

SUMMARY
Inflammatory responses are crucial for controlling infections and initiating tissue repair. However, excessive and uncontrolled inflammation causes inflammatory disease. Processing and release of the pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18 depend on caspase-1 activation within inflammasomes. Assembly of inflammasomes is initiated upon activation of cytosolic pattern recognition receptors (PRRs), followed by sequential polymerization of pyrin domain (PYD)-containing and caspase recruitment domain (CARD)-containing proteins mediated by homotypic PYD and CARD interactions. Small PYD- or CARD-only proteins (POPs and COPs, respectively) evolved in higher primates to target these crucial interactions to limit inflammation. Here, we show the ability of COPs to regulate inflammasome activation by modulating homotypic CARD-CARD interactions in vitro and in vivo. CARD16, CARD17, and CARD18 displace crucial CARD interactions between caspase-1 proteins through competitive binding and ameliorate uric acid crystal-mediated NLRP3 inflammasome activation and inflammatory disease. COPs therefore represent an important family of inflammasome regulators and ameliorate inflammatory disease.
Graphical Abstract
In brief
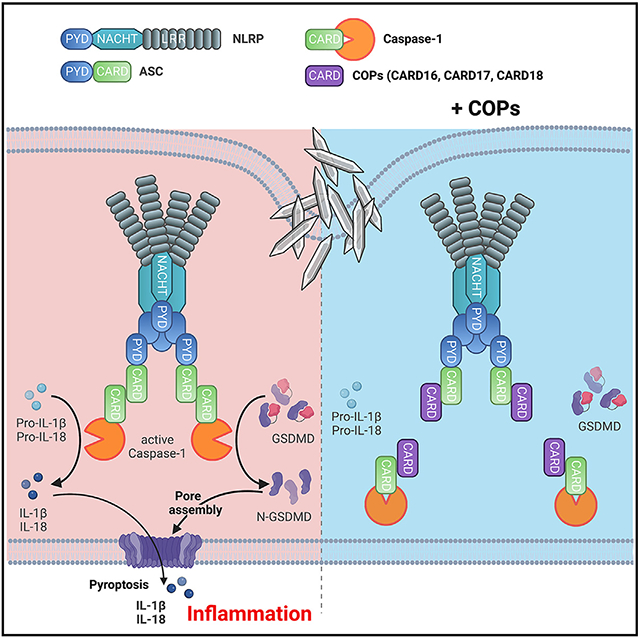
Devi et al. show that the three human caspase recruitment domain (CARD)-only proteins (COPs), CARD16, CARD17, and CARD18, prevent inflammasome assembly. COPs bind to the CARDs of ASC and caspase-1, thereby preventing caspase-1 activation, IL-1β and IL-18 release, and pyroptosis. COP transgenic mice exhibit ameliorated NLRP3 inflammasome-dependent inflammatory disease.
INTRODUCTION
Members of the interleukin-1 (IL-1) family regulate diverse immune-cell-mediated processes, including pathogen defense and inflammation as well as tissue repair and tissue homeostasis. In particular, IL-1β and IL-18, which require proteolytic maturation, play a central role.1 While these cytokines are crucial for innate immune responses, their uncontrolled presence is detrimental and has been linked to autoinflammatory diseases. Consequently, blocking IL-1β, which is primarily produced by phagocytes, including macrophages, monocytes, and dendritic cells (DCs), is an effective treatment strategy for many inflammatory diseases.2 Cellular or microbial danger and stress signals induce inflammatory caspase-1 activation in the inflammasome complex, which is crucial for inflammatory responses promoting tissue repair and wound healing.3-5 Active caspase-1 is responsible for the proteolytic maturation and release of IL-1β and IL-18 6,7 as well as proteolytic cleavage of gasdermin D (GSDMD),8-10 plasma membrane pore formation, induction of pyroptotic cell death, and release of danger-associated molecular patterns (DAMPs).11-18 Inflammasome assembly occurs by sequential polymerization of pyrin domain (PYD)- and/or caspase recruitment domain (CARD)-containing inflammasome components and is mediated by homotypic PYD-PYD and CARD-CARD interactions. While recruitment of the CARD-containing caspase-1 to PYD containing pattern recognition receptors (PRRs), including Nod-like receptors (NLRs) containing a PYD (NLRPs), Pyrin, and AIM2, requires the PYD and CARD-containing adapter, apoptosis-associated speck-like protein containing a CARD (ASC), caspase-1 can be directly recruited to select NLRs containing a CARD (NLRCs). The process of caspase-1 activation in inflammasomes requires dimerization/polymerization of pro-caspase-1 and proteolysis through a proximity-induced autoactivation mechanism.19-22 However, oligomerization of upstream PRRs and/or ASC polymerization is a prerequisite for caspase-1 polymerization.
We and others previously discovered a family of three PYD-only proteins (POPs), POP1/PYDC1, POP2/PYDC2, and POP3/PYDC5, which regulate inflammasome responses through competitive binding between the PYDs of PRRs and ASC.23-26 Thereby, POPs interfere with the polymerization of inflammasome components and consequently prevent inflammasome assembly and inflammation in vivo.23,27-30 A comparable competitive binding mechanism has been proposed for the three human COPs, CARD16/Cop/Pseudo-ICE, CARD17/Inca, and CARD18/Iceberg, but targeting the CARD in caspase-1 or ASC instead of the PYD.31-34 However, most of these studies have been performed by overexpression in epithelial cells before the discovery of the inflammasome, and consequently any relevance of COPs in regulating inflammasome responses and inflammatory disease in vivo remains unknown. Intriguingly, POPs and COPs are lacking from most species, including mice, but evolved in higher primates to potentially enable a more fine-tuned inflammasome response and likely aid the resolution of inflammasome responses.23,35,36 Nevertheless, transgenic expression of human POP1, POP2, and POP3 in macrophages and mice recapitulates their inflammasome inhibitory function observed in humans and ameliorates inflammation in vivo.23,27,29,30 Hence, core PYD- and CARD-containing proteins are sufficiently conserved,37 which allows human proteins to interact with structurally conserved mouse proteins and enables in vivo studies of human inflammasome regulators in mice.
Gout and pseudogout are the most common forms of inflammatory arthritis and are characterized by recurrent inflammatory flares, mostly in a single joint. Inflammation is caused by crystal formation and deposition into joints, which are taken up by resident macrophages and trigger NLRP3 inflammasome activation.38 IL-1β and other IL-1 family cytokines play pivotal roles in the pathology of gout.39 Consequently, blocking IL-1β is an effective treatment strategy in clinical practice.40,41 While monosodium urate (MSU) crystals are characteristic for gout, calcium pyrophosphate dihydrate (CPP) crystals are linked to pseudogout.42 However, not all patients with hyperuricemia and crystal deposition will develop gout. Hence, other regulatory factors that modify acute inflammatory responses can alter the susceptibility for developing disease symptoms.
Here we report the generation of three transgenic mouse lines expressing CARD16, CARD17, or CARD18 in macrophages and demonstrate that all three COPs impair inflammasome responses in mouse macrophages by targeting the recruitment and activation of caspase-1 comparably with human macrophages. Consequently, all COPs ameliorate MSU crystal-induced and NLRP3 inflammasome-mediated inflammatory disease in vivo. Our results provide crucial insights into the molecular mechanisms regulating inflammasome assembly in humans and emphasize species-specific differences and the evolution of a more complex inflammasome regulation in humans.
RESULTS
COPs ameliorate MSU-induced inflammation in mice
Little is known about COP expression, and we therefore analyzed existing expression datasets in silico to determine whether COPs may show differential expression in inflammatory disease. However, very few datasets contained COP expression except for a microarray dataset (GEO: GSE36700) of synovial biopsies from patients with rheumatologic diseases (rheumatoid arthritis, systemic lupus erythematosus, osteoarthritis, and psoriatic arthritis) and crystal-induced arthritis (CIA).43,44 Interestingly, CARD16 expression was lower in CIA than in other forms of arthritis and only narrowly missed significance (p = 0.0506), while CARD18 expression trended higher but was not significant (Figure 1A). A CARD17 probe was not present in this dataset. CIA refers to patients with MSU or CPP crystal deposition in the joints and in extra-articular space, which act as DAMPs that can activate the NLRP3 inflammasome and are responsible for synovial inflammation in gout and pseudogout patients, respectively.38,40,45-48 MSU crystals cause epigenetic changes of myeloid cells and affect transcription and inflammatory and metabolic reprogramming of macrophages.49,50 To further evaluate COP expression in the presence of MSU crystals, we treated human THP-1 macrophages with MSU crystals and determined COP expression by qPCR. Reminiscent of CIA patients, we observed a significant decrease in CARD16 and CARD17 transcript levels and a slight increase of CARD18 transcripts (Figure 1B). Hence, the presence of MSU crystals caused distinct changes in COP gene expression. To further investigate the role of COPs in regulating macrophage inflammasome responses and inflammation in vivo, particularly in response to crystalline DAMPs, we generated transgenic (TG) mice as previously established for analyzing the in vivo function of POPs.23,27,29,51,52 We utilized the human CD68 promoter in combination with the macrophage-specific IVS-1 enhancer for gene expression in monocytes, tissue-resident macrophages, and DCs but not in other leukocyte populations.29,51 Accordingly, we detected CARD16, CARD17, and CARD18 expression in CD115+ blood monocytes from CARD16TG, CARD17TG, and CARD18TG mice, respectively (Figure S1A). MSU crystal deposits in articular joints and bursal tissue promote activation of resident macrophages and neutrophil influx during acute joint inflammation in gout. A murine subcutaneous air pouch resembles this bursa-like space with a synovial-like membrane synonymous to the synovial lining.27,53 Hence, air pouch injection with MSU crystals recapitulates MSU responses in the joints of gout patients.54 While injection of MSU crystals into the air pouch of wild-type (WT) mice resulted in inflammation as quantified by myeloperoxidase (MPO) activity of infiltrating neutrophils by in vivo imaging, it was significantly ameliorated in all three COPTG mice and closely matched the lack of inflammation observed in Casp1−/− mice (Figure 1C). We confirmed reduced cellular infiltrates and, in particular, a reduced number of Ly6G+ neutrophils by flow cytometry of the air pouch lavage (Figure 1D). Sterile neutrophil influx is driven by IL-1β55 and accordingly, lavage fluids contained significantly reduced IL-1β in all three COPTG and Casp1−/− mice compared with WT mice (Figure 1E). The CXCR2 ligands CXCL1 and CXCL2 are produced by tissue-resident cells during local inflammation and control the early stage of neutrophil recruitment in gout.56-58 Accordingly, we detected increased CXCL1 and CXCL2 levels in lavage fluids after MSU crystal injection but, since these chemokines are produced independently of inflammasome activation, we did not observe significant differences between WT, COPTG, and Casp1−/− mice (Figure 1E). Inflammasome activation not only causes cytokine release but also the release of danger signals by pyroptosis, including the release of inflammasome particles, which are phagocytized by neighboring cells and propagate inflammasome responses to bystander cells to perpetuate inflammation.59,60 COPTG mice also showed reduced levels of caspase-1 and NLRP3 in the air pouch lavage, similar to Casp1−/− mice (Figure 1F). This ameliorated MSU crystal-mediated inflammation in COPTG mice was not exclusive to the air pouch. MSU crystal injection into the peritoneal cavity also resulted in a diminished inflammatory response, as also determined by MPO activity (Figure S1B). Hence, our results indicated a regulatory role of COPs in ameliorating NLRP3 inflammasome-mediated inflammatory disease pathology in vivo.
Figure 1. COPs ameliorate MSU-induced inflammation in mice.

(A) CARD16 (p = 0.0506) and CARD18 mRNA expression was determined by high-density oligonucleotide spotted microarrays (GEO: GSE36700) from synovial biopsies from patients with rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), osteoarthritis (OA), and psoriatic arthritis (PA), as well as crystal-induced arthritis (CIA). n = 4–7.43,44
(B) THP-1 cells were left untreated or were treated with MSU crystals (90 μg mL−1) for 5 h, and mRNA expression of CARD16, CARD17, and CARD18 was determined by qPCR and presented as fold expression compared with untreated cells (n = 6, mean ± SD). *p < 0.05.
(C) In vivo imaging of MPO activity correlating with MSU-induced neutrophil infiltration into air pouches 7 h after MSU crystal injection (3 mg per air pouch) in wild-type (WT), CARD16TG, CARD17TG, CARD18TG, and Casp1−/− mice (left) and average radiance (right) presented as photons/s/cm2/sr (n = 4–5, mean ± SD). *p < 0.05.
(D) Total air pouch lavage cells (left) and Ly6G+ neutrophils (right) were determined by flow cytometry in mice injected with either PBS or MSU crystals (n = 4–7, mean ± SD). *p < 0.05.
(E and F) Air pouch lavage fluids were analyzed by ELISA for (E) IL-1β, CXCL1, and CXCL2 and (F) caspase-1 and NLRP3 (n = 4–7, mean ± SD). *p < 0.05. The in vivo gout model was performed twice. See also Figure S1.
COPs inhibit inflammasome-mediated cytokine release in mouse macrophages
To determine the function of COPs in mouse macrophages, we first confirmed CARD16, CARD17, and CARD18 expression by immunoblot in bone-marrow-derived macrophages (BMDM) isolated from COPTG mice (Figure 2A). Since MSU crystal responses are mediated by the NLRP3 inflammasome38 but functional differences have been reported for crystalline and soluble NLRP3 inflammasome agonists,61,62 we analyzed responses to several crystalline (MSU, silica, CPP) and soluble (ATP, nigericin) NLRP3 inflammasome activators. We determined IL-1β, IL-18, and tumor necrosis factor (TNF) release by ELISA in primed and activated BMDM. Expression of any of the COPs significantly reduced the release of IL-1β and IL-18, which were also impaired in Casp1−/− BMDM (Figure 2B). However, TNF release, which is independent of inflammasome activation, was not affected (Figure 2B). Since COPs are thought to block inflamma-ome activation by binding to and inhibiting caspase-1, COPs consequently should affect other inflammasomes besides the NLRP3 inflammasome. Indeed, IL-1β and IL-18 release was also reduced by COPs and in Casp1−/− BMDM in response to AIM2 inflammasome activation by poly(dA:dT) transfection, NLRP1b inflammasome activation by Bacillus anthracis lethal toxin, and NLRC4 inflammasome activation by transfection of Salmonella Typhimurium flagellin (Figure 2C). However, inflammasome-independent TNF release was not affected under these conditions. Hence, while MSU mediated NLRP3 inflammasome activation in BMDM was impaired by COPs and recapitulated the reduced inflammation observed in response to MSU crystal injection in vivo, COPs were also capable of preventing inflammasome-mediated cytokine release by other inflammasome sensors.
Figure 2. COPs inhibit inflammasome-mediated cytokine release in mouse macrophages.

(A) Total cell lysates (TCL) from COPTG transgenic (CARD16, CARD17, CARD18) and wild-type (WT) BMDM were analyzed for GFP-COP expression by immunoblot and immunoprecipitation (IP).
(B and C) WT, COPTG, and Casp1−/− BMDM were primed with Pam3CSK4 (Pam) (1 μg mL−1, 4 h) and (B) treated with MSU crystals (180 μg mL−1, 6 h), silica (200 μg mL−1, 6 h), CPP (200 μg mL−1, 6 h), ATP (5 mM, 30 min), or nigericin (Nig) (5 μM, 45 min), or (C) transfected with p(dA:dT) (1 μg mL−1, 6 h), LeTx (0.5 μg mL−1, 6 h), or flagellin (0.5 μg mL−1, 6 h), and supernatants (SN) were analyzed for secreted IL-1β, IL-18, and TNF by ELISA (n = 3, mean ± SD). *p < 0.05. The dotted line indicates that Casp1−/− BMDM are only present in the primed and activated treatment group. Untreated and primed control cells are the same for different treatment groups.
COPs inhibit cytokine release in human macrophages
To ensure comparable responses to MSU crystals in mouse and human macrophages, we generated human THP-1 cells with stable expression of CARD16, CARD17, and CARD18 and confirmed protein expression (Figure 3A). Reminiscent of mouse BMDM, release of IL-1β and IL-18, but not of TNF, was strongly diminished in COP-expressing as well as CASP1KO THP-1 cells in response to Toll-like receptor (TLR) priming and NLRP3 inflammasome activation with different crystalline or soluble stimuli (Figure 3B). The inflammasome inhibitory function of COPs in human macrophages was also not limited to NLRP3, since release of IL-1β and IL-18, but not release of TNF, was impaired in cells transfected with Salmonella Typhimurium flagellin, poly(dA:dT), FSL-1, and poly(I:C) to activate human NLRC4, AIM2, NLRP7, and NLRP1 inflammasomes, respectively (Figure 3C). Hence, COPs comparably regulate inflammasome-mediated cytokine release in human and mouse macrophages, which suggested sufficiently conserved inflammasome components in humans and mice.
Figure 3. COPs inhibit cytokine release in human macrophages.

(A) Total cell lysates (TCL) from THP-1 cells expressing GFP control (Ctrl) or GFP-tagged CARD16, CARD17, and CARD18 were analyzed by immunoblot.
(B and C) Ctrl, CARD16, CARD17, CARD18, and CASP1KO THP-1 were primed with Pam3CSK4 (Pam) (1 μg mL−1, 4 h) and (B) treated with MSU crystals (180 μg mL−1, 6 h), silica (200 μg mL−1, 6 h), CPP (200 μg mL−1, 6 h), ATP (5 mM, 30 min), or nigericin (Nig) (5 μM, 45 min), or (C) transfected with flagellin (0.5 μg mL−1, 4 h), p(dA:dT) (1 μg mL−1, 4 h), FSL-1 (0.2 μg mL−1, 4 h), or p(I:C) (10 μg mL−1, 4 h), and SN were analyzed for secreted IL-1β, IL-18, and TNF by ELISA (n = 3, mean ± SD). *p < 0.05. The dotted line indicates that CASP1KO THP-1 are only present in the primed and activated treatment group. Untreated and primed control cells are the same for different treatment groups.
COPs inhibit caspase-1 activation and pyroptosis in macrophages
Inflammasome-dependent cytokine release is contingent on caspase-1 activation.6,7 To determine caspase-1 activation, we activated primed BMDM with MSU crystals and analyzed caspase-1 by immunoblot, as previously described.59,60,63 MSU crystal-, as well as nigericin-induced processing, and release of caspase-1 (p20/p10) into cell-culture supernatants was strongly reduced in BMDM expressing CARD16, CARD17, or CARD18 without affecting expression of full-length caspase-1 in total cell lysates (Figures 4A and S2A). In agreement with our ELISA results, CARD16, CARD17, and CARD18 expression also impaired the release of mature IL-1β p17 in response to MSU crystal stimulation (Figure 4A). To directly quantify the activity of caspase-1, we utilized the flow cytometry fluorochrome inhibitor of caspases (FLICA) assay. Caspase-1 activity was strongly reduced in CARD16TG, CARD17TG, and CARD18TG BMDM compared with WT BMDM in response to MSU crystals (Figure 4B) or nigericin (Figure S2B). Besides IL-1β and IL-18, active caspase-1 also cleaves GSDMD.9,10 The p30 N-terminal GSDMD fragment (GSDMD-N) inserts into the plasma membrane and causes pore formation and pyroptosis.11,13-16 As expected, GSDMD processing and pyroptosis was prevented or greatly diminished in CARD16TG, CARD17TG, CARD18TG, and Casp1−/− BMDM in response to MSU crystals (Figures 4C and 4D) and nigericin (Figures S2C and S2D). We earlier demonstrated that POPs interfere with ASC polymerization and inhibit inflammasome responses by competitive PYD binding.23,27,29 In contrast, neither CARD16, CARD17, nor CARD18 affected nigericin-induced ASC polymerization (Figure 4E). Overexpression studies in non-immune cancer cells provided some evidence for a transcriptional effect of COPs, particularly CARD16, in nuclear factor κB (NF-κB) activation.32,33 However, expression of core inflammasome components and IL-1β in response to macrophage priming with a TLR2 agonist was not affected by COP expression (Figure S2E), indicating that the reduced release of IL-1β and IL-18 in COPTG macrophages was caused by effects on inflammasome responses rather than transcription.
Figure 4. COPs inhibit caspase-1 activation and pyroptosis in macrophages.

(A–E) Wild-type (WT), COPTG (CARD16, CARD17, CARD18) and Casp1−/− BMDM were (A) primed with Pam3CSK4 (Pam) (1 μg mL−1, 4 h) and activated with MSU crystals (180 μg mL−1, 2 h) and culture supernatants (SN) and total cell lysates (TCL) analyzed by SDS-PAGE and immunoblot; (B) primed with Pam (1 μg mL−1, 1 h), activated with MSU crystals (180 μg mL−1, 3.5 h), and incubated with an FLICA substrate (10 μM) to determine caspase-1 activity by flow cytometry (FLICA+ cells from a representative experiment are presented, n = 3); (C) primed with Pam (1 μg mL1, 4 h) and activated with MSU crystals (180 μg mL−1, 2 h) (TCL were analyzed by SDS-PAGE and immunoblot); (D) treated with Pam (1 μg mL−1, 4 h) and activated with MSU crystals (180 μg mL−1, 2 h). SN were analyzed for lactate dehydrogenase (LDH) release, presented as percent cytotoxicity compared with maximum LDH release (n = 3, mean ± SD). *p < 0.05. The dotted line indicates that Casp1−/− BMDMare only present in the primed and activated treatment group; (E) primed with Pam (1 μg mL−1, 4 h) and activated with nigericin (Nig) (10 μM 45 min). Disuccinimidyl suberate (DSS) crosslinked and total ASC was analyzed by SDS-PAGE and immunoblot. Arrows indicate mono- and oligomeric ASC. (F–K) Control (Ctrl), CARD16, CARD17, and CARD18 expressing THP-1 cells were (F) primed with Pam (1 μg mL−1, 4 h), activated with MSU crystals (180 μg mL−1, 2 h), and TCL and SN were analyzed by SDS-PAGE and immunoblot; (G) primed with Pam (1 μg mL−1,1 h), activated with Nig (5 μM, 2 h), and incubated with an FLICA substrate (10 μM) to determine caspase-1 activity by flow cytometry (FLICA+ cells from a representative experiment are presented, n = 3); (H) primed with Pam (1 μg mL−1, 4 h), activated with MSU crystals (180 μg mL−1, 1 h), and TCL were analyzed by SDS-PAGE and immunoblot together with CASP1KO THP-1 cells; (I) primed with Pam (1 μg mL−1, 4 h), activated with MSU crystals (180 μg mL−1, 1 h), and LDH release was quantified and presented as percent cytotoxicity compared with maximum LDH release (n = 3, mean ± SD, *p < 0.05). CASP1KO THP-1 cells are separated by a dotted line, indicating that they are only present in the primed and MSU-activated treatment group; (J) primed with Pam (1 μg mL−1) and incubated with Cytotox Red (250 nM) for 3 h prior to activation with MSU crystals (100 μg mL−1) and live cell imaging for 16 h. Cell death was quantified from 9 images/well (n = 4–5, mean ± SD, *p < 0.05); (K) primed with Pam (1 μg mL−1, 4 h) and activated with Nig (10 μM, 45 min). DSS crosslinked and total ASC were analyzed by SDS-PAGE and immunoblot. Arrowheads indicate mono- and oligomeric ASC.
See also Figures S2 and S3.
Similar to BMDM, TLR2-primed and MSU crystal-activated THP-1 cells expressing COPs also showed impaired caspase-1 processing into the active p10 subunit and IL-1β p17 maturation, but total caspase-1 p45 and pro-IL-1β p34 expression in total cell lysates was not impaired (Figure 4F). Furthermore, CARD16, CARD17, and CARD18 expression in THP-1 also showed diminished caspase-1 activity in the FLICA assay after NLRP3 inflammasome stimulation (Figure 4G). Consequently, GSDMD processing and pyroptosis were also impaired after MSU crystal (Figures 4H and 4I) and nigericin (Figures S3A and S3B) activation. We further analyzed pyroptosis in WT and CARD16-, CARD17-, and CARD18-expressing THP-1 cells by real-time Cytotox Red uptake, which only stains cells with compromised plasma membranes. While WT cells continuously took up Cytotox Red after Pam3CSK4 priming and MSU crystal activation, CARD16-, CARD17-, and CARD18-expressing THP-1 cells showed significantly reduced Cytotox Red uptake (Figure 4J). The kinetics of Cytotox Red uptake is shown by video in WT (Video S1), CARD16 (Video S2), CARD17 (Video S3), and CARD18 (Video S4) cells and by selecting representative frames at 30 min and 16 h after MSU crystal activation (Figure S3C). As observed in BMDM, COPs did not affect ASC polymerization, which was determined by immunoblot of non-reversible crosslinked lysates (Figure 4K). Reminiscent of BMDM, COP expression in THP-1 cells also did not affect expression levels of core inflammasome components (Figure S3D). Overall, these results indicate a conserved response of human COPs in human and mouse macrophages.
Expression of COPs specifically inhibits active caspase-1 in macrophages
To exclude that COPs non-specifically affect any CARD interaction and signaling pathway, we investigated several key CARD-CARD-mediated signaling events. Release of cytochrome c from mitochondria triggers CARD-mediated interactions between caspase-9 and the apoptotic protease activating factor 1 (APAF1), assembly of the pro-caspase-9-activating apoptosome, caspase-3 activation, and eventually poly-ADP ribose polymerase (PARP) cleavage.64-68 Stimulation of WT, CARD16TG, CARD17TG, and CARD18TG BMDM with the broad kinase inhibitor staurosporine, which causes mitochondrial damage and cytochrome c release,69 equally resulted in the proteolytic cleavage of pro-caspase-9, pro-caspase-3, and PARP (Figure 5A). Similarly, COPs did not affect staurosporine-mediated caspase-3 and PARP cleavage in THP-1 cells (Figure 5B). Proteolytic cleavage of initiator caspases is not necessarily a requirement for activation,70-72 and proteolytic cleavage may even be a mechanism to eliminate the active caspases.73 We therefore utilized a cell-permeable pan-caspase activity probe, biotin-VAD-FMK, to specifically purify active caspases with immobilized streptavidin.73,74 We detected active caspase-1 p10 and p20 in Pam3CSK4-primed and nigericin-activated WT cells. However, we could not purify any active caspase-1 p10 and p20 in CARD16TG, CARD17TG, and CARD18TG BMDM, using Casp1−/− BMDM as specificity control (Figure 5C). No active caspase-3 or caspase-8 was bound to biotin-VAD-FMK after 3 h of nigericin treatment, but pro-caspase-1 and some caspase-1 p20, pro-caspase-3, and pro-caspase-8 were present in unbound fractions, and expression levels were not influenced by COPs (Figure 5C). Furthermore, we directly quantified caspase-9 and caspase-3 activities in staurosporine- treated THP-1 cell lysates with the specific fluorescent caspase substrates Ac-LEHD-AFC and Ac-DEVD-AFC, respectively. We also used the caspase-1 substrate Ac-YVAD-AFC in Pam3CSK4-primed and MSU-activated cells. While caspase-1 activity was reduced in CARD16-, CARD17-, and CARD18-expressing THP-1 cells (Figure 5D), neither caspase-9 (Figure 5E) nor caspase-3 (Figure 5F) activity was affected by CARD16, CARD17, or CARD18, indicating that COPs are specific for caspase-1 and do not affect the CARD-CARD interaction between pro-caspase-9 and APAF1 under these conditions.
Figure 5. Specificity of COPs for regulating caspase-1 activation.

(A) Wild-type (WT) and COPTG (CARD16, CARD17, CARD18) BMDM were left untreated or treated with staurosporine (STS) (1 μg mL−1, 6 h), and total cell lysates (TCL) were analyzed by SDS-PAGE and immunoblot.
(B) Control (Ctrl), CARD16, CARD17, and CARD18 expressing THP-1 cells were left untreated or treated with STS (1 μg mL−1) for the indicated times, and TCL were analyzed by SDS-PAGE and immunoblot.
(C) WT, COPTG, and Casp1−/− BMDM were primed with Pam3CSK4 (Pam) (1 μg mL−1, 4 h) and activated with nigericin (Nig) (5 μM mL−1, 3 h) in the presence of biotin-VAD-FMK (10 μM), and combined culture supernatants and TCL were purified with immobilized neutravidin and analyzed by SDS-PAGE and immunoblot alongside the unbound flowthrough. Asterisk indicates a cross-reactive protein.
(D–F) Ctrl, CARD16, CARD17, and CARD18 expressing THP-1 cells were left untreated or were (D) primed with Pam (1 μg mL−1, 4 h) and activated with MSU crystals (90 μg mL−1, 6 h) or (E and F) treated with STS (1 μg mL−1, 60 min) and caspase-1, −9, and −3 activity determined with (D) Ac-YVAD-AFC, (E) Ac-LEHD-AFC, and (F) Ac-DEVD-AFC, respectively (n = 3, mean ± SD). *p < 0.05.
(G–I) MHC class I and MHC class II cell surface expression in the presence or absence of COPs was analyzed by flow cytometry in (G) bone-marrow-derived, (H) peritoneal, and (I) splenic macrophages (Mac). Results are presented as median relative fluorescence intensity (RFU) (n = 5, mean ± SD). *p < 0.05.
(J) WT and COPTG BMDM were left untreated or primed with IFN-γ (25 U, 12 h) and activated with dispersible whole glucan particles (WGP, 100 μg mL−1, 24 h), and culture supernatants analyzed for TNF by ELISA (n = 3, mean ± SD).
Major histocompatibility complex (MHC) class I and MHC class II gene expression is regulated by CARD-containing NLRC5 and a myeloid-specific CIITA isoform, respectively.75-80 We therefore tested MHC class I and MHC class II expression by flow cytometry in bone marrow and tissue-resident macrophages. WT, CARD16, CARD17, and CARD18 BMDM showed comparable MHC class I expression, but CARD17 and CARD 18 expression slightly altered MHC class II expression (Figure 5G). MHC class I expression was also comparable in peritoneal macrophages, but MHC class II expression was slightly increased in CARD16- and CARD18-expressing cells (Figure 5H). Splenic macrophages did not show any significant difference in MHC class I or MHC class II expression in the presence of COPs (Figure 5I), suggesting that NLRC5 signaling is not affected by COPs, while CIITA or another aspect of MHC class II expression may be modestly impacted by some COPs in a tissue-dependent manner, which will require further studies.
CARD9 links activation of Dectin-1 to Bcl10/Malt1-mediated NF-κB activation, and this pathway is controlled by CARD-CARD interactions.81-84 BMDM require priming with granulocyte macrophage colony-stimulating factor or interferon-γ (IFN-γ) for Dectin-1 ligand-mediated TNF production.85 We therefore performed a TNF ELISA from IFN-γ-primed and particulate β-glucan-treated cells. WT and CARD16-, CARD17-, and CARD18-expressing BMDM produced comparable TNF (Figure 5J), indicating that the CARD-CARD interaction between CARD9 and Bcl10 is not affected by COPs. These experiments support the specificity of COPs to primarily regulate caspase-1 activation in macrophages.
COP gene knockout augments caspase-1 activation and IL-1β release
To confirm the inflammasome inhibitory function of endogenous COPs, we generated THP-1 cells with CRISPR-Cas9-mediated knockout (KO) of CARD16, CARD17, and CARD18. CARD16KO, CARD17KO, and CARD18KO THP-1 cells showed a deletion of the start ATG for CARD16 and truncation and premature stop of CARD17 and CARD18 after 11 or 76 amino acids, respectively (Figure S4). While TLR-primed CARD16KO, CARD17KO, and CARD18KO THP-1 cells responded with augmented release of IL-1β and IL-18 in response to crystalline MSU and soluble nigericin when compared with Cas9 control (Ctrl) THP-1 cells, TNF release was not significantly altered (Figure 6A). CASP1KO THP-1 cells were included as a control. We also detected increased caspase-1 p20 and IL-1β p17 in culture supernatants of CARD16KO, CARD17KO, and CARD18KO compared with control THP-1 cells, but equal pro-caspase-1 and pro-IL-1β expression in total cell lysates (Figures 6B and 6C). We again included CASP1KO THP-1 cells as control. Hence, endogenous COPs inhibit inflammasome responses.
Figure 6. COP knockout in human macrophages enhances cytokine release and caspase-1 activation.

(A) Control (Ctrl), CARD16KO, CARD17KO, CARD18KO, and CASP1KO THP-1 cells were primed with Pam3CSK4 (Pam) (1 μg mL−1, 4 h) and activated with MSU crystals (180 μg mL−1, 6 h) or nigericin (Nig) (5 μM, 45 min), and supernatants (SN) were analyzed for secreted IL-1β, IL-18, and TNF by ELISA (n = 3, mean ± SD). *p < 0.05. The dotted line indicates that CASP1KO THP-1 are only present in the primed and activated treatment group. Untreated and primed control cells are the same for both treatment groups.
(B and C) Ctrl, CARD16KO, CARD17KO, CARD18KO, and CASP1KO THP-1 cells and CASP1KO THP-1 cells were primed with Pam (1 μg mL−1, 4 h) and activated with MSU crystals (180 μg mL−1, 2 h), and SN and total cell lysates (TCL) were analyzed by SDS-PAGE and immunoblot. Asterisk indicates a cross-reactive protein.
See also Figure S4.
COPs selectively interact with caspase-1 and ASC
To provide further insights into the mechanism by which COPs regulate inflammasome responses, we interrogated the binding of COPs to inflammasome components. All COPs bind to caspase-1 and CARD16 also to ASC in epithelial cancer cells,31-33,86,87 but these cells do not reflect the complex and sequential inflammasome regulation that occurs in resting, primed, and activated macrophages. We immunoprecipitated caspase-1 from resting, primed, or activated THP-1 cells and determined its interaction with CARD16, CARD17, and CARD18. Surprisingly, each COP displayed a unique binding characteristic during different activation steps in macrophages, despite the functional outcome on blocking caspase-1 activation having no apparent differences. While priming induced a weak interaction between CARD16 and caspase-1, which was further strengthened after nigericin-mediated NLRP3 inflammasome activation, CARD17 only interacted with caspase-1 after NLRP3 inflammasome activation. In contrast, CARD18 constitutively bound to caspase-1, independent of the cellular activation state (Figure 7A). COPs not only showed activation-specific interactions with caspase-1 but also with ASC. CARD16 bound to ASC most strongly after priming and to a lesser extent after nigericin-mediated NLRP3 inflammasome activation. CARD17 constitutively bound to ASC under all tested conditions, and CARD18 did not bind to ASC in any of the tested conditions (Figure 7B). Hence, COPs regulate caspase-1 activation in a well-orchestrated manner. Since ASC polymerization results in the clustering, autocatalytic processing, and activation of caspase-1,20,21,87 we hypothesized that COPs may interfere with caspase-1 dimerization/polymerization, reminiscent of POP1 and POP2 interfering with ASC polymerization.27,29 However, previous in vitro studies found that CARD16 and CARD18 promote caspase-1 oligomerization into filaments, which is in disagreement with their inhibitory function.86,87 To further elucidate the mechanism by which COPs inhibit caspase-1 activation, we analyzed caspase-1 polymerization in a cell-based assay. We established a green/red intensiometric Förster resonance energy transfer (FRET) assay based on the monomeric red mScarlet-I fluorescence protein (FP) as an acceptor for the bright, monomeric, green-yellow mNeonGreen FP donor to quantify the intermolecular binding between two caspase-1CARD proteins.88 Compared with transfection of either caspase-1CARD fused at its C-terminal end to mNeonGreen (caspase-1 CARD-mNeonGreen) or caspase-1CARD fused at its C-terminal end to mScarlet-I (caspase-1CARD-mScarlet-I) alone, co-transfection of caspase-1CARD-mNeonGreen with caspase-1cCARD-mScarlet-I resulted in increased fluorescent intensity upon excitation for mNeonGreen and emission wavelength scanning for mScarlet-I, indicating FRET activity and close proximity of two caspase-1CARD molecules (Figure 7C). Co-transfection of CARD16, CARD17, or CARD18 prevented the FRET signal (Figure 7C), demonstrating that COPs interfered with caspase-1CARD interactions, thereby inhibiting the necessary dimerization and subsequent higher-order polymerization that was crucial for proximity-mediated caspase-1 trans-activation. Hence, all three COPs were able to inhibit inflammasome-mediated caspase-1 activation by a competitive binding mechanism.
Figure 7. COPs selectively interact with caspase-1 and ASC by a competitive binding mechanism.

(A and B) GFP, GFP-tagged CARD16, CARD17, and CARD18 expressing, and (A) CASP1KO or (B) ASCKD THP-1 cells were left untreated, primed with Pam3CSK4 (Pam) (1 μg mL−1, 3 h), activated with nigericin (Nig) (2.5 μM, 5 min) as indicated, and were subjected to immunoprecipitation (IP) with immobilized (A) anti-caspase-1 or (B) anti-ASC antibodies and analyzed alongside total cell lysates (TCL) by SDS-PAGE and immunoblot.
(C) HEK293 cells were transfected with caspase-1CARD-mNeonGreen, caspase-1CARD-mScarlet-I, CARD16, CARD17, and CARD18 as indicated, and emission scans were collected from 490 to 750 nm in 2-nm increments using a 470-nm excitation wavelength and presented as fluorescence intensity (n = 2).
(D) PMA-differentiated control (Ctrl), CARD16, CARD17, CARD18 expressing, and CASP1KO and ASCKO THP-1 cells were left untreated, primed with Pam (1 μg mL−1, 3 h), activated with nigericin (Nig) (10 μM, 15 min) as indicated, and subjected to proximity ligation assay (PLA) between caspase-1 and ASC and analyzed by fluorescence microscopy. Results are presented as fold PLA+ cells compared with primed Ctrl cells and normalized to DAPI+ cells (n = 9–15, mean ± SD). *p < 0.05.
See also Figure S5.
To further address whether COPs affect inflammasome assembly, we performed a proximity ligation assay (PLA) between caspase-1 and ASC in intact THP-1 cells using CASP1KO and ASCKO cells as specificity controls. The nigericin-induced interaction between ASC and caspase-1 was only slightly reduced in the presence of COPs, although the effect of CARD17 was significant. Inclusion of CASP1KO and ASCKO cells verified the specificity of this approach (Figures 7D and S5). Hence, while the constitutive interaction of CARD17 with ASC together with the strongly induced interaction between CARD17 and caspase-1 in response to inflammasome activation seems to disrupt the recruitment of caspase-1 to the inflammasome scaffold, all COPs similarly disrupt caspase-1 dimerization and oligomerization to abolish caspase-1 activation and attenuate inflammasome responses.
DISCUSSION
ASC-containing inflammasome assembly and activation is based on sequential nucleated polymerization of ASC and caspase-1 filaments that facilitate the close proximity necessary for caspase-1 activation.20,21,87 We previously demonstrated that this first step of nucleated ASC polymerization is regulated by POP family members by competitive binding to the PYD of either ASC or upstream sensors, there by ameliorating inflammatory disease.23,27,29,30 Reminiscent of the three POP family members, the three COPs also evolved in higher primates.36,89 However, their precise role is less well described, largely due to inconclusive and inconsistent studies, including CARD18 inhibiting IL-1β release in one but not another study,34,87 studies of non-immune cells,31-33,86 or studies of primed but not inflammasome-activated macrophages before the discovery of the inflammasome.31-34 Focusing on macrophages by restoring COP expression in mouse macrophages and in mice, expressing COPs in human THP-1 macrophage-like cells, and CRISPR-Cas9-mediated KO of COP-encoding genes in THP-1 cells, we provide evidence that COPs have an important role in regulating inflammasome responses and ameliorating inflammatory disease in vivo. Our study also provides novel insights into the competitive binding mechanism by which COPs disrupted caspase-1CARD interactions responsible for clustering caspase-1 downstream of ASC polymerization and necessary for activation of caspase-1. Therefore, two protein families of six proteins evolved in higher primates to specifically target these two sequential polymerization processes of inflammasome assembly, emphasizing their importance for balanced inflammasome responses and the crucial role of regulating these key processes for maintaining immune homeostasis.
Previous studies did not investigate all three COPs in macrophages. COP expression in macrophages is low and required metabolic labeling for detection,34 and available antibodies unfortunately cross-react with caspase-1, including efforts from our laboratories to generate COP-specific antibodies. We therefore stably expressed GFP-tagged COPs in THP-1 cells and demonstrate that all three COPs interact with caspase-1 and that this interaction was differently regulated during inflammasome priming and activation for CARD16, CARD17, and CARD18. Constitutive interaction of CARD18 with caspase-1 may already prevent any inflammasome responses, while CARD16 and CARD17 first required inflammasome priming and activation, respectively. Hence, we provide evidence of a non-redundant role of COPs, which was further supported by functional data on cytokine release and caspase-1 activation from individual CARD16, CARD17, and CARD18 KO cells. These distinct mechanisms of interaction between COPs and caspase-1 may have important ramifications for specific inflammasome responses, such as priming-independent rapid inflammasome activation.90 Furthermore, interaction of CARD1686 and CARD17 with ASCCARD in addition to caspase-1CARD enables these COPs to simultaneously target both binding partners necessary for caspase-1 polymerization and activation, which may provide a fail-safe mechanism. COPs readily interact with caspase-1 and some also with ASC when expressed in HEK293 and other epithelial cells, but the signals that regulate these interactions in macrophages in a priming and activation-dependent manner are still unknown. Significantly, ASC as well as caspase-1 undergo sequential post-translational modifications that regulate inflammasome assembly in macrophages.91 An earlier in vitro study demonstrated that CARD17 but not CARD18 terminated caspase-1CARD filaments because it lacks two of the six complementary binding interfaces present in the caspase-1CARD required for self-interaction.87 However, in agreement with the constitutive binding of COPs with caspase-1CARD in HEK293 cells, our FRET assays measuring caspase-1CARD dimerization demonstrate that in these cells all three COPs prevent this key step of caspase-1 activation by competitive binding. Therefore, expression of COPs prevents caspase-1 activation, as determined by quantifying caspase-1 conversion into the active caspase-1 p10 and p20 subunits and by specific affinity purification of active caspases. However, based on these interactions, the question remained as to whether COPs only interfere with caspase-1 dimerization or whether COPs could also be capable of disrupting inflammasome assembly by preventing the recruitment of caspase-1 to inflammasomes. We only observed a modest COP-mediated reduction of caspase-1 recruitment to ASC, as determined by PLA, to capture even weak interactions, despite COPs targeting the crucial CARD motifs in both proteins and the very potent functional consequences on inflammasome responses and caspase-1 activation. A likely reason is that PLA measures the presence or absence of these interactions without accounting for qualitative binding differences. Although COPs may cause a loss of these interactions and consequent loss of PLA+ signals in cells with weak activation, in most cells COP expression only reduces these interactions without completely disrupting it and therefore still yielding a PLA+ signal. Such a threshold regulation mechanism is also sensitive to COP expression levels, which would be consistent with the reduced expression of CARD16 in gout patients. Our interpretation is that a main role of COPs is to destabilize and weaken the CARD-CARD interaction between caspase-1 itself as well as its interaction with ASC to raise the threshold required for caspase-1 dimerization and oligomerization and to inhibit activation of caspase-1. Consequently, CARD16, CARD17, and CARD18 significantly inhibit NLRP3 inflammasome-mediated IL-1β and IL-18 release regardless of activation with crystalline or soluble agonists. COPs similarly also inhibit other, non-NLRP3 inflammasome responses.
Earlier studies in HEK293T cells also reported CARD16-mediated NF-κB activation.32,33 However, COPs neither affected the NF-κB-dependent TNF release nor expression of core inflammasome components. Furthermore, we provide evidence that COP expression does not affect all CARD-CARD-mediated cellular responses, including the pro-caspase-9-APAF-1 interaction in the apoptosome,64-68 the CARD9-BCL10-MALT1 interaction downstream of Dectin-1,81-84 and the NLRC5-mediated MHC class I expression.79,80 However, we observed modest effects on MHC class II expression in a tissue-specific manner, which may indicate effects on the myeloid-specific CIITA isoform and will require further investigation.75-78
Reminiscent of PYD interactions, these CARD-mediated events also were sufficiently conserved between humans and mice, as human COPs also inhibit caspase-1 activation downstream of all tested inflammasomes in mouse macrophages. This approach allowed us to investigate the function of COPs in inflammatory disease. Since we observed that MSU crystals affect the expression of COPs, we investigated their role in gout. Gout is caused by chronic hyperuricemia and, consequently, crystal deposition into joints and inflammation that can lead to permanent joint damage and cardiovascular and renal comorbidities. Despite low levels of CARD16, CARD17, and CARD18 expression in COPTG mice, all three COPs almost completely prevent MSU crystal-induced inflammatory responses, emphasizing the potent anti-inflammatory function of COPs. These findings provide important insights into disease mechanisms, as dysregulated COP expression observed in gout patients may influence NLRP3 inflammasome responses and the severity of gout flares.
In summary, we provide evidence for the caspase-1 inhibitory function of human CARD16, CARD17, and CARD18 in macrophages and the functionality of human COPs in mice, and that COPs may have a role in ameliorating inflammatory responses and the severity of inflammatory disease. We further identified that all three COPs have partially non-redundant roles and act at different steps of caspase-1 activation. These results provide mechanistic details about the function of COPs and important insights into the mechanisms that regulate and fine-tune the activation of caspase-1 in response to inflammasome activation.
Limitations of the study
The main limitation of our study is the expression of human CARD16, CARD17, and CARD18 in mice, and this should be considered when interpreting our results. This approach was necessary because COPs are absent in mice. To minimize this potential caveat, we demonstrate a comparable response of COPs in human THP-1 macrophages and BMDM. Furthermore, we demonstrate that COPs show specificity for caspase-1 among other tested caspases and did not affect CARD9 and NLRC5 responses, but we observe a modest effect on MHC class II expression. This may indicate that COPs potentially may target the myeloid-specific CARD-containing CIITA isoform and therefore potentially also other CARD-containing proteins. Another limitation is that we observe decreased CARD16 but increased CARD18 expression in gout patients, but all COPs were similarly affected by MSU crystal responses. Hence, it is unclear whether changes in COP expression are contributing to the gout pathology in human patients. However, we demonstrate that the expression of COPs alleviated the severity of gout in mice. Therefore, future studies will need to address these limitations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Andrea Dorfleutner (Andrea.Dorfleutner@cshs.org).
Materials availability
Unique reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
This paper analyzes existing, publicly available data. Accession numbers for the datasets are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI link is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-caspase-1_p20, clone D7F10 | Cell Signaling Technology | Cat# 3866; RRID:AB_2069051 |
| Rabbit monoclonal cleaved anti-caspase-1, clone D57A2 | Cell Signaling Technology | Cat# 4199; RRID: AB_1903916 |
| Mouse monoclonal cleaved anti-caspase-1, clone casper-1 | AdipoGen | Cat# AG-20B0042; RRID: AB_2490248 |
| Mouse monoclonal anti-caspase-1, clone 1021323 | R&D Systems | Cat# MAB62152 |
| Mouse monoclonal anti-caspase-1 (p10), clone casper-2 | AdipoGen | Cat# AG-20B-0044-C100; RRID:AB_2490253 |
| Rabbit monoclonal cleaved anti-gasderminD | Abcam | Cat# ab209845; RRID:AB_2783550 |
| Rabbit monoclonal cleaved anti-gasderminD, Clone E7H9G | Cell Signaling Technology | Cat# 36425; RRID:AB_2799099 |
| Rabbit monoclonal anti-gasderminD, clone L60 | Cell Signaling Technology | Cat# 93709; RRID:AB_2800210 |
| Mouse monoclonal anti-NLRP3, clone Cryo-2 | AdipoGen | Cat# AG20B0014; RRID:AB_2885199 |
| Rabbit polyclonal anti-NLRP3, clone D4D8T | Cell Signaling Technology | Cat# 15101; RRID:AB_2722591 |
| Mouse monoclonal anti-GFP, clone B-2 | Santa Cruz | Cat# SC9996; RRID:AB_627695 |
| Rabbit monoclonal anti-GFP, clone D5.1 | Cell Signaling Technology | Cat# 2956; RRID:AB_1196615 |
| Rabbit polyclonal anti-ASC, clone AL177 | AdipoGen | Cat# AG25B0006; RRID:AB_2490440 |
| Mouse monoclonal anti-β-tubulin, clone DSHB, AA12.1 | Developmental Studies Hybridoma Bank | Cat# AA12.1 |
| Rabbit polyclonal anti-IL-1β | Santa Cruz | Cat# SC7884; RRID:AB_2124476 |
| Rabbit monoclonal cleaved anti-IL-1b, clone D3A3Z | Cell Signaling Technology | Cat# 83186; RRID: AB_2800010 |
| Rabbit monoclonal anti-IL-1β | Cell Signaling Technology | Cat# 52719; RRID:AB_2799421 |
| Mouse monoclonal anti-IL-1β | Cell Signaling Technology | Cat# 12242; RRID:AB_2715503 |
| HRP conjugated secondary anti-rabbit | Invitrogen | Cat# 31460 |
| HRP conjugated secondary anti-mouse | Invitrogen | Cat# 32430 |
| HRP conjugated secondary anti-goat | Invitrogen | Cat# A15999 |
| Rabbit monoclonal cleaved anti- caspase-3, clone 5A1E | Cell Signaling Technology | Cat# 9664; RRID:AB_2070042 |
| Rabbit monoclonal anti-caspase-3, clone D3R6Y | Cell Signaling Technology | Cat# 14220; RRID:AB_2798429 |
| Anti-Caspase-8 | Cell Signaling Technology | Cat# 4927; RRID:AB_2068301 |
| Rabbit monoclonal cleaved caspase-8, clone D5B2 | Cell signaling Technology | Cat# 8592; RRID:AB_10891784 |
| Anti-caspase-9 | Cell Signaling Technology | Cat# 9504; RRID:AB_2275591 |
| Anti-cleaved caspase-9 | Cell signaling Technology | Cat# 9509; RRID:AB_2073476 |
| Mouse monoclonal cleaved anti-PARP | Cell signaling Technology | Cat# 9548; RRID:AB_2160592 |
| cleaved anti-Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661; RRID:AB_2341188 |
| Rabbit monoclonal cleaved PARP (Asp214) | Cell Signaling Technology | Cat#5625; RRID:AB_10699459 |
| Anti-mouse CD45.2-BV570, clone 104 | Biolegend | Cat# 109833 |
| Anti-mouse Ly6G-BV785, clone 1A8 | Biolegend | Cat#127645 |
| Anti-mouse CD3e-BV785, clone 145-2C11 | Biolegend | Cat# 100355 |
| Anti-mouse CD19-BV785, clone 6D5 | Biolegend | Cat# 115543 |
| Anti-mouse NK-1.1-BV785, clone PK136 | Biolegend | Cat# 108749 |
| PE-CF594 Rat anti-mouse SiglecF, clone E50-2440 | BD Biosciences | Cat# 562757 |
| BB700 Rat Anti-Cd11b, clone M1/70 | BD Biosciences | Cat# 566417 |
| Alexa Fluor 700 anti-mouse Cd11c, clone N418 | Biolegend | Cat# 117320 |
| APC anti-mouse Cd11c, clone N418 | Biolegend | Cat# 117310 |
| PE-Cy7 anti-mouse F4/80, clone BM8 | Biolegend | Cat# 123114 |
| APC-Cy7 anti-mouse Ly6C, clone HK1.4 | Biolegend | Cat# 128026 |
| BV421 anti-mouse I-A/I-E, clone M5/114.15.2 | Biolegend | Cat# 107632 |
| BV650 anti-mouse H-2Kb/H-2Db, Clone 28-8-6 | BD Biosciences | Cat# 745287 |
| APC anti-mouse CD115, clone AFS98 | eBioscience | Cat# 17-1152-82 |
| APC/Fire750 anti-mouse H-2Kb/H-2Db | Biolegend | Cat# 114618 |
| Purified anti-mouse CD16/32, clone 93 | Biolegend | Cat# 101302 |
| Mouse Monoclonal anti-IL-18, clone 74 | MBL | Cat# D047-3;RRID:AB_592016 |
| Mouse Monoclonal anti-IL-18, Biotin conjugated | MBL | Cat# D048-6;RRID:AB_592021 |
| Chemicals, peptides, and recombinant proteins | ||
| Fetal bovine serum | Thermo Scientific | Cat# 16140071 |
| Penicillin and Streptomycin | Gibco | Cat# 15140122 |
| DPBS | Corning | Cat# 21-031-CV |
| RPMI | Cytiva Hyclone, Fisher Scientific | Cat# SH30027FS |
| DMEM | Corning | Cat# 10-013-CV |
| Opti-MEM | Gibco | Cat# 31985088 |
| LPS | Invivogen | Cat# Tlrl-3pelps |
| Pam3csk4 | Invivogen | Cat# Tlrl-pms |
| MSU | Invivogen | Cat# Tlrl-msu |
| Silica/NanoSiO2 | Invivogen | Cat# Tlrl-SiO-2 |
| Nigericin | Invivogen | Cat# Tlrl-nig |
| ATP | Sigma-Aldrich | Cat# A6419 |
| CPPD | Invivogen | Cat# Tlrl-cppd |
| Flagellin | Invivogen | Cat# Tlrl-epstfla-5 |
| Lipofectamine | Thermo Scientific | Cat# 11668019 |
| DOTAP | Roche | Cat# 11202375001 |
| PMA | Invitrogen | Cat# J63916.MCR |
| Protease inhibitor | Pierce | Cat# A329631 |
| Trizol | Invitrogen | Cat# 15596026 |
| Protein agarose A/G beads | Santa Cruz | Cat# SC2003 |
| Super Signal West Femto | Thermo Scientific | Cat# 34096 |
| Staurosporin | Sigma-Aldrich | Cat# S6942 |
| WGP dispersible | Invivogen | Cat# Tlrl-wgp |
| Ac-YVAD-AFC | Enzo Life Science | Cat# ALX-260-108-M005 |
| Ac-DEVD-AFC | Enzo Life Science | Cat# ALX-260-032-M001 |
| Ac-IETD-AFC | Enzo Life Science | Cat# ALX-260-110-M005 |
| Ac-LEHD-AFC | Enzo Life science | Cat# ALX-260-116-M005 |
| Biotin-VAD-FMK | Santa Cruz | Cat# Sc311290 |
| Biotin-YVAD-CMK | Anaspec | Cat# AS-60841 |
| Neutravidin | Pierce | Cat# 29200 |
| FSL-1 | Invivogen | Cat# tlrl-fsl |
| Bacillus anthracis lethal factor | List labs | Cat# 104 |
| Protective antigen | List labs | Cat# 104 |
| Poly(dA:dT) | Invivogen | Cat# Tlrl-patn |
| Brilliant Stain Buffer | BD Horizon | Cat# 566349 |
| eBioscience™ Fixable Viability Dye eFluor™ 506 | ThermoFisher | Cat# 65-0866-14 |
| MACS Buffer | Miltenyi | Cat# 130-091-221 |
| FcBlock | BD Biosciences | Cat# 553141 |
| Cytofix/Cytoperm | BD Biosciences | Cat# 554714 |
| Cytotox Red | Essen Bioscience | Cat# 4632 |
| XenoLight Rediject Inflammation probe | Perkin Elmer | Cat# 760536 |
| Luminol sodium salt | Sigma Aldrich | Cat# A4685 |
| FLICA Caspase-1 inhibitor II Biotinylated | Anaspec | Cat# AS-60841 |
| DSS Disuccinimidyl suberate | Thermo Scientific | Cat# A39267 |
| recombinant murine IFN-γ | Peprotech | Cat# 315-05 |
| MISSION ExpressMag magnetic beads | Millipore-Sigma | Cat# SHM03 |
| Puromycin | Santa Cruz Biotechnology | Cat# 53-79-2 |
| Critical commercial assays | ||
| IL-1β ELISA (human) | Invitrogen | Cat# 88-7261-22 |
| IL-1β ELISA (mouse) | Invitrogen | Cat# 88-7013-22 |
| IL-18 ELISA (human) | R&D Systems | Cat# DY318-05 |
| TNF ELISA (human) | Invitrogen | Cat# 887346-76 |
| TNF ELISA (mouse) | Invitrogen | Cat# 88-7324-88 |
| TNF ELISA (human) | R&D Systems | Cat# DY410 |
| TNF ELISA (mouse) | R&D Systems | Cat# DY210 |
| CXCL1 ELISA (mouse) | R&D Systems | Cat# DY453-05 |
| CXCL2 ELISA (mouse) | R&D Systems | Cat# DY452-05 |
| Caspase-1 ELISA (mouse) | Adipogen | Cat# AG-45B-0002-KI01 |
| NLRP3 ELISA (mouse) | LSBio | Cat# LS-F17336-1 |
| GFP Taqman assay | Invitrogen | Cat# Mr04329676_mr |
| CARD16 Taqman assay | Invitrogen | Cat# Hs03008439_s1 |
| CARD17 Taqman assay | Invitrogen | Cat# Hs01910190_s1 |
| CARD18 Taqman assay | Invitrogen | Cat# Hs01043258_m1 |
| MycoAlert Mycoplasma Detection Kit | Lonza | Cat# LT07 |
| LDH Cytotoxicity Assay (CyQUANT) | Invitrogen | Cat# C203021 |
| Verso cDNA Synthesis Kit | Thermo | Cat# 75650200RXN |
| Duolink Proximity Ligation Assay | Millipore Sigma | Cat# DUO92101-1KT |
| Deposited data | ||
| Affymetrix Human Genome U133 Plus 2.0 Array (HG-U133_Plus_2) | (Lauwerys et al., 2015;43 Nzeusseu Toukap et al., 2007)44 | GEO: GSE36700 |
| original, unprocessed western blot images | this manuscript | https://doi.org/10.17632/gz8rc92m38.1 |
| Experimental models: Cell lines | ||
| THP-1 | American Type Culture Collection | Cat# TIB-202 |
| HEK293 | American Type Culture Collection | Cat# CRL-3216 |
| HEK293 lenti-X | Takara Bio | Cat# 632180 |
| L292 | American Type Culture Collection | Cat# CCL-1 |
| CASP1KO THP-1 | (Chu et al., 2018)92 | N/A |
| ASCKD THP-1 | (Bryan et al., 2009)93 | N/A |
| Ctrl GFP THP-1 | this manuscript | N/A |
| GFP-CARD16 THP-1 | this manuscript | N/A |
| GFP-CARD17 THP-1 | this manuscript | N/A |
| GFP-CARD18 THP-1 | this manuscript | N/A |
| ASCKO THP-1 | Invivogen | thp-koascz |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory, bred in house | JAX#:00066; RRID:IMSR_JAX:000664 |
| Mouse: B6.Tg(CD68-CARD16) | this manuscript | N/A |
| Mouse: B6.Tg(CD68-CARD17) | this manuscript | N/A |
| Mouse: B6.Tg(CD68-CARD18) | this manuscript | N/A |
| Mouse: Casp1−/− | V. M. Dixit (Kayagaki et al., 20159) | N/A |
| Oligonucleotides | ||
| Please see Table S1 | N/A | |
| Recombinant DNA | ||
| pCAT-Basic-CARD16 | this manuscript | N/A |
| pCAT-Basic-CARD17 | this manuscript | N/A |
| pCAT-Basic-CARD18 | this manuscript | N/A |
| pLex-GFP-CARD16 | this manuscript | N/A |
| pLex-GFP-CARD17 | this manuscript | N/A |
| pLex-GFP-CARD18 | this manuscript | N/A |
| pCDNA3-caspase-1 CARD mNeonGreen | this manuscript | N/A |
| pCDNA3-caspase-1 CARD mScarlet-I | this manuscript | N/A |
| pCDNA3- mNeonGreen-mScarlet-I | this manuscript | N/A |
| pMD2.G | Addgene | Cat# 12259; RRID:Addgene_12259 |
| psPAX2 | Addgene | Cat# 12260; RRID:Addgene_12260 |
| lentiCRISPRv1 | Addgene | Cat# 49535 |
| Software and algorithms | ||
| FlowJo v10 | BD Biosciences | N/A |
| Cytek acquisition software | Cytek | N/A |
| SkanIt | Thermo | N/A |
| IncuCyte Base Analysis Software | Sartorius | N/A |
| NIS-Elements AR with ai | Nikon | N/A |
| Fiji (Image J) | Schindelin et al., 201294 | N/A |
| Living Image software 4.7.3 | Perkin Elmer | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo animal studies
Plasmids pCD68-CARD16, pCD68-CARD17 and pCD68-CARD18 were generated by replacing the sequence encoding chloramphenicol acetyltransferase in the plasmid pCAT-Basic, which contains the promoter of the gene encoding human CD68 and the macrophage-specific IVS-1 enhancer, with the sequence encoding human CARD16, CARD17 or CARD18 fused to EGFP, respectively and flanking of that cassette with AatII restriction sites.23,27,29,51 The AatII fragment was excised and purified and B6.Tg(CD68-CARD16) [CARD16TG], B6.Tg(CD68-CARD17) [CARD17TG] and B6.Tg(CD68-CARD18) [CARD18TG] mice were generated by pronuclear injection of that fragment into C57BL/6J embryos by the Northwestern University Transgenic and Targeted Mutagenesis Laboratory. Mice were genotyped for the presence of EGFP (Fwd: 5′-cctacggcgtgcagtgcttcagc-3′; Rev: 5′-cggcgagctgcacgctgcgtcctc-3′) and by TaqMan gene expression assay (Invitrogen) from mRNA isolated from peripheral blood or by flow cytometry (Northern Lights, Cytek), gating on CD115+ cells. Two lines were initially analyzed, and subsequently a single line was used for most experiments. C57BL/6J (#000066) were obtained from the Jackson Laboratories and bred in house. Casp1−/− mice were described previously.9 All mice were bred and housed in specific pathogen-free animal facilities at Northwestern University and Cedars Sinai. Mice were group housed and allowed water and normal diet chow ad libitum. Light on/off cycles were controlled in a 12h/12h shift, while the temperature of the animal room was kept steady at 22 ± 2°C. All experiments used age- and sex-matched, randomly assigned mice 12-16 weeks of age and were conducted according to procedures approved by the Northwestern University and Cedars Sinai Committees on Use and Care of Animals and adherence to the NIH Guide for the Care and Use of Laboratory Animals. WT mice were compared to CARD16TG, CARD17TG and CARD18TG mice, using Casp1−/− mice as a control. Age and sex matched animals were randomly assigned to groups from multiple cages and all animals with positive genotyping were included in the study. Male and female WT: n=6, CARD16: n=4, CARD17: n=5, CARD18: n=5, Casp1 KO: n=3 (MSU-induced peritonitis); Male and female WT: n=5, CARD16: n=5, CARD17: n=4, CARD18: n=5, Casp1 KO: n=4 (MSU air pouch IVIS imaging) and Male and female WT: n=7, CARD16: n=6, CARD17: n=7, CARD18: n=6, Casp1 KO: n=4 (MSU air pouch neutrophil infiltration and cytokine analysis). The investigators were not blinded to the genotype of mice.
Human studies
Existing data (GEO accession GSE36700)43,44 from an Affymetrix Human Genome U133 Plus 2.0 Array (HG-U133_Plus_2) consisting of synovial biopsies of untreated patients with Rheumatoid Arthritis (RA, n=7), Systemic Lupus Erythematosus (SLE, n=4), Osteoarthritis (OA, n=5), Psoriatic Arthritis (SA, n=4) and microcrystalline arthritis (MIC, n=5) were mined for CARD16 (Affymetrix Probe Set ID: 1552701_a_at, 1552703_s_at) and CARD18 (Affymetrix Probe Set ID: 231733_at) expression. No demographic patient data were available in the original study. Informed consent was obtained from individuals for the work described.43,44
Cell lines
Authenticated THP-1 cells were obtained from the American Type Culture Collection (ATCC, #TIB-202) and maintained in RPMI media supplemented with 10% Fetal bovine serum (Gibco), 1 mM sodium pyruvate, 0.05 mM 2-mercaptoethanol, 1 mM HEPES buffer and 1% penicillin and streptomycin. THP-1 cells were originally derived from the peripheral blood of a 1-year-old human male with acute monocytic leukemia. Mouse bone marrow cells were flushed from femurs of male and female mice, differentiated into bone marrow-derived macrophages (BMDM) with L929 cell-conditioned medium (25% v/v; ATCC, #CCL-1), supplemented with 10% heat-inactivated FBS (Gibco) and were analyzed after 6-7 days. L929 cells were originally derived from a male C3H/An mouse. Authenticated human embryonic kidney (HEK) 293 cells (CRL-3216, ATCC) and Lenti-X HEK293 cells (632180, Takara Bio) were maintained in DMEM high glucose media containing 10% FBS, 100 IU mL−1 penicillin and 1 mg mL−1 streptomycin. HEK293 cells were originally derived from a female fetus. Cells were grown at 37°C in 5% CO2 and were routinely tested for mycoplasma contamination (MycoAlert, Lonza).
METHOD DETAILS
MSU-induced peritonitis
12-16 week old mice had their abdomen shaved under anesthesia and were randomly selected for i.p. injection with PBS or MSU crystals (3 mg) MSU.27,30 After 4h, mice were i.p. injected with XenoLight Rediject Inflammation probe (200 mg kg−1, #760536, PerkinElmer) or luminol (200 mg kg−1) from a 50 mg mL−1 stock solution of luminol sodium salt (#A4685, Sigma), dissolved in sterile PBS and stored at −20°C and in vivo bioluminescence was captured by imaging (IVIS Spectrum, PerkinElmer) 10 min post injection with a 5 min exposure on anesthetized mice. Images were quantified with Living Image software (PerkinElmer).
MSU air pouch
To generate subcutaneous air pouches,27,30,53 anesthetized 1-16-weeks old mice were shaven and injected with 5 mL and 3 mL of sterile air into the subcutaneous tissue of the back on day 0 and 3, respectively. MSU crystals (3 mg) in 1 mL of sterile PBS or 1 mL PBS were randomly injected into the pouch 5 days after the first air injection and IVIS imaging for MPO activity was performed as described above 4h post-injection. For pouch lavage a separate group of mice were treated as above and euthanized mice were injected with 1 mL of PBS containing 5 mM EDTA for analysis of the cellular infiltrate by flow cytometry and cytokines by ELISA.
Gene expression and gene knock out
CARD16, CARD17 and CARD18 cDNAs were synthesized as gBlocks (IDT) and cloned into modified pcDNA3 and pLex expression plasmids in frame with EGFP. For FRET assay, caspase-1CARD (aa1-112) in pcDNA395 was fused at its C-terminal end in frame with either mNeonGreen or mScarlet-I synthetic DNA fragments and a synthetic mNeonGreen-mScarlet-I fusion protein (GeneWiz) as FRET control.96,97 All expression constructs were sequence verified. To express CARD16, CARD17 and CARD18 in THP-1 cells, recombinant lentivirus was produced in Lenti-X HEK293 cells by Xfect-based transfection (Takara Bio) using modified pLEX expression plasmids with the viral packaging plasmids pMD2.G (a gift from Didier Trono, Addgene plasmid # 12259, http://n2t.net/addgene:12259) and psPAX2 (a gift from Didier Trono, Addgene plasmid # 12260, http://n2t.net/addgene:12260), followed by 0.45 μm filtration of virus-containing culture supernatants. THP-1 cells were transduced with lentiviral particles in the presence of polybrene (0.45 μg mL−1) and MISSION ExpressMag magnetic beads (Millipore-Sigma, SHM03). Cells were Puromycin-selected (1 μg mL−1, Santa Cruz Biotechnology) 48h post infection for 2 weeks, sorted by flow cytometry and CARD16, CARD17 and CARD18 expression was verified by immunoblot.
CARD16KO, CARD17KO and CARD18KO THP-1 cells were generated by CRISPR/Cas9. gRNAs were designed with E-CRISP (http://www.e-crisp.org/E-CRISP) and CHOPCHOP (https://chopchop.cbu.uib.no) and cloned into plentiCRISPRv1 (Addgene Plasmid 49,535).98 CARD16 gRNA: 5′-agggtaggagagaaaagccatgg-3′; CARD17 gRNA: 5′-aaagcagtttatccgttcagtgg-3′; CARD18 gRNA: 5′-cctcaacttgcctcaaagatgg-3′; Ctrl gRNA: 5′-acggaggctaagcgtcgcaa-3′. Recombinant lentivirus was produced in Lenti-X HEK293 cells as described above. Single cell clones were sequence analyzed following PCR amplification of the targeting sequence: CARD16 (5′-tgggtgtttcccaatatgtgta-3′, 5′-tgccttttctttctaaagcctg-3′), CARD17 (5′-aatgaggttgccttctggtg-3′, 5′-tcgggccttatccataactg-3′) and CARD18 (5′-ggctcgagtcttgattgacc-3′, 5′-ggaatggagtctgaggctaaaa-3′). CASP1KO THP-1 cells were earlier generated by CRISPR/Cas9 and ASCKD cells by shRNA92,93 and CRISPR/Cas9 generated ASCKO THP-1 cells were obtained from Invivogen (thp-koascz).
Cell stimulation
Human monocytic THP-1 cells were seeded in 1% FBS and antibiotic free media at a density of 1×106 cells in a 6-well plate. THP-1 cells were primed with Pam3CSK4 (1 μg mL−1, InvivoGen, tlrl-pms) or ultrapure LPS from E. coli, Serotype 0111:B4 (InvivoGen, tlrl-3pelps) for 4h followed by activation with nigericin (5 μM, InvivoGen, tlrl-nig) treatment for 30 min, MSU crystals (180 μg mL−1) for 6h, ATP (5 mM, Sigma-Aldrich, A6419) for 30 min, silica (200 μg mL−1, InvivoGen, tlrl-sio-2) for 4h, CPP (50 μg mL−1 InvivoGen, tlrl-cppd) for 4h. Ultrapure flagellin from Salmonella enterica subsp. enterica serovar Typhimurium (0.5 mg mL−1, InvivoGen, tlrl-epstfla), FSL-1 (0.2 mg mL−1, InvivoGen, tlrl-fsl), Bacillus anthracis Lethal Factor (0.5 mg mL−1) and Protective Antigen (0.5 μg mL−1) (Lethal toxin, List Labs, 104) and poly(dA:dT) (1 μg mL−1, Invivogen, tlrl-patn) were diluted in OptiMEM (Invitrogen), mixed with 3 μL lipofectamine (Invitrogen), incubated for 40 min and added to THP-1 cells in 2 mL Opti-MEM for 5-6h. BMDM were seeded in antibiotic-free media at a density of 1 × 106 cells in 6-well plates and incubated overnight before treatment or transfection as above, except that DOTAP (Biontex) was used for transfection. For CARD9 responses, BMDM were primed with recombinant murine IFN-γ (25U, Peprotech, 315–05) 12h and activated with dispersible Whole Glucan Particles (WGP, 100 μg mL−1, InvivoGen, tlrl-wgp) for 24h.85 Cell culture supernatants were harvested for enzyme-linked immunosorbent assays (ELISAs) or immunoblot after stimulation.
ELISA
Cells were seeded into 6 well plates (106 cells per well), treated as indicated and cell culture supernatants or lavage fluids were analyzed for IL-1β (Invitrogen), IL-18 (R&D), TNF (Invitrogen, R&D Systems), CXCL1 (R&D Systems), CXCL2 (R&D Systems), NLRP3 (LSBio), Caspase-1 (Adipogen) secretion by ELISA as per the manufacturer’s instructions.
Lactate dehydrogenase (LDH) cytotoxicity assay
LDH activity was determined using the LDH Cytotoxicity Assay (CyQUANT, Invitrogen) in freshly collected culture supernatants. Cytotoxicity was defined as a percentage of released LDH compared to total LDH activity upon cell lysis with 1% Triton X-100.
IncuCyte live cell analysis - Cytotox Red cell death assay
8 × 104 GFP, GFP-CARD16, GFP-CARD17 and GFP-CARD18 THP-1 cells/well were seeded in a 96-well flat bottom plate and incubated o/n, primed with Pam3CSK4 (1 μg mL−1, 3h) in the presence of Cytotox Red (250 nM, Essen Bioscience) prior activation with MSU crystals (100 μg mL−1) and imaging over 16h (IncuCyte SX5 Live-Cell Analysis System). 9 images per well were taken every 30 min using an acquisition time of 100 ms (green) and 400 ms (red) with a 20x objective. To analyze cell death, red and green fluorescent cells were masked, counted (IncuCyte) and dead cell count (red) over green cell count (total cells) determined.
Proximity ligation assay (PLA)
PLA (Duolink PLA, Millipore Sigma) was performed as per the manufacturer’s instructions. Briefly, THP-1 cells were seeded into 12 well plates (3 × 105 cells/well) on coverslips, differentiated with phorbol 12-myristate 13-acetate (50 nM, 16h), washed in PBS and then rested for 48h. After priming with Pam3CSK4 (1 μg mL−1, 3h) and activation with nigericin (10 μM, 15 min), cells were washed and fixed with 4% paraformaldehyde for 10 min at room temperature, permeabilized with ice-cold 0.1% Triton X-100 for 10 min at room temperature and washed with PBS. Unless specified otherwise, all incubations were performed at 37°C in a humidified chamber. Cells were blocked with the Duolink Blocking Solution for 1h, incubated with mouse/rabbit combinations of primary antibodies, rabbit polyclonal anti-ASC (5 μg mL−1, AL177; AG-25B-0006-C100) and mouse monoclonal anti-caspase-1 (8 μg mL−1, R&D Systems, 1021323, MAB62152-100) for 12h at 4°C in a humidified chamber. Coverslips were then washed and incubated with PLUS and MINUS PLA probes, washed, incubated with the ligase for 30 min, washed and incubated with the polymerase for 100 min. After washing, samples were mounted on slides using the Duolink In Situ Mounting Medium with DAPI, imaged by fluorescence microscopy (Nikon Eclipse TE2000-E2) and the data processed and analyzed using the NIS-Elements AR with ai (Nikon) and Fiji (Image J) software.94
Caspase activity assay
THP-1 cells were seeded into black, flat bottom 96 well plates at a density of 4 × 104 cells per well, primed with Pam3CSK4 (1 μg mL−1, 4h), followed by activation with MSU crystals (90 μg mL−1, 6h). The fluorogenic caspase-1 substrate (Ac-YVAD-AFC, 100 μM, Enzo Life Sciences) was added at the time of activation. Cells were lysed (200 mM NaCl, 50 mM Hepes, pH 8.0, 50 mM KCl, 100 μg mL−1 digitonin, 10 mM DTT and 100 μM Ac-YVAD-AFC). To induce apoptosis, THP-1 cells were treated with Staurosporine (1 μg mL−1, 60 min, Sigma Aldrich), lysed as above and incubated with the fluorogenic caspase-3 substrate Ac-DEVD-AFC (20 μM, Enzo Life Sciences) or the caspase-9 fluorogenic substrate Ac-LEHD-AFC (100 μM, Enzo Life Sciences). Ac-YVAD/DEVD/LEHD-AFC fluorescence was measured at 400 nm excitation and 505 nm emission.
Immunoblotting
THP-1 and BMDM were washed once with ice cold PBS and lysed in RIPA or Laemmli buffers. Proteins were separated on polyacrylamide gels and transferred onto PVDF membrane (Millipore). Membranes were blocked (5% non-fat dry milk, 0.1M Tris-buffered saline, pH 7.4, 0.1 % Tween 20) for 1h at room temperature followed by incubation with primary antibodies for 12-16h at 4°C as indicated with rabbit monoclonal anti-human caspase-1 (Cell Signaling Technology, D7F10), rabbit monoclonal anti-cleaved human caspase-1 (Cell Signaling Technology, D57A2), mouse monoclonal anti-mouse caspase-1 (p20) (AdipoGen, Casper-1, AG-20B-0042), mouse monoclonal anti-mouse caspase-1 (p10) (AdipoGen, Casper-2, AG-20B-0044-C100), rabbit monoclonal anti-mouse gasderminD (Abcam, Ab209845), rabbit monoclonal anti-cleaved human gasdermin D (Cell Signaling Technology, E7H9G), rabbit monoclonal anti-gasdermin D (Cell Signaling Technology, L60), rabbit monoclonal anti-cleaved mouse gasdermin D (Abcam, Ab209845), mouse monoclonal anti-NLRP3 (Adipogen, Cryo-2), rabbit polyclonal anti-NLRP3 (Cell Signaling Technology, D4D8T), mouse monoclonal anti-GFP (Santa Cruz Biotechnology, B-2, SC-9996), rabbit monoclonal anti-GFP (Cell Signaling Technology, D5.1, 2956S), alpaca single-domain GFP Selector beads (NanoTag Biotechnologies, N0310), rabbit polyclonal anti-ASC (AdipoGen, AL177; AG-25B-0006-C100), mouse monoclonal anti-β-tubulin (DSHB, AA12.1), rabbit polyclonal anti-human total IL-1β (Santa Cruz Biotechnology, SC7884), rabbit monoclonal anti-human cleaved IL-1β (Cell Signaling Technology, D3A3Z), rabbit monoclonal anti-mouse cleaved IL-1β (Cell Signaling Technology, 52718S), mouse monoclonal anti-mouse mature IL-1β (Cell Signaling Technology, 12242S), rabbit monoclonal anti-cleaved PARP (Cell Signaling Technology, #5625), rabbit polyclonal anti-caspase-9 (Cell Signaling Technology, #9504), rabbit polyclonal anti-caspase-8 (Cell Signaling Technology, #4927). rabbit monoclonal anti-caspase-3 (Cell Signaling Technology, #14220) and rabbit polyclonal cleaved anti-caspase-3 (Cell Signaling Technology, #9661). Membranes were washed and incubated with HRP-conjugated secondary antibodies, including goat anti-Rabbit IgG (H+L) HRP (Invitrogen, 31460), goat anti-Mouse IgG (H+L) HRP (Invitrogen, 32430) or donkey anti-Goat IgG (H+L) HRP (Invitrogen, A15999) when necessary for 1h at room temperature and proteins were visualized by ECL detection (Super Signal West Femto, ThermoFisher Scientific) and digital image acquisition (Thermo iBright and Ultralum Omega 14vR).
Co-immunoprecipitation
Cells were lysed in lysis buffer (10 mM Tris-HCl, pH 7.6, 5 mM EDTA, 50 mM sodium chloride, 30 mM sodium pyrophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail (Millipore-Sigma)) and lysates were cleared by centrifugation at 12,500 x g for 30 min.99 Total cell lysates were incubated with 1 μg of primary antibody as indicated on a rocking platform for 2h at 4°C. Protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology, sc-2003) were then added to the samples and incubated for 12h on a rocking platform at 4°C. Beads were collected by centrifugation or magnets, washed three times with the lysis buffer and bound proteins were eluted in sample buffer and analyzed by SDS/PAGE and immunoblotting.
Active caspase pulldown
BMDM were seeded at a density of 5 × 105 cells per 60 mm dish, primed with Pam3CSK4 (1 μg mL−1, 4h) and activated with nigericin (5 μM) for 3h. The biotinylated cell-permeable pan caspase activity probe biotin-VAD-FMK (10 μM) was added to cells during activation.73,74 Cells were directly lysed (1% NP-40, 50 mM Tris pH 7.6, 50 mM NaCl, protease inhibitors) in dish and incubated on ice for 15 min. Lysates were cleared by centrifugation and cleared total cell lysates were incubated with Neutravidin beads under gentle rotation at 4°C for 12-16h. Neutravidin unbound fractions were collected, beads were washed 3 times with lysis buffer and resuspended in sample buffer, boiled at 95°C for 10 min and analyzed by SDS/PAGE and immunoblotting.
Flow cytometry
For Caspase-1 fluorochrome inhibitor of caspases (FLICA), cells were pretreated with biotin-conjugated YVAD-CMK (10 μM) (Anaspec) for 1h and then primed with Pam3CSK4 (1 μg mL−1) for 30 min prior to treatment with nigericin (5 μM) for 20 min or MSU crystals (180 μg ml−1) for 3h. Cells were fixed and permeabilized using Cytofix/Cytoperm and Permeabilization Solution (BD Biosciences). AlexaFluor 647-conjugated streptavidin (Invitrogen) was used to quantify active caspase-1 by flow cytometry (BD LSR II, BD Biosciences). For MHC staining, BMDM were harvested after 7d of differentiation using ice-cold PBS 0.5 mM EDTA. BMDM, freshly isolated spleen and peritoneal lavage cells were stained with the fixable viability dye eFluor506 (1:2000,10 min, eBiosciences) in HBS on ice, washed twice with MACS buffer, incubated with FcBlock (1:50, 10 min, BD Biosciences) and were stained in MACS buffer (Miltenyi)/Brilliant stain buffer (BD Biosciences) with the specific antibody panel for BMDM (F4/80 PE-Cy7, MHC-I APC-Fire750, MHC-II BV421), peritoneal lavage (CD45 BV570, Ly6G BV785, SigF PE-CF594, CD11b BB700, CD11c AF700, F4/80 PE-Cy7, CD115 APC, Ly6c APC-Cy7, MHC-I BV650, MHC-II BV421), and spleen (CD45 BV570, CD3 BV785, CD19 BV785, Ly6G BV785, NK1.1 BV785, SigF PE-CF594, CD11b BB700, CD11c APC, F4/80 PE-Cy7, Ly6c APC-Cy7, MHC-I BV650, MHC-II BV421) in a total volume of 100 μl for 30 min. Cells were washed twice with ice-cold MACS buffer. To detect GFP-COP expression, mice were anesthetized and whole blood was collected into EDTA containing tubes. The blood cells were centrifuged and washed once with PBS, incubated with FcBlock (1:50, 10 min, BD Biosciences) for 10 min and stained with a CD115-APC conjugated antibody (e-Biosciences) for 20 min in dark. All samples were acquired on a Northern Lights 3000 V/B/R spectral flow cytometer (Cytek) and data were analyzed with FlowJo v10 (BD Biosciences).
Spectral FRET assay
HEK293 cells were transiently transfected with Caspase-1CARD-mNeonGreen, Caspase-1CARD-mScarlet-I, CARD16, CARD17 and CARD18 and a mNeonGreen-mScarlet-I fusion protein control in optical 96-well plates as indicated. 48h post transfection, emission scans were collected from 490–750 nm in 2 nm increments using a 470 nm excitation wavelength (Varioskan LUX, ThermoFisher Scientific) using a bottom fluorescence scanning module and an environmental chamber. mNeonGreen (Ex: 506 nm, Em: 517 nm), mScarlet-I (Ex: 569 nm, Em: 593 nm). Data were collected and processed with SkanIt Application software package (ThermoFisher Scientific) and regression analysis and curve fitting was performed with Prism 9 (GraphPad).
ASC crosslinking
4×106 BMDM or THP-1 cells were seeded in 60 mmdishes and subjected to cross-linking.27,29,100 Cells were either left untreated or primed with Pam3CSK4 (1 μg mL−1) for 4h and treated with nigericin (5 μM) for 45 min and culture supernatants were removed, cells rinsed with ice-cold PBS and lysed (20 mM Hepes pH 7.4, 100 mM NaCl, 1% NP-40, 1 mM sodium orthovanadate, supplemented with protease inhibitors) and further lysed by shearing. Cleared lysates were stored for immunoblot analysis and the insoluble pellets were resuspended in 500 μL PBS, supplemented with 2 mM disuccinimidyl suberate (DSS, Pierce) and incubated with rotation at room temperature for 30 min. Samples were centrifuged at 5,000 rpm for 10 min at 4°C and the cross-linked pellets were resuspended in 50 μL Laemmli sample buffer and analyzed by SDS/PAGE and immunoblot.
Quantitative real time PCR
Total RNA was isolated from cells using the E.Z.N.A. total RNA isolation Kit (Omega Bio-tek), as per the manufacture’s protocol. RNA samples (1 μg) were subjected to RNase free DNase I digestion and reverse-transcribed (Verso cDNA Synthesis Kit, ThermoFisher Scientific). Taqman multiplexed gene expression analysis was performed on a QuantStudio 3 (ThermoFisher Scientific) and presented as relative expression compared to ACTB, using FAM and VIC labelled exon-spanning primers (Invitrogen).
MSU crystal preparation
1.68 gm of uric acid was added in 25 mM sodium hydroxide solution and left for mixing on a rotator for 18-20h at room temperature. MSU crystals were harvested by filtration through a 0.22-μM filter, washed 3 times with sterile ice cold PBS, followed by one ethanol (70% v/v) wash.27,30,101 MSU crystals were air dried and UV sterilized.
QUANTIFICATION AND STATISTICAL ANALYSIS
All in vitro experiments have been repeated at least 3 times and the in vivo experiments 2 times and graphs were prepared in Prism 9 (GraphPad) and represent the mean ± SD. A standard parametric two-tailed unpaired t-test was used for statistical analysis of two groups with all data points showing a normal distribution. Values of P < 0.05 were considered significant and marked by an asterisk.
Supplementary Material
Highlights.
Caspase recruitment domain (CARD)-only proteins (COPs) regulate caspase-1 recruitment to the inflammasome
COPs inhibit caspase-1 activation
COPs inhibit caspase-1-mediated IL-1β and IL-18 release and prevent pyroptosis
COP transgenic mice are protected from inflammatory disease
ACKNOWLEDGMENTS
psPAX2 and pMD.2G were a gift from Didier Trono, plentiCRISPRv1 was a gift from Feng Zhang, and Casp1−/− mice were a gift from Vishva Dixit (Genentech). This work was supported by the National Institutes of Health (AI099009 and AR064349 to C.S., AI134030, AI140702, AI165797 and AI120625 to C.S. and A.D.) and the American Heart Association (grant 834502 to J.C.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research. We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112265.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Mantovani A, Dinarello CA, Molgora M, and Garlanda C (2019). Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 50, 778–795. 10.1016/j.immuni.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dinarello CA (2018). Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev 281, 8–27. 10.1111/imr.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broz P, and Dixit VM (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16, 407–420. 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 4.Guo H, Callaway JB, and Ting JPY (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med 21, 677–687. 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kesavardhana S, Malireddi RKS, and Kanneganti TD (2020). Caspases in cell death, inflammation, and pyroptosis. Annu. Rev. Immunol 38, 567–595. 10.1146/annurev-immunol-073119-095439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, and Flavell RA (1995). Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267, 2000–2003. 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 7.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. (1995). Mice deficient in IL-1b-converting enzyme are defective in production of mature IL-1b and resistant to endotoxic shock. Cell 80, 401–411. [DOI] [PubMed] [Google Scholar]
- 8.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 25, 1285–1298. 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 10.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 11.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, and Dueber EC (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 113, 7858–7863. 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X, He WT, Hu L, Li J, Fang Y, Wang X, Xu X, Wang Z, Huang K, and Han J (2016). Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26, 1007–1020. 10.1038/cr.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, and Dubyak GR (2016). Active caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J. Immunol 197, 1353–1367. 10.4049/jimmunol.1600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, and Hiller S (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778. 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, and Kagan JC (2018). The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48, 35–44.e6. 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, and Broz P (2018). The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur. J. Immunol 48, 584–592. 10.1002/eji.201747404. [DOI] [PubMed] [Google Scholar]
- 19.Salvesen GS, and Dixit VM (1999). Caspase activation: the induced-proximity model. Proc. Natl. Acad. Sci. USA 96, 10964–10967. 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schröder GF, Fitzgerald KA, Wu H, and Egelman EH (2014). Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206. 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, and Chen ZJ (2014). Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156, 1207–1222. 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Fu TM, Lu A, Witt K, Ruan J, Shen C, and Wu H (2018). Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc. Natl. Acad. Sci. USA 115, 10845–10852. 10.1073/pnas.1810524115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khare S, Ratsimandresy RA, de Almeida L, Cuda CM, Rellick SL, Misharin AV, Wallin MC, Gangopadhyay A, Forte E, Gottwein E, et al. (2014). The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat. Immunol 15, 343–353. chu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorfleutner A, Bryan NB, Talbott SJ, Funya KN, Rellick SL, Reed JC, Shi X, Rojanasakul Y, Flynn DC, and Stehlik C (2007). Cellular pyrin domain-only protein 2 is a candidate regulator of inflammasome activation. Infect. Immun 75, 1484–1492. 10.1128/IAI.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bedoya F, Sandler LL, and Harton JA (2007). Pyrin-only protein 2 modulates NF-kappaB and disrupts ASC:CLR interactions. J. Immunol 178, 3837–3845. [DOI] [PubMed] [Google Scholar]
- 26.Stehlik C, Krajewska M, Welsh K, Krajewski S, Godzik A, and Reed JC (2003). The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated NF-kB and pro-Caspase-1 regulation. Biochem. J 373, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratsimandresy RA, Chu LH, Khare S, de Almeida L, Gangopadhyay A, Indramohan M, Misharin AV, Greaves DR, Perlman H, Dorfleutner A, and Stehlik C (2017). The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat. Commun 8, 15556. 10.1038/ncomms15556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Periasamy S, Porter KA, Atianand MK, T Le H, Earley S, Duffy EB, Haller MC, Chin H, and Harton JA (2017). Pyrin-only protein 2 limits inflammation but improves protection against bacteria. Nat. Commun 8, 15564. 10.1038/ncomms15564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Almeida L, Khare S, Misharin AV, Patel R, Ratsimandresy RA, Wallin MC, Perlman H, Greaves DR, Hoffman HM, Dorfleutner A, and Stehlik C (2015). The PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 43, 264–276. 10.1016/j.immuni.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Almeida L, Devi S, Indramohan M, Huang Q-Q, Ratsimandresy RA, Pope RM, Dorfleutner A, and Stehlik C (2022). POP1 inhibits MSU-induced inflammasome activation and ameliorates Gout. Front. Immunol 13, 912069–912113. 10.3389/fimmu.2022.912069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SH, Stehlik C, and Reed JC (2001). Cop, a caspase recruitment domain-containing protein and inhibitor of caspase-1 activation processing. J. Biol. Chem 276, 34495–34500. 10.1074/jbc.M101415200. [DOI] [PubMed] [Google Scholar]
- 32.Lamkanfi M, Denecker G, Kalai M, D’Hondt K, Meeus A, Declercq W, Saelens X, and Vandenabeele P (2001). INCA, a novel human caspase recruitment domain protein that inhibits interleukin-1beta generation. J. Biol. Chem 279, 51729–51738. [DOI] [PubMed] [Google Scholar]
- 33.Druilhe A, Srinivasula SM, Razmara M, Ahmad M, and Alnemri ES (2001). Regulation of IL-1beta generation by Pseudo-ICE and ICEBERG, two dominant negative caspase recruitment domain proteins. Cell Death Differ. 8, 649–657. 10.1038/sj.cdd.4400881. [DOI] [PubMed] [Google Scholar]
- 34.Humke EW, Shriver SK, Starovasnik MA, Fairbrother WJ, and Dixit VM (2000). ICEBERG: a novel inhibitor of interleukin-1beta generation. Cell 103, 99–111. 10.1016/s0092-8674(00)00108-2. [DOI] [PubMed] [Google Scholar]
- 35.Atianand MK, Fuchs T, and Harton JA (2011). Recent evolution of the NF-kappaB and inflammasome regulating protein POP2 in primates. BMC Evol. Biol 11, 56. 10.1186/1471-2148-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stehlik C, and Dorfleutner A (2007). COPs and POPs: modulators of inflammasome activity. J. Immunol 179, 7993–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reed JC, Doctor K, Rojas A, Zapata JM, Stehlik C, Fiorentino L, Damiano J, Roth W, Matsuzawa SI, Newman R, et al. (2003). Comparative analysis of apoptosis and inflammation genes of mice and humans. Genome Res. 13, 1376–1388. 10.1101/gr.1053803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinon F, Pétrilli V, Mayor A, Tardivel A, and Tschopp J (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 39.Klüuck V, Liu R, and Joosten LAB (2021). The role of interleukin-1 family members in hyperuricemia and gout. Joint Bone Spine 88, 105092. 10.1016/j.jbspin.2020.105092. [DOI] [PubMed] [Google Scholar]
- 40.So A, De Smedt T, Revaz S, and Tschopp J (2007). A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther 9, R28. 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.So A, Dumusc A, and Nasi S (2018). The role of IL-1 in gout: from bench to bedside. Rheumatology 57, i12–i19. 10.1093/rheumatology/kex449. [DOI] [PubMed] [Google Scholar]
- 42.Desai J, Steiger S, and Anders HJ (2017). Molecular pathophysiology of gout. Trends Mol. Med 23, 756–768. 10.1016/j.molmed.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 43.Lauwerys BR, Hernández-Lobato D, Gramme P, Ducreux J, Dessy A, Focant I, Ambroise J, Bearzatto B, Nzeusseu Toukap A, Van den Eynde BJ, et al. (2015). Heterogeneity of synovial molecular patterns in patients with arthritis. PLoS One 10, e0122104. 10.1371/journal.pone.0122104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nzeusseu Toukap A, Galant C, Theate I, Maudoux AL, Lories RJU, Houssiau FA, and Lauwerys BR (2007). Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 56, 1579–1588. 10.1002/art.22578. [DOI] [PubMed] [Google Scholar]
- 45.Jin C, Frayssinet P, Pelker R, Cwirka D, Hu B, Vignery A, Eisenbarth SC, and Flavell RA (2011). NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc. Natl. Acad. Sci. USA 108, 14867–14872. 10.1073/pnas.1111101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pazár B, Ea HK, Narayan S, Kolly L, Bagnoud N, Chobaz V, Roger T, Lioté F, So A, and Busso N (2011). Basic calcium phosphate crystals induce monocyte/macrophage IL-1beta secretion through the NLRP3 inflammasome in vitro. J. Immunol 186, 2495–2502. 10.4049/jimmunol.1001284. [DOI] [PubMed] [Google Scholar]
- 47.Kono H, Chen CJ, Ontiveros F, and Rock KL (2010). Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Invest 120, 1939–1949. 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rock KL, Kataoka H, and Lai JJ (2013). Uric acid as a danger signal in gout and its comorbidities. Nat. Rev. Rheumatol 9, 13–23. 10.1038/nrrheum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Badii M, Gaal OI, Cleophas MC, Klück V, Davar R, Habibi E, Keating ST, Novakovic B, Helsen MM, Dalbeth N, et al. (2021). Urate-induced epigenetic modifications in myeloid cells. Arthritis Res. Ther 23, 202. 10.1186/s13075-021-02580-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cobo I, Cheng A, Murillo-Saich J, Coras R, Torres A, Abe Y, Lana AJ, Schlachetzki J, Liu-Bryan R, Terkeltaub R, et al. (2022). Monosodium urate crystals regulate a unique JNK-dependent macrophage metabolic and inflammatory response. Cell Rep. 38, 110489. 10.1016/j.celrep.2022.110489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iqbal AJ, McNeill E, Kapellos TS, Regan-Komito D, Norman S, Burd S, Smart N, Machemer DEW, Stylianou E, McShane H, et al. (2014). Human CD68 promoter GFP transgenic mice allow analysis of monocyte to macrophage differentiation in vivo. Blood 124, e33–e44. 10.1182/blood-2014-04-568691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gough PJ, Gordon S, and Greaves DR (2001). The use of human CD68 transcriptional regulatory sequences to direct high-level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology 103, 351–361. 10.1046/j.1365-2567.2001.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edwards JC, Sedgwick AD, and Willoughby DA (1981). The formation of a structure with the features of synovial lining by subcutaneous injection of air: an in vivo tissue culture system. J. Pathol 134, 147–156. [DOI] [PubMed] [Google Scholar]
- 54.Gordon TP, Kowanko IC, James M, and Roberts-Thomson PJ (1985). Monosodium urate crystal-induced prostaglandin synthesis in the rat subcutaneous air pouch. Clin. Exp. Rheumatol 3, 291–296. [PubMed] [Google Scholar]
- 55.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, Beck PL, Muruve DA, and Kubes P (2010). Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330, 362–366. 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 56.García-Ramallo E, Marques T, Prats N, Beleta J, Kunkel SL, and Godessart N (2002). Resident cell chemokine expression serves as the major mechanism for leukocyte recruitment during local inflammation. J. Immunol 169, 6467–6473. 10.4049/jimmunol.169.11.6467. [DOI] [PubMed] [Google Scholar]
- 57.De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, and Hogg N (2013). Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 121, 4930–4937. 10.1182/blood-2013-02-486217. [DOI] [PubMed] [Google Scholar]
- 58.Torres R, Macdonald L, Croll SD, Reinhardt J, Dore A, Stevens S, Hylton DM, Rudge JS, Liu-Bryan R, Terkeltaub RA, et al. (2009). Hyperalgesia, synovitis and multiple biomarkers of inflammation are suppressed by interleukin 1 inhibition in a novel animal model of gouty arthritis. Ann. Rheum. Dis 68, 1602–1608. 10.1136/ard.2009.109355. [DOI] [PubMed] [Google Scholar]
- 59.Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, Barberà-Cremades M, Yagüe J, Ruiz-Ortiz E, Antón J, et al. (2014). The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol 15, 738–748. 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 60.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al-Amoudi A, et al. (2014). The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol 15, 727–737. 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen KW, Bezbradica JS, Groß CJ, Wall AA, Sweet MJ, Stow JL, and Schroder K (2016). The murine neutrophil NLRP3 inflammasome is activated by soluble but not particulate or crystalline agonists. Eur. J. Immunol 46, 1004–1010. 10.1002/eji.201545943. [DOI] [PubMed] [Google Scholar]
- 62.Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, and MacMicking JD (2012). GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336, 481–485. 10.1126/science.1217141. [DOI] [PubMed] [Google Scholar]
- 63.Keller M, Rüegg A, Werner S, and Beer HD (2008). Active caspase-1 is a regulator of unconventional protein secretion. Cell 132, 818–831. 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 64.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, and Alnemri ES (1998). Autoactivation of procaspase-9 by apaf-1-mediated oligomerization. Mol. Cell 1, 949–957. 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 65.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, and Wang X (1997). Cytochrome c and dATP-dependent formation of apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–89. 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez J, and Lazebnik Y (1999). Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 13, 3179–3184. 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zou H, Li Y, Liu X, and Wang X (1999). An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem 274, 11549–11556. 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 68.Zou H, Henzel WJ, Liu X, Lutschg A, and Wang X (1997). Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90, 405–413. 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 69.Stepczynska A, Lauber K, Engels IH, Janssen O, Kabelitz D, Wesselborg S, and Schulze-Osthoff K (2001). Staurosporine and conventional anticancer drugs induce overlapping, yet distinct pathways of apoptosis and caspase activation. Oncogene 20, 1193–1202. 10.1038/sj.onc.1204221. [DOI] [PubMed] [Google Scholar]
- 70.Boatright KM, and Salvesen GS (2003). Mechanisms of caspase activation. Curr. Opin. Cell Biol 15, 725–731. 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 71.Chang DW, Ditsworth D, Liu H, Srinivasula SM, Alnemri ES, and Yang X (2003). Oligomerization is a general mechanism for the activation of apoptosis initiator and inflammatory procaspase. J. Biol. Chem 278, 16466–16469. 10.1074/jbc.C300089200. [DOI] [PubMed] [Google Scholar]
- 72.Shi Y (2004). Caspase activation: revisiting the induced proximity model. Cell 117, 855–858. 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 73.Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, Gomez GA, Holley CL, Bierschenk D, Stacey KJ, et al. (2018). Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med 215, 827–840. 10.1084/jem.20172222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faleiro L, Kobayashi R, Fearnhead H, and Lazebnik Y (1997). Multiple species of CPP32 and Mch2 are the major active caspases present in apoptotic cells. EMBO J. 16, 2271–2281. 10.1093/emboj/16.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nickerson K, Sisk TJ, Inohara N, Yee CS, Kennell J, Cho MC, Yannie PJ 2nd, Nunez G, and Chang CH (2001). Dendritic cell-specific MHC class II transactivator contains a caspase recruitment domain that confers potent transactivation activity. J. Biol. Chem 276, 19089–19093. 10.1074/jbc.M101295200. [DOI] [PubMed] [Google Scholar]
- 76.Chang CH, Fontes JD, Peterlin M, and Flavell RA (1994). Class II transactivator (CIITA) is sufficient for the inducible expression of major histocompatibility complex class II genes. J. Exp. Med 180, 1367–1374. 10.1084/jem.180.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steimle V, Otten LA, Zufferey M, and Mach B (1993). Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 75, 135–146. 10.1016/s0092-8674(05)80090-x. [DOI] [PubMed] [Google Scholar]
- 78.Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, and Mach B (1994). Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science 265, 106–109. 10.1126/science.8016643. [DOI] [PubMed] [Google Scholar]
- 79.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, Iliopoulos D, van den Elsen PJ, and Kobayashi KS (2010). NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. USA 107, 13794–13799. 10.1073/pnas.1008684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Staehli F, Ludigs K, Heinz LX, Seguín-Estévez Q, Ferrero I, Braun M, Schroder K, Rebsamen M, Tardivel A, Mattmann C, et al. (2012). NLRC5 deficiency selectively impairs MHC class I- dependent lymphocyte killing by cytotoxic T cells. J. Immunol 188, 3820–3828. 10.4049/jimmunol.1102671. [DOI] [PubMed] [Google Scholar]
- 81.Gross O, Gewies A, Finger K, Schäfer M, Sparwasser T, Peschel C, Förster I, and Ruland J (2006). Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442, 651–656. 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- 82.Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, et al. (2007). The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol 8, 619–629. 10.1038/ni1466. [DOI] [PubMed] [Google Scholar]
- 83.Uren AG, O’Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, and Dixit VM (2000). Identification of paracaspases and metacaspases two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol. Cell 6, 961–967. 10.1016/s1097-2765(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 84.Lucas PC, Yonezumi M, Inohara N, McAllister-Lucas LM, Abazeed ME, Chen FF, Yamaoka S, Seto M, and Nunez G (2001). Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J. Biol. Chem 276, 19012–19019. 10.1074/jbc.M009984200. [DOI] [PubMed] [Google Scholar]
- 85.Goodridge HS, Shimada T, Wolf AJ, Hsu YMS, Becker CA, Lin X, and Underhill DM (2009). Differential use of CARD9 by dectin-1 in macrophages and dendritic cells. J. Immunol 182, 1146–1154. 10.4049/jimmunol.182.2.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karasawa T, Kawashima A, Usui F, Kimura H, Shirasuna K, Inoue Y, Komada T, Kobayashi M, Mizushina Y, Sagara J, and Takahashi M (2015). Oligomerized CARD16 promotes caspase-1 assembly and IL-1beta processing. FEBS Open Bio 5, 348–356. 10.1016/j.fob.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu A, Li Y, Schmidt FI, Yin Q, Chen S, Fu TM, Tong AB, Ploegh HL, Mao Y, and Wu H (2016). Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol 23, 416–25. 10.1038/nsmb.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fekete S, Beck A, Veuthey JL, and Guillarme D (2014). Theory and practice of size exclusion chromatography for the analysis of protein aggregates. J. Pharm. Biomed. Anal 101, 161–173. 10.1016/j.jpba.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 89.Devi S, Stehlik C, and Dorfleutner A (2020). An update on CARD only proteins (COPs) and PYD only proteins (POPs) as inflammasome regulators. Int. J. Mol. Sci 21, 6901. 10.3390/ijms21186901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin KM, Hu W, Troutman TD, Jennings M, Brewer T, Li X, Nanda S, Cohen P, Thomas JA, and Pasare C (2014). IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. USA 111, 775–780. 10.1073/pnas.1320294111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Swanson KV, Deng M, and Ting JPY (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol 19, 477–489. 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM, Dorfleutner A, and Stehlik C (2018). The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat. Commun 9, 996. 10.1038/s41467-018-03409-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bryan NB, Dorfleutner A, Rojanasakul Y, and Stehlik C (2009). Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J. Immunol 182, 3173–3182. 10.4049/jimmunol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, and Reed JC (2003). Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J. Immunol 171, 6154–6163. [DOI] [PubMed] [Google Scholar]
- 96.Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MA, and Gadella TWJ Jr. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14, 53–56. 10.1038/nmeth.4074. [DOI] [PubMed] [Google Scholar]
- 97.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, et al. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407–09. 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Udhane V, Maranto C, Hoang DT, Gu L, Erickson A, Devi S, Talati PG, Banerjee A, Iczkowski KA, Jacobsohn K, et al. (2020). Enzalutamide-induced feed-forward signaling loop promotes therapy-resistant prostate cancer growth providing an exploitable molecular target for Jak2 inhibitors. Mol. Cancer Therapeut 19, 231–246. 10.1158/1535-7163.MCT-19-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, and Alnemri ES (2007). The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604. 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schiltz C, Lioté F, Prudhommeaux F, Meunier A, Champy R, Callebert J, and Bardin T (2002). Monosodium urate monohydrate crystal-induced inflammation in vivo: quantitative histomorphometric analysis of cellular events. Arthritis Rheum. 46,1643–1650. 10.1002/art.10326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. Accession numbers for the datasets are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI link is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-caspase-1_p20, clone D7F10 | Cell Signaling Technology | Cat# 3866; RRID:AB_2069051 |
| Rabbit monoclonal cleaved anti-caspase-1, clone D57A2 | Cell Signaling Technology | Cat# 4199; RRID: AB_1903916 |
| Mouse monoclonal cleaved anti-caspase-1, clone casper-1 | AdipoGen | Cat# AG-20B0042; RRID: AB_2490248 |
| Mouse monoclonal anti-caspase-1, clone 1021323 | R&D Systems | Cat# MAB62152 |
| Mouse monoclonal anti-caspase-1 (p10), clone casper-2 | AdipoGen | Cat# AG-20B-0044-C100; RRID:AB_2490253 |
| Rabbit monoclonal cleaved anti-gasderminD | Abcam | Cat# ab209845; RRID:AB_2783550 |
| Rabbit monoclonal cleaved anti-gasderminD, Clone E7H9G | Cell Signaling Technology | Cat# 36425; RRID:AB_2799099 |
| Rabbit monoclonal anti-gasderminD, clone L60 | Cell Signaling Technology | Cat# 93709; RRID:AB_2800210 |
| Mouse monoclonal anti-NLRP3, clone Cryo-2 | AdipoGen | Cat# AG20B0014; RRID:AB_2885199 |
| Rabbit polyclonal anti-NLRP3, clone D4D8T | Cell Signaling Technology | Cat# 15101; RRID:AB_2722591 |
| Mouse monoclonal anti-GFP, clone B-2 | Santa Cruz | Cat# SC9996; RRID:AB_627695 |
| Rabbit monoclonal anti-GFP, clone D5.1 | Cell Signaling Technology | Cat# 2956; RRID:AB_1196615 |
| Rabbit polyclonal anti-ASC, clone AL177 | AdipoGen | Cat# AG25B0006; RRID:AB_2490440 |
| Mouse monoclonal anti-β-tubulin, clone DSHB, AA12.1 | Developmental Studies Hybridoma Bank | Cat# AA12.1 |
| Rabbit polyclonal anti-IL-1β | Santa Cruz | Cat# SC7884; RRID:AB_2124476 |
| Rabbit monoclonal cleaved anti-IL-1b, clone D3A3Z | Cell Signaling Technology | Cat# 83186; RRID: AB_2800010 |
| Rabbit monoclonal anti-IL-1β | Cell Signaling Technology | Cat# 52719; RRID:AB_2799421 |
| Mouse monoclonal anti-IL-1β | Cell Signaling Technology | Cat# 12242; RRID:AB_2715503 |
| HRP conjugated secondary anti-rabbit | Invitrogen | Cat# 31460 |
| HRP conjugated secondary anti-mouse | Invitrogen | Cat# 32430 |
| HRP conjugated secondary anti-goat | Invitrogen | Cat# A15999 |
| Rabbit monoclonal cleaved anti- caspase-3, clone 5A1E | Cell Signaling Technology | Cat# 9664; RRID:AB_2070042 |
| Rabbit monoclonal anti-caspase-3, clone D3R6Y | Cell Signaling Technology | Cat# 14220; RRID:AB_2798429 |
| Anti-Caspase-8 | Cell Signaling Technology | Cat# 4927; RRID:AB_2068301 |
| Rabbit monoclonal cleaved caspase-8, clone D5B2 | Cell signaling Technology | Cat# 8592; RRID:AB_10891784 |
| Anti-caspase-9 | Cell Signaling Technology | Cat# 9504; RRID:AB_2275591 |
| Anti-cleaved caspase-9 | Cell signaling Technology | Cat# 9509; RRID:AB_2073476 |
| Mouse monoclonal cleaved anti-PARP | Cell signaling Technology | Cat# 9548; RRID:AB_2160592 |
| cleaved anti-Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661; RRID:AB_2341188 |
| Rabbit monoclonal cleaved PARP (Asp214) | Cell Signaling Technology | Cat#5625; RRID:AB_10699459 |
| Anti-mouse CD45.2-BV570, clone 104 | Biolegend | Cat# 109833 |
| Anti-mouse Ly6G-BV785, clone 1A8 | Biolegend | Cat#127645 |
| Anti-mouse CD3e-BV785, clone 145-2C11 | Biolegend | Cat# 100355 |
| Anti-mouse CD19-BV785, clone 6D5 | Biolegend | Cat# 115543 |
| Anti-mouse NK-1.1-BV785, clone PK136 | Biolegend | Cat# 108749 |
| PE-CF594 Rat anti-mouse SiglecF, clone E50-2440 | BD Biosciences | Cat# 562757 |
| BB700 Rat Anti-Cd11b, clone M1/70 | BD Biosciences | Cat# 566417 |
| Alexa Fluor 700 anti-mouse Cd11c, clone N418 | Biolegend | Cat# 117320 |
| APC anti-mouse Cd11c, clone N418 | Biolegend | Cat# 117310 |
| PE-Cy7 anti-mouse F4/80, clone BM8 | Biolegend | Cat# 123114 |
| APC-Cy7 anti-mouse Ly6C, clone HK1.4 | Biolegend | Cat# 128026 |
| BV421 anti-mouse I-A/I-E, clone M5/114.15.2 | Biolegend | Cat# 107632 |
| BV650 anti-mouse H-2Kb/H-2Db, Clone 28-8-6 | BD Biosciences | Cat# 745287 |
| APC anti-mouse CD115, clone AFS98 | eBioscience | Cat# 17-1152-82 |
| APC/Fire750 anti-mouse H-2Kb/H-2Db | Biolegend | Cat# 114618 |
| Purified anti-mouse CD16/32, clone 93 | Biolegend | Cat# 101302 |
| Mouse Monoclonal anti-IL-18, clone 74 | MBL | Cat# D047-3;RRID:AB_592016 |
| Mouse Monoclonal anti-IL-18, Biotin conjugated | MBL | Cat# D048-6;RRID:AB_592021 |
| Chemicals, peptides, and recombinant proteins | ||
| Fetal bovine serum | Thermo Scientific | Cat# 16140071 |
| Penicillin and Streptomycin | Gibco | Cat# 15140122 |
| DPBS | Corning | Cat# 21-031-CV |
| RPMI | Cytiva Hyclone, Fisher Scientific | Cat# SH30027FS |
| DMEM | Corning | Cat# 10-013-CV |
| Opti-MEM | Gibco | Cat# 31985088 |
| LPS | Invivogen | Cat# Tlrl-3pelps |
| Pam3csk4 | Invivogen | Cat# Tlrl-pms |
| MSU | Invivogen | Cat# Tlrl-msu |
| Silica/NanoSiO2 | Invivogen | Cat# Tlrl-SiO-2 |
| Nigericin | Invivogen | Cat# Tlrl-nig |
| ATP | Sigma-Aldrich | Cat# A6419 |
| CPPD | Invivogen | Cat# Tlrl-cppd |
| Flagellin | Invivogen | Cat# Tlrl-epstfla-5 |
| Lipofectamine | Thermo Scientific | Cat# 11668019 |
| DOTAP | Roche | Cat# 11202375001 |
| PMA | Invitrogen | Cat# J63916.MCR |
| Protease inhibitor | Pierce | Cat# A329631 |
| Trizol | Invitrogen | Cat# 15596026 |
| Protein agarose A/G beads | Santa Cruz | Cat# SC2003 |
| Super Signal West Femto | Thermo Scientific | Cat# 34096 |
| Staurosporin | Sigma-Aldrich | Cat# S6942 |
| WGP dispersible | Invivogen | Cat# Tlrl-wgp |
| Ac-YVAD-AFC | Enzo Life Science | Cat# ALX-260-108-M005 |
| Ac-DEVD-AFC | Enzo Life Science | Cat# ALX-260-032-M001 |
| Ac-IETD-AFC | Enzo Life Science | Cat# ALX-260-110-M005 |
| Ac-LEHD-AFC | Enzo Life science | Cat# ALX-260-116-M005 |
| Biotin-VAD-FMK | Santa Cruz | Cat# Sc311290 |
| Biotin-YVAD-CMK | Anaspec | Cat# AS-60841 |
| Neutravidin | Pierce | Cat# 29200 |
| FSL-1 | Invivogen | Cat# tlrl-fsl |
| Bacillus anthracis lethal factor | List labs | Cat# 104 |
| Protective antigen | List labs | Cat# 104 |
| Poly(dA:dT) | Invivogen | Cat# Tlrl-patn |
| Brilliant Stain Buffer | BD Horizon | Cat# 566349 |
| eBioscience™ Fixable Viability Dye eFluor™ 506 | ThermoFisher | Cat# 65-0866-14 |
| MACS Buffer | Miltenyi | Cat# 130-091-221 |
| FcBlock | BD Biosciences | Cat# 553141 |
| Cytofix/Cytoperm | BD Biosciences | Cat# 554714 |
| Cytotox Red | Essen Bioscience | Cat# 4632 |
| XenoLight Rediject Inflammation probe | Perkin Elmer | Cat# 760536 |
| Luminol sodium salt | Sigma Aldrich | Cat# A4685 |
| FLICA Caspase-1 inhibitor II Biotinylated | Anaspec | Cat# AS-60841 |
| DSS Disuccinimidyl suberate | Thermo Scientific | Cat# A39267 |
| recombinant murine IFN-γ | Peprotech | Cat# 315-05 |
| MISSION ExpressMag magnetic beads | Millipore-Sigma | Cat# SHM03 |
| Puromycin | Santa Cruz Biotechnology | Cat# 53-79-2 |
| Critical commercial assays | ||
| IL-1β ELISA (human) | Invitrogen | Cat# 88-7261-22 |
| IL-1β ELISA (mouse) | Invitrogen | Cat# 88-7013-22 |
| IL-18 ELISA (human) | R&D Systems | Cat# DY318-05 |
| TNF ELISA (human) | Invitrogen | Cat# 887346-76 |
| TNF ELISA (mouse) | Invitrogen | Cat# 88-7324-88 |
| TNF ELISA (human) | R&D Systems | Cat# DY410 |
| TNF ELISA (mouse) | R&D Systems | Cat# DY210 |
| CXCL1 ELISA (mouse) | R&D Systems | Cat# DY453-05 |
| CXCL2 ELISA (mouse) | R&D Systems | Cat# DY452-05 |
| Caspase-1 ELISA (mouse) | Adipogen | Cat# AG-45B-0002-KI01 |
| NLRP3 ELISA (mouse) | LSBio | Cat# LS-F17336-1 |
| GFP Taqman assay | Invitrogen | Cat# Mr04329676_mr |
| CARD16 Taqman assay | Invitrogen | Cat# Hs03008439_s1 |
| CARD17 Taqman assay | Invitrogen | Cat# Hs01910190_s1 |
| CARD18 Taqman assay | Invitrogen | Cat# Hs01043258_m1 |
| MycoAlert Mycoplasma Detection Kit | Lonza | Cat# LT07 |
| LDH Cytotoxicity Assay (CyQUANT) | Invitrogen | Cat# C203021 |
| Verso cDNA Synthesis Kit | Thermo | Cat# 75650200RXN |
| Duolink Proximity Ligation Assay | Millipore Sigma | Cat# DUO92101-1KT |
| Deposited data | ||
| Affymetrix Human Genome U133 Plus 2.0 Array (HG-U133_Plus_2) | (Lauwerys et al., 2015;43 Nzeusseu Toukap et al., 2007)44 | GEO: GSE36700 |
| original, unprocessed western blot images | this manuscript | https://doi.org/10.17632/gz8rc92m38.1 |
| Experimental models: Cell lines | ||
| THP-1 | American Type Culture Collection | Cat# TIB-202 |
| HEK293 | American Type Culture Collection | Cat# CRL-3216 |
| HEK293 lenti-X | Takara Bio | Cat# 632180 |
| L292 | American Type Culture Collection | Cat# CCL-1 |
| CASP1KO THP-1 | (Chu et al., 2018)92 | N/A |
| ASCKD THP-1 | (Bryan et al., 2009)93 | N/A |
| Ctrl GFP THP-1 | this manuscript | N/A |
| GFP-CARD16 THP-1 | this manuscript | N/A |
| GFP-CARD17 THP-1 | this manuscript | N/A |
| GFP-CARD18 THP-1 | this manuscript | N/A |
| ASCKO THP-1 | Invivogen | thp-koascz |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory, bred in house | JAX#:00066; RRID:IMSR_JAX:000664 |
| Mouse: B6.Tg(CD68-CARD16) | this manuscript | N/A |
| Mouse: B6.Tg(CD68-CARD17) | this manuscript | N/A |
| Mouse: B6.Tg(CD68-CARD18) | this manuscript | N/A |
| Mouse: Casp1−/− | V. M. Dixit (Kayagaki et al., 20159) | N/A |
| Oligonucleotides | ||
| Please see Table S1 | N/A | |
| Recombinant DNA | ||
| pCAT-Basic-CARD16 | this manuscript | N/A |
| pCAT-Basic-CARD17 | this manuscript | N/A |
| pCAT-Basic-CARD18 | this manuscript | N/A |
| pLex-GFP-CARD16 | this manuscript | N/A |
| pLex-GFP-CARD17 | this manuscript | N/A |
| pLex-GFP-CARD18 | this manuscript | N/A |
| pCDNA3-caspase-1 CARD mNeonGreen | this manuscript | N/A |
| pCDNA3-caspase-1 CARD mScarlet-I | this manuscript | N/A |
| pCDNA3- mNeonGreen-mScarlet-I | this manuscript | N/A |
| pMD2.G | Addgene | Cat# 12259; RRID:Addgene_12259 |
| psPAX2 | Addgene | Cat# 12260; RRID:Addgene_12260 |
| lentiCRISPRv1 | Addgene | Cat# 49535 |
| Software and algorithms | ||
| FlowJo v10 | BD Biosciences | N/A |
| Cytek acquisition software | Cytek | N/A |
| SkanIt | Thermo | N/A |
| IncuCyte Base Analysis Software | Sartorius | N/A |
| NIS-Elements AR with ai | Nikon | N/A |
| Fiji (Image J) | Schindelin et al., 201294 | N/A |
| Living Image software 4.7.3 | Perkin Elmer | N/A |