

Abstract
Background & Aims:
Detailed investigation of the biological pathways leading to hepatic fibrosis and identification of liver fibrosis biomarkers may facilitate early interventions for pediatric cholestasis.
Approach & Results:
A targeted ELISA-based panel of 9 biomarkers (lysyl oxidase (LOX), tissue inhibitor matrix metalloproteinase 1 (TIMP1), connective tissue growth factor (CTGF), interleukin-8 (IL-8), endoglin, periostin, mac-2-binding protein (mac2-BP), matrix metalloproteinase-3 and −7 (MMP-3, MMP-7) was examined in children with biliary atresia (BA, n=187), alpha-1 antitrypsin deficiency (A1AT, n=78) and Alagille syndrome (ALGS, n=65) and correlated with liver stiffness (LSM) and biochemical measures of liver disease. Median age and LSM were 9 years and 9.5 kPa. After adjusting for covariates, there were positive correlations between LSM and endoglin (p=0.04), IL-8 (p<0.001) and MMP-7 (p<0.001) in BA. The best prediction model for LSM in BA using clinical and lab measurements had an R2=0.437; adding IL-8 and MMP7 improved R2 to 0.523 and 0.526 (both p<0.0001). In A1AT, CTGF and LSM were negatively correlated (p=0.004); adding CTGF to a LSM prediction model improved R2 from 0.524 to 0.577 (p=0.0033). Biomarkers did not correlate with LSM in ALGS. A significant number of biomarker/lab correlations were found in BA but not A1AT or ALGS.
Conclusions:
Endoglin, IL-8, and MMP-7 significantly correlate with increased LSM in children with BA, while CTGF inversely correlates with LSM in A1AT; these biomarkers appear to enhance prediction of LSM beyond clinical tests. Future disease-specific investigations of change in these biomarkers over time and as predictors of clinical outcomes will be important.
Keywords: Alagille syndrome, alpha-1 antitrypsin deficiency, biliary atresia, cholestasis, cirrhosis, non-invasive, pediatric liver disease
Background
Detailed investigation of the biological pathways that lead to early and severe fibrosis and identification of non-invasive biomarkers of liver fibrosis can facilitate early interventions for pediatric cholestatic liver disorders. Biomarkers identified by genomic, proteomic, and metabolomic technologies offer new strategies to study etiopathogenesis, inform clinical decision-making or predict outcomes in pediatric liver diseases (1–4). However, each liver disease is unique with different contributory factors and the likely interplay of anatomy, epigenetics, fibrosis, inflammation, and vasculopathy.
Biliary atresia (BA), alpha-1 antitrypsin deficiency (A1AT) and Alagille syndrome (ALGS) represent an important group of congenital cholestatic disorders. BA is a fibro-obliterative disease of the biliary system that presents in the early neonatal period with aggressive progression to cirrhosis within the first year of life (5–9) and continues to be the leading indication for liver transplantation (LT) in children. Inherited liver disorders such as A1AT and ALGS, have a different natural history and distinct pathophysiology from BA and one another. Children with A1AT who are homozygous for the classic mutant Z allele have misfolding and defective secretion of the A1AT protein, with potential to progress to early cirrhosis with or without lung disease (10). However, it is unknown what factors influence the highly variable disease progression and risk of fibrosis seen in A1AT (11). ALGS, a developmental disorder manifest by bile duct paucity and cholestasis often with severe pruritus and extrahepatic involvement, including cardiac, vascular, and renal abnormalities (12) also has unpredictable progression of fibrosis, although the majority of patients are transplanted by 18 years of age for persistent cholestasis or complications of portal hypertension (13).
Single center studies in children with a variety of fibrotic liver diseases have identified specific markers reflecting matrix deposition, hepatic stellate cell activation, collagen turnover, and chemoattractant expression (2–4, 14) that correlate with histologic measures of fibrosis. Meanwhile, imaging modalities have advanced and may further augment our ability to monitor the progression and regression of liver fibrosis. Vibration-controlled transient elastography (TE, FibroScan®) is a non-invasive, painless alternative to liver biopsy that utilizes shear wave velocity to measure liver stiffness, an indirect measure of fibrosis. Use of TE to detect significant fibrosis in children has been validated by liver biopsy in single center studies of both hepatocellular and hepatobiliary diseases (15–21), however the availability of TE as a clinical tool in the pediatric setting is relatively expensive and as such currently limited. Identification of relevant commercially available serum biomarkers of fibrosis and their correlation with transient elastography may inform progression or severity of fibrosis where TE is unavailable and translate into novel markers/endpoints for clinical trials of evolving anti-fibrogenic therapies.
The FibroScan in Pediatric Cholestatic Liver Disease (FORCE) study is an ongoing, prospective, multi-center study (NCT02922751) of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)/National Institutes of Health (NIH)-supported Childhood Liver Disease Research Network (ChiLDReN). This rich resource positioned the network to conduct investigations with the primary objective of identifying serum biomarkers associated with FibroScan™-based liver stiffness measurement (LSM) in children with BA, A1AT, and ALGS. The scientific rationale for the selection of the targeted nine biomarker panel is described in detail (Supplemental Document 1). These biomarkers potentially assess a diverse array of pathways and physiologic mechanisms that may interact, leading to fibrosis. Secondary objectives include investigating the association of these biomarkers with clinical characteristics, biomarker indices, and laboratory determinants of liver disease and ability to enhance prediction of LSM beyond standard clinical tests.
Methods
FORCE includes the collection of serum at the time of FibroScan™ in participants whose clinical status has been well characterized in three distinct prospective longitudinal databases (Prospective Database of Infants with Cholestasis [PROBE, NCT00061828], Biliary Atresia Study in Infants and Children [BASIC, NCT00345553], and Longitudinal Observational Study of Genetic Causes of Intrahepatic Cholestasis [LOGIC, NCT00571272]). This study focused on baseline evaluation of FORCE participants less than 21 years of age with BA, A1AT, or ALGS with their native liver who had a valid LSM and serum available for biomarker investigation.
Clinical data including medical history, physical examination, interval events, and laboratory values, along with biosamples and LSM were collected in FORCE and the parent longitudinal database. Serum samples were collected on the same day as LSM and immediately processed and stored at −80 C. Inclusion and exclusion criteria in FORCE as well as LSM requirements have been described previously in detail (22).
Briefly, BA participants all had a confirmed diagnosis of BA determined by chart review including review of pertinent diagnostic biopsy reports, or radiologic reports and were all post Kasai hepatoportoenterostomy with native liver. ALGS participants in this study met strict diagnostic criteria in which there was a combination of a family history of ALGS, the presence of paucity of interlobular bile ducts on liver biopsy, the identification of a JAGGED1 or NOTCH2 mutation, and clinical criteria (cardiac, ocular, vertebral, renal, facial, cholestasis) with evidence of clinical, biochemical or histological liver disease. Patients with A1AT were defined as having low serum A1AT concentrations (< lower limit of normal for laboratory) with PIZZ or PISZ phenotype or genotype for participants prior to liver transplantation with liver disease associated with A1AT. Children with known polysplenia or asplenia, situs inversus, clinically significant ascites, an implantable active medical device (such as a pacemaker or defibrillator), an open wound near the testing site, current pregnancy, or who had undergone liver transplantation were not eligible.
Liver stiffness (reported in kPa) was measured by vibration-controlled TE in nonfasted and nonsedated participants, using FibroScan according to the manufacturer’s instructions (Echosens, Waltham, MA). All operators were trained and certified at each site by a designated trainer from Echosens to perform FibroScan™ measurements to ensure consistent and standardized acquisition of complete data. A valid LSM required at least ten valid measurements using the appropriate probe (S or M) and exam type (S1 or S2) based on thoracic perimeter and skin capsule distance with interquartile kPa range/median kPa value <30% (23).
LSM was categorized using previously described liver biopsy supported thresholds derived from children with cholestatic or chronic liver diseases and a meta-analysis derived reference range for normal liver stiffness in healthy children (20, 24–29). For analyses, the following categories were applied: <6 (no or mild fibrosis, F0/F1), 6 to <10 (significant fibrosis, ≥F2) 10 to <15 (bridging fibrosis, F3/4), and 15–75 (cirrhosis, F4) kPa. To address the potential impact of cardiac and venous congestion on LSM, for participants with ALGS only, severe cardiac disease was defined as having ≥1 of the following: pulmonary valve stenosis, tetralogy of Fallot, ventricular/atrial septal defect, pulmonary atresia, and/or aortic coarctation. All other cardiac defects were considered mild.
A PubMed search using the following terms: hepatic[All Fields] AND (“liver cirrhosis”[MeSH Terms] OR (“liver”[All Fields] AND “cirrhosis”[All Fields]) OR “liver cirrhosis”[All Fields] OR (“liver”[All Fields] AND “fibrosis”[All Fields]) OR “liver fibrosis”[All Fields]) AND (“biomarkers”[MeSH Terms] OR “biomarkers”[All Fields]) AND (“child”[MeSH Terms] OR “child”[All Fields] OR “children”[All Fields])) on November 1, 2018 yielded 30 peer reviewed articles providing the biological basis, scientific and clinical rationale for the targeted nine biomarker panel. Supplemental Document 1 summarizes what is known about each biomarker and its relationship to fibrosis or liver stiffness, in the context of pediatric liver disease when available.
ELISA-based measurement of these nine targeted biomarkers (lysyl oxidase (LOX, Mybiosource, Cat #MBS039099, San Diego, CA), tissue inhibitor matrix metalloproteinase 1 (TIMP1, R&D, Cat #DTM100, Minneapolis, MN), connective tissue growth factor (CTGF, Mybiosource, Cat # MBS266000, San Diego, CA), interleukin-8 (IL-8, Millipore, Cat # HCYTOMAG-60K-01, Burlington, MA), endoglin (Sigma-Aldrich, Cat # RAB0171, Burlington, MA), periostin (Abcam, Cat # AB213816, Cambridge, UK), mac-2-binding protein (mac2-BP, Mybiosource, Cat # MBS108990, San Diego, CA), matrix metalloproteinase-3 (MMP-3, R&D, Cat# DMP300, Minneapolis, MN) and matrix metalloproteinase-7 (MMP-7, R&D, Cat# DMP700, Minneapolis, MN) were performed according to the manufacturer’s instructions. Serum samples were run in duplicate and randomly aliquoted onto 96-well plates with six internal controls (frozen sera from healthy controls with normal liver biochemistries stored at −80 C). Measurements of each analyte were in the linear portion of the response curve for each biomarker.
Concentrations for selected biomarkers were displayed visually using boxplots. Scatter plots with LOESS (locally estimated scatterplot smoothing) lines were used to visually inspect associations between biomarker concentrations and clinical characteristics. Differences among disease groups were compared using Kruskal-Wallis tests for continuous variables and Chi-Square test for categorical variables. Biomarker differences in participants with clinically evident portal hypertension status (CEPH, absent, possible, definite) using a previously described research definition (30) were also analyzed and tested with Kruskal-Wallis tests. Correlations with LSM were estimated using Spearman’s rank correlation for biomarkers and other clinical parameters; multivariable linear regression was used to examine these associations controlling for age, total bilirubin (TB), albumin, GGT and AST. Residuals were used to evaluate regression model assumptions. Log transformations were applied to all biomarkers except TIMP-1 and MMP-7 to account for skewed distributions prior to regression modeling.
To evaluate whether the targeted biomarkers improve the ability to predict LSM in each diagnosis group beyond clinical characteristics and laboratory measures, a linear regression with forward step-wise selection (inclusion criteria; p≤0.10) was performed first to develop a parsimonious model including all clinical labs and features as candidate predictors for predicting LSM; the targeted biomarkers were then added one at a time to this model to evaluate which biomarker(s) further explain remaining variance in LSM and significantly improve the model fit. For all three disease groups, 50 multiply imputed data sets were utilized to complete any missing values. Rubin’s rule was used to provide pooled results based on parameters obtained from each of the 50 data sets.
Serum samples from 100 children with similar distributions of age- and sex-matched children with normal liver biochemistries and without known liver disease served as case controls (Discovery Life Sciences, Los Osas, CA) for biomarker comparisons only. TE data was not available for case controls. Regression models adjusted for age, sex and race were performed to compare biomarkers levels in controls vs liver disease groups. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc, Cary, NC, USA).
Written informed consent was obtained from caregivers or the participant, and assent was obtained from the child when appropriate according to local Institutional Review Board (IRB) rules. This study was approved by local IRBs and complied with the Declaration of Helsinki and Good Clinical Practice Guidelines.
Results
Participants
The FORCE study obtained valid baseline LSM exams on 458 participants enrolled from November 2016 to August 2019 (22). The current study included a subset of 330 (72%) FORCE participants (n=187 with BA, n=78 with A1AT, n=65 with ALGS) that had baseline serum samples available for biomarker ELISA analysis. Age was significantly greater in FORCE participants with available serum compared to those without (median 9.2 vs 6.1 years, p<0.001); sex, race, and ethnicity were not significantly different (Supplemental Table 1). Demographic characteristics of the study participants are shown in Supplemental Table 2. Ethnicity was not available for controls. BA participants had a higher percentage of females; ALGS participants were older; A1AT had the lowest percentage of Hispanic/Latino participants and highest percentage of White participants. Mean age at Kasai for 182 BA participants was 57 days (± 24).
Clinical and laboratory characteristics
Overall, median age was nine years, LSM 9.5 kPa, platelet count 216X103/ uL, TB 0.6 mg/dL, albumin 4.3 g/d, AST 57 U/L and GGT 78 U/L. Significant differences in all clinical variables except albumin were observed amongst the three disease groups (Table 1). Median LSM was highest (12.8 kPa) and platelet count was lowest (145 ×103/uL) in BA participants. A higher percentage of BA had splenomegaly (40%) compared to A1AT (4%) or ALGS (17%). A1AT had the lowest median LSM (6.3 kPa). Among ALGS, mean LSM between those with a severe cardiac disease (n=22) vs. mild cardiac disease (n=43) were not different (10.7 kPa vs. 11.5; median 9.2 vs. 9.0, data not shown). TB, GGTP, AST and ALT were highest in ALGS. Only ALGS demonstrated growth impairment, with median z-scores below zero in height, weight, and BMI.
Table 1.
Clinical characteristics of study participants by diagnosis
| Characteristic | BA (n=187) |
A1AT (n=78) |
ALGS (n=65) |
p-value* |
|---|---|---|---|---|
| FibroScan LSM (kPa) | ||||
| n | 187 | 78 | 65 | |
| Median (Q1, Q3) | 12.8 (7.3, 20.0) | 6.3 (5.1, 8.5) | 9.0 (6.9, 12.5) | <0.001 |
| Height Z-score | ||||
| n | 186 | 76 | 65 | |
| Median (Q1, Q3) | 0.3 (−0.6, 0.7) | 0.3 (−0.3, 1.0) | −1.2 (−2.0, −0.6) | <0.001 |
| Weight Z-score | ||||
| n | 186 | 76 | 65 | |
| Median (Q1, Q3) | 0.4 (−0.3, 1.0) | 0.5 (−0.1, 0.9) | −1.1 (−2.1, −0.5) | <0.001 |
| BMI Z-score | ||||
| n | 186 | 76 | 65 | |
| Median (Q1, Q3) | 0.5 (−0.1, 1.1) | 0.3 (−0.3, 0.9) | −0.5 (−1.3, 0.2) | <0.001 |
| Total bilirubin (mg/dl) | ||||
| n | 177 | 78 | 64 | |
| Median (Q1, Q3) | 0.6 (0.4, 1.1) | 0.4 (0.2, 0.6) | 1.1 (0.6, 2.9) | <0.001 |
| GGTP | ||||
| n | 164 | 72 | 56 | |
| Median (Q1, Q3) | 79.5 (29.5, 175.5) | 26.0 (17.0, 50.0) | 352.5 (185.0, 797.0) | <0.001 |
| AST | ||||
| n | 177 | 78 | 64 | |
| Median (Q1, Q3) | 54 (34, 94) | 46 (31, 60) | 110 (74, 178) | <0.001 |
| ALT | ||||
| n | 178 | 78 | 64 | |
| Median (Q1, Q3) | 54 (35, 94) | 53 (35, 77) | 140 (88, 243) | <0.001 |
| Albumin (g/dl) | ||||
| n | 176 | 76 | 63 | |
| Median (Q1, Q3) | 4.3 (3.9, 4.5) | 4.4 (4.2, 4.6) | 4.4 (3.9, 4.5) | 0.08 |
| n (%) <3.0 | 3 (2%) | 1 (1%) | 2 (3%) | 0.70 |
| INR | ||||
| n | 148 | 60 | 56 | |
| Median (Q1, Q3) | 1.1 (1.0, 1.2) | 1.1 (1.0, 1.1) | 1.0 (1.0, 1.1) | <0.001 |
| Spleen size (cm below costal margin) | ||||
| n | 187 | 78 | 65 | |
| Median (Q1, Q3) | 1 (0, 5) | 0 (0, 0) | 0 (0, 0) | <0.001 |
| n (%) >2 cm | 75 (40%) | 3 (4%) | 11 (17%) | <0.001 |
| Platelet count | ||||
| n | 183 | 75 | 62 | |
| Median (Q1, Q3) | 145 (84, 242) | 269 (220, 332) | 251 (198, 325) | <0.001 |
| n (%) <150 | 92 (50%) | 7 (9%) | 9 (15%) | <0.001 |
| APRI | ||||
| n | 176 | 75 | 62 | |
| Median (Q1, Q3) | 0.9 (0.4, 2.4) | 0.4 (0.3, 0.6) | 1.2 (0.6, 2.1) | <0.001 |
| n (%) <1.5 | 106 (60%) | 66 (88%) | 39 (63%) | <0.001 |
| PELD | ||||
| n | 141 | 58 | 55 | |
| Median (Q1, Q3) | −10.3 (−12.7, −5.6) | −13.2 (−16.0, −10.7) | −6.3 (−10.1, 0.8) | <0.001 |
Kruskal-Wallis test for continuous variables. Chi-Square tests for binary variables.
Biomarker profiles
MMP-7 and endoglin levels were significantly higher in BA (both p<0.001) than controls (Table 2). Compared to controls, TIMP-1 concentrations were similar in ALGS but lower in both BA and A1AT (p<0.001). Among the disease groups, MMP-7 and endoglin levels were higher in BA than A1AT and ALGS (Figure 1). Among the nine biomarkers studied, the concentrations of all except TIMP-1, MMP-7 and endoglin were higher in age and sex-matched healthy controls with no known liver disease than in one or more of the three disease groups, BA, A1AT, or ALGS. (Table 2).
Table 2.
Biomarker Comparisons* between disease groups and controls
| Control | BA | A1AT | ALGS | ||
|---|---|---|---|---|---|
|
| |||||
|
LOX
(ng/mL) |
Mean | 5.0 | 3.9 | 3.8 | 3.1 |
| p-value | Ref. | 0.045 | 0.22 | 0.004 | |
|
| |||||
|
MMP-3
(ng/mL) |
Mean | 12.1 | 9.9 | 7.2 | 9.3 |
| p-value | Ref. | 0.003 | <0.001 | 0.4 | |
|
| |||||
|
Endoglin
(pg/mL) |
Mean | 7,958 | 11,117 | 9,295 | 10,118 |
| p-value | Ref. | <0.001 | 0.2 | 0.23 | |
|
| |||||
|
TIMP-1
(ng/mL) |
Mean | 171 | 134 | 135 | 176 |
| p-value | Ref. | <0.001 | <0.001 | 0.29 | |
|
| |||||
|
Mac-2BPGi
(ng/mL) |
Mean | 5.5 | 3.9 | 4.0 | 3.5 |
| p-value | Ref. | 0.002 | 0.003 | <0.001 | |
|
| |||||
|
Periostin
(pg/mL) |
Mean | 6,968 | 3,002 | 2,785 | 3,895 |
| p-value | Ref. | <0.001 | <0.001 | <0.001 | |
|
| |||||
|
IL-8
(pg/ML) |
Mean | 99 | 44 | 39 | 45 |
| p-value | Ref. | <0.001 | <0.001 | 0.006 | |
|
| |||||
|
CTGF
(Pg/mL) |
Mean | 695 | 518 | 601 | 434 |
| p-value | Ref. | 0.01 | 0.6 | 0.001 | |
|
| |||||
|
MMP-7
(ng/mL) |
Mean | 3.4 | 6.8 | 3.5 | 3.4 |
| p-value | Ref. | <0.001 | 0.26 | 0.41 | |
p-values based on Kruskal-Wallis tests
Figure 1.
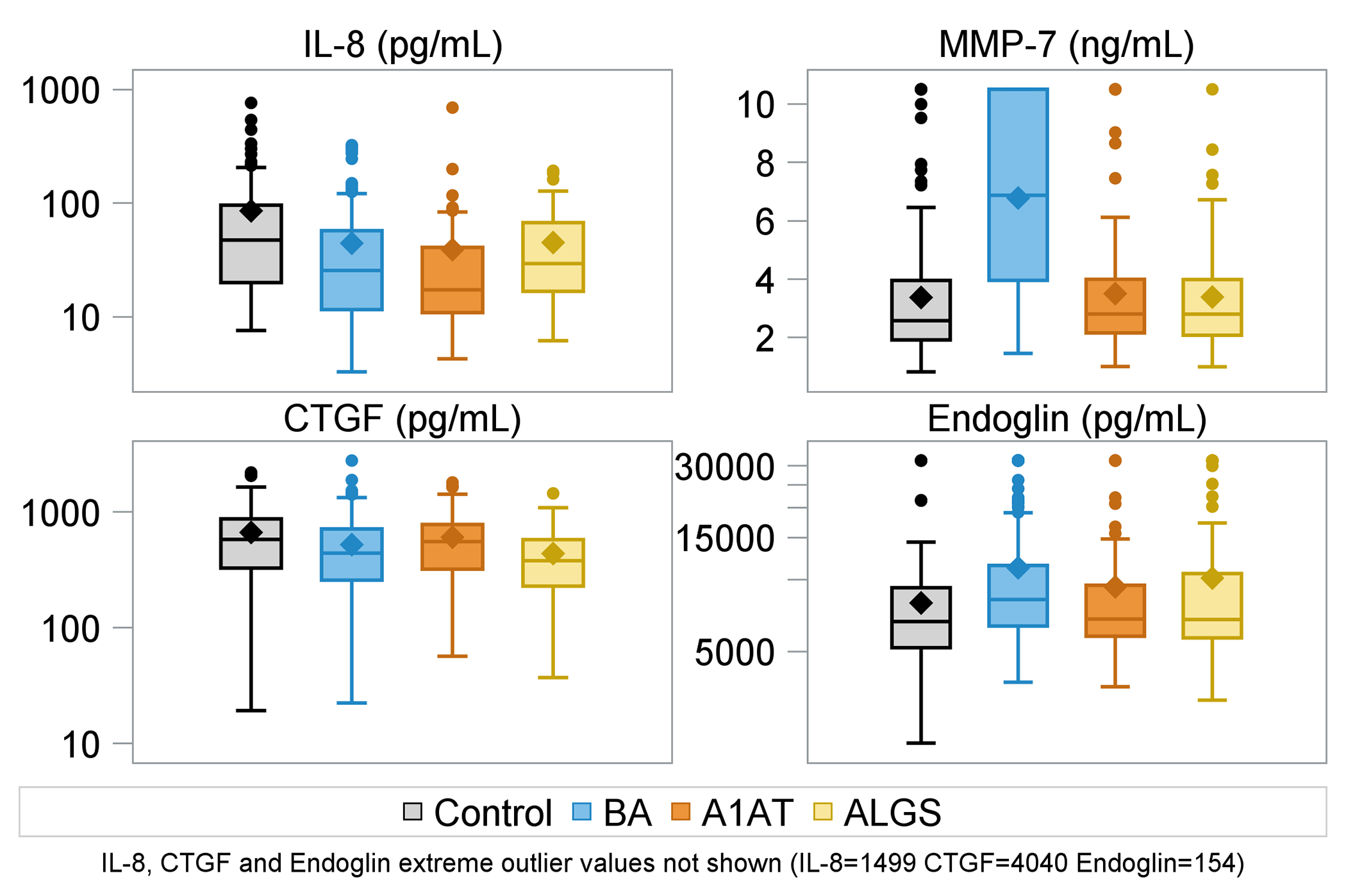
Boxplots of selected biomarker concentrations (IL-8, MMP-7, CTGF, Endoglin) by diagnosis (control, BA, A1AT, ALGS).
Biomarker correlations with participant age varied by liver disease group (Supplemental Document 2). In BA, MMP-7 had the strongest negative correlation with age (r=−0.38, p<0.001); in A1AT, TIMP-1 (r=−0.34, p=0.003) and CTGF (r=−0.37, p=0.001) most strongly correlated with age.
Biomarker and LSM correlations in BA
In BA, LSM correlated with MMP-3 (r =0.19, p=0.02), endoglin (r =0.27, p<0.001), periostin (r=0.22, p=0.01), IL-8 (r=0.47, p<0.001) and MMP-7 (r=0.45, p<0.001) (Figure 2). After adjusting for covariates (age, TB, albumin, GGTP, and AST), the positive association between LSM and IL-8 (p<0.001), MMP-7 (p<0.001), and endoglin (p=0.02) remained significant. Periostin (p=0.051) and MMP-3 (p=0.21) were not significant after adjusting for other covariates. The best parsimonious prediction model for LSM in BA based on clinical characteristics and laboratory measurements (spleen size, PELD, platelets, AST) had an R2=0.437. Adding IL8 and MMP7 further improved the R2 to 0.523 (<0.0001) and 0.526 (p<0.0001), respectively (Table 3). IL-8 and MMP-7 concentrations were higher in BA participants with higher LSM (Figure 3). Endoglin concentrations were also higher in in BA with higher LSM. CTGF had no clear relationship with LSM in BA.
Figure 2.
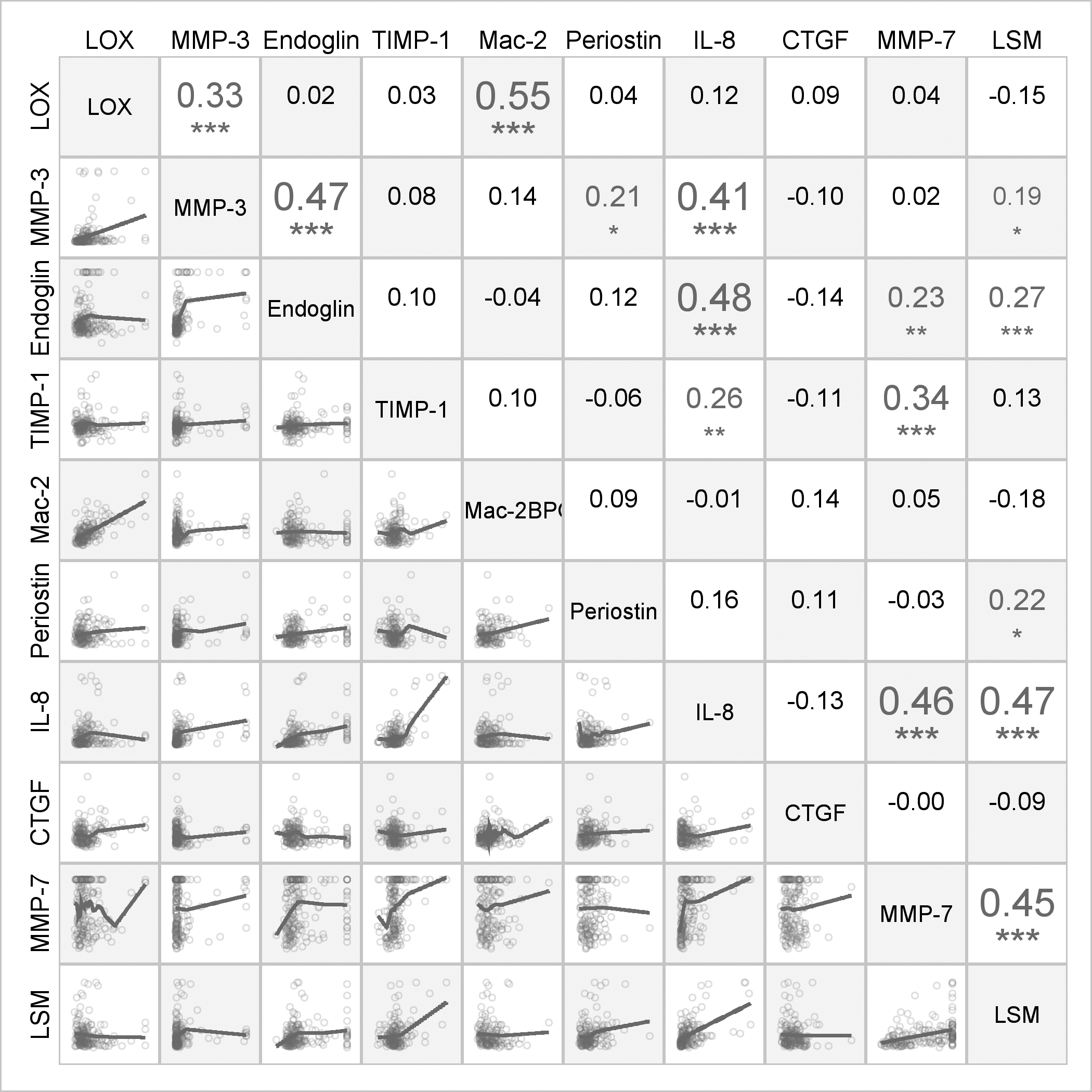
Spearman correlations and scatter plots among biomarker concentrations and liver stiffness measurements for participants with BA. The numbers in the upper triangle of the grid display the Spearman correlation coefficients for pairs of variables. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05; **p<0.01;***p<0.001
The lower triangle of the grid displays scatter plots with LOESS lines.
Table 3.
Linear regression models predicting LSM in BA (n=187) with biomarkers added individually to conventional laboratory measurements and clinical features
|
Best parsimonious model using conventional labs and clinical factors R2= 0.473 (95% CI: 0.361, 0.574); Adj-R2=0.461(0.349, 0.564) | |||||
| Term | Estimate | p-value | |||
| Spleen Size | 0.0108 | 0.06 | |||
| PELD | 0.0165 | <0.001 | |||
| Platelets | −0.00079 | <0.001 | |||
| AST | 0.0014 | <0.001 | |||
| Separate linear regression models predicting LSM with best parsimonious model based on conventional labs + target biomarker one at a time | |||||
| Term | R2 (95% CI) | Adj-R2 (95% CI) | p-value | ||
| Log10(LOX) | 0.478 (0.367, 0.579) | 0.463 (0.351, 0.566) | 0.19 | ||
| Log10(MMP3) | 0.474 (0.363, 0.575) | 0.459 (0.347, 0.562) | 0.55 | ||
| Log10(Endoglin) | 0.474 (0.363, 0.576) | 0.460 (0.348, 0.563) | 0.48 | ||
| TIMP1 | 0.505 (0.397, 0.602) | 0.491 (0.383, 0.590) | <0.001 | ||
| Log10(Mac2) | 0.477 (0.366, 0.578) | 0.462 (0.351, 0.565) | 0.25 | ||
| Log10(Periostin) | 0.480 (0.369, 0.581) | 0.465 (0.353, 0.568) | 0.15 | ||
| Log10(IL8) | 0.523 (0.416, 0.617) | 0.509 (0.402, 0.606) | <0.001 | ||
| Log10(CTGF) | 0.475 (0.364, 0.577) | 0.461 (0.349, 0.564) | 0.35 | ||
| MMP7 | 0.526 (0.419, 0.621) | 0.513 (0.404, 0.610) | <0.001 | ||
Figure 3.
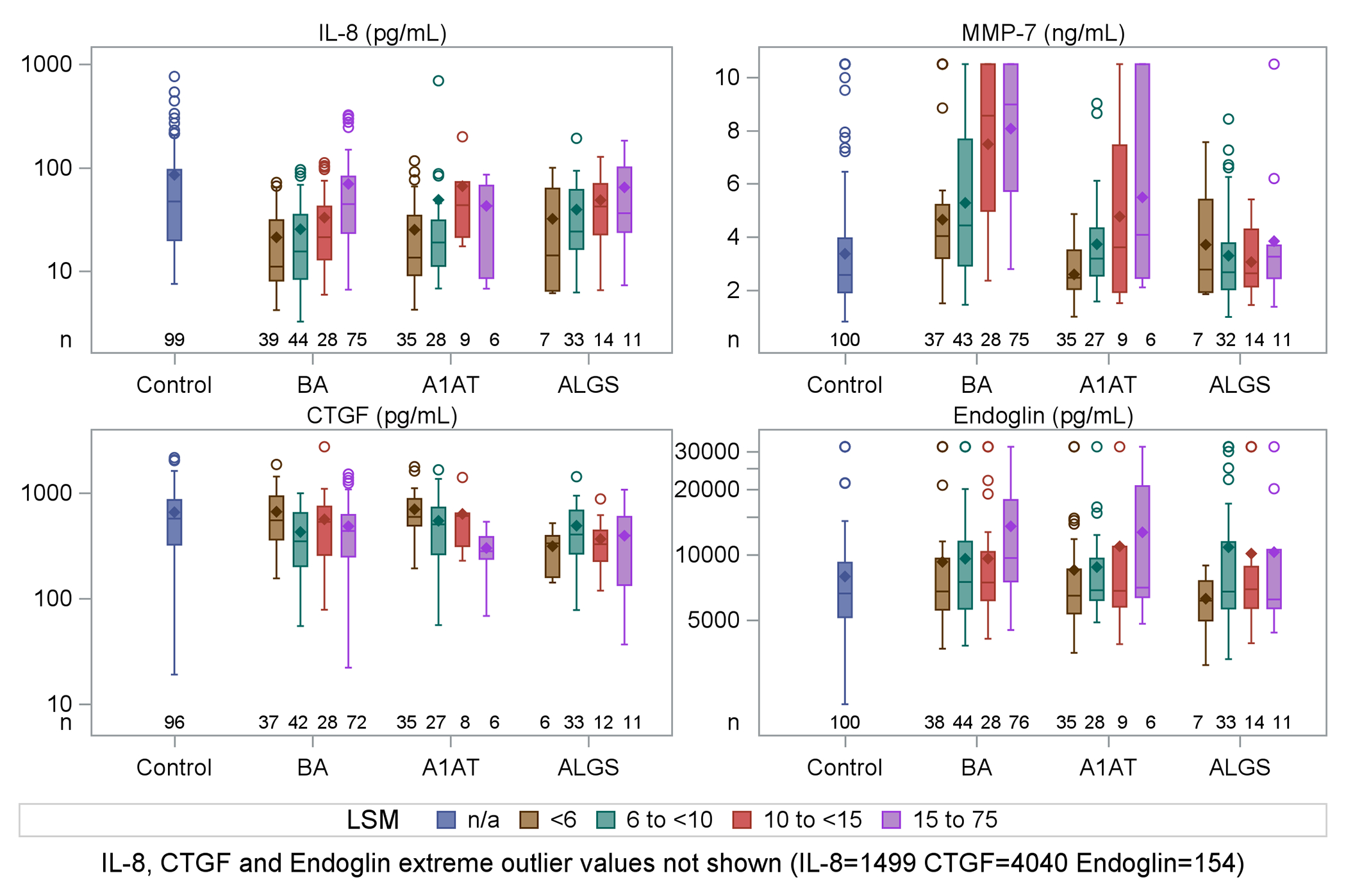
Boxplots for selected biomarkers (IL-8, MMP-7, CTGF, Endoglin) by diagnosis (control, BA, A1AT, ALGS) and liver stiffness measurement category.
BA had the largest sample size and highest number of significant biomarker/biomarker correlations (10 pairs) and significant biomarker correlations with LSM (Figure 2). Specifically, in BA, lysyl oxidase (LOX) and Mac-2BP were correlated with each other (r=0.55, p<0.001), and IL-8 was strongly correlated with MMP-7 (r=0.46, p<0.001), endoglin (r=0.48, p<0.001) and MMP-3 (r=0.41, p<0.001).
Biomarker and LSM correlations in A1AT
Only CTGF was correlated with LSM in A1AT (R = −0.38, p=0.01) (Supplemental Figure 1); CTGF was lower in A1AT participants with higher LSM (Figure 3). The negative association between LSM and CTGF remained significant (p=0.004) after adjusting for covariates. The best parsimonious prediction model for LSM based on clinical and lab measurements (APRI, total bilirubin, spleen size) had an R2=0.524. Adding CTGF (log transformed) improved R2 to 0.577 (p=0.0033) (Supplemental Table 3).
While correlations were not significant, MMP-7 and endoglin concentrations trended higher in A1AT participants with higher categories of LSM (Figure 3). Among A1AT, LOX was correlated with Mac-2BP (r=0.51, p=0.001, Supplemental Figure 1).
Biomarker and LSM correlations in ALGS
No biomarkers were significantly correlated with LSM in ALGS (Figure 3 and Supplemental Figure 2). However, among ALGS, LOX and Mac-2BP were correlated (r=0.48, p<0.05) with one another; similarly, IL-8 and endoglin were strongly correlated with each other (r=0.45, p<0.01), as were MMP-7 and TIMP1 (r=0.52, p<0.001) (Supplemental Figure 2).
Biomarker correlations with clinical features and laboratory measurements in BA
BA had the most significant biomarker/laboratory (40 pairs) correlations. Among BA, endoglin, TIMP-1, periostin, IL-8, and MMP-7 were associated with almost all clinical and laboratory markers of liver disease progression (Figure 4). Endoglin, TIMP1, IL-8 and MMP-7 were all significantly and negatively correlated with age (r= −0.22 to −0.38, p<0.01 for all) and albumin (r= −0.18 to −0.29, p<0.05 or less for all). GGTP correlated with TIMP-1, IL-8, and MMP-7 (r=0.38–0.41, p<0.001), as did AST (r= 0.28–0.53, p<0.001 for all). In BA, platelets had the strongest correlation with TIMP-1, while AST to platelet ratio index (APRI) had the strongest correlation with IL-8. Among BA, endoglin (p=0.003) and IL-8 (p=0.02) were highest in participants with definite clinically evident portal hypertension (CEPH) vs absent or possible CEPH (Supplemental Table 4). TIMP-1 was lowest in BA participants with definite CEPH (p<0.001) vs absent or possible CEPH; LOX and Mac-2BPGi were also lowest in BA with definite CEPH (p=0.04, p=0.008, respectively). MMP-7 was highest (7.8 ng/mL) in BA with definite CEPH vs possible (6.0 ng/mL) and absent (5.2 ng/mL) CEPH but not significant (p=0.19)
Figure 4.
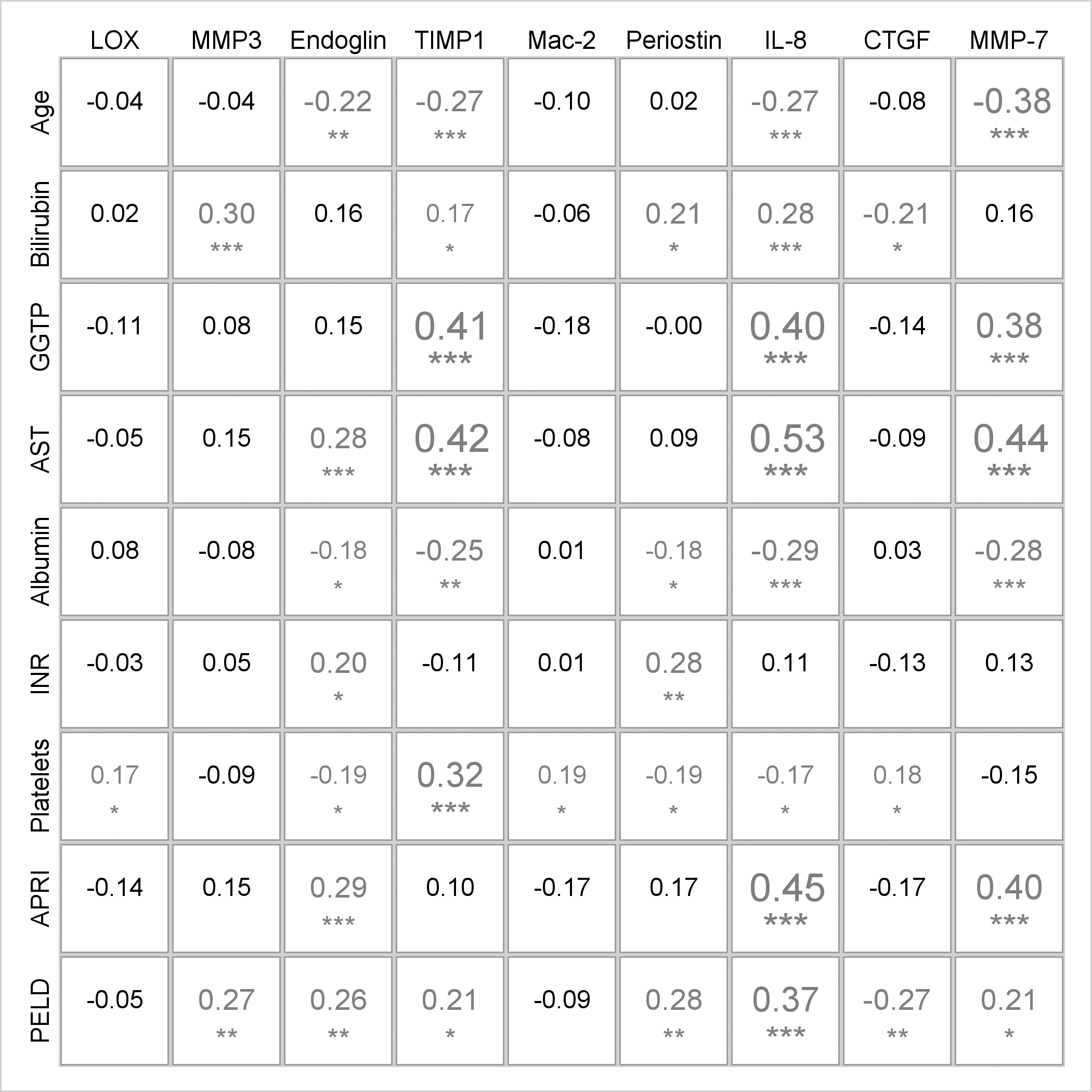
Spearman correlations between biomarker concentrations and laboratory measurements for BA participants. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05;**p<0.01; ***p<0.001
Biomarker correlations with clinical features and laboratory measurements in A1AT
A1AT had the least statistically significant biomarker/laboratory (7 pairs) correlations (Supplemental Figure 3). Periostin was not associated with any laboratory associated with liver injury. Among A1AT, TIMP-1 and CTGF were negatively associated with age. Platelet count was correlated with TIMP-1 and CTGF. AST correlated the highest with IL-8 (r=0.36) and MMP-7 (r=0.32, both p<0.05). APRI had the strongest correlation with IL-8 (r=0.42, p<0.05). Among A1AT, CTGF was the lowest in participants with definite CEPH (288.6 pg/mL) vs absent CEPH (572.5 ng/mL, p=0.05), consistent with its correlation with LSM (Supplemental Table 4). Only 5 patients with A1AT had definite CEPH. Similar to BA, TIMP-1 was lowest among A1AT with definite CEPH (108.7 ng/mL, p=0.04) vs possible (114 ng/mL) and absent (136.6 ng/mL).
Biomarker correlations with clinical features and laboratory measurements in ALGS
ALGS also had few statistically significant biomarker/laboratory (9 pairs) correlations (Supplemental Figure 4). Among ALGS, TIMP-1 was correlated with bilirubin, AST, APRI, and PELD (r=0.41–0.54, all p<0.01). IL-8 also correlated with these same factors (r=0.44–0.50, all p<0.01).
Discussion
Discovery and validation of disease-specific serum and imaging biomarkers of liver fibrosis may inform the unique etiopathogenesis of BA, A1AT, and ALGS and identify distinct targets as well as clinical endpoints for future anti-fibrotic therapies. The mechanisms of liver disease are numerous and include genetic etiologies impacting development or protein trafficking, inflammation, biliary obstruction, vascular abnormalities and congestion, cell death, and fibrogenesis; all could impact liver stiffness. Overall, the study cohort was quite young (median <10 years); yet median LSM was 9.5 kPa, consistent with significant fibrosis, despite preserved liver function with only mildly elevated biochemistries. TE capability is not ubiquitous among most pediatric hospitals, however commercially available serum biomarkers validated by TE may offer additional non-invasive alternatives to assess or predict worsening liver stiffness or disease progression.
Biomarker correlation with LSM and clinical parameters of liver disease varied widely among BA, A1AT, and ALGS highlighting potential disease-specificity of serum and imaging biomarkers of liver injury. BA is biologically distinct and characterized by extrahepatic biliary obstruction with progression to cirrhosis much earlier and more frequently than A1AT and ALGS. As such, it was not surprising that BA participants post Kasai had the highest median LSM (12.8 kPa), lowest platelet count, and highest incidence of splenomegaly despite not having the highest TB, GGT, AST, or ALT (ALGS had the highest levels). In a prior study of 30 infants at the time of Kasai, a LSM of 15.1 kPa or higher predicted cirrhosis (Metavir ≥ F4 by liver biopsy), however this cohort was much younger (mean 76 days) than our study cohort (27). Interestingly, mean MMP-7 levels were also two-fold higher in BA compared to controls, A1AT, or ALGS. This is consistent with other studies showing that serum MMP-7 is significantly higher in BA while also offering high diagnostic accuracy in distinguishing BA from other cholestatic liver disorders (31–33) and correlation with liver fibrosis (34). Notably, in our study, BA had highest MMP-7 concentrations in the first year of life and decreased to control concentration levels by early adulthood. Among BA, MMP-7 was also highly correlated with GGT, AST, and LSM. The addition of MMP-7 improved clinical and lab-based model prediction of LSM in BA by explaining an additional 9% of the variation in LSM. Based on these important biomarker and liver stiffness associations, MMP-7 appears to be a reliable biomarker of worsening fibrosis in BA. Matrix metalloproteinases (MMP) have been found to be involved in the activation of hepatic stellate cells and increased extracellular matrix, both of which are associated with the fibrogenic mechanisms of BA (35, 36). Notably, in a large public domain single cell human RNA data set (37), MMP-7 was highly expressed in cholangiocytes and enriched in individuals with cirrhosis. MMP-7 also appears to be the most consistently expressed marker in animal models of BA (38–40).
In BA, endoglin and IL-8 were positively correlated with LSM; in addition to MMP-7, these biomarkers correlated with at least seven of the 10 clinical parameters of interest. These three biomarkers are involved in hepatic stellate cell differentiation (36), biliary epithelial tissue remodeling (41), and chemoattraction in cholangiocytes (42, 43), respectively, and represent pathways leading to increased liver stiffness and potential targets for anti-fibrotic therapies. Interestingly, endoglin, a glycoprotein co-receptor for TGF-β involved in cytoskeletal organization was the only biomarker that was universally higher in all liver disease groups vs controls, though only significantly higher in BA, likely due to its larger sample size. Our reported mean endoglin level of 11.1 ng/mL in BA post-Kasai is higher than previously reported in a younger post-Kasai cohort (7.8 ng/mL) (44).
The negative correlation between LSM and CTGF among A1AT is intriguing and paradoxical in the context of anticipated liver fibrosis in A1AT. Interestingly, CTGF also had a significant negative correlation with age and portal hypertension severity. CTGF is tightly regulated by HNF4alpha and YAP. HNF4alpha is a transcription factor known for its roles in maintaining balance between hepatocyte differentiation versus quiescence and organization of the sinusoidal endothelium during development (45, 46). YAP, a downstream effector of the Hippo pathway, is known to promote liver regeneration (47, 48) in response to injury or parenchymal loss. It is possible that serum CTGF levels may reflect the real-time state of the hepatocyte, in this case, quiescence, in A1AT. Low CTGF may reflect a defective compensatory hepatocyte proliferation in response to hepatocyte injury in A1AT despite increased fibrosis or liver stiffness. Of note, CTGF is also positively correlated with platelet count in A1AT and seems to be a consistent biomarker of worsening liver disease or emerging portal hypertension in A1AT. The addition of CTGF also improved clinical and lab-based prediction model of LSM in A1AT.
With the exception of endoglin (highest in BA), TIMP-1 (highest in ALGS), and MMP-7 (highest in BA), biomarker concentrations of our targeted panel were surprisingly higher in age-matched healthy controls compared to those with liver disease. Given what is known about the putative role of these biomarkers in liver fibrosis, this was an unexpected but important finding not previously described. This suggests that levels of some collagen/matrix markers may be confounded by factors inherent to liver disease such as cholestasis or suboptimal nutrition. For example, periostin levels were nearly two-fold higher in controls than any of the liver disease groups. Children with cholestatic liver disease commonly have growth stunting, manifest vitamin D deficiency due to poor absorption of fat soluble vitamins, and have osteopenia or fractures (49, 50). Hence, it is critical to acknowledge that some collagen based biomarkers may be more challenging to interpret in the context of specific pediatric liver disorders and age groups. Alternatively, it is possible that some of these collagen/matrix markers may have less biological relevance in pediatric liver disease than previously thought.
This study has some limitations. Although the panel was selected based on a rigorous review of peer-reviewed literature supporting their putative value as liver fibrosis biomarkers, only nine targeted biomarkers were studied. Changes in biomarker serum levels may reflect altered clearance rather than decreased or increased production; they may also not directly reflect changes within the liver. LSMs were not validated by liver biopsy in this study. Ideally, immunostaining of liver tissues to assess the intensity and distribution of biomarker expression can inform hypotheses about reasons for changes in biomarker levels in serum; however, liver biopsy was not considered feasible in the FORCE study. Our cross-sectional study design also does not indicate causation or allow us to measure the biomarkers at multiple time points to correlate with fibrosis progression as measured by Fibroscan and lab parameters. Mean serum concentration of MMP-7 levels among our much older BA cohort in this study (median age of 8.8 years) are lower than other studies reporting median MMP-7 levels at the time of BA diagnosis (median age 54–59 days) (33, 34). Post-Kasai status, age, duration of liver disease, and brand of ELISA kit are likely important factors. Lastly, there were fewer ALGS and A1AT participants than BA, resulting in a limited number of ALGS and A1AT participants with high LSM. This likely increased the risk of a Type II error for detecting significant associations between biomarkers and disease severity.
The correlations between biomarkers may shed light on mechanistic pathways that contribute to increased liver stiffness in specific pediatric liver disorders. Correlation of IL-8 and endoglin was demonstrated in both BA and ALGS participants. In BA, IL-8 also independently correlated with LSM and improved model prediction of LSM among BA. IL-8 is a chemokine involved in angiogenesis and liver inflammation while endoglin is overexpressed after liver or arterial injury. It is plausible that vascular abnormalities, common in BA (e.g., aberrant portal vein or obliterative venopathy) (51) and ALGS (e.g., cerebrovascular and renovascular malformations) (52, 53), may play a role in liver fibrosis or liver stiffness. While the correlation of IL-8 with LSM among ALGS was not significant (p=0.09) in a multivariable model, a larger disease population may have better elucidated this correlation.
Conclusion
Biomarker correlation with LSM and other clinical parameters of liver disease varied by type of pediatric cholestatic liver disease. This study provides evidence that endoglin, IL-8, and MMP-7 significantly correlate with increased LSM in children with BA, while CTGF inversely correlates with LSM in A1AT; these biomarkers also appear to enhance clinical characteristic and laboratory- based prediction of LSM and represent distinct but related mechanistic pathways of liver fibrosis and potential targets for antifibrotic therapies. As LSM via transient elastography is not yet clinically available or a standard of care in most pediatric hospitals, these commercially available biomarkers may offer additional non-invasive tools to guide decision making. However, disease type and age appear to be important factors in biomarker and LSM interpretation. Future investigations of imaging and serum biomarkers in pediatric cholestasis should use disease specific thresholds and would benefit from evaluation of change in biomarker levels over time as predictors of clinical outcomes.
Supplementary Material
Supplemental Figure 1. Spearman correlations and scatter plots among biomarker concentrations and liver stiffness measurements for participants with A1AT. The numbers in the upper triangle of the grid display the Spearman correlation coefficients for pairs of variables. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05; **p<0.01; ***p<0.001
The lower triangle of the grid displays scatter plots with LOESS lines.
Supplemental Figure 2. Spearman correlations and scatter plots among biomarker concentrations and liver stiffness measurements for participants with ALGS. The numbers in the upper triangle of the grid display the Spearman correlation coefficients for pairs of variables. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05; **p<0.01; ***p<0.001
The lower triangle of the grid displays scatter plots with LOESS lines.
Supplemental Figure 3. Spearman correlations between biomarker concentrations and laboratory measurements for A1AT participants. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05; **p<0.01; ***p<0.001
Supplemental Figure 4. Spearman correlations between biomarker concentrations and laboratory measurements for ALGS participants. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05; **p<0.01; ***p<0.001
Acknowledgements
Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL (supported by NIDDK DK62436 and NCATS UL1TR001422): Estella Alonso, MD; Lee Bass, MD; Susan Kelly, RN, BSN; Mary Riordan, CCRP; Hector Melin-Aldana, MD
Cincinnati Children’s Hospital Medical Center, Cincinnati, OH (supported by NIDDK DK62497 and NCATS UL1TR000077): Jorge Bezerra, MD; Kevin Bove, MD; James Heubi, MD; Alexander Miethke, MD; Greg Tiao, MD; Julie Denlinger, BSN, RN; Erin Chapman
Children’s Hospital Colorado, Aurora, CO (supported by NIDDK DK62453 and NCATS UL1TR001082): Ronald Sokol, MD; Amy Feldman, MD; Cara Mack, MD; Michael Narkewicz, MD; Frederick Suchy, MD; Shikha Sundaram, MD; Johan Van Hove, MD; Benigno Garcia; Mikaela Kauma; Kendra Kocher, CCRP; Matthew Steinbeiss, MS, CCRP; Mark Lovell, MD
The Children’s Hospital of Philadelphia, Philadelphia, PA (supported by NIDDK DK62481 ): Kathleen Loomes, MD; David Piccoli, MD; Elizabeth Rand, MD; Pierre Russo, MD; Nancy Spinner, PhD; Jessi Erlichman, MPH; Samantha Stalford, MPH; Dina Pakstis; Sakya King
Children’s Hospital of Pittsburgh, Pittsburgh, PA (supported by NIDDK DK62466 and NCATS UL1TR000005): Robert Squires, MD; Rakesh Sindhi, MD; Veena Venkat, MD; Kathy Bukauskas, RN, CCRC; Patrick McKiernan, MD; Lori Haberstroh, RN, BSN; James Squires, MD, MS
UCSF Children’s Hospital, San Francisco, CA (supported by NIDDK DK62500 and NCATS UL1TR000004): Philip Rosenthal, MD; Laura Bull, PhD; Joanna Curry; Camille Langlois, MS; Grace Kim, MD
Saint Louis University School of Medicine, St. Louis, MO (supported by NIDDK DK62453): Jeffery Teckman, MD; Vikki Kociela, BSN, CCRC; Rosemary Nagy, RDN, MBA; Shraddha Patel, PhD; Jacqueline Cerkoski, BSN
Riley Hospital for Children, Indiana University School of Medicine, Indianapolis, IN (supported by NIDDK DK84536 and NCATS UL1TR001108): Jean P. Molleston, MD; Molly Bozic, MD; Girish Subbarao, MD; Ann Klipsch, RN; Cindy Sawyers, BSRT; Oscar Cummings, MD
Seattle Children’s Hospital, Seattle WA (supported by NIDDK DK84575 and NCATS UL1TR000423): Simon Horslen, MB, ChB, FRCPCH; Karen Murray, MD; Evelyn Hsu, MD; Kara Cooper, CCRC; Melissa Young, CCRC; Laura Finn, MD
The Hospital for Sick Children, Toronto, Ontario, CANADA (supported by NIDDK DK103135): Binita Kamath, MD; Vicky Ng, MD; Claudia Quammie, CCRP; Juan Putra, MD; Deepika Sharma, MSc; Aishwarya Parmar, BSc
University of Utah, Salt Lake City, UT (supported by NIDDK DK103140): Stephen Guthery, MD; Kyle Jensen, MD; Ann Rutherford; Amy Lowichik, MD, PhD; Linda Book, MD; Rebecka Meyers, MD; Tyler Hall
Children’s Hospital Los Angeles, Los Angeles, CA (supported by NIDDK DK84538 and NCATS UL1TR000130): Kasper Wang, MD; Sonia Michail, MD; Danny Thomas, MD; Catherine Goodhue, CPNP; Rohit Kohli, MBBS, MS; Larry Wang, MD, PhD; Nisreen Soufi, MD; Daniel Thomas, MD
Children’s Healthcare of Atlanta, Atlanta, GA (supported by NIDDK DK062470 and NCATS UL1TR000454): Saul Karpen, MD, PhD; Nitika Gupta, MD, DCH, DNB, MRCPH; Rene Romero, Jr., MD; Miriam B. Vos, MD, MSPH; Rita Tory, MS, CCRP; John-Paul Berauer, MD; Carlos Abramowsky, MD; Jeanette McFall, MPH
Texas Children’s Hospital, Houston, TX (supported by NIDDK DK103149): Benjamin Shneider, MD; Sanjiv Harpavat, MD; Paula Hertel, MD; Daniel Leung, MD; Mary Tessier, MD; Deborah Schady, MD; Laurel Cavallo; Diego Olvera; Christina Banks; Cynthia Tsai
King’s College Hospital, London, UK: Richard Thompson, BM, BCh, MRCP, MRCPCH
National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD: Edward Doo, MD; Jay Hoofnagle, MD; Averell Sherker, MD, FRCP; Rebecca Torrance, RN, MSN;
Sherry Hall, MS
Scientific Data Coordinating Center, Ann Arbor, MI (supported by NIDDK DK62456): John Magee, MD; Robert Merion, MD, FACS; Cathie Spino, DSc; Wen Ye, PhD
Financial Support
This work was supported by U01 grants from the National Institute of Diabetes, Digestive and Kidney Diseases (DK 62445 [Mt. Sinai School of Medicine], DK 62497 [Cincinnati Children’s Hospital Medical Center], DK 62470 [Children’s Healthcare of Atlanta], DK 62481 [The Children’s Hospital of Philadelphia], DK 62456 [The University of Michigan], DK 84536 [Riley Hospital for Children], DK 84575 [Seattle Children’s Hospital], DK 62500 [UCSF Children’s Hospital], DK 62466 [Children’s Hospital of Pittsburgh of UPMC], DK 62453 [Children’s Hospital Colorado], DK 84538 [Children’s Hospital Los Angeles], DK 62436 [Ann & Robert H Lurie Children’s Hospital of Chicago], DK103149 [Texas Children’s Hospital], DK103135 [The Hospital for Sick Children], DK103140 [University of Utah]).
In addition, the project described was supported by National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award (NCATS CTSA) grants: University of Colorado UL1 TR002535, UCSF Children’s Hospital UL1 TR001872, Children’s Hospital of Pittsburgh of UPMC UL1 TR001857, The Children’s Hospital of Philadelphia UL1 TR001878, Seattle Children’s Hospital UL1 TR000423 and UL1 RR025014, Children’s Healthcare of Atlanta UL1TR002378, Children’s Hospital of Los Angeles UL1TR00130.
Abbreviations
- A1AT
alpha-1 antitrypsin deficiency
- ALGS
Alagille syndrome
- APRI
AST to platelet ratio index
- AST
aspartate aminotransferase
- BA
biliary atresia
- BASIC
Biliary Atresia Study in Infants and Children
- CEPH
clinically evident portal hypertension
- ChiLDReN
Childhood Liver Disease Research Network
- CI
confidence intervals
- CTGF
connective tissue growth factor
- FORCE
FibroScan− in Pediatric Cholestatic Liver Disease
- GPR
gamma glutamyl transpeptidase to platelet ratio
- GGT
gamma glutamyl transpeptidase
- INR
International Normalized Ratio
- IQR
Interquartile Range
- IRB
Institutional Review Board
- LOESS
locally estimated scatterplot smoothing
- LOGIC
Longitudinal Observational Study of Genetic Causes of Intrahepatic Cholestasis
- LOX
Lysyl Oxidase
- LSM
liver stiffness measurement
- LT
liver transplant
- Mac2-BP
mac-2-binding protein
- MMP
Matrix metalloproteinases
- NIH
National Institutes of Health
- PELD
Pediatric End-Stage Liver Disease
- PROBE
Prospective Database of Infants with Cholestasis
- TB
total bilirubin
- TE
transient elastography
- TIMP1
tissue inhibitor matrix metalloproteinase 1
Footnotes
Disclosures: DHL receives grant/research support from Abbvie, Gilead, and Mirum and has served as medical advisor for Merck, Gilead and Vertex. JPM receives grant/research support from Abbvie, Albireo, Mirum, Gilead. KML is a consultant for Mirum, Albireo and Travere Therapeutics and receives grant/research support from Mirum and Albireo. BMK is a Consultant for Mirum, Albireo, Third Rock Ventures and Audentes, BMK receives unrestricted educational funding from Mirum and Albireo. PR receives grant/research support from Abbvie, Albireo, Arrowhead, Gilead, Merck, Mirum, Takeda/Vertex, Travere and has served as a consultant for Albireo, Ambys, Audentes, BioMarin, Dicerna, Encoded, Gilead, MedinCell, Mirum, Takeda/Vertex, Travere.
References
- 1.Feldstein AE, Nobili V. Biomarkers in nonalcoholic fatty liver disease: a new era in diagnosis and staging of disease in children. J Pediatr Gastroenterol Nutr;51:378–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song Z, Dong R, Fan Y, Zheng S. Identification of Serum Protein Biomarkers in Biliary Atresia by Mass Spectrometry and ELISA. J Pediatr Gastroenterol Nutr. [DOI] [PubMed] [Google Scholar]
- 3.Nobili V, Alisi A, Torre G, De Vito R, Pietrobattista A, Morino G, De Ville De Goyet J, et al. Hyaluronic acid predicts hepatic fibrosis in children with nonalcoholic fatty liver disease. Transl Res;156:229–234. [DOI] [PubMed] [Google Scholar]
- 4.Pereira TN, Lewindon PJ, Smith JL, Murphy TL, Lincoln DJ, Shepherd RW, Ramm GA. Serum markers of hepatic fibrogenesis in cystic fibrosis liver disease. J Hepatol 2004;41:576–583. [DOI] [PubMed] [Google Scholar]
- 5.Gunadi, Sirait DN, Budiarti LR, Paramita VMW, Fauzi AR, Ryantono F, Afandy D, et al. Histopathological findings for prediction of liver cirrhosis and survival in biliary atresia patients after Kasai procedure. Diagn Pathol 2020;15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lakshminarayanan B, Davenport M. Biliary atresia: A comprehensive review. J Autoimmun 2016;73:1–9. [DOI] [PubMed] [Google Scholar]
- 7.Starzl TE, Giles G, Lilly JR, Takagi H, Martineau G, Schroter G, Halgrimson CG, et al. Indications for orthotopic liver transplantation: with particular reference to hepatomas, biliary atresia, cirrhosis, Wilson’s disease and serum hepatitis. Transplant Proc 1971;3:308–312. [PMC free article] [PubMed] [Google Scholar]
- 8.Myers B Case of Persistent Jaundice in an Infant; Atresia of the Common Bile-duct and Biliary Cirrhosis. Proc R Soc Med 1923;16:17–18. [PMC free article] [PubMed] [Google Scholar]
- 9.Okuyama K Primary Liver Cell Carcinoma Associated with Biliary Cirrhosis Due to Congenital Bile Duct Atresia. J Pediatr 1965;67:89–93. [DOI] [PubMed] [Google Scholar]
- 10.Perlmutter DH. Alpha-1-antitrypsin deficiency. Semin Liver Dis 1998;18:217–225. [DOI] [PubMed] [Google Scholar]
- 11.Patel D, McAllister SL, Teckman JH. Alpha-1 antitrypsin deficiency liver disease. Transl Gastroenterol Hepatol 2021;6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spinner NB, Leonard LD, Krantz ID: Alagille syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. Gene Reviews. Seattle WA., 1993. [Google Scholar]
- 13.Kamath BM, Ye W, Goodrich NP, Loomes KM, Romero R, Heubi JE, Leung DH, et al. Outcomes of Childhood Cholestasis in Alagille Syndrome: Results of a Multicenter Observational Study. Hepatol Commun 2020;4:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nobili V, Marcellini M, Giovannelli L, Girolami E, Muratori F, Giannone G, Devito R, et al. Association of serum interleukin-8 levels with the degree of fibrosis in infants with chronic liver disease. J Pediatr Gastroenterol Nutr 2004;39:540–544. [DOI] [PubMed] [Google Scholar]
- 15.Alkhouri N, Sedki E, Alisi A, Lopez R, Pinzani M, Feldstein AE, Nobili V. Combined paediatric NAFLD fibrosis index and transient elastography to predict clinically significant fibrosis in children with fatty liver disease. Liver Int 2013;33:79–85. [DOI] [PubMed] [Google Scholar]
- 16.Aqul A, Jonas MM, Harney S, Raza R, Sawicki GS, Mitchell PD, Fawaz R. Correlation of Transient Elastography With Severity of Cystic Fibrosis-related Liver Disease. J Pediatr Gastroenterol Nutr 2017;64:505–511. [DOI] [PubMed] [Google Scholar]
- 17.Hwang JY, Yoon HM, Kim JR, Lee JS, Jung AY, Kim KM, Cho YA. Diagnostic Performance of Transient Elastography for Liver Fibrosis in Children: A Systematic Review and Meta-Analysis. AJR Am J Roentgenol 2018;211:W257–W266. [DOI] [PubMed] [Google Scholar]
- 18.Lee CK, Mitchell PD, Raza R, Harney S, Wiggins SM, Jonas MM. Validation of Transient Elastography Cut Points to Assess Advanced Liver Fibrosis in Children and Young Adults: The Boston Children’s Hospital Experience. J Pediatr 2018;198:84–89 e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewindon PJ, Puertolas-Lopez MV, Ramm LE, Noble C, Pereira TN, Wixey JA, Hartel GF, et al. Accuracy of Transient Elastography Data Combined With APRI in Detection and Staging of Liver Disease in Pediatric Patients With Cystic Fibrosis. Clin Gastroenterol Hepatol 2019;17:2561–2569 e2565. [DOI] [PubMed] [Google Scholar]
- 20.Shin NY, Kim MJ, Lee MJ, Han SJ, Koh H, Namgung R, Park YN. Transient elastography and sonography for prediction of liver fibrosis in infants with biliary atresia. J Ultrasound Med 2014;33:853–864. [DOI] [PubMed] [Google Scholar]
- 21.Teufel-Schafer U, Flechtenmacher C, Fichtner A, Hoffmann GF, Schenk JP, Engelmann G. Transient elastography correlated to four different histological fibrosis scores in children with liver disease. Eur J Pediatr 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shneider BL, Goodrich NP, Ye W, Sawyers C, Molleston JP, Merion RM, Leung DH, et al. Nonfasted Liver Stiffness Correlates with Liver Disease Parameters and Portal Hypertension in Pediatric Cholestatic Liver Disease. Hepatol Commun 2020;4:1694–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castera L, Foucher J, Bernard PH, Carvalho F, Allaix D, Merrouche W, Couzigou P, et al. Pitfalls of liver stiffness measurement: a 5-year prospective study of 13,369 examinations. Hepatology 2010;51:828–835. [DOI] [PubMed] [Google Scholar]
- 24.Friedrich-Rust M, Ong MF, Martens S, Sarrazin C, Bojunga J, Zeuzem S, Herrmann E. Performance of transient elastography for the staging of liver fibrosis: a meta-analysis. Gastroenterology 2008;134:960–974. [DOI] [PubMed] [Google Scholar]
- 25.Tsochatzis EA, Gurusamy KS, Ntaoula S, Cholongitas E, Davidson BR, Burroughs AK. Elastography for the diagnosis of severity of fibrosis in chronic liver disease: a meta-analysis of diagnostic accuracy. J Hepatol 2011;54:650–659. [DOI] [PubMed] [Google Scholar]
- 26.Li DK, Khan MR, Wang Z, Chongsrisawat V, Swangsak P, Teufel-Schafer U, Engelmann G, et al. Normal liver stiffness and influencing factors in healthy children: An individual participant data meta-analysis. Liver Int 2020;40:2602–2611. [DOI] [PubMed] [Google Scholar]
- 27.Shen QL, Chen YJ, Wang ZM, Zhang TC, Pang WB, Shu J, Peng CH. Assessment of liver fibrosis by Fibroscan as compared to liver biopsy in biliary atresia. World J Gastroenterol 2015;21:6931–6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teufel-Schafer U, Flechtenmacher C, Fichtner A, Hoffmann GF, Schenk JP, Engelmann G. Transient elastography correlated to four different histological fibrosis scores in children with liver disease. Eur J Pediatr 2021;180:2237–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu JF, Lee CS, Lin WH, Jeng YM, Chen HL, Ni YH, Hsu HY, et al. Transient elastography is useful in diagnosing biliary atresia and predicting prognosis after hepatoportoenterostomy. Hepatology 2018;68:616–624. [DOI] [PubMed] [Google Scholar]
- 30.Bass LM, Shneider BL, Henn L, Goodrich NP, Magee JC, Childhood Liver Disease Research N. Clinically Evident Portal Hypertension: An Operational Research Definition for Future Investigations in the Pediatric Population. J Pediatr Gastroenterol Nutr 2019;68:763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakaguchi H, Konishi KI, Yasuda R, Sasaki H, Yoshimaru K, Tainaka T, Fukahori S, et al. Serum matrix metalloproteinase-7 in biliary atresia: A Japanese multicenter study. Hepatol Res 2022. [DOI] [PubMed] [Google Scholar]
- 32.Wu JF, Jeng YM, Chen HL, Ni YH, Hsu HY, Chang MH. Quantification of Serum Matrix Metallopeptide 7 Levels May Assist in the Diagnosis and Predict the Outcome for Patients with Biliary Atresia. J Pediatr 2019;208:30–37 e31. [DOI] [PubMed] [Google Scholar]
- 33.Yang L, Zhou Y, Xu PP, Mourya R, Lei HY, Cao GQ, Xiong XL, et al. Diagnostic Accuracy of Serum Matrix Metalloproteinase-7 for Biliary Atresia. Hepatology 2018;68:2069–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang J, Wang J, Shen Z, Lu X, Chen G, Huang Y, Dong R, et al. Serum MMP-7 in the Diagnosis of Biliary Atresia. Pediatrics 2019;144. [DOI] [PubMed] [Google Scholar]
- 35.Hsieh CS, Chuang JH, Huang CC, Chou MH, Wu CL, Lee SY, Chen CL. Evaluation of matrix metalloproteinases and their endogenous tissue inhibitors in biliary atresia-associated liver fibrosis. J Pediatr Surg 2005;40:1568–1573. [DOI] [PubMed] [Google Scholar]
- 36.Huang CC, Chuang JH, Chou MH, Wu CL, Chen CM, Wang CC, Chen YS, et al. Matrilysin (MMP-7) is a major matrix metalloproteinase upregulated in biliary atresia-associated liver fibrosis. Mod Pathol 2005;18:941–950. [DOI] [PubMed] [Google Scholar]
- 37.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019;575:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iordanskaia T, Malesevic M, Fischer G, Pushkarsky T, Bukrinsky M, Nadler EP. Targeting Extracellular Cyclophilins Ameliorates Disease Progression in Experimental Biliary Atresia. Mol Med 2015;21:657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nadler EP, Li X, Onyedika E, Greco MA. Differential expression of hepatic fibrosis mediators in sick and spontaneously recovered mice with experimental biliary atresia. J Surg Res 2010;159:611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nadler EP, Patterson D, Violette S, Weinreb P, Lewis M, Magid MS, Greco MA. Integrin alphavbeta6 and mediators of extracellular matrix deposition are up-regulated in experimental biliary atresia. J Surg Res 2009;154:21–29. [DOI] [PubMed] [Google Scholar]
- 41.Finnson KW, Philip A. Endoglin in liver fibrosis. J Cell Commun Signal 2012;6:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bessho K, Mourya R, Shivakumar P, Walters S, Magee JC, Rao M, Jegga AG, et al. Gene expression signature for biliary atresia and a role for interleukin-8 in pathogenesis of experimental disease. Hepatology 2014;60:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zimmermann HW, Seidler S, Gassler N, Nattermann J, Luedde T, Trautwein C, Tacke F. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS One 2011;6:e21381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Preativatanyou K, Honsawek S, Chongsrisawat V, Vejchapipat P, Theamboonlers A, Poovorawan Y. Correlation of circulating endoglin with clinical outcome in biliary atresia. Eur J Pediatr Surg 2010;20:237–241. [DOI] [PubMed] [Google Scholar]
- 45.Battle MA, Konopka G, Parviz F, Gaggl AL, Yang C, Sladek FM, Duncan SA. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc Natl Acad Sci U S A 2006;103:8419–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet 2003;34:292–296. [DOI] [PubMed] [Google Scholar]
- 47.Grijalva JL, Huizenga M, Mueller K, Rodriguez S, Brazzo J, Camargo F, Sadri-Vakili G, et al. Dynamic alterations in Hippo signaling pathway and YAP activation during liver regeneration. Am J Physiol Gastrointest Liver Physiol 2014;307:G196–204. [DOI] [PubMed] [Google Scholar]
- 48.Lu L, Finegold MJ, Johnson RL. Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp Mol Med 2018;50:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kindler JM, Mitchell EL, Piccoli DA, Grimberg A, Leonard MB, Loomes KM, Zemel BS. Bone geometry and microarchitecture deficits in children with Alagille syndrome. Bone 2020;141:115576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loomes KM, Spino C, Goodrich NP, Hangartner TN, Marker AE, Heubi JE, Kamath BM, et al. Bone Density in Children With Chronic Liver Disease Correlates With Growth and Cholestasis. Hepatology 2019;69:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel KR, Harpavat S, Khan Z, Dhingra S, Quintanilla N, Firan M, Goss J. Biliary Atresia Patients With Successful Kasai Portoenterostomy Can Present With Features of Obliterative Portal Venopathy. J Pediatr Gastroenterol Nutr 2020;71:91–98. [DOI] [PubMed] [Google Scholar]
- 52.Emerick KM, Krantz ID, Kamath BM, Darling C, Burrowes DM, Spinner NB, Whitington PF, et al. Intracranial vascular abnormalities in patients with Alagille syndrome. J Pediatr Gastroenterol Nutr 2005;41:99–107. [DOI] [PubMed] [Google Scholar]
- 53.Kamath BM, Spinner NB, Emerick KM, Chudley AE, Booth C, Piccoli DA, Krantz ID. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation 2004;109:1354–1358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Spearman correlations and scatter plots among biomarker concentrations and liver stiffness measurements for participants with A1AT. The numbers in the upper triangle of the grid display the Spearman correlation coefficients for pairs of variables. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05; **p<0.01; ***p<0.001
The lower triangle of the grid displays scatter plots with LOESS lines.
Supplemental Figure 2. Spearman correlations and scatter plots among biomarker concentrations and liver stiffness measurements for participants with ALGS. The numbers in the upper triangle of the grid display the Spearman correlation coefficients for pairs of variables. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05; **p<0.01; ***p<0.001
The lower triangle of the grid displays scatter plots with LOESS lines.
Supplemental Figure 3. Spearman correlations between biomarker concentrations and laboratory measurements for A1AT participants. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05; **p<0.01; ***p<0.001
Supplemental Figure 4. Spearman correlations between biomarker concentrations and laboratory measurements for ALGS participants. The size of the font corresponds to the magnitude of the correlation and the symbols below the numbers indicate significance levels after using multiple comparisons correction for false discovery rate: * p<0.05; **p<0.01; ***p<0.001