

Abstract
Phosphatidylinositol 5-phosphate 4-kinases (PI5P4Ks), a family of three members in mammals (α, β and γ), have emerged as potential therapeutic targets due to their role in regulating many important cellular signaling pathways. In comparison to the PI5P4Kα and PI5P4Kβ, which usually have similar expression profiles across cancer cells, PI5P4Kγ exhibits distinct expression patterns, and pathological functions for PI5P4Kγ have been proposed in the context of cancer and neurodegenerative diseases. PI5P4Kγ has very low kinase activity and has been proposed to inhibit the PI4P5Ks through scaffolding function, providing a rationale for developing a selective PI5P4Kγ degrader. Here, we report the development and characterization of JWZ-1–80, a first-in-class PI5P4Kγ degrader. JWZ-1–80 potently degrades PI5P4Kγ via the ubiquitin-proteasome system and exhibits proteome-wide selectivity and is therefore a useful tool compound for further dissecting the biological functions of PI5P4Kγ.
Keywords: PI5P4Kγ, PROTAC, Protein degrader, Selective
1. Introduction
Phosphatidylinositol 5-phosphate (PI-5-P) is one of seven phosphoinositides that regulate a wide range of cellular functions through their ability to recruit a wide array of proteins to different intracellular membranes [1,2]. The phosphorylation of the phosphoinositide PI-5-P at the 4-position yields phosphatidylinositol-4,5-bisphosphate (PI-4, 5-P2), which is the precursor for inositol-1,4,5-trisphosphate, diacylglycerol and phosphatidylinositol-3,4,5-trisphosphate [3]. The transformation of PI-5-P to PI-4,5-P2 is catalyzed by the phosphatidylinositol 5-phosphate 4-kinase (PI5P4K) family (Fig. 1A), which consists of three members, α, β and γ, encoded by the genes PIP4K2A, PIP4K2B, and PIP4K2C, respectively [4]. Although all three members are ubiquitously expressed, tissue-specific differences in expression between the three enzymes have been identified [5]. Moreover, PI5P4Kα/β/γ can form heterodimers, which has been shown to regulate the kinase activity, localization, and even introduce kinase-independent functionality [5]. Previous studies that focused on PI5P4Kα and PI5P4Kβ have demonstrated that they are involved in various cell processes, including the stress response, autophagy, and immunological responses; overexpression of these two kinases have been attributed to several malignancies, including breast and pancreatic cancer [6–14]. In contrast to PI5P4Kα/β, which have similar expression profiles in multiple cancers, PI5P4Kγ has a distinct expression profile, suggesting that PI5P4Kγ might exhibit different functions in specific cancer types [15]. Moreover, while the kinase activities of PI5P4Kα/β are well defined, studies of PI5P4Kγ have suggested that it possesses minimal kinase activity compared to the other two enzymes, indicating that PI5P4Kγ may have more dominant scaffolding functions [16]. For example, PI5P4Kγ was reported to suppress PI4P5K-mediated PI-4,5-P2 synthesis through forming an inactive hetero-tetramer with PI4P5K [17]. Nevertheless, PI5P4Kγ has been linked to multiple signaling pathways. For example, PI5P4Kγ has been reported to be a substrate of mTORC1 and in turn negatively regulates mTORC1 activity, thereby forming a feedback loop to tightly control the activity of mTORC1 under starvation conditions [18]. PI5P4Kγ also positively regulates Notch pathway through promoting recycling of the Notch receptor [19]. In addition, PI5P4Kγ plays a central role in mitotic spindle assembly, as deletion of PI5P4Kγ leads to instability of the mitotic spindle microtubules [20]. Finally, a single nucleotide polymorphism near the human PIP4K2C gene is associated with increased susceptibility to the autoimmune disease rheumatoid arthritis [21], and correspondingly, deletion of PI5P4Kγ in mice resulted in immune hyperactivation in aged mice, including elevated levels of proinflammatory cytokines, increased abundance of T cells and a concomitant reduction in the number of regulatory T cells. Therefore, degradation of PI5P4Kγ could serve as a therapeutic strategy to enhance cancer immunotherapy [22].
Fig. 1.

(A) PI-4,5-P2 is generated by phosphorylation of PI-4-P by PI4P5K and PI5P4K-catalyzed phosphorylation of PI-5-P. (B) Chemical structure of pan-PIP4K inhibitor THZ-P1–2 and noncovalent compound Ac-THZ-P1–2. (C) Illustration of potential exit vector from co-crystal structure of THZ-P1–2 and PIP4K2A (PDB-ID: 6OSP).
In addition to cancers, PI5P4Kγ is also a potential therapeutical target for Huntington’s disease, as genetic knockdown of PI5P4Kγ leads to a reduction of mutant huntingtin protein in human patient fibroblasts and diminished aggregation in neurons [23]. Thus, PI5P4Kγ could be a valuable drug target for a variety of diseases, although potential autoimmune effects due to loss of PI5P4Kγ could limit its therapeutic window in non-cancer settings. Nevertheless, developing small molecules that selectively antagonize PI5P4Kγ function is paramount to investigate the role of PI5P4Kγ in these diseases. However, the PI5P4K inhibitors reported to date either lack selectivity against its isoforms or suffer from low potency with a sub-micromolar biochemical IC50 values [16,23–31]. For example, the compounds NIH-12848 and NCT-504 have been reported as selective PI5P4Kγ binders through binding in the allosteric pocket [16,23,31]. However, difficulties in optimizing these leads to improve their binding affinity has diminished the utility of these compounds as cellular probes for PI5P4Kγ.
Proteolysis targeting chimeras (or PROTACs), heterobifunctional molecules that conjugate ligands for a specific protein and an E3 ubiquitin ligase with a linker, has emerged as a promising therapeutic approach to induce protein degradation. Given the additional constraints imposed by successful degradation, PROTACs can be exploited to engender selectivity, even within a family or related kinases [32,33]. For example, Thal-SNS-032, a PROTAC based on the pan-cyclin-dependent kinase (CDK) ligand SNS032, selectively induces CDK9 degradation while sparing other CDKs [34]. Similarly, CDK4 and CDK6 selective degraders have been developed based off of the dual-CDK4/6 inhibitor Palbociclib [35]. Here, we employ PROTACs to develop JWZ-1–80, a potent and selective PI5P4Kγ degrader that serves as the first specific PI5P4K chemical degrader.
2. Results and discussion
We based our initial design on THZ-P1–2, a covalent pan-PI5P4Ks inhibitor recently developed in our lab [26]. Based on the co-crystal structure with PI5P4Kα (PDB: 6OSP), the acrylamide of THZ-P1–2 is exposed to an open surface region and could be used as an exit vector for linker connection (Fig. 1C). Our first series of compounds incorporated the CRBN-binding ligand thalidomide and employed linkers of varied length and composition, as represented by compounds 1–3. We also designed a truncated version of THZ-P1–2 by removing the terminal aminobenzamide to generate compounds 4–9 (Table 1).
Table 1.
Chemical Structures and binding affinity to PI5P4Kγ of Compounds
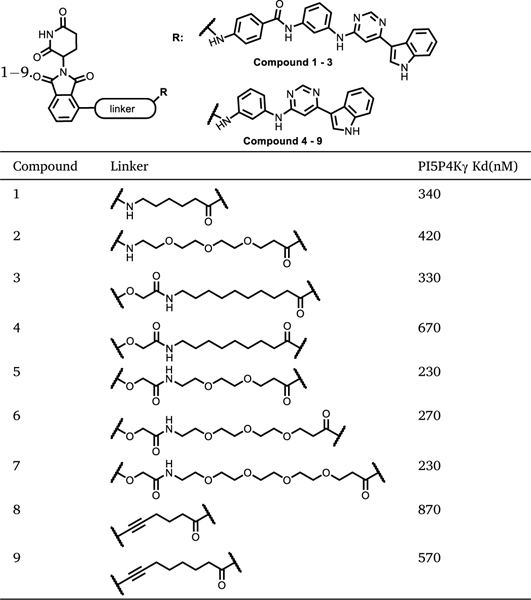
|
The general synthetic route of compounds 1–9 is illustrated in Scheme 1. Briefly, Suzuki coupling of commercially available indole boronic acid A1 and 4,6-dichloropyrimidine A2 generated intermediate A3 (6-chloropyrimidin-4-yl)-1-(phenylsulfonyl)-1H-indole, which subsequently underwent a nucleophilic aromatic substitution (SNAr) reaction with m-phenylenediamine at 150 °C for 4h to give intermediate A4. Condensation of A4 with 4-((tert-butoxycarbonyl)amino)benzoic acid produced intermediate A5, and subsequent deprotection of Boc and phenylsulfonyl afforded A6. Hydrolysis of A4 under basic condition provided another intermediate A7. The final products were synthesized following amide coupling of either intermediate A6 or A7 with CRBN binder linker conjugates.
Scheme 1.

Synthesis of Compounds 1−9.
Reagents and conditions: (a) Pd(PPh3)2Cl2, K2CO3, MeCN, 100 °C; (b) m-phenylenediamine, DIPEA, NMP, 150 °C, 4 h; (c) 4-((tert-butoxycarbonyl)amino)benzoic acid, HATU, DIPEA, DMF, rt; (d) TFA/DCM, rt (e) 2 M NaOH, Dioxane, 80 °C; (f) HATU, DIPEA, ligand, DMF, rt.
The final compounds were first evaluated for PI5P4Kγ binding affinity using the commercial KdELECT assay as shown in Table 1. The parental compound THZ-P1–2 has a Kd of 4.8 nM for PI5P4Kγ binding. In contrast, Ac-THZ-P1–2, the reversible analog of THZ-P1–2, was approximately 7-fold less potent for PI5P4Kγ binding, with a Kd of 34 nM (Fig. 1B). Attaching pomalidomide through alkyl or poly(ethylene glycol) (PEG) linkers resulted in a 10-fold loss of binding affinity (compounds 1–3). Interestingly, PROTACs incorporating the truncated PI5P4K ligand (compounds 4–9) had overall similar binding affinity as the non-truncated compounds, with Kd values ranging from 230 to 870 nM.
Next, we evaluated the ability of this series of compounds to induce cellular degradation of PI5P4Kγ in Molt4 cells. As shown in Fig. 2A, 6 h treatment with either 1 or 10 μM of each compound did not lead to a significant decrease of PI5P4Kγ levels, and 24 h and 48 h treatment failed to reduce PI5P4Kγ(Fig. S1), which suggested that these compounds may not be able to induce a productive ternary complex between PI5P4Kγ and CRBN. Therefore, we next sought to employ VHL, a different E3 ligase substrate adaptor validated for use in PROTACs. We synthesized a set of bivalent compounds with the same PI5P4K ligand used in compounds 1–3 conjugated to VHL recruiting ligands L1– L4 through a variety of exit vectors and linkers (Table 2).
Fig. 2.

(A) Immunoblot analysis of PI5P4Kγ in Molt4 cells treated with CRBN-based compounds for 6 h. (B) Immunoblot analysis of PI5P4Kγ in Molt4 cells treated with VHL-based compounds for 6 h. Quantitation on the right. Quantified data represents mean ± SEM from two independent biological replicates. * p < 0.05; ** p < 0.01; *** p < 0.005 compared with the DMSO -treated control in a t-test.
Table 2.
Chemical Structures and binding affinity to PI5P4Kγ of Compounds 10–20.
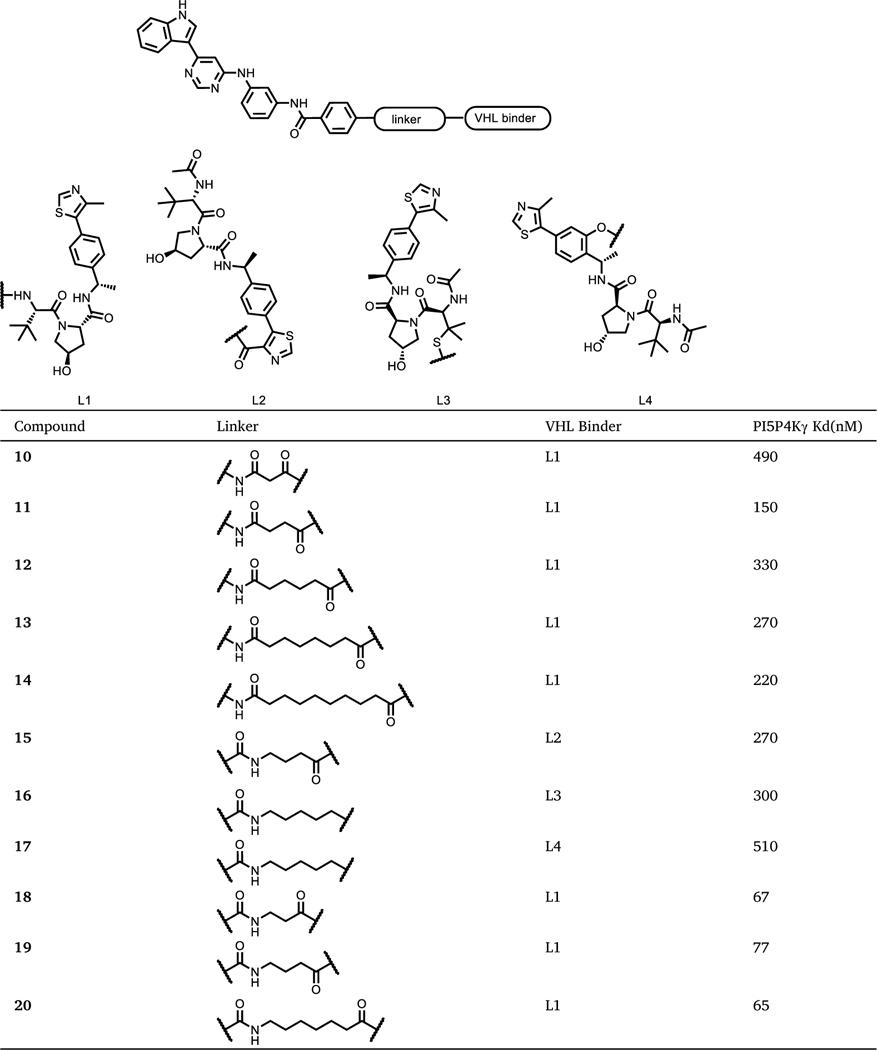
|
The synthesis of VHL-recruiting bivalent compounds is described in Scheme 2. Briefly, HATU-mediated amide coupling of intermediate A6 with VHL ligand L1 linker conjugates afforded compounds 10–14. Intermediate A8 was obtained from the coupling of A4 with methyl 4-(chlorocarbonyl)benzoate, subsequent hydrolysis of A8 under basic condition provided acid A9. Amide coupling of A9 with VHL ligand linker conjugates afforded bivalent molecules 15–20.
Scheme 2.

Synthesis of Compounds 10−20.
Reagents and conditions: (a) HATU, DIPEA, ligand, DMF, rt; (b) methyl 4-(chlorocarbonyl)benzoate, Et3N, DCM 0 °C; (c) 2 M NaOH, Dioxane, 80 °C; (d) HATU, DIPEA, DMF, rt.
The binding affinity of these VHL-recruiting bivalent molecules for PI5P4Kγ ranged from 65 nM to 510 nM (Table 2). Interestingly, the attachment site on the VHL ligands had significant impact on binding affinity to PI5P4Kγ. For example, compound 17 with a phenolic linkage displayed a Kd of 510 nM, which is > 100-fold higher than the parental compound THZ-P1–2, while compounds 18–20 with a linkage to the tert-leucine moiety only exhibited approximately 2-fold loss of binding affinity.
Regardless of binding affinity, all compounds were screened for degradation of PI5P4Kγ in Molt4 cells. As shown in Fig. 2B, while compound 10 induced mild degradation, compounds 11 which possesss slightly longer linkers, induced significant 67% reduction of PI5P4Kγ protein levels with 1 μM treatment. However, further increasing the linker length did not improve degradation potency, as shown by compounds 12 and 13. Compounds 15 and 16, which were designed with a linkage from the thiazole or thioether moieties, respectively, failed to induce any degradation. 6 h treatment of 1 μM compound 17 (JWZ-1–80), which links to the VHL ligand at its phenolic group, induced 75% reduction of PI5P4Kγ. Notably, even though compound 17 was the weakest PI5P4Kγ binder in this second set of compounds, its unique L4 attachment side led to the most efficient degradation effects. This highlights the importance of ternary complex formation ability over potent ligand interactions for the development of degrader molecules.
Next, we performed dose titration and time course experiments with two best compounds 11 and 17. As shown in Fig. 3, degradation was evident after treatment with 500 nM compound 17, and the degradation was observed as early as 2h and sustained up to 24 h. To verify that the observed degradation is VHL-dependent, we synthesized a negative control compound 11-Neg, featuring an analog of the VHL ligand with substantially reduced VHL binding affinity (Fig. 4A). As expected, PI5P4Kγ degradation was not detected using compound 11-Neg (Fig. 4B). In addition, pretreatment with either Ac-THZ-P1–2 or the VHL ligand rescued PI5P4Kγ degradation in the presence of 17 (JWZ-1–80). The rescue was also observed upon co-treatment with a proteasome inhibitor (bortezomib) and the neddylation inhibitor MLN4924, which prevents activation of cullin-RING E3 ligases such as CRL2VHL (Fig. 4C). Taken together, these data support a VHL-dependent mechanism for compound-induced degradation of PI5P4Kγ.
Fig. 3.

(A) Chemical structures of compounds 11 and 17. (B) Immunoblot analysis of PI5P4Kγ in Molt4 cells treated with indicated concentrations of selected compounds 11 and 17. Data in the bar graphs are the means ± SEM (n = 3). * p < 0.05; ** p < 0.01; *** p < 0.005 **** p < 0.0001 compared with the DMSO-treated control in a t-test. (C) Immunoblot analysis of PI5P4Kγ in Molt4 treated with 1 μM of compounds 11 and 17 at the indicated time points.
Fig. 4.

(A) Chemical structure of 11-Neg. (B) Immunoblot analysis of PI5P4Kγ in Molt4 cells treated with 11 or 11-Neg for 6 h. (C) Immunoblot analysis of PI5P4Kγ in Molt4 cells pretreated for 2 h with Ac-THZ-P1–2, VHL ligand, bortezomib or MLN4924, and then treated with 17 for 6 h.
We next inspected PI5P4Ks selectivity for all active degraders (compounds 11–14 and 17). Interestingly, immunoblot analysis indicated that all our PI5P4Kγ-degrading compounds are highly selective for PI5P4Kγ and did not induce degradation of PI5P4Kα or PI5P4Kβ (Fig. 5A). To determining whether the bivalent compounds still bind to PI5P4Kα and PI5P4Kβ, we used commercial KinaseProfiler™ platform for PI5P4Kα and KdELECT for PI5P4Kβ, all the selected compounds lost the binding affinity which explains the selectivity (Table S1). To more broadly assess degrader selectivity, we performed unbiased, multiplexed mass spectrometry (MS)-based proteomics analysis in Molt4 cells, which express all three PI5P4K members. As shown in Fig. 5B, proteomic analysis demonstrated that PI5P4Kγ was the only protein that was significantly degraded. Since the degradation could be cell-type dependent, we examined PI5P4Kγ degradation in four additional cell lines (HEK293T, Jurkat, K562 and MCF7), which demonstrated that robust degradation of PI5P4Kγ can be translated across different cellular models (Fig. 6).
Fig. 5.

(A) Immunoblot analysis of PI5P4Ks in Molt4 cells treated with compounds 11, 12, 13, 14, 17 for 6 h. (B) Quantitative proteomics of compound 17 (JWZ-1–80) showing relative abundance of proteins in Molt4 cells treated for 5 h at 1 μM.
Fig. 6.

(A) Immunoblot analysis of PI5P4Kγ in HEK293 cells treated with compound 17 for 24 h. (B) Immunoblot analysis of PI5P4Kγ in Jurkat cells treated with compound 17 for 24 h. (C) Immunoblot analysis of PI5P4Kγ in K562 cells treated with compound 17 for 24 h. (D) Immunoblot analysis of PI5P4Kγ in MCF7 cells treated with compound 17 for 24 h.
3. Conclusion
In summary, we converted the previously discovered pan-PI5P4K ligand THZ-P1–2 into a potent and selective PI5P4Kγ degrader JWZ-1–80 via conjugation with VHL recruiting ligand. Interestingly, none of the CRBN-based PROTACs synthesized for this study were competent for inducing PI5P4Kγ degradation. We were further able to show that the degradation effects of compound 17 are dependent on VHL and the ubiquitin-proteasome pathway and could be fully rescued by competition with either the parental PI5P4K or VHL ligands. Importantly, JWZ-1–80 exhibited exquisite selectivity for degrading PI5P4Kγ amongst the PI5P4K family as well as across the entire proteome, credentialing JWZ-1–80 as the first selective PI5P4Kγ probe. Finally, we found that PI5P4Kγ degradation could be induced in multiple cell lines, which enables the use of JWZ-1–80 as a selective chemical probe in a variety of biological contexts to investigate the functions of PI5P4Kγ.
4. Experimental section
4.1. Chemical synthesis
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on 500 MHz (Bruker A500), and chemical shifts are reported in parts per million (ppm, d) downfield from tetra-methylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Sunfire C18 column (19 × 50 mm, 5 μM) using a gradient of 15–95% methanol in water containing 0.05% trifluoroacetic acid (TFA) over 22 min (28 min run time) at a flow rate of 20 mL/min. Purities of assayed compounds were in all cases greater than 95%, as determined by reverse-phase HPLC analysis.
4.1.1. Synthesis of 3-(6-chloropyrimidin-4-yl)-1-(phenylsulfonyl)-1H-indole (A3)
4,6-Dichloropyrimidine (300 mg, 2.0 mmol, 1.0 eq), (1-(phenylsulfonyl)-1H-indol-3-yl)boronic acid (602 mg, 2.0 mmol, 1.0 eq), K2CO3 (828 mg, 3.0 eq) and Pd(PPh3)2Cl2 (70 mg, 0.05 eq) were dissolved in acetonitrile (10 mL). The reaction mixture was heated to 100 °C for 12 h under N2 protection. After cooling to room temperature, the solvent was removed under reduced pressure and purified by flash chromatography to give A3 (339 mg) as a brown solid. LC-MS (ESI) m/z: [M+H]+: calcd 370.04, found 370.21.
4.1.2. Synthesis of N 1-(6-(1-(phenylsulfonyl)-1H-indol-3-yl)pyrimidin-4-yl)benzene-1,3-diamine (A4)
A3 (1.0 g, 2.7 mmol, 1.0 eq) and phenylenediamine (1.46 g, 5.0 eq) were dissolved in NMP (10 mL). DIEA (4.7 mL, 10 eq) was added and the reaction mixture was stirred at 150 °C for 4 h. The resulting mixture was cooled to room temperature, diluted with ethyl acetate and water and the organic layer was washed with water and brine. The organic layer was further dried over Na2SO4 and purified by flash chromatography to give A4 (1.3 g) as a brown solid. LC-MS (ESI) m/z: [M+H]+: calcd 442.13, found 442.19.
4.1.3. Synthesis of tert-butyl (4-((3-((6-(1-(phenylsulfonyl)-1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl) carbamoyl)phenyl)carbamate (A5)
To a mixture of A4 (88 mg, 1.0 eq), 4-((tert-butoxycarbonyl)amino) benzoic acid (71 mg, 1.5 eq) and DIPEA (4.0 eq) in DMF (2 mL) was added HATU (114 mg, 1.5 eq). The resultant mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure and purified by flash chromatography to give A5 (101 mg) as a yellow solid. LC-MS (ESI) m/z: [M+H]+: calcd 661.22, found 661.40.
4.1.4. Synthesis of N1-(6-(1H-indol-3-yl)pyrimidin-4-yl)benzene-1,3-diamine(A6)
A5 (101 mg, 1 eq) dissolved in DCM (2 mL) was added TFA (1 mL) and the reaction mixture was stirred at rt for 3 h. The solvent was removed under reduced pressure to give crude product 4-amino-N-(3-((6-(1-(phenylsulfonyl)-1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl) benzamide which was used in next step without further purification. LC-MS (ESI) m/z: [M+H]+: calcd 561.17, found 561.14. 4-amino-N-(3-((6-(1-(phenylsulfonyl)-1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)benzamidewas dissolved in dioxane (2 mL) and then 2 M NaOH (1 mL) was added. The mixture was stirred at 80 °C for 3 h. The resulting mixture was cooled to room temperature, acidified to pH = 5–7 with 2 M HCl, exacted with EtOAc and the organic layer was washed with water and brine. The organic layer was removed and purified by flash chromatography to give. A6 (57 mg) as a yellow solid. LC-MS (ESI) m/z: [M+H]+: calcd 421.18, found 421. 07.
4.1.5. Synthesis of N1-(6-(1H-indol-3-yl)pyrimidin-4-yl)benzene-1,3-diamine (A7)
To A4 (300 mg, 0.67 mmol, 1 eq) in dioxane (3 mL) was added 2 M NaOH (2 mL) and the reaction mixture was stirred at 80 °C for 3 h. The mixture was cooled to room temperature, acidified to pH =5–7 with 2 M HCl, extracted with EtOAc and the organic layer was washed with water and brine. The organic layer was removed under reduced pressure and purified by flash chromatography to give A7 (167 mg) as a yellow solid. LC-MS (ESI) m/z: [M+H]+: calcd 302.14, found 302.23.
4.1.6. Synthesis of methyl 4-((3-((6-(1-(phenylsulfonyl)-1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)carbamoyl)benzoate (A8)
A4 (100 mg, 0.23 mmol, 1.0 eq) and Et3N (63 μL, 3.0 eq) were added to methyl 4-(chlorocarbonyl)benzoate (50 mg, 2.0 eq) in DCM (2 mL) at 0 °C. The reaction mixture was stirred at rt overnight. The solvent was removed under reduce pressure and diluted with water, extracted with ethyl acetate and the organic layer was washed with water and brine. The organic layer was further dried over Na2SO4 and purified by flash chromatography to give A8(120 mg) as a yellow solid. LC-MS (ESI) m/z: [M+H]+: calcd 604.17, found 604.28.
4.1.7. Synthesis of 4-((3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)carbamoyl)benzoic acid(A9)
To A8 (120 mg 1.0 eq) in dioxane (2 mL) was added 2 M NaOH (1 mL) and the reaction mixture was stirred at 80 °C for 3 h. The resulting mixture was cooled to room temperature, acidified to pH =5–7 with 2 M HCl, exacted with EtOAc and the organic layer was washed with water and brine. The organic layer was further dried over Na2SO4 and purified by flash chromatography to give A9 (57 mg) as yellow solid. LC-MS (ESI) m/z: [M+H]+: calcd 450.16, found 450.23.
4.2. General synthesis of compound 1 – 20
To a mixture of A6 (or A7)(1.0 eq), carboxylic acid linker (1.5 eq.) and DIPEA (4.0 eq) in DMF (1 mL) was added HATU (1.5 eq). The resultant mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure and purified by HPLC to give final compounds.
4.2.1. N-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-4-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexanamido) benzamide (compound 1)
8 mg as a white solid. LC-MS (ESI) m/z: [M+H]+: calcd 790.31, found 790.37.
1H NMR (500 MHz, DMSO-d6) δ 12.08 (s, 1H), 11.09 (s, 1H),10.32 (s, 1H), 10.23 (s, 1H), 9.49 (s, 1H), 8.76 (s, 1H), 8.27 (d, J = 3.0 Hz, 1H), 8.24 (d, J = 2.1 Hz, 1H), 8.12 (d, J = 7.7 Hz,1H), 7.97 (d, J = 8.6 Hz, 2H), 7.75 (d, J = 8.8 Hz, 2H), 7.60–7.54 (m, 2H), 7.52 (d, J = 8.1 Hz, 1H), 7.43 (d, J = 8.2 Hz, 1H), 7.41–7.34 (m, 2H), 7.26 (p, J = 7.0 Hz, 2H) 7.08 (d, J = 8.8 Hz, 1H), 7.02 (d, J = 7.2 Hz, 1H), 6.53 (t, J = 6.0 Hz, 1H), 5.06 (dd, J = 5.6, 12.8 Hz, 1H), 3.31 (q, J = 6.8 Hz, 2H), 2.94–2.81 (m, 1H), 2.62–2.49 (m, 2H), 2.32 (t, J = 7.2 Hz, 2H), 2.08–2.01 (m, 1H), 1.62–1.51 (m, 4H), 1.40–1.32 (m, 2H).
4.2.2. N-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-4-(3-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)propanamido)benzamide (compound 2)
9 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 880.34, found 880.40.
1H NMR (500 MHz, DMSO-d6) δ 12.07 (s, 1H), 11.10 (s, 1H), 10.32 (s, 1H), 10.22.
(s, 1H), 9.48 (s, 1H), 8.76 (s, 1H), 8.27 (d, J = 3.0 Hz, 1H), 8.24 (d, J = 2.1 Hz, 1H), 8.12 (d, J = 7.7 Hz,1H), 7.97 (d, J = 8.6 Hz, 2H), 7.75 (d, J = 8.8 Hz, 2H), 7.59–7.54 (m, 2H), 7.52 (d, J =8.1 Hz, 1H), 7.43 (d, J = 8.2 Hz, 1H), 7.41–7.34 (m, 2H), 7.28–7.20 (m, 2H), 7.15 (d, J = 8.6 Hz, 1H), 7.02 (d, J =7.0 Hz, 1H), 6.62 (t, J = 5.8 Hz, 1H), 5.05 (dd, J = 12.8, 5.4 Hz, 1H), 3.64–3 0.47 (m, 14H), 2.93–2.82 (m, 1H), 2.66–2.51 (m, 2H), 2.18 (t, J = 8.1 Hz, 2H), 2.07–1.99 (m, 1H).
4.2.3. N-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-4-(10-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido) decanamido)benzamide (compound 3)
7 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 904.38, found 904.45.
1H NMR (500 MHz, DMSO-d6) δ 12.13 (s, 1H), 11.12 (s, 1H), 10.48 (s, 1H), 10.20 (d, J = 35.8 Hz, 2H), 8.80 (s, 1H), 8.37–8.18 (m, 2H), 8.09 (d, J = 7.7 Hz, 1H), 7.95 (t, J = 8.3 Hz, 3H), 7.85–7.78 (m, 1H), 7.75 (d, J = 8.7 Hz, 2H), 7.58 (d, J = 7.8 Hz, 1H), 7.52 (dd, J = 15.5, 7.5 Hz, 2H), 7.45 (d, J = 8.2 Hz, 1H), 7.42–7.35 (m, 3H), 7.28 (p, J = 7.1 Hz, 2H), 5.12 (dd, J = 12.7, 5.4 Hz, 1H), 4.77 (s, 2H), 3.14 (q, J = 6.7 Hz, 3H), 2.90 (ddd, J = 16.9, 13.7, 5.5 Hz, 2H), 2.64–2.53 (m, 2H), 2.35 (t, J = 7.5 Hz, 2H), 2.10–1.99 (m, 1H), 1.60 (t, J = 7.2 Hz, 2H), 1.45–1.40 (m, 3H), 1.30–1.25 (m, 10H).
4.2.4. N-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-9-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido) nonanamide (compound 4)
7.8 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 771.33, found 771.46.
1H NMR (500 MHz, DMSO-d6) δ 12.24 (s, 1H), 11.12 (s, 1H), 10.68 (s, 1H), 10.01 (s, 1H), 8.83 (s, 1H), 8.33 (d, J = 3.1 Hz, 1H), 8.09 (d, J = 2.2 Hz, 1H), 8.03 (d, J = 7.5 Hz, 1H), 7.94 (t, J = 5.8 Hz, 1H), 7.84–7.78 (m, 1H), 7.63–7.57 (m, 1H), 7.50 (d, J =7.2 Hz, 2H), 7.42–7.22 (m, 6H), 5.12 (dd, J = 12.8, 5.4 Hz, 1H), 4.77 (s, 2H), 3.14 (q, J = 6.7 Hz, 2H), 2.98–2.82 (m, 1H), 2.60 (dt, J = 17.1, 3.0 Hz, 1H), 2.33 (t, J = 7.4 Hz, 2H), 2.09–1.99 (m, 1H), 1.60 (t, J =7.2 Hz, 2H), 1.44 (t, J =6.8 Hz, 2H), 1.36–1.17 (m, 8H).
4.2.5. N-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-3-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido) ethoxy)ethoxy)propanamide (compound 5)
5.8 mg solid. LC-MS (ESI) m/z: [M+H]+: calcd 775.28, found 775.35.
1H NMR (500 MHz, DMSO-d6) δ 12.24 (s, 1H), 11.12 (s, 1H), 10.68 (s, 1H), 10.09 (s, 1H), 8.83 (s, 1H), 8.33 (d, J = 3.1 Hz, 1H), 8.09 (d, J = 2.1 Hz, 1H), 8.07–7.95 (m, 2H), 7.79 (dd, J = 8.5, 7.3 Hz, 1H), 7.65–7.57 (m, 1H), 7.48 (d, J = 7.2 Hz, 2H), 7.43–7.25 (m, 6H), 5.11 (dd, J = 12.8, 5.5 Hz, 1H), 4.78 (s, 2H), 3.56–3.52 (m, 8H), 3.47 (t, J = 5.7 Hz, 2H), 3.30 (d, J = 5.7 Hz, 2H), 2.97–2.84 (m, 1H), 2.74 (s, 1H), 2.59 (t, J = 6.3 Hz, 2H), 2.09–1.96 (m, 2H).
4.2.6. N-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-3-(2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido) ethoxy)ethoxy)ethoxy)propanamide (compound 6)
7.3 mg as a white solid. LC-MS (ESI) m/z: [M+H]+: calcd 819.31, found 819.29.
1H NMR (500 MHz, DMSO-d6) δ 12.25 (s, 1H), 11.12 (s, 1H), 10.69 (s, 1H), 10.09 (s, 1H), 8.84 (s, 1H), 8.33 (d, J = 3.1 Hz, 1H), 8.10 (d, J = 2.2 Hz, 1H), 8.06–7.96 (m, 2H), 7.83–7.75 (m, 1H), 7.64–7.56 (m, 1H), 7.49 (d, J = 7.3 Hz, 2H), 7.45–7.25 (m, 6H), 5.11 (dd, J = 12.8, 5.5 Hz, 1H), 4.78 (s, 2H), 3.58–3.48 (m, 12H), 3.44 (t, J = 5.7 Hz, 2H), 3.30 (q, J = 5.7 Hz, 2H), 2.89 (ddd, J = 17.1, 13.6, 5.4 Hz, 1H), 2.58 (q, J = 5.0, 3.8 Hz, 2H), 2.11–1.97 (m, 1H).
4.2.7. N-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-1-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)-3,6,9,12-tetraoxapentadecan-15-amide (compound 7)
7.3 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 863.34, found 863.51.
1H NMR (500 MHz, DMSO-d6) δ 12.21 (s, 1H), 11.12 (s, 1H), 10.61 (s, 1H), 10.08 (s, 1H), 8.82 (s, 1H), 8.31 (d, J = 3.1 Hz, 1H), 8.10 (d, J = 2.2 Hz, 1H), 8.04 (d, J = 7.4 Hz, 1H), 8.00 (t, J = 5.7 Hz, 1H), 7.80 (dd, J = 8.5, 7.3 Hz, 1H), 7.60 (dd, J = 6.7, 2.3 Hz, 1H), 7.49 (d, J = 7.2 Hz, 2H), 7.42–7.23 (m, 6H), 5.11 (dd, J = 12.8, 5.5 Hz, 1H), 4.78 (s, 2H), 3.72 (t, J = 6.2 Hz, 2H), 3.54–3.41 (m, 16H), 3.31 (q, J = 5.7 Hz, 2H), 2.89 (ddd, J = 17.0, 13.8, 5.4 Hz, 1H), 2.59 (t, J = 6.2 Hz, 2H), 2.11–2.04 (m, 1H).
4.2.8. N-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-6-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)hex-5-ynamide (compound 8)
7.7 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 652.23, found 652.29.
1H NMR (500 MHz, DMSO-d6) δ 12.18 (s, 1H), 11.14 (s, 1H), 10.54 (s, 1H), 10.12 (s, 1H), 8.81 (s, 1H), 8.31 (d, J = 3.1 Hz, 1H), 8.13–8.01 (m, 2H), 7.87 (hept, J = 4.8, 4.2 Hz, 3H), 7.62–7.55 (m, 1H), 7.53–7.43 (m, 1H), 7.41–7.20 (m, 5H), 5.15 (dd, J = 12.9, 5.4 Hz, 1H), 2.88 (ddd, J = 17.1, 13.8, 5.4 Hz, 1H), 2.59 (dt, J = 19.3, 7.1 Hz, 6H), 2.05 (dtd, J = 13.0, 5.2, 2.2 Hz, 1H), 2.01–1.90 (m, 2H).
4.2.9. N-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-8-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oct-7-ynamide (compound 9)
6.1 mg white solid.LC-MS (ESI) m/z: [M+H]+: calcd 680.26, found 680.37.
1H NMR (500 MHz, DMSO-d6) δ 12.18 (s, 1H), 11.14 (s, 1H), 10.55 (s, 1H), 10.04 (s, 1H), 8.81 (s, 1H), 8.31 (d, J = 3.1 Hz, 1H), 8.14–7.96 (m, 2H), 7.91–7.77 (m, 3H), 7.63–7.56 (m, 1H), 7.49 (s, 1H), 7.41–7.24 (m, 5H), 5.15 (dd, J = 12.9, 5.4 Hz, 1H), 2.88 (ddd, J = 17.1, 13.9, 5.4 Hz, 1H), 2.39 (t, J = 7.3 Hz, 2H), 2.09–1.99 (m, 1H), 1.66 (dq, J = 24.8, 7.3 Hz, 6H), 1.51 (ddt, J = 15.1, 9.8, 5.8 Hz, 2H).
4.2.10. N1-(4-((3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl) carbamoyl)phenyl)-N4-((2S)-1-((4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)succinamide (compound 10)
6.2 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 933.39, found 933.45.
1H NMR (500 MHz, DMSO-d6) δ 12.36 (s, 1H), 10.95 (s, 1H), 10.42 (s, 1H), 10.33 (s, 1H), 8.99 (s, 1H), 8.89 (s, 1H), 8.47–8.35 (m, 2H), 8.31–8.19 (m, 2H), 8.00 (dd, J = 20.4, 8.1 Hz, 3H), 7.76 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 7.8 Hz, 1H), 7.55–7.48 (m, 2H), 7.48–7.37 (m, 6H), 7.36–7.27 (m, 2H), 4.94 (t, J = 7.2 Hz, 1H), 4.57 (d, J = 9.3 Hz, 1H), 4.46 (t, J = 8.1 Hz, 1H), 3.71–3.55 (m, 2H), 3.54–3.28 (m, 2H), 2.46 (s, 3H), 2.10–1.98 (m, 1H), 1.39 (d, J = 7.0 Hz, 3H), 0.98 (s, 9H).
4.2.11. N1-(4-((3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl) carbamoyl)phenyl)-N4-((2S)-1-((4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)succinamide (compound 11)
10.6 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 947.40, found 947.55.
1H NMR (500 MHz, DMSO-d6) δ 12.34 (s, 1H), 10.95 (s, 1H), 10.29 (s, 1H), 10.26 (s, 1H), 9.00 (s, 1H), 8.89 (s, 1H), 8.44–8.34 (m, 2H), 8.29 (d, J = 2.2 Hz, 1H), 8.02 (d, J = 7.7 Hz, 1H), 7.95 (dd, J = 13.5, 9.0 Hz, 3H), 7.75 (d, J = 8.8 Hz, 1H), 7.68–7.58 (m, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.46–7.41 (m, 3H), 7.39 (d, J = 8.4 Hz, 2H), 7.31 (td, J = 14.4, 6.3 Hz, 2H), 4.96–4.89 (m, 1H), 4.54 (d, J = 9.3 Hz, 1H), 4.44 (t, J = 8.0 Hz, 1H), 3.66–3.55 (m, 2H), 2.71–2.54 (m, 2H), 2.46 (s, 3H), 2.11–1.94 (m, 2H), 1.80 (ddd, J = 12.9, 8.5, 4.7 Hz, 1H), 1.39 (d, J = 7.0 Hz, 3H), 0.95 (s, 9H).
4.2.12. N1-(4-((3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)carbamoyl)phenyl)-N4-((S)-1-((2S,4S)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)succinamide(compound 11-NEG)
11.3 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 947.40, found 947.46.
1H NMR (500 MHz, DMSO-d6) δ 12.30 (s, 1H), 10.85 (s, 1H), 10.27 (s, 1H), 10.24 (s, 1H), 8.97 (s, 1H), 8.87 (s, 1H), 8.37 (d, J = 3.2 Hz, 1H), 8.28 (d, J = 2.2 Hz, 1H), 8.09–8.03 (m, 3H), 7.98–7.91 (m, 2H), 7.72 (d, J = 8.6 Hz, 2H), 7.64–7.58 (m, 1H), 7.57–7.36 (m, 8H), 7.35–7.25 (m, 2H), 4.94–4.88 (m, 1H), 4.45 (d, J = 8.2 Hz, 1H), 4.40 (dd, J = 8.2, 5.9 Hz, 1H), 4.34–4.30 (m, 1H), 3.81–3.77 (m, 1H), 2.71–2.54 (m, 3H), 2.46 (s, 3H), 2.42 (d, J = 4.9 Hz, 1H), 2.05–1.91 (m, 2H), 1.34 (d, J = 7.0 Hz, 3H), 0.98 (s, 9H).
4.2.13. N1-(4-((3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)carbamoyl)phenyl)-N6-((2S)-1-((4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)adipamide (compound 12)
11.3 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 975.43, found 975.45.
1H NMR (500 MHz, DMSO-d6) δ 12.25 (s, 1H), 10.77 (s, 1H), 10.26 (d, J = 8.3 Hz, 1H), 10.18 (s, 1H), 8.99 (s, 1H), 8.86 (s, 1H), 8.40–8.31 (m, 2H), 8.28 (d, J =2.2 Hz, 1H), 8.04 (d, J =7.8 Hz, 1H), 8.00–7.91 (m, 2H), 7.83 (d, J = 9.2 Hz, 1H), 7.76 (d, J = 8.6 Hz, 2H), 7.61 (d, J = 7.8 Hz, 1H), 7.53 (dd, J = 8.3, 3.8 Hz, 1H), 7.48 (d, J = 8.3 Hz, 1H), 7.46–7.36 (m, 6H), 7.32 (q, J = 8.5, 7.6 Hz, 2H), 4.98–4.85 (m, 1H), 4.53 (d, J = 9.3 Hz, 1H), 4.43 (t, J = 8.0 Hz, 1H), 4.29 (q, J = 3.6 Hz, 1H), 3.64–3.62 (m, 1H), 2.46 (s, 3H), 2.36 (q, J = 8.2, 7.4 Hz, 2H), 2.33–2.26 (m, 1H), 2.18 (dt, J = 13.9, 6.9 Hz, 1H), 2.08 (s, 1H), 2.02 (ddd, J = 11.0, 7.8, 2.8 Hz, 1H), 1.80 (ddd, J = 12.9, 8.5, 4.6 Hz, 1H), 1.57 (ddd, J = 26.6, 14.1, 7.5 Hz, 4H), 1.38 (d, J = 7.0 Hz, 3H), 0.94 (d, J = 7.7 Hz, 9H).
4.2.14. N1-(4-((3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)carbamoyl)phenyl)-N8-((2S)-1-((4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)octanediamide (compound 13)
9.1 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 1003.47, found 1003.61.
1H NMR (500 MHz, DMSO-d6) δ 12.30 (s, 1H), 10.84 (s, 1H), 10.23 (d, J = 46.7 Hz, 2H), 8.93 (d, J = 59.4 Hz, 2H), 8.41–8.32 (m, 2H), 8.28 (d, J = 2.2 Hz, 1H), 8.04 (d, J = 7.7 Hz, 1H), 7.99–7.92 (m, 2H), 7.82–7.72 (m, 3H), 7.64–7.58 (m, 1H), 7.57–7.47 (m, 2H), 7.46–7.35 (m, 7H), 7.36–7.27 (m, 3H), 4.99–4.83 (m, 2H), 4.57–4.36 (m, 4H), 4.29 (d, J = 5.3 Hz, 2H), 2.46 (s, 4H), 2.35 (t, J = 7.4 Hz, 2H), 2.31–2.22 (m, 2H), 2.19–2.08 (m, 2H), 2.01 (d, J = 10.2 Hz, 1H), 1.80 (ddd, J = 12.9, 8.5, 4.6 Hz, 2H), 1.66–1.43 (m, 6H), 1.38 (d, J = 7.0 Hz, 4H), 1.34–1.23 (m, 5H), 0.94 (d, J = 2.6 Hz, 9H).
4.2.15. N1-(4-((3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)carbamoyl)phenyl)-N10-((2S)-1-((4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)decanediamide (compound 14)
6.6 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 1031.50, found 1031.55.
1H NMR (500 MHz, DMSO-d6) δ 12.29 (s, 1H), 10.82 (s, 1H), 10.28 (s, 1H), 10.18 (s, 1H), 8.98 (s, 1H), 8.86 (s, 1H), 8.43–8.32 (m, 2H), 8.28 (d, J = 2.2 Hz, 1H), 8.04 (d, J = 7.6 Hz, 1H), 8.01–7.88 (m, 2H), 7.82–7.69 (m, 3H), 7.65–7.57 (m, 1H), 7.57–7.46 (m, 1H), 7.48–7.20 (m, 8H), 4.91 (q, J = 7.3 Hz, 1H), 4.52 (d, J = 9.3 Hz, 1H), 4.43 (t, J = 8.0 Hz, 1H), 4.29 (q, J = 4.2, 3.6 Hz, 1H), 3.65–3.45 (m, 1H), 2.46 (s, 3H), 2.35 (t, J = 7.4 Hz, 2H), 2.26 (dt, J = 14.7, 7.7 Hz, 1H), 2.11 (ddd, J = 14.1, 8.0, 6.2 Hz, 1H), 2.06–1.95 (m, 1H), 1.80 (ddd, J = 12.9, 8.5, 4.6 Hz, 1H), 1.61 (t, J = 7.2 Hz, 2H), 1.56–1.41 (m, 1H), 1.38 (d, J = 7.0 Hz, 2H), 1.28 (d, J = 13.7 Hz, 10H), 0.93 (d, J = 5.8 Hz, 9H).
4.2.16. N1-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-N4-(5-(5-(4-((S)-1-((2S,4R)-1-((S)-2-acetamido-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2 carboxamido)ethyl)phenyl)thiazole-4-carboxamido) pentyl)terephthalamide (compound 15)
7.2 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 1032.46, found 1032.49.
1H NMR (500 MHz, DMSO-d6) δ 12.22 (s, 1H), 10.68 (s, 1H), 10.50 (s, 1H), 9.10 (s, 1H), 8.84 (s, 1H), 8.62 (t, J = 5.6 Hz, 1H), 8.45 (t, J = 5.9 Hz, 1H), 8.39 (d, J = 7.8 Hz, 1H), 8.33 (d, J = 3.1 Hz, 1H), 8.29 (d, J = 2.1 Hz, 1H), 8.05 (d, J = 8.3 Hz, 3H), 7.99 (d, J = 8.3 Hz, 2H), 7.89 (d, J = 9.3 Hz, 1H), 7.64–7.54 (m, 2H), 7.53–7.46 (m, 3H), 7.46–7.38 (m, 2H), 7.36–7.22 (m, 4H), 4.92 (p, J = 6.9 Hz, 2H), 4.56–4.40 (m, 2H), 4.28 (q, J = 3.6 Hz, 1H), 3.25 (dq, J = 32.4, 6.7 Hz, 5H), 2.07–1.98 (m, 1H), 1.88 (s, 3H), 1.81 (ddd, J = 12.9, 8.5, 4.7 Hz, 2H), 1.62–1.45 (m, 5H), 1.36 (dd, J = 15.1, 7.3 Hz, 5H), 0.93 (d, J = 5.0 Hz, 9H).
4.2.17. N1-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-N4-(5-(((R)-3-acetamido-4-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-2-methyl-4-oxobutan-2-yl)thio)pentyl)terephthalamidede (compound 16)
7.0 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 1021.42, found 1021.51.
1H NMR (500 MHz, DMSO-d6) δ 12.32 (s, 1H), 10.90 (s, 1H), 10.52 (s, 1H), 8.98 (s, 1H), 8.88 (s, 1H), 8.62 (t, J = 5.6 Hz, 1H), 8.37 (d, J = 3.1 Hz, 1H), 8.30 (d, J = 2.1 Hz, 1H), 8.21 (d, J = 7.9 Hz, 1H), 8.12 (d, J = 9.5 Hz, 1H), 8.09–7.95 (m, 5H), 7.66–7.24 (m, 11H), 5.00–4.78 (m, 3H), 4.41 (t, J = 8.0 Hz, 2H), 4.30 (s, 2H), 3.28 (q, J = 6.6 Hz, 3H), 2.56 (q, J = 7.2 Hz, 2H), 2.45 (s, 3H), 2.10–2.00 (m, 1H), 1.89 (s, 3H), 1.52 (dt, J = 29.6, 7.4 Hz, 4H), 1.43–1.31 (m, 8H), 1.28 (s, 3H).
4.2.18. N1-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-N4-(5-(2-((S)-1-((2S,4R)-1-((S)-2-acetamido-3,3-dimethylbutanoyl)-4-hydroxypyrrolidine-2-carboxamido)ethyl)-5-(4-methylthiazol-5-yl)phenoxy)pentyl)terephthalamide (compound 17)
10.3 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 1019.46, found 1019.55.
1H NMR (500 MHz, DMSO-d6) δ 12.28 (s, 1H), 10.82 (s, 1H), 10.51 (s, 1H), 8.98 (d, J = 3.4 Hz, 1H), 8.87 (s, 1H), 8.65 (t, J = 5.7 Hz, 1H), 8.41 (d, J = 7.3 Hz, 1H), 8.36 (d, J = 3.1 Hz, 1H), 8.32–8.25 (m, 1H), 8.09–7.85 (m, 6H), 7.66–7.37 (m, 6H), 7.36–7.24 (m, 2H), 7.05–6.94 (m, 1H), 6.89 (dd, J = 7.9, 1.6 Hz, 1H), 4.98 (t, J = 7.0 Hz, 1H), 4.58–4.40 (m, 3H), 4.35 (t, J = 3.9 Hz, 2H), 4.07 (t, J = 6.3 Hz, 3H), 3.64–3.60 (m, 5H), 3.34 (d, J = 6.5 Hz, 3H), 2.47 (d, J = 7.0 Hz, 3H), 2.00 (dd, J = 8.1, 2.7 Hz, 1H), 1.95–1.77 (m, 6H), 1.65 (q, J = 7.0 Hz, 2H), 1.56 (q, J = 8.1, 7.7 Hz, 2H), 1.29 (dd, J = 16.1, 6.9 Hz, 3H), 0.93 (d, J = 2.6 Hz, 2H), 0.85 (s, 9H). 13C NMR (126 MHz, DMSO) δ 171.52, 170.82, 170.09, 169.62, 165.86, 165.60, 161.73, 159.12, 158.86, 158.59, 155.83, 154.11, 151.88, 148.31, 148.10, 140.16, 138.63, 137.79, 137.74, 137.31, 132.92, 131.98, 130.97, 130.38, 129.73, 128.17, 127.68, 126.98, 124.45, 123.60, 122.18, 121.11, 119.91, 117.32, 115.86, 113.85, 113.61, 112.08, 69.35, 67.83, 59.03, 56.88, 44.50, 38.30, 35.60, 29.22, 28.98, 26.88, 26.77, 23.67, 23.56, 22.79, 21.26, 16.51, 16.44.
4.2.19. N1-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-N4-(3-(((2S)-1-((4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-3-oxopropyl)terephthalamide (compound 18)
7.9 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 947.40, found 947.42.
1H NMR (500 MHz, DMSO-d6) δ 12.30 (s, 1H), 10.86 (s, 1H), 10.51 (d, J = 14.1 Hz, 1H), 8.99 (s, 1H), 8.87 (s, 1H), 8.63 (t, J = 5.6 Hz, 1H), 8.41–8.34 (m, 2H), 8.32–8.26 (m, 1H), 8.09–7.94 (m, 5H), 7.63–7.59 (m, 1H), 7.56 (s, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.47–7.41 (m, 3H), 7.41–7.36 (m, 2H), 7.35–7.27 (m, 2H), 4.91 (dt, J = 15.6, 7.7 Hz, 1H), 4.55 (d, J =9.3 Hz, 1H), 4.44 (t, J =8.0 Hz, 1H), 4.30 (t, J =3.8 Hz, 1H), 2.59 (dt, J = 14.8, 7.5 Hz, 1H), 2.46 (s, 3H), 2.42 (s, 1H), 2.03 (ddd, J = 11.2, 7.7, 2.7 Hz, 1H), 1.81 (ddd, J = 13.0, 8.5, 4.7 Hz, 1H), 1.38 (d, J = 7.0 Hz, 3H), 0.93 (d, J = 7.7 Hz, 9H).
4.2.20. N1-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-N4-(4-(((2S)-1-((4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-4-oxobutyl)terephthalamide (compound 19)
6.1 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 961.42, found 961.46.
1H NMR (500 MHz, DMSO-d6) δ 12.32 (s, 1H), 10.90 (s, 1H), 10.53 (s, 1H), 8.99 (s, 1H), 8.88 (s, 1H), 8.66 (t, J = 5.6 Hz, 1H), 8.41–8.28 (m, 3H), 8.10–7.95 (m, 5H), 7.91 (d, J = 9.3 Hz, 1H), 7.66–7.25 (m, 11H), 4.93 (t, J = 7.2 Hz, 2H), 4.62–4.39 (m, 4H), 4.29 (q, J = 3.5 Hz, 2H), 3.68–3.57 (m, 4H), 3.29 (t, J = 6.6 Hz, 3H), 2.46 (s, 3H), 2.29 (ddd, J = 49.1, 14.4, 7.1 Hz, 3H), 2.02 (t, J = 10.3 Hz, 1H), 1.85–1.71 (m, 3H), 1.38 (d, J = 7.0 Hz, 3H), 0.95 (d, J = 7.0 Hz, 9H).
4.2.21. N1-(3-((6-(1H-indol-3-yl)pyrimidin-4-yl)amino)phenyl)-N4-(7-(((2S)-1-((4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-7-oxoheptyl)terephthalamide (compound 20)
9.2 mg white solid. LC-MS (ESI) m/z: [M+H]+: calcd 1003.47, found 1003.49.
1H NMR (500 MHz, DMSO-d6) δ 12.30 (s, 1H), 10.86 (s, 1H), 10.51 (d, J = 6.0 Hz, 1H), 8.99 (s, 1H), 8.87 (s, 1H), 8.62 (t, J = 5.7 Hz, 1H), 8.41–8.33 (m, 2H), 8.30 (d, J = 2.2 Hz, 1H), 8.09–7.95 (m, 5H), 7.80 (d, J = 9.3 Hz, 1H), 7.64–7.48 (m, 3H), 7.49–7.40 (m, 5H), 7.38 (d, J = 8.2 Hz, 2H), 7.36–7.25 (m, 3H), 4.92 (t, J = 7.2 Hz, 2H), 4.60–4.41 (m, 3H), 4.29 (t, J = 3.7 Hz, 2H), 3.28 (q, J = 6.9 Hz, 4H), 2.46 (s, 4H), 2.32–2.20 (m, 2H), 2.14 (dt, J = 14.4, 7.3 Hz, 2H), 2.01 (td, J = 9.1, 7.7, 4.4 Hz, 1H), 1.80 (ddd, J = 12.9, 8.5, 4.6 Hz, 1H), 1.53 (dp, J = 14.1, 6.7 Hz, 6H), 1.34 (dd, J = 32.8, 6.6 Hz, 9H), 0.94 (d, J = 6.5 Hz, 9H).
4.3. Cell culture
Molt-4 (CRL-1582), Jurkat (clone E6–1, Lot Number 60628582) and HEK293T cells (CRL-3216) were purchased from ATCC. All media was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. MOLT-4 cells were cultured in RPMI-1640 media containing L-glutamine. Jurkat (male) cells were cultured in RPMI containing 10% fetal bovine serum and 1% Penicillin/Streptomycin. HEK293T cells were cultured in DMEM media without L-glutamine. Mycoplasma testing was performed on a monthly basis using the MycoAlert mycoplasma detection kit (Lonza) and all lines were negative.
4.4. Kd evaluation for PI5P4Kγ, PI5P4Kβ and enzymatic inhibition for PI5P4Kα
The Kd values for PI5P4Kγ, PI5P4Kβ binding were determined by the commercially available KdELECT. Enzymatic inhibition for PI5P4Kα were determined by commercially available KinaseProfiler™ and respective protocols are available from Eurofins DiscoverX.
4.5. Immunoblotting analysis
Cells were lysed in RIPA buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) (Sigma, cat# R0278) with protease inhibitor (Sigma, cat# 11836153001)/phosphatase (Sigma, cat# 04906837001) inhibitor. The protein concentrations were measured by BCA analysis (Thermo Fisher Scientific, cat # PI23225). Equal amounts of protein were resolved by 4–12% Tris-Base gels (Life Technologies, cat# NW04125BOX), and then transferred to the Immuno-Blot PVDF membrane (BioRad, cat# 1620177). Proteins were probed with appropriate primary antibodies at 4 °C overnight and then with IRDye®800-labeled goat anti-rabbit IgG (LICOR Biosciences, cat # 926–32211), IRDye®800-labeled goat anti-mouse IgG (LICOR Biosciences, cat# 926–32210) or IRDye 680RD goat anti-Mouse IgG (LICOR Biosciences, cat# 926–68070) secondary antibodies at room temperature for 1 h. The membranes were detected on Odyssey CLx system. Antibodies used in this study include anti-following proteins:
β-Actin (Cell Signaling Technology, cat#3700).
PI5P4Kα (Cell Signaling Cat# 5527) PI5P4Kβ (Cell Signaling Technologies Cat# 9694), PI5P4Kγ (Sigma-Aldrich Cat# HPA058551); Vinculin(Cell Signaling Technologies Cat# 4650).Sample preparation TMT LC-MS3 mass spectrometry.
Molt4 cells were plated in fresh media in 10 cm plates and treated for 5 h with DMSO or 1 μM JWZ-1–80, harvested by centrifugation, washed twice with PBS and snap frozen in liquid nitrogen. Cell lysis and Tandem Mass Tagged (TMT) tryptic peptides were prepared for LCMS analysis following published procedures [36].
The data were acquired using a mass range of m/z 340–1350, resolution 120,000, AGC target 5 × 105, maximum injection time 100 ms, dynamic exclusion of 120 s for the peptide measurements in the Orbitrap. Data dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 35%, AGC target set to 1.8 × 104 and a maximum injection time of 120 ms. MS3 scans were acquired in the Orbitrap with a HCD collision energy set to 55%, AGC target set to 2 × 105, maximum injection time of 150 ms, resolution at 50,000 and with a maximum synchronous precursor selection (SPS) precursors set to 10.
4.6. LC-MS data analysis
Proteome Discoverer 2.4 (Thermo Fisher Scientific) was used for. RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides. MS/MS spectra were searched against a Swissprot human database (February 2020) with both the forward and reverse sequences. Database search criteria are as follows: tryptic with two missed cleavages, a precursor mass tolerance of 20 ppm, fragment ion mass tolerance of 0.6 Da, static alkylation of cysteine (57.0215 Da), static TMT labelling of lysine residues and N-termini of peptides (304.2071 Da), and variable oxidation of methionine (15.9949 Da). TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. Peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (summed signal-to-noise across 16 channels <100 and precursor isolation specificity <0.5), and resulting data was filtered to only include proteins that had a minimum of 2 unique peptides identified. Reporter ion intensities were normalized and scaled using in-house scripts in the R framework [37]. Statistical analysis was carried out using the limma package within the R framework [38].
Supplementary Material
Acknowledgment
The authors would like to thank the National Institutes of Health for their generous financial support through grants R01 CA197329-03 and R01 CA218278-02.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: PI5P4Kγ degraders developed in this manuscript are licensed to Larkspur where N.S.G. and L.C.C. have a financial interest. W.J., E.S.W., T.D. M., K.A.D., E.S.F., T.Z., L.C.C. and N.S.G are inventors on PI5P4Kγ patent (WO 2022140554A1). N.S.G. is a founder, science advisory board member (SAB) and equity holder in Syros, C4, Allorion, Lighthorse, Voronoi, Inception, Matchpoint, CobroVentures, GSK, Larkspur (board member) and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Deerfield, Springworks, Interline and Sanofi. T.Z. is a scientific funder, equity holder and consultant of Matchpoint. L.C.C. is a founder and scientific advisory board member of Agios Pharmaceuticals, Faeth Therapeutics, Petra Pharma Corporation, Larkspur Therapeutics and Volastra Pharmaceuticals, and scientific advisory board member for Scorpion Therapeutics. E.S.F. is a founder, scientific advisory board (SAB) member, and equity holder of Civetta Therapeutics, Lighthorse Therapeutics, Proximity Therapeutics, and Neomorph, Inc. (board member). E.S.F. is an equity holder and SAB member for Avilar Therapeutics and Photys Therapeutics and a consultant to Novartis, Sanofi, EcoR1 Capital, and Deerfield. The Fischer lab receives or has received research funding from Novartis, Ajax, and Astellas. K.A. D. is a consultant to Kronos Bio and Neomorph Inc.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2022.115027.
Data availability
Data will be made available on request.
References
- [1].Balla T, Szentpetery Z, Kim YJ, Phosphoinositide signaling: new tools and insights, Physiology 24 (2009) 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bulley SJ, Clarke JH, Droubi A, Giudici ML, Irvine RF, Exploring phosphatidylinositol 5-phosphate 4-kinase function, Adv. Biol. Regul 57 (2015) 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rameh LE, Tolias KF, Duckworth BC, Cantley LC, A new pathway for synthesis of phosphatidylinositol-4, 5-bisphosphate, Nature 390 (6656) (1997) 192–196. [DOI] [PubMed] [Google Scholar]
- [4].Rameh LE, Cantley LC, The role of phosphoinositide 3-kinase lipid products in cell function, J. Biol. Chem 274 (13) (1999) 8347–8350. [DOI] [PubMed] [Google Scholar]
- [5].Wang M, Bond NJ, Letcher AJ, Richardson JP, Lilley KS, Irvine RF, Clarke JH, Genomic tagging reveals a random association of endogenous PtdIns5 P 4-kinases IIα and IIβ and a partial nuclear localization of the IIα isoform, Biochem. J 430 (2) (2010) 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hu A, Zhao X-T, Tu H, Xiao T, Fu T, Wang Y, Liu Y, Shi X-J, Luo J, Song B-L, PIP4K2A regulates intracellular cholesterol transport through modulating PI (4, 5) P2 homeostasis, JLR (J. Lipid Res.) 59 (3) (2018) 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lamia KA, Peroni OD, Kim Y-B, Rameh LE, Kahn BB, Cantley LC, Increased insulin sensitivity and reduced adiposity in phosphatidylinositol 5-phosphate 4-kinase β−/− mice, Mol. Cell Biol 24 (11) (2004) 5080–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lundquist MR, Goncalves MD, Loughran RM, Possik E, Vijayaraghavan T, Yang A, Pauli C, Ravi A, Verma A, Yang Z, Phosphatidylinositol-5-phosphate 4-kinases regulate cellular lipid metabolism by facilitating autophagy, Mol. Cell 70 (3) (2018) 531–544, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bulley SJ, Droubi A, Clarke JH, Anderson KE, Stephens LR, Hawkins PT, Irvine RF, In B cells, phosphatidylinositol 5-phosphate 4-kinase–α synthesizes PI (4, 5) P2 to impact mTORC2 and Akt signaling, Proc. Natl. Acad. Sci. USA 113 (38) (2016) 10571–10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Keune W-J, Jones DR, Divecha N, PtdIns5P and Pin1 in oxidative stress signaling, Adv. Biol. Regul 53 (2) (2013) 179–189. [DOI] [PubMed] [Google Scholar]
- [11].Burke JE, Structural basis for regulation of phosphoinositide kinases and their involvement in human disease, Mol. Cell 71 (5) (2018) 653–673. [DOI] [PubMed] [Google Scholar]
- [12].Jude JG, Spencer GJ, Huang X, Somerville TD, Jones DR, Divecha N, Somervaille TC, A targeted knockdown screen of genes coding for phosphoinositide modulators identifies PIP4K2A as required for acute myeloid leukemia cell proliferation and survival, Oncogene 34 (10) (2015) 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf G, Bell EL, Shim H-S, Lamia KA, Rameh LE, Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors, Cell 155 (4) (2013) 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ma G, Duan Y, Huang X, Qian CX, Chee Y, Mukai S, Cui J, Samad A, Matsubara JA, Kazlauskas A, Prevention of proliferative vitreoretinopathy by suppression of phosphatidylinositol 5-phosphate 4-kinases, Investig. Ophthalmol. Vis. Sci 57 (8) (2016) 3935–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Arora GK, Palamiuc L, Emerling BM, Expanding role of PI5P4Ks in cancer: a promising druggable target, FEBS Lett. 596 (1) (2022) 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Clarke JH, Giudici ML, Burke JE, Williams RL, Maloney DJ, Marugan J, Irvine RF, The function of phosphatidylinositol 5-phosphate 4-kinase gamma (PI5P4Kgamma) explored using a specific inhibitor that targets the PI5P-binding site, Biochem. J 466 (2) (2015) 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang DG, Paddock MN, Lundquist MR, Sun JY, Mashadova O, Amadiume S, Bumpus TW, Hodakoski C, Hopkins BD, Fine M, PIP4Ks suppress insulin signaling through a catalytic-independent mechanism, Cell Rep. 27 (7) (2019) 1991–2001, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mackey AM, Sarkes DA, Bettencourt I, Asara JM, Rameh LE, PIP4kγ is a substrate for mTORC1 that maintains basal mTORC1 signaling during starvation, Sci. Signal 7 (350) (2014) ra104-ra104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zheng L, Conner SD, PI5P4Kγ functions in DTX1-mediated Notch signaling, Proc. Natl. Acad. Sci. USA 115 (9) (2018) E1983–E1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lin T-C, Kuo H-H, Wu Y-C, Pan TS, Yih L-H, Phosphatidylinositol-5-phosphate 4-kinase gamma accumulates at the spindle pole and prevents microtubule depolymerization, Cell Div. 14 (1) (2019) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Raychaudhuri S, Remmers EF, Lee AT, Hackett R, Guiducci C, Burtt NP, Gianniny L, Korman BD, Padyukov L, Kurreeman FA, Common variants at CD40 and other loci confer risk of rheumatoid arthritis, Nat. Genet 40 (10) (2008) 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shim H, Wu C, Ramsamooj S, Bosch KN, Chen Z, Emerling BM, Yun J, Liu H, Choo-Wing R, Yang Z, Deletion of the gene Pip4k2c, a novel phosphatidylinositol kinase, results in hyperactivation of the immune system, Proc. Natl. Acad. Sci. USA 113 (27) (2016) 7596–7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Al-Ramahi I, Giridharan SSP, Chen Y-C, Patnaik S, Safren N, Hasegawa J, de Haro M, Gee AKW, Titus SA, Jeong H, Inhibition of PIP4Kγ ameliorates the pathological effects of mutant huntingtin protein, Elife 6 (2017), e29123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kitagawa M, Liao P-J, Lee KH, Wong J, Shang SC, Minami N, Sampetrean O, Saya H, Lingyun D, Prabhu N, Dual blockade of the lipid kinase PIP4Ks and mitotic pathways leads to cancer-selective lethality, Nat. Commun 8 (1) (2017) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Voss MD, Czechtizky W, Li Z, Rudolph C, Petry S, Brummerhop H, Langer T, Schiffer A, Schaefer H-L, Discovery and pharmacological characterization of a novel small molecule inhibitor of phosphatidylinositol-5-phosphate 4-kinase, type II, beta, Biochem. Biophys. Res. Commun 449 (3) (2014) 327–331. [DOI] [PubMed] [Google Scholar]
- [26].Sivakumaren SC, Shim H, Zhang T, Ferguson FM, Lundquist MR, Browne C, Seo H-S, Paddock MN, Manz TD, Jiang B, Targeting the PI5P4K lipid kinase family in cancer using covalent inhibitors, Cell Chem. Biol 27 (5) (2020) 525–537, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Manz TD, Sivakumaren SC, Yasgar A, Hall MD, Davis MI, Seo H-S, Card JD, Ficarro SB, Shim H, Marto JA, Structure–activity relationship study of covalent pan-phosphatidylinositol 5-phosphate 4-kinase inhibitors, ACS Med. Chem. Lett 11 (3) (2019) 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Manz TD, Sivakumaren SC, Ferguson FM, Zhang T, Yasgar A, Seo HS, Ficarro SB, Card JD, Shim H, Miduturu CV, Simeonov A, Shen M, Marto JA, Dhe-Paganon S, Hall MD, Cantley LC, Gray NS, Discovery and structure-activity relationship study of (Z)-5-Methylenethiazolidin-4-one derivatives as potent and selective pan-phosphatidylinositol 5-phosphate 4-kinase inhibitors, J. Med. Chem 63 (9) (2020) 4880–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Davis MI, Sasaki AT, Shen M, Emerling BM, Thorne N, Michael S, Pragani R, Boxer M, Sumita K, Takeuchi K, A homogeneous, high-throughput assay for phosphatidylinositol 5-phosphate 4-kinase with a novel, rapid substrate preparation, PLoS One 8 (1) (2013), e54127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen S, Chandra Tjin C, Gao X, Xue Y, Jiao H, Zhang R, Wu M, He Z, Ellman J, Ha Y, Pharmacological inhibition of PI5P4Kα/β disrupts cell energy metabolism and selectively kills p53-null tumor cells, Proc. Natl. Acad. Sci. USA 118 (21) (2021), e2002486118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Boffey HK, Rooney TP, Willems HM, Edwards S, Green C, Howard T, Ogg D, Romero T, Scott DE, Winpenny D, Development of selective phosphatidylinositol 5-phosphate 4-kinase γ inhibitors with a non-ATP-competitive, allosteric binding mode, J. Med. Chem 65 (4) (2022) 3359–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li K, Crews CM, PROTACs: Past, Present and Future, Chemical Society Reviews, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu Z, Crews CM, Recent developments in PROTAC-mediated protein degradation: from bench to clinic, Chembiochem 23 (2) (2022), e202100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, Zhang T, Kwiatkowski N, Boukhali M, Green JL, Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation, Nat. Chem. Biol 14 (2) (2018) 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, Gray NS, Development of dual and selective degraders of cyclin-dependent kinases 4 and 6, Angew. Chem. Int. Ed 58 (19) (2019) 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, Ebert BL, Fischer ES, Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome, Elife 7 (2018), e38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].R.C. Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2013. http://www.R-project.org/. [Google Scholar]
- [38].Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, Limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic Acids Res. 43 (7) (2015) e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.