

Abstract

CCT251236 1, a potent chemical probe, was previously developed from a cell-based phenotypic high-throughput screen (HTS) to discover inhibitors of transcription mediated by HSF1, a transcription factor that supports malignancy. Owing to its activity against models of refractory human ovarian cancer, 1 was progressed into lead optimization. The reduction of P-glycoprotein efflux became a focus of early compound optimization; central ring halogen substitution was demonstrated by matched molecular pair analysis to be an effective strategy to mitigate this liability. Further multiparameter optimization led to the design of the clinical candidate, CCT361814/NXP800 22, a potent and orally bioavailable fluorobisamide, which caused tumor regression in a human ovarian adenocarcinoma xenograft model with on-pathway biomarker modulation and a clean in vitro safety profile. Following its favorable dose prediction to human, 22 has now progressed to phase 1 clinical trial as a potential future treatment for refractory ovarian cancer and other malignancies.
Introduction
Ovarian cancer is the most lethal and the second most common gynecological malignancy in the developed world, and only a modest decrease in mortality has been achieved over the past three decades.1 Approximately 80% of patients are diagnosed at an advanced stage, leading to poor prognosis with little prospect of cure.1 The combination of surgical cytoreduction and the administration of platinum complexes and taxanes remains the standard of care for advanced ovarian cancer.2 Although initial treatment is effective in ∼70% of patients, and the introduction of the anti-VEGF receptor monoclonal antibody bevacizumab and PARP inhibitors provides a welcome addition to initial therapy, the majority develop drug resistance and relapse, resulting in a 5 year survival rate of only ∼30%.3 Clearly, there is a high unmet medical need in the treatment of ovarian cancer.
Multidrug resistance (MDR) in relapsed ovarian cancer is observed in 50–75% of patients following first-line chemotherapy.4 MDR is often the result of the overexpression of ABC-transporter proteins at the cancer cell surface, which efflux compounds and reduce their intracellular free concentrations.5 Various ABC-transporter proteins have been linked to MDR with oncology drugs,6 but the most commonly encountered is the overexpression of multidrug resistance protein 1 (MDR1), also known as the P-glycoprotein (P-gp) efflux pump.7 Consequently, the reduction of P-gp-mediated efflux in lead optimization is important for the successful development of novel and effective ovarian anticancer therapies.7
Heat shock transcription factor 1 (HSF1) is the master regulator of the canonical heat shock stress response.8 In cancer, HSF1 is important for tumorigenesis and progression and is activated by various elements of the cancer state.9 HSF1 reprograms the transcriptome in a manner overlapping with, but distinct from, the classical heat shock response.10 Also, HSF1 is amplified, overexpressed, or activated in multiple human cancers; these features, combined with the oncogenic HSF1 gene signature, predict adverse clinical outcomes, including in ovarian cancer.11,12 Moreover, in ovarian cancer cells, the shRNA knockdown of HSF1 leads to decreased proliferation and increased apoptosis.12 In contrast, the knockout of HSF1 is tolerated in flies and mice.12 Together with a range of other data, these results support the inhibition of HSF1 as a “nononcogene addiction” approach to exploit tumor stress with the potential to antagonize multiple hallmark cancer traits.12 Unfortunately, HSF1 is a ligandless transcription factor and is predicted to be very difficult-to-drug directly. Therefore, we sought HSF1 pathway inhibitors that could indirectly inhibit HSF1-mediated transcription.
We previously reported the discovery of a new chemical probe, CCT251236 1 (Figure 1), which was developed from a low solubility hit identified using a proximal but mechanism-agnostic phenotypic screen to detect inhibitors of the HSF1 stress pathway.13 Bisamide 1 displayed potent cellular activity in the human ovarian cancer cell line SK-OV-3, blocking HSP72 induction by an HSP90 inhibitor, which was used as a surrogate biomarker of HSF1 pathway inhibition (IC50 = 19 nM). Also, consistent with the sensitivity to HSF1 RNAi knockdown both in vitro and in vivo,121 displayed excellent antiproliferative activity against cancer cells (GI50 = 2.2 nM) as a single agent. Furthermore, we demonstrated that bisamide 1 is a potent ligand for the putative transcription factor regulator pirin (KD = 44 nM), with clear in vitro antimigratory activity, the phenotype previously associated with pirin binding, in the melanoma cell line WM266.4, at low free concentrations (<100 nM).13 Subsequently, intracellular target engagement with pirin by 1 in intact cancer cells was confirmed via a CRBN-mediated PROTAC probe.14 However, the molecular mechanism of action for the wide-ranging antiproliferative activity of this chemotype still remains to be confirmed. Finally, in the in vivo SK-OV-3 human ovarian cancer solid tumor xenograft model in nude mice, bisamide 1 was shown to be orally bioavailable and displayed clear efficacy (tumor growth inhibition (TGI) = 70%), driven by a low free drug exposure (free Cav0–24h = 1.2 nM)15 achieved using a well-tolerated intermittent 20 mg/kg po dosing regimen.13
Figure 1.
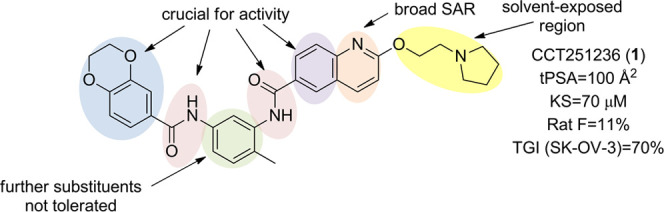
Complex cellular structure–activity relationships (SARs) of the bisamide chemotype and poor rat-free exposure from an oral dose of the lead compound 1.
We now report the development of the probe HSF1 pathway inhibitor 1 into a clinical candidate, which shows future potential for the treatment of relapsed ovarian cancer and other malignancies. We developed the compound using only cell-based structure–activity relationships (SARs) and focused on improving the oral absorption while reducing in vivo unbound clearance and the P-gp-efflux-mediated multidrug resistance risk.
Results and Discussion
Targeting Ovarian Cancer
It has been proposed that HSF1 pathway inhibition could be an effective treatment in relapsed ovarian cancer,12 as well as other malignancies;16 therefore, to assess the development potential of probe bisamide 1, the compound was screened against a panel of genetically diverse human ovarian cancer cell lines (Table S1). Compared to the standard-of-care drug carboplatin (pGI50 < 6, N = 4 cell lines), bisamide 1 displayed significantly more potent antiproliferative activity against this panel (8.7 > pGI50 > 7.3, N = 9 cell lines). Given the challenges in treating relapsed ovarian cancer and the clear treatment potential of this chemotype, bisamide 1 was progressed into lead optimization.
Rodent Pharmacokinetic (PK) Optimization
A pharmacokinetic (PK) study in Sprague-Dawley (SD) rats (Table S13) revealed that bisamide 1 possessed poor oral bioavailability (F = 11%) from moderate total blood clearance (CLtb = 20 mL/min/kg, extraction ratio = 29%, Fmax = 71%) with moderate in vivo unbound clearance (CLu = 1100 mL/min/kg, fub = 0.019). Unbound clearance, when acting as a suitable estimate for unbound intrinsic clearance, is an important target parameter owing to its relationship with free drug exposure and unbound average concentration.17,18 In vitro, low-to-moderate passive permeability was observed in the Caco-2 assay, which is commonly used to predict absorption19 (A–B = 2.4 × 10–6 cm/s, efflux ratio = 16; Table S8). The high efflux ratio indicated that 1 is likely to suffer from P-gp-mediated efflux, which can present a challenge for clinical development. Therefore, we began a medicinal chemistry campaign to improve the preclinical PK profile of this highly potent and effective chemotype to increase oral absorption, reduce unbound clearance, and mitigate the risk of P-gp-mediated efflux.
Bisamide 1 represents a challenging start-point for lead optimization, as the cellular SAR of this chemotype is complex, with steep activity cliffs from small structural changes and few clear patterns to drive compound development.13 The incorporation of a solubilizing group had been crucial for the favorable mouse PK profile of bisamide 1, so to carry out multiparameter optimization on the chemotype, we focused on structural changes to this region. The solubilizing group of each analogue was inferred to be solvent-exposed due to its tolerance to a broad range of structural changes in the cell-based assays.13 To expedite analogue evaluation, a new synthetic route was developed incorporating a late-stage selenium dioxide-mediated benzylic oxidation (Scheme 1).14,20
Scheme 1. Generic Synthesis of Bisamide Lead Optimization Analogues.

2-Methylquinoline-6-carboxylic acid 2 was converted to the acid chloride using oxalyl chloride and catalytic N,N-dimethylformamide (DMF), before reacting with 1,3-nitroaniline 3 to give nitroamide 4. Iron(0)-mediated reduction of the nitro group gave 5, which was then subjected to a second amide bond formation reaction following the in situ generation of the acid chloride of the dihydrobenzodioxine-carboxylic acid 6 to give bisamide 7. Subsequent selenium dioxide-mediated oxidation of the quinolinic methyl group of 7 gave aldehyde 8 in low to moderate yields, as both the optimal reaction temperature and ratio of 1,4-dioxane/DMF cosolvents were dependent on the benzylic substituent, X. This optimization was also critical owing to the poor solubility of bisamide 7 in 1,4-dioxane and the tendency of the aldehyde to overoxidize to the carboxylic acid under the reaction conditions. Aldehyde 8 then underwent reductive amination with various amine bases 9 under standard conditions and in moderate yields to give analogues 10. Variations on this general route were also carried out to synthesize specific analogues, and details are available in the Experimental Section and Supporting Information.
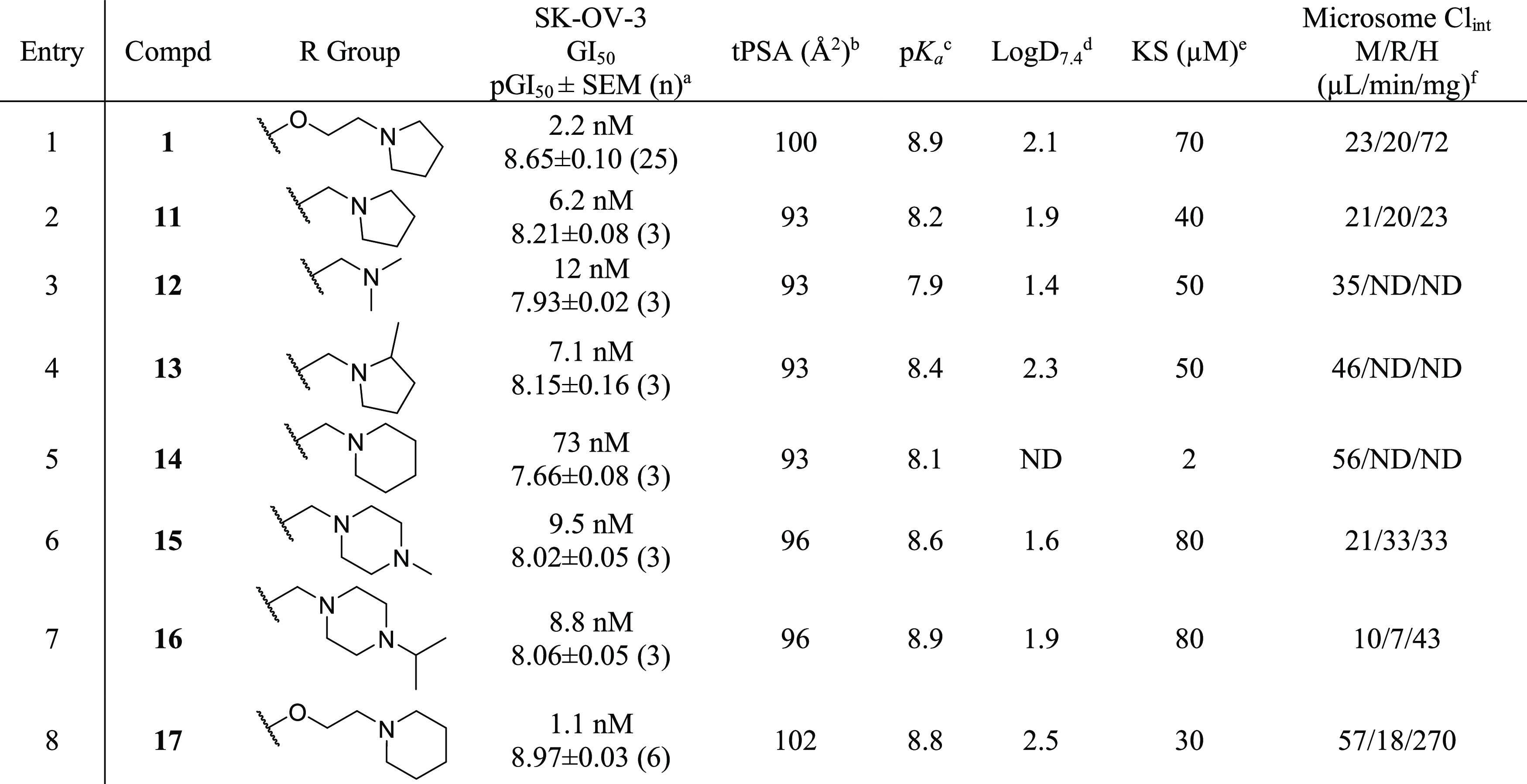
Our first target for lead optimization was to improve oral absorption in a manner that would be tolerated as part of the multiparameter optimization and would maintain the potent antiproliferative activity of this chemotype. Therefore, we replaced the linker to the solubilizing group with a shorter chain and removed the oxygen in bisamide 1, reducing both topological polar surface area (tPSA) and basicity without significantly increasing lipophilicity (Table 1).
Table 1. Solubilizing Group In Vitro Optimization.

All data were reprocessed using GraphPad Prism 7.01. Growth inhibition was measured after 96 h of treatment and compared to the vehicle control using the CellTiter-Blue assay. GI50 values were estimated by fitting a log[Inhibitor] vs response—variable slope (four-parameter) model. The number of repeats (n) is given in parentheses. All results are quoted as the geometric mean ± standard error of the mean (SEM). pGI50 = −log GI50 (M).
Calculated using ChemDraw 16.0.1.4 and quoted to 0 dp.
Calculated using MoKa version 2.5.2; all values quoted to 2 SF.
Measured using a previously described high-performance liquid chromatography (HPLC) method, n = 1; all values quoted to 2 SF.13
Kinetic solubility (KS) measured via an HPLC method from phosphate buffer at pH 7.4; all values quoted to 1 SF; the dynamic range of the assay is 1–100 μM, n = 1.
Mouse (M), rat (R), and human (H) liver microsome (MLM, RLM, and HLM) assays were carried out at Cyprotex, n = 1. In vitro Clint values are calculated from the half-lives using standard procedures. Assumes the fraction unbound in the assay is 1.
ND, not determined.
The one-carbon linker analogue, methylene 11 (Table 1, entry 2), displayed a 2.8-fold decrease in antiproliferative activity but only a 1.7-fold decrease in kinetic solubility (KS, used as a crude estimate of thermodynamic solubility), despite the predicted decrease in pKa compared to 1 (8.2–8.9, respectively). Both analogues displayed similar lipophilicity (Log D7.4), but 11 exhibited a 3.1-fold reduction in human liver microsome (HLM) intrinsic clearance (CLint) (23 vs 72 μL/min/mg) while maintaining comparable microsomal stability to 1 in both rodent species (RLM CLint = 20 μL/min/mg).21
The reduced tPSA (93 Å2), combined with the improved metabolic stability profile, led us to investigate this structural change further with a series of solubilizing group analogues (Table 1, entries 3–7), with compound design focusing on maintaining antiproliferative activity while improving metabolic stability. The potential for progressing compounds to in vivo mouse PK evaluation was assessed through changes in physicochemical properties and microsomal clearance data.
Comparing subsequent analogues to 11: the acyclic dimethylamino analogue 12 (Table 1, entry 3) displayed a 1.3-fold increase in KS (50 μM), but unfortunately, also a 1.6-fold increase in MLM CLint, presumably due to CYP450-mediated N-demethylation. Therefore, all subsequent analogues were limited to cyclic structures that should display increased resistance to oxidation.
The more lipophilic analogues, methylpyrrolidine 13 and piperidine 14 (Table 1, entries 4 and 5), suffered a decrease in metabolic stability and KS (2.0–50 μM), so to balance their physicochemical properties, an additional nitrogen was introduced to the six-membered ring of the solubilizing group. The N-methylpiperazine derivative 15 (Table 1, entry 6) displayed a favorable 2.0-fold increase in KS (80 μM), combined with metabolic stability comparable to 11. Hypothesizing that the N-methyl moiety of 15 remained an oxidative metabolic liability, the N-isopropylpiperazine analogue 16 was synthesized (Table 1, entry 7) and, consistent with our design strategy, the metabolic stability was compared favorably to that of lead bisamide 1 across all species (RLM CLint = 7.0 μL/min/mg, HLM CLint = 43 μL/min/mg) while maintaining improved KS (80 μM). Compound 16 was therefore selected for further evaluation in mouse PK studies (Table 2, entry 1).
Table 2. Selected In Vivo Mouse Blood PK Parameters of Lead Compoundsa.
| Entry | Compd | Dose po/iv (mg/kg)b | Tmax (h) | po AUC0–6h (h*nM) | iv Cltb (mL/min/kg)c | t1/2 (h)d | F (%)e | fubf | AUCu0–6h (h*nM)g | Free Cav0–24h (nM)h | CLu (mL/min/kg)i |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 16 | 5/5 | 1.7 | 430 (890–210) | 34 (36–32) | 1.2 | 11 | 0.032 | 14 | 0.66 | 1100 |
| 2 | 17 | 5/5 | 2.3 | 830 (910–750) | 40 (48–32) | 1.7 | 24 | 0.015 | 12 | 0.83 | 2700 |
| 3 | 18 | 5/1 | 1.7 | 2400 (2800–2000) | 33 (35–31) | 2.0 | 63 | 0.011 | 26 | 1.6 | 3000 |
| 4 | 21 | 5/5 | 1 7 | 3900 (4900–3000) | 2.8 (33–25) | 2.3 | 96 | 0.0053 | 20 | 1.3 | 5300 |
All graphs were plotted using GraphPad Prism 7.01. PK parameters were derived from the blood concentration/time using noncompartmental analysis (Model 200 and 201) (Phoenix, version 6.1). All results are quoted to two significant figures as the geometric mean of n = 3 individual BALB/c mice. The 90% confidence intervals are in parentheses.
The po and iv dosing vehicles are described in the Supporting Information.
CLtb = total blood clearance.
Terminal half-life calculated from the iv dose.
Assumes linear PK.
fub = fraction unbound in blood, fub = fup/B:P, fup = fraction unbound in plasma using equilibrium dialysis, B:P = blood-to-plasma ratio and quoted as the geometric mean from n = 3 statistical repeats from pooled samples; see the Supporting Information for details.
AUCu = AUC*fub.
Free Cav0–24h = AUCinf/24*fub.
CLu = CLtb/fub.
P-gp-Mediated Efflux
N-Isopropylpiperazine 16 was dosed in BALB/c mice, both as an oral solution and iv bolus, and blood concentrations were measured over a 24 h period. Unfortunately, the in vivo PK profile of 16 was disappointing, with low oral bioavailability (11%) from moderate total blood clearance (CLtb = 34 mL/min/kg, extraction ratio = 38%, Fmax = 62%),22 corresponding to an unbound clearance of 1100 mL/min/kg (Table S4). The predicted CLu from the MLM assay was only 39 mL/min/kg,23 28-fold lower than the experimental result (Table 3, entry 1 and Table S5). We hypothesized that this disconnection between predicted and experimentally determined CLu was due to P-gp-mediated efflux increasing biliary excretion in vivo, which could not be predicted by the MLM assay.24
Table 3. Multiparameter Optimization of the Piperazine Subseriesa.

| Entry | Compd | R group | X | SK-OV-3 GI50 pGI50 ± SEM (n)b | CH1doxR/CH1WTc | Log D7.4d | KS (μM)e | MLM (μL/min/mg)f | Pred. in vivo CLu from MLM (mL/min/kg)j | Mouse Heps (μL/min/106)g | Pred. in vivo CLu from Heps (mL/min/kg)j |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 16 | iPr | Me | 8.8 nM 8.06 ± 0.05 (3) | 8.4 | 1.9 | 80 | 10 | 39 | 20 | 460h |
| 2 | 18 | iPr | Cl | 15 nM 7.83 ± 0.08 (8) | 1.7 | 2.3 | 70 | 17 | 65 | 28 | 930i |
| 3 | 19 | H | Cl | 12 nM 7.94 ± 0.01 (3) | 5.2 | ND | ND | 12 | ND | ND | ND |
| 4 | 20 | Me | Cl | 19 nM 7.72 ± 0.16 (3) | 1.2 | 2.1 | 30 | 110 | ND | ND | ND |
| 5 | 21 | Et | Cl | 11 nM 7.96 ± 0.09 (10) | 1.4 | 2.2 | 50 | 26 | 100 | 71 | 1400i |
| 6 | 22 | Et | F | 8.5 nM 8.07 ± 0.02 (45) | 1.8 | 1.7 | 50 | 15 | 58 | 53 | 900h |
All data were reprocessed using GraphPad Prism Version 7.01. ND = not determined. All results are quoted to two significant figures unless otherwise stated.
The number of repeats (n) are described in parentheses; growth inhibition was measured after 96 h of treatment and compared to the vehicle control; all results are quoted as the geometric mean ± SEM, pGI50 = −log GI50 (M).
The fold resistance is determined by the ratio of geometric mean GI50 values in CH1doxR cells and CH1WT cells in the CellTiter-Blue growth inhibition assay.
Measured via an HPLC method, n = 1.
Measured via an HPLC method from phosphate buffer at pH 7.4; all values quoted to 1 SF; the dynamic range of the assay is 1–100 μM, n = 1.
Mouse liver microsome (MLM) assay was carried out at Cyprotex, n = 1; in vitro CLint (μL/min/mg of protein) is calculated from the half-life using standard procedures and assumes that the fraction unbound in the assay is 1.
Mouse hepatocyte assay was carried out at Cyprotex, n = 1; in vitro CLint (μL/min/106 cells) is calculated from the half-life using standard procedures.
Assumes that the fraction unbound in the assay is 0.4.
Assumes that the fraction unbound in the assay is 0.2.
Calculated from the in vitro CLint using scaling factors and applying the well-stirred model; see the Supporting Information for details.
To assess the effect of P-gp-mediated efflux on in vivo clearance, 16 was submitted for comparative studies in wild-type (CF1WT) and P-gp-knockout (CF1PGP-KO) mice (Figure S8).25 The PK data revealed that the CLtb in CF1WT (35 mL/min/kg) was significantly higher than in the CF1PGP-KO (24 mL/min/kg, p = 0.024, Student’s t-test), indicating that P-gp-efflux did contribute to the unfavorable mouse PK profile.26 Interestingly, the volume of distribution (Vss) remained unchanged.27
Once efflux was highlighted as a concern for further compound optimization, both for preclinical PK optimization and for future clinical success against refractory ovarian cancer, we required a medium-throughput assay to rapidly determine efflux-mediated SAR. Cellular P-gp-mediated efflux acquired MDR to the cytotoxic anthracycline, doxorubicin is well established and can be used as both a P-gp efflux model and MDR-resistance model.28 We therefore proposed that doxorubicin resistance could be exploited to establish a surrogate assay for P-gp-mediated efflux with appropriate throughput in matched pair ovarian cancer cell lines. An acquired doxorubicin-resistant human cancer cell line, CH1doxR, was previously obtained in-house through exposure of the wild-type cell line CH1WT to doxorubicin.29 After several passages, the CH1doxR cell line demonstrated >100-fold resistance. The P-gp-dependent MDR properties of CH1doxR were confirmed by the rescue of the antiproliferative activity of doxorubicin by cotreatment with the P-gp-inhibitor verapamil,30 resulting in a shift in GI50 in CH1doxR from 310 to 1.9 nM, now within 5.0-fold of the antiproliferative activity observed in CH1WT (Figure S4).
After demonstrating that the antiproliferative activity of the bisamide chemical probe 1 in CH1doxR cells could also be increased by cotreatment with the P-gp inhibitor (Figure S4), we aimed to validate the use of CH1WT and CH1doxR cells as a viable approach to establish useful SAR by carrying out a screen of selected bisamide analogues (N = 43; Table S3). The comparative antiproliferative activity of each analogue against the CH1WT and CH1doxR cells was measured, and the fold-differences between their respective geometric mean GI50 values were used as a surrogate for their respective CH1doxR/CH1WT P-gp-mediated efflux ratios.31 The statistical significance of the ratio was determined using Student’s t-test from the respective pGI50 values, and only ratios of analogues where p < 0.05 were considered to be P-gp-substrates. Using this approach, compound 16 gave a moderately high CH1doxR/CH1WT ratio (8.4), consistent with its poor in vivo mouse PK profile. In contrast, the oxygen-linked piperidine analogue 17 displayed no significant difference in the comparison of their respective GI50 values (CH1doxR/CH1WT = 1.7), indicating that this compound is likely only a weak P-gp substrate. Owing to its acceptable in vitro and wild-type Balb/c mouse in vivo profile (Table 2, entry 2), 17 was selected for in vivo study in P-gp-knockout mice (Figure S8). The PK data revealed that there was no significant difference in the CLtb of 17 between P-gp-knockout and wild-type mice, consistent with our in vitro prediction from the CH1 cell-based assay.
Unfortunately, the in vitro cell-based MDR assay revealed no clear SAR or patterns relating to the physicochemical properties that are typically used to remove P-gp-mediated efflux.32 Critical structural features likely to be important for passive permeability, such as the two amide moieties, could not be replaced in a manner consistent with the cellular SAR to retain activity and there was no clear correlation with compound lipophilicity.13 To carry out the necessary optimization to improve compound PK profiles across multiple species, a general method to eliminate P-gp-efflux was needed. Levatic et al. have reported the empirical observation that molecular density is an important feature in P-gp-drug recognition and compounds with high specific volumes are less likely to suffer from P-gp-mediated efflux.33 We hypothesized that as halogens possess high van der Waals volumes,34 they could be used to reduce the molecular density of the bisamide chemotype and mitigate this liability (Table 3).
It was important that we introduced the halogen on the bisamide chemotype distal from the solubilizing group to allow for further orthogonal PK optimization. We hypothesized that the benzylic methyl on the central ring of the bisamide, which was critical to cellular activity of this chemotype, was suitable for substitution, as halogens have been shown to act as good bioisosteric replacements for small lipophilic groups.35 A matched molecular pair (MMP)36 of piperazine 16 was synthesized, replacing the methyl with chlorine to give 18 (Table 3, entry 2). Chlorobisamide 18 maintained excellent antiproliferative activity, and importantly, the efflux-mediated CH1doxR/CH1WT ratio was reduced from 8.4-fold to 1.7-fold, with respect to 16. To assess whether this effect was general to this chemotype, we synthesized halogenated MMPs of the compounds that exhibited significant efflux in the CH1doxR/CH1WT assay (Table S3). In all cases, halogen substitution strongly reduced efflux compared to their respective methyl MMPs (Figure 2, colored lines) and by comparing the average effect of each halogen (Figure 2, gray bars). In particular, the larger halogens had an apparently greater impact (Br > Cl > F), consistent with their effect on molecular density or possibly through more efficient steric shielding of the proximal amide moiety, although the exact mechanism for the change in P-gp recognition with this chemotype remains unclear (Figure 2).34,38
Figure 2.
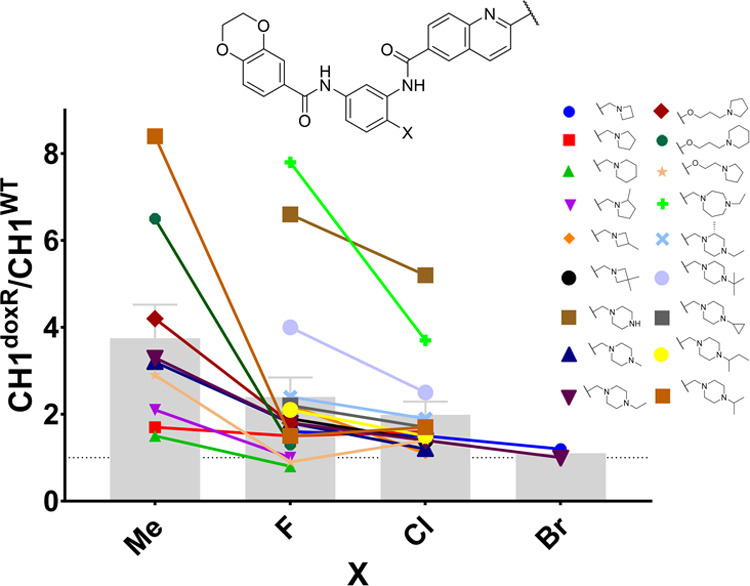
Matched molecular pair analysis on the effect of halogen substitution on multidrug resistance. The efflux ratio is calculated from the ratio of geometric mean GI50 values in the CH1WT and CH1doxR cell lines; each GI50 is calculated from at least n = 3 biological repeats. Each colored line represents an MMP varying only at the central ring substituent. The gray bars represent the arithmetic mean ± SEM of the grouped central ring substituents. The gray dotted line is the efflux ratio = 1. See Table S3 for details.
In Vitro/In Vivo Correlation Disconnection
Following our discovery of a general strategy to mitigate the P-gp-mediated efflux liability of the bisamide chemotype, we then sought to complete the multiparameter optimization necessary to deliver a clinical candidate. Further in vitro analysis of chlorobisamide 18 revealed that the compound possessed low MLM CLint and good KS (Table 3, entry 2) and so was selected for an in vivo PK study in BALB/c mice (Table 2, entry 2). Chlorobisamide 18 displayed an impressive improvement in oral bioavailability (F = 63%, CLtb = 33 mL/min/kg, extraction ratio = 37%, Fmax = 63%), resulting from high absorption consistent with its in vitro property profile. However, 18 still displayed disappointingly high in vivo CLu (3000 mL/min/kg, fub = 0.011), despite the predicted CLu from the MLM being 51-fold lower. No significant degradation was observed following the incubation of bisamide analogues in mouse plasma, so plasma instability was considered unlikely to be contributing to the discrepancy (Table S7). From these data, it was clear that there was still another component contributing to the in vivo clearance that needed to be addressed.
Hepatocyte Clearance
Microsomes can underpredict in vivo CLu from either the loss of metabolic enzyme integrity during tissue handling. the effect of uptake transporters on intracellular free concentrations or under-representation of cytosolic enzymes and cofactors.24 Therefore, chlorobisamide 18 was screened in vitro using mouse hepatocytes (MHeps), which revealed a predicted in vivo CLu now within 3.5-fold of the measured value.21 The improved correlation could be due to better representation of amide hydrolysis degradation pathways in hepatocytes compared to that in microsomes. The MHeps assay, combined with our CH1doxR/CH1WT ratio assay, finally gave us an appropriate in vitro triage of compounds for in vivo PK assessment.
Although introducing the chlorine substituent had mitigated the P-gp-mediated efflux risk, it had also significantly increased the lipophilicity of 18 relative to its MMP, 16, potentially leading to the increased in vivo CLu. To improve the metabolic stability of 18, we aimed to decrease the lipophilicity of the chloro-series closer to the value obtained with methyl analogue 16, which showed lower in vivo CLu. Removal of the N-alkyl moiety to afford the free piperazine 19 was not tolerated, resulting in a large CH1WT/CH1doxR ratio for predicted P-gp-mediated efflux (Table 3, entry 3), while N-methylpiperazine 20 displayed high MLM CLint (110 μL/min/mg) and so was not investigated further. However, N-ethylpiperazine 21 displayed a good balance of physicochemical properties (Table 3, entry 5), which translated into excellent mouse oral bioavailability (Table 2, entry 4) from moderate total blood clearance (CLtb = 28 mL/min/kg, extraction ratio = 31%, Fmax = 69%). Unfortunately, the high CLu persisted and no other changes to the solubilizing group of the chloro-series were able to significantly improve the metabolic stability predicted from in vitro analysis (see Table S2 for details).
To further reduce lipophilicity, we hypothesized that we could replace the benzylic chlorine substituent with fluorine, but we were concerned that this decrease could have a detrimental effect on passive permeability. However, analysis of the Cambridge Structural Database of small molecules37 revealed that ortho-fluorobenzamides tend to adopt more planar conformations than their methyl counterparts (Figure S3). The amide–NH bond can eclipse the fluorine–carbon bond, forming a dipole–dipole interaction and masking the hydrogen bond donor, hence mitigating concerns of decreased lipophilicity on passive permeability.38 The fluorine MMP, CCT361814/NXP800 22, pleasingly displayed the desired reduction in lipophilicity (Table 3, entry 6), which correlated with reduced in vitro MLM (15 μL/min/mg) and mouse hepatocyte CLint; while maintaining excellent antiproliferative activity (free GI50 = 3.7 nM, fua = 0.43; Table S4)39 and acceptable KS (50 μM). Fluorobisamide 22 was therefore submitted for an in vivo mouse PK study (Table 4, entry 1).
Table 4. In Vivo Blood PK Profiles of 22 in Rodent and Nonrodent Speciesa.
| Entry | Speciesb | Dose po/iv (mg/kg)e | Tmax (h) | po AUC0–t (h*nM) | iv Cltb (mL/min/kg)h | t1/2 (h)i | F (%)j | fubk | AUCu0–t (h*nM)l | Free Cav0–24h (nm)m | iv Clu (mL/min/kg)n |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | mouse | 5/5 | 2.0 | 6000 (7800–4600)f | 10 (10–9.7) | 4.0 | 42 | 0.012 | 72 | 3.3 | 830 |
| 2 | ratc | 5/1 | 6.0 | 2600f | 24 | 3.1 | 45 | 0.033 | 86 | 3.7 | 730 |
| 3 | dogd | 2.5/0.5 | 2.0 | 250g | 21 | 1.4 | 9.1h | 0.14 | 35 | 1.9 | 150 |
All values are quoted to two SFs and are the geometric mean of n = 3 individual animals unless otherwise stated. PK parameters are calculated from the blood concentration/time curve using noncompartmental analysis model 201 and 202 Phoenix version.6.1. The 90% confidence intervals are in parentheses.
Immunocompetent BALB/c mice, SD rats, and beagle dogs.
The rat blood PK was determined as a composite profile of six animals.
Dog live phase was carried out at Charles River Laboratories; data are derived from the geometric mean of n = 4 individual dogs (two males and two females); PK parameters in the dog study were calculated from the plasma concentration/time curve and were converted to blood PK parameters using the blood-to-plasma ratio determined in vitro at Cyprotex.
The po and iv dosing vehicles are described in the Supporting Information.
t = 24 h.
t = 6 h.
CLtb = total blood clearance.
Terminal half-life calculated from the iv dose.
Assumes linear PK.
fub = fraction unbound in blood, fub = fup/B:P, fup = fraction unbound in plasma, B:P = blood-to-plasma ratio, measured in vitro using dialysis and quoted as the geometric mean from n = 3 statistical repeats from pooled samples; see the Supporting Information for details.
AUCu = AUC*fub.
Free Cav0–24h = AUCinf/24*fub.
CLu = CLtb/fub.
The mouse in vivo CLu for compound 22 was consistent with the predicted value from the MHeps assay and comparable to methyl analogue 16 (Table 2, entry 1). Despite the decreased lipophilicity, the CH1doxR/CH1WT-predicted P-gp-mediated efflux ratio was low and fluorobisamide 22 displayed good mouse oral bioavailability (42%) from moderate total blood clearance (CLtb = 10 mL/min/kg, extraction ratio = 11%, Fmax = 89%).22 Owing to these favorable data, fluorobisamide 22 was selected for evaluation of its in vivo efficacy against established SK-OV-3 human ovarian cancer solid tumor xenografts in athymic immunodeficient mice (Table 5).
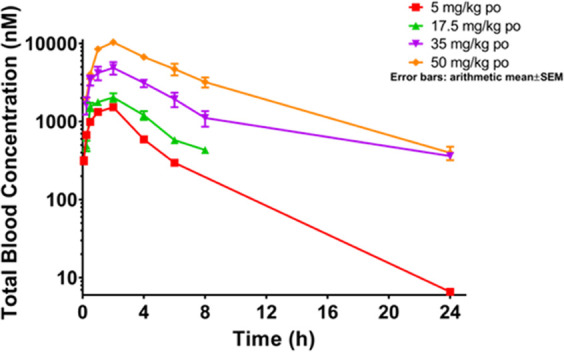
Table 5. Blood Pharmacokinetic Profiles of Fluorobisamide 22 in Athymic Mice with Increasing Oral Dosea.
| Entry | Dose (mg/kg)b | Tmax (h) | AUC0–t (h*nM) | iv Cltb (mL/min/kg)f | t1/2 (h)g | F (%)h | fubi | AUCu0–t (h*nM)j | Free Cav0–24h (nM)k | iv CLu (mL/min/kg)l |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5 iv | NA | 6700 (7800–5700)c | 21 (24–18) | 1.5 | NA | 0.0070 | 47 | 2.1 | 3000 |
| 2 | 5 po | 1.7 | 1300 (1800–990)c | 20 | 9.1 | 0.59 | ||||
| 3 | 17.5 po | 1.7 | 9000 (12 000–7000)d | 39 | 63 | 3.1 | ||||
| 4 | 35 po | 2 | 34 000 (57 000–21 000)d | 72 | 240 | 10 | ||||
| 5 | 50 po | 1.7 | 78 000 (100 000–60 000)e | 120 | 550 | 24 |
All graphs were plotted using GraphPad Prism Version 7.01. NA = not applicable. Each point on the PK curve is the arithmetic mean ± SEM of n = 3 individual animals. All values are quoted to two SFs and are the geometric mean of n = 3 individual mice. PK parameters are calculated from the blood concentration/time curve using noncompartmental analysis Phoenix version 6.1. The 90% confidence intervals are in parentheses.
The po and iv dosing vehicles are described in the Supporting Information.
t = 6 h.
t = 8 h.
t = 24 h.
CLtb = total blood clearance from the 5 mg/kg iv dose.
Terminal half-life calculated from the iv dose.
Assumes linear PK.
fub = fraction unbound in blood, fub = fup/B:P, fup = fraction unbound in plasma, B:P = blood-to-plasma ratio, measured in vitro using dialysis and quoted as the geometric mean from n = 3 statistical repeats from pooled samples; see the Supporting Information for details.
AUCu = AUC*fub.
Free Cav0–24h = AUCinf/24*fub.
CLu = CLtb/fub.
Efficacy and PD
The assessment of the blood PK profiles of fluorobisamide 22 in immunodeficient athymic mice revealed a much higher CLu (3000 mL/min/kg) following an iv dose than had been observed in immunocompetent BALB/c mice, consistent with our previous observations with chemical probe 1,13 and overproportional exposure with increasing po dose. Owing to the nonlinear PK, high CLu in this mouse strain, and following a multidose tolerability study (Figure S9), a 35 mg/kg po qd dose was selected for the efficacy study, which should give a 2.7-fold coverage (AUCu0–24h = 240 h*nM, free Cav0–24h = 10 nM) of the in vitro free GI50 (Figure 3A).
Figure 3.

Antitumor, PK, and pharmacodynamic (PD) activity of fluorobisamide 22 in immunodeficient athymic mice. All data were analyzed and plotted using GraphPad Prism 7.01. NS p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. A: Antitumor activity of fluorobisamide 22 following a 35 mg/kg po qd dose against established SK-OV-3 human ovarian cancer xenografts in immunodeficient athymic mice. Each point represents the arithmetic mean ± SEM of n = 10 mice. Analysis by standard two-way ANOVA: interaction p<0.001, time p=0.0032, treatment p<0.0001. Comparison of vehicle vs treated arms at each time point using Sidak’s multiple comparison test. See the Supporting Information for details. B: PK/PD analysis following a single 50 mg/kg po dose in athymic mice with established SK-OV-3 xenografts. The CHAC1 and HSPA1A biomarker responses in the tumor were quantified using an MSD and quantitative polymerase chain reaction (qPCR) assays, respectively, and were correlated to the relative free concentration of 22 in both the plasma (n = 4) and the tumor (n = 4 except 4 and 16 h when n = 3). The free concentration in the plasma was estimated by Cu = CT*fup; fup was measured in vitro using standard dialysis methods in pooled athymic mouse plasma. Tumor-free concentrations were estimated by calculating the free fraction in the tumor post distribution equilibrium (>8 h). The significance of all CHAC1 and HSPA1A biomarker responses in treated samples (CHAC1: n = 4 at each time point except 6 h where n = 3, HSPA1A: n = 4 at each time point except 4 and 6 h where n = 3) is described relative to the vehicle control (n = 23, 0–24 h) and was analyzed using a one-way ANOVA (CHAC1: p < 0.0001, HSPA1A: p < 0.0001) and Dunnett’s multiple comparison test of the log-transformed data. C: Structure of the clinical candidate fluorobisamide 22. D: Clinical development candidate profile of fluorobisamide 22 with predicted human parameters; aheps = human hepatocytes, CLu calculated from in vitro CLint using scaling factors and the well-stirred model; brat SSS = rat single-species scaling (SSS) CLuhuman = CLurat (body weighthuman/body weightrat) ∧ 0.75; cCLtb = CLu*fub, fub = fup/B:P, fup and B:P were determined in vitro by dialysis of pooled plasma and blood samples; ddetermined from the rat t1/2 using an empirical approach log t1/2(human) = 0.906*log t1/2(rat) + 0.723; emeasured at Pharmidex, the dynamic range of the assay is 0–1 mg/mL, FaSSIIF and FeSSIF pH6.5, FaSSGF pH1.6; festimated from Cav(free) = 10 nM in the mouse model, dose = (Ceff(free)*CLu*τ)/F, where τ = dose frequency (24 h) and F is assumed to be 0.4.
The mice maintained acceptable condition and body weight (Figure S10; <10% loss) when dosed at 35 mg/kg po continuously with a solution of fluorobisamide 22 for 20 days without the need for dose breaks. Excellent efficacy was observed against established SK-OV-3 human ovarian cancer solid tumor xenografts grown subcutaneously, with tumor growth inhibition (TGI) of 120% relative to control40 and 8 out of 10 tumors displaying regression. T/C values, based on the final mean tumor weights, also showed a significant reduction to 37% of control (p = 0.0008, Student’s t-test; Figure S11).
Discovering pharmacodynamic (PD) biomarkers using compounds developed from phenotypic screens of transcription inhibitors can be challenging, as to observe an effect the biological pathways of interest must commonly be potentiated by an external stimulus.41a In vitro, we utilized as a PD biomarker the blocked induction of the protein product HSP72, which is encoded by the canonical HSF1-regulated gene HSPA1A in human cancer cell lines, following activation with the HSP90 inhibitor 17-AAG, to confirm that fluorobisamide 22 antagonized the pathway (SK-OV-3, HSP72 cell-based ELISA, pIC50 = 7.03 ± 0.07, IC50 = 94 nM, n = 40).42 However, for in vivo studies, we required protein PD biomarkers that would not need an HSP90 inhibitor to activate HSF1. Also, modulation at the mRNA rather than protein level would provide more proximal biomarkers for the inhibition of HSF1-mediated transcription.43 Therefore, we carried out gene expression profiling of cancer cell lines and tumor xenograft tissues following treatment with fluorobisamide 22; this demonstrated the inhibition of heat shock response gene signature and activation of the related integrated stress response signature, which we then exploited for PD biomarker development.44−46
qPCR analysis of the end-of-study tumor samples from the treated and untreated arms of the efficacy study revealed a clear increased expression of the mRNA of CHAC1 (Figure S12), a gene involved in the integrated stress response and activation of which would be consistent with the inhibition of the HSF1 stress pathway.45CHAC1 is known to be downstream of the HSF1-regulated gene, ATF3.(10,44) At the protein level, the induction of CHAC1 was confirmed by immunoblot, from both in vivo tumor samples and in vitro data in SK-OV-3 cells treated with fluorobisamide 22 (19 nM, 5 × free GI50; Figure S13).47
To investigate the relationship between the free exposure of 22 and HSF1 pathway inhibition PD biomarker modulation, a single dose (50 mg/kg), single agent PK/PD study was designed and carried out on athymic mice bearing established SK-OV-3 human ovarian cancer solid subcutaneous tumor xenografts, as were used in the efficacy study (Figure 3B). The PK/PD data revealed a tumor Tmax at 6 h, 4 h later than the dose escalation study blood Tmax in nontumor-bearing mice.48 Distribution equilibrium was achieved after 8 h, and by comparison with free plasma concentrations at these later time points, we estimated the free tumor concentrations49 to achieve a tumor-free Cmax = 21 nM and free Cmin = 2.8 nM after 24 h. At the dose of fluorobisamide 22 used, free tumor concentrations were greater than the in vitro free GI50 for 21 h. CHAC1 protein expression was confirmed using immunoblotting (Figure S14) and was quantified by an MSD assay we developed in-house50 (Figure S15). In vivo, CHAC1 induction by fluorobisamide 22 correlated well with the free concentration in the tumor, with CHAC1 induction Tmax (PD) also occurring at the fluorobisamide 22 tumor Tmax (PK) and at a free concentration comparable to those for the in vitro induction (Figure 3B).
To assess a more direct biomarker for the antagonism of HSF1-mediated transcription, we turned to the inhibition of expression of the canonical heat shock HSPA1A gene, which encodes the HSP72 protein. As expected, given its long degradation half-life,43 no clear changes to basal HSP72 protein levels were observed over the 24 h of the PK/PD study. However, analysis of the short half-life HSPA1A mRNA tumor levels using qPCR revealed significant depletion by fluorobisamide 22 that also correlated well with the tumor-free concentrations (Figure 3B).
Clinical Candidate
The optimized fluorobisamide 22 clearly displayed improved mouse PK and efficacy in the SK-OV-3 human ovarian xenograft model compared to our earlier chemical probe 1. Screening 22 against our panel of genetically diverse human ovarian cancer cell lines demonstrated that the compound retained excellent antiproliferative activity (Table S1). To further assess the potential of 22 as a clinical candidate, the compound was assayed in the Cerep in vitro safety screen of 87 potentially high-risk off-target proteins (Table S9).51 From this screen, only adenosine A2A receptor antagonism (IC50 = 2.0 μM; Figure S4) was confirmed by a functional assay but at a value ∼100-fold higher than the efficacious free concentrations.52 Fluorobisamide 22 also displayed no hERG (IC50 > 30 μM)53 or CYP (IC50 > 10 μM) inhibition (Table S10 and Figure S7) liability and was clean in kinase screening panels (data not shown), consistent with our previous analysis.13 Analysis of rat PK data for 22 (Table 4, entry 2) revealed a clear improvement with respect to the lead compound 1, with reduced CLu, consistent with the rat hepatocyte prediction of 410 mL/min/kg, good oral bioavailability (45%) from a moderate total blood clearance (CLtb = 24 mL/min/kg, extraction ratio = 34%, Fmax = 66%),17,57 and acceptable half-life (t1/2) (Table 4, entry 2, and Table S12).
To select a higher species for further PK study, 22 was submitted to minipig and dog hepatocyte assays (Table S6). The minipig hepatocyte CLint (860 μL/min/106 cells) was very high, consistent with a previous study, which showed that the bisaryl amide motif found in the bisamide chemotype is particularly susceptible to hydrolysis by minipig liver amidases.54 Given the confidence we had gained in the predictive value of in vitro hepatocyte metabolism for in vivo CLu prediction, minipig was not considered further. The dog hepatocytes gave CLint = 31 μL/min/106 cells, which predicted an in vivo CLu = 96 mL/min/kg. However, in contrast to the very low blood-free fraction (fub; Table S4) of 22 in mice, rats, and humans (0.0053–0.033), the dog blood-free fraction was surprisingly high (fub = 0.14). Despite this contrast, the dog was selected as our higher species for further study (Table 4, entry 3).55 The dog PK data revealed a lower CLu compared to the rodent species and were consistent with the hepatocyte prediction, but oral bioavailability (9.1%) was also low, from a moderate total blood clearance (CLtb = 21 mL/min/kg, extraction ratio = 52%, Fmax = 48%).56,57 The low absorption in the dog compared to that in the rat for 22 could be due to solubility-limited absorption of this basic compound in the relatively high pH of the dog’s stomach.58 Nevertheless, the free exposure from the 2.5 mg/kg po dose in the dog was still comparable to the efficacy study free exposure due to the much lower CLu (AUCu0–6h = 40 h*nM, free Cav0–6h = 6.6 nM).
Furthermore, 22 displayed improved in vitro permeability in the Caco-2 assay (A–B = 7.7 × 10–6 cm/s, efflux ratio = 2.8; Table S8), despite low compound recovery (∼50%, consistent with intracellular compound retention), which can limit passive permeability.59 Thermodynamic solubility assays in simulated human biorelevant simulated fluids showed that compound 22 (Figure 3D) was highly soluble in simulated gastric fluid, possibly due to the low pH, and modestly soluble in intestinal fluid (Table S11).60 From our studies and the excellent efficacy in the SK-OV-3 human ovarian xenograft model driven by a low free concentration, the calculated dose predictions for 22 to human based on the efficacious AUCu0–24h were favorable, using both single-species scaling (SSS)61 and scaling from human hepatocytes, at less than 210 mg/person/day.62 These data led to the nomination of fluorobisamide 22 as our clinical candidate (Figure 3).63
Conclusions
We carried out multiparameter lead optimization of the bisamide chemical probe 1, discovered from an HSF1 stress pathway inhibitor phenotypic screen, using cell-based SAR to maintain the excellent antiproliferative activity. During our PK optimization, minimizing P-gp-mediated efflux became an early focus. We developed a medium-throughput cell-based antiproliferation sensitivity assay as a surrogate to assess P-gp-mediated efflux and demonstrated that incorporating halogens into our analogue design reduced this liability in all examples across a wide range of substrates. This led to an empirical but effective strategy to mitigate P-gp-mediated efflux that could potentially be applicable to other chemotypes. A further multiparameter optimization gave us our clinical candidate, fluorobisamide 22. This compound displayed a good PK profile across different species and excellent therapeutic efficacy, including tumor regression, from a low free exposure in an in vivo human ovarian cancer xenograft mouse model, and demonstrated biomarker modulation in tumor tissue consistent with the HSF1 pathway inhibition—representing overall a strong pharmacological audit trail.41
We have carried out numerous studies to determine the molecular mechanism of action of fluorobisamde 22, including transcriptional profiling and use of multiple orthogonal chemoproteomic technologies. Demonstration of the increased expression of CHAC1 mRNA and the reduced expression of HSPA1A mRNA, which represent useful PD markers, is consistent with the activation of the integrated stress response and inhibition of HSF1-mediated transcription. Further mechanistic follow-up studies are underway.
Following successful preclinical development, CCT361814/NXP800 22 entered phase 1 clinical trial (NCT05226507) in cancer patients in 2022 as a potential future treatment for refractory ovarian cancer and other malignancies.64
Experimental Section
All experiments using animals were performed in accordance with the local Animal Welfare and Ethical Review Board, the U.K. Home Office Animals Scientific Procedures Act 1986, and the U.K. National Cancer Research Institute Guidelines for the Welfare of Animals in Cancer Research.65 The ICR does not undertake research in nonrodent species and requires internal ethical review when such studies are sponsored by organizations with whom we collaborate. Collaborator-sponsored nonrodent pharmacology studies of compound 22 necessary for the prediction of therapeutic window and application to the clinic were approved by the ICR Animal Welfare and Ethics Review Board and were conducted in full compliance with national regulations at AAALAC accredited R&D sites.
General Procedures (Chemistry)
All final compounds were screened through our in-house computational PAINS filter and gave no structural alerts as potential assay interference compounds.66 Unless otherwise stated, reactions were conducted in oven-dried glassware under an atmosphere of nitrogen or argon using anhydrous solvents. All commercially obtained reagents and solvents were used as received. Thin-layer chromatography (TLC) was performed on precoated aluminum sheets of silica (60 F254 nm, Merck) and visualized using short-wave UV light. Flash column chromatography was carried out on Merck silica gel 60 (particle size 40–65 μm). Column chromatography was also performed on Biotage SP1 or Isolera 4 purification systems using Biotage Flash silica cartridges (SNAP KP-Sil). Ion-exchange chromatography was performed using acidic Biotage Isolute Flash SCX-2 columns. All compounds are >95% pure by HPLC analysis. HPLC traces of the clinical candidate 22 and all in vivo compounds are included in the Supporting Information.
Semipreparative HPLC
500 μL standard injections (with needle wash) of the sample were made on a Phenomenex Gemini C18 column (5 μ, 250 mm × 21.2 mm, Phenomenex, Torrence). Chromatographic separation at room temperature was carried out using a 1200 Series Preparative HPLC (Agilent, USA) over a 15 min gradient elution from 90:10 to 0:100 water:methanol (both modified with 0.1% formic acid) at a flow rate of 20 mL/min. UV–vis spectra were acquired at 254 nm on a 1200 Series Prep Scale diode array detector (Agilent). Post-UV and pre-MS splitting were achieved using an Active Split (Agilent) before being infused into a 6120 Series Quad mass spectrometer fitted with an ESI/APCI Multimode ionization source (Agilent). Collection was triggered by UV signal and collected on a 1200 Series Fraction Collector (Agilent). 1H NMR spectra were recorded on Bruker Avance 500 (500 MHz) spectrometers using an internal deuterium lock. Chemical shifts are quoted in parts per million (ppm) using the following internal references: CDCl3 (δH 7.26), MeOD (δH 3.31), and DMSO-d6 (δH 2.50). Signal multiplicities are recorded as singlet (s), doublet (d), triplet (t), quartet (q), quintet (qn), and multiplet (m), doublet of doublets (dd), doublet of doublet of doublets (ddd), broad (br), obscured (obs) or apparent (app). Coupling constants, J, are measured to the nearest 0.1 Hz. 13C NMR spectra were recorded on Bruker Avance 500 spectrometers at 126 MHz using an internal deuterium lock. Chemical shifts are quoted to 0.01 ppm, unless greater accuracy was required, using the following internal references: CDCl3 (δC 77.0), MeOD (δC 49.0), and DMSO-d6 (δC 39.5). High-resolution mass spectra were recorded on an Agilent 1200 series HPLC and a diode array detector coupled to a 6210 time-of-flight mass spectrometer with a dual multimode APCI/ESI source (methods I–IV) or on a Waters Acquity UPLC and a diode array detector coupled to a Waters G2 QToF mass spectrometer fitted with a multimode ESI/APCI source (methods V–VI). Analytical separation was carried out according to the methods listed below. The mobile phase was a mixture of methanol (solvent A) and water (solvent B), both containing formic acid at 0.1%; UV detection was at 254 nm.
Method I: analytical separation was carried out at 30 °C on a Merck Purospher STAR column (RP-18e, 30 mm × 4 mm) using a flow rate of 1.5 mL/min in a 4 min gradient elution. Gradient elution was as follows: 10:90 (A/B) to 90:10 (A/B) over 2.5 min, 90:10 (A/B) for 1 min, and then reversion back to 10:90 (A/B) over 0.3 min, finally 10:90 (A/B) for 0.2 min. Method II: analytical separation was carried out at 30 °C on a Merck chromolith flash column (RP-18e, 25 mm × 2 mm) using a flow rate of 0.75 mL/min in a 4 min gradient elution. Gradient elution was as follows: 5:95 (A/B) to 100:0 (A/B) over 2.5 min, 100:0 (A/B) for 1 min, and then reversion back to 5:95 (A/B) over 0.1 min, finally 5:95 (A/B) for 0.4 min. Method III: analytical separation was carried out at 40 °C on a Merck Purospher STAR column (RP-18e, 30 mm × 4 mm) using a flow rate of 3 mL/min in a 2 min gradient elution. Gradient elution was as follows: 10:90 (A/B) to 90:10 (A/B) over 1.25 min, 90:10 (A/B) for 0.5 min, and then reversion back to 10:90 (A/B) over 0.15 min, finally 10:90 (A/B) for 0.1 min. Method IV: analytical separation was carried out at 40 °C on a Merck Purospher STAR column (RP-18e, 30 mm × 4 mm) using a flow rate of 1.5 mL/min in a 2 min gradient elution. Gradient elution was as follows: 5:95 (A/B) to 100:0 (A/B) over 1.25 min, 100:0 (A/B) for 0.5 min, and then reversion back to 5:95 (A/B) over 0.05 min, finally 5:95 (A/B) for 0.2 min. Method V: Waters Acquity UPLC, Phenomenex Kinetex XB-C18 column (30 mm × 2.1 mm, 1.7 μ, 100 Å) at 30 °C using a flow rate of 0.3 mL/min in a 4 min gradient elution. Gradient elution was as follows: 10:90 (A/B) to 90:10 (A/B) over 3 min, 90:10 (A/B) for 0.5 min, and then reversion back to 10:90 (A/B) over 0.3 min, finally 10:90 (A/B) for 0.2 min. Method VI: Waters Acquity UPLC, Phenomenex Kinetex C18 column (30 mm × 2.1 mm, 2.6 μ, 100 Å), flow rate and gradient elution according to Method V. The following reference masses were used for HRMS analysis: Agilent 1200 series: caffeine [M + H]+ 195.087652; hexakis(1H,1H,3H-tetrafluoropentoxy)phosphazene [M + H]+ 922.009798 and hexakis(2,2-difluoroethoxy)phosphazene [M + H]+ 622.02896 or reserpine [M + H]+ 609.280657; Waters Acquity UPLC: Leucine Enkephalin fragment ion [M + H]+ 397.1876. All compounds were >95% purity by liquid chromatography-mass spectrometry (LCMS) analysis unless otherwise stated.
Synthetic Route I
N-(3-Amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide
Oxalyl chloride (1.40 mL, 16.6 mmol) was added dropwise to a solution of 1,4-benzodioxane-6-carboxylic acid (2.49 g, 13.8 mmol) and DMF (0.027 mL, 0.340 mmol) in anhydrous dichloromethane (DCM) (34 mL). The reaction mixture was stirred at room temperature for 3.5 h and then concentrated. The residue was dissolved in DCM and concentrated again. This residue was dissolved in anhydrous DCM (12 mL) and added dropwise to a solution of 4-methyl-3-nitroaniline (2.10 g, 13.8 mmol) and pyridine (2.23 mL, 27.6 mmol) in anhydrous DCM (25 mL). The reaction mixture was stirred at room temperature for 2 h and then concentrated. The resulting solid was suspended in MeOH, diluted with water, and then isolated by filtration and washed with water to afford the title compound (4.24 g, 98%) as a pale tan-colored amorphous solid. 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.54 (d, J = 2.2 Hz, 1H), 7.99 (dd, J = 8.4, 2.3 Hz, 1H), 7.55 (d, J = 2.1 Hz, 1H), 7.52 (dd, J = 8.4, 2.2 Hz, 1H), 7.47 (dd, J = 8.4, 0.8 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 4.34–4.29 (m, 4H), 2.49 (s, 3H). HRMS (ESI+): calcd for C16H15N2O5 (M + H)+, 315.0976; found 315.0982.
Palladium (10% on activated carbon, 0.567 g, 5.33 mmol) was added to a suspension of N-(4-methyl-3-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (4.24 g, 13.5 mmol) in ethanol (90 mL) and ethyl acetate (90 mL). The reaction mixture was stirred under hydrogen (1 atm) at 28 °C overnight, filtered through celite with EtOAc, and concentrated to afford the title compound (3.80 g, 99%) as a pale yellow amorphous solid. 1H NMR (500 MHz, DMSO-d6) δ 9.70 (s, 1H), 7.49 (d, J = 2.2 Hz, 1H), 7.46 (dd, J = 8.3, 2.2 Hz, 1H), 7.10 (d, J = 2.0 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 8.1 Hz, 1H), 6.79 (dd, J = 8.1, 2.0 Hz, 1H), 4.81 (s, 2H), 4.32–4.26 (m, 4H), 2.01 (s, 3H). HRMS (ESI+): calcd for C16H17N2O3 (M + H)+, 285.1234; found 285.1233.
Methyl 2-Formylquinoline-6-carboxylate
To a solution of 2-methylquinoline-6-carboxylic acid (3.00 g, 16.0 mmol) in anhydrous MeOH (40 mL) under argon at room temperature, 4 M HCl in 1,4-dioxane (16.0 mL, 64.1 mmol) was added dropwise and the resulting mixture was heated at 85 °C for 4 h. Then, the reaction mixture was allowed to cool to room temperature, concentrated under reduced pressure, diluted with EtOAc (40 mL), and washed with 1 M NaOH (2 × 40 mL), water (1 × 40 mL), and brine (1 × 40 mL). The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure to afford a light tan-colored solid as a crude product, which was carried onto the next step without purification (2.36 g, 73%). 1H NMR (500 MHz, CDCl3) δ 8.54 (d, J = 1.9 Hz, 1H), 8.27 (dd, J = 8.8, 1.9 Hz, 1H), 8.16–8.12 (m, 1H), 8.04 (dt, J = 8.8, 0.7 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 3.98 (s, 3H), 2.77 (s, 3H). HRMS (ESI+): calcd for C12H12NO2 (M + H)+, 202.0868; found 202.0863.
To a suspension of selenium dioxide (0.873 g, 7.87 mmol) in anhydrous 1,4-dioxane (11 mL) under argon at room temperature, methyl 2-methylquinoline-6-carboxylate (1.44 g, 7.16 mmol) was added in one portion and the resulting suspension was allowed to stir at 80 °C for 18 h. The reaction was allowed to cool to room temperature, filtered through celite, and concentrated under vacuo to afford an orange solid as a crude product, which was purified by column chromatography on silica gel using a gradient of 10–20% EtOAc in petroleum ether to afford the clean product as a pale yellow amorphous solid (1.28 g, 83%). 1H NMR (500 MHz, CDCl3) δ 10.26 (d, J = 0.6 Hz, 1H), 8.68 (d, J = 1.6 Hz, 1H), 8.45 (d, J = 8.7 Hz, 1H), 8.42 (dd, J = 8.7, 1.6 Hz, 1H), 8.32 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 4.04 (s, 3H). HRMS (ESI+): calcd for C12H10NO3 (M + H)+, 216.0660; found 216.0658.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(pyrrolidin-1-ylmethyl)quinoline-6-carboxamide (11)
Pyrrolidine (0.144 mL, 1.74 mmol) was added to a suspension of methyl 2-formylquinoline-6-carboxylate (0.250 g, 1.16 mmol) in anhydrous DCM (5 mL). The reaction mixture was allowed to stir at room temperature for 6 h. Then, sodium triacetoxyborohydride (0.369 g, 1.74 mmol) was added in one portion and the reaction mixture was stirred overnight at room temperature. The reaction mixture was diluted with DCM (5 mL) and washed with NaHCO3 saturated aqueous solution (1 × 10 mL). The two layers were separated, and the aqueous phase was extracted with DCM (1 × 10 mL). The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography using a gradient of 2–5% MeOH in DCM to afford the title compound as a light brown amorphous solid (225 mg, 71%). 1H NMR (500 MHz, CDCl3) δ 8.58 (d, J = 1.9 Hz, 1H), 8.32–8.24 (m, 2H), 8.10 (d, J = 8.8 Hz, 1H), 7.85 (br s, 1H), 4.14 (br s, 2H), 4.00 (s, 3H), 2.83 (br s, 4H), 1.94 (br s, 4H). HRMS (ESI+): calcd for C16H19N2O2 (M + H)+, 271.1441; found 271.1444.
Aqueous NaOH solution (1.02 M) (2.29 mL, 2.33 mmol) was added to a solution of methyl 2-(pyrrolidin-1-ylmethyl)quinoline-6-carboxylate (0.210 g, 0.777 mmol) in tetrahydrofuran (THF, 3 mL), followed by MeOH (1 mL) to ensure a homogeneous solution. The reaction mixture was stirred at room temperature overnight. Then, the reaction mixture was heated to 35 °C and a further 1.25 mL of NaOH aqueous solution (1.02 M) was added and the reaction mixture was allowed to stir overnight. The reaction mixture was concentrated to remove THF and MeOH. The remaining aqueous layer was washed with EtOAc (1 × 5 mL) and acidified to pH 3 with 2 M aqueous HCl. A precipitate was formed and filtered off. The filtrate was then concentrated to dryness to afford the title compound as a brown solid, which was carried onto the next step without purification (630 mg, contains NaCl, quantitative yield assumed for the next synthetic step). LCMS (ESI+): tR = 1.42 min, m/z = 257 (M + H)+.
2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) (0.323 g, 0.850 mmol) was added to a solution of 2-(pyrrolidin-1-ylmethyl)quinoline-6-carboxylic acid (0.199 g, 0.680 mmol) and N,N-diisopropylethylamine (0.594 mL, 3.40 mmol) in anhydrous DMF (4 mL). The reaction mixture was stirred for 5 min before N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (0.193 g, 0.680 mmol) was added. The reaction mixture was allowed to stir at room temperature overnight. Then, a further portion of N,N-diisopropylethylamine (263 μL) and HATU (258 mg) was added and the resulting mixture was allowed to stir for 6 h. The reaction mixture was diluted with water (8 mL), and the resulting precipitate was isolated by filtration and washed with water. The residue was purified by column chromatography using a gradient of 5–12% MeOH in DCM to afford 65 mg of a semicrude product as an orange-brown solid. Repurification by semipreparative HPLC afforded the title compound as a pale yellow amorphous solid (27 mg, 6.7% over two steps). 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H), 10.08 (s, 1H), 8.63 (d, J = 2.0 Hz, 1H), 8.48 (d, J = 8.5 Hz, 1H), 8.26 (dd, J = 8.8, 2.1 Hz, 1H), 8.16 (s, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.88 (d, J = 2.2 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.59 (dd, J = 8.2, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.25 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.34–4.27 (m, 4H), 3.94 (s, 2H), 2.61–2.52 (m, 4H), 2.25 (s, 3H), 1.77–1.73 (m, 4H) (formic acid salt). 13C NMR (126 MHz, DMSO-d6) δ 165.39, 164.82, 163.84, 162.24, 148.59, 146.81, 143.39, 137.96, 137.78, 136.69, 132.39, 130.60, 129.22, 129.16, 128.64, 128.45, 128.14, 126.67, 122.22, 121.66, 119.06, 118.65, 117.30, 117.12, 65.37, 64.86, 64.48, 62.29, 54.29, 23.74, 17.94. HRMS (ESI+): calcd for C31H31N4O4 (M + H)+, 523.2340; found 523.2342.
Racemic-N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-((2-methylpyrrolidin-1-yl)methyl)quinoline-6-carboxamide (13)
To a solution of methyl 2-formylquinoline-6-carboxylate (150 mg, 0.697 mmol) in anhydrous DCM (7 mL), 2-methylpyrrolidine (0.213 mL, 2.09 mmol) was added dropwise at room temperature and the resulting mixture was stirred for 2.5 h. Then, sodium triacetoxyborohydride (443 mg, 2.09 mmol) was added in one portion and the resulting mixture was stirred overnight at room temperature. The reaction mixture was diluted with DCM (10 mL) and washed with a NaHCO3 saturated aqueous solution (20 mL). The aqueous phase was extracted with DCM (3 × 10 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to afford a yellow oil as a crude product, which was carried onto the next step without purification (194 mg). 1H NMR (500 MHz, CDCl3) δ 8.51 (d, J = 2.0 Hz, 1H), 8.22 (dd, J = 8.8, 2.0 Hz, 1H), 8.19–8.12 (m, 1H), 8.05 (dt, J = 8.9, 0.8 Hz, 1H), 7.67 (d, J = 8.5 Hz, 1H), 4.23 (d, J = 14.1 Hz, 1H), 3.94 (s, 3H), 3.57 (d, J = 14.1 Hz, 1H), 2.99–2.91 (m, 1H), 2.61–2.50 (m, 1H), 2.27 (q, J = 8.9 Hz, 1H), 2.07–1.86 (m, 1H), 1.79–1.57 (m, 2H), 1.45 (dddd, J = 12.5, 10.7, 8.5, 6.1 Hz, 1H), 1.13 (d, J = 6.0 Hz, 3H). LCMS (ESI+): tR = 0.88 min, m/z = 285, (M + H)+.
To a solution of methyl 2-((2-methylpyrrolidin-1-yl)methyl)quinoline-6-carboxylate (194 mg, 0.682 mmol) in anhydrous THF (3.2 mL), 2 M aqueous NaOH solution (1.70 mL, 3.41 mmol) was added dropwise and MeOH (1.3 mL) was added to increase the miscibility of the two layers. The resulting red/brown solution was allowed to stir at 20 °C for 3 h. The reaction mixture was concentrated under reduced pressure, and the remaining aqueous layer was acidified to pH 3 with 1 M aqueous HCl and then washed with EtOAc (1 × 5 mL). The organic phase was discarded, and the aqueous phase was concentrated under reduced pressure to afford a beige amorphous solid as a crude product, which was carried onto the next step without purification. HRMS (ESI+): calcd for C16H19N2O2 (M + H)+, 272.1472; found 272.1468.
To a solution of 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) (176 mg, 0.462 mmol), 2-((2-methylpyrrolidin-1-yl)methyl)quinoline-6-carboxylic acid hydrochloride (100 mg, 0.326 mmol) in anhydrous DMF (2.5 mL) with N,N-diisopropylethylamine (0.322 mL, 1.85 mmol), N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (105 mg, 0.370 mmol) was added in one portion and the resulting mixture was allowed to stir at 20 °C under an inert argon atmosphere for 18 h. The reaction mixture was poured onto water to afford a light brown precipitate, which was washed with water. The crude product was purified by column chromatography using a gradient of 0–20% EtOAc in DCM. A second purification by column chromatography using a gradient of 0–10% MeOH in DCM + 1% NH3 in MeOH afforded the title compound as a light brown amorphous solid (24 mg, ∼12%). 1H NMR (500 MHz, MeOD) δ 8.69–8.55 (m, 1H), 8.49 (d, J = 8.5 Hz, 1H), 8.31 (dd, J = 8.8, 1.8 Hz, 1H), 8.17 (d, J = 8.8 Hz, 1H), 7.87–7.70 (m, 2H), 7.59–7.42 (m, 3H), 7.31 (d, J = 8.4 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 4.43 (d, J = 15.1 Hz, 1H), 4.34–4.24 (m, 4H), 3.75 (s, 1H), 3.11 (s, 1H), 2.77 (s, 1H), 2.51 (s, 1H), 2.34 (s, 3H), 2.12 (s, 1H), 1.83 (s, 2H), 1.64–1.49 (m, 1H), 1.27 (d, J = 5.9 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.39, 164.83, 148.55, 146.81, 143.39, 137.88, 137.79, 136.69, 132.43, 130.61, 129.22, 129.15, 128.65, 128.50, 128.14, 126.68, 122.41, 121.67, 119.07, 118.66, 117.31, 117.14, 64.86, 64.49, 60.40, 60.30, 60.08, 54.50, 32.72, 21.94, 19.24, 17.95. HRMS (ESI+): calcd for C32H32N4NaO4 (M + Na)+, 559.2316; found 559.2308.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxamide
To a solution of methyl 2-formylquinoline-6-carboxylate (150 mg, 0.697 mmol) in anhydrous DCM, 1-ethylpiperazine (0.266 mL, 2.09 mmol) was added dropwise at room temperature and the resulting mixture was allowed to stir under an inert argon atmosphere for 2.5 h. Then, sodium triacetoxyborohydride (443 mg, 2.09 mmol) was added in one portion and the resulting mixture was allowed to stir overnight at room temperature. The reaction mixture was diluted with DCM (20 mL) and quenched with a NaHCO3 saturated aqueous solution (20 mL). The aqueous phase was extracted with DCM (3 × 10 mL), and the combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to afford a yellow amorphous solid as a crude product, which was carried onto the next step without purification (197 mg). 1H NMR (500 MHz, CDCl3) δ 8.55 (d, J = 1.9 Hz, 1H), 8.31–8.15 (m, 2H), 8.08 (dt, J = 8.9, 0.7 Hz, 1H), 7.69 (d, J = 8.5 Hz, 1H), 3.97 (s, 3H), 3.85 (s, 2H), 2.57 (d, J = 47.4 Hz, 8H), 2.42 (q, J = 7.2 Hz, 2H), 1.08 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 166.67, 162.22, 149.53, 137.47, 130.66, 129.33, 128.90, 127.62, 126.49, 121.86, 65.10, 53.37, 52.76, 52.36, 52.29, 11.94.
To a solution of methyl 2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxylate (197 mg, 0.629 mmol) in THF (3.0 mL), 2 M aqueous NaOH (1.57 mL, 3.14 mmol) was added dropwise at 20 °C and MeOH (1.2 mL) was added to increase the miscibility of the two layers. The resulting red/brown solution was allowed to stir at 20 °C for 2 h. The reaction mixture was concentrated under reduced pressure to remove THF and MeOH; then, the aqueous layer was acidified to pH 3 with 1 M aqueous HCl and washed with EtOAc (3 × 5 mL). The aqueous layer was concentrated under vacuo to afford a salmon solid as a crude product, which was carried onto the next step without purification. 1H NMR (500 MHz, DMSO-d6) δ 13.37 (br s, 1H), 8.80–8.62 (m, 2H), 8.27 (dd, J = 8.9, 2.0 Hz, 1H), 8.16 (d, J = 8.8 Hz, 1H), 7.84 (d, J = 8.4 Hz, 1H), 4.56 (s, 2H), 3.62 (br s, 8H), 3.18 (br s, 2H), 1.26 (t, J = 7.3 Hz, 3H). HRMS (ESI+): calcd for C17H22N3O2 (M + H)+, 302.1764; found 302.1762.
To a solution of 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) (159 mg, 0.418 mmol) and 2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxylic acid hydrochloride salt (100 mg, 0.298 mmol) in anhydrous DMF (2.3 mL) with N,N-diisopropylethylamine (0.291 mL, 1.67 mmol), N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (95.0 mg, 0.334 mmol) was added in one portion and the resulting mixture was allowed to stir at 20 °C for 18 h. The reaction mixture was poured onto water (3 mL) to afford a pale yellow precipitate, which was washed with water (3 × 5 mL). Then, the solid was purified by flash column chromatography eluting with 20% EtOAc in DCM and then a gradient of 0–10% MeOH in DCM + 1% 7 N NH3 in MeOH to afford the title compound as a pale yellow amorphous solid (48 mg, 25%). 1H NMR (500 MHz, MeOD) δ 8.58 (d, J = 2.0 Hz, 1H), 8.45 (d, J = 8.5 Hz, 1H), 8.29 (dd, J = 8.9, 2.1 Hz, 1H), 8.13 (d, J = 8.8 Hz, 1H), 7.85–7.73 (m, 2H), 7.59–7.39 (m, 3H), 7.29 (dd, J = 8.3, 0.9 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 4.40–4.17 (m, 4H), 3.89 (s, 2H), 2.93–2.37 (m, 10H), 2.33 (s, 3H), 1.11 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 166.87, 166.54, 161.28, 148.33, 147.01, 143.45, 138.07, 136.96, 135.72, 132.24, 130.35, 130.19, 128.13, 128.01, 127.92, 127.59, 126.83, 122.13, 120.71, 119.45, 119.21, 116.76, 116.61, 64.52, 64.13, 64.01, 52.60, 52.20, 51.87, 48.44, 16.39, 10.37. HRMS (ESI+): calcd for C33H36N5O4 (M + H)+, 567.2793; found 567.2789.
N-(3-Amino-4-fluorophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide
Oxalyl chloride (18.5 mL, 211 mmol) was added to a stirred solution of 1,4-benzodioxane-6-carboxylic acid (34.6 g, 192 mmol) and pyridine (31.1 mL, 384 mmol) in anhydrous DCM (400 mL) at 0 °C. After 1 h, the reaction mixture was concentrated in vacuo. The remaining residue was redissolved in anhydrous DCM (200 mL) and concentrated in vacuo. The remaining residue was redissolved in DCM (40 mL) and added to a stirred solution of 4-fluoro-3-nitroaniline (30 g, 192 mmol) and pyridine (31.1 mL, 384 mmol) in DCM (400 mL) at 0 °C. After stirring for 16 h, the reaction mixture was concentrated in vacuo and diluted with MeOH (400 mL) and water (400 mL). A precipitate was formed, which was isolated by filtration and washed with water. The solid was dried under vacuum to afford the desired product as a yellow amorphous solid (52.2 g, 85%). 1H NMR (500 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.69 (dd, J = 6.9, 2.8 Hz, 1H), 8.13 (ddd, J = 9.1, 4.0, 2.8 Hz, 1H), 7.66–7.44 (m, 3H), 7.01 (d, J = 8.4 Hz, 1H), 4.32 (td, J = 5.3, 3.6 Hz, 4H). 13C NMR (126 MHz, DMSO-d6) δ 165.30, 150.98 (d, J = 252.69 Hz), 147.28, 143.49, 136.69 (d, J = 7.65 Hz), 136.57 (d, J = 2.77 Hz), 127.95 (d, J = 8.74 Hz), 127.19, 121.83, 119.10 (d, J = 22.18 Hz), 117.50, 117.21, 117.03 (d, J = 2.17 Hz), 64.91, 64.49. HRMS (ESI+): calcd for C15H12FN2O5 (M + H)+ 319.0725; found 319.0729.
Ammonium chloride (10.3g, 192 mmol) and iron (10.7 g, 192 mmol) were added to a mixture of N-(4-fluoro-3-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (12.228 g, 38.4 mmol) in ethanol (120 mL) and water (40 mL). The reaction was refluxed at 90 °C for 1 h. The reaction was cooled to room temperature and diluted with DCM (30 mL) and MeOH (30 mL). The resulting mixture was filtered through celite and washed with MeOH. The filtrate was concentrated under reduced pressure. The crude solid was diluted in an aqueous saturated NaHCO3 solution (150 mL) to make a slurry, which was filtered. The solid was collected, washed with water, and then diluted with toluene and dried in vacuo to afford the crude product as a beige amorphous solid, used as a crude in the next synthetic step (6.25 g). 1H NMR (500 MHz, DMSO-d6) δ 9.81 (s, 1H), 7.55–7.41 (m, 2H), 7.28 (d, J = 7.8 Hz, 1H), 7.02–6.78 (m, 3H), 5.14 (s, 2H), 4.30 (d, J = 5.6 Hz, 4H). 13C NMR (126 MHz, DMSO-d6) δ 164.63, 147.57 (d, J = 227.57 Hz), 146.62, 143.34, 136.56 (d, J = 14.52 Hz), 136.06 (d, J = 2.88 Hz), 128.37, 121.59, 117.24, 117.08, 114.86 (d, J = 21.65 Hz), 109.02 (d, J = 2.54 Hz), 108.65 (d, J = 5.94 Hz), 64.84, 64.48. HRMS (ESI+): calcd for C15H14FN2O3 (M + H)+ 289.0988; found 289.0992.
2-((4-Isopropylpiperazin-1-yl)methyl)quinoline-6-carboxylic Acid
Pyrrolidine (0.399 mL, 2.79 mmol) was added to a suspension of methyl 2-formylquinoline-6-carboxylate (0.200 g, 0.929 mmol) in anhydrous DCM (9 mL). The reaction mixture was stirred at room temperature for 2.5 h. Then, sodium triacetoxyborohydride (0.591 g, 2.79 mmol) was added in one portion and the reaction mixture was stirred overnight at room temperature. The reaction mixture was diluted with DCM (20 mL) and washed with a NaHCO3 saturated aqueous solution (1 × 20 mL). The two layers were separated, and the aqueous layer was extracted with DCM (3 × 10 mL). The combined organic layers were dried over MgSO4 and concentrated to afford the crude product as an amorphous orange solid (322 mg). 1H NMR (500 MHz, CDCl3) δ 8.54 (d, J = 1.9 Hz, 1H), 8.25 (dd, J = 8.8, 1.9 Hz, 1H), 8.18 (dd, J = 8.6, 0.7 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 7.69 (d, J = 8.5 Hz, 1H), 3.96 (s, 3H), 3.84 (s, 2H), 2.67–2.52 (m, 9H), 1.04 (s, 3H), 1.03 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 166.81, 162.43, 149.68, 137.55, 130.78, 129.46, 129.00, 127.71, 126.60, 122.00, 77.42, 77.16, 76.91, 65.25, 54.56, 53.88, 52.48, 48.78, 18.77. HRMS (ESI+): calcd for C19H26N3O2 (M + H)+ 328.2020; found 328.2031.
To a solution of methyl 2-((4-isopropylpiperazin-1-yl)methyl)quinoline-6-carboxylate (320 mg, 0.977 mmol) in THF (6.0 mL), 2 M aqueous NaOH (2.44 mL, 4.89 mmol) was added dropwise at 20 °C and MeOH (2.4 mL) was added to increase the miscibility of the two layers. The resulting red/brown solution was allowed to stir at 20 °C for 2 h. The reaction mixture was concentrated under reduced pressure to remove THF and MeOH; then, the aqueous layer was acidified to pH 3 with 1 M aqueous HCl and washed with EtOAc (3 × 5 mL). The aqueous layer was concentrated under vacuum to afford a salmon solid as a crude product, which was carried onto the next step without purification (306 mg, contains NaCl). 1H NMR (500 MHz, DMSO-d6) δ 8.73 (d, J = 1.9 Hz, 1H), 8.71 (d, J = 8.6 Hz, 1H), 8.27 (dd, J = 8.7, 1.9 Hz, 1H), 8.18 (d, J = 8.8 Hz, 1H), 7.89 (dd, J = 8.5, 1.8 Hz, 1H), 3.74–3.41 (m, 11H), 1.30 (m, 6H). LCMS (ESI+): tR = 0.70 min, m/z = 314, (M + H)+.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((4-isopropylpiperazin-1-yl)methyl)quinoline-6-carboxamide
N-(3-Amino-4-fluorophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (100 mg, 0.347 mmol), 2-((4-isopropylpiperazin-1-yl)methyl)quinoline-6-carboxylic acid hydrochloride (130 mg, 0.372 mmol), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 166 mg, 0.867 mmol) were dissolved in anhydrous DMF (2.5 mL); then, pyridine (0.140 mL, 1.73 mmol) was added dropwise and the resulting mixture was allowed to stir at 20 °C for 20 h. A further portion of 2-((4-isopropylpiperazin-1-yl)methyl)quinoline-6-carboxylic acid (130 mg, 0.416 mmol), EDC (166 mg, 0.867 mmol), and pyridine (0.140 mL, 1.73 mmol) was added, and the resulting mixture was allowed to stir for a total of 72 h at 20 °C. The reaction was quenched with water (5 mL) and extracted with DCM/MeOH 9/1 (3 × 5 mL). Purification by column chromatography using a gradient of 0–10% MeOH in DCM + 1% 7 N NH3 in MeOH followed by trituration in diethyl ether afforded the title compound as an orange amorphous solid (50 mg, 9.2% over three steps). 1H NMR (500 MHz, DMSO-d6) δ 10.41 (br s, 1H), 10.21 (br s, 1H), 8.65 (s, 1H), 8.49 (d, J = 8.78 Hz, 1H), 8.25 (dd, J = 8.78, 1.88 Hz, 1H), 8.14 (dd, J = 6.90, 2.51 Hz, 1H), 8.08 (d, J = 8.78 Hz, 1H), 7.73 (d, J = 8.78 Hz, 1H), 7.68–7.63 (m, 1H), 7.55 (d, J = 2.51 Hz, 1H), 7.52 (dd, J = 8.78, 1.88 Hz, 1H), 7.29 (app t, J = 9.28 Hz, 1H), 6.99 (d, J = 8.78 Hz, 1H), 4.36–4.27 (m, 4H), 3.79 (br s, 2H), 3.15–2.34 (m, 9H), 0.99 (br s, 6H). 13C NMR (126 MHz, MeOD) δ 166.69, 166.62, 161.22, 152.52 (d, J = 245.41 Hz), 151.49, 148.39, 147.12, 143.49, 138.19, 134.88 (d, J = 3.22 Hz), 131.99, 128.21, 127.99, 127.37, 126.82, 125.27 (d, J = 11.96 Hz), 122.18, 120.76, 119.62 (d, J = 7.97 Hz), 119.01, 116.80, 116.64, 115.28 (d, J = 21.26 Hz), 64.54, 64.15, 63.80, 55.27, 52.30, 29.34, 16.98. HRMS (ESI+): calcd for C33H35FN5O4 (M + H)+, 584.2668; found 584.2636.
Synthetic Route II
Ethyl 2-((Tosyloxy)methyl)quinoline-6-carboxylate
To a stirred solution of 2-methylquinoline-6-carboxylic acid (2.00 g, 10.7 mmol) in ethanol (50 mL) was added sulfuric acid (0.4 mL, 10.7 mmol). The reaction was heated to 80 °C under argon for 22 h. The solvent was removed in vacuo. The resulting residue was taken up in water (100 mL). The solution was basified (∼pH 10) by the addition of 2 M aqueous NaOH solution. The resulting precipitate was collected by filtration and washed with copious water and then dried under vacuum to afford a pale pink amorphous solid (1.57 g, 68%). 1H NMR (500 MHz, CDCl3) δ 8.56 (d, J = 1.9 Hz, 1H), 8.29 (dd, J = 8.8, 1.9 Hz, 1H), 8.19–8.13 (m, 1H), 8.05 (dt, J = 8.8, 0.7 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 4.46 (q, J = 7.1 Hz, 2H), 2.79 (s, 3H), 1.46 (t, J = 7.1 Hz, 3H). LCMS (ESI+): tR = 2.24 min, m/z 216 (M + H)+.
3-Chloroperbenzoic acid (0.695 g, 3.02 mmol) was added to a solution of ethyl 2-methylquinoline-6-carboxylate (0.5 g, 2.32 mmol) in anhydrous DCM (7 mL) at 0 °C. The reaction mixture was then allowed to warm to room temperature and stirred overnight. The orange reaction mixture was washed with 10% aqueous Na2SO3 solution (1 × 10 mL) and saturated aqueous NaHCO3 solution (1 × 10 mL). The two layers were separated, and the aqueous layer was diluted with brine and extracted with DCM (3 × 10 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated in vacuo. The crude orange oil was crystallized from EtOAc/PE. The solid was isolated by filtration and washed with PE/EtOAc (3/1 mixture). A second product fraction was isolated after the concentration of the filtrate. This solid was triturated with PE/EtOAc (∼4/1) and isolated by filtration. The title compound was obtained as a pale orange amorphous solid (381 mg, 71%). 1H NMR (500 MHz, CDCl3) δ 8.82 (d, J = 9.1 Hz, 1H), 8.58 (d, J = 1.7 Hz, 1H), 8.33 (dd, J = 9.1, 1.8 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.39 (d, J = 8.5 Hz, 1H), 4.46 (q, J = 7.1 Hz, 2H), 2.74 (s, 3H), 1.45 (t, J = 7.1 Hz, 3H). LCMS (ESI+): tR = 2.32 min, m/z 232.10 (M + H)+.
To a solution of ethyl 2-methylquinoline-6-carboxylate N-oxide (0.274 g, 1.19 mmol) in anhydrous acetonitrile (10 mL) at 0 °C, K2CO3 (0.246 g, 1.78 mmol) was added in one portion, followed by p-toluenesulfonyl chloride (0.271 g, 1.42 mmol). The reaction mixture was stirred at 0 °C for 4 h. The reaction mixture was diluted with saturated aqueous NaHCO3 solution and extracted with EtOAc (2 × 10 mL). The organic layer was washed with water (1 × 10 mL) and brine (1 × 10 mL), dried over MgSO4, filtered, and concentrated in vacuo. The crude (dark blue-green solid) was purified by column chromatography using a gradient of 16–40% EtOAc in petroleum ether to afford an orange amorphous solid (186 mg, 41%). 1H NMR (500 MHz, CDCl3) δ 8.58 (d, J = 1.9 Hz, 1H), 8.34–8.25 (m, 2H), 8.01 (d, J = 8.8 Hz, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.5 Hz, 1H), 7.33 (d, J = 8.1 Hz, 2H), 5.32 (s, 2H), 4.45 (q, J = 7.1 Hz, 2H), 2.42 (s, 3H), 1.45 (t, J = 7.1 Hz, 3H). LCMS (ESI+): tR = 3.11 min, m/z 386.22 (M + H)+.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-((dimethylamino)methyl)quinoline-6-carboxamide (12)
A solution of ethyl 2-((tosyloxy)methyl)quinoline-6-carboxylate (62.0 mg, 0.161 mmol) in dimethylamine (2 M in THF) (0.080 mL, 0.161 mmol) was heated under microwave irradiation at 60 °C for 1 h. The reaction mixture was concentrated under reduced pressure, diluted with EtOAc, and washed with water (1 × 1 mL) and saturated aqueous NaHCO3 solution (1 × 1 mL). The aqueous phase was extracted with EtOAc (1 × 1 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford an orange oil (42 mg). 1H NMR (500 MHz, CDCl3) δ 8.57 (d, J = 1.8 Hz, 1H), 8.29 (dd, J = 8.8, 1.9 Hz, 1H), 8.23 (d, J = 8.5 Hz, 1H), 8.11 (d, J = 8.8 Hz, 1H), 7.67 (d, J = 8.5 Hz, 1H), 4.45 (q, J = 7.2 Hz, 2H), 3.79 (s, 2H), 2.35 (s, 6H), 1.45 (t, J = 7.1 Hz, 3H).
Aqueous NaOH solution (1.04 M, 0.304 mL, 0.317 mmol) was added to a solution of ethyl 2-((dimethylamino)methyl)quinoline-6-carboxylate (41.0 mg, 0.159 mmol) in THF (1 mL) and MeOH (0.3 mL). The reaction mixture was stirred at room temperature overnight. A further portion of water (0.5 mL) and aqueous NaOH (1.15 M, 0.276 mL, 0.317 mmol) was added, and the resulting mixture was allowed to stir overnight. The reaction mixture was concentrated to remove the organic solvents, diluted with water, and washed with EtOAc (1 × 1 mL). The aqueous phase was acidified to ∼pH 3 with aqueous HCl solution (2 M) and then concentrated to afford the title compound as a crude pale yellow solid, which was carried onto the next step without purification. 1H NMR (500 MHz, DMSO-d6) δ 12.11 (br s, 1H), 8.70 (d, J = 1.8 Hz, 1H), 8.65 (d, J = 8.5 Hz, 1H), 8.25 (dd, J = 8.8, 1.9 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 7.84 (d, J = 8.5 Hz, 1H), 4.42 (br s, 2H), 2.70 (s, 6H). LCMS (ESI+): tR = 0.82 min, m/z 231.11 (M + H)+.
HATU (72.0 mg, 0.189 mmol) was added to a solution of 2-((dimethylamino)methyl)quinoline-6-carboxylic acid (0.106 g, 0.151 mmol) and N,N-diisopropylethylamine (0.111 mL, 0.634 mmol) in anhydrous DMF (1.5 mL). The reaction mixture was stirred for 5 min before N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (32.0 mg, 0.113 mmol) was added. The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with water, and the resulting precipitate was isolated by filtration and washed with water. The residue was purified by column using a gradient of 4–10% MeOH in DCM to afford the title compound as an off-white amorphous solid (24 mg, 30% over 3 steps). 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H), 10.07 (s, 1H), 8.63 (d, J = 1.9 Hz, 1H), 8.48 (d, J = 8.5 Hz, 1H), 8.26 (dd, J = 8.8, 2.0 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.88 (d, J = 2.1 Hz, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.62–7.48 (m, 3H), 7.24 (d, J = 8.5 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.30 (td, J = 5.1, 3.6 Hz, 4H), 3.74 (s, 2H), 2.25 (d, J = 6.0 Hz, 9H). 13C NMR (126 MHz, DMSO-d6) δ 165.39, 164.82, 162.29, 148.58, 146.81, 143.39, 137.90, 137.78, 136.69, 132.42, 130.60, 129.22, 129.18, 128.63, 128.42, 128.14, 126.70, 122.25, 121.65, 119.06, 118.65, 117.30, 117.12, 66.13, 64.86, 64.48, 45.88, 17.93. HRMS (ESI+): calcd for C29H29N4O4 (M + H)+, 497.2183; found 497.2183.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-((4-methylpiperazin-1-yl)methyl)quinoline-6-carboxamide (15)
1-Methylpiperazine (0.058 mL, 0.519 mmol) was added to a solution of ethyl 2-((tosyloxy)methyl)quinoline-6-carboxylate (80.0 mg, 0.208 mmol) in anhydrous THF (1.5 mL). The reaction mixture was heated to reflux for 1.5 h. The reaction mixture was cooled to room temperature, stirred for 2 h, and concentrated under reduced pressure. The residue was diluted with EtOAc (2 mL) and washed with water (1 × 2 mL) and saturated aqueous NaHCO3 solution (1 × 2 mL). The aqueous phase was extracted with EtOAc (1 × 2 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated in vacuo to afford the title compound as a yellow amorphous solid, which was carried onto the next step without purification (64 mg). 1H NMR (500 MHz, CDCl3) δ 8.56 (d, J = 1.9 Hz, 1H), 8.29 (dd, J = 8.8, 1.9 Hz, 1H), 8.22 (d, J = 8.5 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 8.5 Hz, 1H), 4.45 (q, J = 7.1 Hz, 2H), 3.86 (s, 2H), 2.61 (br s, 4H), 2.49 (br s, 4H), 2.31 (s, 3H), 1.45 (t, J = 7.1 Hz, 3H). HRMS (ESI+): calcd for C18H24N3O2 (M + H)+, 314.1863; found 314.1871.
Aqueous NaOH solution (0.82 M, 0.735 mL, 0.603 mmol) was added to a solution of ethyl 2-((4-methylpiperazin-1-yl)methyl)quinoline-6-carboxylate (63.0 mg, 0.201 mmol) in THF (1 mL) and MeOH (0.25 mL), and the reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated to remove the organic solvents, diluted with water, and washed with EtOAc (1 × 1 mL). The aqueous phase was acidified with aqueous HCl solution (2 M) to pH 2–3 and then concentrated to dryness. The crude product (light brown amorphous solid) was carried onto the next step without further purification (114 mg). 1H NMR (500 MHz, DMSO-d6) δ 13.33 (br s, 1H), 11.77 (br s, 1H), 8.72 (d, J = 1.9 Hz, 1H), 8.69 (d, J = 8.5 Hz, 1H), 8.26 (dd, J = 8.8, 1.9 Hz, 1H), 8.16 (d, J = 8.9 Hz, 1H), 7.85 (d, J = 8.5 Hz, 1H), 4.53 (br s, 2H), 3.83–3.45 (br m, 8H), 2.80 (s, 3H) (hydrochloric acid salt). HRMS (ESI+): calcd for C16H20N3O2 (M + H)+, 286.1550; found 286.1553.
HATU (68.0 mg, 0.178 mmol) was added to a suspension of 2-((4-methylpiperazin-1-yl)methyl)quinoline-6-carboxylic acid (81.0 mg, 0.142 mmol) and N,N-diisopropylethylamine (0.131 mL, 0.748 mmol) in anhydrous DMF (1 mL). The reaction mixture was stirred for 4 min before N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (34.0 mg, 0.121 mmol) was added. The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with water, and the resulting precipitate was isolated by filtration and washed with water. The residue was purified by column chromatography using a gradient of 5–18% MeOH in DCM to afford the title compound as an off-white amorphous solid (36 mg, 31% over three steps). 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H), 10.08 (s, 1H), 8.63 (d, J = 1.9 Hz, 1H), 8.48 (d, J = 8.4 Hz, 1H), 8.26 (dd, J = 8.8, 2.0 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.88 (d, J = 2.1 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.59 (dd, J = 8.3, 2.2 Hz, 1H), 7.56–7.48 (m, 2H), 7.25 (d, J = 8.5 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.31 (q, J = 5.1 Hz, 4H), 3.79 (s, 2H), 2.50–2.32 (m, 8H), 2.24 (s, 3H), 2.17 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.37, 164.82, 161.97, 148.63, 146.81, 143.39, 137.93, 137.78, 136.69, 132.39, 130.61, 129.23, 129.16, 128.64, 128.42, 128.14, 126.69, 122.22, 121.66, 119.07, 118.65, 117.30, 117.12, 64.86, 64.49, 55.18, 53.35, 46.19, 40.89, 17.93. HRMS (ESI+): calcd for C32H34N5O4 (M + H)+, 552.2605; found 552.2591.
Synthetic Route III
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(2-(piperidin-1-yl)ethoxy)quinoline-6-carboxamide (17)
4-(2-Hydroxyethyl)piperidine (0.197 mL, 1.49 mmol) was added to a suspension of NaH (60%, 57.0 mg, 1.42 mmol) in anhydrous THF at 0 °C. The reaction mixture was stirred for 5 min and then allowed to warm to room temperature and stirred for 35 min before 6-bromo-2-chloroquinoline (300 mg, 1.24 mmol) was added. The reaction mixture was then heated to reflux. After 4.5 h, the reaction mixture was cooled to room temperature and diluted with first water and then saturated aqueous NaHCO3 solution. This mixture was extracted with DCM three times. The combined organic layers were washed with water, dried over MgSO4, and concentrated. The crude product was purified by column chromatography using a gradient of 4–5% MeOH in DCM to give 6-bromo-2-(2-(piperidin-1-yl)ethoxy)quinoline as a pale yellow oil (334 mg, 81%). 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 8.9 Hz, 1H), 7.87–7.84 (m, 1H), 7.72–7.65 (m, 2H), 6.95 (d, J = 8.8 Hz, 1H), 4.62 (t, J = 6.1 Hz, 2H), 2.83 (t, J = 6.1 Hz, 2H), 1.63 (m, 6H), 1.53–0.46 (m, 4H). HRMS (ESI+): calcd for C16H2079BrN2O (M + H)+ 335.0754, found 335.0787.
n-BuLi (1.62 M in hexanes, 0.742 mL, 1.21 mmol) was added dropwise to a solution of 6-bromo-2-(2-(piperidin-1-yl)ethoxy)quinoline (325 mg, 0.969 mmol) in anhydrous THF (3.25 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 50 min before solid CO2 was added. After stirring for 5 min, the reaction mixture was allowed to warm to room temperature. The reaction was quenched with water and reduced in vacuo to remove the THF. The remaining residue was diluted with water and washed with ethyl acetate. The aqueous layer was then acidified to pH 3 by the addition of aqueous 2 M HCl and concentrated to dryness to give the product as an off-white amorphous solid. The product, 2-(2-(piperidin-1-yl)ethoxy)quinoline-6-carboxylic acid hydrochloride, was used in the next synthetic step without further purification (301 mg, contains LiCl). 1H NMR (500 MHz, DMSO-d6) δ 8.59 (d, J = 2.0 Hz, 1H), 8.47 (d, J = 8.9 Hz, 1H), 8.16 (dd, J = 8.7, 2.0 Hz, 1H), 7.84 (d, J = 8.7 Hz, 1H), 7.16 (d, J = 8.9 Hz, 1H), 5.08–4.70 (m, 2H), 3.75–3.36 (m, 4H), 3.01 (tdd, J = 12.3, 9.1, 3.3 Hz, 2H), 1.92–1.75 (m, 4H), 1.74–1.55 (m, 1H), 1.38 (ddt, J = 12.7, 8.1, 4.0 Hz, 1H). HRMS (ESI+): calcd for C17H21N2O3 (M + H)+ 301.1547, found 301.1534.
HATU (111 mg, 0.293 mmol) was added to a solution of 2-(2-(piperidin-1-yl)ethoxy)quinoline-6-carboxylic acid hydrochloride (93.0 mg, contains LiCl, purity 85%) and DIPEA (0.174 mL, 0.997 mmol) in anhydrous DMF (1.5 mL). The reaction mixture was stirred for 5 min before N-(3-amino-4-methylphenyl)2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (50.0 mg, 0.176 mmol) was added. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was then diluted with water, and the resulting precipitate was isolated by filtration and washed with water. The crude product was purified by column chromatography using a gradient of 3.5–10% MeOH in DCM to afford the title compound as a white amorphous solid (75.0 mg, 75%). 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 8.56 (d, J = 2.1 Hz, 1H), 8.38 (d, J = 8.8 Hz, 1H), 8.22 (dd, J = 8.8, 2.1 Hz, 1H), 7.90–7.79 (m, 2H), 7.58 (dd, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.57 (s, 2H), 4.37–4.24 (m, 4H), 2.75 (s, 2H), 2.50 (p, J = 1.8 Hz, 4H), 2.23 (s, 3H), 1.51 (d, J = 7.6 Hz, 4H), 1.38 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.39, 164.81, 163.15, 148.00, 146.80, 143.39, 140.71, 137.75, 136.79, 130.57, 130.52, 129.23, 128.88, 128.57, 128.15, 127.21, 124.48, 121.65, 119.08, 118.58, 117.30, 117.12, 114.47, 64.86, 64.48, 63.73, 57.50, 54.76, 25.95, 24.30, 17.95. HRMS (ESI+): calcd for C33H35N4O5 (M + H)+ 567.2602, found 567.2635.
2-(2-(Piperidin-1-yl)propoxy)quinoline-6-carboxylic Acid
1-Piperidinepropanol (0.235 mL, 1.55 mmol) was added to a suspension of NaH (60%, 59.0 mg, 1.49 mmol) in anhydrous THF (4 mL) at 0 °C. The reaction mixture was stirred for 5 min and then allowed to warm to room temperature and stirred for 40 min before 6-bromo-2-chloroquinoline (300 mg, 1.24 mmol) was added. The reaction mixture was then heated to reflux. After 6 h, the reaction mixture was cooled to room temperature and concentrated to remove the THF. The remaining residue was diluted with water first and then saturated aqueous NaHCO3 solution. This mixture was extracted with DCM three times. The combined organic layers were washed with water, dried over MgSO4, and concentrated. The crude product was purified by column chromatography using a gradient of 2.5–6% of MeOH in DCM to give 6-bromo-2-(2-(piperidin-1-yl)propoxy)quinoline as a pale yellow oil that solidified (329 mg, 76%). 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 8.9 Hz, 1H), 7.86 (d, J = 1.9 Hz, 1H), 7.73–7.61 (m, 2H), 6.91 (d, J = 8.8 Hz, 1H), 4.50 (t, J = 6.5 Hz, 2H), 2.63–2.35 (m, 6H), 2.05 (p, J = 6.7 Hz, 2H), 1.75–1.55 (m, 4H), 1.47 (m, 2H). HRMS (ESI+): calcd for C17H2279BrN2O (M + H)+ 351.0893, found 351.0883.
n-BuLi (1.56 M in hexanes, 0.732 mL, 1.14 mmol) was added dropwise to a solution of 6-bromo-2-(2-(piperidin-1-yl)propoxy)quinoline (319 mg, 0.913 mmol) in anhydrous THF (3.1 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 45 min before solid CO2 was added. After stirring for 5 min, the reaction mixture was allowed to warm to room temperature. The reaction was quenched with water and reduced in vacuo to remove the THF. The remaining residue was diluted with water and washed with ethyl acetate. The aqueous layer was then acidified to pH 3 by the addition of aqueous 2 M HCl and concentrated to dryness to give the product as an off-white amorphous solid. The product, 2-(2-(piperidin-1-yl)propoxy)quinoline-6-carboxylic acid hydrochloride, was used in the next synthetic step without further purification (347 mg, contains LiCl). 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 8.9 Hz, 1H), 7.86 (d, J = 1.9 Hz, 1H), 7.73–7.61 (m, 2H), 6.91 (d, J = 8.8 Hz, 1H), 4.50 (t, J = 6.5 Hz, 2H), 2.63–2.35 (m, 6H), 2.05 (p, J = 6.7 Hz, 2H), 1.75–1.55 (m, 4H), 1.47 (s, 2H). HRMS (ESI+): calcd for C18H23N2O3 (M + H)+ 315.1703, found 315.1686.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(2-(piperidin-1-yl)propoxy)quinoline-6-carboxamide
HATU (89.0 mg, 0.234 mmol) was added to a solution of 2-(2-(piperidin-1-yl)propoxy)quinoline-6-carboxylic acid hydrochloride (76.0 mg, 0.188 mmol, contains LiCl) and DIPEA (0.139 mL, 0.797 mmol) in anhydrous DMF (1.35 mL). The reaction mixture was stirred for 5 min before N-(3-amino-4-methylphenyl)2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (40.0 mg, 0.141 mmol) was added. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was then diluted with water, and the resulting precipitate was isolated by filtration and washed with water. The crude product was purified by column chromatography using a gradient of 2.5–15% MeOH in DCM to give the title compound as an off-white amorphous solid (53 mg, 65%). 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 8.57 (d, J = 2.1 Hz, 1H), 8.38 (d, J = 8.9 Hz, 1H), 8.22 (dd, J = 8.7, 2.1 Hz, 1H), 7.94–7.80 (m, 2H), 7.58 (dd, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.49 (t, J = 6.5 Hz, 2H), 4.35–4.26 (m, 4H), 2.50 (m, 6H), 2.23 (s, 3H), 2.01 (m, 2H), 1.56 (m, 4H), 1.42 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.38, 164.81, 163.24, 148.04, 146.80, 143.39, 140.69, 137.75, 136.78, 130.57, 130.51, 129.23, 128.89, 128.60, 128.14, 127.17, 124.47, 121.65, 119.09, 118.59, 117.30, 117.12, 114.41, 64.86, 64.66, 64.48, 55.57, 54.13, 25.76, 25.45, 23.47, 17.94. HRMS (ESI+): calcd for C34H37N4O5 (M + H)+ 581.2759, found 581.2712.
1-(3-(Pyrrolidin-1-yl)propoxy)quinoline-6-carboxylic Acid
Pyrrolidine (1.00 mL, 12.0 mmol) was added to a suspension of potassium carbonate (1.29 g, 9.35 mmol) and 3-bromopropanol (0.65 mL, 7.19 mmol) in anhydrous THF (3 mL) at 0 °C. The reaction mixture was then allowed to warm to room temperature and stirred overnight. The reaction was then diluted with ethyl acetate and filtered through a pad of silica gel. The filtrate was concentrated to give 3-(pyrrolidin-1-yl)propan-1-ol as a colorless oil (645 mg, 69%). 1H NMR (500 MHz, CDCl3) δ 3.88–3.73 (m, 2H), 2.82–2.67 (m, 2H), 2.66–2.51 (m, 4H), 1.81–1.75 (m, 4H), 1.75–1.69 (m, 2H).
NaH (60%, 59 mg, 1.476 mmol) was added to a solution of 3-(pyrrolidin-1-yl)propan-1-ol (199 mg, 1.540 mmol) in anhydrous THF at 0 °C. The reaction mixture was stirred for 5 min, then allowed to warm to room temperature and stirred for 35 min before 6-bromo-2-chloroquinoline (311 mg, 1.28 mmol) was added. The reaction mixture was then heated to reflux. After 3.5 h, the reaction mixture was cooled to room temperature and concentrated to remove the THF. The remaining residue was diluted with water first and then saturated aqueous NaHCO3 solution. This mixture was extracted with DCM three times. The combined organic layers were washed with water, dried over MgSO4, and concentrated. The crude product was purified by column chromatography using a gradient of 3–6% MeOH in DCM to give 6-bromo-2-(3-(pyrrolidin-1-yl)propoxy)quinoline as an off-white amorphous solid (290 mg, 67%). 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 8.8 Hz, 1H), 7.87–7.84 (m, 1H), 7.73–7.63 (m, 2H), 6.92 (d, J = 8.8 Hz, 1H), 4.53 (t, J = 6.5 Hz, 2H), 2.70–2.62 (m, 2H), 2.57 (s, 5H), 2.14–2.02 (m, 2H), 1.82 (m, 4H). HRMS (ESI+): calcd for C16H2079BrN2O (M + H)+ 335.0754, found 335.0763.
n-BuLi (2.22 M in hexanes, 0.472 mL, 1.048 mmol) was added dropwise to a solution of 6-bromo-2-(3-(pyrrolidin-1-yl)propoxy)quinoline (281 mg, 0.838 mmol) in anhydrous THF (6 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 40 min before solid CO2 was added. After stirring for 5 min, the reaction mixture was allowed to warm to room temperature. The reaction was quenched with water and reduced in vacuo to remove the THF. The remaining residue was diluted with water and washed with ethyl acetate. The precipitate was carried through in the aqueous layer. The aqueous layer was then acidified to pH 3 by the addition of aqueous 2 M HCl. At this point, the precipitate dissolved and the solution was concentrated to dryness. The crude product was triturated with acetonitrile and dried to give the product as a dull yellow solid. The product, 2-(3-(pyrrolidin-1-yl)propoxy)quinoline-6-carboxylic acid hydrochloride, was used in the next synthetic step without further purification (298 mg, contains LiCl). 1H NMR (500 MHz, DMSO-d6) δ 8.57 (d, J = 2.0 Hz, 1H), 8.52–8.35 (m, 1H), 8.15 (dd, J = 8.7, 2.0 Hz, 1H), 7.82 (dd, J = 8.7, 0.7 Hz, 1H), 7.10 (d, J = 8.9 Hz, 1H), 4.53 (t, J = 6.2 Hz, 2H), 3.55 (q, J = 5.3 Hz, 2H), 3.36–3.24 (m, 2H), 3.08–2.89 (m, 2H), 2.29–2.13 (m, 2H), 1.99 (q, J = 7.3, 6.5 Hz, 2H), 1.93–1.79 (m, 2H). HRMS (ESI+): calcd for C17H21N2O3 (M + H)+ 301.15467, found 301.1501.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(3-(pyrrolidin-1-yl)propoxy)quinoline-6-carboxamide
HATU (121 mg, 0.319 mmol) was added to a solution of 2-(3-(pyrrolidin-1-yl)propoxy)quinoline-6-carboxylic acid hydrochloride (100 mg, contains LiCl) and DIPEA (0.190 mL, 1.085 mmol) in anhydrous DMF (10 mL). The reaction mixture was stirred for 5 min before N-(3-amino-4-methylphenyl)2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (54.0 mg, 0.192 mmol) was added followed by anhydrous DMF (1.5 mL) to rinse the vial. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was then diluted with water, and the resulting precipitate was isolated by filtration and washed with water. The crude product was purified by column chromatography using a gradient of 5–18% MeOH in DCM to afford the title compound as an off-white amorphous solid (65 mg, 60%). 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 8.57 (d, J = 2.0 Hz, 1H), 8.38 (d, J = 8.8 Hz, 1H), 8.22 (dd, J = 8.8, 2.1 Hz, 1H), 7.88–7.84 (m, 2H), 7.58 (dd, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.2 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.24 (d, J = 8.3 Hz, 1H), 7.10 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.49 (t, J = 6.6 Hz, 2H), 4.33–4.28 (m, 4H), 2.65 (br s, 2H), 2.54 (br s, 4H), 2.23 (s, 3H), 1.99 (p, J = 6.8 Hz, 2H), 1.72 (br s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 164.95, 164.36, 162.86, 147.62, 146.35, 142.94, 140.21, 137.31, 136.34, 130.12, 130.03, 128.78, 128.42, 128.15, 127.69, 126.73, 124.01, 121.21, 118.63, 118.13, 116.85, 116.67, 113.98, 64.40, 64.33, 64.03, 53.59, 52.24, 27.57, 23.07, 17.49. HRMS (ESI+): calcd for C33H35N4O5 (M + H)+ 567.2602, found 567.2698.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxamide
2-Fluoro-5-nitroaniline (27.3 mg, 0.175 mmol), 2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxylic acid hydrochloride (50 mg, 0.175 mmol), and EDC (67.0 mg, 0.349 mmol) were dissolved in anhydrous DMF (1.0 mL) and pyridine (0.070 mL, 0.873 mmol) was added dropwise. The mixture was stirred at 20 °C for 5 h. The reaction mixture was diluted with DCM/MeOH and washed with saturated aqueous NaHCO3. Purification by column chromatography using a gradient of 0–50% MeOH in DCM afforded the desired product N-(2-fluoro-5-nitrophenyl)-2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxamide as a pale yellow solid (40 mg). LCMS (ESI+): tR = 1.13 min, m/z 425.16 (M + H)+.
N-(2-fluoro-5-nitrophenyl)-2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxamide (40 mg, 0.094 mmol), ammonium chloride (35.3 mg, 0.66 mmol), and iron powder (36.8 mg, 0.66 mmol) were suspended in a mixture of ethanol (1.5 mL) and water (0.5 mL), and the resulting mixture was heated at 90 °C for 1 h. Then, the reaction mixture was cooled to room temperature and filtered through celite. The solvents were removed under vacuo to afford N-(5-amino-2-fluorophenyl)-2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxamide as a light beige solid (37.2 mg). LCMS (ESI+): tR = 0.79 min, m/z 395.19 (M + H)+.
N-(5-Amino-2-fluorophenyl)-2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxamide (37.2 mg, 0.094 mmol), 2,3-dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (16.90 mg, 0.094 mmol), and EDC (45.0 mg, 0.235 mmol) were dissolved in anhydrous DMF (0.6 mL); then, pyridine (0.038 mL, 0.469 mmol) was added dropwise and the resulting mixture was stirred at 20 °C for 18 h. The reaction mixture was diluted with DCM/MeOH and washed with water (5 mL) to afford a pale yellow solid as a crude product, which was purified by column chromatography using a gradient of 0–10% MeOH in DCM. This residue was then repurified via semipreparative TLC (DCM/MeOH 9/1) to afford the title compound as a white amorphous solid (5 mg, 9.3% over three steps). 1H NMR (500 MHz, MeOD) δ 8.48 (d, J = 2.04 Hz, 1H), 8.29 (d, J = 8.85 Hz, 1H), 8.20 (dd, J = 8.17, 2.04 Hz, 1H), 8.17 (dd, J = 6.81, 2.72 Hz, 1H), 7.92 (d, J = 8.17 Hz, 1H), 7.62–7.56 (m, 1H), 7.50 (d, J = 2.04 Hz, 1H), 7.48 (dd, J = 8.17, 2.04 Hz, 1H), 7.23 (app t, J = 9.79 Hz, 1H), 7.09 (d, J = 8.85 Hz, 1H), 6.96 (d, J = 8.17 Hz, 1H), 4.70 (t, J = 5.32 Hz, 2H), 4.36–4.28 (m, 4H), 3.08 (t, J = 5.25 Hz, 2H), 2.84–2.76 (m, 4H), 1.91–1.85 (m, 4H). 13C NMR (126 MHz, MeOD) δ 166.91, 166.59, 163.24, 152.49 (d, J = 249.95 Hz), 148.40, 147.13, 143.51, 139.86, 134.85 (d, J = 2.13 Hz), 129.65, 127.92, 127.43, 127.03, 125.60, 125.41 (d, J = 11.60 Hz), 124.46, 120.76, 119.54 (d, J = 9.28 Hz), 119.03, 116.81, 116.64, 115.23 (d, J = 18.56 Hz), 113.86, 64.54, 64.15, 64.08, 54.37, 54.13, 22.80. HRMS (ESI+): calcd for C31H30FN4O5 (M + H)+, 557.2195; found 557.2196.
N-(3-Amino-4-chlorophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide
Oxalyl chloride (0.141 mL, 1.67 mmol) was added dropwise to a solution of 1,4-benzodioxane-6-carboxylic acid (250 mg, 1.39 mmol) and N,N-dimethylformamide (3 μL, 0.035 mmol) in anhydrous DCM (7 mL) under an inert atmosphere at room temperature. Effervescence was observed, and the reaction was stirred for 2 h. The reaction mixture was concentrated, anhydrous DCM was added (10 mL), and the reaction was concentrated again. The residue was redissolved in anhydrous DCM (3 mL, followed by 3 mL, then 1 mL to rinse out the flask) and added dropwise to a solution of 4-chloro-3-nitroaniline (239 mg, 1.39 mmol) and pyridine (0.22 mL, 2.78 mmol) in anhydrous DCM (7 mL). The reaction was stirred for 4 h. The solvent was removed in vacuo, and the resulting residue was taken up in a small volume of MeOH. The solid was precipitated by the addition of water. The precipitate was isolated by filtration, washed well with water, and dried under high vacuum to afford the product as a dark yellow amorphous solid (417 mg, 90%).
A mixture of N-(4-chloro-3-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (188 mg, 0.562 mmol), ammonium chloride (210 mg, 3.93 mmol), and iron powder (220 mg, 3.93 mmol) in ethanol (2.9 mL) and water (0.95 mL) was heated to reflux overnight. The reaction was allowed to cool to room temperature and filtered through celite, eluting with a mixture of EtOH in EtOAc. The reaction mixture was concentrated in vacuo, and the resulting residue was partitioned between saturated aqueous NaHCO3 solution and EtOAc. The organic layer was washed with water and brine, dried over MgSO4, and concentrated in vacuo to afford the crude product as a light brown amorphous solid (156 mg, 91%).
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxamide
2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxylic acid hydrochloride (75.0 mg, 0.232 mmol) was suspended in thionyl chloride (2 mL), and the reaction mixture was heated to 60 °C for 4 h. The reaction mixture was allowed to cool, and the thionyl chloride was removed in vacuo. The residue was redissolved in anhydrous DCM, and then the solvent was removed in vacuo. This procedure was repeated twice. The acid chloride was resuspended in anhydrous DCM (2 mL), then N-(3-amino-4-chlorophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (78.0 mg, 0.256 mmol), followed by triethylamine (0.16 mL, 1.16 mmol) was added. Not all of the reagents were fully solubilized; therefore, anhydrous dioxane was added (1 mL); however, this did not lead to an improvement. The reaction mixture was allowed to stir at room temperature overnight. The reaction mixture was concentrated in vacuo, and the resulting residue was purified by Isolute SCX-II chromatography (eluting with MeOH, followed by 10% 2 M NH3 in MeOH). The crude product was further purified by column chromatography using a gradient of 0–10% MeOH in DCM, followed by purification using preparative TLC eluting with 5% MeOH in DCM. The preparative TLC elution was carried out twice to afford the title compound as a white amorphous solid (0.9 mg, 0.7%). 1H NMR (500 MHz, MeOD) δ 8.54 (d, J = 2.0 Hz, 1H), 8.37 (d, J = 8.9 Hz, 1H), 8.27–8.24 (m, 2H), 7.96 (d, J = 8.8 Hz, 1H), 7.64 (dd, J = 8.8, 2.5 Hz, 1H), 7.52–7.47 (m, 3H), 7.17 (d, J = 8.9 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 4.35–4.29 (m, 4H), 3.73–3.67 (m, 2H), 3.49–3.41 (m, 2H), 2.15–2.10 (s, 4H), 1.36–1.30 (m, 4H). HRMS (ESI+): calcd for C31H3035ClN4O5 (M + H)+ 573.1905, found 573.1997.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-(3-(pyrrolidin-1-yl)propoxy)quinoline-6-carboxamide
N-(3-Amino-4-fluorophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (100 mg, 0.347 mmol), 2-(3-(pyrrolidin-1-yl)propoxy)quinoline-6-carboxylic acid (156 mg, 0.520 mmol), and EDC (166 mg, 0.867 mmol) were dissolved in anhydrous DMF (2 mL), and pyridine (0.140 mL, 1.73 mmol) was added dropwise. The reaction mixture was allowed to stir at room temperature for 72 h, then it was poured onto water, and the resulting precipitate was washed with water. The residue was purified by column chromatography using a gradient of 0–10% MeOH in DCM, washed with water, and triturated in diethyl ether to afford the title compound as a beige amorphous solid (30.0 mg, 15%). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (br s, 1H), 10.19 (br s, 1H), 8.59 (d, J = 2.21 Hz, 1H), 8.41 (d, J = 8.84 Hz, 1H), 8.23 (dd, J = 8.84, 2.21 Hz, 1H), 8.13 (dd, J = 8.84, 2.21 Hz, 1H), 7.87 (d, J = 8.84 Hz, 1H), 7.66–7.61 (m, 1H), 7.55 (d, J = 2.21 Hz, 1H), 7.52 (dd, J = 8.11, 2.21 Hz, 1H), 7.29 (app t, J = 10.23 Hz, 1H), 7.12 (d, J = 8.84 Hz, 1H), 6.99 (d, J = 8.84 Hz, 1H), 4.53 (t, J = 5.57 Hz, 2H), 4.34–4.28 (m, 4H), 3.41 −2.66 (m, 6H), 2.21–2.09 (m, 2H), 1.93–1.80 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 165.50, 164.95, 163.36, 151.38 (d, J = 244.33 Hz), 148.19, 146.91, 143.41, 140.75, 135.88 (d, J = 2.31 Hz), 129.87, 128.95, 128.90, 127.93, 127.25, 125.88 (d, J = 12.81 Hz), 124.44, 121.72, 119.30, 119.12 (d, J = 9.61 Hz), 117.36, 117.16, 116.03 (d, J = 19.22 Hz), 114.49, 64.88, 64.63, 64.49, 53.88, 52.50, 27.57, 23.47. HRMS (ESI+): calcd for C32H32FN4O5 (M + H)+, 571.2351; found 571.2321.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-(3-(piperidin-1-yl)propoxy)quinoline-6-carboxamide
N-(3-Amino-4-fluorophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (100 mg, 0.347 mmol), 2-(2-(piperidin-1-yl)propoxy)quinoline-6-carboxylic acid (164 mg, 0.52 mmol), and EDC (166 mg, 0.867 mmol) were dissolved in anhydrous DMF (2 mL), and pyridine (0.140 mL, 1.734 mmol) was added dropwise. The resulting mixture was allowed to stir at room temperature for 72 h, then it was poured onto water, and the resulting precipitate was washed with water and purified by column chromatography using a gradient of 0–10% MeOH in DCM, followed by washing in water (5 mL) and trituration in diethyl ether to afford the title compound as a beige amorphous solid (40 mg, 20%). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (br s, 1H), 10.19 (br s, 1H), 8.59 (d, J = 1.76 Hz, 1H), 8.39 (d, J = 8.79 Hz, 1H), 8.22 (dd, J = 8.79, 1.76 Hz, 1H), 8.13 (dd, J = 7.03 Hz, 2.34 Hz, 1H), 7.86 (d, J = 8.79 Hz, 1H), 7.67–7.62 (m, 1H), 7.55 (d, J = 1.76 Hz, 1H), 7.52 (dd, J = 8.21, 1.76 Hz, 1H), 7.28 (app t, J = 9.96 Hz, 1H), 7.11 (d, J = 8.90 Hz, 1H), 6.99 (d, J = 8.90 Hz, 1H), 4.51 (t, J = 6.29 Hz, 2H), 4.34–4.28 (m, 4H), 3.16–2.65 (m, 4H), 2.36–1.30 (m, 10H). 13C NMR (126 MHz, DMSO-d6) δ 165.51, 164.97, 163.26, 152.37 (d, J = 242.81 Hz), 148.18, 146.91, 143.41, 140.77, 135.87 (d, J = 2.19 Hz), 129.87, 128.96, 128.89, 127.92, 127.25, 125.88 (d, J = 14.51 Hz), 124.45, 121.72, 119.33, 119.14 (d, J = 8.29 Hz), 117.36, 117.16, 116.01 (d, J = 20.73 Hz), 114.48, 64.87, 64.59, 64.49, 55.03, 53.82, 25.40, 24.99, 23.67. HRMS (ESI+): calcd for C33H34FN4O5 (M + H)+, 585.2508; found 585.2485.
Synthetic Route IV
2-Methyl-N-(2-methyl-5-nitrophenyl)quinoline-6-carboxamide
To a suspension of 2-methylquinoline-6-carboxylic acid (3.69 g, 19.72 mmol) in anhydrous DCM (35 mL), oxalyl chloride (1.8 mL, 20.15 mmol) and DMF (310 μL, 4.00 mmol) were added dropwise and the resulting green solution was allowed to stir at 20 °C for 3 h after which it was concentrated under vacuum to afford a dry pale green solid. The solid was dissolved in pyridine (35 mL), and 2-methyl-5-nitroaniline (3.0 g, 19.72 mmol) was added in one portion and was allowed to stir for 2 h. The reaction mixture was reduced in vacuo until dryness. The remaining residue was triturated with diethyl ether. The crude product was purified by column chromatography on silica gel in gradient DCM/EtOH 0–50% to afford the title compound as a yellow amorphous solid (5.57 g, 88%). 1H NMR (500 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.63 (d, J = 1.8 Hz, 1H), 8.43 (d, J = 8.4 Hz, 1H), 8.40 (d, J = 2.4 Hz, 1H), 8.25 (dd, J = 8.7, 2.0 Hz, 1H), 8.07 (d, J = 2.9 Hz, 1H), 8.05 (d, J = 3.0 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 2.71 (s, 3H), 2.44 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.54, 161.05, 146.20, 144.90, 143.38, 142.15, 137.76, 132.04, 130.08, 129.93, 129.31, 126.62, 126.09, 124.16, 121.00, 120.94, 23.98, 18.82. HRMS (ESI+): calcd for C18H15NaN3O3 (M + Na)+, 344.1006; found 344.0999.
N-(5-Amino-2-methylphenyl)-2-methylquinoline-6-carboxamide
2-Methyl-N-(2-methyl-5-nitrophenyl)quinoline-6-carboxamide (4.0 g, 12.45 mmol), iron powder (6.95 g, 124.0 mmol), and ammonium chloride (2.12 g, 124.0 mmol) in ethanol (50 mL) and water (12.5 mL) were allowed to stir at 90 °C for 1 h. Then, the reaction mixture was allowed to cool to room temperature and was filtered through a short pad of celite. The eluate was concentrated in vacuo to afford the title compound as a light yellow amorphous solid, which was carried onto the next step without purification (1.95 g, 54%). 1H NMR (500 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.61–8.51 (m, 1H), 8.38 (d, J = 8.4 Hz, 1H), 8.21 (dd, J = 8.8, 1.7 Hz, 1H), 8.00 (d, J = 8.8 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.65 (d, J = 2.0 Hz, 1H), 6.42 (dd, J = 8.1, 2.2 Hz, 1H), 4.94 (s, 2H), 2.70 (s, 3H), 2.09 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.18, 161.09, 148.86, 147.31, 137.58, 137.01, 132.25, 130.87, 128.72, 128.49, 128.38, 125.84, 123.38, 120.72, 112.75, 112.61, 25.49, 17.50. HRMS (ESI+): calcd for C18H18N3O (M + H)+, 292.1444; found 292.1446.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-methylquinoline-6-carboxamide
2-Methyl-6-quinolinecarboxylic acid (600 mg, 3.21 mmol), HATU (1.46 g, 3.85 mmol), and N-(5-amino-2-methylphenyl)-2-methylquinoline-6-carboxamide (910 mg, 3.21 mmol) were suspended in anhydrous DMF (25 mL), and N,N-diisopropylethylamine (1.12 mL, 6.41 mmol) was added dropwise. The resulting solution was allowed to stir at room temperature under an inert atmosphere overnight. The reaction mixture was poured onto water, and the resulting precipitate was filtered and washed with water to afford the title compound as an off-white amorphous solid (1.38 g, 95%). 1H NMR (500 MHz, DMSO-d6) δ 10.14 (s, 1H), 10.08 (s, 1H), 8.61 (s, 1H), 8.41 (br d, J = 7.3 Hz, 1H), 8.24 (d, J = 8.7 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.87 (d, J = 2.1 Hz, 1H), 7.64–7.56 (m, 2H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.24 (d, J = 8.5 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.35–4.26 (m, 4H), 2.71 (s, 3H), 2.24 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.44, 164.82, 161.21, 148.93, 146.80, 143.39, 137.77, 137.62, 136.74, 131.97, 130.59, 129.23, 128.81, 128.64, 128.38, 128.14, 125.86, 123.44, 121.66, 119.07, 118.63, 117.30, 117.12, 64.86, 64.48, 25.51, 17.94. HRMS (ESI+): calcd for C27H24N3O4 (M + H)+, 454.1761; found 454.1733.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-formylquinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-methylquinoline-6-carboxamide (0.165 g, 0.364 mmol) and selenium dioxide (0.444 g, 0.400 mmol) in anhydrous 1,4-dioxane (0.6 mL) and anhydrous DMF (0.6 mL) was heated at 150 °C for 1 h after which the reaction mixture was allowed to cool to room temperature, diluted with DCM, and filtered through a pad of celite. The filtrate was concentrated under vacuum to afford the crude product as a brown solid, which was taken directly onto the next step without purification (0.17 g). 1H NMR (500 MHz, DMSO-d6) δ 10.29 (s, 1H), 10.17 (s, 1H), 10.09 (s, 1H), 8.81–8.77 (m, 1H), 8.41 (dd, J = 8.29, 1.66 Hz, 1H), 8.36 (d, J = 8.29 Hz, 1H), 8.17–8.12 (m, 1H), 8.08 (d, J = 9.12 Hz, 1H), 7.90 (d, J = 2.49 Hz, 1H), 7.58 (dd, J = 8.29, 2.49 Hz, 1H), 7.54 (d, J = 2.49 Hz, 1H), 7.52 (dd, J = 8.29, 2.49 Hz, 1H), 7.25 (d, J = 8.29 Hz, 1H), 6.98 (d, J = 8.29 Hz, 1H), 4.35–4.28 (m, 4H), 2.31 (s, 3H).
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(piperidin-1-ylmethyl)quinoline-6-carboxamide (14)
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-formylquinoline-6-carboxamide (58 mg, 0.124 mmol) and piperidine (31.7 mg, 0.372 mmol) in anhydrous DCM (1.2 mL) was allowed to stir at room temperature for 7 h. Then, sodium triacetoxyborohydride (79 mg, 0.372 mmol) was added in one portion at room temperature and the resulting mixture was allowed to stir under an inert argon atmosphere for 2 h at room temperature. The reaction mixture was diluted with DCM (5 mL), washed with brine (1 × 5 mL), and the aqueous phase was extracted with DCM/MeOH 9:1 mixture (3 × 5 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by column chromatography using a gradient of 0–10% MeOH in DCM afforded the title compound as a pale yellow solid (20 mg, 30%). 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H), 10.08 (s, 1H), 8.63 (s, 1H), 8.48 (d, J = 8.1 Hz, 1H), 8.26 (d, J = 9.0 Hz, 1H), 8.08 (d, J = 8.6 Hz, 1H), 7.91–7.85 (m, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.61–7.56 (m, 1H), 7.56–7.47 (m, 2H), 7.25 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.31 (q, J = 4.7 Hz, 4H), 3.77 (s, 2H), 2.43 (s, 4H), 2.24 (s, 3H), 1.55 (s, 4H), 1.43 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.46, 164.94, 163.18, 153.31, 151.37, 148.11, 146.91, 143.41, 140.84, 135.86, 129.96, 129.00, 128.92, 127.92, 127.24, 125.92, 125.81, 124.50, 121.72, 119.34, 119.11, 117.34, 117.17, 116.07, 115.91, 114.41, 64.87, 64.49, 63.99, 53.66, 52.05, 23.28. HRMS (ESI+): calcd for C32H32N4NaO4 (M + Na)+, 559.2316; found 559.2325.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-((4-isopropylpiperazin-1-yl)methyl)quinoline-6-carboxamide (16)
To a solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-formylquinoline-6-carboxamide (170 mg, 0.364 mmol) in anhydrous DCM, 1-isopropylpiperazine (0.16 mL, 1.09 mmol) was added dropwise at room temperature and the resulting mixture was allowed to stir under an inert argon atmosphere for 2.5 h. Then, sodium triacetoxyborohydride (231 mg, 1.09 mmol) was added in one portion and the resulting mixture was allowed to stir overnight at room temperature. The reaction was diluted with DCM (5 mL) and washed with a NaHCO3 saturated aqueous solution (5 mL). The aqueous phase was extracted with DCM (3 × 5 mL), and the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to afford a brown oil as a crude product. Purification by column chromatography using a gradient of 0–10% MeOH in DCM afforded the title compound as a beige solid (30 mg, 14%). 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H), 10.08 (s, 1H), 8.65–8.61 (m, 1H), 8.48 (d, J = 8.6 Hz, 1H), 8.29–8.23 (m, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.90–7.86 (m, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.59 (dd, J = 8.3, 2.0 Hz, 1H), 7.56–7.48 (m, 2H), 7.25 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.31 (q, J = 5.0 Hz, 4H), 3.78 (s, 2H), 2.65–2.59 (m, 1H), 2.47 (s, 8H), 2.24 (s, 3H), 0.96 (d, J = 6.5 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 165.37, 164.82, 162.08, 148.63, 146.81, 143.39, 137.89, 137.78, 136.69, 132.37, 130.60, 129.22, 129.15, 128.63, 128.41, 128.14, 128.09, 126.69, 122.21, 121.66, 119.06, 118.64, 117.30, 117.12, 64.92, 64.86, 64.49, 54.05, 53.94, 48.47, 31.16, 28.11, 18.71, 17.93. HRMS (ESI+): calcd for C34H38N5O4 (M + H)+, 580.2918; found 580.2896.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide
To a suspension of 2-methylquinoline-6-carboxylic acid (1.5 g, 8.01 mmol) in anhydrous DCM (40 mL), DMF (1.40 μL, 0.018 mmol) and oxalyl chloride (0.74 mL, 8.74 mmol) were added dropwise and the resulting green solution was allowed to stir at 20 °C for 3 h, after which it was concentrated under vacuum to afford a dry pale green solid. The solid was dissolved in pyridine (40 mL), and 2-chloro-5-nitroaniline (1.26 g, 7.28 mmol) was added in one portion. The resulting dark yellow suspension was allowed to stir for 2 h, after which it was poured onto water and the yellow precipitate was filtered and washed several times with water, diethyl ether, and finally with a minimum amount of DCM to afford the product as a yellow amorphous solid, which was used without further purification (2.20 g, 88%). 1H NMR (500 MHz, DMSO-d6) δ 10.59 (s, 1H), 8.65 (d, J = 2.0 Hz, 1H), 8.60 (d, J = 2.7 Hz, 1H), 8.44 (d, J = 8.6 Hz, 1H), 8.25 (dd, J = 8.6, 2.0 Hz, 1H), 8.15 (dd, J = 8.6, 2.7 Hz, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 8.6 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 2.71 (s, 3H). HRMS (ESI+): calcd for C17H1335ClN3O3 (M + H)+, 342.0640; found 342.0646.
N-(2-Chloro-5-nitrophenyl)-2-methylquinoline-6-carboxamide was suspended in water (7 mL) and EtOH (21 mL). Ammonium chloride (2.41 g, 45.1 mmol) and iron powder (2.52 g, 45.1 mmol) were added, and the resulting suspension was allowed to stir at 90 °C for 1 h. The reaction mixture was allowed to cool to room temperature, diluted with MeOH and DCM, and filtered through a pad of celite. The resulting filtrate was concentrated under vacuum to afford a light brown amorphous solid as a crude product, which was used directly in the next step without purification (2.00 g, 100%). 1H NMR (500 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.58 (d, J = 2.2 Hz, 1H), 8.41 (d, J = 8.7 Hz, 1H), 8.21 (dd, J = 8.7, 2.2 Hz, 1H), 8.02 (d, J = 8.7 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.15 (d, J = 8.7 Hz, 1H), 6.87 (d, J = 2.2 Hz, 1H), 6.50 (dd, J = 8.7, 2.2 Hz, 1H), 5.41 (bs, 2H), 2.70 (s, 3H). HRMS (ESI+): calcd for C17H1535ClN3O (M + H)+, 312.0898; found 312.0902.
2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (1.27 g, 7.06 mmol) was suspended in anhydrous DCM (20 mL), and DMF (1.23 μL, 0.016 mmol) and oxalyl chloride (0.65 mL, 7.70 mmol) were added dropwise, and the resulting green solution was allowed to stir at 20 °C for 3 h after which it was concentrated under vacuum to afford a dry pale green solid. The solid was dissolved in pyridine (20.0 mL), and N-(5-amino-2-chlorophenyl)-2-methylquinoline-6-carboxamide (2.00 g, 6.42 mmol) was added in one portion. The resulting dark yellow suspension was allowed to stir for 2 h after which it was poured onto water, and the yellow precipitate was filtered and washed several times with water, diethyl ether, and finally with a minimum amount of DCM to afford the crude product as a pale yellow amorphous solid, which was carried onto the next step without purification (1.86 g, 61%). 1H NMR (500 MHz, DMSO-d6) δ 10.31 (s, 1H), 10.27 (s, 1H), 8.63 (d, J = 1.5 Hz, 1H), 8.43 (d, J = 8.8 Hz, 1H), 8.25 (dd, J = 8.8, 2.2 Hz, 1H), 8.14 (d, J = 2.2 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.75 (dd, J = 8.8, 2.9 Hz, 1H), 7.58 – 7.49 (m, 4H), 7.00 (d, J = 8.8 Hz, 1H), 4.37–4.26 (m, 4H), 2.71 (s, 3H). HRMS (ESI+): calcd for C26H2135ClN3O4 (M + H)+, 474.1215; found 474.1210.
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-methylquinoline-6-carboxamide (0.500 g, 1.06 mmol) and selenium dioxide (0.129 g, 1.16 mmol) in anhydrous DMF (12.0 mL) and 1,4-dioxane (12.0 mL) was heated at 150 °C for 2 h. A further portion of selenium dioxide (0.129 g, 1.16 mmol) was added to the reaction mixture and stirred at 150 °C for a further 1 h. The reaction mixture was allowed to cool to room temperature, diluted with DCM, and filtered through a pad of celite. The filtrate was concentrated under vacuum to afford the crude product as a yellow amorphous solid, which was carried onto the next step without purification (0.515 g). 1H NMR (500 MHz, DMSO-d6) δ 10.45 (s, 1H), 10.40 (s, 1H), 10.17 (s, 1H), 8.81–8.79 (m, 2H), 8.42–8.34 (m, 2H), 8.19 (app t, J = 7.47 Hz, 1H), 8.09 (d, J = 8.13 Hz, 1H), 7.79–7.74 (m, 2H), 7.55 (d, J = 1.99 Hz, 1H), 7.52 (dd, J = 8.13, 1.99 Hz, 1H), 6.99 (d, J = 9.04 Hz, 1H), 4.36–4.26 (m, 4H). HRMS (ESI+): calcd for C26H1935ClN3O5 (M + H)+, 488.1013; found 488.1012.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((4-isopropylpiperazin-1-yl)methyl)quinoline-6-carboxamide (18)
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (2.00 g, 4.10 mmol) and 1-isopropylpiperazine (1.58 g, 12.3 mmol) in anhydrous DCM (35 mL) was allowed to stir at 20 °C for 6 h, after which sodium triacetoxyborohydride (2.61 g, 12.3 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (35 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 35 mL). The crude product (pale yellow solid) was purified by column chromatography using a gradient of 0–10% MeOH in DCM, followed by trituration in diethyl ether to afford the title compound as a pale yellow amorphous solid (0.830 g, 34%). 1H NMR (500 MHz, DMSO-d6) δ 10.33 (s, 1H), 10.28 (s, 1H), 8.66 (s, 1H), 8.50 (d, J = 8.5 Hz, 1H), 8.27 (d, J = 8.8 Hz, 1H), 8.20–8.04 (m, 2H), 7.81–7.64 (m, 2H), 7.62–7.46 (m, 3H), 7.00 (d, J = 8.4 Hz, 1H), 4.31 (q, J = 5.1 Hz, 4H), 3.81 (s, 2H), 2.59 (d, J = 43.9 Hz, 5H), 2.45 (br s, 4H), 0.99 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 165.45, 165.11, 161.95, 148.75, 147.03, 143.43, 139.06, 138.02, 135.35, 131.83, 129.81, 129.29, 128.89, 128.37, 127.76, 126.70, 124.04, 122.31, 121.80, 120.31, 119.72, 117.37, 117.23, 64.88, 64.49, 60.05, 53.76, 48.34, 18.44. HRMS (ESI+): calcd for C33H3535ClN5O4 (M + H)+, 600.2372; found 600.2336.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-(piperazin-1-ylmethyl)quinoline-6-carboxamide (19)
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (0.300 g, 0.615 mmol) and 1-(tert-butyl)piperazine (0.262 g, 1.85 mmol) in anhydrous DCM (5 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (0.391 g, 1.85 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (5 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 5 mL). The crude product (pale yellow solid) was purified by column chromatography 0–10% MeOH in DCM, followed by trituration in diethyl ether to afford tert-butyl 4-((6-((2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)carbamoyl)quinolin-2-yl)methyl)piperazine-1-carboxylate as a beige solid (0.060 g, 16%). 1H NMR (500 MHz, DMSO-d6): δ 10.34 (s, 1H), 10.29 (s, 1H), 8.66 (d, J = 1.6 Hz, 1H), 8.50 (d, J = 8.6 Hz, 1H), 8.26 (dd, J = 8.6, 2.4 Hz, 1H), 8.14 (d, J = 2.4 Hz, 1H), 8.09 (d, J = 8.6 Hz, 1H), 7.77–7.72 (m, 2H), 7.56 (d, J = 2.4 Hz, 1H), 7.54 (d, J = 1.6 Hz, 1H), 7.53 (dd, J = 7.8, 2.4 Hz, 1H), 7.00 (d, J = 8.6 Hz, 1H), 4.35–4.27 (m, 4H), 3.79 (s, 2H), 2.79–2.29 (m, 8H), 1.03 (br s, 9H). To a suspension of tert-butyl 4-((6-((2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)carbamoyl)quinolin-2-yl)methyl)piperazine-1-carboxylate (674 mg, 1.02 mmol) in anhydrous DCM (10 mL), trifluoroacetic acid (TFA, 0.78 mL, 10.2 mmol) was added dropwise, and the resulting mixture was allowed to stir at 20 °C for 4 h. The reaction mixture was concentrated under vacuum to afford the crude product as a light brown oil. The crude was purified by column chromatography using a gradient of 0–15% MeOH in DCM, followed by trituration in diethyl ether to afford the title compound as a yellow amorphous solid (39 mg, 6.8%). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 10.27 (s, 1H), 8.65 (d, J = 2.0 Hz, 1H), 8.49 (d, J = 8.4 Hz, 1H), 8.26 (dd, J = 8.8, 2.0 Hz, 1H), 8.15 (d, J = 2.5 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.76–7.71 (m, 2H), 7.58–7.47 (m, 3H), 7.00 (d, J = 8.4 Hz, 1H), 4.40–4.23 (m, 4H), 3.76 (s, 2H), 2.74 (t, J = 4.8 Hz, 4H), 2.47–2.42 (br m, 4H) (1 proton missing). 13C NMR (126 MHz, DMSO-d6) δ 165.00, 164.64, 161.79, 148.29, 146.57, 142.97, 138.58, 137.45, 134.89, 131.32, 129.36, 128.81, 128.39, 127.85, 127.31, 126.21, 123.54, 121.85, 121.32, 119.81, 119.23, 116.92, 116.74, 65.11, 64.42, 64.03, 54.26, 45.54. HRMS (ESI+): calcd for C30H2935ClN5O4 (M + H)+ 558.1903, found 558.1885.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((4-methylpiperazin-1-yl)methyl)quinoline-6-carboxamide (20)
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (0.100 g, 0.205 mmol) and 1-methylpiperazine (61.6 mg, 0.615 mmol) in anhydrous DCM (2 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (0.130 g, 0.615 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (5 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 5 mL). The crude product (brown oil) was purified by column chromatography using a gradient of 0–10% MeOH in DCM to afford the desired product as a white amorphous solid (0.010 g, 8.5%). 1H NMR (500 MHz, DMSO-d6) δ 10.31 (s, 1H), 10.23 (s, 1H), 8.65 (s, 1H), 8.48 (d, J = 8.7 Hz, 1H), 8.30–8.26 (m, 1H), 8.15 (s, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.71 (dd, J = 8.7, 4.2 Hz, 2H), 7.57–7.47 (m, 3H), 6.99 (d, J = 8.4 Hz, 1H), 4.31 (q, J = 4.9 Hz, 4H), 3.79 (s, 2H), 2.35 (br s, 8H), 2.16 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.48, 165.11, 162.14, 148.72, 147.03, 143.44, 139.02, 138.00, 129.80, 129.24, 128.83, 128.41, 127.79, 126.70, 124.01, 122.27, 121.80, 120.25, 119.56, 117.39, 117.22, 117.22, 64.89, 64.84, 64.49, 55.21, 53.40, 46.24. HRMS (ESI+): calcd for C31H3135ClN5O4 (M + H)+, 572.2059; found 572.2031.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxamide (21)
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (1.03 g, 2.11 mmol) and 1-ethylpiperazine (723 mg, 6.33 mmol) in anhydrous DCM (20 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (1.34 g, 6.33 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 3 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (20 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 20 mL). The crude product was purified by column chromatography using a gradient of 0–10% MeOH in DCM, followed by purification by Isolute SCX-II chromatography eluting with MeOH/NH3 to afford the title compound as a white amorphous solid (0.431 g, 35%). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 10.27 (s, 1H), 8.65 (d, J = 2.0 Hz, 1H), 8.49 (d, J = 8.4 Hz, 1H), 8.26 (dd, J = 8.8, 2.0 Hz, 1H), 8.20–8.04 (m, 2H), 7.80–7.66 (m, 2H), 7.62–7.46 (m, 3H), 7.00 (d, J = 8.4 Hz, 1H), 4.39–4.23 (m, 4H), 3.79 (s, 2H), 2.51–2.29 (m, 10H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.45, 165.11, 162.24, 148.74, 147.03, 143.43, 139.05, 138.01, 135.34, 131.83, 129.82, 129.29, 128.88, 128.36, 127.77, 126.70, 124.02, 122.32, 121.79, 120.29, 119.71, 117.38, 117.21, 64.88, 64.49, 54.09, 53.86, 48.38, 18.71. HRMS (ESI+): calcd for C32H3335ClN5O4 (M + H)+, 586.2216; found 586.2189.
2-((4-(tert-Butyl)piperazin-1-yl)methyl)-N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)quinoline-6-carboxamide
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (300 mg, 0.615 mmol) and 1-(tert-butyl)piperazine (262 mg, 1.85 mmol) in anhydrous DCM (5 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (391 mg, 1.85 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (5 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 5 mL). The crude product (pale yellow solid) was purified by column chromatography using a gradient of 0–10% MeOH in DCM followed by washing in water and trituration in diethyl ether to afford the title compound as a beige amorphous solid (60 mg, 16%). 1H NMR (500 MHz, DMSO-d6) δ 10.34 (s, 1H), 10.29 (s, 1H), 8.66 (d, J = 1.56 Hz, 1H), 8.50 (d, J = 8.60 Hz, 1H), 8.26 (dd, J = 8.60, 2.35 Hz, 1H), 8.14 (d, J = 2.35 Hz, 1H), 8.09 (d, J = 8.60 Hz, 1H), 7.77–7.72 (m, 2H), 7.56 (d, J = 2.35 Hz, 1H), 7.54 (d, J = 1.56 Hz, 1H), 7.53 (dd, J = 7.82, 2.35 Hz, 1H), 7.00 (d, J = 8.60 Hz, 1H), 4.35–4.27 (m, 4H), 3.79 (s, 2H), 2.79–2.29 (m, 8H), 1.03 (br s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 165.46, 165.11, 162.21, 148.75, 147.03, 143.43, 139.05, 137.95, 135.35, 131.79, 129.82, 129.28, 128.86, 128.33, 127.77, 126.69, 124.00, 122.29, 121.79, 120.27, 119.69, 117.38, 117.21, 64.88, 64.76, 64.49, 54.13, 45.77, 26.05. HRMS (ESI+): calcd for C34H3735ClN5O4 (M + H)+, 614.2529; found 614.2502.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((4-ethyl-1,4-diazepan-1-yl)methyl)quinoline-6-carboxamide
A suspension of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (103 mg, 0.211 mmol) and 1-ethyl-1,4-diazepane (0.12 mL, 108 mg, 0.844 mmol) in anhydrous DCM (4.5 mL sonication and a large volume of DCM were used in an attempt to fully solubilize the starting material) was allowed to stir at room temperature overnight. Then, sodium triacetoxyborohydride (179 mg, 0.844 mmol) was added and the reaction mixture was stirred at room temperature for 40 h. The reaction was quenched with saturated aqueous NaHCO3 solution. The aqueous layer was extracted three times with a mixture of 10% MeOH in DCM; the combined organic layer was dried (MgSO4) and concentrated in vacuo. This crude product was purified by Biotage chromatography using a gradient of 0–10% MeOH in DCM with a KPNH2 column to afford a yellow oil. This material was further purified by Isolute SCX-II column chromatography (eluting with MeOH, followed by 10% of 2 M NH3 in MeOH) to give a yellow gum. Finally, the gum was triturated with diethyl ether to afford a yellow amorphous solid (9 mg, 7%). 1H NMR (500 MHz, DMSO-d6) δ 10.31 (s, 1H), 10.26 (s, 1H), 8.65 (s, 1H), 8.49 (d, J = 8.5 Hz, 1H), 8.25 (d, J = 8.8 Hz, 1H), 8.14 (d, J = 2.0 Hz, 2H), 8.08 (d, J = 8.6 Hz, 1H), 7.75 (dd, J = 8.9, 2.2 Hz 1H), 7.57–7.48 (m, 3H), 7.00 (d, J = 8.4 Hz, 1H), 4.36–4.26 (m, 4H), 3.94 (s, 2H), 2.78–2.60 (m, 6H), 2.55–2.45 (m, 2H–hidden under DMSO peak, observed by HSQC), 1.75 (p, J = 5.8 Hz, 4H), 0.99 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.47, 165.11, 163.34, 148.72, 147.03, 143.43, 139.04, 137.91, 135.36, 131.73, 129.82, 129.26, 128.84, 128.29, 127.77, 126.67, 124.00, 122.20, 121.79, 120.27, 119.68, 117.38, 117.21, 64.88, 64.71, 64.49, 55.71, 54.81, 54.60, 53.77, 51.92, 27.75, 13.03. HRMS (ESI+): calcd for C33H3535ClN5O4 (M + H)+ 600.2372, found 600.2351.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((4-cyclopropylpiperazin-1-yl)methyl)quinoline-6-carboxamide
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (255 mg, 0.523 mmol) and 1-cyclopropylpiperazine (0.189 mL, 1.568 mmol) in anhydrous DCM (5 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (332 mg, 1.568 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (5 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 5 mL). The crude product (brown solid) was purified by two rounds of column chromatography using a gradient of 0–10% MeOH in DCM followed by trituration in diethyl ether to afford the title compound as a pale yellow amorphous solid (44 mg, 14%). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 10.27 (s, 1H), 8.65 (s, 1H), 8.50 (d, J = 7.87 Hz, 1H), 8.26 (d, J = 7.87 Hz, 1H), 8.15 (d, J = 1.83 Hz, 1H), 8.09 (d, J = 7.87 Hz, 1H), 7.77–7.71 (m, 2H), 7.57–7.49 (m, 3H), 7.00 (d, J = 7.87 Hz, 1H), 4.36–4.24 (m, 4H), 3.78 (s, 2H), 2.57 (br s, 4H), 2.43 (br s, 4H), 1.63–1.58 (m, 1H), 0.42–0.37 (m, 2H), 0.30–0.24 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.47, 165.12, 162.22, 148.75, 147.05, 143.45, 139.06, 138.01, 135.36, 131.81, 129.84, 129.30, 128.87, 128.34, 127.78, 126.70, 124.02, 122.31, 121.80, 120.29, 119.70, 117.39, 117.22, 64.88, 64.49, 53.46, 53.25, 30.89, 6.09. HRMS (ESI+): calcd for C33H3335ClN5O4 (M + H)+, 598.2216; found 598.2210.
2-((4-(sec-Butyl)piperazin-1-yl)methyl)-N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)quinoline-6-carboxamide
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (255 mg, 0.523 mmol) and 1-(sec-butyl)piperazine (223 mg, 1.57 mmol) in anhydrous DCM (5 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (332 mg, 1.57 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (5 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 5 mL). The crude product (brown solid) was purified by two rounds of column chromatography using a gradient of 0–10% MeOH in DCM, followed by trituration in diethyl ether to afford the title compound as a pale yellow amorphous solid (35 mg, 11%). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 10.27 (s, 1H), 8.66 (d, J = 1.6 Hz, 1H), 8.50 (d, J = 8.4 Hz, 1H), 8.26 (dd, J = 8.4, 1.60 Hz, 1H), 8.15 (d, J = 2.3 Hz, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.75 (dd, J = 8.4, 2.36 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.55 (d, J = 2.3 Hz, 1H), 7.54 (d, J = 1.6 Hz, 1H), 7.52 (dd, J = 7.8, 2.3 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 4.35–4.27 (m, 4H), 3.81 (s, 2H), 2.87–2.17 (m, 9H), 1.51 (br s, 1H), 1.28 (br s, 1H), 0.95 (br s, 3H), 0.85 (t, J = 7.65 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.46, 165.11, 162.20, 148.75, 147.04, 143.44, 139.05, 137.98, 135.35, 131.80, 129.83, 129.29, 128.88, 128.34, 127.77, 126.69, 124.01, 122.30, 121.80, 120.28, 119.70, 117.39, 117.22, 64.89, 64.49, 60.25, 53.91, 48.15, 26.05, 14.08, 11.51. HRMS (ESI+): calcd for C34H3735ClN5O4 (M + H)+, 614.2529; found 614.2495.
(R)-N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((4-ethyl-2-methylpiperazin-1-yl)methyl)quinoline-6-carboxamide
A suspension of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (1.21 g, 2.48 mmol) and (R)-tert-butyl 3-methylpiperazine-1-carboxylate (0.993 g, 4.96 mmol) in anhydrous DCM (24 mL) was allowed to stir at room temperature overnight. Then, sodium triacetoxyborohydride (1.05 mg, 4.96 mmol) was added and the reaction mixture was stirred at room temperature overnight. The reaction was quenched with saturated aqueous NaHCO3 solution. The aqueous layer was extracted three times with a mixture of 10% MeOH in DCM; the combined organic layer was dried (Na2SO4) and concentrated in vacuo. The crude product was purified by Biotage chromatography using a gradient of 0–10% MeOH in DCM to afford the product as a yellow semisolid (560 mg, 34%). (R)-tert-Butyl 4-((6-((2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)carbamoyl)quinolin-2-yl)methyl)-3-methylpiperazine-1-carboxylate (560 mg, 0.83 mmol) was taken up in anhydrous DCM (8 mL), and TFA (0.319 mL, 4.17 mmol) was added dropwise. The reaction was allowed to stir at room temperature for 48 h, after which time the starting material was still observed by LCMS. Therefore, further TFA (0.319 mL, 4.17 mmol) was added, and the reaction mixture was left to stir for 18 h. The solvents were removed in vacuo, and the resulting solid was taken up in 10% MeOH in DCM. The organic layer was washed twice with saturated aqueous NaHCO3 solution, followed by water, dried (Na2SO4), and then concentrated in vacuo. The material was partially purified by Isolute SCX-II column chromatography (eluting with MeOH, followed by 10% of 2 M NH3 in MeOH) to afford the crude product as a yellow solid (272 mg, ∼73% purity).
To a solution of (R)-N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((2-methylpiperazin-1-yl)methyl)quinoline-6-carboxamide (100 mg, 0.128 mmol) in anhydrous MeOH (1.5 mL) at 0 °C was added sodium cyanoborohydride (8.82 mg, 0.140 mmol) in one portion. Acetaldehyde (5.02 μL, 0.089 mmol) was then added as a cooled solution (0 °C) in MeOH (0.5 mL) dropwise for 2 min. The reaction was allowed to warm slowly to room temperature and left to stir for 48 h. The MeOH was removed in vacuo, and the resulting residue was taken up in 10% MeOH in DCM. The organic layer was washed with 1 M NaOH aqueous solution and dried (Na2SO4). The resulting residue was purified by Biotage chromatography using a gradient of 0–10% MeOH in DCM and then further purified by Isolute SCX-II chromatography (eluting with MeOH, followed by 10% of 2 M NH3 in MeOH) to afford the product as a yellow amorphous solid (33 mg, 43%). 1H NMR (500 MHz, CDCl3) δ 8.63 (s, 1H), 8.60 (d, J = 2.5 Hz, 1H), 8.41 (d, J = 1.8 Hz, 1H), 8.26 (d, J = 8.5 Hz, 1H), 8.19 (d, J = 8.8 Hz, 1H), 8.16 (dd, J = 8.8, 2.0 Hz, 1H), 8.07 (s, 1H), 7.99 (dd, J = 8.8, 2.5 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 7.46 (d, J = 1.8 Hz, 1H), 7.45 (d, J = 4.8 Hz, 1H), 7.39 (dd, J = 8.4, 2.2 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 4.35 (d, J = 14.5 Hz, 1H), 4.34–4.29 (m, 4H), 3.63 (d, J = 14.6 Hz, 1H), 2.83 (br. d, J = 10.9 Hz, 1H), 2.81–2.75 (m, 2H), 2.72–2.63 (m, 1H), 2.53–2.37 (m, 3H), 2.21–2.14 (m, 1H), 2.01 (br t, J = 10.5 Hz, 1H), 1.19 (d, J = 6.2 Hz, 3H), 1.11 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.05, 164.92, 163.62, 149.06, 146.96, 143.58, 137.94, 137.18, 134.67, 131.65, 130.02, 129.51, 127.79, 127.74, 126.72, 122.27, 120.48, 117.81, 117.46, 116.89, 116.79, 112.45, 77.23, 64.58, 64.19, 60.73, 60.66, 55.85, 53.00, 52.58, 52.26, 17.84, 11.91. HRMS (ESI+): calcd for C33H3535ClN5O4 (M + H)+ 600.2327, found 600.2352.
2-(Azetidin-1-ylmethyl)-N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)quinoline-6-carboxamide
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (300 mg, 0.615 mmol) and azetidine (0.124 mL, 1.84 mmol) in anhydrous DCM (2.0 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (391 mg, 1.84 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction mixture was quenched with a NaHCO3 saturated aqueous solution (2 mL) and extracted with a mixture of DCM/MeOH 9/1 (3 × 2 mL). The crude product was purified by column chromatography on silica gel using a gradient of 0–15% MeOH in DCM and then washed with water and triturated with diethyl ether to afford the title compound as a pale yellow amorphous solid (20 mg, 6%). 1H NMR (500 MHz, DMSO-d6) δ 10.35 (s, 1H), 10.31 (s, 1H), 8.66 (d, J = 2.2 Hz, 1H), 8.49 (d, J = 8.7 Hz, 1H), 8.27 (dd, J = 8.7, 2.2 Hz, 1H), 8.14 (d, J = 2.2 Hz, 1H), 8.08 (d, J = 8.7 Hz, 1H), 7.76 (dd, J = 8.7, 2.2 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.58–7.51 (m, 3H), 6.99 (d, J = 8.7 Hz, 1H), 4.35–4.27 (m, 4H), 3.91 (s, 2H), 3.39–3.27 (m, 4H), 2.06 (qn, J = 7.10 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.47, 165.11, 161.47, 148.75, 147.04, 143.43, 139.05, 138.09, 135.35, 131.80, 129.83, 129.27, 128.90, 128.41, 127.77, 126.62, 124.02, 121.91, 121.80, 120.28, 119.70, 117.39, 117.22, 65.18, 64.89, 64.49, 55.42, 49.06, 17.92. HRMS (ESI+): calcd for C29H2635ClN4O4 (M + H)+, 529.1637; found 529.1616.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-(pyrrolidin-1-ylmethyl)quinoline-6-carboxamide
A solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (300 mg, 0.615 mmol) and pyrrolidine (131 mg, 1.84 mmol) in anhydrous DCM (2 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (391 mg, 1.84 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The resulting mixture was quenched with a NaHCO3 saturated aqueous solution (2 mL) and extracted with a mixture of DCM/MeOH 9/1 (3 × 2 mL). The crude product was purified by column chromatography using a gradient of 0–15% MeOH in DCM, then washed with water and triturated with diethyl ether to afford the title compound as a pale yellow amorphous solid (20 mg, 6%). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 10.27 (s, 1H), 8.65 (s, 1H), 8.49 (d, J = 7.98 Hz, 1H), 8.26 (dd, J = 7.98, 2.28 Hz, 1H), 8.14 (d, J = 2.28 Hz, 1H), 8.09 (d, J = 7.98 Hz, 1H), 7.77–7.70 (m, 2H), 7.56–7.50 (m, 3H), 7.00 (d, J = 7.98 Hz, 1H), 4.35–4.28 (m, 4H), 3.92 (s, 2H), 2.57–2.52 (m, 4H), 1.77–1.70 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 165.49, 165.11, 162.75, 148.72, 147.04, 143.44, 139.05, 137.98, 135.36, 131.77, 129.83, 129.30, 128.86, 128.33, 127.77, 126.66, 124.01, 122.29, 121.79, 120.27, 119.69, 117.39, 117.22, 64.89, 64.49, 62.45, 54.29, 23.77. HRMS (ESI+): calcd for C30H2835ClN4O4 (M + H)+, 543.1794; found 543.1778.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((3-methylazetidin-1-yl)methyl)quinoline-6-carboxamide
A solution of 3-methylazetidine hydrochloride (33.1 mg, 0.307 mmol) in anhydrous DCM (0.5 mL) was added to a stirring solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (50 mg, 0.102 mmol) in anhydrous DCM (1.5 mL) at room temperature. The reaction was stirred for 5.5 h, then NaBH(OAc)3 (65.2 mg, 0.307 mmol) was added, and the reaction mixture was stirred for 48 h. The reaction was quenched with saturated aqueous NaHCO3 solution. The aqueous layer was extracted with 3 × 10% MeOH in DCM. The combined organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by Biotage chromatography using a gradient of 0–10% MeOH in DCM to afford a yellow gum. This gum was further purified by Isolute SCX-II chromatography (eluting with MeOH and then 10% of 2 M NH3 in MeOH) to afford the title product as a yellow amorphous solid (16.5 mg, 30%). 1H NMR (500 MHz, CDCl3) δ 8.63 (br s, 1H), 8.58 (d, J = 2.5 Hz, 1H), 8.41 (d, J = 1.8 Hz, 1H), 8.28 (d, J = 8.5 Hz, 1H), 8.20 (d, J = 8.8 Hz, 1H), 8.16 (dd, J = 8.8, 2.0 Hz, 1H), 7.98 (dd, J = 8.8, 2.4 Hz, 2H), 7.64 (d, J = 8.5 Hz, 1H), 7.46 (s, 1H), 7.45 (d, J = 7.2 Hz, 1H), 7.39 (dd, J = 8.4, 2.2 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 4.38–4.29 (m, 4H), 3.99 (s, 2H), 3.64 (t, J = 7.5 Hz, 2H), 2.95 (t, J = 7.2 Hz, 2H), 2.70 (dt, J = 14.0, 7.0 Hz, 1H), 1.21 (d, J = 6.8 Hz, 3H). HRMS (ESI+): calcd for C30H2835ClN4O4 (M + H)+ 543.1794, found 543.1769.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((3,3-dimethylazetidin-1-yl)methyl)quinoline-6-carboxamide
3,3-Dimethylazetidine (55.0 mg, 0.646 mmol) was added to a stirring solution of N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (210 mg, 0.431 mmol) in anhydrous DCM (1.5 mL) at room temperature under an inert atmosphere. The reaction was stirred for 1 h. Then, NaBH(OAc)3 (137 mg, 0.646 mmol) was added, and the reaction mixture was stirred for 18 h. The reaction was quenched with saturated aqueous NaHCO3 solution (4 mL). The aqueous layer was extracted with DCM (3 × 4 mL); the combined organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by column chromatography using a gradient of 0–10% MeOH in DCM to afford a yellow gum. This gum was further purified by Isolute SCX-II chromatography (eluting with MeOH and then 10% NH3 in MeOH) to afford the title product as a yellow amorphous solid (16.0 mg, 7%). 1H NMR (500 MHz, DMSO-d6) δ 10.35 (s, 1H), 10.27 (s, 1H), 8.70–8.66 (m, 1H), 8.55 (d, J = 8.5 Hz, 1H), 8.29 (dd, J = 9.0, 2.0 Hz, 1H), 8.17–8.09 (m, 2H), 7.74 (dd, J = 8.8, 2.6 Hz, 1H), 7.65 (d, J = 8.5 Hz, 1H), 7.57–7.49 (m, 3H), 7.00 (d, J = 8.4 Hz, 1H), 4.36–4.27 (m, 4H), 3.45–3.20 (m, 6H), 1.27 (s, 6H). δ HRMS (ESI+): calcd for C31H3037ClN4O4 (M + H)+ 559.1937, found 559.1921.
2-(Azetidin-1-ylmethyl)-N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)quinoline-6-carboxamide
To a solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (100 mg, 0.212 mmol) in anhydrous DCM (2 mL), azetidine (43 μL, 0.636 mmol) was added dropwise at room temperature, and the resulting mixture was allowed to stir for 12 h. Then, sodium triacetoxyborohydride (135 mg, 0.636 mmol) was added in one portion and the resulting mixture was allowed to stir for 2 h at room temperature. The reaction mixture was washed with brine (2 mL) and purified by column chromatography using a gradient of 0–10% MeOH in DCM + 1% 7 N NH3 in MeOH to afford the title compound as a white amorphous solid (10 mg, 9%). 1H NMR (500 MHz, MeOD) δ 8.59 (d, J = 2.1 Hz, 1H), 8.48 (d, J = 8.5 Hz, 1H), 8.29 (dd, J = 8.8, 2.1 Hz, 1H), 8.24–8.10 (m, 2H), 7.71–7.55 (m, 2H), 7.55–7.44 (m, 2H), 7.24 (dd, J = 10.1, 8.9 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 4.38–4.26 (m, 4H), 3.99 (s, 2H), 3.47 (t, J = 7.1 Hz, 4H), 2.21 (qn, J = 7.2 Hz, 2H). 13C NMR (126 MHz, MeOD) δ 166.69, 166.60, 160.42, 152.36 (d, J = 248.54 Hz), 148.53, 147.10, 143.47, 138.18, 134.84 (d, J = 2.22 Hz), 131.94, 128.36, 128.18, 127.96, 127.38, 126.66, 125.28 (d, J = 11.11 Hz), 121.58, 120.75, 119.56 (d, J = 6.67 Hz), 118.95, 116.79, 116.64, 115.23 (d, J = 20.01 Hz), 64.53, 64.13, 55.13, 29.36, 17.33. HRMS (ESI+): calcd for C29H26FN4O4 (M + H)+, 513.1933; found 513.1930.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-(pyrrolidin-1-ylmethyl)quinoline-6-carboxamide
To a solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (100 mg, 0.212 mmol) in anhydrous DCM (2 mL), pyrrolidine (53 μL, 0.636 mmol) was added dropwise at room temperature, and the resulting mixture was allowed to stir for 12 h. Then, sodium triacetoxyborohydride (135 mg, 0.636 mmol) was added in one portion and the resulting mixture was allowed to stir for 2 h at room temperature. The reaction mixture was washed with brine (2 mL) and purified by column chromatography using a gradient of 0–10% MeOH in DCM + 1% 7 N NH3 in MeOH to afford the title compound as a pale yellow amorphous solid (55 mg, 49%). 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 10.18 (s, 1H), 8.64 (d, J = 1.8 Hz, 1H), 8.48 (d, J = 8.5 Hz, 1H), 8.24 (dd, J = 8.8, 1.9 Hz, 1H), 8.14 (dd, J = 7.0, 2.6 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.66 (ddd, J = 8.9, 4.1, 2.8 Hz, 1H), 7.57–7.49 (m, 2H), 7.30 (t, J = 9.5 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 4.31 (q, J = 5.1 Hz, 4H), 3.92 (s, 2H), 2.50 (br s, 4H), 1.75 (br s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 165.52, 164.96, 162.79, 152.32 (d, J = 245.73 Hz), 148.72, 146.93, 143.43, 137.98, 135.93 (d, J = 2.12 Hz), 131.72, 129.23, 128.97, 128.44, 127.93, 126.62, 125.83 (d, J = 13.69 Hz), 122.28, 121.73, 119.27, 119.17 (d, J = 7.12 Hz), 117.36, 117.16, 116.03 (d, J = 20.87 Hz), 64.88, 64.50, 62.47, 54.29, 23.77. HRMS (ESI+): calcd for C30H28FN4O4 (M + H)+, 527.2089; found 527.2074.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-(piperidin-1-ylmethyl)quinoline-6-carboxamide
To a solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (100 mg, 0.212 mmol) in anhydrous DCM (2 mL), piperidine (63 μL, 0.636 mmol) was added dropwise at room temperature, and the resulting mixture was allowed to stir under an inert argon atmosphere for 7 h. Then, sodium triacetoxyborohydride (135 mg, 0.636 mmol) was added in one portion and the resulting mixture was allowed to stir for 2 h at room temperature. The reaction mixture was washed with brine (2 mL) and purified by column chromatography using a gradient of 0–10% MeOH in DCM + 1% 7 N NH3 in MeOH to afford the title compound as a pale yellow amorphous solid (50 mg, 44%). 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 10.18 (s, 1H), 8.64 (s, 1H), 8.48 (d, J = 8.6 Hz, 1H), 8.24 (d, J = 8.7 Hz, 1H), 8.11 (dd, J = 30.3, 9.0 Hz, 2H), 7.74 (d, J = 8.7 Hz, 1H), 7.67–7.59 (m, 1H), 7.58–7.46 (m, 2H), 7.30 (t, J = 9.8 Hz, 1H), 6.99 (d, J = 9.5 Hz, 1H), 4.31 (d, J = 5.6 Hz, 4H), 3.75 (s, 2H), 2.42 (br s, 4H), 1.53 (br s, 4H), 1.42 (br s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.49, 164.94, 162.63, 152.34 (d, J = 248.13 Hz), 148.72, 146.91, 143.41, 137.92, 135.89 (d, J = 2.18 Hz), 131.68, 129.18, 128.96, 128.41, 127.92, 126.62, 125.81 (d, J = 10.90 Hz), 122.19, 121.71, 119.35, 119.18 (d, J = 8.17 Hz), 117.35, 117.15, 116.03 (d, J = 21.79 Hz), 65.65, 64.87, 64.49, 54.79, 26.09, 24.29. HRMS (ESI+): calcd for C31H30FN4O4 (M + H)+, 541.2246; found 541.2242.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((2-methylpyrrolidin-1-yl)methyl)quinoline-6-carboxamide
2-Fluoro-5-nitroaniline (31.7 mg, 0.203 mmol), 2-((2-methylpyrrolidin-1-yl)methyl)quinoline-6-carboxylic acid hydrochloride (62.4 mg, 0.203 mmol), and EDC (78 mg, 0.406 mmol) were dissolved in anhydrous DMF (2 mL), and pyridine (0.082 mL, 1.02 mmol) was added dropwise. The mixture was allowed to stir at 20 °C for 17 h. The reaction mixture was washed with NaHCO3 saturated aqueous solution, extracted with DCM/MeOH 9/1 mixture, and dried over Na2SO4. The crude product was purified by column chromatography using a gradient of 0–10% MeOH in DCM + 1% 7 N NH3 in MeOH to afford the product as a yellow solid, which was carried directly onto the next step (83 mg). LCMS (ESI+): m/z = 409.1655, (M + H)+. N-(2-Fluoro-5-nitrophenyl)-2-((2-methylpyrrolidin-1-yl)methyl)quinoline-6-carboxamide (83 mg, 0.203 mmol), ammonium chloride (76 mg, 1.42 mmol), and iron powder (79 mg, 1.42 mmol) were combined and suspended in EtOH (3 mL) and water (1 mL) at room temperature, affording a beige suspension, which was heated at 90 °C for 1 h. The reaction mixture was cooled to room temperature and filtered through a pad of celite to remove the iron (eluting with EtOH/DCM/MeOH). The solvents were then removed in vacuo. The resulting residue was dried to afford a pale beige solid as a crude product, which was taken onto the next step without purification (77 mg). LCMS (ESI+), m/z = 379.1918, (M + H)+.
N-(5-Amino-2-fluorophenyl)-2-((2-methylpyrrolidin-1-yl)methyl)quinoline-6-carboxamide (77 mg, 0.203 mmol), 2,3-dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (36.7 mg, 0.203 mmol), and EDC (98 mg, 0.509 mmol) were dissolved in anhydrous DMF (1.5 mL); then, pyridine (82 μL, 1.02 mmol) was added dropwise and the resulting mixture was allowed to stir at 20 °C for 72 h. The reaction mixture was washed with water (2 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 5 mL) to afford a pale yellow solid as a crude product, which was purified by flash column chromatography using a gradient of 0–10% MeOH in DCM + 1% 7 N NH3 in MeOH to afford a yellow solid as a semicrude product, which was then repurified by semipreparative TLC (10% MeOH in DCM) to afford the title compound as a pale yellow amorphous solid (30 mg, 27%). 1H NMR (500 MHz, MeOD) δ 8.58 (d, J = 1.9 Hz, 1H), 8.46 (d, J = 8.5 Hz, 1H), 8.27 (dd, J = 8.8, 2.0 Hz, 1H), 8.22–8.11 (m, 2H), 7.78 (d, J = 8.5 Hz, 1H), 7.58 (ddd, J = 8.9, 4.2, 2.7 Hz, 1H), 7.51–7.44 (m, 2H), 7.26–7.18 (m, 1H), 6.94 (d, J = 8.3 Hz, 1H), 4.37–4.26 (m, 5H), 3.63 (d, J = 13.6 Hz, 1H), 3.00 (ddd, J = 10.1, 7.6, 3.5 Hz, 1H), 2.64 (dq, J = 13.5, 6.2 Hz, 1H), 2.37 (q, J = 9.0 Hz, 1H), 2.11–2.00 (m, 1H), 1.77 (dtd, J = 12.5, 9.1, 7.9, 3.2 Hz, 2H), 1.52 (tdd, J = 9.9, 7.3, 4.6 Hz, 1H), 1.22 (d, J = 6.1 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 166.70, 166.61, 165.65, 162.27, 152.47 (d, J = 244.5 Hz), 148.38, 147.12, 143.49, 137.99, 134.88 (d, J = 2.5 Hz), 131.93, 128.19, 127.94, 127.40, 126.73, 125.31 (d, J = 12.4 Hz), 122.37, 120.76, 119.54 (d, J = 9.1 Hz), 118.93, 116.80, 116.65, 115.23 (d, J = 19.1 Hz), 64.53, 64.14, 60.40, 59.89, 54.17, 32.13, 21.14, 17.67. HRMS (ESI+): calcd for C31H30FN4O4 (M + H)+, 541.2246; found 541.2236.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((3-methylazetidin-1-yl)methyl)quinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (0.25 g, 0.530 mmol) and 3-methylazetidine (113 mg, 1.591 mmol) in anhydrous DCM (4 mL) was allowed to stir at 20 °C for 18 h, after which sodium triacetoxyborohydride (0.337 g, 1.591 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 3 h. The reaction was quenched with a NaHCO3 saturated aqueous solution and extracted with a mixture of DCM/MeOH 9/1 (3 × 5 mL). Purification by column chromatography using a gradient of 0–10% MeOH in DCM, followed by further purification by Isolute SCX-II cartridge with MeOH/NH3, afforded the title compound as a pale yellow amorphous solid (114 mg, 41%). 1H NMR (500 MHz, DMSO-d6) δ 10.46 (s, 1H), 10.20 (s, 1H), 8.71 (d, J = 1.7 Hz, 1H), 8.59 (d, J = 8.5 Hz, 1H), 8.32 (dd, J = 8.8, 1.9 Hz, 1H), 8.21–8.08 (m, 2H), 7.64 (ddd, J = 7.6, 4.3, 2.8 Hz, 2H), 7.58–7.44 (m, 2H), 7.38–7.23 (m, 1H), 6.99 (d, J = 8.4 Hz, 1H), 4.66 (s, 2H), 4.43–4.22 (m, 4H), 4.27–4.02 (m, 2H), 3.71 (s, 2H), 2.95–2.76 (m, 1H), 1.23 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.48, 164.95, 161.35, 152.37 (d, J = 244.0 Hz), 148.71, 146.91, 143.41, 138.17, 135.90 (d, J = 1.6 Hz), 131.77, 129.20, 129.02, 128.56, 127.93, 126.60, 125.80 (d, J = 14.0 Hz), 121.89, 121.72, 119.28, 119.23 (d, J = 7.8 Hz), 117.35, 117.17, 116.07 (d, J = 20.4 Hz), 66.85, 64.88, 64.49, 25.90, 21.53, 19.29. HRMS (ESI+): calcd for C30H28FN4O4 (M + H)+, 527.2068; found 527.2089.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((3,3-dimethylazetidin-1-yl)methyl)quinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (0.25 g, 0.530 mmol) and 3,3-dimethylazetidine (135 mg, 1.59 mmol) in anhydrous DCM (4 mL) was allowed to stir at 20 °C for 18 h, after which sodium triacetoxyborohydride (0.337 g, 1.59 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 3 h. The reaction was quenched with a NaHCO3 saturated aqueous solution and extracted with a mixture of DCM/MeOH 9/1 (3 × 5 mL). Purification by column chromatography using a gradient of 0–10% MeOH in DCM, followed by further purification by Isolute SCX-II cartridge with MeOH/NH3, afforded the title compound as a pale yellow solid (156 mg, 54%). 1H NMR (500 MHz, DMSO-d6) δ 10.40 (s, 1H), 10.19 (s, 1H), 8.66 (d, J = 2.0 Hz, 1H), 8.51 (d, J = 8.5 Hz, 1H), 8.26 (dd, J = 8.7, 2.1 Hz, 1H), 8.18–8.06 (m, 2H), 7.69–7.62 (m, 2H), 7.57–7.49 (m, 2H), 7.29 (dd, J = 10.1, 9.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 4.35–4.27 (m, 4H), 4.07 (br s, 2H), 3.22 (br s, 4H), 1.24 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 165.46, 164.95, 161.71, 152.28 (d, J = 250.1 Hz), 148.68, 146.92, 143.41, 138.23, 135.90, 135.88 (d, J = 2.1 Hz), 131.83, 129.20, 129.03, 128.61, 127.92, 126.62, 125.80 (d, J = 15.0 Hz), 121.88, 121.72, 119.28, 119.21 (d, J = 6.0 Hz), 117.35, 117.16, 116.10 (d, J = 21.0 Hz), 66.80, 64.88, 64.49, 32.19, 27.33. HRMS (ESI+): calcd for C31H30FN4O4 (M + H)+, 541.2246; found 541.2240.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-(piperazin-1-ylmethyl)quinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (255 mg, 0.541 mmol) and tert-butyl piperazine-1-carboxylate (302 mg, 1.62 mmol) in anhydrous DCM (5 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (344 mg, 1.62 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (5 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 5 mL). The crude product (pale yellow solid) was purified by column chromatography using a gradient of 0–6% MeOH in DCM, followed by trituration in diethyl ether to afford the desired product as a pale beige amorphous solid (325 mg, 94%). 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 10.18 (s, 1H), 8.65 (d, J = 2.0 Hz, 1H), 8.50 (d, J = 8.6 Hz, 1H), 8.25 (dd, J = 8.8, 2.1 Hz, 1H), 8.19–8.06 (m, 2H), 7.75 (d, J = 8.5 Hz, 1H), 7.65 (ddd, J = 9.0, 4.3, 2.6 Hz, 1H), 7.57–7.49 (m, 2H), 7.30 (dd, J = 10.1, 9.0 Hz, 1H), 6.99 (d, J = 8.5 Hz, 1H), 4.35–4.27 (m, 4H), 3.82 (s, 2H), 3.33 (br s, 4H), 2.44 (br s, 4H), 1.40 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 165.46, 164.94, 161.81, 154.27, 152.26 (d, J = 245.8 Hz), 148.76, 146.91, 143.41, 138.07, 135.89 (d, J = 2.9 Hz), 131.80, 129.23, 128.99, 128.49, 127.92, 126.66, 125.83 (d, J = 12.1 Hz), 122.30, 121.71, 119.27, 119.18 (d, J = 7.89 Hz), 117.35, 117.15, 116.02 (d, J = 21.0 Hz), 79.26, 64.87, 64.69, 64.49, 53.18, 28.52.
To a suspension of tert-butyl 4-((6-((5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)carbamoyl)quinolin-2-yl)methyl)piperazine-1-carboxylate (300 mg, 0.468 mmol) in anhydrous DCM (5 mL), TFA (0.179 mL, 2.338 mmol) was added dropwise, and the resulting mixture was allowed to stir at 20 °C for 3 h. The reaction mixture was concentrated under reduced pressure to afford the crude product as a light brown oil. The crude was purified by column chromatography using a gradient of 0–15% MeOH in DCM followed by trituration in diethyl ether to afford the title compound as an amorphous white solid (174 mg, 68.7%). 1H NMR (500 MHz, DMSO-d6) δ 10.41 (s, 1H), 10.20 (s, 1H), 8.73 (br s, 1H), 8.67 (d, J = 1.7 Hz, 1H), 8.52 (d, J = 8.7 Hz, 1H), 8.27 (dd, J = 8.7, 1.7 Hz, 1H), 8.15 (dd, J = 7.1, 2.7 Hz, 1H), 8.09 (d, J = 8.7 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.66–7.61 (m, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.52 (dd, J = 8.4, 2.1 Hz, 1H), 7.30 (app t, J = 10.1 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 4.35–4.27 (m, 4H), 3.89 (s, 2H), 3.19–3.09 (m, 4H), 2.76–2.66 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 164.98, 164.50, 160.59, 151.87 (d, J = 243.8 Hz), 148.27, 146.47, 142.96, 137.76, 135.45 (d, J = 2.7 Hz), 131.45, 128.79, 128.57, 128.13, 127.46, 126.25, 125.30 (d, J = 13.2 Hz), 121.95, 121.26, 118.86, 118.78 (d, J = 7.8 Hz), 116.90, 116.70, 115.59 (d, J = 20.9 Hz), 64.42, 64.04, 63.67, 49.45, 43.03. HRMS (ESI+): calcd for C30H29FN5O4 (M + H)+, 542.2198; found 542.2190.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((4-methylpiperazin-1-yl)methyl)quinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (5.15 g, 10.92 mmol) and 1-methylpiperazine (3.28 g, 32.8 mmol) in anhydrous DCM (90 mL) was allowed to stir at 20 °C for 18 h, after which sodium triacetoxyborohydride (6.95 g, 32.8 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 1.5 h. The reaction was quenched with NaHCO3 aqueous saturated solution (50 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 50 mL). Purification by column chromatography on silica gel using a gradient of 0–20% MeOH in DCM followed by washing in water and trituration in diethyl ether afforded the title compound as an amorphous white solid (2.85 g, 47%). 1H NMR (500 MHz, DMSO-d6) δ 10.40 (s, 1H), 10.19 (s, 1H), 8.65 (d, J = 1.7 Hz, 1H), 8.49 (d, J = 8.4 Hz, 1H), 8.25 (dd, J = 8.4, 1.7 Hz, 1H), 8.14 (dd, J = 7.6, 2.5 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.72 (d, J = 8.4 Hz, 1H), 7.68–7.63 (m, 1H), 7.55 (d, J = 2.5 Hz, 1H), 7.52 (dd, J = 8.4, 1.7 Hz, 1H), 7.29 (app t, J = 9.2 Hz, 1H), 6.99 (d, J = 9.2 Hz, 1H), 4.37–4.27 (m, 4H), 3.79 (s, 2H), 2.54–2.27 (m, 8H), 2.16 (br s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.49, 164.96, 162.20, 152.38 (d, J = 243.33 Hz), 148.74, 146.93, 143.42, 138.01, 135.89 (d, J = 2.33 Hz), 131.75, 129.22, 128.99, 128.46, 127.93, 126.66, 125.80 (d, J = 13.1 Hz), 122.28, 121.73, 119.31, 119.18 (d, J = 6.7 Hz), 117.36, 117.17, 116.04 (d, J = 20.6 Hz), 64.87, 64.49, 55.21, 53.41, 46.24. HRMS (ESI+): calcd for C31H31FN5O4 (M + H)+, 556.2355; found 556.2329.
2-((4-(sec-Butyl)piperazin-1-yl)methyl)-N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)quinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (100 mg, 0.212 mmol) and 1-(sec-butyl)piperazine (0.103 mL, 0.636 mmol) in anhydrous DCM (2 mL) was allowed to stir at 20 °C for 6 h, after which sodium triacetoxyborohydride (135 mg, 0.636 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with a NaHCO3 (5 mL) aqueous saturated solution and extracted with a DCM/MeOH 9/1 mixture (3 × 5 mL). Purification by column chromatography on silica gel using a gradient of 0–10% MeOH in DCM followed by trituration in diethyl ether afforded the title compound as an amorphous pale pink solid (51 mg, 40%). 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 10.19 (s, 1H), 8.65 (d, J = 1.7 Hz, 1H), 8.49 (d, J = 8.5 Hz, 1H), 8.25 (dd, J = 8.5, 1.7 Hz, 1H), 8.14 (dd, J = 6.8, 2.6 Hz, 1H), 8.08 (d, J = 9.1 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.68–7.63 (m, 1H), 7.54 (d, J = 1.70 Hz, 1H), 7.52 (d, J = 8.5, 2.5 Hz, 1H), 7.29 (app t, J = 9.9 Hz, 1H), 6.99 (d, J = 8.5 Hz, 1H), 4.36–4.27 (m, 4H), 3.79 (s, 2H), 2.61–2.28 (m, 9H), 1.55–1.38 (m, 1H), 1.35–1.16 (m, 1H), 0.91 (br s, 3H), 0.84 (t, J = 7.97 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.49, 164.96, 162.30, 152.30 (d, J = 247.7 Hz), 148.75, 146.93, 143.43, 137.97, 135.89 (d, J = 2.0 Hz), 131.75, 129.22, 128.99, 128.45, 127.93, 126.66, 125.82 (d, J = 13.6 Hz), 122.29, 121.73, 119.28, 119.19 (d, J = 7.5 Hz), 117.36, 117.16, 116.05 (d, J = 21.1 Hz), 64.88, 64.50, 60.14, 54.07, 48.14, 26.26, 14.13, 11.56. HRMS (ESI+): calcd for C34H37FN5O4 (M + H)+, 598.2824; found 598.2808.
2-((4-(tert-Butyl)piperazin-1-yl)methyl)-N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)quinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (100 mg, 0.212 mmol) and 1-(tert-butyl)piperazine (91 mg, 0.636 mmol) in anhydrous DCM (2.0 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (135 mg, 0.636 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (5 mL) and extracted with a mixture of DCM/MeOH 9/1 (3 × 5 mL). The crude product was purified by column chromatography on silica gel using a gradient of 0–15% MeOH in DCM and then washed with water and triturated with diethyl ether to afford the title compound as an amorphous beige solid (63 mg, 50%). 1H NMR (500 MHz, DMSO-d6) δ 10.50 (s, 1H), 10.29 (s, 1H), 8.69 (d, J = 1.5 Hz, 1H), 8.50 (d, J = 8.4 Hz, 1H), 8.27 (dd, J = 8.4, 1.5 Hz, 1H), 8.13 (dd, J = 7.3, 2.4 Hz, 1H), 8.09 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.61–7.66 (m, 1H), 7.57 (d, J = 2.2 Hz, 1H), 7.55 (d, J = 8.5, 2.2 Hz, 1H), 7.28 (app t, J = 9.8 Hz, 1H), 6.98 (d, J = 8.5 Hz, 1H), 4.35–4.27 (m, 4H), 3.85 (s, 2H), 3.09–2.54 (m, 8H), 1.20 (br s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 165.48, 164.96, 161.49, 152.55 (d, J = 247.7 Hz) 148.73, 146.92, 143.41, 138.04, 135.94 (d, J = 2.3 Hz), 131.78, 129.21, 129.00, 128.50, 127.91, 126.66, 125.74 (d, J = 12.4 Hz), 122.33, 121.73, 119.63, 119.42 (d, J = 7.1 Hz), 117.35, 117.23, 116.02 (d, J = 20.7 Hz), 64.87, 64.43, 56.26, 53.11, 45.83, 25.59. HRMS (ESI+): calcd for C34H37FN5O4 (M + H)+, 598.2824; found 598.2809.
(R)-N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((4-ethyl-2-methylpiperazin-1-yl)methyl)quinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (0.200 g, 0.424 mmol) and (R)-tert-butyl 3-methylpiperazine-1-carboxylate (255 mg, 1.27 mmol) in anhydrous DCM (2.00 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (0.270 g, 1.27 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with NaHCO3 aqueous saturated solution (5 mL) and extracted with a mixture of DCM/MeOH 9/1 (3 × 5 mL). The organic layers were dried over Na2SO4 and concentrated under reduced pressure to afford the crude product (R)-tert-butyl 4-((6-((5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)carbamoyl)quinolin-2-yl)methyl)-3-methylpiperazine-1-carboxylate (278 mg), which was dissolved in anhydrous DCM (2.5 mL) and treated with TFA (0.162 mL, 2.12 mmol). The resulting mixture was allowed to stir for 18 h, after which it was concentrated under vacuum to afford the crude product, which was taken onto the next step without purification (185 mg).
To a solution of (R)-N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((2-methylpiperazin-1-yl)methyl)quinoline-6-carboxamide (0.185 g, 0.333 mmol) in anhydrous MeOH (3 mL) at 0 °C, sodium cyanoborohydride (23.02 mg, 0.366 mmol) was added in one portion, followed by the dropwise addition of acetaldehyde (0.013 mL, 0.233 mmol), and the resulting solution was allowed to warm to 20 °C and stirred under argon for 18 h. The solvent was removed under reduced pressure, and the crude was redissolved in DCM and washed with NaOH aqueous solution (1 M). It was purified by column chromatography on silica gel using a gradient of 0–20% MeOH in DCM and then washed with water and triturated with diethyl ether to afford the title compound as an amorphous white solid (27 mg, 14%). 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H), 10.18 (s, 1H), 8.64 (d, J = 2.2 Hz, 1H), 8.48 (d, J = 8.8 Hz, 1H), 8.24 (dd, J = 8.8, 2.2 Hz, 1H), 8.14 (dd, J = 6.6, 2.2 Hz, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.68–7.63 (m, 1H), 7.54 (d, J = 2.2 Hz, 1H), 7.52 (d, J = 8.8, 2.2 Hz, 1H), 7.29 (app t, J = 10.3 Hz, 1H), 6.99 (d, J = 8.8 Hz, 1H), 4.35–4.28 (m, 4H), 4.23 (d, J = 14.2 Hz, 1H), 3.53 (d, J = 14.1 Hz, 1H), 2.74–2.57 (m, 3H), 2.34–2.21 (m, 4H), 2.12–1.99 (m, 1H), 1.95–1.82 (m, 1H), 1.09 (d, J = 6.5 Hz, 3H), 0.98 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.49, 164.95, 163.37, 153.31, 152.41 (d, J = 241.12 Hz), 146.91, 143.41, 137.80, 135.91 (d, J = 2.70 Hz), 131.65, 129.15, 128.99, 128.44, 127.92, 126.58, 125.81 (d, J = 13.49 Hz), 122.32, 121.75, 119.36, 119.21 (d, J = 6.28 Hz), 117.34, 117.19, 116.02 (d, J = 20.16 Hz), 64.87, 64.49, 60.70, 55.68, 53.09, 52.34, 52.02, 29.47, 17.61, 12.44. HRMS (ESI+): calcd for C33H35FN5O4 (M + H)+, 584.2668; found 584.2665.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((4-ethyl-1,4-diazepan-1-yl)methyl)quinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (250 mg, 0.530 mmol) and 1-ethyl-1,4-diazepane (204 mg, 1.59 mmol) in anhydrous DCM (5 mL) was allowed to stir at 20 °C for 18 h, after which sodium triacetoxyborohydride (337 mg, 1.59 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 3 h. The reaction was quenched with a NaHCO3 saturated aqueous solution (5 mL) and extracted with a mixture of DCM/MeOH 9/1 (3 × 5 mL). Purification by column chromatography on silica gel using a gradient of 0–10% MeOH in DCM, followed by elution through an Isolute SCX-II cartridge using MeOH/NH3, and trituration with MeOH, afforded the title compound as an amorphous pale yellow solid (40 mg, 13%). 1H NMR (500 MHz, DMSO-d6) δ 10.43 (s, 1H), 10.23 (s, 1H), 8.68 (d, J = 1.7 Hz, 1H), 8.51 (d, J = 8.5 Hz, 1H), 8.27 (dd, J = 8.8, 1.9 Hz, 1H), 8.14 (dd, J = 7.0, 2.4 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.80 (d, J = 8.5 Hz, 1H), 7.66 (ddd, J = 8.7, 4.1, 2.7 Hz, 1H), 7.58–7.50 (m, 2H), 7.29 (t, J = 9.5 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 4.31 (q, J = 5.0 Hz, 5H), 4.00 (s, 2H), 3.07 (s, 4H), 2.94 (s, 2H), 2.82–2.73 (m, 2H), 1.97 (s, 2H), 1.20 (s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 165.47, 164.96, 162.47, 152.56 (d, J = 242.7 Hz), 148.71, 146.92, 143.42, 138.11, 134.86 (d, J = 2.5 Hz), 131.79, 129.22, 129.04, 128.53, 127.92, 126.70, 125.81 (d, J = 13.9 Hz), 122.28, 121.75, 119.41, 119.28 (d, J = 8.8 Hz), 117.34, 117.20, 116.01 (d, J = 20.2 Hz), 64.88, 64.49, 64.17, 54.26, 53.74, 52.32, 51.78, 49.58, 26.29, 10.79. HRMS (ESI+): calcd for C33H35FN5O4 (M + H)+, 584.2673; found 584.2697.
2-((4-Cyclopropylpiperazin-1-yl)methyl)-N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)quinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (100 mg, 0.212 mmol) and 1-cyclopropylpiperazine (0.077 mL, 0.636 mmol) in anhydrous DCM (2 mL) was allowed to stir at 20 °C for 6 h, after which sodium triacetoxyborohydride (135 mg, 0.636 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with NaHCO3 aqueous saturated solution (5 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 5 mL). Purification by column chromatography on silica gel using a gradient of 0–10% MeOH in DCM, then triturated with diethyl ether, afforded the desired product as a pale yellow amorphous solid (47 mg, 38.1%). 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H), 10.18 (s, 1H), 8.65 (d, J = 1.7 Hz, 1H), 8.49 (d, J = 8.5 Hz, 1H), 8.25 (dd, J = 9.1, 2.6 Hz, 1H), 8.14 (dd, J = 6.8, 2.6 Hz, 1H), 8.08 (d, J = 9.15 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.68–7.63 (m, 1H), 7.54 (d, J = 1.7 Hz, 1H), 7.52 (dd, J = 7.7, 1.7 Hz, 1H), 7.30 (app t, J = 9.9 Hz, 1H), 6.99 (d, J = 8.5 Hz, 1H), 4.34–4.28 (m, 4H), 3.78 (s, 2H), 2.57 (br s, 4H), 2.44 (br s, 4H), 1.61 (app heptet, J = 3.3 Hz, 1H), 0.42–0.37 (m, 2H), 0.29–0.25 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.48, 164.95, 162.19, 152.33 (d, J = 243.7 Hz), 148.73, 146.92, 143.42, 138.00, 135.91 (d, J = 2.4 Hz), 131.74, 129.22, 128.98, 128.45, 127.93, 126.66, 125.81 (d, J = 12.2 Hz), 122.28, 121.72, 119.27, 119.18 (d, J = 7.4 Hz), 117.36, 117.16, 116.05 (d, J = 21.2 Hz), 64.88, 64.49, 53.47, 53.30, 53.26, 38.48, 6.10. HRMS (ESI+): calcd for C33H33FN5O4 (M + H)+, 582.2511; found 582.2487.
N-(2-Bromo-5-nitrophenyl)-2-methylquinoline-6-carboxamide
To a suspension of 2-methylquinoline-6-carboxylic acid (1.50 g, 8.01 mmol) in anhydrous DCM (20 mL), DMF (1.11 μL, 0.014 mmol) followed by oxalyl chloride (0.588 mL, 6.95 mmol) was added dropwise, and the resulting green solution was allowed to stir at 20 °C for 3 h, after which it was concentrated under vacuum to afford a dry pale green solid. The solid was dissolved in pyridine (20 mL), and 2-bromo-5-nitroaniline (1.26 g, 5.79 mmol) was added in one portion. The resulting dark yellow suspension was allowed to stir for 2 h, after which it was poured into water. The resulting yellow precipitate was filtered and washed several times with water, diethyl ether, and finally with a minimum amount of DCM to afford the crude product as an amorphous yellow solid, which was used without further purification (2.47 g). 1H NMR (500 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.65 (d, J = 1.9 Hz, 1H), 8.52 (app t, J = 1.8 Hz, 1H), 8.44 (d, J = 8.3 Hz, 1H), 8.26 (dd, J = 8.9, 1.88 Hz, 1H), 8.09–8.05 (m, 3H), 7.55 (d, J = 8.3 Hz, 1H), 2.71 (s, 3H). HRMS (ESI+): calcd for C17H1379BrN3O3 (M + H)+, 386.0135; found 386.0129.
N-(5-Amino-2-bromophenyl)-2-methylquinoline-6-carboxamide
To a solution of N-(2-bromo-5-nitrophenyl)-2-methylquinoline-6-carboxamide (2.00 g, 5.18 mmol) in water (7 mL) and EtOH (21 mL), ammonium chloride (1.939 g, 36.3 mmol) and iron powder (2.03 g, 36.3 mmol) were added, and the resulting suspension was allowed to stir at 90 °C for 1 h. The reaction mixture was allowed to cool to room temperature, diluted with MeOH and DCM, and filtered through a pad of celite. The resulting filtrate was concentrated under vacuum to afford a light brown solid as a crude product, which was taken onto the next step without purification (1.80 g, 98%). 1H NMR (500 MHz, DMSO-d6) δ 9.94 (s, 1H), 8.59 (d, J = 1.8 Hz, 1H), 8.40 (d, J = 8.8 Hz, 1H), 8.22 (dd, J = 8.8, 1.8 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 8.8 Hz, 1H), 7.28 (d, J = 8.8 Hz, 1H), 6.86 (d, J = 1.8 Hz, 1H), 6.45 (dd, J = 8.8, 1.8 Hz, 1H), 5.40 (br s, 2H), 2.70 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.22, 161.30, 149.30, 148.96, 137.63, 136.84, 132.78, 131.74, 128.87, 128.64, 128.23, 125.84, 123.47, 114.07, 105.01, 49.06, 25.51. HRMS (ESI+): calcd for C17H1579BrN3O (M + H)+, 358.0393; found 358.0386.
N-(2-Bromo-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-methylquinoline-6-carboxamide
To a suspension of 2,3-dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (1.00 g, 5.56 mmol) in anhydrous DCM (20 mL), DMF (0.972 μL, 0.013 mmol) and oxalyl chloride (0.513 mL, 6.06 mmol) were added dropwise, and the resulting green solution was allowed to stir at 20 °C for 3 h, after which it was concentrated under vacuum to afford a dry pale green solid. The solid was dissolved in pyridine (20 mL), and N-(5-amino-2-bromophenyl)-2-methylquinoline-6-carboxamide (1.80 g, 5.05 mmol) was added in one portion. The resulting dark yellow suspension was allowed to stir for 72 h, after which it was poured into water. The resulting yellow precipitate was filtered and washed several times with water, diethyl ether, and finally with a minimum amount of DCM to afford the crude product as a pale yellow amorphous solid, which was used without further purification (2.11 g, 81%). 1H NMR (500 MHz, DMSO-d6) δ 10.29 (s, 1H), 10.27 (s, 1H), 8.63 (s, 1H), 8.42 (d, J = 9.1 Hz, 1H), 8.25 (d, J = 7.3 Hz, 1H), 8.10 (s, 1H), 8.05 (d, J = 8.2 Hz, 1H), 7.69 (br s, 2H), 7.58–7.49 (m, 3H), 6.99 (d, J = 9.1 Hz, 1H), 4.37–4.27 (m, 4H), 2.71 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.49, 165.13, 161.43, 149.03, 147.04, 143.43, 139.67, 137.68, 136.89, 132.90, 131.43, 128.95, 128.81, 128.25, 127.76, 125.86, 123.54, 121.79, 120.72, 120.18, 117.39, 117.21, 114.50, 64.88, 64.49, 25.51. HRMS (ESI+): calcd for C26H2179BrN3NaO4 (M + Na)+, 540.0529; found 520.0542.
N-(2-Bromo-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide
A solution of N-(2-bromo-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-methylquinoline-6-carboxamide (1.00 g, 1.93 mmol) and selenium dioxide (0.235 g, 2.12 mmol) in anhydrous DMF (4 mL) and anhydrous 1,4-dioxane (12 mL) was heated at 150 °C for 1 h. The reaction mixture was allowed to cool to room temperature, and it was diluted with DCM and filtered through a pad of celite. The filtrate was concentrated under vacuum to afford the crude product as a yellow amorphous solid, which was used without purification (1.00 g, 97%). 1H NMR (500 MHz, DMSO-d6) δ 10.47 (s, 1H), 10.28 (s, 1H), 10.17 (d, J = 0.5 Hz, 1H), 8.81–8.77 (m, 2H), 8.42–8.36 (m, 2H), 8.13–8.11 (m, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 1.1 Hz, 2H), 7.55 (d, J = 2.2 Hz, 1H), 7.52 (dd, J = 8.6, 2.2 Hz, 1H), 7.00 (d, J = 8.6 Hz, 1H), 4.36–4.23 (m, 4H). HRMS (ESI+): calcd for C26H1979BrN3O5 (M + H)+ 532.0503; found 532.0513.
2-(Azetidin-1-ylmethyl)-N-(2-bromo-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)quinoline-6-carboxamide
A solution of N-(2-bromo-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (0.500 g, 0.939 mmol) and azetidine (161 mg, 2.82 mmol) in anhydrous DCM (8 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (0.597 g, 2.82 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 1 h. The reaction was quenched with NaHCO3 aqueous saturated solution (10 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 10 mL). Purification by column chromatography on silica gel using a gradient of 0–20% MeOH in DCM, followed by washing with water, trituration with diethyl ether and elution through an Isolute SCX-II cartridge using MeOH/NH3, afforded the title compound as a pale beige amorphous solid (54 mg, 10%). 1H NMR (500 MHz, DMSO-d6) δ 10.33 (s, 1H), 10.27 (s, 1H), 8.67 (d, J = 1.5 Hz, 1H), 8.53 (d, J = 8.0 Hz, 1H), 8.29 (dd, J = 8.8, 1.5 Hz, 1H), 8.13–8.09 (m, 2H), 7.69 (d, J = 1.5 Hz, 1H), 7.65 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 2.4 Hz, 1H), 7.52 (dd, J = 8.0, 1.5 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 4.35–4.28 (m, 4H) 4.19 (br s, 2H), 3.59 (br s, 4H), 2.18 (br s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.31, 165.13, 157.8, 148.47, 147.05, 143.43, 140.01, 139.71, 138.62, 136.81, 132.90, 132.34, 129.29, 128.99, 128.82, 127.74, 126.77, 121.81, 120.80, 120.29, 117.38, 117.23, 114.57, 64.89, 64.48, 61.17, 55.23, 17.44. HRMS (ESI+): calcd for C29H2681BrN4O4 (M + H)+, 575.1116; found 575.1088.
N-(2-Bromo-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxamide
A solution of N-(2-bromo-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (0.500 g, 0.939 mmol) and 1-ethylpiperazine (322 mg, 2.82 mmol) in anhydrous DCM (8 mL) was allowed to stir at 20 °C for 12 h, after which sodium triacetoxyborohydride (0.597 g, 2.82 mmol) was added in one portion, and the resulting mixture was allowed to stir at 20 °C for 1 h. The reaction was quenched with NaHCO3 aqueous saturated solution (10 mL) and extracted with a DCM/MeOH 9/1 mixture (3 × 10 mL). Purification by column chromatography on silica gel using a gradient of 0–20% MeOH in DCM, followed by elution through an Isolute SCX-II cartridge using MeOH/NH3, afforded the title compound as a bright yellow amorphous solid (110 mg, 18%). 1H NMR (500 MHz, DMSO-d6) δ 10.31 (s, 1H), 10.27 (s, 1H), 8.66 (d, J = 2.18 Hz, 1H), 8.50 (d, J = 8.70 Hz, 1H), 8.27 (dd, J = 8.70, 2.18 Hz, 1H), 8.13–8.08 (m, 2H), 7.73 (d, J = 8.70 Hz, 1H), 7.70–7.68 (m, 2H), 7.55 (d, J = 2.18 Hz, 1H), 7.52 (dd, J = 8.70, 2.18 Hz, 1H), 7.00 (d, J = 8.70 Hz, 1H), 4.35–4.28 (m, 4H), 3.81 (br s, 2H), 2.51 (br s, 10 H), 1.02 (br t, J = 6.68 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.39, 165.12, 162.02, 148.74, 147.05, 143.44, 139.69, 138.01, 136.85, 132.90, 131.89, 129.30, 128.83, 128.32, 127.75, 126.71, 122.32, 121.79, 120.73, 120.20, 117.39, 117.21, 114.49, 64.88, 64.68, 64.49, 53.13, 52.61, 51.94, 12.18. HRMS (ESI+): calcd for C32H3379BrN5O4 (M + H)+, 630.1710; found 632.1694.
Synthesis of the Clinical Candidate CCT361814
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxamide (22)
Step 1. N-(2-Fluoro-5-nitrophenyl)-2-methylquinoline-6-carboxamide
Oxalyl chloride (3.25 mL, 38.4 mmol) was added dropwise to a solution of 2-methylquinoline-6-carboxylic acid (6.59 g, 35.2 mmol) and DMF (0.0062 mL, 0.080 mmol) in anhydrous DCM (80 mL). The reaction mixture was stirred at room temperature for 3 h and then concentrated under reduced pressure. The residue was dissolved in DCM (30 mL) and concentrated again under reduced pressure. The resulting dry residue was dissolved in pyridine (80 mL), and 2-fluoro-5-nitroaniline (5.00 g, 32.0 mmol) was added in one portion. The reaction mixture was stirred at room temperature for 18 h and then poured into water (100 mL). The green precipitate was filtered and washed with water (3 × 20 mL), diethyl ether (3 × 20 mL), and DCM (10 mL) to afford the title compound as a light green solid, which was carried onto the next step without further purification (10.4 g). 1H NMR (500 MHz, DMSO-d6) δ 10.70 (s, 1H), 8.72 (dd, J = 6.45, 2.93 Hz, 1H), 8.63 (d, J = 2.02 Hz, 1H), 8.43 (d, J = 8.46 Hz, 1H), 8.23 (dd, J = 8.48, 2.02 Hz, 1H), 8.21–8.16 (m, 1H), 8.05 (d, J = 8.86 Hz, 1H), 7.65 (app t, J = 9.25 Hz, 1H), 7.54 (d, J = 8.46 Hz, 1H), 2.71 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.53, 161.22, 158.67 (d, J = 258.2 Hz), 148.65, 143.72, 137.32, 130.36, 128.88, 128.48, 128.00, 127.08 (d, J = 13.9 Hz), 125.33, 123.18, 122.14 (d, J = 9.6 Hz), 121.25 (d, J = 3.8 Hz), 117.19 (d, J = 22.8 Hz), 25.07. 19F NMR (470 MHz, DMSO-d6) δ −110.20. HRMS (ESI+): calcd for C17H13FN3O3 (M + H)+, 326.0935; found 326.0931.
Step 2. N-(5-Amino-2-fluorophenyl)-2-methylquinoline-6-carboxamide
To a solution of N-(2-fluoro-5-nitrophenyl)-2-methylquinoline-6-carboxamide (10.4 g, 32.0 mmol) in ethanol (120 mL) and water (40 mL), ammonium chloride (12.0 g, 224 mmol) and iron powder (12.5 g, 224 mmol) were added in one portion, and the resulting suspension was allowed to stir at 90 °C for 1 h. The reaction mixture was allowed to cool to room temperature, diluted with MeOH (20 mL) and DCM (20 mL), and filtered through a pad of celite. The resulting filtrate was concentrated under vacuum to afford a light brown solid, which was redissolved in a mixture of DCM/MeOH (9:1, 150 mL) and washed with saturated aqueous NaHCO3 (150 mL). The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure to afford a yellow solid as a crude product, which was taken directly onto the next step without further purification (9.46 g). 1H NMR (500 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.57 (d, J = 1.67 Hz, 1H), 8.39 (d, J = 8.74 Hz, 1H), 8.19 (dd, J = 8.74, 1.67 Hz, 1H), 8.01 (d, J = 8.74 Hz, 1H), 7.52 (d, J = 8.33 Hz, 1H), 6.94 (dd, J = 9.78, 8.28 Hz, 1H), 6.89 (dd, J = 6.58, 2.74 Hz, 1H), 6.46–6.39 (m, 1H), 5.05 (br s, 2H), 2.70 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 164.93, 160.84, 148.49, 147.72 (d, J = 233.9 Hz), 145.08 (d, J = 1.9 Hz), 137.19, 131.19, 128.44, 128.33, 127.95, 125.50 (d, J = 13.1 Hz), 125.34, 122.99, 115.54 (d, J = 20.6 Hz), 111.52, 111.39 (d, J = 6.6 Hz), 25.05. 19F NMR (470 MHz, DMSO-d6) δ −138.12. HRMS (ESI+): calcd for C17H15FN3O (M + H)+, 296.1194; found 296.1191.
Step 3. N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-methylquinoline-6-carboxamide
To a suspension of 2,3-dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (12.7 g, 70.5 mmol) in anhydrous DCM (100 mL) under an inert atmosphere was dropwise added a catalytic amount of anhydrous DMF (6.16 μL, 0.080 mmol) and oxalyl chloride (6.51 mL, 77.0 mmol) and the resulting green solution was allowed to stir at room temperature for 3 h. After which time, the reaction mixture was concentrated under vacuum to afford a dry pale green solid. The solid was dissolved in pyridine (100 mL), and N-(5-amino-2-fluorophenyl)-2-methylquinoline-6-carboxamide (9.46 g, 32.0 mmol) was added in one portion. The resulting dark yellow suspension was allowed to stir for 2 h and was then poured onto water (100 mL). The yellow precipitate was filtered and washed with water (3 × 20 mL), diethyl ether (3 × 20 mL), and DCM (10 mL) to afford the crude product as a pale yellow solid, which was taken directly onto the next step without further purification (12.5 g). 1H NMR (500 MHz, DMSO-d6): δ 10.37 (s, 1H), 10.18 (s, 1H), 8.62 (d, J = 1.65 Hz, 1H), 8.41 (d, J = 8.77 Hz, 1H), 8.23 (dd, J = 8.77, 2.19 Hz, 1H), 8.13 (dd, J = 7.01, 2.63 Hz, 1H), 8.03 (d, J = 8.51 Hz, 1H), 7.68–7.63 (m, 1H), 7.55–7.53 (m, 2H), 7.52 (dd, J = 8.51, 2.09 Hz, 1H), 7.29 (dd, J = 9.98, 8.69 Hz, 1H), 6.99 (d, J = 8.51 Hz, 1H), 4.34–4.28 (m, 4H), 2.71 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.09, 164.48, 160.95, 151.83 (d, J = 243.4 Hz), 148.57, 146.45, 142.95, 137.22, 135.42 (d, J = 2.0 Hz), 130.87, 128.50, 128.41, 127.94, 127.47, 125.39 (d, J = 9.5 Hz), 125.35, 123.05, 121.25, 118.81, 118.75 (d, J = 13.0 Hz), 116.89, 116.69, 115.56 (d, J = 21.2 Hz), 64.41, 64.03, 25.06. 19F NMR (470 MHz, DMSO-d6) δ −126.65. HRMS (ESI+): calcd for C26H21FN3O4 (M + H)+, 458.1511; found 458.1499.
Step 4. N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-methylquinoline-6-carboxamide (5.00 g, 10.9 mmol) and selenium dioxide (1.33 g, 12.0 mmol) in anhydrous DMF (40 mL) and 1,4-dioxane (120 mL) was heated at reflux for 1 h under an inert atmosphere. After which time, the reaction mixture was allowed to cool to room temperature, diluted with DCM (20 mL), and filtered through a pad of celite. The filtrate was concentrated under vacuum (using a heptane/EtOAc azeotrope to remove DMF) to afford the crude product as a yellow solid, which was carried onto the next step without further purification (5.15 g). 1H NMR (500 MHz, DMSO-d6) δ 10.54 (s, 1H), 10.19 (s, 1H), 10.17 (s, 1H), 8.81–8.77 (m, 2H), 8.39 (dd, J = 8.73, 1.95 Hz, 1H), 8.36 (d, J = 8.73 Hz, 1H), 8.17 (dd, J = 6.93, 2.57 Hz, 1H), 8.09 (d, J = 9.26 Hz, 1H), 7.69–7.64 (m, 1H), 7.55 (d, J = 1.99 Hz, 1H), 7.52 (dd, J = 8.30, 1.99 Hz, 1H), 7.31 (app t, J = 9.97 Hz, 1H), 6.99 (d, J = 8.30 Hz, 1H), 4.36–4.27 (m, 4H). HRMS (ESI+): calcd for C26H19FN3O5 (M + H)+, 472.1303; found 472.1286.
Step 5. N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxamide (22)
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (1.19 g, 2.52 mmol) and 1-ethylpiperazine (7.57 mmol, 0.87 g, 0.96 mL) in anhydrous DCM (20 mL) was allowed to stir at 20 °C for 6 h. After which time, sodium triacetoxyborohydride (1.61 g, 7.57 mmol) was added in one portion and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with saturated aqueous NaHCO3 (20 mL) and extracted with a mixture of DCM/MeOH (9:1, 3 × 20 mL). Purification by column chromatography on silica gel in gradient with DCM/MeOH (0–10%) afforded a yellow solid, which was redissolved in DCM/MeOH (9:1, 100 mL) and washed with water (100 mL). The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The final trituration of the resulting residue with diethyl ether (20 mL) afforded the title compound as a white solid (0.95 g, 66%). 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H), 10.18 (s, 1H), 8.65 (d, J = 1.9 Hz, 1H), 8.49 (d, J = 8.4 Hz, 1H), 8.25 (dd, J = 8.8, 2.0 Hz, 1H), 8.14 (dd, J = 7.1, 2.6 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.67–7.64 (m, 1H), 7.55 (d, J = 1.8 Hz, 1H), 7.53 (dd, J = 8.5, 1.8 Hz, 1H),7.29 (app t, J = 9.2 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 4.39–4.18 (m, 4H), 3.79 (s, 2H), 2.48–2.41 (m, 8H), 2.32 (q, J = 7.2 Hz, 2H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 165.02, 164.50, 161.69, 151.85 (d, J = 242.7 Hz), 148.28, 146.46, 142.96, 137.53, 135.44 (d, J = 2.4 Hz), 131.29, 128.76, 128.53, 128.00, 127.47, 126.20, 125.35 (d, J = 12.9 Hz), 121.81, 121.27, 118.83, 118.72 (d, J = 7.3 Hz), 116.89, 116.71, 115.57 (d, J = 21.0 Hz), 64.42, 64.38, 64.04, 52.93, 52.34, 51.60, 11.91. 19F NMR (470 MHz, DMSO-d6) δ −126.63. HRMS (ESI+): calcd for C32H33FN5O4 (M + H)+, 570.2511; found 570.2532.
Acknowledgments
This work was supported by Cancer Research U.K. grant numbers C309/A11566 and C2739/A22897. The authors acknowledge support from the Cancer Research Technology Pioneer Fund and Battle Against Cancer Investment Trust. Paul Workman is a Cancer Research U.K. Life Fellow. The clinical candidate CCT36184 has been licensed by Nuvectis Pharma as NXP800. The authors would like to thank Meirion Richards, Maggie Liu, and Amin Mirza of the ICR Structural Chemistry team for their expertise and assistance. The authors also thank Professors Stan Kaye and Udai Banerji of the ICR/Royal Marsden Drug Development Unit for clinical advice.
Glossary
Abbreviations Used
- MDR
multidrug resistance
- MDR1
multidrug resistance protein 1
- PGP
p-glycoprotein
- CRBN
cereblon
- TGI
tumor growth inhibition
- SD
Sprague-Dawley rats
- HSF1
heat shock factor protein 1
- SAR
structure–activity relationship
- HSP72
heat shock 70 kDa protein 1
- HTS
high-throughput screen
- nM
nanomolar
- μM
micromolar
- IC50
half-minimal (50%) inhibitory concentration
- GI50
concentration for 50% of maximal inhibition of cell proliferation
- SEM
standard error of the mean
- tPSA
topological polar surface area
- MLMs
mouse liver microsomes
- KS
kinetic solubility
- HBF
hepatic blood flow
- PK
pharmacokinetics
- AUC
area under the curve
- AUCu
unbound area under the curve
- Cav0–24h
average concentration over a 24 hour period
- po
per os, oral dose
- qd
quaque die, once-a-day dose
- CLtb
total blood clearance
- CLu
unbound clearance
- CLint
intrinsic clearance
- Vss
volume of distribution at steady state
- Vdu
unbound volume of distribution
- %F
percentage oral bioavailability
- fup
fraction unbound in plasma
- fub
fraction unbound in blood
- fua
fraction unbound in the assay
- B:P
blood-to-plasma ratio
- CYP450
cytochrome P450
- DMF
dimethylformamide
- DCM
dichloromethane
- ND
not determined
- SF
significant figure
- HLMs
human liver microsomes
- RLMs
rat liver microsomes
- WT
wild-type
- KO
knockout
- MMP
matched molecular pair
- n
number of repeats
- N
number of distinct samples
- MHeps
mouse hepatocytes
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00156.
In vitro compound optimization data, in vivo prediction from in vitro data, pharmacokinetic and in vivo efficacy data, ER-stress in vivo biomarker discovery, full chemistry experimental, NMR spectra of key compounds, physicochemical property experimental, full biology experimental, and pharmacokinetics experimental (PDF)
SMILES molecular formula strings (CSV)
Author Contributions
All authors have given approval to the final version of the manuscript. S.Y.S. and M.P. carried out in vitro cellular biology experiments. A.E.P., N.E.A.C., M.J.T., B.W., L.E.E., C.S.R., and M.D.C. synthesized compounds. A.E.P., N.E.A.C., M.J.T., B.W., L.E.E., C.S.R., N.Y.M., M.D.C., and K.J. contributed to the design of compounds. M.L. ran and designed the physicochemical analysis of compounds. A.H., A.M., A.T.H., and F.I.R. carried out internal in vitro and in vivo pharmacokinetic studies. L.P., S.G., and S.A.E. carried out in vivo efficacy experiments. R.t.P. carried out the biomarker qPCR analysis. A.E.P., S.Y.S., N.E.A.C., A.H., L.P., M.J.T., A.M., B.W., L.E.E., C.S.R., N.Y.M., F.I.R., S.G., E.D.B., R.t.P., M.P., S.A.E., P.A.C., P.W., M.D.C., and K.J. designed studies and interpreted results. A.E.P., P.W., and M.D.C. wrote the manuscript.
The authors declare the following competing financial interest(s): The Institute of Cancer Research has a potential financial interest in inhibitors of the HSF1 pathway. CCT36184/NXP800 22 has been licensed to Nuvectis Pharma. All authors are current or previous employees of The Institute of Cancer Research (ICR), which has a commercial interest in a range of drug targets and operates a Rewards to Discoverers scheme, through which employees may receive financial benefits following the commercial licensing of a project. PW is a consultant/scientific advisory board member for Alterome Therapeutics, Astex Pharmaceuticals, Black Diamond Therapeutics, CHARM Therapeutics, CV6 Therapeutics, Epicombi Therapeutics, Merck KGaA, NextechInvest, Nuvectis Pharma, STORM Therapeutics and Vividion Therapeutics (acquired by Bayer AG); holds stock/options in Alterome, Black Diamond, CHARM, Chroma Therapeutics, Epicombi, NextInvest, Nuvectis and STORM; is a non-executive director of STORM; is a former employee of AstraZeneca; and received research funding from Astex, AstraZeneca, Battle Against Cancer Investment Trust (BACIT), Cyclacel Pharmaceuticals, Merck KGaA, Nuvectis, Sixth Element Capital/CRT Pioneer Fund and Vernalis. PAC has received research funding from Astex Pharmaceuticals, Merck KGaA and Nuvectis Pharma.
Supplementary Material
References
- a Banerjee S.; Bookman M. A.; Gore M.. Emerging Therapeutic Targets in Ovarian Cancer; Kaye S.; Brown R.; Gabra H.; Gore M., Eds.; Springer, 2011; pp 1–33. [Google Scholar]; b Torre L. A.; Bray F.; Siegel R. L.; Ferlay J.; Lortet-Tieulent J.; Jemal A. Global cancer statistics, 2012. Cancer J. Clin. 2015, 65, 87–108. 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]; c National Cancer Institute, B. M. SEER Cancer Stat Facts: Ovarian Cancer. Retrieved from National Cancer Institute . B. M. SEER Cancer Stat Facts: Ovarian Cancer. Retrieved from http://seer.cancer.gov/statfacts/html/ovary.html, accessed May 2022.
- a Sudo T. Molecular-targeted therapies for ovarian cancer: prospects for the future. Int. J. Clin. Oncol. 2012, 17, 424–429. 10.1007/s10147-012-0461-1. [DOI] [PubMed] [Google Scholar]; b Ledermann J. A.; Raja F. A.; Fotopoulou C.; Gonzalez-Martin A.; Colombo N.; Sessa C. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi24–32. 10.1093/annonc/mdt333. [DOI] [PubMed] [Google Scholar]
- a Colombo N.; Sessa C.; Bois A. D.; Ledermann J.; McCluggage W. G.; McNeish I.; Morice P.; Pignata S.; Ray-Coquard I.; Vergote I.; Baert T.; Belaroussi I.; Dashora A.; Olbrecht S.; Planchamp F.; Querleu D. ESMO-ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Int. J. Gynecol. Cancer 2019, 29, 728–760. 10.1136/ijgc-2019-000308. [DOI] [PubMed] [Google Scholar]; b Tew W. P.; Lacchetti C.; Kohn E. C. Poly(ADP-ribose) polymerase inhibitors in the management of ovarian cancer: ASCO guideline rapid recommendation update. J. Clin. Oncol. 2022, 40, 3878–3881. 10.1200/JCO.22.01934. [DOI] [PubMed] [Google Scholar]; Online ahead of print. PMID: 36150092
- a Yakirevich E.; Sabo E.; Naroditsky I.; Sova Y.; Lavie O.; Resnick M. B. Multidrug resistance-related phenotype and apoptosis-related protein expression in ovarian serous carcinomas. Gynecol. Oncol. 2006, 100, 152–159. 10.1016/j.ygyno.2005.08.050. [DOI] [PubMed] [Google Scholar]; b Davis A.; Tinker A. V.; Friedlander M. ″Platinum resistant″ ovarian cancer: what is it, who to treat and how to measure benefit?. Gynecol. Oncol. 2014, 133, 624–631. 10.1016/j.ygyno.2014.02.038. [DOI] [PubMed] [Google Scholar]
- Schinkel A. H.; Jonker J. W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Delivery Rev 2012, 64, 138–153. 10.1016/j.addr.2012.09.027. [DOI] [PubMed] [Google Scholar]
- Holohan C.; Van Schaeybroeck S.; Longley D. B.; Johnston P. G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- a Gottesman M. M.; Fojo T.; Bates S. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]; b Freimund A. E.; Beach J. A.; Christie E. L.; Bowtell D. D. L. Mechanisms of drug resistance in high-grade serous ovarian cancer. Hematol. Oncol. Clin. North Am. 2018, 32, 983–996. 10.1016/j.hoc.2018.07.007. [DOI] [PubMed] [Google Scholar]
- a Anckar J.; Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 2011, 80, 1089–1115. 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]; b Li J.; Labbadia J.; Morimoto R. I. Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol. 2017, 27, 895–905. 10.1016/j.tcb.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Whitesell L.; Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Ther. Targets 2009, 13, 469–478. 10.1517/14728220902832697. [DOI] [PubMed] [Google Scholar]; b Moore C. L.; Dewal M. B.; Nekongo E. E.; Santiago S.; Lu N. B.; Levine S. S.; Shoulders M. D. Transportable, chemical genetic methodology for the small molecule-mediated inhibition of heat shock factor 1. ACS Chem. Biol. 2016, 11, 200–210. 10.1021/acschembio.5b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jiang S.; Tu K.; Fu Q.; Schmitt D. C.; Zhou L.; Lu N.; Zhao Y. Multifaceted roles of HSF1 in cancer. Tumor Biol 2015, 36, 4923–4931. 10.1007/s13277-015-3674-x. [DOI] [PubMed] [Google Scholar]; d Pincus D.Regulation of HSF1 and the heat shock response. In Advances in Experimental Medicine and Biology; Springer International Publishing, 2020; Vol. 1243, pp 41–50. [DOI] [PubMed] [Google Scholar]
- a Dai C.; Whitesell L.; Rogers A. B.; Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mendillo M. L.; Santagata S.; Koeva M.; Bell G. W.; Hu R.; Tamimi R. M.; Fraenkel E.; Ince T. A.; Whitesell L.; Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Santagata S.; Hu R.; Lin N. U.; Mendillo M. L.; Collins L. C.; Hankinson S. E.; Schnitt S. J.; Whitesell L.; Tamimi R. M.; Lindquist S.; Ince T. A. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 18378–18383. 10.1073/pnas.1115031108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fang F.; Chang R.; Yang L. Heat shock factor 1 promotes invasion and metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer 2012, 118, 1782–1794. 10.1002/cncr.26482. [DOI] [PubMed] [Google Scholar]; c de Billy E.; Travers J.; Workman P. Shock about heat shock in cancer. Oncotarget 2012, 3, 741–743. 10.18632/oncotarget.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Powell C. D.; Paullin T. R.; Aoisa C.; Menzie C. J.; Ubaldini A.; Westerheide S. D. The heat shock transcription factor HSF1 induces ovarian cancer epithelial-mesenchymal transition in a 3D spheroid growth model. PLoS One 2016, 11, e0168389 10.1371/journal.pone.0168389. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen Y. F.; Wang S. Y.; Yang Y. H.; Zheng J.; Liu T.; Wang L. Targeting HSF1 leads to an antitumor effect in human epithelial ovarian cancer. Int. J. Mol. Med. 2017, 39, 1564–1570. 10.3892/ijmm.2017.2978. [DOI] [PubMed] [Google Scholar]; c Dong B.; Jaeger A. M.; Thiele D. J. Inhibiting heat shock factor 1 in cancer: A unique therapeutic opportunity. Trends Pharmacol. Sci. 2019, 40, 986–1005. 10.1016/j.tips.2019.10.008. [DOI] [PubMed] [Google Scholar]; d Cyran A. M.; Zhitkovich A. Heat shock proteins and HSF1 in cancer. Front. Oncol. 2022, 12, 860320 10.3389/fonc.2022.860320. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Jedlicka P.; Mortin M. A.; Wu C. Multiple functions of Drosophila heat shock transcription factor in vivo. EMBO J. 1997, 16, 2452–2462. 10.1093/emboj/16.9.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Xiao X.; Zuo X.; Davis A. A.; McMillan D. R.; Curry B. B.; Richardson J. A.; Benjamin I. J. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 1999, 18, 5943–5952. 10.1093/emboj/18.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Luo J.; Solimini N. L.; Elledge S. J. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009, 136, 823–837. 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Nagel R.; Semenova E. A.; Berns A. Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016, 17, 1516–1531. 10.15252/embr.201643030. [DOI] [PMC free article] [PubMed] [Google Scholar]; i de Billy E.; Powers M. V.; Smith J. R.; Workman P. Drugging the heat shock factor 1 pathway: exploitation of the critical cancer cell dependence on the guardian of the proteome. Cell Cycle 2009, 8, 3806–3808. 10.4161/cc.8.23.10423. [DOI] [PubMed] [Google Scholar]; j Workman P.Reflections and outlook on targeting HSP90, HSP70 and HSF1 in cancer: A personal perspective. In Advances in Experimental Medicine and Biology; Springer International Publishing, 2020; Vol. 1243, pp 163–179. [DOI] [PubMed] [Google Scholar]
- a Cheeseman M. D.; Chessum N. E. A.; Rye C. S.; Pasqua A. E.; Tucker M. J.; Wilding B.; Evans L. E.; Lepri S.; Richards M.; Sharp S. Y.; Ali S.; Rowlands M.; O’Fee L.; Miah A.; Hayes A.; Henley A. T.; Powers M.; Poele R.; De Billy E.; Pellegrino L.; Raynaud F.; Burke R.; van Montfort R. L. M.; Eccles S. A.; Workman P.; Jones K. discovery of a chemical probe bisamide (CCT251236): An orally bioavailable efficacious pirin ligand from an HSF1 phenotypic screen. J. Med. Chem. 2017, 60, 180–201. 10.1021/acs.jmedchem.6b01055. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jones K.; Rye C.; Chessum N.; Cheeseman M.; Pasqua A. E.; Pike K. G.; Faulder P. F.. Fused 1,4-dihydrodioxin derivatives as inhibitors of heat shock transcription factor 1. WO2015/049535A1, April 9, 2015.; c Rye C. S.; Chessum N. E. A.; Lamont S.; Pike K. G.; Faulder P.; Demeritt J.; Kemmitt P.; Tucker J.; Zani L.; Cheeseman M. D.; Isaac R.; Goodwin L.; Boros J.; Raynaud F.; Hayes A.; Henley A. T.; de Billy E.; Lynch C. J.; Sharp S. Y.; Te Poele R.; O’Fee L.; Foote K. M.; Green S.; Workman P.; Jones K. Discovery of 4,6-disubstituted pyrimidines as potent inhibitors of the heat shock factor 1 (HSF1) stress pathway and CDK9. Med. Chem. Commun. 2016, 7, 1580–1586. 10.1039/C6MD00159A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessum N. E. A.; Sharp S. W.; Caldwell J. J.; Pasqua A. E.; Wilding B.; Colombano G.; Collins I.; Ozer B.; Richards M.; Rowlands M.; Stubbs M.; Burke R.; McAndrew P. C.; Clarke P. A.; Workman P.; Cheeseman M. D.; Jones K. Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766). J. Med. Chem. 2018, 61, 918–933. 10.1021/acs.jmedchem.7b01406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. A.; Morgan P.; Vogel W. M.; Walker D. K. The use of C(av) rather than AUC in safety assessment. Regul. Toxicol. Pharmacol. 2010, 57, 70–73. 10.1016/j.yrtph.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Fok J. H. L.; Hedayat S.; Zhang L.; Aronson L. I.; Mirabella F.; Pawlyn C.; Bright M. D.; Wardell C. P.; Keats J. J.; De Billy E.; C S; Rye C. S.; Chessum N. E. A.; Chessum N. E.; Jones K.; Jones K.; Morgan G. J.; Morgan G. J.; Eccles S. A.; Eccles S. A.; Workman P.; Workman P.; Davies F. E. HSF1 is essential for myeloma cell survival and a promising therapeutic target. Clin. Cancer Res 2018, 24, 2395–2407. 10.1158/1078-0432.CCR-17-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLu is assumed to be an acceptable estimate of CLint,u for low to moderate CLtb compounds: since CLint,u = CL(Qh/(Qh – CLtb))*1/fub. The hepatic blood flow in a rat is assumed to be 70 mL/min/kg. Fmax = (70 – CLtb)*1.42%.
- Smith D. A.; Di L.; Kerns E. H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discovery 2010, 9, 929–939. 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- Sun H.; Pang K. S. Permeability, transport, and metabolism of solutes in Caco-2 cell monolayers: A theoretical study. Drug Metab. Dispos. 2008, 36, 102–123. 10.1124/dmd.107.015321. [DOI] [PubMed] [Google Scholar]
- Coa J. C.; Castrillon W.; Cardona W.; Carda M.; Ospina V.; Munoz J. A.; Velez I. D.; Robledo S. M. Synthesis, leishmanicidal, trypanocidal and cytotoxic activity of quinoline-hydrazone hybrids. Eur. J. Med. Chem. 2015, 101, 746–753. 10.1016/j.ejmech.2015.07.018. [DOI] [PubMed] [Google Scholar]
- All microsomal and hepatocyte stability assays were run at Cyprotex (www.cyprotex.com September 31, 2017). For assay details and scaling factors and methods used for in vivo predictions, see Supporting Information.
- Hepatic blood flow in a mouse was assumed to be 90 mL/min·kg. Fmax = (90 – CLtb)*0.9%.
- a Riley R. J.; McGinnity D. F.; Austin R. P. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab. Dispos. 2005, 33, 1304–1311. 10.1124/dmd.105.004259. [DOI] [PubMed] [Google Scholar]; b Obach R. S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [PubMed] [Google Scholar]
- Chiba M.; Ishii Y.; Sugiyama Y. Prediction of hepatic clearance in human from in vitro data for successful drug development. AAPS J. 2009, 11, 262–276. 10.1208/s12248-009-9103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel A. H.; Wagenaar E.; van Deemter L.; Mol C. A. A. M.; Borst P. Absence of the MDR1A p-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J. Clin. Invest. 1995, 96, 1698–1705. 10.1172/JCI118214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostendorp R. L.; Buckle T.; Beijnen J. H.; van Tellingen O.; Schellens J. H. M. The effect of P-GP (MDR1A/1B), BCRP (BCRP1) and P-GP/BCRP inhibitors on the in vivo absorption, distribution, metabolism and excretion of imatinib. Invest. New Drugs 2009, 27, 31–40. 10.1007/s10637-008-9138-z. [DOI] [PubMed] [Google Scholar]
- Schinkel A. H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Delivery Rev. 1999, 36, 179–194. 10.1016/S0169-409X(98)00085-4. [DOI] [PubMed] [Google Scholar]
- Kartner N.; Riordan J. R.; Ling V. Cell surface p-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 1983, 221, 1285–1288. 10.1126/science.6137059. [DOI] [PubMed] [Google Scholar]
- Sharp S. Y.; Rowlands M. G.; Jarman M.; Kelland L. R. Effects of a new antioestrogen, idoxifene, on cisplatin- and doxorubicin-sensitive and -resistant human ovarian carcinoma cell lines. Br. J. Cancer 1994, 70, 409–414. 10.1038/bjc.1994.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr. Med. Chem. 2006, 13, 1859–1876. 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- For examples of the use of cancer cell resistance to define efflux see:; a Szakács G.; Annereau J.-P.; Lababidi S.; Shankavaram U.; Arciello A.; Bussey K. J.; Reinhold W.; Guo Y.; Kruh G. D.; Reimers M.; Weinstein J. N.; Gottesman M. M. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell 2004, 6, 129–137. 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]; b Alvarez M.; Paull K.; Monks A.; Hose C.; Lee J. S.; Weinstein J.; Grever M.; Bates S.; Fojo T. Generation of a drug resistance profile by quantitation of MDR-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. J. Clin. Invest. 1995, 95, 2205–2214. 10.1172/JCI117910. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wallqvist A.; Rabow A. A.; Shoemaker R. H.; Sausville E. A.; Covell D. G. Linking the growth inhibition response from the National Cancer Institute’s anticancer screen to gene expression levels and other molecular target data. Bioinformatics 2003, 19, 2212–2224. 10.1093/bioinformatics/btg302. [DOI] [PubMed] [Google Scholar]
- a Hitchcock S. A. Structural modifications that alter the p-glycoprotein efflux properties of compounds. J. Med. Chem. 2012, 55, 4877–4895. 10.1021/jm201136z. [DOI] [PubMed] [Google Scholar]; b Waghray D.; Zhang Q. Inhibit or evade multidrug resistance p-glycoprotein in cancer treatment. J. Med. Chem. 2018, 61, 5108–5121. 10.1021/acs.jmedchem.7b01457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levatić J.; Curak J.; Kralj M.; Smuc T.; Osmak M.; Supek F. Accurate models for P-gp drug recognition induced from a cancer cell line cytotoxicity screen. J. Med. Chem. 2013, 56, 5691–5708. 10.1021/jm400328s. [DOI] [PubMed] [Google Scholar]
- Zhao Y. H.; Abraham M. H.; Zissimos A. M. Fast calculation of van der Waals volume as a sum of atomic and bond contributions and its application to drug compounds. J. Org. Chem. 2003, 68, 7368–7373. 10.1021/jo034808o. [DOI] [PubMed] [Google Scholar]
- Patani G. A.; LaVoie E. Bioisosterism: A rational approach to drug design. Chem. Rev. 1996, 96, 3147–3176. 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- Griffen E.; Leach A. G.; Robb G. R.; Warner D. J. Matched molecular pairs as a medicinal chemistry tool. J. Med. Chem. 2011, 54, 7739–7750. 10.1021/jm200452d. [DOI] [PubMed] [Google Scholar]
- Groom C. R.; Bruno I. J.; Lightfoot M. P.; Ward S. C. The Cambridge Structural Database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72, 171179 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gerebtzoff G.; Li-Blatter X.; Fischer H.; Frentzel A.; Seelig A. Halogenation of drugs enhances membrane binding and permeation. ChemBioChem 2004, 5, 676–684. 10.1002/cbic.200400017. [DOI] [PubMed] [Google Scholar]; b Meanwell N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]; c Raub T. J. P-Glycoprotein Recognition of Substrates and Circumvention through Rational Drug Design. Mol. Pharmaceutics 2006, 3, 3–25. 10.1021/mp0500871. [DOI] [PubMed] [Google Scholar]; d Pettersson M.; Hou X.; Kuhn M.; Wager T. T.; Kauffman G. W.; Verhoest P. R. Quantitative assessment of the impact of fluorine substitution on p-glycoprotein (p-gp) mediated efflux, permeability, lipophilicity, and metabolic stability. J. Med. Chem. 2016, 59, 5284–5296. 10.1021/acs.jmedchem.6b00027. [DOI] [PubMed] [Google Scholar]
- The geometric mean total GI50 was corrected for free concentration in the cell assay using the assay free fraction (fua) measured in separate dialysis study (free GI50=GI50*fua), see Table S4 for details.
- The Tumor Growth Inhibition is defined as %TGI = (((Ct – C0) – (Tt – T0))/(Ct – C0))*100; Tt = arithmetic mean volume of treated tumors at time = t; T0 = arithmetic mean volume of treated tumors at time = 0; Ct = arithmetic mean volume of control tumors at time = t; C0 = arithmetic mean volume control tumors at time = 0.
- a Swinney D. C. The value of translational biomarkers to phenotypic assays. Front. Pharmacol. 2014, 5, 1–4. 10.3389/fphar.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Banerji U.; Workman P. Critical parameters in targeted drug development: the pharmacological audit trail. Semin. Oncol. 2016, 43, 436–445. 10.1053/j.seminoncol.2016.06.001. [DOI] [PubMed] [Google Scholar]; c Rossanese O.; Eccles S.; Springer C.; Swain A.; Raynaud F. I.; Workman P.; Kirkin V. The pharmacological audit trail (PhAT): Use of tumor models to address critical issues in the preclinical development of targeted anticancer drugs. Drug Discovery Today: Dis. Models 2016, 21, 23–32. 10.1016/j.ddmod.2017.07.002. [DOI] [Google Scholar]
- See ref 13 for assay details.
- Schwanhäusser B.; Busse D.; Li N.; Dittmar G.; Schuchhardt J.; Wolf J.; Chen W.; Selbach M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Chang A. Heat shock response relieves ER stress. EMBO J. 2008, 27, 1049–1059. 10.1038/emboj.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensen S. M. M.; Heldens L.; van Enckevort C. M. W.; van Genesen S. T.; Pruijn G. J. M.; Lubsen N. H. Heat shock factor 1 is inactivated by amino acid deprivation. Cell Stress Chaperones 2012, 17, 743–755. 10.1007/s12192-012-0347-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Data not shown.
- Mungrue I. N.; Pagnon J.; Kohannim O.; Gargalovic P. S.; Lusis A. J. CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J. Immunol. 2009, 182, 466–476. 10.4049/jimmunol.182.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. Volume of distribution in drug design. J. Med. Chem. 2015, 58, 5691–5698. 10.1021/acs.jmedchem.5b00201. [DOI] [PubMed] [Google Scholar]
- a Yamazaki S.; Nguyen L.; Vekich S.; Shen Z.; Yin M-J.; Mehta P. P.; Kung P-P.; Vicini P. Pharmacokinetic-pharmacodynamic modeling of biomarker response and tumor growth inhibition to an orally available heat shock protein 90 inhibitor in a human tumor xenograft mouse model. J. Pharmacol. Exp. Ther. 2011, 338, 964–973. 10.1124/jpet.111.181339. [DOI] [PubMed] [Google Scholar]; b Tuntland T.; Ethell B.; Kosaka T.; Blasco F.; Zang R. X.; Jain M.; Gould T.; Hoffmaster K. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front. Phamacol. 2014, 5, 174. 10.3389/fphar.2014.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mariappan T. T.; Mandlekar S.; Marathe P. Insight into tissue unbound concentration: Utility in drug discovery and development. Curr. Drug Metab. 2013, 14, 324–340. 10.2174/1389200211314030008. [DOI] [PubMed] [Google Scholar]
- www.mesoscale.com, accessed May 2022. See Supporting Information for details.
- Bowes J.; Brown A. J.; Hamon J.; Jarolimek W.; Sridhar A.; Waldron G.; Whitebread S. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat. Rev. Drug Discovery 2012, 11, 909–922. 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- Carried out at Eurofins. https://www.eurofinsdiscoveryservices.com/services/safety-and-efficacy/safety-pharmacology/safety-panels, accessed May, 2022.
- Carried out at Cyprotex. http://www.cyprotex.com/toxicology/cardiotoxicity/hergsafety/, accessed May, 2022.
- Jones R.; Marschmann M.; Keller M.; Qiu N. H.; Fowler S.; Singer T.; Schuler F.; Funk C.; Schadt S. Shedding light on minipig drug metabolism - elevated amide hydrolysis in vitro. Xenobiotica 2015, 1–12. [DOI] [PubMed] [Google Scholar]
- The dog PK study was carried out at Charles River Laboratories. http://www.criver.com/products-services/drug-discovery/capabilities/dmpk/in-vivo-pk-screening, accessed May, 2022.
- Hepatic blood flow in a dog was assumed to be 40 mL/min/kg Fmax = (40 – CLtb)*2.5%.
- Thummel K.; Kunze K. L.; Shen D. D. Enzyme-catalyzed processes of first-pass hepatic and intestinal drug extraction. Adv. Drug Delivery Rev 1997, 27, 99–127. 10.1016/S0169-409X(97)00039-2. [DOI] [PubMed] [Google Scholar]; HBF: Hepatic blood flow. For a table of assumed HBF values in preclinical species and human see Supporting Information.
- a Sagawa K.; Li F.; Liese R.; Sutton S. C. Fed and fasted gastric pH and gastric residence time in conscious beagle dogs. J. Pharm. Sci. 2009, 98, 2494–2500. 10.1002/jps.21602. [DOI] [PubMed] [Google Scholar]; b Lui C. Y.; Amidon G. L.; Berardi R. R.; Fleisher D.; Youngberg C.; Dressman J. B. Comparison of gastrointestinal pH in dogs and humans: Implications on the use of the beagle dog as a model for oral absorption in humans. J. Pharm. Sci. 1986, 75, 271–274. 10.1002/jps.2600750313. [DOI] [PubMed] [Google Scholar]
- Heikkinen A. T.; Mönkkönen J.; Korjamo T. Kinetics of cellular retention during caco-2 permeation experiments: role of lysosomal sequestration an impact on permeability estimates. J. Pharmacol. Exp. Ther. 2009, 328, 882–892. 10.1124/jpet.108.145797. [DOI] [PubMed] [Google Scholar]
- Carried out at Pharmidex. http://www.pharmidex.com/service/physiocochemical-properties, accessed May, 2022.
- a Hosea N. A.; Collard W. T.; Cole S.; Maurer T. S.; Fang R. X.; Jones H.; Kakar S. M.; Nakai Y.; Smith B. J.; Webster R.; Beaumont K. Prediction of human pharmacokinetics from preclinical information: Comparative accuracy of quantitative prediction approaches. J. Clin. Pharmacol. 2009, 49, 513–533. 10.1177/0091270009333209. [DOI] [PubMed] [Google Scholar]; Human oral bioavailability was assumed to be the same as rat, see:; b Cao X.; Gibbs S. T.; Fang L.; Miller H. A.; Landowski C. P.; Shin H.-C.; Lennernas H.; Zhong Y.; Amidon G. L.; Yu L. X.; Sun D. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm. Res. 2006, 23, 1675–1686. 10.1007/s11095-006-9041-2. [DOI] [PubMed] [Google Scholar]
- Di L.; Atkinson K.; Orozco C. C.; Funk C.; Zhang H.; McDonald T. S.; Tan B.; Lin J.; Chang C.; Obach R. S. In vitro-in vivo correlation for low-clearance compounds using hepatocyte relay method. Drug Metab. Dispos. 2013, 41, 2018–2023. 10.1124/dmd.113.053322. [DOI] [PubMed] [Google Scholar]
- a Zou P.; Yu Y.; Zheng N.; Yang Y.; Paholak H. J.; Yu L. X.; Sun D. Applications of human pharmacokinetic prediction in first-in-human dose estimation. AAPS J. 2012, 14, 262–281. 10.1208/s12248-012-9332-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Obach R. S.; Baxter J. G.; Liston T. E.; Silber B. M.; Jones B. C.; MacIntyre F.; Rance D. J.; Wastall P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [PubMed] [Google Scholar]
- a Workman P. Abstract ND08: NXP800: A first-in-class orally active, small-molecule HSF1 pathway inhibitor. Cancer Res. 2022, 82, ND08. 10.1158/1538-7445.AM2022-ND08. [DOI] [Google Scholar]; b https://clinicaltrials.gov/ct2/show/NCT05226507, accessed November 2022.
- Workman P.; Aboagye E.; Balkwill F.; Balmain A.; Bruder G.; Chaplin D. J.; Double J. A.; Everitt J.; Farningham D. A. H.; Glennie M. J.; Kelland L. R.; Robinson V.; Stratford I. J.; Tozer G. M.; Watson S.; Wedge S. R.; Eccles S. A. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In-house PAINS filter protocol: Biovia Pipeline Pilot Version 9.5.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.