

Abstract
Plasma soluble prorenin receptor (sPRR) displays sexual dimorphism and is higher in women with type 2 diabetes mellitus (T2DM). However, the contribution of plasma sPRR to the development of vascular complications in T2DM remains unclear. We investigated if plasma sPRR contributes to sex differences in the activation of the systemic renin-angiotensin-aldosterone system (RAAS) and vascular damage in a model of high-fat diet (HFD)-induced T2DM. Male and female C57BL/6J mice were fed either a normal fat diet (NFD) or an HFD for 28 wk to assess changes in blood pressure, cardiometabolic phenotype, plasma prorenin/renin, sPRR, and ANG II. After completing dietary protocols, tissues were collected from males to assess vascular reactivity and aortic reactive oxygen species (ROS). A cohort of male mice was used to determine the direct contribution of increased systemic sPRR by infusion. To investigate the role of ovarian hormones, ovariectomy (OVX) was performed at 32 wk in females fed either an NFD or HFD. Significant sex differences were found after 28 wk of HFD, where only males developed T2DM and increased plasma prorenin/renin, sPRR, and ANG II. T2DM in males was accompanied by nondipping hypertension, carotid artery stiffening, and aortic ROS. sPRR infusion in males induced vascular thickening instead of material stiffening caused by HFD-induced T2DM. While intact females were less prone to T2DM, OVX increased plasma prorenin/renin, sPRR, and systolic blood pressure. These data suggest that sPRR is a novel indicator of systemic RAAS activation and reflects the onset of vascular complications during T2DM regulated by sex.
NEW & NOTEWORTHY High-fat diet (HFD) for 28 wk leads to type 2 diabetes mellitus (T2DM) phenotype, concomitant with increased plasma soluble prorenin receptor (sPRR), nondipping blood pressure, and vascular stiffness in male mice. HFD-fed female mice exhibiting a preserved cardiometabolic phenotype until ovariectomy revealed increased plasma sPRR and blood pressure. Plasma sPRR may indicate the status of systemic renin-angiotensin-aldosterone system (RAAS) activation and the onset of vascular complications during T2DM in a sex-dependent manner.
Keywords: cardiovascular complications, circadian rhythms, reactive oxygen species, renin-angiotensin-aldosterone system, vascular dysfunction
INTRODUCTION
Obesity is a multifactorial disorder with an increased risk for hypertension and type 2 diabetes mellitus (T2DM) (1, 2). Obese women with T2DM experience decreased estrogenic protection and have a higher risk of cardiovascular (CV) events than diabetic men (3). In addition, many obese individuals with hypertension and T2DM exhibit inappropriate activation of the circulating and tissue renin-angiotensin-aldosterone system (RAAS), which accelerates metabolic disorders and the progression of cardiovascular disease (CVD) and chronic kidney disease (CKD) in a sex-dependent manner (4, 5). Thus, identifying novel indicators of systemic RAAS activation during obesity-induced hypertension and T2DM is critical for reducing the progression of CVD in both women and men.
The prorenin receptor (PRR), a novel component of the RAAS, is upregulated during obesity-associated hypertension (6–8). PRR is expressed in three molecular forms, a cell membrane-bound PRR (or full-length PRR), a truncated form, and a soluble PRR (sPRR) that is processed intracellularly by serine proteases and secreted to the extracellular space (9–11). Both PRR and sPRR fully activate renin and its inactive precursor, prorenin (10, 12, 13). The nonproteolytic activation of prorenin by either PRR or sPRR catalyzes the conversion of angiotensinogen to angiotensin I, promoting ANG II formation, which are critical processes involved in the regulation of the RAS (ANG II-dependent effects). The binding of prorenin to PRR triggers intracellular signaling linked to the upregulation of profibrotic genes (ANG II-independent effects) (14). The binding of prorenin either to the PRR or to the sPRR enables a conformational change in prorenin that allows enzymatic activity without proteolytic removal of the prosegment (15); however, whether prorenin elicits a different signaling pathway when bound to PRR versus sPRR remains unknown. PRR is expressed in the brain, heart, placenta, lung, kidney, and adipose tissue (13), where the soluble form is detected in biological fluids such as plasma and urine (10, 16). Growing evidence supports significant relationships among PRR, obesity, and T2DM. Blocking PRR reduces weight gain in obese mice by 20%, reduces insulinemia by 34%, and normalizes hypertriglyceridemia (17). Although the role of PRR has expanded our knowledge of the activation of RAAS in tissue, the function of plasma sPRR still needs to be better understood.
Several studies demonstrate that plasma sPRR levels are elevated in patients with CVD, including obesity (17), hypertension (8, 18), preeclampsia (19, 20), and T2DM (21). Sex also influences plasma sPRR concentrations, where obese women with T2DM display higher plasma renin activity (PRA) and sPRR levels than men (21). Equally important, circulating prorenin is most of the total plasmatic renin and presents sexual dimorphism (22–25). Patients with T2DM have high levels of prorenin, which is associated with increased risk and onset of microvascular complications (26). Furthermore, we previously showed that sPRR levels are increased in the urine of hypertensive chronic infused SD rats since recombinant prorenin substantially increased urine renin activity (16). This raises the possibility that plasma sPRR activates circulating prorenin in the extracellular space and may contribute to increased systemic RAAS activation and, consequently the development of CV in patients with T2DM. Yet, it remains uncertain whether systemic interactions between sPRR and prorenin increase the risk of CV complications during T2DM in a sex-dependent fashion.
The effects of sPRR are actively investigated in hypertensive, diabetic, and obese animal models (27–30). However, the contribution of circulating sPRR to the development of CV complications during HFD-induced obesity and T2DM remains unclear. We previously demonstrated that male mice chronically fed an HFD develop an obesity and T2DM phenotype, concomitantly with increased systolic blood pressure (SBP) and activation of intrarenal RAAS (31). To test the hypothesis that HFD elevates plasma sPRR levels and contributes to systemic RAAS activation and vascular damage during T2DM in a sex-dependent manner, we examined whether plasma sPRR and blood pressure (BP) exhibit sex differences in mice with HFD-induced T2DM. Second, because only male mice displayed changes in BP patterns along with the augmentation of plasma sPRR, we further examined the direct effects of exogenous sPRR infusion on vascular function in male mice. Finally, as sex hormones status and age are significant risk factors for hypertension and may affect systemic RAAS levels differently in men and women and, we further assessed the consequences of ovariectomy on sPRR levels in female mice with HFD-induced T2DM.
MATERIAL AND METHODS
Experimental Protocol 1: Sex Differences in HFD
Male and female C57BL/6J mice were randomly divided into two groups at 4–5 wk of age to receive either a normal fat diet (NFD) or high-fat diet (HFD) for 33–36 wk. The NFD (PicoLab Rodent Diet 20 EXT IRR 5R53) consisted of 25% kcal from protein, 13% from fat, and 62% from carbohydrates. The HFD (TestDiet, DIO 58V8-45 kcal% Fat, consisted of 18% kcal from protein, 46% from fat, and 36% from carbohydrates. The n number for the metabolic study was estimated upon sample size calculation and as previously described (31). These male and female mice were further randomly distributed in two cohorts: one used for metabolic phenotyping and blood collection by submandibular plexus bleeding (n = 5–7 mice/group), and the other for BP recordings by radiotelemetry (n = 9–10 mice/group). Every 4 wk, plasma sPRR concentrations were measured. At 28 wk of HFD, body weight, 12-h fasting plasma glucose (ONETOUCH Ultra glucometer, LifeScan, Cat. No. ZJZ8158JT), intraperitoneal glucose tolerance test (ipGTT), plasma insulin (Millipore, Cat. No. EZRMI-13K), plasma total prorenin/renin and ANG II were measured as previously described (31). After 33 wk on NFD or HFD, male mice were euthanized, and tissues were collected to assess carotid pressure myography, mesenteric artery reactivity, and aortic reactive oxygen species (ROS).
Experimental Protocol 2: Chronic Infusion of sPRR in Male Mice
To mimic the sPRR increase observed in the HFD-T2DM model, we infused exogenous sPRR in male mice and examined the direct effect of elevated plasma sPRR on the vasculature. Human recombinant sPRR was cloned in pET30a vector (17-244 aa, COOH-terminal-His tag, Genescript) and expressed in bacterial expression system (Escherichia coli, BL21-DE3). A different cohort of male C57Bl/6J mice (12 wk of age) fed a standard diet was implanted with osmotic minipumps (Alzet, model 2004) containing human recombinant sPRR-His-Tag (30 µg/kg/day) for 4 wk, following a protocol previously used (7). Baseline BP values were then taken for five consecutive days the week before sPRR initiation and during every week of sPRR infusion. Intracarotid pulse wave velocity (ic-PWV) was measured via ultrasound using the transit time method in the common carotid artery as previously described (32). Mice were euthanized at 16 wk of age, and carotid pressure myography, mesenteric artery reactivity, and aortic ROS were assessed as previously described (32, 33).
Experimental Protocol 3: Impact of Ovariectomy in Female Mice Treated with HFD
To investigate the protective effects of ovarian hormones, bilateral ovariectomy (OVX) was performed at 32 wk in all females fed either with NFD or HFD. After surgery, the mice were maintained on their respective diet. We initiated the current study using radiotelemetry in four animals/group. Because of prolonged feeding HFD protocol, we are unable to continue the BP recording by radiotelemetry on these female mice. Therefore, in this cohort, BP was measured by tail-cuff plethysmography using the Visitech BP-2000 Blood Pressure Analysis System. Baseline BP values were taken for seven consecutive days after OVX then weekly between 10:00 am and 1:00 pm. At 44 wk on HFD (12 wk after OVX), body weight, 12-h fasting glucose, ipGTT, plasma prorenin/renin, ANG II, and sPRR were measured. Female mice were euthanized at 12 wk after OVX and tissues were collected to assess mesenteric artery reactivity and aortic ROS. Plasma circulating estradiol was measured by ELISA (Abcam, Cat. No. ab108667). All animal procedures were performed in the 12-h:12-h light/dark cycle and were approved by the Tulane University Animal Care and Use Committee (Protocol No. 1467).
Quantification of sPRR, ANG II, and Plasma Prorenin/Renin
Plasma sPRR concentrations were measured by ELISA (IBL, Cat. No. 27782), as previously reported (10, 13, 34). To determine plasma ANG II concentrations at the study endpoint, blood samples were collected into mixed inhibitor solution chilled tubes. Blood was processed as previously described (35), and ANG II concentrations were measured by ELISA (Angiotensin II EIA Kit, Cayman, Cat. No. A05880). Plasma total prorenin/renin was measured using mouse prorenin and renin total antigen assay by ELISA kit following the manufacturer’s recommendations (Molecular Innovations, Cat. No. MPREKT-TOT).
SBP, MAP, and Dipping Blood Pressures Measurements
At 10–11 wk of age, mice were anesthetized with isoflurane to implant a telemetry probe (Data Sciences, TA11-PAC10) into the left carotid artery as previously described (31). Blood pressure recordings started in conscious mice after 2 wk of recovery. Data were collected and analyzed every 4 wk using Ponemah v. 6.0 software. Measurements are representative of 8 h of active phase (dark cycle) or inactive phase (light cycle) of SBP and mean arterial pressure (MAP). The percentage of dipping (nocturnal blood pressure fall) was calculated as (active BP − inactive BP)/active BP for both SBP and MAP. Mice were defined as dippers if BP decreased by ≥10%, whereas values <10% were considered nondippers.
Vascular Stiffness in Carotid Artery
At the study endpoint, common carotid arteries were harvested and cannulated for assessment of biaxial mechanical properties under passive conditions using a Danish Myotechnology 114P pressure myograph with Krebs–Henseleit solution, consisting of (in mM) 118 NaCl, 4.8 KCl, 2.5 CaCl2, 1.2 MgSO4, 25 NaHCO3, 1.2 KH2PO4, and 11 glucose at 37°C and oxygenated with 95% O2-5% CO2, as previously described (32). After equilibration and preconditioning, in vivo carotid artery length was estimated by finding the length at which there was minimal change in longitudinal force when pressures was changed from 70 to 150 mmHg (36). Continuous pressure-diameter data were collected during unloading and binned at each 10 ± 2 mmHg. Distensibility was calculated as the percent change in outer diameter from the starting diameter at 10 mmHg, and all other parameters were calculated as previously described (37).
Vascular Reactivity in Mesenteric Arteries
Mesenteric arteries were isolated and mounted on a wire myograph as previously described (38). After normalizing to optimal resting tension, vessels were exposed to 10−5 M phenylephrine (PE) and 10−6 M acetylcholine (ACh) to test the viability and endothelial integrity. Vessels that did not relax at least 50% were only used for endothelium-independent testing. Increasing concentrations of PE (10−7 to 10−3 M; MP Biomedicals, Santa Ana, CA) and prostaglandin F2α (PGF2α; 10−8 to 10−5 M; Tocris) were used to assess vascular contractility. Vessels were preconstricted with PGF2α (10−5 M) before assessing relaxation to increasing concentrations of sodium nitroprusside (SNP; 10−10 to 10−5 M; Sigma) and ACh (10−10 to 10−5 M; Enzo Life Sciences).
Quantification of ROS in the Aorta
Aortas were collected to measure reactive oxygen species (ROS) production by electron spin resonance (ESR) spectroscopy, as described previously (39). Briefly, diethyldithiocarbamate (2.5 μmol/L) and desferoxamine (25 μmol/L) were dissolved under oxygen bubbling in ice-cold modified Krebs-HEPES (KH) buffer at pH 7.3. Aortas from NFD and HFD mice were rapidly placed in cold PBS. Subsequently, 3–5 mm of vessel samples were placed in KH buffer. ESR measurements were performed using an EMX ESR eScan BenchTop spectrometer (Bruker, Germany) and a spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl-pyrrolidine. Time-dependent formation of ROS was determined using previous ESR settings. The amplitude measurements in arbitrary units were normalized to the dry weight of the corresponding aortic samples (33).
Histology
Aortic segments were fixed in 10% formalin overnight, followed by embedding in paraffin. The samples were serially cut into 5 µm sections and stained with Masson’s trichrome (MTC) using Trichrome, Gomori One-Step, Aniline Blue Stain Kit (Cat. No. 9176A). The images were obtained under brightfield using a Nikon 90i microscope and NIS-Element Software (Nikon Corporation). A custom MATLAB (MathWorks) code quantified the area fraction of smooth muscle, collagen, large diameter (thick) collagen fibers, and small diameter (thin) collagen fibers by determining the ratio of stained constituent pixels to total pixels (40). All quantifications were performed on ×40 images displaying the full cross section.
Statistical Analyses
All variables were screened for outliers using means ± 2SD and removed from analyses. The Kolmogorov–Smirnov and Bartlett tests were used to test the normality and homogeneity of the data. Differences between NFD and HFD groups were compared using unpaired t test. Two-way ANOVA was used to compare the interaction of sex and diet, followed by Sidak test for multiple comparisons. Two-way repeated-measures ANOVA was used to compare the timeline course of iGTT response and plasma sPRR, with Geiser–Greenhouse corrections and no assumptions of sphericity, and Sidak’s test for multiple comparison. Three-way repeated-measures ANOVA was used to compare the interaction of BP diurnal variation versus sex or diet, followed by Sidak test for multiple comparison. Power calculation was used for the sensibility analysis of the n number in radiotelemetry data. All data are presented as means ± SD and were analyzed using Prism v.9 software (GraphPad Software).
RESULTS
Metabolic Phenotyping Shows Sex Differences in HFD-Induced T2DM Model
Body weight was increased by HFD in mice from both sexes, but significantly greater in males compared with females (Pint < 0.001, Psex < 0.001, Pdiet < 0.001). At the end of 28 wk of HFD, fasting plasma glucose was elevated in males but did not reach statistical significance in females (P = 0.06; two-way ANOVA: Pint = 0.15, Psex < 0.001, Pdiet = 0.004). However, Fig. 1, D and E, shows that glycemia at 60 min in response to a glucose overload of 2 g/kg (ip) was elevated in both HFD groups (Males: Pint < 0.001, Ptime < 0.001, Pdiet = 0.005; NFD vs. HFD: 60 min P = 0.008 and 90 min P = 0.007; Females: Pint < 0.001, Ptime < 0.001, Pdiet = 0.003; NFD vs. HFD: 30 min P = 0.03 and 60 min P = 0.02). Similarly, the area under the curve (AUC) for ipGTT was elevated in HFD groups but not impacted by sex (Pint = 0.66, Psex = 0.32, Pdiet < 0.001). Plasma insulin was significantly increased only in HFD males resulting in both a sex and diet effect (Pint < 0.001, Psex < 0.001, Pdiet < 0.001, Fig. 1C).
Figure 1.
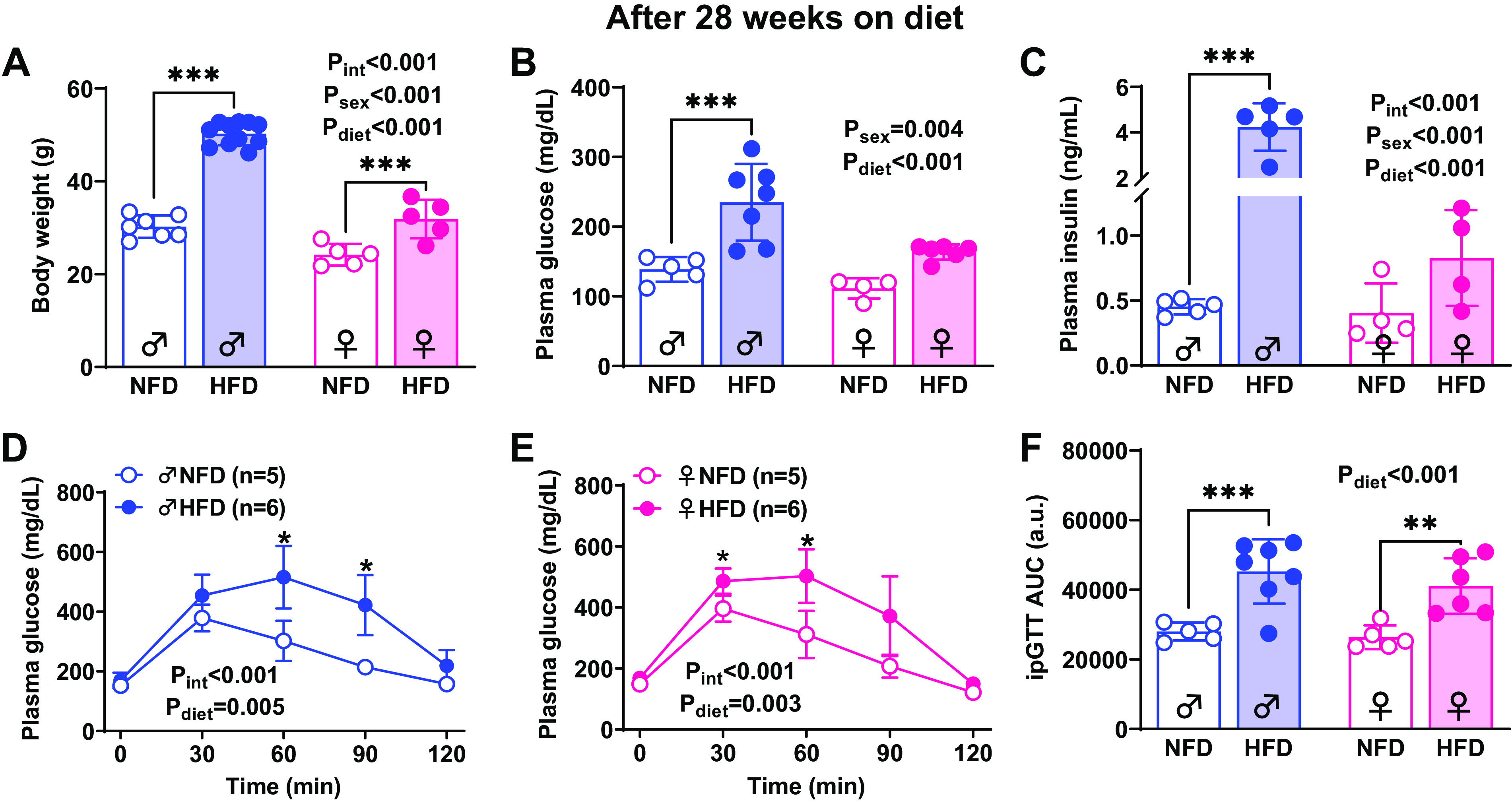
Effect of high-fat diet (HFD) in males and females. Body weight (A), fasting glycemia (B), plasma insulin (C), and glucose intolerance test (D–F) in 28 wk on normal fat diet (NFD) and HFD. No significant findings from two-way ANOVA are listed on each graph along with results of Sidak’s post hoc tests. ***P < 0.0001, **P < 0.001, *P < 0.05. Values are represented as means ± SD.
Male but Not Female HFD-Induced T2DM Mice Display Elevated Plasma sPRR Concomitantly with High SBP and MAP and Nondipping Pattern
The temporal changes in plasma levels of sPRR, plasma total prorenin/renin, and ANG II after 28 wk of HFD are illustrated in Fig. 2. At the study’s endpoint, plasma concentrations of plasma total prorenin/renin were significantly higher in male HFD mice compared with age-matched NFD mice (P < 0.001). In contrast, female mice experienced no changes in plasma total prorenin/renin (P = 0.13) and no sex differences or interaction were detected (Pint = 0.34, Psex = 0.41, Pdiet = 0.002, Fig. 2A). Plasma ANG II was increased only in male HFD mice compared with age-matched NFD mice (P = 0.01) but not in female mice (P = 0.64). No sex differences or diet effect were observed in plasma ANG II levels (Pint = 0.01, Psex = 0.95, Pdiet = 0.17, Fig. 2B). In male NFD mice, sPRR did not change over time. However, it was significantly augmented after 24 wk on HFD compared with aged-matched NFD mice (P = 0.03; two-way ANOVA: Pint < 0.016, Ptime < 0.001, Pdiet = 0.011, Fig. 2C). Female mice exhibited changes over time in both groups (Pint = 0.39, Ptime = 0.005, Pdiet = 0.36); however, no changes were detected when compared with NFD versus HFD (Fig. 2D).
Figure 2.

Plasma prorenin/renin, ANG II, and soluble prorenin receptor (sPRR) in normal fat diet (NFD) and high-fat diet (HFD) mice. Plasma total renin/prorenin was increased in male HFD compared with NFD (A). Plasma ANG II levels were significantly increased in male HFD compared with NFD mice (B). In male HFD mice, plasma sPRR was higher than in age-matched NFD mice after 24 wk on diet (C). No changes were observed in females compared with NFD vs. HDF (D). Significant findings from two-way ANOVA are listed on each graph along with results of Sidak’s post hoc tests. **P < 0.001, *P < 0.05. Values are represented as means ± SD.
Although 9–10 males and females were surgically implanted with a telemetry probe, three animals died unexpectedly, and because of the long protocol of HFD, some devices stopped recording the data. Consequently, eight males and six females were included in the BP analysis. Power analysis revealed that a sample size of three animals per treatment is sufficient to detect changes in 13 mmHg in BP of both groups, considering a power of 90% and an α level of 5%. Figure 3 shows BP rhythms and nondipping BP over 28 wk on HFD. As expected, normal diurnal SBP and MAP variations were similar after 12 wk on diet in males and females, and no sex, diet, or interaction effects were detected (SBP: Pint = 0.74, Psex × diet = 0.69, Plight-phase × sex = 0.89, Plight-phase × diet = 0.37; MAP: Pint = 0.57, Psex × diet = 0.01, Plight-phase × sex = 0.18, Plight-phase × diet = 0.65; Fig. 3, A, B, E, and F). After 20 wk on the diet regimen, this diurnal pattern in SBP and MAP was lost in males fed an HFD, exacerbating the sex differences, diet, and light-phase interactions at this time point on HFD (Pint = 0.03, Psex × diet = 0.04, Plight-phase × sex = 0.004; Plight-phase × diet = 0.15; MAP: Pint = 0.01, Psex × diet < 0.001, Plight-phase × sex = 0.01; Plight-phase × diet = 0.01; Fig. 3, A and E). In addition, SBP was significantly higher during both light periods at 20 wk in male HFD compared with NFD mice (dark phase: NFD 115 ± 2 vs. HFD 130 ± 2, P < 0.0001; light phase: NFD 102 ± 1 vs. 122 ± 4 mmHg; P < 0.0001). BP continued to increase in HFD males through 28 wk (dark phase: NFD 117 ± 8 vs. HFD 138 ± 10, P = 0.02; light phase: NFD 103 ± 7 vs. 131 ± 7 mmHg, P < 0.0001). Equally important, MAP was elevated after 20 wk of HFD (dark phase: NFD 105 ± 3 vs. HFD 116 ± 2, P = 0.001; light phase: NFD 94 ± 3 vs. 111 ± 1.2 mmHg; P = 0.002) and continued to be elevated at 28 wk (dark phase: NFD 104 ± 3 vs. HFD 125 ± 11, P = 0.01; light phase: NFD 93 ± 3 vs. 118 ± 9 mmHg; P < 0.0001, Fig. 3E) (Fig. 3, B, D, and F). In contrast to these differences in SBP and MAP in males on HFD, females on HFD preserved the levels of SBP and MAP and the diurnal pattern over 28 wk on diet, intensifying the sex differences, diet, and light-phase effect interactions (Pint = 0.52, Psex × diet = 0.008, Plight-phase × sex = 0.008; Plight-phase × diet = 0.02; MAP: Pint = 0.77, Psex × diet = 0.06, Plight-phase × sex = 0.01; Plight-phase × diet = 0.008; Fig. 3, B and F). Hence, male HFD mice developed nondipping BP status by 20 wk on HFD, which was maintained through 28 wk (Fig. 3, C and E, SBP: Pint < 0.001, Psex = 0.002, Pdiet = 0.001; MAP: Pint < 0.001, Psex = 0.002, Pdiet = 0.001). The only difference detected in response to the HFD in females was significantly less BP dipping at 28 wk, although this level was still within the expected range of 10%–20% (P = 0.03, Fig. 3H).
Figure 3.

Impact of high-fat diet (HFD) and sex on dark/light phases averages of SBP and mean arterial pressure (MAP) and dipping pattern. Temporal changes of SBP (A and B) and MAP (E and F) and nondipping BP (C, D, G, and H) over 28 wk. Open bars, light phase; hatched bars, dark phase. No changes in SBP and MAP were observed in female mice at a different time point (B and F). Dipping was calculated as a percentage of SBP and MAP. Mice were defined as dippers when BP decline 10%–20%, and nondippers were defined when <10% decline in blood pressure. Significant findings from three-way and two-way ANOVA with results of Sidak’s post hoc tests are listed on each graph. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. ns, not significant. Values are represented as means ± SD.
Vascular Reactivity and Damage Are Associated with HFD-Induced T2DM and Increased Plasma sPRR in Male Mice
Since female mice were less prone to the T2DM phenotype (Fig. 1, A–C) and were protected from elevated BP and systemic RAAS activation, we assessed vascular damage only in male mice. Carotid distensibility showed a significant interaction between diet and pressure with multiple comparisons indicating the greatest differences at pressures above 110 mmHg (Pint = 0.021, Ppress < 0.0001, Pdiet = 0.156, Fig. 4A). Wall thickness (Pint = 0.28, Ppress < 0.0001, Pdiet < 0.001, Fig. 4B) and wall/lumen ratio (Pint < 0.001, Ppress < 0.001, Pdiet = 0.09, Fig. 4C) were lower in mice fed the HFD. The stress-strain relationship was significantly shifted to the left in HFD carotids (P < 0.001, Fig. 4D), indicating increased stiffness, whereas incremental modulus calculations confirmed a significantly higher slope in HFD at higher pressures.
Figure 4.
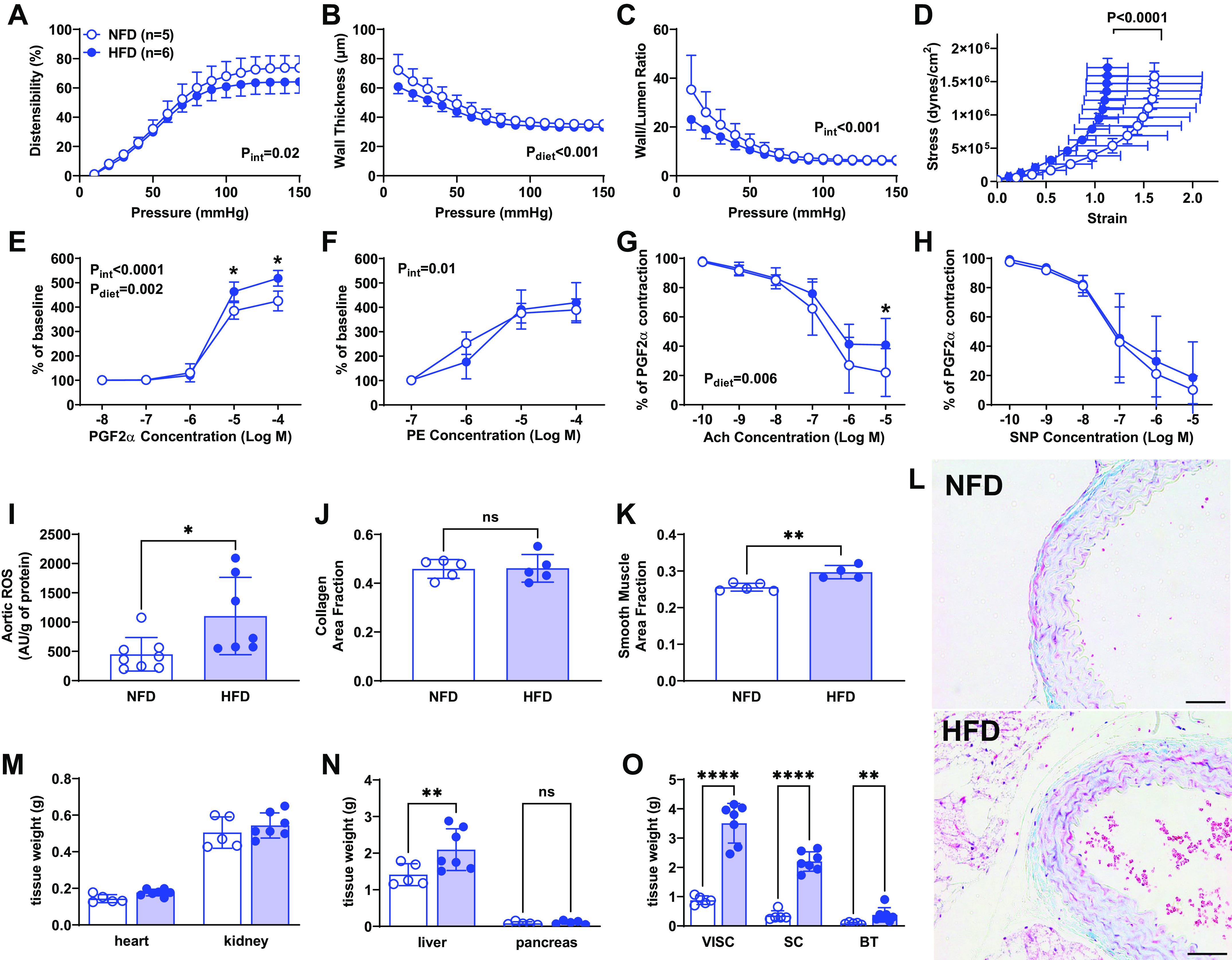
Impact of high-fat diet (HFD) on vascular damage in male mice. Ex vivo arterial stiffness was assessed by pressure myography in HFD and normal fat diet (NFD) mice. Distensibility, calculated as percentage of baseline diameter, was significantly lower in HFD vessels, especially at higher pressure (A). Wall thickness (B) and wall-to-lumen ratio (C) were both lower in HFD males. The stress-strain curve significantly shifted to the left, indicating increased arterial stiffness due to HFD (D). Vascular contractility in response to prostaglandin F2α (PGF2α) was higher in HFD group (E). Vasocontraction to phenylephrine (PE) showed an interaction effect (F). Acetylcholine (ACh) relaxation was significantly suppressed in HFD vessels (G). Endothelium-independent relaxation assessed using sodium nitroprusside (SNP) was unaltered by HFD (H). Aortic reactive oxygen species (ROS) production was elevated in HFD mice compared with NFD mice (I). Quantitative analyses of the smooth muscle (J) and collagen (K) from the NFD and HFD groups, *P < 0.05 vs. NFD at same age. Histological images of aortic ring in NFD and HFD mice: scale bar = 50 µM (L). Significant findings from two-way ANOVA are listed on each graph along with results of Sidak’s post hoc tests. ****P < 0.0001, **P < 0.01, *P < 0.05. D was analyzed using nonlinear regression, and I–L were analyzed by t test. Heart, liver, kidneys, and pancreas weight in NFD and HFD mice after 33 wk on diet are shown in M and N. Visceral, subcutaneous, and brown fat tissue weights were significantly higher in male mice fed a HFD than in those on NFD (O). Paired t tests, *P < 0.05, ****P < 0.0001. ns, not significant. Data are represented as means ± SD.
In mesenteric resistance arteries, the concentration-response curve to PGF2α was significantly impacted by HFD in males (Pint < 0.001, Pconc < 0.001, Pdiet = 0.002, Fig. 4E), with the maximum constriction to PGF2α significantly greater in HFD-treated animals compared with controls (518 ± 14 vs. 425 ± 18%, P < 0.01). The concentration-response curve to PE was not impacted by an HFD (Pint = 0.01, Pconc < 0.001, Pdiet = 0.74, Fig. 4F), and similarly, the maximum response to PE was not different between groups (419 ± 41 vs. 390 ± 17%, P = 0.45). In rings preconstricted with PGF2α, acetylcholine relaxation was significantly greater in NFD vessels at the maximum dose (41 ± 7 vs. 22 ± 6%; P = 0.03; Fig. 4G). However, endothelium-independent relaxation to SNP was not different between NFD and HFD vessels (19 ± 8 vs. 10 ± 3%, P = 0.33; Fig. 4H).
As shown in Fig. 4, after 28 wk on diet, ROS production in the aorta was significantly increased in HFD compared with NFD mice (P = 0.02; Fig. 4I). To determine whether the increase in ROS and the increased stiffness was associated with augmentation in vascular collagen deposition, we assessed MTC staining in aortic rings but found no difference between NFD and HFD (P = 0.9; Fig. 4J). However, there was an increase in smooth muscle in HFD (P = 0.004; Fig. 4K). Representative images show collagen in blue and smooth muscle in red (Fig. 4L). Liver, heart, and fat tissue weights were significantly higher in mice fed a HFD than in those on NFD (Fig. 4, M–O). No differences were obtained in kidney and pancreas weights (Fig. 4, M and N).
sPRR Chronic Infusion in Male Mice
Given that female mice were less prone to the T2DM phenotype and were protected from elevated BP, systemic RAAS activation, and did not exhibit increases in plasma sPRR, we analyzed the effects of chronic sPRR infusions on vascular function only in males. Compared with control mice, there were no significant changes in body weight, nor SBP during the infusion periods (Fig. 5, A and B). The infusion of exogenous sPRR significantly increased plasma sPRR levels, as observed in the HFD-induced T2DM model (P < 0.0001, Fig. 5C). PWV was elevated after chronic sPRR infusion (P = 0.03, Fig. 5D), and changes were not observed in aortic ROS in mice treated with sPRR (P = 0.51, Fig. 5E).
Figure 5.
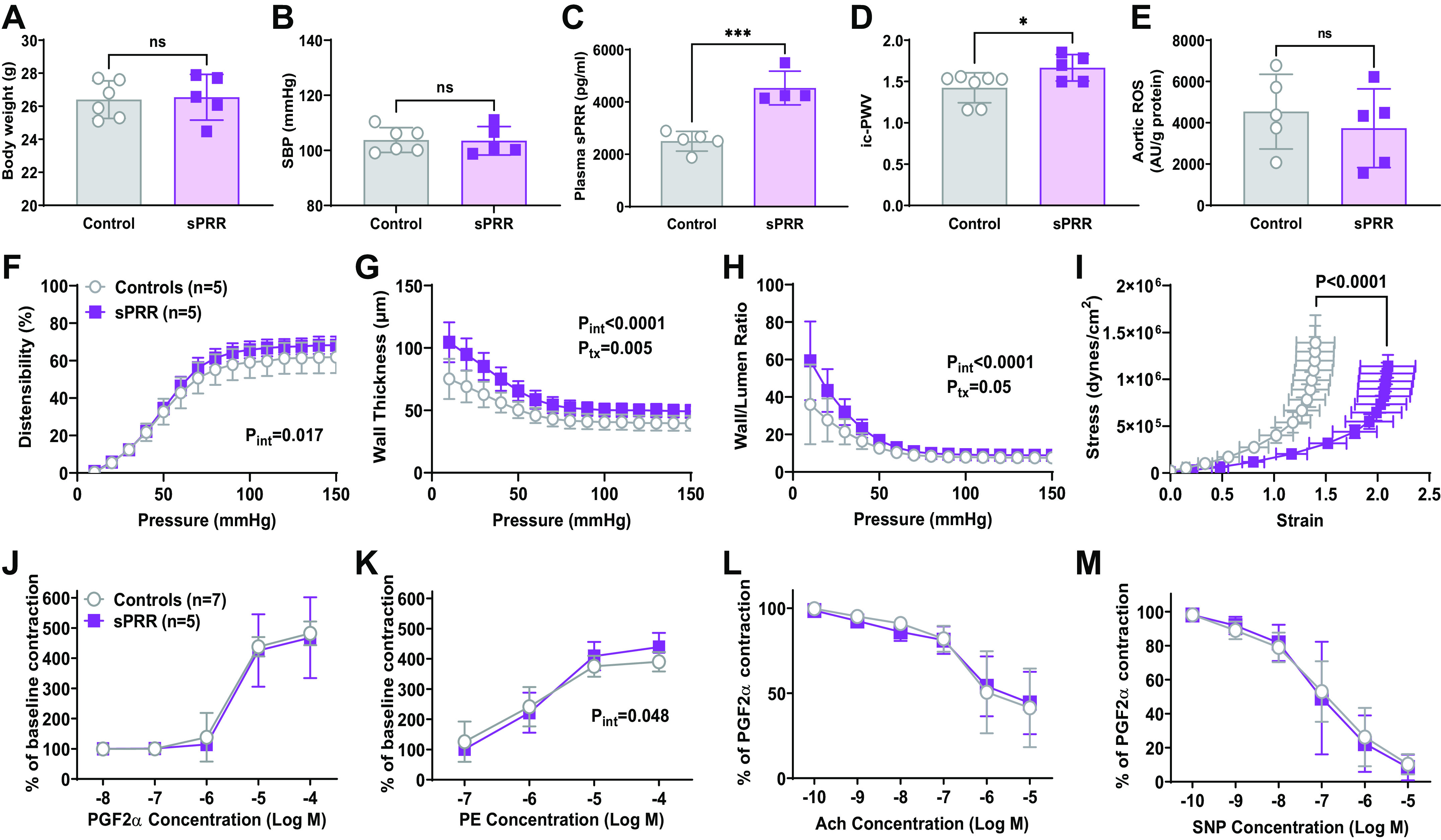
Impact of soluble prorenin receptor (sPRR) infusion on vascular male mice fed normal fat diet (NFD). Body weight (A), SBP (B), and reactive oxygen species (ROS) (E) production were not impacted by sPRR treatment, t test. The infusion of exogenous sPRR significantly increased plasma sPRR levels (C). D: pulse wave velocity (PWV) was significantly increased, unpaired t test, *P < 0.05. Distensibility (F), wall thickness (G), and wall-to-lumen ratio (H) were significantly higher in high-fat diet (HFD) male mice. The stress-strain curve significantly shifted to the right, indicating decreased material stiffness (I). Extra sum-of-squares F test after nonlinear regression, P < 0.0001. Vascular contractility in response to prostaglandin F2α (PGF2α) was not impacted by HFD (J), whereas vasocontraction to phenylephrine (PE) was increased by sPRR infusion (K). Neither ACh-induced relaxation (L) nor endothelium-independent relaxation (M) assessed using sodium nitroprusside (SNP) was impacted by HFD. Significant findings from two-way ANOVA are listed on each graph, no post hoc tests reached statistical significance. ***P < 0.001, *P < 0.05. ns, not significant. Values are represented as means ± SD.
Ex vivo analysis of the carotid artery showed that distensibility was slightly better in sPRR mice (Pint = 0.017, Ppress < 0.0001, Ptx = 0.19, Fig. 5F). The most striking data were in wall thickness (Pint < 0.0001, Ppress < 0.0001, Ptx = 0.005, Fig. 5G) and wall/lumen ratio (Pint < 0.0001, Ppress < 0.0001, Ptx = 0.05, Fig. 5H), which were significantly increased by treatment, indicating vascular thickening due to sPRR infusion alone. Since the stress is normalized by the amount of tissue, it was not surprising that the stress-strain curve was shifted to the right because of the larger wall area (P < 0.0001, Fig. 5I). Wire myography of mesenteric arteries revealed a significant increase in PE contraction response in males treated with sPRR (Pint = 0.048, Pconc < 0.0001, Ptx = 0.68, Fig. 5K). However, the sPRR treatment did not impact the responses to PGF2a, ACh, or SNP in the mesenteric arteries (Fig. 5, J, L, and M).
Impact of Ovariectomy on Female HFD Mice
At 32 wk of age, females fed either a NFD or HFD were ovariectomized and followed for an additional 12 wk to determine the impact of ovarian hormones loss on the development of a T2D phenotype. After 12 wk, ovariectomy (OVX)-HFD female mice displayed increased body weight, hyperglycemia, and glucose intolerance but not insulin resistance in comparison with OVX-NFD females (Fig. 6, A–E).
Figure 6.
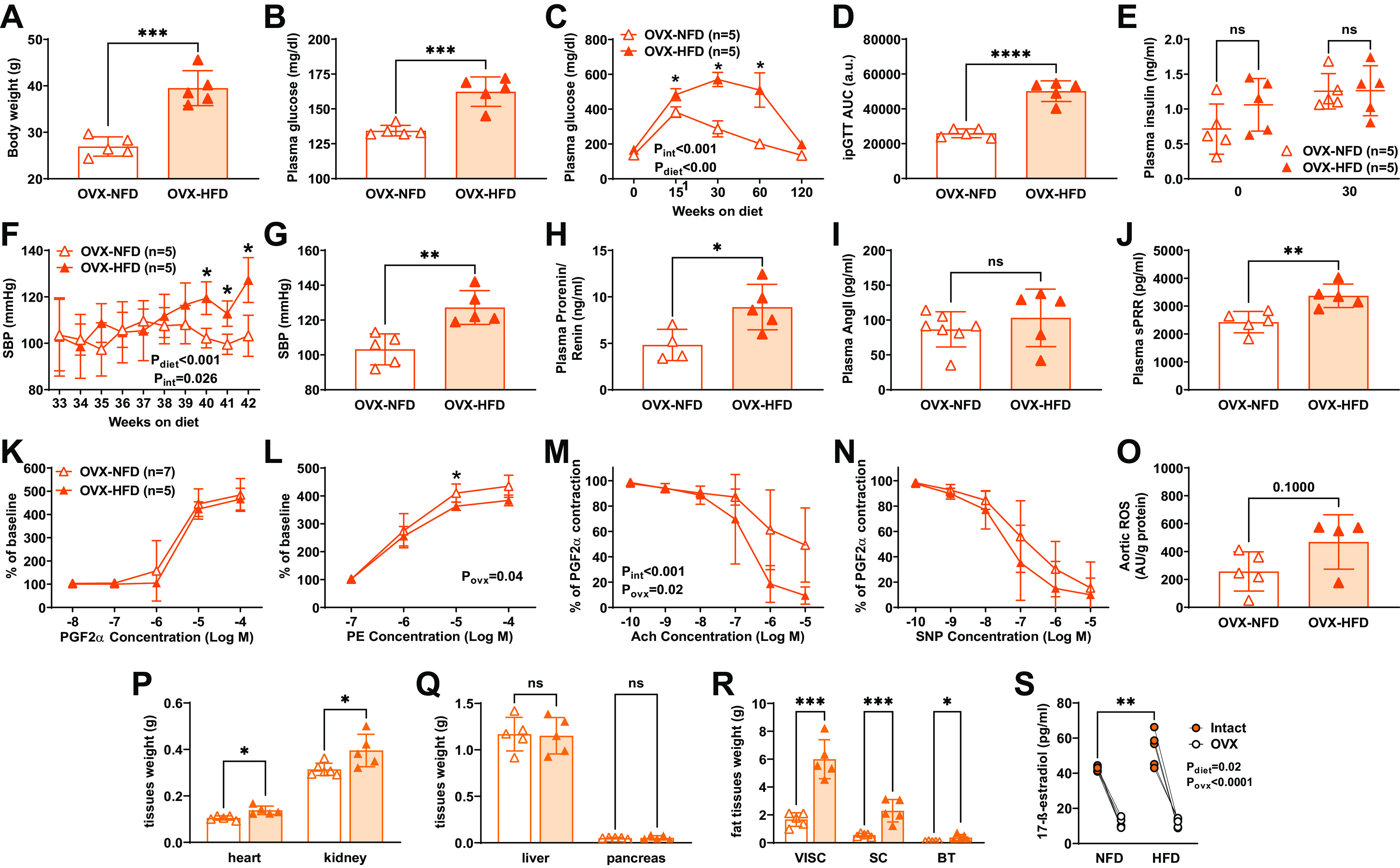
Impact of ovarian hormones loss on diabetic phenotype, SBP and soluble prorenin receptor (sPRR) components, tissue weights, and plasma estradiol. Body weight (A) and fasting glycemia (B) were elevated in ovariectomy (OVX)-high-fat diet (HFD) compared with OVX- normal fat diet (NFD) female mice, paired t test, ***P < 0.001. Fasting glycemia (C) at 30, 60, and 90 min in response to a glucose overload was elevated in HFD group, two-way ANOVA results listed on graph with Sidak’s post hoc tests, *P < 0.05, **P < 0.01, ****P < 0.0001. The area under the curve (AUC) for intraperitoneal glucose tolerance test (ipGTT) (D) was elevated in the OVX-HFD group, t test, ****P < 0.0001. No changes were observed in plasma insulin levels, two-way ANOVA (E). SBP was higher in OVX-HFD females starting at 40 wk of HFD (8 wk post-OVX) (F). Two-way ANOVA results listed on graph with Sidak’s post hoc tests, *P < 0.05. Final SBP was higher in OVX-HFD mice, unpaired t test (G). Total plasma prorenin/renin (H) and sPRR (J) were elevated in OVX-HFD, unpaired t test, *P < 0.05, **P < 0.01. Plasma ANG II levels were not elevated in female mice, unpaired t test (I). Vascular contractility in response to PGF2α was not impacted by HFD (K). Vasocontraction to phenylephrine (PE) was decreased in the OVX-HFD group (L). ACh relaxation in PGF2α preconstricted rings was significantly reduced in OVX-NFD vessels (M). Endothelium-independent relaxation assessed using sodium nitroprusside (SNP) was unchanged by HFD, all two-way ANOVA with results listed on graph and Sidak’s post hoc tests, *P < 0.05 (N). Aortic reactive oxygen species (ROS) production was not impacted by HFD (O). Heart and kidneys were increased in OVX-HFD (P), and no changes were observed in liver and pancreas weight (Q). Paired t tests, *P < 0.05, ***P < 0.0001. Visceral, subcutaneous, and brown fat tissue weights were significantly higher in female mice fed a HFD than in those on NFD (R). ns, not significant. Two-way ANOVA with results listed on graph and Sidak’s post hoc tests. **P < 0.001. Plasma estradiol concentrations were reduced after 12 wk of OVX in all groups tissue (S). Data are represented as means ± SD.
SBP was significantly higher in OVX-HFD mice by 8 wk post-OVX or 40 wk on diet (Pint = 0.026, Pdiet < 0.001, Ptime = 0.114, Fig. 6F) and maintained elevated at 44 wk on HFD (P = 0.0035, Fig. 6G). Total prorenin/renin was augmented in the OVX-HFD group compared with OVX-NFD mice (P = 0.025, Fig. 6H), but no changes were observed in ANG II levels (P = 0.40; Fig. 6I). Importantly, at 12 wk post-OVX and 44 wk on the HFD, we observed significantly increased plasma sPRR levels (P = 0.006, Fig. 6J). In mesenteric resistance arteries, the concentration-response curve to PGF2α was not impacted by HFD in OVX mice (Pint = 0.97, Pconc < 0.0001, Povx = 0.64, Fig. 6K). However, the concentration-response curve to PE was decreased in the OVX-HFD group (Pint = 0.04, Pconc < 0.0001, Povx = 0.68, Fig. 6L), and similarly, the maximum response to PE was not different between groups (351 ± 39 vs. 351 ± 63%, P = 0.99). In rings preconstricted with PGF2α, ACH relaxation was significantly greater in NFD vessels (Pint < 0.001, Pconc < 0.0001, Povx = 0.02, Fig. 6M). Endothelium-independent relaxation to SNP was not different between NFD and HFD vessels (Pint = 0.37, Pconc < 0.0001, Povx = 0.26; Fig. 6N). No differences were observed in ROS production in the aortas of OVX females (P = 0.10; Fig. 6O). Moreover, heart, kidney, and fat tissue weights were significantly higher in OVX-HFD female mice than in OVX-NFD mice (Fig. 6, P–R). In addition, plasma estradiol levels were significantly lower after 12 wk of OVX in NFD and HFD female groups (Fig. 6S).
DISCUSSION
In the present study, a T2DM phenotype was induced by HFD only in males and OVX female mice. Despite weight gain and glucose intolerance in intact females fed a HFD, hyperglycemia, hyperinsulinemia, increased BP, and markers of RAAS activation including circulating sPRR were not detected, except after OVX. The fact that many of these phenotypic changes became evident in females on a HFD only after OVX indicate protective effects of ovarian hormones (41–43). The HFD phenotype in males also included a loss in nocturnal BP dipping, carotid artery stiffness, small artery dysfunction, and aortic ROS. Even though the chronic sPRR infusion in male NFD mice did not impact body weight or BP, sPRR chronic administration enhanced wall thickness and wall/lumen ratio, suggesting direct vascular damage due to sPRR alone. Taken together, this study indicates that circulating sPRR accompanies the development of a T2DM phenotype and may play a role in functional and structural changes in the vasculature.
The current study reveals relevant findings of the sex differences in the development of a cardiometabolic T2DM phenotype and systemic RAAS activation in response to an HFD. Although both male and female mice had increased body weight and ipGTT, plasma glucose, and insulin were not significantly higher in HFD females compared with NFD controls. Moreover, only HFD males displayed significantly higher circulating levels of sPRR, prorenin/renin, and ANG II, along with SBP and MAP, and vascular dysfunction. In contrast, HFD females only exhibited these phenotypic changes after ovarian hormones were reduced via OVX. Multiple studies have shown that estrogen is beneficial to glucose metabolism, controlling insulin resistance, and confers vascular protection in experimental models (44, 45). Intact female mice are protected from weight gain, vascular damage, increased BP, and changes in plasma ANG II concentrations compared with males, and this protection is abolished after OVX and recovered by estrogen treatment (46). Not only does estrogen change in OVX, but testosterone is also affected and ratio between testosterone and estrogen may impact the vasculature (47). The mechanisms by which estrogen and/or testosterone precisely enhance sPRR levels were not investigated in this study. However, emerging evidence shows that treatment with resveratrol, which activates the G protein-coupled estrogen receptor (GPER) (48), suppresses activity of the PRR-ACE-ANG II axis and subsequently decreases fibrosis and vascular senescence (49). The fact that sex steroids are essential modulators of RAAS activity in obese women (4) suggests that plasma sPRR might be a good indicator for vascular damage in postmenopausal women. Age is also an important variable in our study and plasma sPRR levels may differ in aged women with hypertension and T2DM (18, 21), where circulating sPRR levels positively correlate with age, body mass index (BMI), and PRA (21). Moreover, CV risks in postmenopausal women also increase, at least in part, to distribution and accumulation of fat in the subcutaneous tissue (50). It is important to note that adipose tissue is a crucial source for elevated levels of plasma sPRR and circulating ANG II during the development of hypertension in female obese mice (8). Although the levels of plasma sPRR in NFD-OVX middle-aged females did not change over time, we cannot exclude that age and adiposity may also affect sPRR levels, increased BP, and vascular damage in HFD-OVX mice. The interplay between sex, hormonal status, adiposity, and age, is certainly complex and expected to involve several other mechanisms and other tools, which were not investigated in this study. However, we cannot rule out the possibility that the protective role of sex hormones observed here may underlie the greater predisposition of middle-aged females to develop CV complications during T2DM (51, 52).
HFD promotes an increase in BP through activation of the RAAS and is associated with increased circulating levels of sPRR (6, 8, 53). Our results using a murine model of HFD-induced T2DM similarly reveal a relationship between elevated plasma sPRR and BP, particularly with nondipping status in male mice. Although male HFD mice exhibited higher SBP and MAP than NFD mice, this elevation did not reach a hypertensive range. Previous clinical studies demonstrate that nondipping BP could be independent of hypertension, which is associated with left ventricular hypertrophy and secondary forms of hypertension, including insulin resistance and fibrosis (54). Blood pressure follows a circadian rhythm that repeats every 24 h, and healthy individuals experience a 10%–20% decrease in BP at night (dipping) during rest (55). Increased sympathetic and decreased parasympathetic activities may impair BP dipping (56). Moreover, it has been shown that obese male mice receiving recombinant sPRR exhibit increased BP through increased sympathetic tone and reduced baroreflex sensitivity (7). Although the investigators did not calculate BP dipping effect, they reported that in male and female HFD mice with specific deletion of PRR in adipocytes, increased circulating sPRR levels paralleled with a lack of 10% fall in resting BP (7, 8). In the current study, increased sPRR in male mice due to the HFD was similarly associated with elevated SBP and loss of BP dipping. The loss of the diurnal pattern in night/day BP in male HFD mice may promote vascular complications during obesity-induced T2DM. Patients with obesity and T2DM often experience nocturnal nondipping hypertension (57, 58) as well as arterial stiffening (59), increasing the risk for mortality and morbidity due to CVD (60). Although growing evidence reveals that blunted nocturnal fall in BP is a better predictor of CV events in women than in men (61), there is a long way to go to understand the mechanisms underlying the impact of sex differences on nondipping BP effect.
Arterial stiffness is also associated with worse CV outcomes in patients with obesity and T2DM (62). Even though we did not observe changes in pulse pressure, an indicator of arterial stiffening (31), male HFD mice showed a decrease in ex vivo carotid artery material stiffness while reducing wall thickness. This finding agrees with other T2DM animal models that demonstrate increases in femoral and mesenteric artery stiffness in db/db mice (63, 64). However, it opposes to what has been reported in the coronary microcirculation of T2DM mice and pigs (37, 64–66), suggesting vascular bed-specific responses to T2DM. Furthermore, it is not known whether the observed increases in vascular stiffness and ROS precede or follow the increase in circulating sPRR and BP (67). We also observed that HFD in males did not alter collagen content but increased vascular smooth muscle content in the aorta. Indeed, collagen deposition is expected in obesity; however, it is possible that the long-term diet, lower fat content, and other collagen-interacting factors not explored in this study may also affect vascular remodeling (68, 69). PRR is abundantly expressed in the vasculature, specifically in vascular smooth muscle and in endothelial cells (13, 70–72). Evidence suggests that vascular smooth muscle-derived sPRR is a paracrine factor that induces endothelial dysfunction and is associated with the onset of hypertension and CV complications during HFD (73, 74). Thus, it is likely that the observed ex vivo material stiffness was a product of increased smooth muscle ROS, wall stress, and shear stress from BP due to the increase in sPRR and ANG II that induce hypertrophic and hyperproliferative events (75).
Since obesity induces plasma sPRR and stimulates systemic RAAS, we further investigated the direct contribution of sPRR to BP regulation and vascular function. Evidence supports that sPRR infusion at a dose of 30 µg/kg/day intensifies BP elevation, dipping phenotype, and adiposity in HFD-fed male mice, allowing a connection with the observed effects described in our study (7). In addition, the infusion of exogenous sPRR significantly increases plasma sPRR to similar levels found in our study and other HFD models (7). Aortic stiffness, as evidenced by increased PWV was significantly elevated in male mice treated with sPRR; however, no changes in BP or aortic ROS were observed. Administration of exogenous sPRR enhanced wall thickness and wall/lumen ratio, suggesting direct vascular remodeling due to sPRR independent of hypertension. Our findings indicate that the impact of sPRR on BP and arterial stiffening is not the same in NFD and HFD male mice. It is still unclear whether arterial stiffening leads to increased BP, but endothelial dysfunction, aging, and metabolic syndrome are risk factors. Data from our laboratory, examining the effect of ANG II-induced hypertension and aging in males and females, showed increased PWV and stiffness in mice without altering the increase in BP (32, 76). The cause of arterial stiffening is complex and includes structural changes in vascular beds, which were not explored in this study. Recent evidence reporting that chronic infusion of recombinant sPRR in HFD-induced obese mice decreases body weight, hyperglycemia, insulin resistance, and albuminuria, thereby suggesting that sPRR exerts beneficial effects in this experimental mouse model, requires further support to reconcile the findings from others (30). Since obesity is a complex metabolic disease with expected CV complications, it is possible that activated compensatory mechanism of obesity-related hypertension may contribute to changes in plasma sPRR levels. The implications of sPRR independent of BP emphasize its clinical relevance in vascular pathology (77, 78). Future studies are warranted to define the properties and different doses of recombinant sPRR chronic infusion to improve our understanding of the mechanism of action on these aspects.
In normal individuals, circulating prorenin is present at higher concentrations than renin. Patients with T2DM show high levels of circulating prorenin and decreases in renin activity, which correlates with the risk of onset for microvascular complications and diabetic nephropathy (79, 80). The fact that increased plasma sPRR in obese female mice is also associated with systemic RAAS activation (8, 21), raises the possibility that sPRR contributes to prorenin activation in the systemic circulation, thus inducing vascular damage. Likewise, sexual dimorphism exists in plasma prorenin concentrations in humans and rodents (22–25), and sex hormones and aging may affect prorenin systemic levels differently in men and women (22). We measured total prorenin and renin in the plasma, and no sexual dimorphism was observed. Unfortunately, we could not distinguish prorenin or renin content because most commercially available ELISA kits provide only prorenin as a reference (81, 82). One mechanistic factor may be ANG II formation, which is elevated in obesity and known to contribute to obesity-induced hypertension differently in both sexes (4, 6, 7, 53, 83). Despite growing evidence showing the implication of sPRR in the development of obesity to BP regulation during HFD, it is still unclear whether circulating prorenin is sufficient to induce physiological effects on sPRR (84). Nevertheless, we cannot rule out the possibility whether augmentation of systemic sPRR in HFD males and OVX-HFD females concomitant with higher plasma prorenin levels are differentially involved with a greater burden of microvascular complications in T2DM in each sex. Sex differences exist in systemic and intrarenal RAAS activation (21). Women with T2DM exhibit elevated plasma sPRR, which parallels systemic RAAS activation, whereas decreased sPRR in urines from men counterpart correlates with intrarenal RAAS stimulation (21). These findings suggest that sPRR is a potential indicator of RAAS activation and kidney dysfunction in women and men with T2DM.
Our study has limitations. In the present study, intact female mice were not examined for vascular damage following HFD as they were less prone to develop the T2DM phenotype and did not exhibit changes in BP and RAAS components. Nonetheless, vascular damage or other alterations may still exist during the development of obesity and T2DM. On the other hand, OVX female mice, when euthanized, were older and gained more body weight than intact females, which might influence the levels of plasma sPRR. Additional limitations of our study were that we did not investigate the direct effects of estrogen on plasma sPRR levels, as the OVX groups did not receive estrogen to examine whether vascular damage can be reversed by estrogen supplementation. Thus, we acknowledge that some vascular damage may be present but not detected with the tools used in the study. In addition, the long-term diet or lower fat content in the current study compared with other studies (46% vs. 60%) may also affect vascular remodeling (68, 69). Furthermore, sex differences, different infusion doses of recombinant sPRR, lower fat content, and other vascular studies, such as histology of different types of collagens, could be relevant to evaluate the mechanisms and impact of CV complications during obesity and T2DM. Current efforts in our laboratory are addressing all these questions toward defining the role of plasma sPRR in the interactions between sex hormones, age, and obesity and its actual contribution to the regulation of BP.
Perspectives and Conclusions
In perspective, the data from this study suggest that increased levels of systemic sPRR may anticipate the development of hypertension and CV complications during HFD in a sex-dependent manner. The fact that females fed a HFD do not develop hyperglycemia or elevations in BP and plasma sPRR to the same extent as male HFD mice suggests a connection between sex and systemic RAAS activation. The uncovering of the described phenotype in middle-aged females when ovarian hormones is reduced via OVX may help to explain the greater predisposition of postmenopausal females to develop CV complications during T2DM, and further support our previous findings of enhanced plasma sPRR in patients with T2DM (21). Disruption of circadian rhythms is also implicated in obese-induced HFD (85). This is relevant because circulating RAAS follows a circadian rhythm and has an important role in BP rhythms regulation (86, 87). In contrast, plasma sPRR did not display circadian rhythm variation in patients with T2DM (88). The mechanisms by which higher levels of sPRR increase diurnal BP and arterial stiffness are clinically relevant. Current efforts are conducted to elucidate the relevance of sPRR as an indicator of systemic RAAS activation and its impact on the development and progression of CV complications in females and males.
In conclusion, male mice fed aN HFD exhibited high plasma sPRR that coincided with nondipping BP, vascular stiffness, arterial remodeling, vascular ROS in the aorta, and T2DM phenotype. In contrast, HFD-fed female mice display a preserved metabolic phenotype but OVX increased plasma sPRR and BP. During T2DM, plasma sPRR levels may reflect the status of systemic RAAS activation and predisposition to the onset of vascular complications in a sex-dependent manner. Thereby, identifying the underlying mechanism of sPRR obesity-related hypertension and the contribution to CV complications will provide better strategies to promote vascular health or slow down the development and progression of vascular complications in patients with T2DM.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work was supported by National Institutes of Health Grants DK104375 and P30GM103337 (to M. C. Prieto), HL133619 (to S. H. Lindsey), NS094834 (to P. V. G. Katakam), and K99HL155841 (to B. O. Ogola); Fundação de Amparo à Pesquisa do Estado de São Paulo, Brazil, Grant 17/17027-0 (to M. C. Prieto); and American Heart Association Grant AHA829713 (to B. Visniauskas).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.V., V.R., P.V.G.K., S.H.L., and M.C.P. conceived and designed research; B.V., V.R., C.M.A., B.O.O., C.B.R., M.G.-P., V.N.S., S.S.V.P.S., N.R.H., I.K.-D., A.B.M., A.C.H., M.Z., P.V.G.K., and S.H.L. performed experiments; B.V., V.R., B.O.O., C.B.R., and S.H.L. analyzed data; B.V., V.R., P.V.G.K., S.H.L., and M.C.P. interpreted results of experiments; B.V. and S.H.L. prepared figures; B.V., B.O.O., S.H.L., and M.C.P. drafted manuscript; B.V., V.R., B.O.O., P.V.G.K., S.H.L., and M.C.P. edited and revised manuscript; B.V., V.R., C.M.A., B.O.O., C.B.R., M.G.-P., V.N.S., S.S.V.P.S., N.R.H., I.K.-D., A.B.M., A.C.H., M.Z., P.V.G.K., S.H.L., and M.C.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Molecular and Analytical Core and the Mouse Phenotyping Core of the Tulane Renal Hypertension Center of Excellence and the technical experience and support of Alexander Castillo (Physiology, Tulane University SOM) for the radiotelemetry blood pressure approach. We also thank Sufen Zheng and Sravya Tahineni (Pharmacology, Tulane University SOM) for assistance with quantification of ROS in the aorta.
REFERENCES
- 1. Haslam DW, James WP. Obesity. Lancet 366: 1197–1209, 2005. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 2. Ley SH, Schulze MB, Hivert M-F, Meigs JB, Hu FB. Risk factors for type 2 diabetes. In: Diabetes in America (3rd ed.). Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases, 2018. [PubMed] [Google Scholar]
- 3. Peters SA, Huxley RR, Woodward M. Diabetes as a risk factor for stroke in women compared with men: a systematic review and meta-analysis of 64 cohorts, including 775,385 individuals and 12,539 strokes. Lancet 383: 1973–1980, 2014. doi: 10.1016/S0140-6736(14)60040-4. [DOI] [PubMed] [Google Scholar]
- 4. White MC, Fleeman R, Arnold AC. Sex differences in the metabolic effects of the renin-angiotensin system. Biol Sex Differ 10: 31, 2019. doi: 10.1186/s13293-019-0247-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shepard BD. Sex differences in diabetes and kidney disease: mechanisms and consequences. Am J Physiol Renal Physiol 317: F456–F462, 2019. doi: 10.1152/ajprenal.00249.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL, Daugherty A, Cassis LA. Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension 60: 1524–1530, 2012. doi: 10.1161/HYPERTENSIONAHA.112.192690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gatineau E, Gong MC, Yiannikouris F. Soluble prorenin receptor increases blood pressure in high fat-fed male mice. Hypertension 74: 1014–1020, 2019. doi: 10.1161/HYPERTENSIONAHA.119.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gatineau E, Cohn DM, Poglitsch M, Loria AS, Gong M, Yiannikouris F. Losartan prevents the elevation of blood pressure in adipose-PRR deficient female mice while elevated circulating sPRR activates the renin-angiotensin system. Am J Physiol Heart Circ Physiol 316: H506–H515, 2019. doi: 10.1152/ajpheart.00473.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakagawa T, Suzuki-Nakagawa C, Watanabe A, Asami E, Matsumoto M, Nakano M, Ebihara A, Uddin MN, Suzuki F. Site-1 protease is required for the generation of soluble (pro)renin receptor. J Biochem 161: 369–379, 2017. doi: 10.1093/jb/mvw080. [DOI] [PubMed] [Google Scholar]
- 10. Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, Nguyen G. Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension 53: 1077–1082, 2009. doi: 10.1161/HYPERTENSIONAHA.108.127258. [DOI] [PubMed] [Google Scholar]
- 11. Yoshikawa A, Aizaki Y, Kusano K, Kishi F, Susumu T, Iida S, Ishiura S, Nishimura S, Shichiri M, Senbonmatsu T. The (pro)renin receptor is cleaved by ADAM19 in the Golgi leading to its secretion into extracellular space. Hypertens Res 34: 599–605, 2011. doi: 10.1038/hr.2010.284. [DOI] [PubMed] [Google Scholar]
- 12. Nabi AH, Kageshima A, Uddin MN, Nakagawa T, Park EY, Suzuki F. Binding properties of rat prorenin and renin to the recombinant rat renin/prorenin receptor prepared by a baculovirus expression system. Int J Mol Med 18: 483–488, 2006. [PubMed] [Google Scholar]
- 13. Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109: 1417–1427, 2002. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krop M, Lu X, Danser AH, Meima ME. The (pro)renin receptor. A decade of research: what have we learned? Pflugers Arch 465: 87–97, 2013. doi: 10.1007/s00424-012-1105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cousin C, Bracquart D, Contrepas A, Nguyen G. Potential role of the (pro)renin receptor in cardiovascular and kidney diseases. J Nephrol 23: 508–513, 2010. [PubMed] [Google Scholar]
- 16. Gonzalez AA, Lara LS, Luffman C, Seth DM, Prieto MC. The soluble form of the prorenin receptor [s(PRR)] is augmented in the collecting ducts and in the urine of angiotensin II (Ang II)-dependent hypertensive rats. Hypertension 57: 859–864, 2011. doi: 10.1161/HYPERTENSIONAHA.110.167957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tan P, Shamansurova Z, Bisotto S, Michel C, Gauthier MS, Rabasa-Lhoret R, Nguyen TM, Schiller PW, Gutkowska J, Lavoie JL. Impact of the prorenin/renin receptor on the development of obesity and associated cardiometabolic risk factors. Obesity (Silver Spring) 22: 2201–2209, 2014. doi: 10.1002/oby.20844. [DOI] [PubMed] [Google Scholar]
- 18. Morimoto S, Ando T, Niiyama M, Seki Y, Yoshida N, Watanabe D, Kawakami-Mori F, Kobori H, Nishiyama A, Ichihara A. Serum soluble (pro)renin receptor levels in patients with essential hypertension. Hypertens Res 37: 642–648, 2014. doi: 10.1038/hr.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Watanabe N, Bokuda K, Fujiwara T, Suzuki T, Mito A, Morimoto S, Jwa SC, Egawa M, Arai Y, Suzuki F, Sago H, Ichihara A. Soluble (pro)renin receptor and blood pressure during pregnancy: a prospective cohort study. Hypertension 60: 1250–1256, 2012. doi: 10.1161/HYPERTENSIONAHA.112.197418. [DOI] [PubMed] [Google Scholar]
- 20. Narita T, Ichihara A, Matsuoka K, Takai Y, Bokuda K, Morimoto S, Itoh H, Seki H. Placental (pro)renin receptor expression and plasma soluble (pro)renin receptor levels in preeclampsia. Placenta 37: 72–78, 2016. doi: 10.1016/j.placenta.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 21. Visniauskas B, Arita DY, Rosales CB, Feroz MA, Luffman C, Accavitti MJ, Dawkins G, Hong J, Curnow AC, Thethi TK, Lefante JJ, Jaimes EA, Mauvais-Jarvis F, Fonseca VA, Prieto MC. Sex differences in soluble prorenin receptor in patients with type 2 diabetes. Biol Sex Differ 12: 33, 2021. doi: 10.1186/s13293-021-00374-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Danser AH, Derkx FH, Schalekamp MA, Hense HW, Riegger GA, Schunkert H. Determinants of interindividual variation of renin and prorenin concentrations: evidence for a sexual dimorphism of (pro)renin levels in humans. J Hypertens 16: 853–862, 1998. doi: 10.1097/00004872-199816060-00017. [DOI] [PubMed] [Google Scholar]
- 23. Fujimoto K, Kawamura S, Bando S, Kamata Y, Kodera Y, Shichiri M. Circulating prorenin: its molecular forms and plasma concentrations. Hypertens Res 44: 674–684, 2021. doi: 10.1038/s41440-020-00610-0. [DOI] [PubMed] [Google Scholar]
- 24. Johannessen A, Nielsen AH, Poulsen K. Sexual dimorphism of inactive renin in rat plasma. Clin Exp Hypertens A 12: 1405–1417, 1990. doi: 10.3109/10641969009073527. [DOI] [PubMed] [Google Scholar]
- 25. Nielsen AH, Johannessen A, Poulsen K. Inactive plasma renin exhibits sex difference in mice. Clin Sci (Lond) 76: 439–446, 1989. doi: 10.1042/cs0760439. [DOI] [PubMed] [Google Scholar]
- 26. Deinum J, Tarnow L, van Gool JM, de Bruin RA, Derkx FH, Schalekamp MA, Parving HH. Plasma renin and prorenin and renin gene variation in patients with insulin-dependent diabetes mellitus and nephropathy. Nephrol Dial Transplant 14: 1904–1911, 1999. doi: 10.1093/ndt/14.8.1904. [DOI] [PubMed] [Google Scholar]
- 27. Ramkumar N, Stuart D, Peterson CS, Hu C, Wheatley W, Cho JM, Symons JD, Kohan DE. Loss of soluble (pro)renin receptor attenuates angiotensin-II induced hypertension and renal injury. Circ Res 129: 50–62, 2021. doi: 10.1161/CIRCRESAHA.120.317532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feng Y, Peng K, Luo R, Wang F, Yang T. Site-1 protease-derived soluble (pro)renin receptor contributes to angiotensin II-induced hypertension in mice. Hypertension 77: 405–416, 2021. doi: 10.1161/HYPERTENSIONAHA.120.15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gatineau E, Arthur G, Poupeau A, Nichols K, Spear BT, Shelman NR, Graf GA, Temel RE, Yiannikouris FB. The prorenin receptor and its soluble form contribute to lipid homeostasis. Am J Physiol Endocrinol Metab 320: E609–E618, 2021. doi: 10.1152/ajpendo.00135.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang F, Luo R, Zou CJ, Xie S, Peng K, Zhao L, Yang KT, Xu C, Yang T. Soluble (pro)renin receptor treats metabolic syndrome in mice with diet-induced obesity via interaction with PPARγ. JCI Insight 5: e128061, 2020. doi: 10.1172/jci.insight.128061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reverte V, Gogulamudi VR, Rosales CB, Musial DC, Gonsalez SR, Parra-Vitela AV, Galeas-Pena M, Sure VN, Visniauskas B, Lindsey SH, Katakam PV, Mc P. Urinary angiotensinogen increases in the absence of overt renal injury in high fat diet-induced type 2 diabetic mice. J Diabetes Complications 34: 107448, 2019. doi: 10.1016/j.jdiacomp.2019.107448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ogola BO, Clark GL, Abshire CM, Harris NR, Gentry KL, Gunda SS, Kilanowski-Doroh I, Wong TJ, Visniauskas B, Lawrence DJ, Zimmerman MA, Bayer CL, Groban L, Miller KS, Lindsey SH. Sex and the G protein-coupled estrogen receptor impact vascular stiffness. Hypertension 78: e1–e14, 2021. doi: 10.1161/HYPERTENSIONAHA.120.16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ogola BO, Zimmerman MA, Sure VN, Gentry KM, Duong JL, Clark GL, Miller KS, Katakam PVG, Lindsey SH. G protein-coupled estrogen receptor protects from angiotensin II-induced increases in pulse pressure and oxidative stress. Front Endocrinol (Lausanne) 10: 586, 2019. doi: 10.3389/fendo.2019.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ichihara A, Hayashi M, Kaneshiro Y, Suzuki F, Nakagawa T, Tada Y, Koura Y, Nishiyama A, Okada H, Uddin MN, Nabi AH, Ishida Y, Inagami T, Saruta T. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest 114: 1128–1135, 2004. doi: 10.1172/JCI21398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shao W, Seth DM, Prieto MC, Kobori H, Navar LG. Activation of the renin-angiotensin system by a low-salt diet does not augment intratubular angiotensinogen and angiotensin II in rats. Am J Physiol Renal Physiol 304: F505–F514, 2013. doi: 10.1152/ajprenal.00587.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferruzzi J, Bersi MR, Humphrey JD. Biomechanical phenotyping of central arteries in health and disease: advantages of and methods for murine models. Ann Biomed Eng 41: 1311–1330, 2013. doi: 10.1007/s10439-013-0799-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Trask AJ, Delbin MA, Katz PS, Zanesco A, Lucchesi PA. Differential coronary resistance microvessel remodeling between type 1 and type 2 diabetic mice: impact of exercise training. Vascul Pharmacol 57: 187–193, 2012. doi: 10.1016/j.vph.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zimmerman MA, Hutson DD, Trimmer EH, Kashyap SN, Duong JL, Murphy B, Grissom EM, Daniel JM, Lindsey SH. Long- but not short-term estradiol treatment induces renal damage in midlife ovariectomized Long-Evans rats. Am J Physiol Renal Physiol 312: F305–F311, 2017. doi: 10.1152/ajprenal.00411.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Katakam PV, Gordon AO, Sure VN, Rutkai I, Busija DW. Diversity of mitochondria-dependent dilator mechanisms in vascular smooth muscle of cerebral arteries from normal and insulin-resistant rats. Am J Physiol Heart Circ Physiol 307: H493–H503, 2014. doi: 10.1152/ajpheart.00091.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 23: 291–299, 2001. [PubMed] [Google Scholar]
- 41. Yakar S, Nunez NP, Pennisi P, Brodt P, Sun H, Fallavollita L, Zhao H, Scavo L, Novosyadlyy R, Kurshan N, Stannard B, East-Palmer J, Smith NC, Perkins SN, Fuchs-Young R, Barrett JC, Hursting SD, LeRoith D. Increased tumor growth in mice with diet-induced obesity: impact of ovarian hormones. Endocrinology 147: 5826–5834, 2006. doi: 10.1210/en.2006-0311. [DOI] [PubMed] [Google Scholar]
- 42. Kumagai S, Holmäng A, Björntorp P. The effects of oestrogen and progesterone on insulin sensitivity in female rats. Acta Physiol Scand 149: 91–97, 1993. doi: 10.1111/j.1748-1716.1993.tb09596.x. [DOI] [PubMed] [Google Scholar]
- 43. Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology 150: 2109–2117, 2009. doi: 10.1210/en.2008-0971. [DOI] [PubMed] [Google Scholar]
- 44. Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 34: 309–338, 2013. doi: 10.1210/er.2012-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Visniauskas B, Kilanowski-Doroh I, Ogola BO, McNally AB, Horton AC, Imulinde Sugi A, Lindsey SH. Estrogen-mediated mechanisms in hypertension and other cardiovascular diseases. J Hum Hypertens, 2022. Online ahead of print. doi: 10.1038/s41371-022-00771-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Y, Shoemaker R, Thatcher SE, Batifoulier-Yiannikouris F, English VL, Cassis LA. Administration of 17β-estradiol to ovariectomized obese female mice reverses obesity-hypertension through an ACE2-dependent mechanism. Am J Physiol Endocrinol Physiol 308: E1066–E1075, 2015. doi: 10.1152/ajpendo.00030.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu A, Gao L, Kang S, Liu Y, Xu C, Sun H, Li D, Yan C. Testosterone enhances estradiol's cardioprotection in ovariectomized rats. J Endocrinol 212: 61–69, 2012. doi: 10.1530/JOE-11-0181. [DOI] [PubMed] [Google Scholar]
- 48. Dong WH, Chen JC, He YL, Xu JJ, Mei YA. Resveratrol inhibits K(v)2.2 currents through the estrogen receptor GPR30-mediated PKC pathway. Am J Physiol Cell Physiol 305: C547–C557, 2013. doi: 10.1152/ajpcell.00146.2013. [DOI] [PubMed] [Google Scholar]
- 49. Kim EN, Kim MY, Lim JH, Kim Y, Shin SJ, Park CW, Kim YS, Chang YS, Yoon HE, Bs C. The protective effect of resveratrol on vascular aging by modulation of the renin-angiotensin system. Atherosclerosis 270: 123–131, 2018. doi: 10.1016/j.atherosclerosis.2018.01.043. [DOI] [PubMed] [Google Scholar]
- 50. Chait A, den Hartigh LJ. Adipose tissue distribution, inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Front Cardiovasc Med 7: 22, 2020. doi: 10.3389/fcvm.2020.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Toledo DP, Akamine E, Nigro D, Passaglia RC, Carvalho MH, Fortes ZB. Microvascular reactivity in experimental diabetes: responses of male and female rats. Inflamm Res 52: 191–198, 2003. doi: 10.1007/s000110300071. [DOI] [PubMed] [Google Scholar]
- 52. Peters SA, Huxley RR, Sattar N, Woodward M. Sex differences in the excess risk of cardiovascular diseases associated with type 2 diabetes: potential explanations and clinical implications. Curr Cardiovasc Risk Rep 9: 36, 2015. doi: 10.1007/s12170-015-0462-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu CH, Mohammadmoradi S, Thompson J, Su W, Gong M, Nguyen G, Yiannikouris F. Adipocyte (pro)renin-receptor deficiency induces lipodystrophy, liver steatosis and increases blood pressure in male mice. Hypertension 68: 213–219, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cuspidi C, Facchetti R, Bombelli M, Sala C, Negri F, Grassi G, Mancia G. Nighttime blood pressure and new-onset left ventricular hypertrophy: findings from the Pamela population. Hypertension 62: 78–84, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00682. [DOI] [PubMed] [Google Scholar]
- 55. Douma LG, Gumz ML. Circadian clock-mediated regulation of blood pressure. Free Radic Biol Med 119: 108–114, 2018. doi: 10.1016/j.freeradbiomed.2017.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kanbay M, Turgut F, Uyar ME, Akcay A, Covic A. Causes and mechanisms of nondipping hypertension. Clin Exp Hypertens 30: 585–597, 2008. doi: 10.1080/10641960802251974. [DOI] [PubMed] [Google Scholar]
- 57. Sun L, Yan B, Gao Y, Su D, Peng L, Jiao Y, Wang Y, Han D, Wang G. Relationship between blood pressure reverse dipping and type 2 diabetes in hypertensive patients. Sci Rep 6: 25053, 2016. doi: 10.1038/srep25053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ayala DE, Moya A, Crespo JJ, Castiñeira C, Domínguez-Sardiña M, Gomara S, Sineiro E, Mojón A, Fontao MJ, Hermida RC; Hygia Project Investigators. Circadian pattern of ambulatory blood pressure in hypertensive patients with and without type 2 diabetes. Chronobiol Int 30: 99–115, 2013. doi: 10.3109/07420528.2012.701489. [DOI] [PubMed] [Google Scholar]
- 59. Bruno RM, Penno G, Daniele G, Pucci L, Lucchesi D, Stea F, Landini L, Cartoni G, Taddei S, Ghiadoni L, Del Prato S. Type 2 diabetes mellitus worsens arterial stiffness in hypertensive patients through endothelial dysfunction. Diabetologia 55: 1847–1855, 2012. doi: 10.1007/s00125-012-2517-1. [DOI] [PubMed] [Google Scholar]
- 60. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation 137: e67–e492, 2018. [Erratum in Circulation 137: e493, 2018]. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 61. Boggia J, Thijs L, Hansen TW, Li Y, Kikuya M, Björklund-Bodegård K, Richart T, Ohkubo T, Jeppesen J, Torp-Pedersen C, Dolan E, Kuznetsova T, Olszanecka A, Tikhonoff V, Malyutina S, Casiglia E, Nikitin Y, Lind L, Maestre G, Sandoya E, Kawecka-Jaszcz K, Imai Y, Wang J, Ibsen H, O'Brien E, Staessen JA; International Database on Ambulatory blood pressure in relation to Cardiovascular Outcomes Investigators. Ambulatory blood pressure monitoring in 9357 subjects from 11 populations highlights missed opportunities for cardiovascular prevention in women. Hypertension 57: 397–405, 2011. doi: 10.1161/HYPERTENSIONAHA.110.156828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Urbina EM, Kimball TR, Khoury PR, Daniels SR, Dolan LM. Increased arterial stiffness is found in adolescents with obesity or obesity-related type 2 diabetes mellitus. J Hypertens 28: 1692–1698, 2010. doi: 10.1097/HJH.0b013e32833a6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Souza-Smith FM, Katz PS, Trask AJ, Stewart JA Jr, Lord KC, Varner KJ, Vassallo DV, Lucchesi PA. Mesenteric resistance arteries in type 2 diabetic db/db mice undergo outward remodeling. PLoS One 6: e23337, 2011. doi: 10.1371/journal.pone.0023337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Katz PS, Trask AJ, Souza-Smith FM, Hutchinson KR, Galantowicz ML, Lord KC, Stewart JA Jr, Cismowski MJ, Varner KJ, Lucchesi PA. Coronary arterioles in type 2 diabetic (db/db) mice undergo a distinct pattern of remodeling associated with decreased vessel stiffness. Basic Res Cardiol 106: 1123–1134, 2011. doi: 10.1007/s00395-011-0201-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Anghelescu M, Tonniges JR, Calomeni E, Shamhart PE, Agarwal G, Gooch KJ, Trask AJ. Vascular mechanics in decellularized aortas and coronary resistance microvessels in type 2 diabetic db/db mice. Ann Biomed Eng 43: 2760–2770, 2015. doi: 10.1007/s10439-015-1333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Trask AJ, Katz PS, Kelly AP, Galantowicz ML, Cismowski MJ, West TA, Neeb ZP, Berwick ZC, Goodwill AG, Alloosh M, Tune JD, Sturek M, Lucchesi PA. Dynamic micro- and macrovascular remodeling in coronary circulation of obese Ossabaw pigs with metabolic syndrome. J Appl Physiol (1985) 113: 1128–1140, 2012. doi: 10.1152/japplphysiol.00604.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lacolley P, Bezie Y, Girerd X, Challande P, Benetos A, Boutouyrie P, Ghodsi N, Lucet B, Azoui R, Laurent S. Aortic distensibility and structural changes in sinoaortic-denervated rats. Hypertension 26: 337–340, 1995. doi: 10.1161/01.HYP.26.2.337. [DOI] [PubMed] [Google Scholar]
- 68. Carroll JF, Zenebe WJ, Strange TB. Cardiovascular function in a rat model of diet-induced obesity. Hypertension 48: 65–72, 2006. doi: 10.1161/01.HYP.0000224147.01024.77. [DOI] [PubMed] [Google Scholar]
- 69. Silva DC, Lima-Leopoldo AP, Leopoldo AS, Campos DH, Nascimento AF, Oliveira Junior SA, Padovani CR, Cicogna AC. Influence of term of exposure to high-fat diet-induced obesity on myocardial collagen type I and III. Arq Bras Cardiol 102: 157–163, 2014. doi: 10.5935/abc.20130232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Obradovic D, Loncar G, Radenovic S, Tahirovic E, Heidecke H, Schulze-Forster K, Muller D, Busjahn A, Buttner P, Veskovic J, Zdravkovic M, Li H, Li S, Savkovic V, Pieske B, Dungen HD, Dechend R. Soluble (pro)renin receptor in elderly chronic heart failure patients. Front Biosci 25: 1839–1853, 2020. doi: 10.2741/4880. [DOI] [PubMed] [Google Scholar]
- 71. Kanda A, Noda K, Saito W, Ishida S. (Pro)renin receptor is associated with angiogenic activity in proliferative diabetic retinopathy. Diabetologia 55: 3104–3113, 2012. doi: 10.1007/s00125-012-2702-2. [DOI] [PubMed] [Google Scholar]
- 72. Patel NR, K CR, Blanks AE, Li Y, Prieto MC, Meadows SM. Endothelial cell polarity and extracellular matrix composition require functional ATP6AP2 during developmental and pathological angiogenesis. JCI Insight 7: e154379, 2022. doi: 10.1172/jci.insight.154379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fu Z, Wang F, Liu X, Hu J, Su J, Lu X, Lu A, Cho JM, Symons JD, Zou CJ, Yang T. Soluble (pro)renin receptor induces endothelial dysfunction and hypertension in mice with diet-induced obesity via activation of angiotensin II type 1 receptor. Clin Sci (Lond) 135: 793–810, 2021. doi: 10.1042/CS20201047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xu C, Liu C, Xiong J, Yu J. Cardiovascular aspects of the (pro)renin receptor: function and significance. FASEB J 36: e22237, 2022. doi: 10.1096/fj.202101649RRR. [DOI] [PubMed] [Google Scholar]
- 75. Berk BC. Vascular smooth muscle growth: autocrine growth mechanisms. Physiol Rev 81: 999–1030, 2001. doi: 10.1152/physrev.2001.81.3.999. [DOI] [PubMed] [Google Scholar]
- 76. Ogola BO, Abshire CM, Visniauskas B, Kiley JX, Horton AC, Clark-Patterson GL, Kilanowski-Doroh I, Diaz Z, Bicego AN, McNally AB, Zimmerman MA, Groban L, Trask AJ, Miller KS, Lindsey SH. Sex differences in vascular aging and impact of GPER deletion. Am J Physiol Heart Circ Physiol 323: H336–H349, 2022. doi: 10.1152/ajpheart.00238.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hennrikus M, Gonzalez AA, Prieto MC. The prorenin receptor in the cardiovascular system and beyond. Am J Physiol Heart Circ Physiol 314: H139–H145, 2018. doi: 10.1152/ajpheart.00373.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kurauchi-Mito A, Ichihara A, Bokuda K, Sakoda M, Kinouchi K, Yaguchi T, Yamada T, Sun-Wada GH, Wada Y, Itoh H. Significant roles of the (pro)renin receptor in integrity of vascular smooth muscle cells. Hypertens Res 37: 830–835, 2014. doi: 10.1038/hr.2014.92. [DOI] [PubMed] [Google Scholar]
- 79. Luetscher JA, Kraemer FB, Wilson DM, Schwartz HC, Bryer-Ash M. Increased plasma inactive renin in diabetes mellitus. A marker of microvascular complications. N Engl J Med 312: 1412–1417, 1985. doi: 10.1056/NEJM198505303122202. [DOI] [PubMed] [Google Scholar]
- 80. Deinum J, Rønn B, Mathiesen E, Derkx FH, Hop WC, Schalekamp MA. Increase in serum prorenin precedes onset of microalbuminuria in patients with insulin-dependent diabetes mellitus. Diabetologia 42: 1006–1010, 1999. [Erratum in Diabetologia 42: 1444, 1999]. doi: 10.1007/s001250051260. [DOI] [PubMed] [Google Scholar]
- 81. Roksnoer LC, Verdonk K, Garrelds IM, van Gool JM, Zietse R, Hoorn EJ, Danser AH. Methodologic issues in the measurement of urinary renin. Clin J Am Soc Nephrol 9: 1163–1167, 2014. doi: 10.2215/CJN.12661213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Prieto MC, Gonzalez AA, Visniauskas B, Navar LG. The evolving complexity of the collecting duct renin-angiotensin system in hypertension. Nat Rev Nephrol 17: 481–492, 2021. doi: 10.1038/s41581-021-00414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cassis LA, Police SB, Yiannikouris F, Thatcher SE. Local adipose tissue renin-angiotensin system. Curr Hypertens Rep 10: 93–98, 2008. doi: 10.1007/s11906-008-0019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Danser AH. The role of the (pro)renin receptor in hypertensive disease. Am J Hypertens 28: 1187–1196, 2015. doi: 10.1093/ajh/hpv045. [DOI] [PubMed] [Google Scholar]
- 85. Rudic RD, Fulton DJ. Pressed for time: the circadian clock and hypertension. J Appl Physiol (1985) 107: 1328–1338, 2009. doi: 10.1152/japplphysiol.00661.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kool MJ, Wijnen JA, Derkx FH, Struijker Boudier HA, Van Bortel LM. Diurnal variation in prorenin in relation to other humoral factors and hemodynamics. Am J Hypertens 7: 723–730, 1994. doi: 10.1093/ajh/7.8.723. [DOI] [PubMed] [Google Scholar]
- 87. Richards AM, Nicholls MG, Espiner EA, Ikram H, Cullens M, Hinton D. Diurnal patterns of blood pressure, heart rate and vasoactive hormones in normal man. Clin Exp Hypertens A 8: 153–166, 1986. doi: 10.3109/10641968609074769. [DOI] [PubMed] [Google Scholar]
- 88. Nguyen G, Blanchard A, Curis E, Bergerot D, Chambon Y, Hirose T, Caumont-Prim A, Tabard SB, Baron S, Frank M, Totsune K, Azizi M. Plasma soluble (pro)renin receptor is independent of plasma renin, prorenin, and aldosterone concentrations but is affected by ethnicity. Hypertension 63: 297–302, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.