

Abstract
Acute myeloid leukemia (AML) is a highly heterogeneous cancer of the hematopoietic system with no cure for most patients. In addition to chemotherapy, treatment options for AML include recently approved therapies that target proteins with roles in AML pathobiology, such as FLT3, BLC2, and IDH1/2. However, due to disease complexity, these therapies produce very diverse responses, and survival rates are still low. Thus, despite considerable advances, there remains a need for therapies that target different aspects of leukemic biology and for associated biomarkers that define patient populations likely to respond to each available therapy. To meet this need, drugs that target different AML vulnerabilities are currently in advanced stages of clinical development. Here, we review proteomics and phosphoproteomics studies that aimed to provide insights into AML biology and clinical disease heterogeneity not attainable with genomic approaches. To place the discussion in context, we first provide an overview of genetic and clinical aspects of the disease, followed by a summary of proteins targeted by compounds that have been approved or are under clinical trials for AML treatment and, if available, the biomarkers that predict responses. We then discuss proteomics and phosphoproteomics studies that provided insights into AML pathogenesis, from which potential biomarkers and drug targets were identified, and studies that aimed to rationalize the use of synergistic drug combinations. When considered as a whole, the evidence summarized here suggests that proteomics and phosphoproteomics approaches can play a crucial role in the development and implementation of precision medicine for AML patients.
Keywords: Cancer, leukemia, cell signaling, therapeutics, mass spectrometry, personalized medicine, kinase inhibitors, epigenetic inhibitors, immunotherapy, BLC-2 inhibitors, machine learning, artificial inteligence
Graphical Abstract
Highlights
-
•
Acute myeloid leukemia (AML) is a disease of unmet clinical need.
-
•
New therapies for AML are becoming available to healthcare systems worldwide.
-
•
Selecting best treatments for a given patient requires biomarkers of drug responses.
-
•
Proteomic are identifying targets and biomarkers for individualizing AML treatments.
In Brief
Proteomic and genomic studies have identified new drug targets for acute myeloid leukemia (AML), leading to new therapeutic options for certain patient subpopulations. In addition, many other drugs are in advanced stages of clinical development and proteomics keeps uncovering targets of interest. Given the large number of new therapies likely to be available for AML in the near future, identification of proteomic signatures of response for each drug will be key to select the best treatment for a given patient.
Introduction to Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is a hematological cancer with an incidence of approximately 18,500 and 20,000 new cases in Europe and USA, respectively, making this the most common form of acute leukemia (1). Contrary to other forms of leukemia, for which targeted therapies have transformed their prognosis (2, 3), curative treatments do not exists for most AML patients, and according to the American Cancer Society, AML caused 11,500 deaths in the US in 2022. The risk of suffering AML at some point in life is 0.5 to 1%, and although it can occur at all ages, it is more frequent in adults with an average age at first diagnosis of 68 years. AML risks factors include exposure to chemicals (like benzene and chemotherapeutic agents) or radiation, suffering from myelodysplastic syndrome or myeloproliferative neoplasms, having germ line mutations in genes linked to familial AML (e.g., CEBPA, DDX4, or RUNX1) or somatic mutations in genes linked to clonal hematopoiesis (e.g., DNMT3A, TET2, or ASXL1) (4, 5). Patients with inherited genetic syndromes such as Fanconi’s anemia, Bloom syndrome, Down syndrome, and others also present an increased risk of AML (6, 7, 8).
AML originates from the malignant transformation of myeloid precursors, leading to the uncontrolled production of undifferentiated, immature, and nonfunctional blasts, causing the displacement of hematopoietic stem cells and leading to bone marrow (BM) failure. This process was postulated to require genomic aberrations or mutations in epigenetic modifiers and/or transcription factors that impair hematopoietic differentiation alongside mutations in signaling pathways that sustain cell survival and proliferation (9). This “two-hit” model, although now thought to be over-simplistic, presents clinical implications for the design of combinational therapies (10).
Recent mutational studies show that frequent genetic alterations in AML include single gene mutations that affect proteins with multiple functions, together with large cytogenetic aberrations that involve chromosome gains, losses, or rearrangements that generate fusion proteins with aberrant functions (Table 1). Another level of complexity in AML pathogenesis was provided by functional studies that showed that AML is initiated and maintained by leukemia stem cells (LSCs, also known as leukemia initiating cells), from where the bulk of AML blast are derived (11, 12). The LSCs are resistant to chemotherapy because they are quiescent and express antiapoptotic molecules at high levels (13, 14). Therefore, after the elimination of the bulk of leukemic cells by therapy, residual LSCs in BM have the potential to replenish the leukemic population, leading to relapse in patients that initially responded to chemotherapy (15).
Table 1.
Frequent genetic alterations in AML
| Cytogenetic alterations | |
|---|---|
| Balanced | Unbalanced |
| KMT2A rearranged | Complex Karyotype |
| DEK-NUP | −5/del(5q) |
| RUNX1-RUNX1T1 | −7/del(7q) |
| PML-RARA | −17/del(17p) |
| CBFB-MYH11 | +8/8q |
| MECOM rearranged | |
| Single gene mutations | |
|---|---|
| Signaling and kinase pathway | FLT3, KRAS, NRAS, KIT, PTPN11, NF1, JAK2, CBL |
| DNA Methylation | DNMT3A, IDH1, IDH2, TET2, |
| Chromatin-Remodeling | ASXL1, EZH2, KMT2A, BCOR, BCORL1 |
| Transcription factor | CEBPA, RUNX1, GATA2, SETBP1 |
| Tumor suppressor | TP53, WT1, PH6 |
| spliceosome–complex | SRSF2, U2AF1, SF3B1, ZRSR2 |
| Cohesin | RAD21, STAG1, STAG2, SMC1A, SMC3 |
| Nucleophosmin | NPM1 |
AML has been historically subdivided by the French-American-British classification system (Table 2), which is based on morphological and immunophenotypic features linked to the differentiation and maturation stage of blasts (16). More recently, the World Health Organization established an AML classification scheme that incorporated genetic abnormalities and other clinicopathological features. In addition, the European LeukemiaNet integrated genetic and cytogenetic alterations into a risk classification system (favorable, intermediate, and adverse) that prognosticates overall survival and response to standard chemotherapy (17, 18, 19) (Table 2).
Table 2.
Classification of AML cases based on FAB, WHO, and ENL
| FAB classification | |
|---|---|
| FAB-M0 | Minimally differentiated AML |
| FAB-M1 | AML without maturation |
| FAB-M2 | AML with maturation |
| FAB-M3 | Acute promyelocytic leukemia |
| FAB-M4 | Acute myelomonocytic leukemia |
| FAB-M4Eo | Acute myelomonocytic leukemia with eosinophils |
| FAB-M5a | Acute monoblastic leukemia |
| FAB-M5b | Acute monocytic leukemia |
| FAB-M6 | Acute erytroleukemia |
| FAB-M7 | Acute megakaryoblastic leukemia |
| WHO classification | |
|---|---|
| 1-AML with recurrent genetic abnormalities | 3-Therapy-related myeloid neoplasms |
| RUNX1-RUNX1T1 fusion | 4-AML, not otherwise specified (NOS) |
| CBFB-MYH11 fusion | AML with minimal differentiation |
| PML-RARA fusion | AML without maturation |
| KMT2A-MLLT3 fusion | AML with maturation |
| DEK-NUP214 fusion | Acute myelomonocytic leukemia |
| GATA2(promotor)-MECOM(coding) fusion | Acute monoblastic and monocytic leukemia |
| RBM15-MKL1 fusion | Pure erythroid leukemia |
| BCR-ABL1 fusion (Provisional) | Acute megakaryoblastic leukemia |
| Mutated NPM1 | Acute basophilic leukemia |
| Biallelic mutation of CEBPA | Acute panmyelosis with myelofibrosis |
| Mutated RUNX1 (Provisional) | 5-Myeloid sarcoma |
| 2-AML with myelodysplasia (MSD) | 6-Myeloid proliferations associated with Down syndrome (DS) |
| Phenotypic changes associated to MSD | Transient abnormal myelopoiesis associated with DS |
| Cytogenetic alterations associated to MSD | Myeloid leukemia associated with DS |
| Unbalanced: −7/del(7q), del(5q)/t(5q), i(17q)/t(17p), −13/del(13q), del(11q), del(12p)/t(12p) and idic(X)(q13) | 7-Blastic plasmacytoid dendritic cell neoplasm |
| Acute leukemias of ambiguous lineage | |
| Acute undifferentiated leukemia | |
| Balanced: t(11;16)(q23.3;p13.3), t(3;21)(q26.2;q22.1), t(1;3)(p36.3;q21.2), t(2;11)(p21;q23.3), t(5;12)(q32;p13.2) | Mixed phenotype acute leukemia with; BCR-ABL1 fusion |
| Mixed phenotype acute leukemia with rearranged KMT2A | |
| Mixed phenotype acute leukemia, B/myeloid, NOS | |
| Mixed phenotype acute leukemia, T/myeloid, NOS | |
| ELN classification | |
| Favorable | t(8;21)(q22;q22.1); RUNX1-RUNX1T1 |
| inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 | |
| Mutated NPM1 without FLT3-ITD or with FLT3-ITD low | |
| Biallelic mutated CEBPA | |
| Intermediate | Mutated NPM1 and FLT3-ITD high |
| WT NPM1 without FLT3-ITD or with FLT3-ITD low (and no adverse-risk genetic lesions) | |
| t(9;11)(p21.3;q23.3); MLLT3-KMT2A | |
| Cytogenetic abnormalities not classified as favorable or adverse | |
| Adverse | t(6;9)(p23;q34.1); DEK-NUP214 |
| t(v;11q23.3); KMT2A rearranged | |
| t(9;22)(q34.1;q11.2); BCR-ABL1 | |
| inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2,MECOM(EVI1) | |
| t (3q26.2;v); MECOM (EVI1)-rearranged | |
| −5 or del(5q); −7; −17/abn(17p) | |
| Complex karyotype, monosomal karyotype | |
| WT NPM1 and FLT3-ITD high | |
| Mutated ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, or ZRSR2 | |
| Mutated TP53 | |
Therapeutic Options for AML
Until recently, essentially all AML patients were treated with an induction therapy regime that aims to achieve a complete morphologic remission and the restoration of normal hematopoiesis, followed by consolidation therapy to minimize the probability of relapse (1). The exception are patients in the acute promyelocytic leukemia subgroup, whose blast cells have an immature phenotype and who therefore respond to differentiating agents such as all-trans retinoic acid. For induction therapy, non-acute promyelocytic leukemia fit AML patients (generally <65 years) receive several cycles of cytarabine and daunorubicin, while unfit patients (usually >65 years) are treated with low dose cytarabine, DNA hypomethylating agents, like azacitidine and decitabine, or palliative care. Consolidation therapy for patients who achieve complete remission includes different dosages of cytarabine and stem cell transplantation depending on the fitness of the patient and the prognosis of the leukemia provided by genetic and other biomarkers. Although most patients initially respond to this standard chemotherapeutic regime, most of them eventually relapse, leading to a 5-year survival of just 20%, and highlighting the need for more targeted approaches to treat AML (1).
The standard of care started to change gradually from 2017, when several targeted therapies were approved to treat AML subpopulations (Fig. 1 and Table 3). These advances were spurred by the increasing understanding of AML genetic landscape as well as of its molecular biology at the proteomic level.
Fig. 1.

Compounds approved or under clinical trials for the treatment of AML and their intended targets. Names in green denote drugs that have been approved by the FDA for the treatment of AML, while those in magenta are compounds currently under clinical trials. Proteins, when mutated or with a fusion partner produce 2HG or interact with DOT1L, are shown in red fonts. 2HG, 2-hydroxyglutarate; AML, acute myeloid leukemia.
Table 3.
Compounds approved or under clinical trials for the treatment of AML and their intended targets
| Target | Compound | Compound class | Clinical trial |
|---|---|---|---|
| Topoisomerase II complex | Daunorubicin | DNA intercalator | Approved |
| DNA polymerase complex | Cytarabine | Antimetabolite | Approved |
| DNMTs | Azacitidine | Pyrimidine nucleoside analog | Approved |
| Decitabine | Pyrimidine nucleoside analog | Approved | |
| FLT3 | Midostaurin | Small molecule inhibitor | Approved |
| Gilteritinib | Small molecule inhibitor | Approved | |
| BCL2 | Venetoclax | Small molecule inhibitor | Approved |
| Mutated IDH1 | Ivosidenib | Small molecule inhibitor | Approved |
| Mutated IDH2 | Enasidenib | Small molecule inhibitor | Approved |
| SMO | Glasdegib | Small molecule inhibitor | Approved |
| CD33 | Gemtuzumab | Conjugated antibody | Approved |
| KIT | Sorafenib | Small molecule inhibitor | NCT05404516 |
| Dasatinib | Small molecule inhibitor | NCT02013648 | |
| MERTK | MRX-2843 | Small molecule inhibitor | NCT03510104 |
| AXL | Bemcentinib | Small molecule inhibitor | NCT03824080 |
| XPO1 | Selinexor | Small molecule inhibitor | NCT02835222, NCT02403310, NCT04898894 |
| MCL1 | AZD5991 | Small molecule inhibitor | NCT03218683 |
| MIK665 | Small molecule inhibitor | NCT02979366 | |
| AMG 176 | Small molecule inhibitor | NCT02675452 | |
| AMG 397 | Small molecule inhibitor | NCT03465540 | |
| CDK9 | Alvocidib | Small molecule inhibitor | NCT03969420 |
| Dinaciclib | Small molecule inhibitor | NCT03484520 | |
| AZD4573 | Small molecule inhibitor | NCT03263637 | |
| Voruciclib | Small molecule inhibitor | NCT03547115 | |
| BRD4 | Mivebresib | Small molecule inhibitor | NCT02391480 |
| FT-1101 | Small molecule inhibitor | NCT02543879 | |
| Birabresib | Small molecule inhibitor | NCT02303782 | |
| ETC complex I | IACS-010759 | Small molecule inhibitor | NCT02882321 |
| CLPP | ONC201 | Allosteric agonist | NCT02392572 |
| CHK1 | Prexasertib | Small molecule inhibitor | NCT02649764 |
| PARP1 | Talazoparib | Small molecule inhibitor | NCT02878785 |
| Veliparib | Small molecule inhibitor | NCT03289910 | |
| MDM2 | Idasanutlin | Small molecule inhibitor | NCT04029688 |
| HDAC | Belinostat | Small molecule inhibitor | NCT03772925 |
| Entinostat | Small molecule inhibitor | NCT01305499 | |
| Pracinostat | Small molecule inhibitor | NCT01912274 | |
| CDK6 | Palbociclib | Small molecule inhibitor | NCT03844997 |
| KMT2A | SNDX-5613 | Small molecule inhibitor | NCT05326516 |
| MENIN | JNJ-75276617 | Small molecule inhibitor | NCT04811560 |
| DOT1L | Pinometostat | Small molecule inhibitor | NCT03724084 |
| CD123 | MGN632 | Conjugated antibody | NCT04086264 |
| Tagraxofusp | Conjugated interleukin | NCT04342962 | |
| CD47 | Magrolimab | Naked antibody | NCT04435691 |
| CD33 | Anti-CD33-CART | CAR-T Cell | NCT05445765 |
| CD123 | Anti-CD123-CART | CAR-T Cell | NCT04318678 |
| CLL-1 | Anti-CLL-1-CART | CAR-T Cell | NCT04219163 |
| CD7 | Anti-CD7-CART | CAR-T Cell | NCT04033302 |
| CD28 | Anti-CD28-CART | CAR-T Cell | NCT04850560 |
| CD38 | Anti-CD38-CART | CAR-T Cell | NCT05239689 |
| CD19 | Anti-CD19-CART | CAR-T Cell | NCT04257175 |
| FLT3 | Anti-FLT3-CART | CAR-T Cell | NCT03904069 |
| NKG2D | Anti-NKG2D | CAR-T Cell | NCT04658004 |
Activating mutations of the receptor tyrosine kinase FLT3, due to internal tandem duplications (ITDs) in the juxtamembrane domain or point mutations in the tyrosine kinase domain (TKD), occur in approximately 30% of patients, making this the most common genetic alteration in AML. WT FLT3, when bound to its ligand, promotes the generation of myeloid cells from precursors. However, ITD and TKD mutations constitutively activate FLT3 and downstream prosurvival pathways, which support tumorigenesis in hematopoietic precursor cells (20). Midostaurin, a multitargeted kinase inhibitor, was approved by the FDA in April 2017 for adult patients with newly diagnosed FLT3-mutated AML, and this was followed in November 2018 by the approval of gilteritinib for the treatment of adult patients with relapsed/refractory (R/R) AML with FLT3 mutations (21, 22). Quizartinib, another FLT3 inhibitor, was rejected by the FDA but was approved in 2019 by the Japan Ministry of Health for the treatment of R/R AML with mutated FLT3 (15).
Other druggable recurrent genetic alterations in AML include gain-of-function mutations in isocitrate dehydrogenases (IDHs), which occur in about 20% of AML patients (23, 24). IDH1 (cytoplasmic) and IDH2 (mitochondrial) catalyze the transformation of isocitrate into α-ketoglutarate and generate NADPH. However, gain-of-function mutated forms or IDHs use NADPH as a cofactor to generate the oncometabolite 2-hydroxyglutarate. Accumulation of 2-hydroxyglutarate together with the depletion of NADPH and α-ketoglutarate produce a series of metabolic and epigenetic alterations that favor the leukemogenic process (24). In August 2017, the IDH2 mutant inhibitor enasidenib and in July 2018, the IDH1 mutant inhibitor ivosidenib were approved for treatment of refractory or relapsed AML patients with gain-of-function mutations in IDH2 and IDH1, respectively (25, 26).
Although the genetic mutational landscape has revealed opportunities for therapeutic intervention, not all new drugs derive from such new knowledge. A critical feature of cancer cells is their ability to evade proapoptotic signals that prevent normal cells to be transformed. BCL2, a key antiapoptotic protein, has roles in leukemogenesis and is overexpressed in leukemia stem and progenitor cells, as well as in AML blasts when compared to normal hematopoietic cells (27, 28). BCL2 uses its BH3 domain to bind and inhibit the proapoptotic proteins BIM, truncated BID and BAX that facilitate the translocation of cytochrome C from the mitochondria to the cytoplasm, which consequently activates caspases that execute the apoptotic process (29). As an antiapoptotic protein, BCL2 has also been shown to play a role in resistance to chemotherapy of AML (29). Venetoclax is a “BH3 mimetic” small molecule inhibitor that blocks BCL2 antiapoptotic activity (30). In November 2018, the FDA approved the use of venetoclax in combination with hypomethylating agent or low dose cytarabine for the treatment of newly diagnosed AML patients aged ≥75 years or unfit for intensive induction chemotherapy (31). In addition, venetoclax is currently being studied in numerous clinical trials as single agent and in combination therapies (15).
Glasdegib, an antagonist of the G protein–coupled receptor Smoothened (SMO), is another drug derived from an increased mechanistic understanding of AML biology. The Hedgehog (HH) signaling pathway is important to normal hematopoiesis, promotes embryogenesis, maintains adult stem cells, and regulates cell proliferation and differentiation (32). In the absence of HH ligands (Sonic HH, Indian HH, and Desert HH), SMO is inhibited by the Patched transmembrane proteins (PTCH-1 and PTCH-2). The union of the HH ligands to the Patched proteins triggers the activation of SMO and its downstream targets, the transcription factors GLI1 and GLI2, that regulate the expression of proteins involved in cell cycle, apoptosis, and differentiation (33). HH signaling is aberrantly activated in AML and associated with poor clinical outcomes, and SMO activity plays a critical role for disease progression in several AML models (34). Glasdegib was approved by the FDA in November 2018 for use in combination with LDAC for the treatment of newly diagnosed AML patients aged ≥75 years or unfitted for intensive induction chemotherapy (35).
Another proteomic feature that has been exploited for drug development is the presence of cluster of differentiation (CD) proteins on the surface of blast cells. These CD markers have roles in cell adhesion, immune recognition, and signaling and are indicative of the maturation state of the myeloid cell type from where AML derived and confer them with a unique immunophenotype. The CD33 protein antigen in particular is frequently expressed on the surface of AML blasts (36). Gemtuzumab is an antibody drug-conjugate (ADC) directed at CD33, where a humanized monoclonal antibody covalently linked to the cytotoxic drug N-acetyl gamma calicheamicin binds to the CD33 antigens. Binding prompts internalization and subsequent calicheamicin release to the nucleus, leading to DNA damage and cell death (36). In September 2017, the FDA approved gemtuzumab for the treatment of adults with CD33-positive AML (37).
Candidate Targets for New AML Therapies
The drugs discussed above are gradually being incorporated into treatment protocols in healthcare systems as monotherapies or in combination. In addition, several other proteins are currently being tested as targets for the treatment of AML. Below, we provide a nonexhaustive summary of those that are currently (end of 2022) undergoing clinical trials (Fig. 1 and Table 3).
Kinase Inhibitors
In addition to FLT3, other RTKs that have a major impact on leukemia biology include KIT (also known as CD117 and SCF receptor), MERTK, and AXL. KIT, which codes for a protein with important roles in self-renewal and differentiation of hematopoietic stem cells (38), is found mutated in about 17% of AML patients, and its expression is increased in 60 to 80% of AML samples when compared to normal hematopoietic cells (39). There are no specific KIT inhibitors, but multiple clinical trials are currently evaluating broad-spectrum RTK inhibitors that also target KIT including midostaurin (already approved for AML treatment), sorafenib (NCT05404516), and dasatinib (NCT02013648). Inhibitors of MERTK and AXL, which are also frequently overexpressed in leukemic cells, reduce the proliferation of AML cell lines and increase survival in animal models of AML (40). Clinical trials with the MERTK inhibitor MRX-2843 (NCT03510104) and the AXL inhibitor bemcentinib (NCT03824080) are ongoing. In addition, the approved FLT3 inhibitor gilteritinib also targets AXL.
Overactive RTKs promote cell proliferation by activating intracellular kinase-driven signaling cascades—including MEK/ERK, PI3K/AKT, and JAK/STAT—which in turn regulate cell cycle kinases. Although inhibitors of MEK and PI3K/AKT have not progressed in the clinic, considerable effort is currently underway to treat cell cycle kinases downstream of these signaling pathways (41, 42). CDK9 is a cyclin-dependent kinase that forms part of the transcription elongation factor b. This factor phosphorylates the CTD of RNA polymerase 2 and stimulates the elongation of the transcription of most protein-coding genes but in particular, those regulated by super enhancers like MYC and MCL1 (43). In in vitro AML models, CDK9 inhibitors decreased the phosphorylation of RNA Pol II and the expression of MYC, MCL1, XIAP, and cyclin D1, induced apoptosis, and reduced cell proliferation. In animal AML models, these compounds reduced tumor growth and prolonged survival (43). These results encouraged the initiation of clinical trials with the CDK9 inhibitors alvocidib (NCT03969420), dinaciclib (NCT03484520), AZD4573 (NCT03263637), and voruciclib (NCT03547115). Another cell cycle regulator that has been targeted in AML is CDK6, a kinase important for the leukemogenic processes triggered by FLT3-ITD and JAK-V617F mutations and KMT2A-MLLT3 and RUNX1-ETO fusion proteins (44). Currently, the CDK4/6 inhibitor palbociclib (NCT03844997) is in clinical trials for the treatment of AML.
Proapoptotic Therapies
In addition to promoting uncontrolled proliferation, overactive RTKs and downstream signaling pathways promote survival of cancer cells by suppressing apoptosis, a process that, in normal cells, regulate cellular homeostasis (15). Therefore, several small molecule inhibitors of antiapoptotic proteins have been tested for AML treatment. Indeed, the success of venetoclax provides proof-of-concept for this inhibitor class. Targeting the MCL1 provides an approach for targeting the process of apoptosis because this protein is an antiapoptotic member of the BCL2 protein family that, similarly to BCL2 (the target of venetoclax), binds and inhibits the proapoptotic activity of BIM, truncated BID, BAK, and BAX (29, 45). MCL1 is highly expressed in patients with untreated AML and is necessary for survival of AML cells and for the development and persistence of the disease (46, 47). Inhibitor screens (48) showed that AML cells were sensitive to MCL1 inhibition leading to the initiation of several phase I studies for the MCL1 inhibitors AZD5991 (NCT03218683), MIK665 (NCT02979366), AMG 176 (NCT02675452), and AMG 397 (NCT03465540) in AML.
DNA Damage Response Modulators
Targeting the DNA damage response (DDR) provides another opportunity for interfering with the antiapoptotic process in AML. DDR is a complex system that maintains genomic integrity through regulation of DNA damage repair, cell cycle progression, and apoptosis. Deregulation of the DDR allows cancer cells to withstand DNA damage, which would normally trigger apoptosis, but this also generates certain vulnerabilities that can be exploited therapeutically. One of such targets is CHK1, a kinase that upon activation by DNA damage blocks cell cycle progression and triggers DNA damage repair (15, 49, 50). The CHK1 inhibitor prexasertib is being tested in combination with chemotherapy in a clinical trial for the treatment of R/R AML (NCT02649764). Similarly, PARP1, a key mediator of various forms of DNA damage repair, is used by AML cells to withstand replicative stress and/or deficiencies on the homologous repair system, features that sensitize cells to PARR1 inhibitors (51). Currently, the PARP1 inhibitors talazoparib (NCT02878785) and veliparib (NCT03289910) are in clinical trials for the treatment of AML. A further protein involved in DDR and apoptosis that is being targeted in AML is MDM2, which is an ubiquitin ligase that negatively regulates the levels and activity of TP53, a key regulator of DNA repair, cell cycle progression, and apoptosis. Impairment of TP53 plays a pivotal role in the process of leukemogenesis, and, although just 10% of AML cases present TP53 inactivating mutations, its impairment is more generally associated to the overexpression of MDM2 (52). This has led to the development of MDM2 inhibitors like idasanutlin, currently in clinical trials for the treatment of AML (NCT04029688).
Epigenetic Targeted Therapies
Epigenetic regulation has also been targeted for the development of AML therapeutics because genes coding for epigenetic proteins are frequently mutated or otherwise deregulated in this disease. One of such protein is the histone deacetylase (HDAC) family of epigenetic erasers that remove acetylation marks from histones, leading to chromatin compaction and gene silencing. HDACs also act on nonhistone proteins to regulate their activities. HDAC inhibitors cause changes in the expression of multiple genes, including downregulation of the key oncogene MYC and induce differentiation, cell cycle arrest, and apoptosis in AML cells (53). Belinostat (NCT03772925), entinostat (NCT01305499), and pracinostat (NCT01912274) are currently in clinical trials for AML treatment.
Another epigenetic protein targeted in AML is the lysine methyltransferase KMT2A, which methylates histone H3K4. In 10% of AML cases, KMT2A fusion proteins generated by chromosomal rearrangements drive the leukemogenic process. In these fusion proteins, the catalytic domain of KMT2A is replaced by a region of the partner protein that facilitates recruitment of the histone H3K79 methyltransferase DOT1L to the KMT2A target genes, a process that requires the KMT2A-MENIN protein–protein interaction (54). Numerous small molecule inhibitors have been designed to target proteins necessary for the activity of KMT2A fusion proteins and currently the KMT2A, MENIN, and DOT1L inhibitors SNDX-5613, JNJ-75276617, and pinometostat, respectively, are in clinical trials for the treatment of AML (NCT05326516, NCT04811560, NCT03724084).
The bromodomain and extraterminal domain-containing protein BRD4 is yet another promising epigenetic target for AML treatment. BRD4 is a reader that binds to acetylated histones and other proteins thus recruiting the TEFb complex that, as previously discussed, stimulates transcription elongation of genes relevant for the leukemogenic process like MYC and MCL1 (55). Currently, the BET inhibitors mivebresib (NCT02391480), FT-1101 (NCT02543879), and birabresib (NCT02303782) have entered clinical trials for the treatment of AML.
Protein Transport Inhibitors
Protein mislocalization is a frequent occurrence in cancer cells and a potential mechanism of oncogenesis. Indeed, several nuclear tumor suppressors cannot serve their functions in cancer cells because they are mislocated to the cytoplasm. TP53, BRCA1/2, NPM1, FOXO, RB1, and other tumor suppressors are substrates of XPO1, an export receptor responsible for the nuclear-cytoplasmic transport of hundreds of proteins and RNA species (56). In addition, XPO1 levels inversely correlate with the overall survival of AML patients (57). Encouraging preclinical studies showing that XPO1 inhibition promoted cell cycle arrest and apoptosis in in vitro and in vivo AML models (58) accelerated the initiation of multiple clinical trials with the XPO1 inhibitor selinexor in AML patients (NCT02835222, NCT02403310, or NCT04898894).
Energy Metabolism Targeting Compounds
Like in other cancer types, targeting metabolic processes open avenues for therapeutic intervention in AML. The electron transport chain (ETC) powers the mitochondrial oxidative phosphorylation (OXPHOS), which has been proved as a main source of energy in AML LSCs and chemotherapy-resistant cells (59). The ETC complex I inhibitor IACS-010759 is a potent antileukemic agent in in vitro and in vivo models and has entered clinical trials for the treatment of AML (NCT02882321) (60). Another way to target OXPHOS is with activators of the ATP-dependent mitochondrial caseinolytic protease P, which regulates OXPHOS by controlling the degradation of the respiratory chain components and triggering the mitochondrial unfolded protein response. ONC201 is an allosteric agonist that hyperactivates caseinolytic protease P impairing the OXPHOS and triggering an atypical integrated stress response mediated by ATF4. This compound has also been effective as an antileukemic agent in in vitro and in vivo AML models and is in clinical trials for the treatment of AML (NCT02392572) (49).
Immunotherapies
In addition to the targeted agents discussed above, new immunotherapy approaches have been developed in AML with the aim to treat the disease using ADCs or immune cells directed against surface markers on LSCs or bulk cell populations. As noted above, gemtuzumab, an approved ADC against CD33, provides proof-of-concept for this approach. The receptor of interleukin 3 CD123 and the antiphagocytic protein CD47 are highly expressed in LSCs (61, 62). Reagents targeting CD123 in clinical trials for AML include MGN632 (NCT04086264), an anti-CD123 antibody linked to a genotoxic compound, and tagraxofusp (NCT04342962), an agent composed of human IL-3 fused to a portion of the diphtheria toxin. In addition, the CD47 antibody magrolimab that facilitates the phagocytosis of LSC is also in clinical trials (NCT04435691). CAR-T cells are cytotoxic T cells with T cell receptors modified to target specific antigens (63). There are currently multiple clinical trials testing the treatment of AML with CAR-T cells design to target CD33 (NCT05445765), CD123 (NCT04318678), CLL-1 (NCT04219163), CD7 (NCT04033302), CD28 (NCT04850560), CD38 (NCT05239689), CD19 (NCT04257175), FLT3 (NCT03904069), and NKG2D (NCT04658004).
The Need to Prioritize Therapies for AML Patients
As outlined above, intense research is producing a large array of therapies that could potentially be used to treat AML. Oncologists are already facing the issue of having to select the drugs or drug combinations, out of the many available, more likely to be efficacious for a given patient, a dilemma that will become even more intricate as some of the new therapies discussed above reach the clinic. Current technologies for precision and personalized medicine are based on the analysis of genomic markers (64). Although these methods can enrich for potential responders, and are thus an improvement over therapies in unselected patient populations, results of clinical trials show that genetic screens often fail to accurately predict therapy outcome. For example, only ∼20% of R/R AML patients positive for NRAS or KRAS mutations responded to the MEK inhibitor trametinib (65). Similarly, ∼40% of FLT3 mutant–positive newly diagnosed AML patients failed to respond to the FLT3 inhibitor midostaurin in combination with chemotherapy (66), while, conversely, 41% to 56% of FLT3 mutant-negative patients responded to FLT3 inhibitors (65, 67, 68).
Focusing only on the genomic and transcriptomic layers of cell function regulation leaves us blind to other important regulators of cell phenotypes and outcomes (64). It is well known that changes in gene expression do not always reflect changes in protein abundance (69) and protein amounts in cells may not predict how enzymatically active they are (70, 71). Proteins are the major effectors of cell functions and their regulation by allosteric and posttranslational modifications, localization, interaction partners, and abundance combine to regulate their enzymatic activity and proximity to their substrates, which together influence pathway fluxes and cell phenotypes. It is therefore critical to also consider proteomics, phosphoproteomics, and other posttranslational modification-‘omics’ datasets to understand disease development and subtypes, as they can better capture the functional state and dynamic properties of a given cell or cell population (64). Therefore, the combination of genomics and transcriptomics with proteomics and phosphoproteomics could complement current approaches for disease classifications (including AML) by defining new pathological subtypes linked to specific therapeutic vulnerabilities.
Overview of Proteomics and Phosphoproteomics Approaches
Proteomics and phosphoproteomics methods based on LC-MS/MS can accurately and simultaneously measure abundances for thousands of proteins and phosphorylation sites. Consequently, LC-MS/MS techniques are contributing to the identification of clinically relevant biomarkers and targets for disease diagnosis and prognosis in precision medicine (72, 73, 74, 75). Quantitative proteomics and phosphoproteomic approaches for profiling cell lines and primary samples could be subdivided into label-based and label-free methods. Labeling approaches put a limit on the number of samples that can be assessed and makes it difficult to compare the results of different experimental batches but are reputed to offer greater analytical precision. Label-free and targeted proteomics methods, such as those based on data-independent analysis, solve many of these issues and renders them suitable for clinical assays (76, 77). Detailed description of quantitative labeling and label-free mass spectrometry methods have been described elsewhere (78, 79, 80, 81, 82, 83) and will not be discussed here.
Quantitative proteomics produces large volumes of data that require specialized computational methods for their biological interpretation. Multiple bioinformatics tools originally designed for the analysis of gene expression, such as term overrepresentation analysis and gene set enrichment analysis, are also useful for obtaining pathway level information from proteins differentially expressed or phosphorylated between groups of interest. Term and ontology enrichment can be computed against different databases (e.g., gene ontology, KEGG, and NCI) utilizing algorithms frequently used in transcriptomic data analysis (84). In addition, specialized software has been developed for the analysis of phosphoproteomics data. The algorithm kinase substrate enrichment analysis (KSEA) uses phosphoproteomics data to estimate kinase activities based on the phosphorylation of their substrates. The first version of the algorithm (85) used databases based on empirically demonstrated associations between kinases and substrates. An improved version of the algorithm uses databases defined by a chemical proteomics approach that codes information on kinase–kinase interactions that can allow the reconstruction of kinase network topologies (71). Several other methods for the inference of kinase network activity from phosphoproteomics data have been developed and discussed in detail elsewhere (86). One of such tool, named integrative inferred kinase activity (INKA), calculates kinase activity form phosphoproteomics data takes by measuring phosphorylation sites on the kinase and on its activation loop, in addition to experimentally established substrates (as in KSEA) and on substrates predicted in silico by NetworKin (87).
Studies that inferred kinase activity from phosphoproteomic data are starting to provide biological insights in the field of AML. For example, Van Alpen et al. showed that INKA analysis of 16 AML cell lines using tyrosine-targeted phosphoproteomics identified hyperphosphorylated, active kinases as candidates for targeted therapies. Validation drug response experiments showed that, in addition to driver kinases corresponding with activating mutations present in these cell lines, INKA analysis also pinpointed driver kinases undetected by standard molecular analyses. Furthermore, INKA detected hyperactivation of FLT3 in two clinical AML samples with an FLT3-ITD mutation (88). Another example is the application of KSEA to primary cells from a cohort of 20 patients (85), which identified MEK1/2, casein kinases, CDKs, and PAKs as the most frequently activated kinases in AML cases compared to periferal blood cells from healthy donors. Furthermore, KSEA showed that substrates of ERK, CDC7 were more phosphorylated in primary cells resistant to a compound targeting the PI3K/MTOR pathway, while substrates of ABL, LCK, SRC, and CDK1 were more phosphorylated in sensitive cells.
Limitation of AML Cell Lines as Clinically Relevant Models for Proteomics and Phosphoproteomics Studies
Cell lines are frequently used as disease models in basic research. However, cell lines may not always resemble the biology of primary cells, and therefore are not useful to model all aspects of cancer biology. In the case of AML, although cytogenetic signatures based on gene expression have been found to be conserved, to some extend at least, between AML primary samples and cell lines, other reports highlight the limitations of cell lines as disease models (89). For example, analysis of the expression of 380 genes linked to multidrug resistance proteins and upregulated genes that facilitate cell survival showed that cell lines from different origins (including AML) were more similar between them than to the primary tumor cells that they were supposed to model (90). Regarding protein expression, Aasebo et al. (91) found, in a dataset comprising five AML cell lines and 27 primary AML samples, that about one third of the proteins quantified were differentially expressed between cell lines and primary cells, with proteins involved in translation overexpressed in cell lines and proteins associated to the mitochondria overexpressed in primary cells. These results imply that, although cells lines can be used to investigate some aspects of tumor biology and for drug development, they do not fully recapitulate the biological complexity of AML (89). Therefore, the focus of this review is on studies that used primary AML cells as the focus of the analysis.
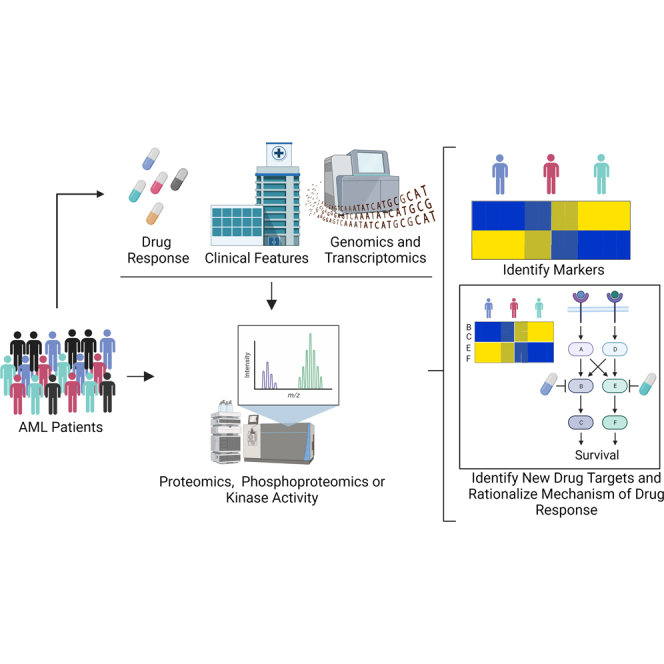
Proteomics and Phosphoproteomics Studies for Target Identification and Drug Response Prediction
Access to large cohorts of primary AML clinical specimens and the availability of appropriate control samples still remains challenging (89). Nevertheless, multiple studies have used mass spectrometry to identify proteins or phosphorylation sites differentially regulated between AML patients and healthy donors, to define subgroups of AML patients, to model drug responses and other features using machine learning (ML) algorithms, and to identify determinants of responses to approved and experimental therapies. Differentially expressed features derived from these studies could potentially be used as new drug targets and/or response markers for precision medicine in AML. Here, we review in a chronological order of publication, proteomics, and phosphoproteomic studies of primary AML that aimed to identify drug targets or mechanisms of drug sensitivity.
Casado et al. (85) profiled the basal phosphoproteomes in a cohort of 20 primary AML samples linked to preclinical responses to kinase inhibitors. A pathway activity signature of phosphosites that decreased by the treatment of cell lines with PI3K and MTOR inhibitors was then evaluated in primary AML cells for its ability to predict responses to kinase inhibitors in preclinical development. Linear regression models were trained using signatures derived from individual phosphopeptides as well as KSEA-estimated kinase activities and values of phosphorylation motifs enrichment. Interestingly, it was found that the activity of the PI3K/MTOR pathway was not the only determinant of sensitivity to inhibitors of this pathway and that the activity of pathways, such as ERK1/2 and PKC, that can compensate for PI3K target inhibition were elevated in resistant cells. Thus, accurate predictive models could be constructed by combining kinase activities in the target kinase pathways relative to those that act in parallel to compensate for target inhibition.
A follow up study from the same group integrated phosphoproteomics, proteomics, genomics, and mass cytometry (immunophenotype) data with ex vivo responses to MEK, PAK, PKC/FLT3, CK2, and MAPK P38 inhibitors (92). The study showed evidence of molecular features associated to response to treatment and identified kinase and differentiation determinants as markers of sensitivity to kinase inhibitors in primary leukemic blasts. The study, performed in primary cells form 30 AML cases, revealed that protein phosphorylation positively correlated with the surface expression of differentiation makers (CDs) associated to myeloid differentiation. AML cases were subsequently separated into more and less differentiated cases (CDs+ and CDs groups, respectively) using the surface expression of myeloid differentiation markers. More differentiated (CDs+) cases expressed higher levels of kinases and signal transduction regulators and increased the activity of kinases downstream of growth factors such as MAPK1, MAPK3, PAK2, and PKCδ. Consistently, CDs+ cases were more sensitive to the MEK, PAK, and FLT3/PKC inhibitors in ex vivo assays. These results established a link between differentiation, kinase activity, and sensitivity to kinase inhibitors.
In the same study, a more integrative and systematic analysis of mutational profiles and mass spectrometry and cytometry data showed that NRAS or BRAF mutations, high MAPK1 activity, or the CDs+ phenotype were associated with sensitivity to trametinib, while FLT3-ITD mutations or high pSTAT5A phosphorylation were linked to resistance. These data suggested two distinct mechanisms of intrinsic resistance to MEK inhibition. In the first one, cells with low RAS/MEK/ERK pathway activity were not addicted to the prosurvival actions of MEK. In the second, cells with highly active RAS/MEK/ERK, as a result of RAS/RAF mutations and/or expression of myeloid differentiation markers, but resistant to MEKi, were found to activate an FLT3/STAT5 axis, which may act in parallel to MEK/ERK to sustain cell survival. Interestingly, the response to midostaurin was not associated to FLT3 mutations; instead, the CDs+ phenotype and the phosphorylation of PKCδ, a known target of midostaurin, were associated to the response to this drug, suggesting that the mode of action of midostaurin involves inhibiting other kinases in addition to FLT3.
In a different study, Alanazi et al. (93) identified misexpressed or mislocalized proteins to the nucleus that could regulate the malignant properties of AML blasts. The comparison of the nuclear proteomes of CD34+ cord blood cells from five healthy donors and blasts from 15 cases with FAB-M2 AML led to the identification of 113 proteins, 11 of which were transcription factors frequently mislocalized in AML blasts. S100A4 was the highest differentially expressed protein in AML nuclei that was not previously implicated in AML. Relevantly, protein but not mRNA levels of S100A4 were overexpressed in the nucleus of a larger cohort of 24 patients. Functional experiments showed that knock down of S100A4 strongly impacted the survival of AML cell lines, but not the survival of normal hematopoietic stem progenitor cells, suggesting that S100A4 could be a new target for AML treatment.
In a follow up from (92), Hijazi et al. (71) analyzed drug response data by multivariate regression using kinase network edge activities (derived by KSEA) as input to predict response to trametinib, midostaurin, silmitasertib, and the PAK4 inhibitor PF-3758309. These models predicted drug response with an accuracy between 20 and 40% and highlighted that the activity of target and parallel pathways contribute to model performance.
Aasebo et al. (94) profiled the proteome and phosphoproteome of AML primary blasts at the time of diagnosis from 41 AML patients that reached complete remission, from where proteins differentially expressed or phosphorylated were identified in patients that relapsed within 5 years relative to those who did not. Relapsed cases presented increased expression of RNA processing proteins and increased phosphorylation of proteins liked to CDKs and CK2, while relapse-free cases increased the expression of V-ATPase proteins. Therefore, this study suggested markers that could help to predict relapse in AML.
Assessing drug response in ex vivo models has provided highly valuable information related to targetable pathways in AML (95). However, ex vivo models are devoid of all signals from stromal components and clearly differ from human in vivo responses. Therefore, proteomics and phosphoproteomics studies in primary AML blast obtained at diagnosis from AML patients with known responses to treatment would provide extremely useful information. In line with this, Hernandez-Valladares et al. (96) performed a proteomics analysis in nontreated samples from 28 AML cases that were subsequently treated with all-trans retinoic acid and valporic (a HDAC inhibitor), a treatment used alone or in combination with low dose chemotherapy for patients unfit for standard chemotherapy. Nonresponders overexpressed the lysosomal protein ARSA, while responders overexpressed proteins linked to myeloid cells, neutrophil degranulation, lysosomes, carcinogenesis (including ANO6, CHI3L1, CTSG, ELANE, and FGR), and M phase of the cell cycle (including CENPE, CENPK, CDK1, NCAPG; and several histones). Interestingly, the proteins differentially expressed between responders and nonresponders presented a low overlap between mRNA and protein levels. Phosphoproteomics analysis showed that responders increased the phosphorylation of proteins liked to the process of apoptosis, such as SPTAN1 and ACIN1, as well as LIMKs and CDKs substrates. In addition, proteomics and phosphoproteomics analysis comparing patient samples before and after 3-day treatment revealed an altered expression and phosphorylation of proteins involved in the regulation of transcription, translation, and RNA metabolism.
Nguyen et al. (97) profiled the proteome of BM cells from 16 pediatric AML patients at diagnosis and found that 117 proteins that were differentially expressed in cases with fusion proteins involving components of core binding factor complex (CBF). Patients with CBF rearrangements deregulate proteins involved in several metabolic pathways such as the TCA cycle and the ATP synthesis coupled to proton transport. In addition, CD34 protein expression was significantly increased in cases with CBF rearrangements, suggesting that CBF AMLs carry unique protein expressions that resemble CD34+ progenitor cells. These data also imply that cases with or without CBF rearrangements could present a differential response to compounds that interfere with the ETC.
Casado et al. (72) described a ML workflow aimed to predict ex vivo patient response to trametinib, a specific inhibitor of MEK1 and MEK2, in primary AML using kinase activity (KSEA) as input. A model using the partial least squares algorithm was used to rank and filter the most relevant kinase activities in the training set, which were subsequently used as input for a random forest regression model. The ML model predicted trametinib response with a root median square error of 0.131 in the validation dataset.
A proteogenomic study by Jayavelu et al. (98), in addition to providing insights into the pathogenesis of AML, also revealed altered molecular features that could influence response to therapy and suggested that a group, termed C-Mito AML, with defined mitochondrial protein expression patterns, tended to respond to venetoclax and electron transport I complex inhibitors.
In a more integrative study, Kramer et al. (99) performed proteomics and phosphoproteomics in BM samples from six healthy donors and 44 patients for which data on DNA and RNA sequencing were available. The patient cohort covered all European LeukemiaNet cytogenetic risk groups and frequent single gene mutations. Protein-mRNA level correlation analysis showed no positive correlation (spearman < 0) for more than a thousand proteins that were mainly linked to spliceosome, OXPHOS, and RNA polymerase processes. On the other hand, the 1198 proteins that showed positive correlation included differentiation markers and other proteins relevant for the AML physiopathology. The authors demonstrated that IDH1 or IDH2 mutations caused increased protein expression of the 2-HG–dependent histone demethylases KDM4A, KDM4B, and KDM4C. NPM1c mutations relocate NPM1 to the cytoplasm and it was shown that cases with NPM1c mutations had increased levels of the nuclear importins KPNA4 and KPNA1 that are able to interact with mutated but not with normal forms of NPM1. Of interest, proteomics data guided the identification of the markers CD180 and CD206 in the plasma membrane of AML blast from some patients but not in the surface of healthy CD34+ cells, suggesting that these markers could represent novel targets for immunotherapy. Clustering analysis using phosphoproteomics data also separated healthy donors from AML cases and segregated patients according to the presence of FLT3 mutations or PML-RARA fusions. When compared to healthy donors, AML cases presented abnormal phosphorylation of specific residues in PTPN11, STAT3, AKT1, and PRKCD. In addition, FLT3-TKD–mutated samples increased the phosphorylation of activating residues in the tyrosine kinases FGR and HCK and other signaling related proteins, while the PML-RARA–initiated samples displayed a unique phosphorylation signature that included increased phosphorylation of JUN at S60 and STK26 at the activation loop. Finally, TP53-mutant samples showed abundant phosphorylation of TP53 at S183. This study shows that the link between mRNA and protein abundance in AML cells is relatively limited, especially for proteins linked to RNA maturation and mitochondrial function, highlighting the relevance of proteomics studies.
In agreement with this, a study by Caplan et al. (100), combining proteomics and transcriptomics in AML mouse models, identified a set of 34 proteins upregulated in AML tumors at the protein but not at the mRNA level that also showed an enrichment of mitochondrial and spliceosome proteins. Relevantly, proteins differentially expressed or phosphorylated were found associated to AML and specific mutations that could constitute new targets or markers for precision medicine. However, no link was established between protein expression or phosphorylation and drug response.
In another study that used ML from mass spectrometry data to derive models of therapeutic response and mechanistic inference in AML cases, Gosline et al. (101) performed proteomics and phosphoproteomics from 38 AML cases for which genomics and transcriptomics data were available. Identified signatures from different omic layers were evaluated for their ability to model ex vivo responses to 26 drugs. Genetic mutations were found to be relatively robust in modeling responses to targeted therapy (e.g., trametinib and quizartinib for NRAS- and FLT3-activating mutations), but models based on protein features showed greater performance when assessed across all drugs. The proteins and phosphopeptides more relevant for quizartinib and trametinib predictive models clustered AML cell lines based on response to these drugs. In addition, these features were integrated to identify putative underlying molecular pathways and provide a biological interpretation of the signatures. The phosphatase SHP-1 (also known as INPP5D) was part of the signature used by LASSO and logistic regression models, and patients sensitive to quizartinib downregulated this phosphatase, a known regulator of signaling downstream of FLT3 (102). An integration algorithm added the expression of SHC1, a protein expressed in AML blasts, and the phosphorylation of SMC3, that synergize with FLT3 in AML, to the SHP-1 network. Patients highly resistant to trametinib expressed proteins associated to mRNA processing and catabolism. Network integration using mRNA and protein expression data highlighted BID, CASP1, GZMB, and other proteins linked to apoptosis, thus suggesting that expression of apoptosis-related proteins and transcripts could predispose patients to trametinib sensitivity.
In another recent study, Casado et al. (103) profiled 74 AML patients from Finland and the UK with poor risk karyotype using genomics, transcriptomics, proteomics, and phosphoproteomics platforms, as well as ex vivo responses to 550 drugs. Integration of the data identified a phosphoproteomics signature that defined two biologically distinct groups of KMT2A-rearranged leukemia, which were termed MLLGA and MLLGB. Increased DOT1L phosphorylation and HOXA gene expression indicated that MLLGA cases have higher activity of DOT1L and TEFb at KMT2A target genes when compared to MLLGB and the no KMT2A rearrangement group. MLLGA cases also increased the activity of CDK1 and the phosphorylation of proteins involved in RNA metabolism, replication, and DNA damage when compared to MLLGB and no KMT2A. Compared to other groups, MLLGA was particularly sensitive to 15 compounds including genotoxic drugs and inhibitors of mitotic kinases and inosine-5-monosphosphate dehydrogenase (IMPDH). Further experiments suggested that the ability of IMPDH inhibitors to interfere with the nucleolar biology is, at least partially, responsible for the higher efficiency of these compounds in MLLGA. This study identifies a signature that classifies AML patients with KMT2A rearrangements into two biologically and functionally different groups, and it provides a rationale for the potential testing of IMPDH inhibitors and potentially mitotic and genotoxic compounds in KMT2A patients positive for the MLLGA signature.
In summary, multiple strategies using phosphoproteomics and proteomics data have been implemented to identify potential new targets and response markers in AML (Fig. 2). The wealth of data that is now generated using proteomic approaches require data science strategies to mine such rich datasets for biological and clinical insights. A frequently used strategy consists of classifying AML cases into groups using previously known labels (e.g., responder versus nonresponder groups to a given drug) for the identification of proteins differentially phosphorylated or expressed between groups using classical statistics (Fig. 2). Differentially expressed or modified features provide biological insights and may reveal candidate drug response markers and drug targets specific for each patient population. Groups have been defined based on patients versus healthy donors (85), relapsed versus no relapsed patients (94), patients with presence versus no presence of chromosome rearrangements (97) or mutations (99), or responders versus nonresponders to a treatment (96).
Fig. 2.

Data science strategies for the identification of new drug targets and response markers for the treatment of AML using proteomics or phosphoproteomics data. Individuals are classified based on known labels (e.g., resistant or sensitive to a given treatment). Differences in protein phosphorylation and expression between groups are determined using classical statistics and utilized to rationalize mechanisms, from which potential drug targets and response markers may be identified. Unsupervised ML methods, like hierarchical clustering or PCA, may use proteomics, phosphoproteomics, or other omics data to define new labels and patient groups. Differences between such groups may again reveal potential treatments for these patient subpopulations, and response markers may in turn be used to refine labels further. Supervised ML is used to generate regression or classification models of drug responses from proteomics and phosphoproteomics data (or values derived thereof). Features (proteins, phosphopeptides, etc.) that define the models may give insights into drug response mechanism. AML, acute myeloid leukemia; ML, machine learning.
Another option is to define new labels using proteomics, phosphoproteomics, or other omics data as input for unsupervised ML algorithms like hierarchical clustering or principal component analysis to generate new labels, which may be used to classify cases and identify potential response markers and drug targets as described above (Fig. 2). This strategy has been used to define the CDs groups and C-Mito cluster (92, 98). The rationalization of response markers and drug targets can also be used to refine the definition of the labels used to classify the patients (92, 93). Other approaches used supervised ML to generate regression or classification models that model and predict drug response or other relevant features (71, 104). While these models may in the future be useful to clinicians when deciding on therapeutic options, it is clear that these will not be used in isolation and will instead complement the information derived from the assessment of classical clinicopathological and genomics features, which together may provide more accurate drug response predictions. A limitation of this approach is that it produces black box models from which is it difficult to derive mechanistic insight, although considerable work to solve this issue is being carried out (105). In some instances (e.g., when explainability is an essential requirement or when sample numbers are limited), it may be preferable to use ML methods based on linear regression or random forest (106), which although simpler and less powerful than deep learning (105), allow inferring feature importance with relative ease (106).
Rationalization of Drug Synergy in AML Cells Using Proteomics and Phosphoproteomics
Tumors depend on a limited number of molecular mechanisms for their survival and proliferation. Therefore, drug combination therapies that simultaneously target several of these crucial mechanisms would produce a synergistic effect that is greater than the sum of the effect of the single treatments. Although attractive, precision medicine for cancer treatment using combinations of drugs with a synergistic effect proves to be highly challenging (107). Proteomics and phosphoproteomics approaches may contribute to the rationalization of drug combinations, and below we summarize illustrative studies that aimed to do so in AML.
Cucchi et al. (67) used an ex vivo drug response analysis in 19 AML cases to identify sensitive and resistant patients to the FLT3 inhibitors gilteritinib and midostaurin. FLT3-ITD–mutated samples were more responsive towards gilteritinib and midostaurin, compared with FLT3 Wt cases, but the presence of FLT3-ITD mutations could not fully explain these responses. To address this, phosphoproteomics was used to investigate associations between response and phosphorylation. Gilteritinib-resistant samples increased the phosphorylation and activity of MAPK1/MAPK3, EGFR1, and KIT. While no difference in responses were found between FLT3 mutant and WT cases, a small fraction of peptides differentially phosphorylated between sensitive and resistant cases to gilteritinib and midostaurin were also differentially phosphorylated between ITD and Wt FLT3 cases, suggesting that most of the differences in phosphorylation between resistant and sensitive cells come from parallel pathways to FLT3 signaling that support survival in the absence of FLT3 activity. Consequently, their targeting could sensitize cells to FLT3 inhibition, a hypothesis that was confirmed with the combination of gilteritinib or midostaurin with the MEK inhibitor trametinib, which showed synergy, although this was only observed in one NRAS-mutated sample. This is in line with studies reporting resistance to midostaurin in NRAS-mutated cases (92).
Murray et al. (108) profiled the phosphoproteome of primary AML blasts from seven patients with Wt or mutant FLT3. A set of 143 peptides were found to be highly phosphorylated in FLT3 mutant patients, and these presented an enrichment of proteins linked to the Non Homologous End Joining DNA repair pathway, while the 90 peptides downphosphorylated in FLT3 mutant cases showed an enrichment of proteins associated to the Base Excision Repair pathway. Mutant cases increased the autophosphorylation DNA protein kinase (PRKDC) at S2612 and the phosphorylation of other key proteins in the Non Homologous End Joining pathway like XRCC4, XRCC5, and TP53BP1. PRKDC phosphorylation at S2612 was sensitive to FLT3 inhibitors. Furthermore, the PRKDC inhibitor M3814, when combined with the FLT3 inhibitor sorafenib, showed a synergistic effect in reducing the survival of primary blast with mutated FLT3 but not of blast with Wt FLT3. Finally, a xerograph mice model of the FLT3-ITD mutant AML cell line MV4-11 survived longer when treated with the combination of M3814 and sorafenib than when subjected to single agent therapy. In summary, phosphoproteome profiling showed that primary AML blast with mutated FLT3 overactivated the PRKDC pathway, and this rationalized a synergistic FLT3 and PRKDC inhibitor combination in FLT3 mutant–positive AML cells.
Zhu et al. carried out a phosphoproteome profiling of primary cells at diagnosis from eight AML patients that did (remission) or did not (refractory) respond to standard chemotherapy. This study showed that refractory patients increased the phosphorylation of proteins linked to ATM, FLT3, and MAPK/ERK signaling (109). Kinase activity inference using NetworKIN showed that refractory samples had an increase in the phosphorylation of putative substrates of CK2 and CDKs. Consistent with these observations, the CK2 inhibitor silmitasertib increased cell death induced by cytarabine in primary AML cases from refractory cases. The CK2 substrate HMGA1 was highly phosphorylated in refractory cases; removal of this protein in AML cell lines reduced their proliferation, and conversely, transduction of a phosphomimetic mutant for HMGA1 at CK2 sites increased the colony formation of a MLL-AF9/FLT3-ITD murine model of AML. HMGA1 and the transcription factor SP1 regulate the expression of BIRC5, a relevant antiapoptotic protein in AML. A functional study showed that only phosphorylable forms of HMGA1 at CK2 sites bind SP1 at the BIRC5 promoter. This work revealed a potential mechanism by which HMGA1 phosphorylation at CK2 sites promotes intrinsic resistance cytarabine-based chemotherapy and how CK2 inhibitors could sensitize chemo-resistant cells.
In another elegant study, Emdal et al. (110) performed a phosphoproteomic analysis of 20 primary AML samples sensitive or resistant to ex vivo treatment with selinexor for 6 h. In patient samples and cell lines sensitive to the drug, selinexor increased the phosphorylation of TP53 at S15, a marker of its transcriptional activity. However, in patients resistant to selinexor treatment, the drug increased the phosphorylation of FOXO3A at S253, an AKT site that sequesters FOXO3A in the cytoplasm and inhibit its proapoptotic transcriptional activity. In AML cell lines with Wt TP53 and sensitive to selinexor, nutlin-3 (an MDM2 inhibitor that increase TP53 transcriptional activity) and selinexor treatment produced a synergistic effect in which nutlin-3 decreased the degradation of TP53, a key protein for the cell death induced by selinexor. In addition, in AML cell lines resistant to selinexor, the combination of the AKT inhibitor MK-220 with selinexor showed a synergistic effect together with an increase in the nuclear localization of FOXO3A where it executes its proapoptotic transcriptional activity.
Inspired by previous studies showing that differentiation is closely linked to kinase signaling and response to kinase inhibitors (92), Pedicona et al. (111) tested the idea that inducing differentiation in AML would reshape kinase networks into topologies that confer sensitivity to kinase-targeted drugs. Phosphoproteomics and proteomics showed that inhibitors of the lysine-specific demethylase 1 (LSD1) rewired kinase signaling in AML cells in a way that increased the activity of the kinase MEK and suppressed the activity of other kinases and feedback loops. Consequently, AML cell lines and about half of the 17 primary AML samples tested were primed for sensitivity to the MEK inhibitor trametinib. Phosphoproteomics analysis showed that cases that responded to sequential treatment with LSD1 inhibitors and trametinib presented KRAS mutations and high MEK activity, whereas those with NRAS mutations and high MTOR activity were poor responders. This study revealed the MEK pathway as a mechanism of resistance to LSD1 inhibitors in AML, and more importantly, it provided a rationale to modulate kinase network circuitry to potentially overcome therapeutic resistance to kinase inhibitors. This approach targets both epigenetics and signaling processes, which are key for the “two-hit” theory of leukemogenesis (10).
Another study used proteomics and phosphoproteomics to identify and rationalize the synergistic effect between FLT3 and autophagy inhibitors in AML cells positive for FLT3-ITD mutations. Koschade et al. (112) used quantification of translation changes by proteomics to reveal global attenuation of translation, but an increase in the transcription and synthesis of proteins involved in autophagy, upon FLT3 inhibition. Phosphoproteomics analysis showed an increase in the phosphorylation of proteins related to autophagy and MTOR signaling after pharmacological inhibition of FLT3. Functional studies demonstrated that FLT3 inhibitors induced autophagy in cell lines with FLT3-ITD mutations, but not in cells with WT FLT3 through a pathway that involves inhibition of MTOR and activation of ULK1. Autophagy is a cytoprotective mechanism that increases survival. Genetic or chemical inhibition of the autophagy induced by FLT3 inhibitors in cell lines with FLT3-ITD increased the sensitivity of these cells to FLT3 inhibitors. Consistently, the combination of FLT3 inhibitors and autophagy inhibitors synergistically reduced the viability of FLT3-ITD primary blast from AML patients ex vivo and reduced the tumor burthen in FLT3-ITD cell line and PDX models.
Single Cell Proteomics
MS-based proteomics and phosphoproteomics are providing valuable information on AML biology. However, these analyses, so far carried out on bulk cell populations, do not capture biological clonality within complex cell systems. Therefore, since AML show highly intratumor heterogeneity, single cell resolution would provide important information for targeting the entire leukemia rather than the dominant AML subclones. While single cell methods based on nucleotide sequencing are increasingly used, very recent improvements in mass spectrometry sensitivity also allow quantifying ∼1000 proteins per single cell across thousands of individual cells (113). This approach was able to recapitulate the hierarchy of AML populations obtained by flow cytometry analysis (114) and therefore it has the potential to provide new information on AML intratumor heterogeneity not attainable with standard proteomic methods.
Implementation of MS-Based Proteomics in Clinical Settings
Despite recent enormous technological and conceptual advances, broad implementation of MS-based proteomics and phosphoproteomics in routine clinical environments has not yet materialized. Issues with analyte stability, complexity of workflows, reproducibility of protein extraction, and cost make translation of these techniques to the clinic challenging. Validation and implementation of candidate markers in clinical settings require precise and specific protein quantification approaches that are harmonized across laboratories and run in a multiplexed manner with sufficient throughput for the information to be actionable (114, 115, 116). These requirements are better addressed by targeted MS approaches, like selected or parallel reaction monitoring, that focus the full analytic capacity of the instrument on a discrete number of analytes of interest previously identified by shoot-gun proteomics. The FDA considers all targeted MS proteomics approaches run in clinical laboratories as in vitro diagnostic medical devices (77). Specific guidelines for LC-MS–based tests are not yet issued, but the studies required for their clearance or approval will likely follow the standards of other in vitro diagnostic medical device tests (77). While to our knowledge no LC-MS–based targeted proteomics have been cleared or approved by the FDA by 2022 (75, 77, 117), a small number of laboratory developed tests based on MS proteomics have been developed, and some of them are implemented as end points in clinical trials in the field of cancer (e.g., NCT04389112) and other diseases (e.g., NCT04049019, NCT04514744).
Conclusions and Outlook
Proteomics and phosphoproteomics provide biological information that cannot always be extracted from genomics and transcriptomics data. Protein expression rarely overlap with mRNA expression, especially for mitochondrial proteins, an organelle targeted by new AML treatments. Similarly, protein expression may not correlate with their extent of activation. Protein phosphorylation is closer to protein function and can be used to infer the activities of kinases, which are enzymes that participate in the regulation of essentially all biological processes in normal and cancer cells (118). Therefore, proteomics and phosphoproteomics approaches—alone or integrated with genomics and transcriptomics—provide new mechanistic insights into disease pathogenesis from which to identify drug targets and biomarkers of drug response, to be used as input of predictive models, and to rationalize drug combinations that synergistically induce AML cell death. Application of these concepts more broadly will advance precision medicine and may also be used for analyzing and interpreting single cell proteomics data, now that instruments with the required sensitivity are becoming available (119), thus contributing to the deconvolution of the intratumor complexity that characterizes AML. Proteomics and phosphoproteomics approaches are therefore likely to play a pivotal role in the design and implementation of precision medicine in AML in the near future.
Conflict of interest
P. R. C. is an academic co-founder of Kinomica Ltd. The other author declares that they have no conflicts of interest with the contents of this article.
Acknowledgments
This work was funded by Higher Education Funding Council for England (HEFCE), Cancer Research UK (C15966/A24375), and Blood Cancer UK (20008).
Author contributions
P. C. and P. R. C. conceptualization; P. C. and P. R. C. writing—original draft.
References
- 1.Khwaja A., Bjorkholm M., Gale R.E., Levine R.L., Jordan C.T., Ehninger G., et al. Acute myeloid leukaemia. Nat. Rev. Dis. Primers. 2016;2:16010. doi: 10.1038/nrdp.2016.10. [DOI] [PubMed] [Google Scholar]
- 2.Quintás-Cardama A., Kantarjian H., Cortes J. Imatinib and beyond—exploring the full potential of targeted therapy for CML. Nat. Rev. Clin. Oncol. 2009;6:535–543. doi: 10.1038/nrclinonc.2009.112. [DOI] [PubMed] [Google Scholar]
- 3.Garber K. Kinase inhibitors overachieve in CLL. Nat. Rev. Drug Discov. 2014;13:162–164. doi: 10.1038/nrd4259. [DOI] [PubMed] [Google Scholar]
- 4.Charrot S., Armes H., Rio-Machin A., Fitzgibbon J. AML through the prism of molecular genetics. Br. J. Haematol. 2020;188:49–62. doi: 10.1111/bjh.16356. [DOI] [PubMed] [Google Scholar]
- 5.Rio-Machin A., Vulliamy T., Hug N., Walne A., Tawana K., Cardoso S., et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat. Commun. 2020;11:1044. doi: 10.1038/s41467-020-14829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caldwell J.T., Ge Y., Taub J.W. Prognosis and management of acute myeloid leukemia in patients with down syndrome. Expert Rev. Hematol. 2014;7:831–840. doi: 10.1586/17474086.2014.959923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alter B.P. Fanconi anemia and the development of leukemia. Best Pract. Res. Clin. Haematol. 2014;27:214–221. doi: 10.1016/j.beha.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bochtler T., Frohling S., Kramer A. Role of chromosomal aberrations in clonal diversity and progression of acute myeloid leukemia. Leukemia. 2015;29:1243–1252. doi: 10.1038/leu.2015.32. [DOI] [PubMed] [Google Scholar]
- 9.Gilliland D.G., Griffin J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 10.Narayan N., Huntly B.J.P. Targeting AML at the intersection of epigenetics and signaling. Sci. Signal. 2022;15 doi: 10.1126/scisignal.abo0059. [DOI] [PubMed] [Google Scholar]
- 11.Bonnet D., Dick J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 12.Bhatia M., Wang J.C., Kapp U., Bonnet D., Dick J.E. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 1997;94:5320–5325. doi: 10.1073/pnas.94.10.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miraki-Moud F., Anjos-Afonso F., Hodby K.A., Griessinger E., Rosignoli G., Lillington D., et al. Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proc. Natl. Acad. Sci. U. S. A. 2013;110:13576–13581. doi: 10.1073/pnas.1301891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griessinger E., Anjos-Afonso F., Pizzitola I., Rouault-Pierre K., Vargaftig J., Taussig D., et al. A niche-like culture system allowing the maintenance of primary human acute myeloid leukemia-initiating cells: a new tool to decipher their chemoresistance and self-renewal mechanisms. Stem Cells Transl. Med. 2014;3:520–529. doi: 10.5966/sctm.2013-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter J.L., Hege K., Yang J., Kalpage H.A., Su Y., Edwards H., et al. Targeting multiple signaling pathways: the new approach to acute myeloid leukemia therapy. Signal Transduct. Target. Ther. 2020;5:288. doi: 10.1038/s41392-020-00361-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett J.M., Catovsky D., Daniel M.T., Flandrin G., Galton D.A., Gralnick H.R., et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976;33:451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 17.Dohner H., Wei A.H., Appelbaum F.R., Craddock C., DiNardo C.D., Dombret H., et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140:1345–1377. doi: 10.1182/blood.2022016867. [DOI] [PubMed] [Google Scholar]
- 18.Estey E.H. Acute myeloid leukemia: 2021 update on risk-stratification and management. Am. J. Hematol. 2020;95:1368–1398. doi: 10.1002/ajh.25975. [DOI] [PubMed] [Google Scholar]
- 19.Hwang S.M. Classification of acute myeloid leukemia. Blood Res. 2020;55:S1–S4. doi: 10.5045/br.2020.S001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daver N., Schlenk R.F., Russell N.H., Levis M.J. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299–312. doi: 10.1038/s41375-018-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim E.S. Midostaurin: first global approval. Drugs. 2017;77:1251–1259. doi: 10.1007/s40265-017-0779-0. [DOI] [PubMed] [Google Scholar]
- 22.Dhillon S. Gilteritinib: first global approval. Drugs. 2019;79:331–339. doi: 10.1007/s40265-019-1062-3. [DOI] [PubMed] [Google Scholar]
- 23.Marcucci G., Maharry K., Wu Y.Z., Radmacher M.D., Mrozek K., Margeson D., et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J. Clin. Oncol. 2010;28:2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montalban-Bravo G., DiNardo C.D. The role of IDH mutations in acute myeloid leukemia. Future Oncol. 2018;14:979–993. doi: 10.2217/fon-2017-0523. [DOI] [PubMed] [Google Scholar]
- 25.Kim E.S. Enasidenib: first global approval. Drugs. 2017;77:1705–1711. doi: 10.1007/s40265-017-0813-2. [DOI] [PubMed] [Google Scholar]
- 26.Dhillon S. Ivosidenib: first global approval. Drugs. 2018;78:1509–1516. doi: 10.1007/s40265-018-0978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J.D., Zhang T.J., Xu Z.J., Gu Y., Ma J.C., Li X.X., et al. BCL2 overexpression: clinical implication and biological insights in acute myeloid leukemia. Diagn. Pathol. 2019;14:68. doi: 10.1186/s13000-019-0841-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andreeff M., Jiang S., Zhang X., Konopleva M., Estrov Z., Snell V.E., et al. Expression of Bcl-2-related genes in normal and AML progenitors: changes induced by chemotherapy and retinoic acid. Leukemia. 1999;13:1881–1892. doi: 10.1038/sj.leu.2401573. [DOI] [PubMed] [Google Scholar]
- 29.Singh R., Letai A., Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell. Biol. 2019;20:175–193. doi: 10.1038/s41580-018-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J., et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 31.DiNardo C.D., Pratz K., Pullarkat V., Jonas B.A., Arellano M., Becker P.S., et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17. doi: 10.1182/blood-2018-08-868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shallis R.M., Bewersdorf J.P., Boddu P.C., Zeidan A.M. Hedgehog pathway inhibition as a therapeutic target in acute myeloid leukemia. Expert Rev. Anticancer Ther. 2019;19:717–729. doi: 10.1080/14737140.2019.1652095. [DOI] [PubMed] [Google Scholar]
- 33.Yao Z., Han L., Chen Y., He F., Sun B., Kamar S., et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9:701. doi: 10.1038/s41419-018-0647-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaudhry P., Singh M., Triche T.J., Guzman M., Merchant A.A. GLI3 repressor determines hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood. 2017;129:3465–3475. doi: 10.1182/blood-2016-05-718585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoy S.M. Glasdegib: first global approval. Drugs. 2019;79:207–213. doi: 10.1007/s40265-018-1047-7. [DOI] [PubMed] [Google Scholar]
- 36.Godwin C.D., Gale R.P., Walter R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia. 2017;31:1855–1868. doi: 10.1038/leu.2017.187. [DOI] [PubMed] [Google Scholar]
- 37.Jen E.Y., Ko C.W., Lee J.E., Del Valle P.L., Aydanian A., Jewell C., et al. FDA approval: gemtuzumab ozogamicin for the treatment of adults with newly diagnosed CD33-positive acute myeloid leukemia. Clin. Cancer Res. 2018;24:3242–3246. doi: 10.1158/1078-0432.CCR-17-3179. [DOI] [PubMed] [Google Scholar]
- 38.Shin J.Y., Hu W., Naramura M., Park C.Y. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J. Exp. Med. 2014;211:217–231. doi: 10.1084/jem.20131128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malaise M., Steinbach D., Corbacioglu S. Clinical implications of c-Kit mutations in acute myelogenous leukemia. Curr. Hematol. Malig. Rep. 2009;4:77–82. doi: 10.1007/s11899-009-0011-8. [DOI] [PubMed] [Google Scholar]
- 40.Brandao L., Migdall-Wilson J., Eisenman K., Graham D.K. TAM receptors in leukemia: expression, signaling, and therapeutic implications. Crit. Rev. Oncog. 2011;16:47–63. doi: 10.1615/critrevoncog.v16.i1-2.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou C., Du J., Zhao L., Liu W., Zhao T., Liang H., et al. GLI1 reduces drug sensitivity by regulating cell cycle through PI3K/AKT/GSK3/CDK pathway in acute myeloid leukemia. Cell Death Dis. 2021;12:231. doi: 10.1038/s41419-021-03504-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee D.J., Zeidner J.F. Cyclin-dependent kinase (CDK) 9 and 4/6 inhibitors in acute myeloid leukemia (AML): a promising therapeutic approach. Expert Opin. Investig. Drugs. 2019;28:989–1001. doi: 10.1080/13543784.2019.1678583. [DOI] [PubMed] [Google Scholar]
- 43.Boffo S., Damato A., Alfano L., Giordano A. CDK9 inhibitors in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018;37:36. doi: 10.1186/s13046-018-0704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uras I.Z., Sexl V., Kollmann K. CDK6 inhibition: a novel approach in AML management. Int. J. Mol. Sci. 2020;21:2528. doi: 10.3390/ijms21072528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Opferman J.T., Letai A., Beard C., Sorcinelli M.D., Ong C.C., Korsmeyer S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 46.Glaser S.P., Lee E.F., Trounson E., Bouillet P., Wei A., Fairlie W.D., et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26:120–125. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiang W., Yang C.Y., Bai L. MCL-1 inhibition in cancer treatment. Onco Targets Ther. 2018;11:7301–7314. doi: 10.2147/OTT.S146228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tron A.E., Belmonte M.A., Adam A., Aquila B.M., Boise L.H., Chiarparin E., et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018;9:5341. doi: 10.1038/s41467-018-07551-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishizawa J., Kojima K., Chachad D., Ruvolo P., Ruvolo V., Jacamo R.O., et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci. Signal. 2016;9 doi: 10.1126/scisignal.aac4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chamoun K., Borthakur G. Investigational CHK1 inhibitors in early stage clinical trials for acute myeloid leukemia. Expert Opin. Investig. Drugs. 2018;27:661–666. doi: 10.1080/13543784.2018.1508448. [DOI] [PubMed] [Google Scholar]
- 51.Padella A., Ghelli Luserna Di Rora A., Marconi G., Ghetti M., Martinelli G., Simonetti G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J. Hematol. Oncol. 2022;15:10. doi: 10.1186/s13045-022-01228-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang D.D., Tang Q., Kong Y., Rong T., Wang Q., Li N., et al. MDM2 inhibitor APG-115 exerts potent antitumor activity and synergizes with standard-of-care agents in preclinical acute myeloid leukemia models. Cell Death Discov. 2021;7:90. doi: 10.1038/s41420-021-00465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.San Jose-Eneriz E., Gimenez-Camino N., Agirre X., Prosper F. HDAC inhibitors in acute myeloid leukemia. Cancers (Basel) 2019;11:1794. doi: 10.3390/cancers11111794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dafflon C., Craig V.J., Mereau H., Grasel J., Schacher Engstler B., Hoffman G., et al. Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia. 2017;31:1269–1277. doi: 10.1038/leu.2016.327. [DOI] [PubMed] [Google Scholar]
- 55.Reyes-Garau D., Ribeiro M.L., Roue G. Pharmacological targeting of BET bromodomain proteins in acute myeloid leukemia and malignant lymphomas: from molecular characterization to clinical applications. Cancers (Basel) 2019;11:1483. doi: 10.3390/cancers11101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan D.S., Bedard P.L., Kuruvilla J., Siu L.L., Razak A.R. Promising SINEs for embargoing nuclear-cytoplasmic export as an anticancer strategy. Cancer Discov. 2014;4:527–537. doi: 10.1158/2159-8290.CD-13-1005. [DOI] [PubMed] [Google Scholar]
- 57.Kojima K., Kornblau S.M., Ruvolo V., Dilip A., Duvvuri S., Davis R.E., et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121:4166–4174. doi: 10.1182/blood-2012-08-447581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ranganathan P., Yu X., Na C., Santhanam R., Shacham S., Kauffman M., et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012;120:1765–1773. doi: 10.1182/blood-2012-04-423160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Beauchamp L., Himonas E., Helgason G.V. Mitochondrial metabolism as a potential therapeutic target in myeloid leukaemia. Leukemia. 2022;36:1–12. doi: 10.1038/s41375-021-01416-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Molina J.R., Sun Y., Protopopova M., Gera S., Bandi M., Bristow C., et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018;24:1036–1046. doi: 10.1038/s41591-018-0052-4. [DOI] [PubMed] [Google Scholar]
- 61.Jordan C.T., Upchurch D., Szilvassy S.J., Guzman M.L., Howard D.S., Pettigrew A.L., et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14:1777–1784. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 62.Majeti R., Chao M.P., Alizadeh A.A., Pang W.W., Jaiswal S., Gibbs K.D., Jr., et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sterner R.C., Sterner R.M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69. doi: 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giudice G., Petsalaki E. Proteomics and phosphoproteomics in precision medicine: applications and challenges. Brief. Bioinform. 2019;20:767–777. doi: 10.1093/bib/bbx141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borthakur G., Popplewell L., Boyiadzis M., Foran J., Platzbecker U., Vey N., et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RAS-mutant relapsed or refractory myeloid malignancies. Cancer. 2016;122:1871–1879. doi: 10.1002/cncr.29986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stone R.M., Mandrekar S.J., Sanford B.L., Laumann K., Geyer S., Bloomfield C.D., et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017;377:454–464. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cucchi D.G.J., Van Alphen C., Zweegman S., Van Kuijk B., Kwidama Z.J., Al Hinai A., et al. Phosphoproteomic characterization of primary AML samples and relevance for response toward FLT3-inhibitors. Hemasphere. 2021;5 doi: 10.1097/HS9.0000000000000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fischer T., Stone R.M., Deangelo D.J., Galinsky I., Estey E., Lanza C., et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 2010;28:4339–4345. doi: 10.1200/JCO.2010.28.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gholami A.M., Hahne H., Wu Z., Auer F.J., Meng C., Wilhelm M., et al. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013;4:609–620. doi: 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 70.Sousa A., Dugourd A., Memon D., Petursson B., Petsalaki E., Saez-Rodriguez J., et al. Pan-cancer landscape of protein activities identifies drivers of signalling dysregulation and patient survival. bioRxiv. 2021 doi: 10.1101/2021.06.09.447741. [preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hijazi M., Smith R., Rajeeve V., Bessant C., Cutillas P.R. Reconstructing kinase network topologies from phosphoproteomics data reveals cancer-associated rewiring. Nat. Biotechnol. 2020;38:493–502. doi: 10.1038/s41587-019-0391-9. [DOI] [PubMed] [Google Scholar]
- 72.Casado P., Hijazi M., Gerdes H., Cutillas P.R. Implementation of clinical phosphoproteomics and proteomics for personalized medicine. Methods Mol. Biol. 2022;2420:87–106. doi: 10.1007/978-1-0716-1936-0_8. [DOI] [PubMed] [Google Scholar]
- 73.Cutillas P.R. Role of phosphoproteomics in the development of personalized cancer therapies. Proteomics Clin. Appl. 2015;9:383–395. doi: 10.1002/prca.201400104. [DOI] [PubMed] [Google Scholar]
- 74.Macklin A., Khan S., Kislinger T. Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clin. Proteomics. 2020;17:17. doi: 10.1186/s12014-020-09283-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He T. Implementation of proteomics in clinical trials. Proteomics Clin. Appl. 2019;13 doi: 10.1002/prca.201800198. [DOI] [PubMed] [Google Scholar]
- 76.Bell A.W., Deutsch E.W., Au C.E., Kearney R.E., Beavis R., Sechi S., et al. A HUPO test sample study reveals common problems in mass spectrometry-based proteomics. Nat. Methods. 2009;6:423–430. doi: 10.1038/nmeth.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang B., Whiteaker J.R., Hoofnagle A.N., Baird G.S., Rodland K.D., Paulovich A.G. Clinical potential of mass spectrometry-based proteogenomics. Nat. Rev. Clin. Oncol. 2019;16:256–268. doi: 10.1038/s41571-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mertins P., Tang L.C., Krug K., Clark D.J., Gritsenko M.A., Chen L., et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018;13:1632–1661. doi: 10.1038/s41596-018-0006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Montoya A., Beltran L., Casado P., Rodriguez-Prados J.C., Cutillas P.R. Characterization of a TiO(2) enrichment method for label-free quantitative phosphoproteomics. Methods. 2011;54:370–378. doi: 10.1016/j.ymeth.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bakalarski C.E., Kirkpatrick D.S. A biologist's field guide to multiplexed quantitative proteomics. Mol. Cell. Proteomics. 2016;15:1489–1497. doi: 10.1074/mcp.O115.056986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu K.H., Snyder M. Omics profiling in precision oncology. Mol. Cell. Proteomics. 2016;15:2525–2536. doi: 10.1074/mcp.O116.059253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ruggles K.V., Krug K., Wang X., Clauser K.R., Wang J., Payne S.H., et al. Methods, tools and current perspectives in proteogenomics. Mol. Cell. Proteomics. 2017;16:959–981. doi: 10.1074/mcp.MR117.000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leutert M., Entwisle S.W., Villen J. Decoding post-translational modification crosstalk with proteomics. Mol. Cell. Proteomics. 2021;20:100129. doi: 10.1016/j.mcpro.2021.100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tyagi P., Bhide M. Development of a bioinformatics platform for analysis of quantitative transcriptomics and proteomics data: the OMnalysis. PeerJ. 2021;9 doi: 10.7717/peerj.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Casado P., Rodriguez-Prados J.C., Cosulich S.C., Guichard S., Vanhaesebroeck B., Joel S., et al. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013;6 doi: 10.1126/scisignal.2003573. [DOI] [PubMed] [Google Scholar]
- 86.Piersma S.R., Valles-Marti A., Rolfs F., Pham T.V., Henneman A.A., Jimenez C.R. Inferring kinase activity from phosphoproteomic data: tool comparison and recent applications. Mass Spectrom. Rev. 2022 doi: 10.1002/mas.21808. [DOI] [PubMed] [Google Scholar]
- 87.Beekhof R., van Alphen C., Henneman A.A., Knol J.C., Pham T.V., Rolfs F., et al. INKA, an integrative data analysis pipeline for phosphoproteomic inference of active kinases. Mol. Syst. Biol. 2019;15 doi: 10.15252/msb.20198981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Alphen C., Cloos J., Beekhof R., Cucchi D.G.J., Piersma S.R., Knol J.C., et al. Phosphotyrosine-based phosphoproteomics for target identification and drug response prediction in AML cell lines. Mol. Cell. Proteomics. 2020;19:884–899. doi: 10.1074/mcp.RA119.001504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aasebo E., Forthun R.B., Berven F., Selheim F., Hernandez-Valladares M. Global cell proteome profiling, phospho-signaling and quantitative proteomics for identification of new biomarkers in acute myeloid leukemia patients. Curr. Pharm. Biotechnol. 2016;17:52–70. doi: 10.2174/1389201016666150826115626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gillet J.P., Calcagno A.M., Varma S., Marino M., Green L.J., Vora M.I., et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. U. S. A. 2011;108:18708–18713. doi: 10.1073/pnas.1111840108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aasebo E., Vaudel M., Mjaavatten O., Gausdal G., Van der Burgh A., Gjertsen B.T., et al. Performance of super-SILAC based quantitative proteomics for comparison of different acute myeloid leukemia (AML) cell lines. Proteomics. 2014;14:1971–1976. doi: 10.1002/pmic.201300448. [DOI] [PubMed] [Google Scholar]
- 92.Casado P., Wilkes E.H., Miraki-Moud F., Hadi M.M., Rio-Machin A., Rajeeve V., et al. Proteomic and genomic integration identifies kinase and differentiation determinants of kinase inhibitor sensitivity in leukemia cells. Leukemia. 2018;32:1818–1822. doi: 10.1038/s41375-018-0032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alanazi B., Munje C.R., Rastogi N., Williamson A.J.K., Taylor S., Hole P.S., et al. Integrated nuclear proteomics and transcriptomics identifies S100A4 as a therapeutic target in acute myeloid leukemia. Leukemia. 2020;34:427–440. doi: 10.1038/s41375-019-0596-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aasebo E., Berven F.S., Bartaula-Brevik S., Stokowy T., Hovland R., Vaudel M., et al. Proteome and phosphoproteome changes associated with prognosis in acute myeloid leukemia. Cancers (Basel) 2020;12:709. doi: 10.3390/cancers12030709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Malani D., Kumar A., Bruck O., Kontro M., Yadav B., Hellesoy M., et al. Implementing a functional precision medicine tumor board for acute myeloid leukemia. Cancer Discov. 2022;12:388–401. doi: 10.1158/2159-8290.CD-21-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hernandez-Valladares M., Wangen R., Aasebo E., Reikvam H., Berven F.S., Selheim F., et al. Proteomic studies of primary acute myeloid leukemia cells derived from patients before and during disease-stabilizing treatment based on all-trans retinoic acid and valproic acid. Cancers (Basel) 2021;13:2143. doi: 10.3390/cancers13092143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nguyen N.H.K., Wu H., Tan H., Peng J., Rubnitz J.E., Cao X., et al. Global proteomic profiling of pediatric AML: a pilot study. Cancers (Basel) 2021;13:3161. doi: 10.3390/cancers13133161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jayavelu A.K., Wolf S., Buettner F., Alexe G., Haupl B., Comoglio F., et al. The proteogenomic subtypes of acute myeloid leukemia. Cancer Cell. 2022;40:301–317.e12. doi: 10.1016/j.ccell.2022.02.006. [DOI] [PubMed] [Google Scholar]
- 99.Kramer M.H., Zhang Q., Sprung R., Day R.B., Erdmann-Gilmore P., Li Y., et al. Proteomic and phosphoproteomic landscapes of acute myeloid leukemia. Blood. 2022;140:1533–1548. doi: 10.1182/blood.2022016033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Caplan M., Wittorf K.J., Weber K.K., Swenson S.A., Gilbreath T.J., Willow Hynes-Smith R., et al. Multi-omics reveals mitochondrial metabolism proteins susceptible for drug discovery in AML. Leukemia. 2022;36:1296–1305. doi: 10.1038/s41375-022-01518-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gosline S.J.C., Tognon C., Nestor M., Joshi S., Modak R., Damnernsawad A., et al. Proteomic and phosphoproteomic measurements enhance ability to predict ex vivo drug response in AML. Clin. Proteomics. 2022;19:30. doi: 10.1186/s12014-022-09367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen P., Levis M., Brown P., Kim K.T., Allebach J., Small D. FLT3/ITD mutation signaling includes suppression of SHP-1. J. Biol. Chem. 2005;280:5361–5369. doi: 10.1074/jbc.M411974200. [DOI] [PubMed] [Google Scholar]
- 103.Casado P., Rio-Machin A., Miettinen J.J., Bewicke-Copley F., Rouault-Pierre K., Krizsan S., et al. Integrative phosphoproteomics defines two biologically distinct groups of KMT2A rearranged acute myeloid leukaemia with different drug response phenotypes. bioRxiv. 2022 doi: 10.1101/2022.11.10.515949. [preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gerdes H., Casado P., Dokal A., Hijazi M., Akhtar N., Osuntola R., et al. Drug ranking using machine learning systematically predicts the efficacy of anti-cancer drugs. Nat. Commun. 2021;12:1850. doi: 10.1038/s41467-021-22170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tjoa E., Guan C. A Survey on explainable artificial intelligence (XAI): toward medical XAI. IEEE Trans. Neural Netw. Learn. Syst. 2021;32:4793–4813. doi: 10.1109/TNNLS.2020.3027314. [DOI] [PubMed] [Google Scholar]
- 106.Volovici V., Syn N.L., Ercole A., Zhao J.J., Liu N. Steps to avoid overuse and misuse of machine learning in clinical research. Nat. Med. 2022;28:1996–1999. doi: 10.1038/s41591-022-01961-6. [DOI] [PubMed] [Google Scholar]
- 107.Narayan R.S., Molenaar P., Teng J., Cornelissen F.M.G., Roelofs I., Menezes R., et al. A cancer drug atlas enables synergistic targeting of independent drug vulnerabilities. Nat. Commun. 2020;11:2935. doi: 10.1038/s41467-020-16735-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murray H.C., Enjeti A.K., Kahl R.G.S., Flanagan H.M., Sillar J., Skerrett-Byrne D.A., et al. Quantitative phosphoproteomics uncovers synergy between DNA-PK and FLT3 inhibitors in acute myeloid leukaemia. Leukemia. 2021;35:1782–1787. doi: 10.1038/s41375-020-01050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhu Y., He X., Li S., Gan Y., Li Z., Wang H., et al. Phosphoproteomics profiling reveals a kinase network conferring acute myeloid leukaemia intrinsic chemoresistance and indicates HMGA1 phosphorylation as a potential influencer. Clin. Transl. Med. 2022;12 doi: 10.1002/ctm2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Emdal K.B., Palacio-Escat N., Wigerup C., Eguchi A., Nilsson H., Bekker-Jensen D.B., et al. Phosphoproteomics of primary AML patient samples reveals rationale for AKT combination therapy and p53 context to overcome selinexor resistance. Cell Rep. 2022;40:111177. doi: 10.1016/j.celrep.2022.111177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pedicona F., Casado P., Hijazi M., Gribben J.G., Rouault-Pierre K., Cutillas P.R. Targeting the lysine-specific demethylase 1 rewires kinase networks and primes leukemia cells for kinase inhibitor treatment. Sci. Signal. 2022;15 doi: 10.1126/scisignal.abl7989. [DOI] [PubMed] [Google Scholar]
- 112.Koschade S.E., Klann K., Shaid S., Vick B., Stratmann J.A., Tholken M., et al. Translatome proteomics identifies autophagy as a resistance mechanism to on-target FLT3 inhibitors in acute myeloid leukemia. Leukemia. 2022;36:2396–2407. doi: 10.1038/s41375-022-01678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kelly R.T. Single-cell proteomics: progress and prospects. Mol. Cell. Proteomics. 2020;19:1739–1748. doi: 10.1074/mcp.R120.002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schoof E.M., Furtwängler B., Üresin N., Rapin N., Savickas S., Gentil C., et al. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021;12:3341. doi: 10.1038/s41467-021-23667-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aasebo E., Mjaavatten O., Vaudel M., Farag Y., Selheim F., Berven F., et al. Freezing effects on the acute myeloid leukemia cell proteome and phosphoproteome revealed using optimal quantitative workflows. J. Proteomics. 2016;145:214–225. doi: 10.1016/j.jprot.2016.03.049. [DOI] [PubMed] [Google Scholar]
- 116.Casado P., Bilanges B., Rajeeve V., Vanhaesebroeck B., Cutillas P.R. Environmental stress affects the activity of metabolic and growth factor signaling networks and induces autophagy markers in MCF7 breast cancer cells. Mol. Cell. Proteomics. 2014;13:836–848. doi: 10.1074/mcp.M113.034751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Smejkal G.B., Rivas-Morello C., Chang J.H., Freeman E., Trachtenberg A.J., Lazarev A., et al. Thermal stabilization of tissues and the preservation of protein phosphorylation states for two-dimensional gel electrophoresis. Electrophoresis. 2011;32:2206–2215. doi: 10.1002/elps.201100170. [DOI] [PubMed] [Google Scholar]
- 118.Boys E.L., Liu J., Robinson P.J., Reddel R.R. Clinical applications of mass spectrometry-based proteomics in cancer: where are we? Proteomics. 2022 doi: 10.1002/pmic.202200238. [DOI] [PubMed] [Google Scholar]
- 119.Gross S., Rahal R., Stransky N., Lengauer C., Hoeflich K.P. Targeting cancer with kinase inhibitors. J. Clin. Invest. 2015;125:1780–1789. doi: 10.1172/JCI76094. [DOI] [PMC free article] [PubMed] [Google Scholar]