascent ssDNA exposure. (B) Experimental scheme of nascent ssDNA detection through Iododeoxyuridine (IdU) incorporation. (C) Analysis of nascent ssDNA after 2 mM hydroxyurea (HU) treatment in presence or absence of RAD52 inhibitors in MRC5 SV40 and MRC5 shRAD52 cell lines. Graph shows the intensity of ssDNA staining for single nuclei. Values are presented as means ± SE (ns = not significant; ***P< 0.001; ****P < 0.0001; Mann–Whitney test). (D) Representative images of nascent ssDNA in MRC5 SV40 cells and in MRC5 cells depleted of RAD52. DAPI-stained nuclei are shown in blue, nascent ssDNA signal is shown in light blue. Insets show enlarged images of representative nuclei. (E) Experimental scheme of Proximity Ligation Assay (PLA) after parental ssDNA labelling with IdU. (F) Analysis of DNA–RAD52 interactions by in situ PLA assay. Graphs show the mean of PLA spots per cell ± SE. Values are presented as mean ± SE (***P< 0.001; ****P < 0.0001; Mann–Whitney test). (G) Representative PLA images. DAPI-stained nuclei are shown in blue, PLA signals are shown in pink.
Since Z56 appeared to interfere with both the functions of RAD52 at perturbed replication forks and reduced association of RAD52 with ssDNA in vitro without interfering with RPA–ssDNA interaction, we evaluated if it affected the RAD52 loading on ssDNA in the cell. To this end, we performed in situ proximity ligation assay (PLA) after treatment with HU to assess the association of RAD52 with parental ssDNA at the fork (Figure 5E). Replication fork arrest induced association of RAD52 with parental ssDNA, which was supressed by EGC (Figure 5F and G). Similar to EGC, Z56 also suppressed binding of RAD52 at parental ssDNA as shown by a strong reduction of the number of PLA spots (Figure 5F and G). Collectively, these results indicate that Z99 and Z56 were able to affect the function of RAD52 at replication forks and that Z56 can prevent RAD52 binding to ssDNA in the cell.
Expansion of the Z99 scaffolds improves activity against RAD52, but not RPA, and efficacy in selective killing of BRCA2-mutant cells
In order to enhance the affinity and specificity of our inhibitors, we selected Z99 as the query in a large search of chemical space using the program InfiniSee (BiosolveIT). This program works not by searching chemical libraries for similarity to the query, but rather by a combinatorial fragment build-up scheme, based on predetermined number of chemical reactions and defined fragments (the rules which circumscribe the chemical space). We searched the REAL (Enamine) chemical space, which is vast, consisting of 31612189569 (∼3.2 × 1010) virtual products; based on 165 Enamine reactions, 100 526 in-stock reagents, and 182 381 selected building blocks. Six compounds (Figure 6A) were selected based on the diversity of the generated scaffolds and were synthesized to >99% purity by Enamine. Figure 6B shows the placement of the compounds (stick model) into the same pocket on RAD52 as Z99 (shown as a semi-transparent surface). Supplementary Figure S5 shows ligand maps for all compounds. In addition to the R55 interaction observed with Z99 compound, the scaffold-expanded compounds also make electrostatic contacts with another key ssDNA binding residue, K152. Binding sites for all scaffold-expanded compounds contained Y65 and/or Y126 aromatic residues. Not surprisingly, binding of these compounds to RAD52 resulted in significant quenching of the RAD52 intrinsic fluorescence confirming a direct ligand-protein interaction (Supplementary Figure S5).
We used our original experimental workflow to evaluate the efficacy of these compounds as RAD52–ssDNA inhibitors. All six compounds showed a marked improvement in their ability to inhibit the RAD52–ssDNA interactions (Figure 6C, Table 2, and Supplementary Figure S5). The IC50 values for these new compounds ranged between ∼2 and ∼8 μM, a 6–40-fold improvement over the original Z99 scaffold (Figure 6C, blue bars). While all of the scaffold-expanded compounds retained their ability to inhibit the RPA–ssDNA binding, three compounds had higher IC50 than Z99, two compounds were similar in potency to Z99 for RPA inhibition, and only one (Z58- (69) had lower IC50 for RPA. Thus, these new compounds can provide a broader therapeutic window in cell-based experiments. One of the compounds, Z58-54, while effective in disrupting the RAD52–ssDNA interaction (IC50 of 7.26 ± 0.61 μM), and slightly more poor disruption of the RPA–ssDNA complex (IC50 of 18.0 ± 1.3 μM), was unable to dislodge RAD52 from the RPA–ssDNA–RAD52 complex. Nevertheless, it was effective and specific in cell viability assays and colony formation assays (Figure 6D and Supplementary Figure S6). The expanded compounds were tested for their activity in five cell lines. The CellTiter-Glo®-based viability experiments (Figure 6D, Supplementary Figure S6) were carried out identically to the experiments that used Z56 and Z99 inhibitors, except at 10 μM of respective scaffold-expanded compound. In contrast to their parental Z99 scaffold, the expanded compounds were non-toxic to normal (MCF10a, grey bars) and BRCA2-complemented (EUFA423F HA, green shaded bars), but displayed selective toxicity towards BRCA1-deficient (MDA-MB-436, blue bars) and BRCA2-deficent (Capan1, orange bars) cells. Compounds Z58-54, Z58-72, Z58-83 and Z58-87 were also effective against BRCA2-deficient EUFA423F cells (green bars). Surprisingly, Z58-62 and Z58-69 did not affect viability of EUFA423F cells. Clonogenic survival experiments confirmed the efficacy and improved specificity of the expanded compounds (Supplementary figure S6). Among all tested compounds, Z58-54 and Z58-87 has the most consisted activity profiles for all methodologies and cells lines.
To understand the nature of the RPA–ssDNA complex disruption by some of our compounds, we carried out their molecular docking alongside EGC and EGCG, a known RPA binder (8). Human RPA interacts with ssDNA through four OB folds, known as DNA binding domains (DBD) A, B, C and D. The four DBDs and their modes of ssDNA binding are structurally similar (18). Therefore, we used the PDB: 1JMC structure of the DBD-A and DBD-B of human RPA in complex with ssDNA (33) to evaluate the interaction between RPA-displacing compounds and RPA (Supplementary Figure S7A). The most favourable binding pose of Z99 vis-à-vis EGCG is shown in Supplementary Figure S7B and C. In this pose, stacking occurs between Z99 and the aromatic residues W212 and F238 of the RPA DBD-A. Note that F238 is one of the key aromatic residues important for RPA function (71). There is also hydrogen bonding donation from R216 and N214 (ssDNA interacting residues) to the carbonyl oxygen of Z99. The same contacts were also present in the EGCG–RPA complex. In addition, the EGCG pocket also contains R210 and R234. Interestingly, among our new compounds Z99 itself was the strongest RPA binder from the docking simulations, and its heavier derivatives did not replicate the same excellent stacking and hydrogen bonding indicated above (Supplementary Figure S7D). While binding in the same area as Z99, compounds Z58-54, Z58-62 and Z58-87, completely lacked stacking with F238 and protruded out of the sub-pocket, also forming poorer stacking with W212. Notably, the binding pocket accommodating Z58-87 also included F269, the second aromatic residue of the ssDNA binding cleft of DBD-A. Z58-69 and Z58-72 could not even bind in the Z99 sub-pocket, binding instead at the DBD-A/DBD-B interface close to the W361, the first aromatic residue of the DBD-B. Z58-83 was able to bind in the same sub-pocket as Z99, with stacking to W212, but weaker interaction with F238.
Thus, it is fortunate that the steric requirements for Z99 derivatives are challenging, and that their larger volumes are not allowing the optimal stacking and hydrogen bonding seen with Z99. This nicely parallels what is seen in the experimental in vitro studies (discussed above), in which, although RAD52 potency is increased with the Z99 derivatives, one does not see this mirrored in terms of disruption of the RPA–ssDNA complex. This bodes well for the continued modification and expansion of the Z99 scaffold.
DISCUSSION
In this study we have successfully overcome several challenges in developing drug-like small molecule inhibitors of an attractive anticancer drug target, human DNA repair protein RAD52. The main challenge rested in the necessity to disrupt an extensive and multivalent interaction between RAD52 and ssDNA, as RAD52 can accommodate approximately 44 nucleotides of ssDNA (4 nt per monomer in an undecameric protein ring) and utilizes two distinct DNA binding sites (3,9). Another important challenge was to develop a strategy that yields inhibitors that are specific to RAD52 over other ssDNA binding proteins, such as RPA, whose inhibition may be generally toxic to cells. Finally, all the specifications mentioned above must be accomplished while remaining in drug-like chemical space (reviewed in (3,9)).
In our previous work (8) we have identified several natural products that inhibit RAD52 in vitro and in cells. Among them, EGC was highly specific to RAD52, while a more promiscuous inhibitor EGCG also inhibited the RPA–ssDNA interaction. Natural products (secondary metabolites) from plants and fungi have a storied history in early phase drug discovery, but for numerous reasons are seldom employed as drugs themselves. It is often the case that the pharmacophore (the specific region of a molecule containing the essential organic functional groups that contributes to activity against the receptor) has been used as a basis for designing novel drugs; only about 5% of new chemical entities (NCEs) that target disease are unmodified natural products, while ∼23% are semisynthetic derivatives of natural products, and about 14% are synthetic compounds inspired directly by natural products (72). This is usually based on the poor ADME properties possessed by many natural product classes. The flavan class, and particularly the epigallocatechins, are well known to have poor ADME properties, largely due to absorbtion in the gut and stability, due to both pH and degradation by gut flora (73). This low absorbance was found to be further reduced when accompanied by food (74). Additionally, the very high hydrophilicity of epigallocatechins creates homogeneity problems in lipid formulations (74). Other RAD52 inhibitors have been shown to have a variety of additional ADME challenges (reviewed in (9)). Our solution to overcoming challenges associated with current RAD52 inhibitors was to exploit the EGC pharmacophore, yet to do it from drug-like chemical space which is additionally synthetically tractable, which will facilitate SAR on the RAD52 system. Our extensive computational analyses suggested that both EGC and EGCG bind within the RAD52–ssDNA binding groove but occupy neighbouring pockets. Based on these observations and on a high confidence about approximate location of these two natural products when bound to RAD52 (8), we have developed the concept of a local hotspot for each of these compounds and have focused our computational efforts on the EGC pocket. Using the Enamine REadily AccessibLe through parallel synthesis (REAL) library (26,27) allowed for scaffold hopping into a drug-like synthetically tractable space. The REAL compounds have high retrosynthetic scores, i.e. a high probability of synthesis and purification of the desired compound.
Our computational workflow (Figure 1A) bifurcated at the scoring function step into using either a docking scoring function (which should be more attentive to exploiting binding features different from EGC) or the more conservative PLIF scoring function. Notably, both scoring functions yielded efficient RAD52 inhibitors (Figure 3, Supplementary Figure S1, and Table 1). The interactions between our new inhibitors and RAD52 were more promising compared to EGC and EGCG, which mainly utilize van der Waals interactions. Among selected inhibitors, Z56 (docking/scoring function hit; N-[o-methoxybenzene]-4-methyl-benzoic sulfonamide) and Z99 (PLIF hit; indole-3-ethanone-N-a-one-b-nitro-pyridine) were especially attractive as they showed enhanced activity in cell viability assays (Figure 3). A wide range of ADME properties for Z99 and Z56 were calculated with the program SWISS-ADME (75). Z99 shows excellent drug-likeness using 6 different approaches (including the filters of Lipinski and Ghose), with zero violations using any of these methods, and is considered a good lead compound, from a drug discovery perspective, with a consensus LogPO/W = 1.36 and solubility (LogS) = –3.7 to –4.2 (Supplementary Figure S8). Other promising drug-like properties of Z99 are that it is predicted to have a high gastrointestinal absorption using the program BOILED – Egg (76). The scaffold, however, could be improved in the future by finding a bioisoteric replacement for the nitro group. We see a similar case for Z56, which is also predicted to be drug-like using six different metrics, with zero violations, and has a consensus LogPO/W of 2.12 and solubility (LogS) ranging from –3.36 to –4.75, and also projected to have a high GI absorption (Supplementary Figure S9).
Z56 was specific to RAD52 over RPA in vitro, and showed toxicity only towards BRCA2 mutant cells. In contrast, Z99 was more promiscuous, inhibited RPA in vitro, and exhibited toxicity towards BRCA2-complemented cells. While the activity of new compounds in vitro and in cells was several-fold poorer than low μM activity of EGC and EGCG, It is remarkable that these two lead compounds, with strong drug-likeness, were obtained by scaffold hopping from a natural product with notoriously poor drug-like properties. These scaffolds can be readily modified, as we showed for the Z99 scaffold.
Docking of Z99 and Z56 to DBD-A/B of the RPA70 subunit of human RPA showed that Z99 strongly binds to a deep pocket on the RPA DBD-A, which is also the location of the top docking pose for EGCG (Supplementary Figure S7). We selected Z99 for scaffold expansion, as it was the compound that we had done extensive and early characterization on. Z99 scaffold expansion also allowed us to demonstrate definitively that increasing the molecular weight of the compounds we can shift specificity towards RAD52 without increasing affinity for RPA. Scaffold expansion was carried out using methodology that accessed ∼1010 theoretical compounds in REAL space. Notably, the fragment build-up scheme we used is not a library search, but navigation of the chemical space, with the search output being a set of specific chemical fragments and reactions to build a desired compound with high probability. The result of the scaffold expansion was a set of six novel compounds with improved potency towards RAD52 and no further increase in activity towards RPA.
Z99 scaffold expansion aimed primarily on enhancing affinity of the derivatives by introducing one or a few additional high quality contacts while maintaining or improving bio-activity. Fortunately, docking studies indicated that all of the top scoring candidates maintained the same orientation on the target as the original Z99 scaffold. A comparison of Supplementary Figure S6 (ligand maps of the expanded scaffolds) to Figure 2 (ligand map of Z99) indicates that two of the most promising derivatives, Z58-54 and Z58-87, have a number of improved contacts. Particularly, Z58-87, which has a strongly enhanced van der Waals contact with the sub-pocket, much better desolvation of the ligand atoms, and a halogen bonding between the Cl atom and D277. Indeed, since halogen bonding is enhanced with heavier halogens, this implies that future derivatives with both I and Br should be synthesized and tested as well. Additionally, since electron-withdrawing substituents on the donor strengthen the halogen bonds (77), an additional strategy should be to add, for example, one or more additional fluoro substituents in adjacent positions. This can be rationalized as causing an expansion of the σ-hole potential on the halogen donor group (77). In terms of Z58-54, it is also more desolvated than the original Z99 scaffold, and benefits from several stacking interactions with F238.
While still relatively small, these compounds inhibit RAD52 with IC50 values in low μM range (Figure 6, Supplementary Figure S6, Table 2), with excellent discrimination between BRCA1 or BRCA2-mutant, and BRCA-proficient cells (Figure 6D, Supplementary Figure S6). We anticipate future studies on the expansion of Z56 in a similar manner. There is no a priori reason that Z56 expansion in similar manner as detailed for Z99 would compromise selectivity, based on RPA docking. Furthermore, additional rounds of expansion of Z99 appear to be promising with respect to developing potent and selective inhibitors of RAD52, as we were able to improve activity towards RAD52 without compromising specificity. Specifically, compounds Z58-54 and Z58-87 showed the most consistent activity against BRCA1 and BRCA2-mutated cells and low toxicity in BRCA proficient cells. These two scaffolds therefore, will be used in further rounds of improvement. Given the emergence of resistance to PARP inhibitors, our new RAD52 inhibitors and the overall approach bode well for the possibility of further rapid development of designer drugs that expand our pharmacopeia for synthetic lethality.
DATA AVAILABILITY
The data underlying this article will be shared on reasonable request to the corresponding author.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Kailey Cash (University of Iowa) for critical reading of the manuscript and valuable discussions.
Notes
Present address: Sarah R. Hengel. Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA.
Contributor Information
Divya S Bhat, Department of Biochemistry, University of Iowa Carver College of Medicine, 51 Newton Road, Iowa City, IA 52242, USA.
Eva Malacaria, Mechanisms, Biomarkers and Models Section, Department of Environment and Health, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy.
Ludovica Di Biagi, Mechanisms, Biomarkers and Models Section, Department of Environment and Health, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy.
Mortezaali Razzaghi, Department of Biochemistry, University of Iowa Carver College of Medicine, 51 Newton Road, Iowa City, IA 52242, USA.
Masayoshi Honda, Department of Biochemistry, University of Iowa Carver College of Medicine, 51 Newton Road, Iowa City, IA 52242, USA.
Kathryn F Hobbs, Department of Biochemistry, University of Iowa Carver College of Medicine, 51 Newton Road, Iowa City, IA 52242, USA; Division of Medicinal and Natural Products Chemistry, Department of Pharmaceutical Sciences and Experimental Therapeutics, The University of Iowa, Iowa City, IA 52242, USA.
Sarah R Hengel, Department of Biochemistry, University of Iowa Carver College of Medicine, 51 Newton Road, Iowa City, IA 52242, USA.
Pietro Pichierri, Mechanisms, Biomarkers and Models Section, Department of Environment and Health, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy.
M Ashley Spies, Department of Biochemistry, University of Iowa Carver College of Medicine, 51 Newton Road, Iowa City, IA 52242, USA; Division of Medicinal and Natural Products Chemistry, Department of Pharmaceutical Sciences and Experimental Therapeutics, The University of Iowa, Iowa City, IA 52242, USA; Naturis Informatika LLC, 401 Mullin Ave., Iowa City, IA 52246, USA.
Maria Spies, Department of Biochemistry, University of Iowa Carver College of Medicine, 51 Newton Road, Iowa City, IA 52242, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
National Institutes of Health (NIH) [R01 CA232425 to M.S., P.P., R01 GM09737, R01 GM138471 to M.A.S.]; D.B. acknowledges support of predoctoral fellowship from the University of Iowa Center for Biocatalysis and Bioprocessing affiliated with the NIH-sponsored Predoctoral Training Program in Biotechnology [T32 GM008365]. MR was supported by a postdoctoral fellowship from the Free Radicals and Radiation Biology training grant [T32 CA078586]. Funding for open access charge: NIH [R01 CA232425 to M.S., P.P.].
Conflict of interest statement. A US provisional patent application no. 63/427759 includes Z56, Z99 and its derivatives.
REFERENCES
- 1. Mortensen U.H., Lisby M., Rothstein R. Rad52. Curr. Biol. 2009; 19:R676–R677. [DOI] [PubMed] [Google Scholar]
- 2. Muris D.F., Bezzubova O., Buerstedde J.M., Vreeken K., Balajee A.S., Osgood C.J., Troelstra C., Hoeijmakers J.H., Ostermann K., Schmidt H. et al. Cloning of human and mouse genes homologous to RAD52, a yeast gene involved in DNA repair and recombination. Mutat Res. 1994; 315:295–305. [DOI] [PubMed] [Google Scholar]
- 3. Bhat D.S., Spies M.A., Spies M. A moving target for drug discovery: structure activity relationship and many genome (de)stabilizing functions of the RAD52 protein. DNA Repair. 2022; 120:103421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cramer-Morales K., Nieborowska-Skorska M., Scheibner K., Padget M., Irvine D.A., Sliwinski T., Haas K., Lee J., Geng H., Roy D. et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013; 122:1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lok B.H., Carley A.C., Tchang B., Powell S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013; 32:3552–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feng Z., Scott S.P., Bussen W., Sharma G.G., Guo G., Pandita T.K., Powell S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rossi M.J., DiDomenico S.F., Patel M., Mazin A.V. RAD52: paradigm of synthetic lethality and new developments. Front. Genet. 2021; 12:780293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hengel S.R., Malacaria E., Folly da Silva Constantino L., Bain F.E., Diaz A., Koch B.G., Yu L., Wu M., Pichierri P., Spies M.A. et al. Small-molecule inhibitors identify the RAD52–ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. Elife. 2016; 5:e14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chheda P., Spies M., Spies M.A. Small-molecule Effectors of DNA Repair Proteins: Applications for Development of Cancer Therapeutics and Research. Burger's Medicinal Chemistry and Drug Discovery. 2021; 1–39. [Google Scholar]
- 10. Treuner K., Helton R., Barlow C. Loss of Rad52 partially rescues tumorigenesis and T-cell maturation in Atm-deficient mice. Oncogene. 2004; 23:4655–4661. [DOI] [PubMed] [Google Scholar]
- 11. Chandramouly G., McDevitt S., Sullivan K., Kent T., Luz A., Glickman J.F., Andrake M., Skorski T., Pomerantz R.T. Small-molecule disruption of RAD52 rings as a mechanism for precision medicine in BRCA-deficient cancers. Chemistry & Biology. 2015; 22:1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sullivan K., Cramer-Morales K., McElroy D.L., Ostrov D.A., Haas K., Childers W., Hromas R., Skorski T. Identification of a small molecule inhibitor of RAD52 by structure-based selection. PLoS One. 2016; 11:e0147230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang F., Goyal N., Sullivan K., Hanamshet K., Patel M., Mazina O.M., Wang C.X., An W.F., Spoonamore J., Metkar S. et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic. Acids. Res. 2016; 44:4189–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rijkers T., Van Den Ouweland J., Morolli B., Rolink A.G., Baarends W.M., Van Sloun P.P., Lohman P.H., Pastink A. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol. 1998; 18:6423–6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamaguchi-Iwai Y., Sonoda E., Buerstedde J.M., Bezzubova O., Morrison C., Takata M., Shinohara A., Takeda S. Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol. 1998; 18:6430–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yanez R.J., Porter A.C. Differential effects of Rad52p overexpression on gene targeting and extrachromosomal homologous recombination in a human cell line. Nucleic. Acids. Res. 2002; 30:740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malacaria E., Honda M., Franchitto A., Spies M., Pichierri P. Physiological and pathological roles of RAD52 at DNA replication forks. Cancers. 2020; 12:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caldwell C.C., Spies M. Dynamic elements of replication protein A at the crossroads of DNA replication, recombination, and repair. Crit. Rev. Biochem. Mol. Biol. 2020; 55:482–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bushweller J.H. Targeting transcription factors in cancer — from undruggable to reality. Nat. Rev. Cancer. 2019; 19:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Radaeva M., Ton A.-T., Hsing M., Ban F., Cherkasov A. Drugging the ‘undruggable’. Therapeutic targeting of protein–DNA interactions with the use of computer-aided drug discovery methods. Drug Discov. Today. 2021; 26:2660–2679. [DOI] [PubMed] [Google Scholar]
- 21. Grimme J.M., Honda M., Wright R., Okuno Y., Rothenberg E., Mazin A.V., Ha T., Spies M. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 2010; 38:2917–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Honda M., Okuno Y., Yoo J., Ha T., Spies M. Tyrosine phosphorylation enhances RAD52-mediated annealing by modulating its DNA binding. EMBO J. 2011; 30:3368–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kagawa W., Kurumizaka H., Ishitani R., Fukai S., Nureki O., Shibata T., Yokoyama S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell. 2002; 10:359–371. [DOI] [PubMed] [Google Scholar]
- 24. Saotome M., Saito K., Yasuda T., Ohtomo H., Sugiyama S., Nishimura Y., Kurumizaka H., Kagawa W. Structural basis of homology-directed DNA repair mediated by RAD52. Iscience. 2018; 3:50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Singleton M.R., Wentzell L.M., Liu Y., West S.C., Wigley D.B. Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:13492–13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grygorenko O.O., Radchenko D.S., Dziuba I., Chuprina A., Gubina K.E., Moroz Y.S. Generating multibillion chemical space of readily accessible screening compounds. Iscience. 2020; 23:101681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shivanyuk A., Ryabukhin S., Tolmachev A., Bogolyubsky A., Mykytenko D., Chupryna A., Heilman W., Kostyuk A. Enamine real database: making chemical diversity real. Chem. Today. 2007; 25:58–59. [Google Scholar]
- 28. Halgren T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996; 17:490–519. [Google Scholar]
- 29. Li Q., Folly da Silva Constantino L., Spies M.A Integrating experimental and In Silico HTS in the discovery of inhibitors of protein-nucleic acid interactions. Methods Enzymol. 2018; 601:243–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naïm M., Bhat S., Rankin K.N., Dennis S., Chowdhury S.F., Siddiqi I., Drabik P., Sulea T., Bayly C.I., Jakalian A. et al. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inform. Model. 2007; 47:122–133. [DOI] [PubMed] [Google Scholar]
- 31. Steinbrecher T., Labahn A. Towards accurate free energy calculations in ligand protein-binding studies. Curr. Med. Chem. 2010; 17:767–785. [DOI] [PubMed] [Google Scholar]
- 32. Wang W., Kollman P.A. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J. Mol. Biol. 2000; 303:567–582. [DOI] [PubMed] [Google Scholar]
- 33. Bochkarev A., Pfuetzner R.A., Edwards A.M., Frappier L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 1997; 385:176–181. [DOI] [PubMed] [Google Scholar]
- 34. Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010; 31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krieger E., Vriend G. YASARA view - molecular graphics for all devices - from smartphones to workstations. Bioinformatics. 2014; 30:2981–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Henricksen L.A., Umbricht C.B., Wold M.S. Recombinant replication protein A: expression, complex formation, and functional characterization. J. Biol. Chem. 1994; 269:11121–11132. [PubMed] [Google Scholar]
- 37. Howlett N.G., Taniguchi T., Olson S., Cox B., Waisfisz Q., De Die-Smulders C., Persky N., Grompe M., Joenje H., Pals G. et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science (New York, N.Y.). 2002; 297:606–609. [DOI] [PubMed] [Google Scholar]
- 38. Goggins M., Schutte M., Lu J., Moskaluk C.A., Weinstein C.L., Petersen G.M., Yeo C.J., Jackson C.E., Lynch H.T., Hruban R.H. et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996; 56:5360–5364. [PubMed] [Google Scholar]
- 39. Holt J.T., Toole W.P., Patel V.R., Hwang H., Brown E.T. Restoration of CAPAN-1 cells with functional BRCA2 provides insight into the DNA repair activity of individuals who are heterozygous for BRCA2 mutations. Cancer Genet. Cytogenet. 2008; 186:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berman D.B., Costalas J., Schultz D.C., Grana G., Daly M., Godwin A.K. A common mutation in BRCA2 that predisposes to a variety of cancers is found in both Jewish Ashkenazi and non-Jewish individuals. Cancer Res. 1996; 56:3409–3414. [PubMed] [Google Scholar]
- 41. Elstrodt F., Hollestelle A., Nagel J.H., Gorin M., Wasielewski M., van den Ouweland A., Merajver S.D., Ethier S.P., Schutte M. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res. 2006; 66:41–45. [DOI] [PubMed] [Google Scholar]
- 42. Soule H.D., Maloney T.M., Wolman S.R., Peterson W.D. Jr, Brenz R., McGrath C.M., Russo J., Pauley R.J., Jones R.F., Brooks S.C Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990; 50:6075–6086. [PubMed] [Google Scholar]
- 43. Malacaria E., Pugliese G.M., Honda M., Marabitti V., Aiello F.A., Spies M., Franchitto A., Pichierri P. Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat. Commun. 2019; 10:1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grimme J.M., Spies M. FRET-based assays to monitor DNA binding and annealing by Rad52 recombination mediator protein. Methods Mol. Biol. 2011; 745:463–483. [DOI] [PubMed] [Google Scholar]
- 45. Yates L.A., Aramayo R.J., Pokhrel N., Caldwell C.C., Kaplan J.A., Perera R.L., Spies M., Antony E., Zhang X. A structural and dynamic model for the assembly of Replication Protein A on single-stranded DNA. Nat. Commun. 2018; 9:5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Subramanyam S., Spies M. Expression, purification, and biochemical evaluation of Human RAD51 protein. Methods Enzymol. 2018; 600:157–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wold M.S. Replication Protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997; 66:61–92. [DOI] [PubMed] [Google Scholar]
- 48. Kagawa W., Kagawa A., Saito K., Ikawa S., Shibata T., Kurumizaka H., Yokoyama S. Identification of a second DNA binding site in the human Rad52 protein. J. Biol. Chem. 2008; 283:24264–24273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lloyd J.A., McGrew D.A., Knight K.L. Identification of residues important for DNA binding in the full-length human Rad52 protein. J. Mol. Biol. 2005; 345:239–249. [DOI] [PubMed] [Google Scholar]
- 50. Stasiak A.Z., Larquet E., Stasiak A., Muller S., Engel A., Van Dyck E., West S.C., Egelman E.H. The human Rad52 protein exists as a heptameric ring. Curr. Biol. 2000; 10:337–340. [DOI] [PubMed] [Google Scholar]
- 51. Asor R., Kukura P. Characterising biomolecular interactions and dynamics with mass photometry. Curr. Opin. Chem. Biol. 2022; 68:102132. [DOI] [PubMed] [Google Scholar]
- 52. Esashi F., Christ N., Gannon J., Liu Y., Hunt T., Jasin M., West S.C. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005; 434:598–604. [DOI] [PubMed] [Google Scholar]
- 53. Feng W., Jasin M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun. 2017; 8:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lomonosov M., Anand S., Sangrithi M., Davies R., Venkitaraman A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003; 17:3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011; 145:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Paul M.W., Sidhu A., Liang Y., van Rossum-Fikkert S.E., Odijk H., Zelensky A.N., Kanaar R., Wyman C. Role of BRCA2 DNA-binding and C-terminal domain in its mobility and conformation in DNA repair. Elife. 2021; 10:e67926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Esashi F., Galkin V.E., Yu X., Egelman E.H., West S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007; 14:468–474. [DOI] [PubMed] [Google Scholar]
- 58. Yang H., Jeffrey P.D., Miller J., Kinnucan E., Sun Y., Thoma N.H., Zheng N., Chen P.L., Lee W.H., Pavletich N.P. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002; 297:1837–1848. [DOI] [PubMed] [Google Scholar]
- 59. Zhao W., Vaithiyalingam S., San Filippo J., Maranon D.G., Jimenez-Sainz J., Fontenay G.V., Kwon Y., Leung S.G., Lu L., Jensen R.B. et al. Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol. Cell. 2015; 59:176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Clark C.C., Weitzel J.N., O’Connor T.R Enhancement of synthetic lethality via combinations of ABT-888, a PARP inhibitor, and carboplatin in vitro and in vivo using BRCA1 and BRCA2 isogenic models. Mol. Cancer Ther. 2012; 11:1948–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stark J.M., Pierce A.J., Oh J., Pastink A., Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell. Biol. 2004; 24:9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kan Y., Batada N.N., Hendrickson E.A. Human somatic cells deficient for RAD52 are impaired for viral integration and compromised for most aspects of homology-directed repair. DNA Repair (Amst.). 2017; 55:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Murfuni I., Basile G., Subramanyam S., Malacaria E., Bignami M., Spies M., Franchitto A., Pichierri P. Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLoS Genet. 2013; 9:e1003910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Llorens-Agost M., Ensminger M., Le H.P., Gawai A., Liu J., Cruz-García A., Bhetawal S., Wood R.D., Heyer W.D., Löbrich M. POLθ-mediated end joining is restricted by RAD52 and BRCA2 until the onset of mitosis. Nat. Cell Biol. 2021; 23:1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bhowmick R., Minocherhomji S., Hickson I.D. RAD52 Facilitates mitotic DNA synthesis following replication stress. Mol. Cell. 2016; 64:1117–1126. [DOI] [PubMed] [Google Scholar]
- 66. Sotiriou S.K., Kamileri I., Lugli N., Evangelou K., Da-Re C., Huber F., Padayachy L., Tardy S., Nicati N.L., Barriot S. et al. Mammalian RAD52 functions in break-induced replication repair of collapsed DNA replication forks. Mol. Cell. 2016; 64:1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Welty S., Teng Y., Liang Z., Zhao W., Sanders L.H., Greenamyre J.T., Rubio M.E., Thathiah A., Kodali R., Wetzel R. et al. RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J. Biol. Chem. 2018; 293:1353–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Panzarino N.J., Krais J.J., Cong K., Peng M., Mosqueda M., Nayak S.U., Bond S.M., Calvo J.A., Doshi M.B., Bere M. et al. Replication gaps underlie BRCA deficiency and therapy response. Cancer Res. 2021; 81:1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Franchitto A., Pirzio L.M., Prosperi E., Sapora O., Bignami M., Pichierri P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 2008; 183:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Iannascoli C., Palermo V., Murfuni I., Franchitto A., Pichierri P. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 2015; 43:9788–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hass C.S., Lam K., Wold M.S. Repair-specific functions of replication protein A. J. Biol. Chem. 2012; 287:3908–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Roche V.F., Zito S.W., Lemke T.L., Williams D.A. Foye's Principles of Medicinal Chemistry. 2020; 8th edn.Philadelphia: Wolters Kluwer. [Google Scholar]
- 73. Zeng L., Ma M., Li C., Luo L. Stability of tea polyphenols solution with different pH at different temperatures. Int. J. Food Properties. 2017; 20:1–18. [Google Scholar]
- 74. Andreu-Fernández V., Almeida Toledano L., Pizarro N., Navarro-Tapia E., Gómez-Roig M.D., de la Torre R., García-Algar Ó. Bioavailability of epigallocatechin gallate administered with different nutritional strategies in healthy volunteers. Antioxidants (Basel). 2020; 9:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports. 2017; 7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Daina A., Zoete V. A BOILED-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016; 11:1117–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Persch E., Dumele O., Diederich F. Molecular recognition in chemical and Biological systems. Angew. Chem. Int. Ed. 2015; 54:3290–3327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.