

Abstract
Mycobacterium tuberculosis (Mtb) infection is initiated by inhalation of bacteria into lung alveoli, where they are phagocytosed by resident macrophages. Intracellular Mtb replication induces the death of the infected macrophages and the release of bacterial aggregates. Here, we show that these aggregates can evade phagocytosis by killing macrophages in a contact‐dependent but uptake‐independent manner. We use time‐lapse fluorescence microscopy to show that contact with extracellular Mtb aggregates triggers macrophage plasma membrane perturbation, cytosolic calcium accumulation, and pyroptotic cell death. These effects depend on the Mtb ESX‐1 secretion system, however, this system alone cannot induce calcium accumulation and macrophage death in the absence of the Mtb surface‐exposed lipid phthiocerol dimycocerosate. Unexpectedly, we found that blocking ESX‐1‐mediated secretion of the EsxA/EsxB virulence factors does not eliminate the uptake‐independent killing of macrophages and that the 50‐kDa isoform of the ESX‐1‐secreted protein EspB can mediate killing in the absence of EsxA/EsxB secretion. Treatment with an ESX‐1 inhibitor reduces uptake‐independent killing of macrophages by Mtb aggregates, suggesting that novel therapies targeting this anti‐phagocytic mechanism could prevent the propagation of extracellular bacteria within the lung.
Keywords: calcium flux, EspB, plasma membrane damage, pyroptosis, time‐lapse fluorescence microscopy
Subject Categories: Autophagy & Cell Death; Membranes & Trafficking; Microbiology, Virology & Host Pathogen Interaction
Mycobacterium tuberculosis aggregates evade phagocytosis by inducing macrophage pyroptosis via a mechanism involving its type VII secretion system ESX‐1 and surface lipid PDIM.
Introduction
The success of Mycobacterium tuberculosis (Mtb) as a human pathogen hinges on the ability to survive attacks by the immune cells that mediate host defenses against lung infections. Tuberculosis infections are initiated by the inhalation and implantation of small numbers of bacteria in the lung alveoli, where they are rapidly phagocytosed by resident alveolar macrophages (Riley et al, 1995; Cohen et al, 2018). Upon phagocytosis, Mtb uses different strategies to adapt to the intracellular milieu, such as prevention of phagolysosome acidification (Sturgill‐Koszycki et al, 1994), metabolic adaptation (Warner, 2014), lysis of the phagosomal membrane, and escape into the cytosol (van der Wel et al, 2007; Simeone et al, 2012). Replication of cytosolic bacteria ultimately leads to lysis of the infected macrophage, which is a key process in spreading the infection to other cells (Davis & Ramakrishnan, 2009).
Lysis of infected macrophages in vitro results in rapid replication of Mtb on the debris of the dead cells and formation of extracellular bacterial aggregates (Lerner et al, 2017). Formation of Mtb bacterial aggregates is also observed in vivo in animal models of infection (Hoff et al, 2011; Repasy et al, 2013; Irwin et al, 2015) and in human patients (Kaplan et al, 2003; Timm et al, 2006; Hunter, 2011; Lerner et al, 2020; Dinkele et al, 2021; Rodel et al, 2021; Wells et al, 2021), and necrotic foci in the lungs are typically associated with a high burden of extracellular bacteria (Kaplan et al, 2003; Hoff et al, 2011; Hunter, 2011). Phagocytosis of extracellular Mtb aggregates by newly‐recruited macrophages triggers cycles of intracellular infection and host‐cell lysis, contributing to the progressive propagation of the bacteria (Dallenga et al, 2017; Mahamed et al, 2017). In the early stages of tuberculosis, immune cells are recruited to the site of infection by the pro‐inflammatory signaling associated with host‐cell death (Davis & Ramakrishnan, 2009; Repasy et al, 2015). At later stages, host‐cell death in granulomas, the multi‐cellular structures that encapsulate the bacteria and limit their propagation, can lead to caseation, cavitation, and dissemination of the infection to the airways, allowing transmission of Mtb to other hosts (Ehlers & Schaible, 2013). Despite the importance of these processes in tuberculosis pathogenesis, the host‐cell pathways that mediate cell death are still controversial and different forms of cell death have been reported (Mohareer et al, 2018), including apoptosis (Aguilo et al, 2013; Augenstreich et al, 2017), necrosis (Lee et al, 2006; Park et al, 2006), necroptosis (Zhao et al, 2017; Pajuelo et al, 2018), pyroptosis (Beckwith et al, 2020), and other non‐canonical forms of death mediated by type I interferon (Zhang et al, 2020) or tumor necrosis factor signaling (Roca et al, 2019).
Induction of host‐cell death by intracellular bacteria requires the Mtb ESX‐1 type VII secretion system (van der Wel et al, 2007; Simeone et al, 2012). This system secretes several proteins, including the EsxA/EsxB heterodimer, EspA, EspB, and EspC, which are required for Mtb virulence (Gröschel et al, 2016). In particular, the ESX‐1‐secreted EsxA protein has been shown to mediate the breakdown of phagolysosomal membrane integrity and translocation of Mtb into the cytosol (van der Wel et al, 2007; Simeone et al, 2012). Phagolysosomal membrane rupture also requires the complex lipid phthiocerol dimycocerosate (PDIM) displayed on the bacterial cell surface (Gröschel et al, 2016; Augenstreich et al, 2017). Rupture of the phagolysosome or cytosolic sensing of Mtb DNA can activate several different death pathways in the host cells (Wassermann et al, 2015; Zhao et al, 2017; Roca et al, 2019), while physical contact between cytosolic bacteria and the inner face of the host‐cell plasma membrane can lead to plasma membrane damage and death by pyroptosis (Beckwith et al, 2020). EsxA has also been shown to be required for the induction of macrophage death after phagocytosis of extracellular Mtb growing on the debris of dead host cells (Dallenga et al, 2017).
The killing of host cells by intracellular bacteria may promote tuberculosis pathogenesis by allowing Mtb to evade macrophage defense mechanisms (MacMicking, 2014) and to grow rapidly on the debris of dead host cells (Lerner et al, 2017). As an alternative to killing macrophages by intracellular bacteria, we considered the possibility that evasion of host‐cell defenses might also be achieved by killing macrophages by extracellular bacteria in an uptake‐independent manner. Previous studies have shown that intracellular Mtb can induce plasma membrane damage and inhibit plasma membrane repair in infected macrophages (Divangahi et al, 2009; Beckwith et al, 2020). Contact with extracellular Mtb also induces host‐cell plasma membrane damage and inflammasome activation, but it was not reported whether these events resulted in host‐cell death (Beckwith et al, 2020). In an earlier study, contact of macrophages with extracellular Mtb did not lead to cell death when uptake was inhibited with cytochalasin D treatment (Lee et al, 2006). In both studies, macrophages were exposed to single‐cell bacterial suspensions rather than aggregates of Mtb formed after the death of infected macrophages and extracellular growth on host‐cell debris.
In this study, we use time‐lapse fluorescence microscopy to investigate the dynamic interaction between macrophages and Mtb at the single‐cell level. We show that contact of macrophages with extracellular aggregates of Mtb results in pyroptotic host‐cell death in an uptake‐independent manner. The killing of macrophages by extracellular Mtb is dependent on aggregation per se because similar numbers of non‐aggregated individual bacteria are less efficient at inducing macrophage death upon contact. Before dying, macrophages exhibit signs of local plasma membrane perturbation at the site of contact with Mtb aggregates and progressive accumulation of calcium in the cytosol. These two processes require the Mtb ESX‐1 secretion system and the surface‐exposed lipid phthiocerol dimycocerosate (PDIM). Unexpectedly, we found that secretion and proteolytic processing of EspB is responsible for driving plasma membrane perturbation, calcium accumulation, and host‐cell death in the absence of EsxA/EsxB secretion, revealing previously unknown roles for this protein in Mtb pathogenesis.
Results
Mycobacterium tuberculosis aggregates induce contact‐dependent, uptake‐independent killing of macrophages
We used time‐lapse fluorescence microscopy to track individual mouse bone marrow‐derived macrophages (BMDMs) infected with Mtb expressing tdTomato (Movie EV1). After phagocytosis, intracellular Mtb replicates (Fig EV1A) and eventually kills and lyses host cells (Fig EV1B; Movies EV1 and EV2). Once released from lysed macrophages, Mtb replicates rapidly on the debris of the dead cells to form extracellular aggregates (Fig EV1A and C; Movies EV1 and EV2). As previously described (Dallenga et al, 2017; Mahamed et al, 2017), we observed that macrophages that interact with these bacterial aggregates eventually die (Fig 1A; Appendix Figs S1A and S2A), leading to uncontrolled bacterial proliferation on the host‐cell debris and formation of large extracellular Mtb aggregates (Movie EV1). Bacterial aggregates from axenic cultures induce macrophage death with comparable timing (Fig 1B and C; Movie EV3), demonstrating that neither a previous intracellular passage of the bacteria in macrophages nor the presence of cellular debris on the aggregates is required to induce macrophage death. Physical interaction between bacterial aggregates and macrophages is required to induce death of the infected cells, as highlighted by the observation that bystander macrophages near infected cells show the same survival as uninfected cells over the course of the infection (Fig 1D; Appendix Fig S2A).
Figure EV1. Intracellular growth of Mtb results in death and lysis of the infected macrophage, rapid extracellular growth on the host‐cell debris, and formation of large extracellular Mtb aggregates.
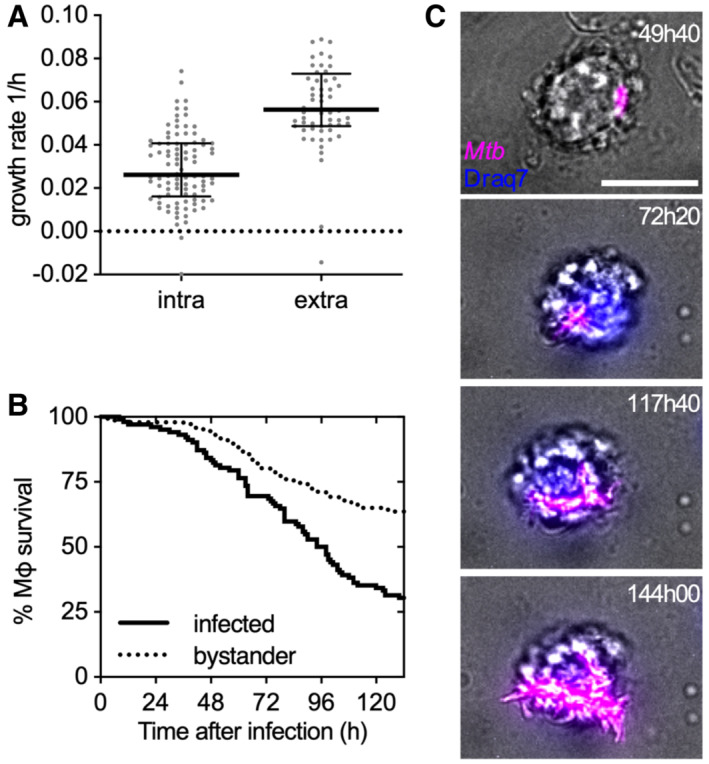
- Growth rate of individual Mtb microcolonies growing inside a macrophage (intra) or on the debris of a lysed macrophage (extra). Black lines indicate median values and interquartile ranges. (n = 96, 47 bacterial microcolonies, respectively)
- Percentage survival over time of infected versus uninfected bystander macrophages. (n = 110 and 102, respectively)
- Representative example of an intracellular Mtb microcolony that after lysis of the host macrophage (Draq7 positive cell) grows on the debris of the dead cell. Scale bar, 10 μm. Representative snapshots from Movie EV2.
Source data are available online for this figure.
Figure 1. Aggregates of Mtb kill macrophages in a contact‐dependent but uptake‐independent manner.
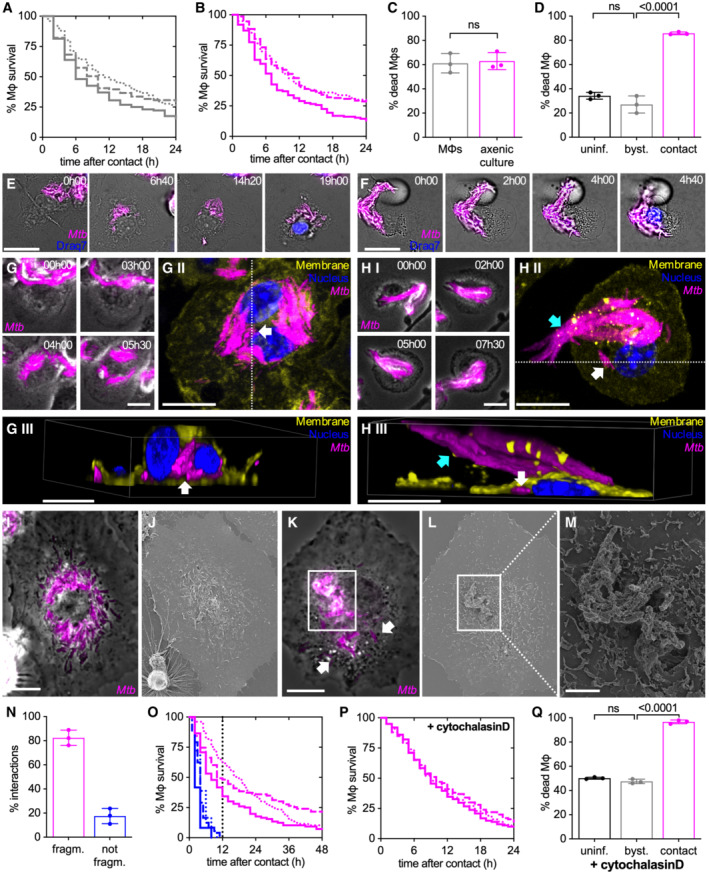
Mouse bone marrow‐derived macrophages (BMDMs) infected with Mtb Erd‐tdTomato and imaged by time‐lapse microscopy at 1‐ or 2‐ intervals for 132 h (A, C, N, O). BMDMs infected with aggregates of Mtb Erd‐tdTomato from axenic culture and imaged by time‐lapse microscopy at 1‐h intervals for 60 h (B–M, P, Q).
-
A, BPercentage of macrophages surviving after interaction with an extracellular Mtb aggregate originating from the debris of dead macrophages (A) or from axenic culture (B). Each line represents an independent experimental replicate (n = 3 replicates with ≥ 100 cells per replicate).
-
CPercentage of macrophages that die within the first 12 h after stable contact with an Mtb aggregate originating either from a dead macrophage (MΦ) or from an axenic culture. Each symbol represents the percentage of events for a single experimental replicate (n ≥ 100 events per replicate). Bars represent means and standard deviations; P‐value calculated using a t‐test ns, P‐values > 0.05.
-
DPercentage of macrophages that die by the end of the experiment (by 60 h post‐infection). Macrophages in “contact” represents the fraction of cells that interact with an Mtb aggregate during the course of the experiment. “Bystander” (byst.) macrophages are in the same sample as the infected ones but they do not establish physical contact with an Mtb aggregate during the course of the experiment. “Uninfected” (uninf.) macrophages are not exposed to Mtb. Each symbol represents the percentage of events for a single experimental replicate (n ≥ 50 events per replicate). Bars represent means and standard deviations; P‐value calculated using a one‐way ANOVA test, ns, P‐values > 0.05.
-
E, F(E) Example of a macrophage that interacts with an Mtb aggregate (00:00 h), fragments it (03:00 h), redistributes the bacteria in a “bullseye” pattern around the nucleus (12:00 h), and dies (16:00 h). (F) Example of a macrophage that stably interacts with an Mtb aggregate without fragmenting it (00:00–09:00 h) and ultimately dies (12:00 h). In (E and F), Draq7 staining of the nucleus (in blue) is used as a marker for cell death. Scale bars, 20 μm.
-
G, HBMDMs infected with aggregates of Mtb were imaged by time‐lapse microscopy (every 30 min for up to 13.5 h) followed by fixation, immunostaining (nuclei stained with Hoechst, membrane staining with anti‐CD45 antibody), and imaging by confocal microscopy. White arrows indicate intracellular bacteria, cyan arrows indicate extracellular bacteria. All scale bars, 10 μM. (G.I, H.I) Time‐lapse microscopy image series of macrophages that interacts with Mtb aggregates and fragment (G) or do not fragment (H) them. (G.II, H.II) Max intensity projection of confocal microscopy images of the same macrophages are shown in panels I. (G.III, H.III) 3‐D reconstruction of the cells imaged in panels II, images are cropped in x or y in the position indicated by the white dotted lines in panels II to show the inside of the cell.
-
I–MBMDMs infected with aggregates of Mtb were imaged by time‐lapse fluorescence microscopy followed by SEM. (I) Example of a macrophage showing the typical “bullseye” pattern of bacterial redistribution around the cell nucleus. Scale bar, 10 μm. (J) Correlative SEM image of (I). (K) Example of a macrophage interacting with an extracellular Mtb aggregate without complete fragmentation and redistribution of bacteria. White arrows indicate intracellular bacteria that have been detached from the main aggregate and internalized. Scale bar, 10 μm. Time‐lapse microscopy image series of these macrophages are shown in Appendix Fig S4A. (L, M) Correlative SEM image of (K). Scale bar, 2 μm (M).
-
NPercentage of macrophage‐Mtb aggregate interactions resulting (“fragm.,” pattern exemplified by panel E) or not resulting in fragmentation (“not fragm.,” pattern exemplified by panel F) of the Mtb aggregate. Quantification was performed by manual annotation of all the macrophage‐Mtb aggregate interactions observed in the time‐lapse microscopy movies. Each symbol represents the percentage of events for a single biological replicate (n ≥ 100 events per replicate). Bars represent means and standard deviations.
-
OPercentage of macrophages surviving over time after uptake and fragmentation of Mtb aggregates or after contact without fragmentation. Each line represents an independent biological replicate. The vertical dotted line marks the 12 h post‐interaction time‐point.
-
P, QBMDMs were treated with cytochalasin D to prevent bacterial uptake, infected with aggregates of Mtb, and imaged by time‐lapse microscopy at 1‐h intervals for 60 h. (P) Percentage survival over time for macrophages in contact with Mtb aggregates. Time 0 represents the time when stable contact with an aggregate begins. Each line represents an independent biological replicate (n = 3 replicates with ≥ 100 cells per replicate). (Q) Percentage of cytochalasin D‐treated macrophages that die by the end of the experiment (by 60 h post‐infection). Each symbol represents the percentage of events for a single biological replicate (n ≥ 100 events per replicate). Bars represent means and standard deviations; P‐value calculated using a one‐way ANOVA test; ns, P‐values > 0.05.
Source data are available online for this figure.
Time‐lapse microscopy reveals two different patterns of long‐term interaction of macrophages with Mtb aggregates. Approximately 80% of interactions between macrophages and extracellular Mtb aggregates result in fragmentation of the Mtb aggregate within 2–3 h after first contact and redistribution of the bacteria in a “bullseye” pattern around the host‐cell nucleus (Fig 1E and N; Movie EV4). In the remaining ~ 20% of interactions, macrophages remain in stable contact with extracellular Mtb aggregates for hours without any sign of fragmentation of the aggregate or perinuclear redistribution of bacteria (Fig 1F and N; Movie EV5). We hypothesized that the first pattern follows the uptake of the Mtb aggregate by the macrophage (Fig 1E), whereas the second pattern represents a stable association of the Mtb aggregate with the macrophage without uptake (Fig 1F). We tested this hypothesis using correlative single‐cell time‐lapse microscopy followed by macrophage plasma membrane immunostaining and confocal imaging or scanning electron microscopy (SEM). Bacterial aggregates that upon interaction with a macrophage get fragmented and acquire the “bullseye” pattern (Fig 1G.I–II; Appendix Fig S3A.I–II and C.I–II) are localized inside the macrophage (Fig 1G.III; Appendix Fig S3A.III and C.III) and are not visible on the surface of the macrophage in SEM images (Fig 1I and J; Appendix Fig S4A and C). Conversely, aggregates that do not undergo fragmentation and perinuclear redistribution (Fig 1H.I–II; Appendix Fig S3B.I–II and D.I–II) remain extracellular (Fig 1H.III; Appendix Fig S3B.III and D.III) and readily visible on the surface of the macrophage by SEM (Fig 1K–M; Appendix Fig S4A and D–I). Occasionally, some individual bacteria can be detached from the main extracellular aggregate visible on the surface of the cell and be internalized (Fig 1H.II–III and K; Appendix Fig S3B.II–III and D.II–III). These results are consistent with the notion that the first pattern represents intracellular bacterial aggregates whereas the second pattern represents extracellular bacterial aggregates.
Both these patterns are observed in macrophages that die upon interaction with a bacterial aggregate (Fig 1E, F and N). Notably, macrophages that do not take up and fragment the Mtb aggregates invariably die within 12 h of first contact with the aggregate (Fig 1O), while macrophages that take up and fragment an Mtb aggregate survive longer (Fig 1O). We thus asked whether extracellular Mtb aggregates may induce macrophage death prior to uptake. By treating macrophages with cytochalasin D, an actin polymerization inhibitor that blocks phagocytosis without affecting Mtb growth (Appendix Fig S5), we confirmed that uptake is not required for killing of macrophages by extracellular Mtb aggregates (Fig 1P; Appendix Figs S1B and S2B; Movies EV6 and EV7). We also observed that uninfected bystander macrophages treated with cytochalasin D survive over the same time course (Fig 1Q; Appendix Fig S2B; Movie EV6), further confirming that killing of macrophages by extracellular Mtb aggregates needs physical contact despite not requiring uptake.
Bacterial aggregation is important for the contact‐dependent, uptake‐independent killing of macrophages
Phagocytosis of single Mtb bacilli or aggregated Mtb induces macrophage death in a dose‐dependent manner (Fig EV2A and B) (Mahamed et al, 2017). Interestingly, as previously reported (Mahamed et al, 2017), we observe that aggregated bacteria kill more efficiently than similar amounts of single bacilli (Fig EV2A and B) leading to faster bacterial propagation (Fig EV2C and D). We thus asked whether bacterial aggregation per se is also important for the contact‐dependent, uptake‐independent killing of macrophages by Mtb. We found that macrophages treated with cytochalasin D to prevent bacterial uptake and challenged with increasing numbers of non‐aggregated bacteria (Fig 2A‐E) are killed in a dose‐dependent manner (Fig 2F). However, comparisons of macrophages challenged with similar numbers of aggregated or non‐aggregated bacteria (Fig 2A–E; cf. “high” and “aggregate”) show that aggregated bacteria are significantly more toxic than non‐aggregated bacteria (Fig 2F). We conclude that both uptake‐dependent and contact‐dependent (uptake‐independent) killing of macrophages by Mtb depends on both the number of bacteria and their aggregation status.
Figure EV2. Bacterial aggregation enhances the uptake‐dependent killing of macrophages and bacteria propagation.
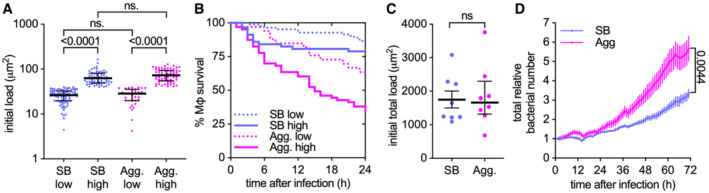
- Infected individual macrophages are binned into low and high initial loads according to the amount of single (SB) or aggregated (Agg) bacteria they internalize. The bacterial load is calculated as fluorescent area per macrophage and gates are set at < 40 μm2 (low) or > 40.0 μm2 (high) per macrophage. The area of one bacterium is included between 0.5 and 2 μm2. Each symbol represents the bacterial load of one individual macrophage (n = 82, 57, 33, and 63 macrophages, respectively). Bars represent the median and interquartile range. P‐values were calculated using a Krustal–Wallis test; ns, P‐values > 0.05.
- Percentage survival over time for individual macrophages with an initial bacterial load as indicated in panel A (n = 82, 57, 33, and 63 macrophages, respectively).
- Total initial bacterial load per microscopy field of view (332.80 × 332.80 μm2, approx. 100 cells/field of view). Each symbol represents one field of view (n = 8). Bars represent the mean and standard errors of the mean. P‐value calculated using an unpaired t‐test; P‐values > 0.05.
- Total relative bacterial load over time per microscopy field of view. Symbols represent the average bacterial load (n = 8) and bars represent standard errors of the mean. P‐values were calculated using an unpaired t‐test.
Source data are available online for this figure.
Figure 2. Bacterial aggregation is important for the contact‐dependent uptake‐independent killing of macrophages.
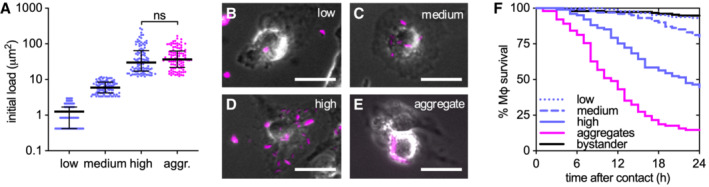
BMDMs were treated with cytochalasin D and infected with aggregated or non‐aggregated Mtb Erd‐tdTomato and imaged by time‐lapse microscopy at 1‐h intervals for 60 h.
-
AInitial bacterial load calculated as the total fluorescent area per macrophage. Infected individual macrophages are binned into low, medium, and high initial loads according to the number of single bacteria they are in contact with. For comparison, we selected macrophages in contact with aggregates (aggr.) of Mtb having a load similar to the high gate. Gates were set at < 3.0 μm2 (low), 3.0–12.0 μm2 (medium), or > 12.0 μm2 (high and aggregates) per macrophage (n ≥ 100 cells per gate). The area of one bacterium is included between 0.5 and 2 μm2. Each symbol represents the bacterial load of one macrophage. Bars represent the median and interquartile range. P‐value calculated using an unpaired Mann–Whitney test.
-
B–EExamples of macrophages infected with low (B), medium (C), or high (D) doses of non‐aggregated bacteria or with bacterial aggregates (E). Scale bars, 20 μm.
-
FPercentage survival over time for macrophages in contact with increasing doses of non‐aggregated bacteria or with bacterial aggregates. Time 0 for cells “in contact” represents the time when stable contact with the bacteria begins. Each line represents a biological replicate (n ≥ 100 cells per condition).
Source data are available online for this figure.
Plasma membrane perturbation in macrophages at the site of contact with extracellular Mtb aggregates
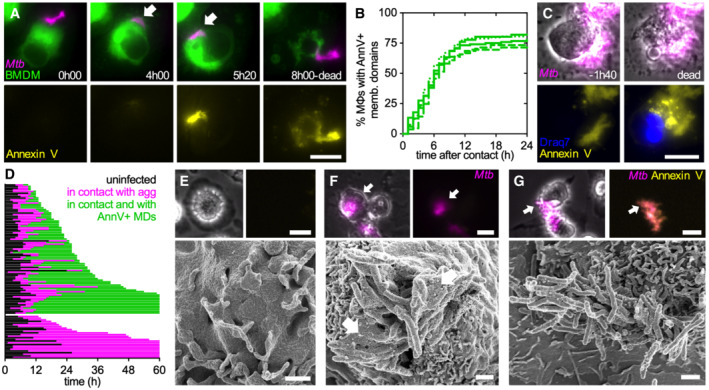
We used single‐cell time‐lapse fluorescence microscopy to identify events leading up to the death of macrophages in contact with Mtb aggregates. Cytochalasin D‐treated BMDMs establish stable interactions with extracellular Mtb aggregates despite being unable to internalize them (Figs 3A and EV3A and B). Staining with fluorescent Annexin V shows that approximately 75% of these macrophages (Fig 3B), display Annexin V‐positive plasma membrane domains at the site of contact with an Mtb aggregate, indicating the presence of exposed phosphatidylserine associated with a local plasma membrane perturbation (Figs 3A and EV3A–C; Movies EV7 and EV8). Affected macrophages become Annexin V‐positive over the entire plasma membrane only after death (Figs 3A and C, and EV3D; Movies EV7 and EV8) which happens within 11 ± 10 h after membrane perturbation (Fig 3D). Notably, the cells that develop local Annexin V‐positive membrane domains seem to die faster than the cells that do not show this pattern (Fig 3D). Correlative SEM revealed that the membrane of cytochalasin D‐treated Annexin V‐negative macrophages extends and partially covers the associated bacterial aggregate (Fig 3E and F). At later stages of the interaction, when the plasma membrane in contact with the Mtb aggregate becomes Annexin V‐positive, the membrane undergoes fragmentation and blebbing (Fig 3G). Aggregates that do not contact macrophages never become Annexin V‐positive (Fig EV3C). However, we observe Annexin V‐positive foci colocalize not only with markers for the macrophage plasma membrane (Fig EV3A and B) but also with more distal areas of the bacterial aggregates that do not stain positive for plasma membrane markers (Fig EV3B). Moreover, we see that upon macrophage death, aggregates in contact with dead cells retain some Annexin V‐positive material on their surface (Fig EV3C; Movie EV8). Vesicle budding and shedding is a common ESCRT III‐mediated membrane repair strategy that allows the removal of damaged portions of the plasma membrane and wound resealing (Jimenez et al, 2014). We therefore think that Annexin V‐positive foci might represent both areas of the damaged membrane as well as macrophage plasma membrane vesicles that were released and which stick to the hydrophobic surface of the bacterial aggregates. Interestingly, the fluorescence intensity of the Annexin V‐positive domains did not increase over time, but we detected several single intensity peaks (Movie EV8), suggesting that multiple discrete damaging events happen upon interaction with an Mtb aggreate.
Figure 3. The uptake‐independent killing of macrophages by Mtb aggregates involves intimate contact between the macrophage membrane and the bacterial aggregate.
-
ABMDMs expressing a membrane‐targeted tdTomato were treated with cytochalasin D, infected with aggregates of Mtb expressing GFP, and imaged by time‐lapse microscopy at 20‐min intervals for 24 h. Example of a BMDM interacting with an extracellular Mtb aggregate. The top panels show the macrophage plasma membrane and the bacterial aggregate. The intimate interaction between the macrophage plasma membrane and the bacterial aggregate begins at 04:00 h (indicated by the white arrow). The bottom panels show the Annexin V‐647 positive plasma membrane domain that appears at 5 h 20 min and co‐localizes with the bacterial aggregate. At 08:00 h, plasma membrane integrity is lost (top panel) and Annexin V accumulates throughout the dead cell (bottom panel). Scale bar, 20 μm.
-
BPercentage of cytochalasin D‐treated macrophages in contact with Mtb aggregates that show local Annexin V‐positive membrane domains at the site of contact with the bacteria over time. Time 0 represents the time when stable contact with an aggregate begins. Cells were imaged at 1‐h intervals for 60 h. Each line represents an independent biological replicate (n = 5 replicates with ≥ 100 cells per replicate).
-
CMacrophages treated and imaged as in (A). Example of a BMDM that dies after interacting with an extracellular Mtb aggregate. The top panels show a macrophage in contact with an extracellular Mtb aggregate 01:40 h before death (left) and just after death (right). The bottom panels show an Annexin V‐FITC positive plasma membrane domain; Draq7 stains the macrophage nucleus only after the cell dies. Scale bar, 10 μm.
-
DBehavior of 105 individual cytochalasin D‐treated macrophages in contact with Mtb aggregates. Cells were imaged at 1‐h intervals for 60 h in the presence of Annexin V‐FITC. Each line represents the life span of an individual cell; the fraction of the line in black represents the time spent as uninfected, the fraction of the line in magenta represents the time spent interacting with an Mtb aggregate and the fraction in green represents the time upon formation of local Annexin V‐positive membrane domains (MDs) at the site of contact with the Mtb aggregate.
-
E–GCytochalasin D‐treated BMDMs were infected with aggregates of Mtb Erd‐tdTomato in the presence of Annexin V‐FITC. Selected cells were imaged by time‐lapse fluorescence microscopy followed by SEM. Arrows in the top panels indicate the cell imaged by SEM. Top left: brightfield image of macrophage. Top left and right: fluorescence image of the Mtb aggregate. Top right: fluorescence image of Annexin V staining. Bottom: correlative SEM image. Scale bars, 20 μm in top panels and 1 μm in bottom panels. (E) Example of a bystander BMDM that is not in contact with a bacterial aggregate. (F) Example of an Annexin V‐negative BMDM interacting with an Mtb aggregate. Arrows in the bottom panel indicate areas of intact plasma membrane interacting with bacterial aggregates. (G) Example of a BMDM with blebbed Annexin V‐positive membrane domains interacting with an Mtb aggregate.
Source data are available online for this figure.
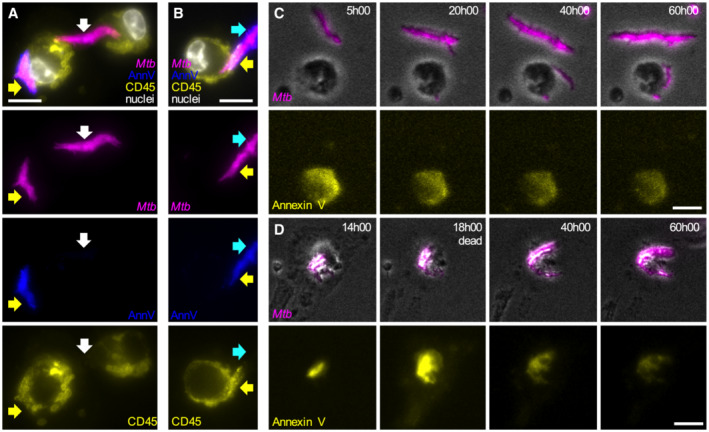
Figure EV3. Formation of Annexin V‐positive membrane domains requires physical contact between Mtb aggregates and live macrophages.
-
A, BRepresentative fluorescence microscopy images of cytochalasin D‐treated BMDMs infected with aggregates of Mtb Erd‐tdTomato in the presence of Annexin V‐FITC and fixed at 8 h post‐infection. The plasma membrane of the cells was stained with an anti‐CD‐45 antibody and nuclei were stained with Hoechst (white). Yellow arrows point at Mtb aggregates (magenta) overlapping with areas that stain positive for Annexin V (blue) and macrophages plasma membrane (yellow). White arrows indicate an Mtb aggregate that do not colocalize neither with the macrophages plasma membrane now with an Annexin V area. Cyan arrows indicate the distal area of an Mtb aggregate that stains positive for Annexin V but does not colocalize with the macrophages plasma membrane. Scale bars, 20 μm.
-
C, DBMDMs treated with cytochalasin D were infected with aggregates of Mtb, incubated with Annexin V, and imaged by time‐lapse microscopy at 1‐h intervals for 60 h. (C) Example of bacterial aggregates (magenta, top panels) that do not interact with macrophages and never become Annexin V‐positive (yellow, bottom panel) during the course of the experiment. (D) Example of bacterial aggregate (magenta, top panels) that induces the formation of a local Annexin V‐positive membrane domain in the interacting macrophage (yellow, bottom panel). After the death of the macrophage (at 18:00 h) the bacterial aggregates gradually lose fluorescence (40:00 h–60:00 h). Scale bars, 20 μm.
These observations suggest that induction of macrophage death does not require an uptake of the bacteria per se but does involve intimate contact between the macrophage membrane and the bacterial aggregate followed by local membrane perturbation.
Contact with extracellular Mtb aggregates induces calcium accumulation in macrophages
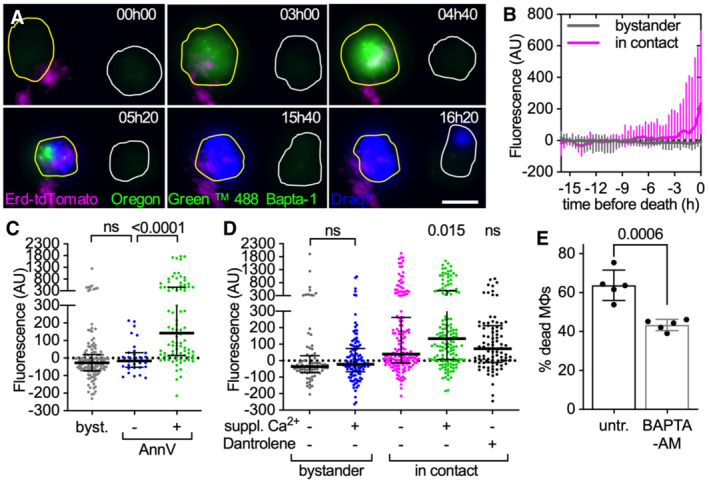
After escaping from the phagosome into the cytosol of infected macrophages, intracellular Mtb has been shown to damage the plasma membrane and affect its permeability to ions (Beckwith et al, 2020). We therefore asked whether the membrane perturbations observed at the site of contact between macrophages and extracellular Mtb aggregates are linked to aberrant ion permeability. We used a Ca2+‐dependent fluorescent dye to monitor intracellular Ca2+ dynamics in cytochalasin D‐treated BMDMs exposed to extracellular Mtb aggregates. Before death, cells interacting with Mtb aggregates accumulate significantly more cytosolic Ca2+ than uninfected bystander cells (Fig 4A and B; Movie EV9). Ca2+ also accumulates in cells interacting with Mtb aggregates that do not die over the course of the experiment (Appendix Fig S6A and B). Cytosolic Ca2+ accumulation is significantly higher in cells with Annexin V‐positive membrane domains at the site of contact with Mtb aggregates, suggesting that local membrane perturbations could make the cells permeable to small ions such as Ca2+ (Fig 4C; Appendix Fig S6C and D). The appearance of Annexin V‐positive local membrane domains and intracellular Ca2+ accumulation was also observed in BMDMs upon contact with extracellular Mtb aggregates in the absence of cytochalasin D (Appendix Fig S7A–D), thereby validating the use of cytochalasin D treatment to study uptake‐independent killing of macrophages by extracellular Mtb aggregates.
Figure 4. Extracellular Mtb aggregates induce cytosolic calcium accumulation in cytochalasin D‐treated macrophages.
-
A–DCytochalasin D‐treated BMDMs were stained with the membrane‐permeable dye Oregon Green 488 Bapta‐1 AM to visualize intracellular Ca2+, infected with aggregates of Mtb Erdman WT and imaged by time‐lapse microscopy at 20‐min intervals for 24 h. Oregon Green 488 Bapta‐1 AM fluorescence values at each time point were normalized to time 0 for uninfected bystander cells and to the time of first contact with an Mtb aggregate for infected cells. In (C, D) values for infected cells correspond to the time of death after first contact or 16 h post‐contact for cells that survive. Values for uninfected bystander cells correspond to the time of death or 16 h. Each symbol represents a single macrophage. Black bars represent the median and interquartile range. (A) Examples of BMDMs dying with (yellow outline) or without (white outline) interacting with extracellular Mtb aggregates. Cell outlines are based on brightfield images (see Movie EV9). Cell death is indicated by Draq7 nuclear staining. Scale bar, 10 μm. (B) Oregon Green 488 Bapta‐1 AM fluorescence over time in dying bystander macrophages (n = 15) and in dying macrophages in contact with an Mtb aggregate (n = 63). Lines represent median fluorescence values for all cells, error bars represent interquartile ranges. The distributions of the fluorescence values at the time of death in bystander and dying macrophages are significantly different, P‐value < 0.0001, calculated using a Welch's t‐test. (C) Oregon Green 488 Bapta‐1 AM fluorescence for uninfected bystander cells (byst.; n = 137) and for infected cells with (+; n = 91) or without (−; n = 32) Annexin V‐positive plasma membrane domains at the site of contact with an Mtb aggregate. P‐values were calculated using a Krustal–Wallis test; ns, P‐values > 0.05. (D) Oregon Green 488 Bapta‐1 AM fluorescence in bystander and infected macrophages incubated in medium with or without 10 mM Ca2+ and dantrolene. Lines represent median fluorescence values for all cells, error bars represent interquartile ranges. P‐values for bystander cells were calculated using an unpaired Mann–Whitney test. P‐values for infected cells were calculated using a Krustal–Wallis test comparing the treated samples to the untreated control; ns, P‐values > 0.05. (n = 81, 122, 165, 152, and 94, respectively, n represents the number of individual cells analyzed per condition; pooled values from ≥2 biological replicates).
-
EBMDMs treated with cytochalasin D were infected with aggregates of Mtb Erd‐tdTomato and imaged by time‐lapse microscopy at 1‐h intervals for 60 h. Percentage of macrophages that die within the first 12 h after stable contact with an Mtb aggregate without (untr.) or with supplementation of BAPTA‐AM. Each symbol represents the percentage of dead macrophages for a single biological replicate (n ≥ 60 cells per replicate). Bars represent means and standard deviations, P‐value calculated using a t‐test.
Source data are available online for this figure.
Cells incubated in a medium supplemented with Ca2+ accumulate more intracellular Ca2+ upon contact with Mtb aggregates in comparison to cells incubated with a regular medium (Fig 4D). Moreover, macrophages treated with dantrolene, a RyR inhibitor that inhibits Ca2+ release from the endoplasmic reticulum, still accumulate Ca2+ in the cytosol (Fig 4D). Ca2+ chelation with the cell‐permeant chelator BAPTA‐AM significantly reduces the percentage of macrophages that die upon contact with an extracellular Mtb aggregate (Fig 4E) without affecting the formation of Annexin V‐positive membrane domains (Appendix Fig S8). These observations suggest that Mtb‐induced plasma membrane perturbation may cause an influx of Ca2+ from the extracellular milieu into the cytosol, leading to cell death.
Extracellular Mtb aggregates induce uptake‐independent pyroptosis in macrophages
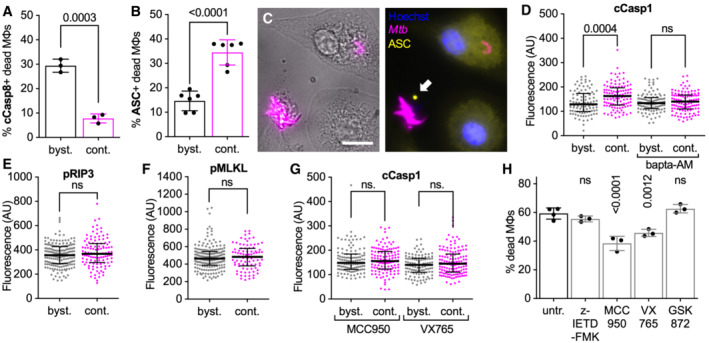
Perturbations in intracellular calcium homeostasis have been linked to different cell death pathways (Nomura et al, 2013; Galluzzi et al, 2018; Roca et al, 2019; Paik et al, 2021). We asked whether the uptake‐independent killing of macrophages by extracellular Mtb aggregates is mediated by one of the known cell death pathways. We observed that uninfected bystander macrophages that die show signs of Caspase‐8 activation, a common marker induced in apoptosis (Fig 5A; Appendix Fig S9A–D) (Galluzzi et al, 2018), but rarely display “specks” of apoptosis‐associated speck‐like protein containing a CARD (ASC) (Fig 5B), a marker for inflammasome activation induced in pyroptosis (Appendix Fig S9E and F; Swanson et al, 2019). Conversely, macrophages that die upon contact with extracellular Mtb aggregates usually display ASC‐specks (Fig 5B and C) but seldom stain positive for cleaved Caspase‐8 (Fig 5A). These observations are consistent with a previous study showing that contact between extracellular Mtb and plasma membrane can induce inflammasome activation (Beckwith et al, 2020) and suggest that contact with extracellular aggregates of Mtb induces a death pathway different from apoptosis. Cytochalasin D‐treated macrophages in contact with Mtb aggregates also show Ca2+‐dependent enhanced activation of Caspase‐1 (Fig 5D; Appendix Fig S9G–K), an effector caspase that is autoproteolytically activated by the inflammasome complex and that promotes pyroptotic cell death by inducing cleavage of gasdermin D (GSDMD; Paik et al, 2021). Macrophages in contact with Mtb aggregates do not show any enhanced activation of the necroptosis effectors RIP3 (Fig 5E; Appendix Fig S9L–P) or MLKL (Fig 5F; Appendix Fig S9Q–U). Taken together, these observations suggest that contact with extracellular Mtb aggregates may induce pyroptotic cell death in macrophages. In agreement with this interpretation, we observed that two different pyroptosis inhibitors (the NLRP3 inhibitor MCC950 and the Caspase‐1 inhibitor VX765) block Caspase‐1 activation (Fig 5G) and partially reduce cell death (Fig 5H) in cytochalasin D‐treated macrophages in contact with Mtb aggregates, whereas inhibition of Caspase‐8 or RIP3 has no effect (Fig 5H). Notably, treatment with pyroptosis inhibitors does not affect Mtb growth (Appendix Fig S10A), formation of Annexin V‐positive membrane domains (Appendix Fig S10B) or Ca2+ accumulation (Appendix Fig S10C) in macrophages in contact with Mtb aggregates, suggesting that the Ca2+ influx depends on contact with the bacteria and not on GSDMD pore formation downstream of Caspase‐1 activation.
Figure 5. Extracellular Mtb aggregates induce inflammasome activation and pyroptosis in cytochalasin D‐treated macrophages.
-
A–GBMDMs treated with cytochalasin D were infected with aggregates of Mtb Erd‐tdTomato, fixed at 24 h post‐infection, and processed for immunofluorescence with antibodies targeting cellular markers for cell death pathways. Controls and representative microscopy images for all the antibodies used are provided in Appendix Fig S9. Macrophages are defined as “in contact” when the body of a macrophage identified in brightfield images overlaps with a bacterial aggregate identified in the fluorescence channel (see Appendix Fig S9 for representative examples). (A) Percentage of dead uninfected bystander macrophages (byst.) or dead macrophages in contact (cont.) with an extracellular Mtb aggregate that stains positive for cleaved Caspase‐8 (cCasp8). Each symbol represents the percentage of positive dead macrophages for a single biological replicate (n ≥ 50 cells per replicate). Bars represent means and standard deviations. P‐values were calculated using a t‐test. (B) Percentage of dead uninfected bystander macrophages (byst.) or dead macrophages in contact with an extracellular Mtb aggregate (cont.) that display ASC‐specks. Each symbol represents the percentage of positive dead macrophages for a single biological replicate (n ≥ 80 cells per replicate). Bars represent means and standard deviations. P‐values were calculated using a t‐test. (C) Representative example of a dead macrophage in contact with an extracellular Mtb aggregate that displays a ASC‐speck (white arrow). Nuclei stained with Hoechst. Scale bar, 10 μm.(D–G) Median fluorescence values for bystander macrophages (byst.) or macrophages in contact with an extracellular Mtb aggregate (cont.) stained with an (D, G) anti‐cleaved Caspase‐1 antibody (cCasp1), an (E) anti‐phosphorylated RIP3 antibody (pRIP3), or (F) anti‐phosphorylated MLKL antibody (pMLKL). In (D) BMDMs were untreated or incubated with Bapta‐AM during the course of the infection. In (G) BMDMs were treated with MCC950 (NLRP3 inhibitor) or with VX765 (Caspase‐1 inhibitor) during the course of the infection. Each symbol represents a single macrophage. Black bars represent the median and interquartile range. P‐values were calculated using an unpaired Mann–Whitney test; ns, P‐values > 0.05. (D: n = 93, 108, 101, and 115, respectively; E: n = 182 and 112, respectively; F: n = 186 and 84, respectively; G: n = 117, 121, 111, and 129, respectively; and n represents the number of individual cells analyzed per condition).
-
HBMDMs treated with cytochalasin D were infected with aggregates of Mtb and imaged by time‐lapse microscopy at 1‐h intervals for 60 h. Percentage of macrophages that die within the first 12 h after stable contact with an Mtb aggregate. Macrophages were treated with the apoptosis inhibitor Z‐IETD‐FMK (Caspase‐8 inhibitor); with pyroptosis inhibitors MCC950 (NLRP3 inhibitor) or VX765 (Caspase‐1 inhibitor); or with necroptosis inhibitor GSK872 (RIP3 inhibitor). Each symbol represents the percentage of dead macrophages for a single biological replicate (n ≥ 95 cells per replicate). Bars represent means and standard deviations. P‐values were calculated using a one‐way ANOVA test comparing treated samples with the untreated control; ns, P‐values > 0.05.
Source data are available online for this figure.
ESX‐1 and PDIM are both required for uptake‐independent killing of macrophages by Mtb aggregates
The Mtb ESX‐1 type VII secretion system and the surface‐exposed lipid phthiocerol dimycocerosate (PDIM) are required for phagosomal membrane damage, bacterial translocation into the cytosol, and induction of host‐cell death by intracellular bacteria (Hsu et al, 2003; van der Wel et al, 2007; Passemar et al, 2014; Augenstreich et al, 2017). We therefore assessed whether these virulence factors also have a role in the uptake‐independent killing of macrophages by extracellular Mtb aggregates.
We found that a strain of Mtb with a large deletion in the ESX‐1 operon (∆RD1) (Fig EV4A; Hsu et al, 2003) does not induce uptake‐independent killing of macrophages (Fig 6A). Killing is largely restored by genetic complementation of the ∆RD1 mutant (Fig 6A), confirming the essential role of the ESX‐1 system. We also observed that a PDIM‐deficient strain of Mtb with a disrupted fadD26 gene (Appendix Fig S11) does not induce uptake‐independent macrophage death (Fig 6B). PDIM production and ESX‐1‐dependent secretion have been suggested to be interdependent (Barczak et al, 2017), however, in line with previous observations (Augenstreich et al, 2017), we observed that the fadD26 mutant shows unaltered secretion of the ESX‐1 secreted proteins (Fig EV4B and G; Appendix Fig S12) and that the ∆RD1 strain is not impaired in PDIM production (Appendix Fig S11), suggesting that mutations in these genes do not affect each other.
Figure EV4. Expression and secretion patterns of ESX‐1 proteins in the different mutant strains used in this study.
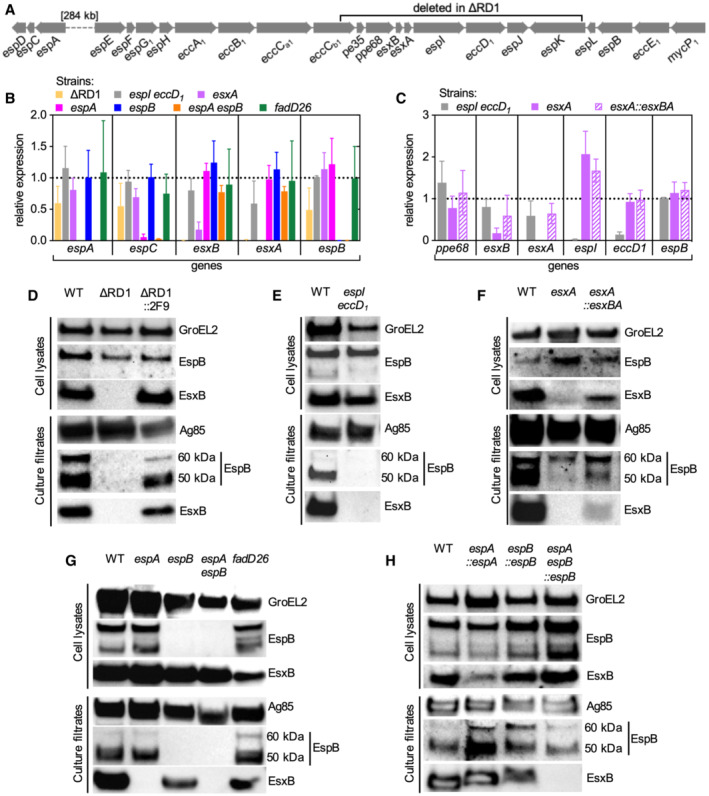
-
ARepresentation of the espACD and esx‐1 loci in the Mtb genome.
-
B, CExpression levels of selected genes of the espACD and esx‐1 loci in different mutants. Relative expression (fold changes) was normalized to the WT strain (WT expression = 1, indicated by the dotted line). Bars represent mean values (n = 3 biological replicates) and error bars represent standard deviation.
-
D–HRepresentative immunoblots of cell lysates and culture filtrates from different Mtb strains were probed with the indicated antibodies. In culture filtrates EspB shows a full‐length 60‐kDa isoform and a truncated 50‐kDa isoform as previously reported (Ohol et al, 2010; Chen et al, 2013a, 2013b). GroEL2 and Ag85 were respectively used as loading controls for cell lysates and culture filtrates.
Figure 6. ESX‐1‐secreted proteins and PDIM are required for uptake‐independent killing of macrophages by Mtb aggregates.
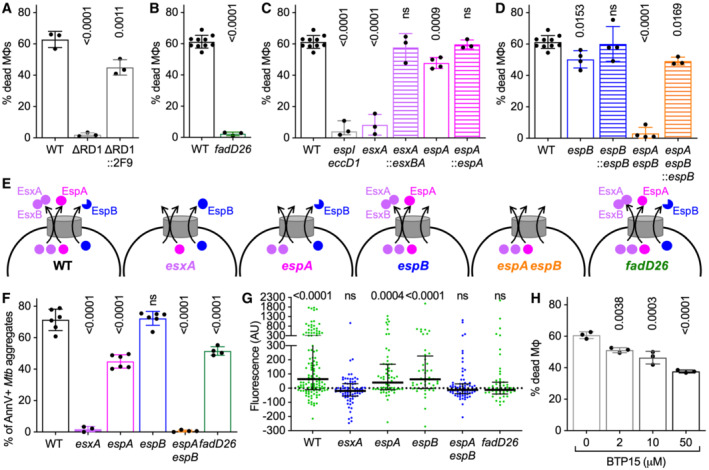
-
A–DBMDMs treated with cytochalasin D were infected with aggregates of different Mtb strains and imaged by time‐lapse microscopy at 1‐h intervals for 60 h. The plots represent the percentage of macrophages that die within the first 12 h after stable contact with an Mtb aggregate. Each symbol represents the percentage of dead macrophages for a single biological replicate (n ≥ 3 replicates with ≥ 70 cells per replicate). Bars represent means and standard deviations. P‐values were calculated using a one‐way ANOVA test (A, C, D) or t‐test (B) comparing each strain to the wild‐type reference strain (A–D); ns, P‐values > 0.05. (A) Macrophages in contact with aggregates of Mtb H37Rv wild‐type (WT), ∆RD1 mutant, and complemented ∆RD1 mutant (∆RD1::2F9). (B) Macrophages in contact with aggregates of Mtb Erdman wild‐type (WT) or PDIM‐deficient (fadD26) strains. (C) Macrophages in contact with aggregates of Mtb Erdman wild‐type (WT) or mutant strains (espI eccD1, esxA, espA) and the complemented strains (esxA::esxBA, espA::espA). (D) Macrophages in contact with aggregates of Mtb Erdman wild‐type (WT) or mutant strains (espB, espA espB) and the espB‐complemented strains (espB::espB, espA espB::espB).
-
ESchematic representation of EsxA, EsxB, EspA, and EspB secretion pattern in the mutant strains used in this study (based on Western Blot and quantitative proteomics data shown in Fig EV4 and in Appendix Figs S13 and S14).
-
FBMDMs treated with cytochalasin D were infected with aggregates of different Mtb strains, incubated with Annexin V, and imaged by time‐lapse microscopy at 1‐h intervals for 48 h. Percentage of macrophages that show Annexin V—positive membrane domains within the first 12 h after entering in contact with Mtb aggregates. Each symbol represents a single biological replicate (n ≥ 3 replicates with ≥ 90 cells per replicate). Bars represent means and standard deviations. P‐values were calculated using a one‐way ANOVA test.
-
GCytochalasin D‐treated BMDMs were infected with aggregates of Mtb Erdman WT, stained with the membrane‐permeable dye Oregon Green 488 Bapta‐1 AM to visualize cytosolic Ca2+ and imaged by time‐lapse microscopy at 20‐min intervals for 24 h. Oregon Green 488 Bapta‐1 AM fluorescence values at each time point were normalized to the time of first contact with an Mtb aggregate for infected cells. Values in the plot correspond to the time of death after first contact or 16 h post‐contact for cells that survive. Each symbol represents a single macrophage. (n = 123, 64, 48, 71, 78, and 55, respectively). Black bars represent the median and interquartile range. P‐values were calculated using a one‐sample Wilcoxon test; ns, P‐values > 0.05.
-
HBMDMs were treated and imaged as in (A–D). Percentage of macrophages that die within the first 12 h after interaction with an Mtb aggregate. Infected cells are incubated with different concentrations of BTP15 (0, 2, 10, and 50 μM) during the course of the experiment. Each symbol represents the percentage of dead macrophages for a single biological replicate (n ≥ 3 replicates with ≥ 70 cells per replicate). Bars represent means and standard deviations. P‐values were calculated using a one‐way ANOVA test comparing the treated vs. untreated samples.
Source data are available online for this figure.
Interestingly, when phagocytosis is not inhibited by cytochalasin D treatment, lack of PDIM has only a minor effect on macrophage killing by Mtb aggregates (Appendix Fig S13), suggesting that this factor may have a less significant role in inducing uptake‐dependent macrophage death.
Expression but not secretion of EsxA/EsxB is required for uptake‐independent killing of macrophages by Mtb aggregates
We used a panel of Mtb mutant strains to investigate how different ESX‐1 components are involved in the induction of uptake‐independent macrophage death. We observed that macrophages in contact with aggregates of an espI‐eccD 1 mutant with disrupted structural components of the ESX‐1 secretion system (Zhang et al, 2014; Fig EV4B, C and E) do not die (Fig 6C), demonstrating that a functional ESX‐1 system is required for uptake‐independent killing of macrophages.
Among the ESX‐1‐secreted proteins, EsxA has been shown to mediate the breakdown of the phagolysosomal membrane (van der Wel et al, 2007). EsxA is co‐secreted in a 1:1 heterodimer with EsxB (Renshaw et al, 2002; Stanley et al, 2003) but the uncoupled secretion of these proteins can occur in the presence of an aberrant ESX‐1 system (Pang et al, 2013; Aguilo et al, 2017). We confirmed that in our hands, loss of EsxA eliminates EsxB expression and secretion (Stanley et al, 2003; Brodin et al, 2006; Augenstreich et al, 2017) (Fig EV4B, C and F; Appendix Fig S14A). We found that deletion of esxA reduces killing to nearly background levels (Fig 6C) and that complementation with both esxA and esxB restores secretion of EsxB (Fig EV4F) and uptake‐independent killing of macrophages (Fig 6C). EsxA secretion requires EspA, another ESX‐1‐secreted protein (Fortune et al, 2005), and deletion of espA abolishes EsxA/EsxB secretion (Fig EV4G; Appendix Fig S14B) without affecting their expression (Fig EV4B and G) (Chen et al, 2013a, 2013b). Unexpectedly, we found that deletion of espA has only a slight impact on macrophage killing (Fig 6C), suggesting that although expression of EsxA/EsxB is required for uptake‐independent killing of macrophages, EspA‐mediated secretion of these proteins is not required.
EspB mediates uptake‐independent killing in the absence of EsxA/EsxB secretion
The unexpected finding that uptake‐independent killing of macrophages is eliminated by deletion of esxA but not espA is not linked to any discernable differences in the mutant strains' production of PDIM (Appendix Fig S11), aggregate morphology (Appendix Fig S15A, B, F and G), or growth rates (Appendix Fig S15H). However, quantitative proteomics revealed that the deletion of esxA also decreases the secretion of EspK and EspB, two ESX‐1‐secreted proteins whose secretion is not affected by the deletion of espA (Appendix Fig S14A and B). In particular, the deletion of esxA eliminates the secretion of the cleaved 50‐kDa isoform of EspB while having little or no impact on the production and secretion of the full‐length 60‐kDa isoform (Fig EV4F). We therefore asked whether Mtb requires EspK or EspB to induce uptake‐independent killing of macrophages.
We excluded the involvement of EspK by showing that an espK mutant strain induces uptake‐independent killing of macrophages at levels comparable to wild‐type Mtb (Appendix Fig S16). On the other hand, we observed that although deletion of espB has only a slight impact on uptake‐independent killing (Fig 6D), similar to deletion of espA (Fig 6C), deletion of both espA and espB reduces killing to background levels (Fig 6D), similar to the impact of esxA deletion (Fig 6C). The espB and espA espB mutants show no alteration in PDIM production (Appendix Fig S11), aggregate morphology (Appendix Fig S15C and D), or growth rates (Appendix Fig S15H), and complementation with espB (Fig EV4H) restores macrophage killing to similar levels as the wild‐type and espA strains (Fig 6D). Quantitative proteomics revealed that espB deletion does not detectably affect the secretion of any other protein in either the wild‐type or espA background (Fig 6E; Appendix Fig S14C and D), indicating that the impact of espB on killing is not due to the loss of another secreted protein. We conclude that secreted EsxA/EsxB and EspB are both involved in the uptake‐independent killing of macrophages by Mtb aggregates because blocking the secretion of only one of these proteins reduces but does not eliminate cell death.
ESX‐1 but not PDIM is required for membrane perturbation in macrophages contacting Mtb aggregates
Since macrophage killing requires both ESX‐1 and PDIM, we asked whether these factors are also involved in triggering local membrane perturbations in macrophages after contact with Mtb aggregates. We found that aggregates of bacteria that secrete either EsxA/EsxB (espB mutant) or the 50‐kDa isoform of EspB (espA mutant) are able to induce local membrane perturbations in contacted macrophages at nearly wild‐type levels (Fig 6E and F). In sharp contrast, blocking the secretion of both EsxA/EsxB and the 50‐kDa isoform of EspB (esxA and espA espB mutants) reduces membrane perturbations in contacted macrophages to nearly background levels (Fig 6E and F). Although aggregates of PDIM‐deficient fadD26 bacteria do not kill macrophages (Fig 6B), they still induce local membrane perturbations (Fig 6F), consistent with the observation that this strain expresses and secretes EsxA/EsxB and EspB normally (Fig EV4B and G; Appendix Fig S12). These results indicate that membrane perturbation per se is not sufficient to cause cell death and further support our conclusion that secreted EsxA/EsxB and EspB have overlapping roles in the uptake‐independent killing of macrophages.
ESX‐1 and PDIM are both required for calcium accumulation in macrophages contacting Mtb aggregates
Since membrane perturbation and calcium accumulation may be causally related, we evaluated the role of ESX‐1 and PDIM in triggering calcium accumulation in macrophages after contact with extracellular Mtb aggregates. We found that aggregates of Mtb strains that do not induce membrane perturbations (esxA and espA espB mutants) also do not induce calcium accumulation in macrophages (Fig 6G). However, we also found that PDIM‐deficient bacteria (fadD26 mutant) also fail to induce calcium accumulation in contacted macrophages (Fig 6G) despite inducing membrane perturbation at nearly normal levels (Fig 6F). Taken together, our results suggest that ESX‐1‐mediated membrane perturbation is not sufficient to induce calcium accumulation and macrophage death in the absence of PDIM.
A small‐molecule inhibitor of ESX‐1 reduces uptake‐independent killing of macrophages by Mtb aggregates
We asked whether BTP15, a small molecule that inhibits the secretion of components of the ESX‐1 system (Rybniker et al, 2014), can inhibit the uptake‐independent killing of macrophages by Mtb aggregates. Treatment with BTP15 reduces the secretion of EsxB and EspB (in particular the 50‐kDa isoform) and the uptake‐independent killing of macrophages in a dose‐dependent manner (Figs 6H and EV5A and B). Mtb aggregates treated with BTP15 for 48 h before infection, induce reduced uptake‐independent killing of macrophages, even upon drug washout, excluding a possible direct effect of BTP15 on macrophages (Fig EV5C). We show that by inhibiting the secretion of components of the ESX‐1 system, BTP15 significantly reduces macrophage membrane perturbation (Fig EV5H), uptake‐independent (Fig 6H), and uptake‐dependent (Fig EV5I) macrophage killing, without affecting bacterial growth or aggregation (Fig EV5D–G). These observations suggest that inhibitors targeting factors involved in macrophage killing by extracellular Mtb aggregates could interfere with bacterial evasion of host defense mechanisms that depend on phagocytic uptake.
Figure EV5. BTP15 treatment reduces ESX‐1 secretion and local plasma membrane perturbation in macrophages in contact with Mtb aggregates, without affecting ESX‐1 expression, aggregates growth dynamics and morphology in Mtb .
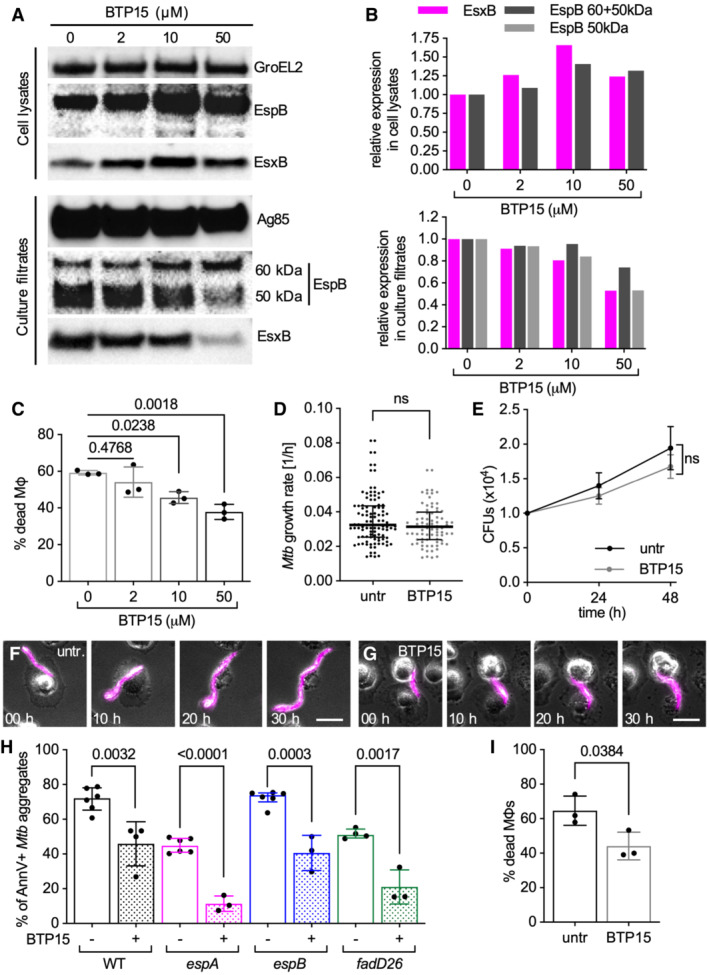
-
AWestern blot showing EspB and EsxB expression (cell lysates) and secretion (culture filtrates) pattern of Mtb Erdman WT cultures treated with different concentrations of BTP15 (0, 2, 10, and 50 μM). GroEL2 and Ag85 were respectively used as loading controls for cell lysates and culture filtrates.
-
BEspB (whole or 50 kDa isoform) and EsxB quantification from the Western blot images. Values were normalized to the loading control first and then to the untreated samples.
-
C–HBMDMs treated with cytochalasin D were infected with aggregates of Mtb and incubated with or without BTP15. Infected cells were imaged by time‐lapse microscopy at 1‐h intervals for 60 h (C, D, F–H), or incubated at 37°C with 5% CO2 for quantification of colony‐forming units (CFU) at 0, 24, and 48 h post‐infection (E). (C) Percentage of macrophages that die within the first 12 h after interaction with an Mtb aggregate. Bacteria are incubated with different concentrations of BTP15 (0, 2, 10, and 50 μM) for 48 h before infection. BTP15 is not added to the medium of the cells during the course of the experiment. Each symbol represents a single biological replicate (> 70 macrophage‐Mtb aggregate interactions per replicate). Bars represent average and standard deviation. P‐values were calculated using a one‐way ANOVA test. (D) Growth rate of individual untreated and BTP15‐treated (50 μM) Mtb aggregates. Each symbol represents a microcolony. Black lines indicate median values and interquartile ranges (n ≥ 40 bacterial aggregates per condition). P‐value calculated using an unpaired Mann–Whitney test; ns, P‐value > 0.05. (E) Total CFU per well at different time points from untreated and BTP15‐treated (50 μM) bacterial cultures. Symbols and bars represent means and standard deviations (n = 4 biological replicates). P‐value calculated using an unpaired t‐test; ns, P‐value > 0.05. (F, G) Representative examples of untreated (F) or BTP15‐treated (50 μM) (G) aggregates of fluorescent Mtb in contact with cytochalasin D‐treated BMDMs at different time‐points post‐infection. Scale bars, 20 μm. (H) BMDMs treated with cytochalasin D were infected with aggregates of different Mtb strains, incubated with Annexin V, and imaged by time‐lapse microscopy at 1‐h intervals for 60 h. Percentage of macrophages that show Annexin V—positive membrane domains within the first 12 h after entering in contact with Mtb aggregates without (−) or with (+) BTP15 treatment (50 μM). Each symbol represents a single biological replicate (> 90 macrophage‐Mtb aggregate interactions per replicate). Bars represent average and standard deviation. P‐values were calculated using an unpaired t‐test comparing the treated samples with their untreated reference.
-
IBMDMs were infected with aggregates of Mtb strains, incubated without (untr) or with (BTP15) 50 μM BTP15 and imaged by time‐lapse microscopy at 1‐h intervals for 60 h. Percentage of macrophages that die within the first 12 h after interaction with an Mtb aggregate. Each symbol represents a single biological replicate (> 100 macrophage‐Mtb aggregate interactions per replicate). Bars represent average and standard deviation. P‐values were calculated using an unpaired t‐test.
Source data are available online for this figure.
Discussion
Death of infected host macrophages is an important process in Mtb infections, as it promotes the spreading of the intracellular bacteria to other host cells, inflammation, recruitment of immune cells, caseation, and disruption of granulomas (Davis & Ramakrishnan, 2009; Ehlers & Schaible, 2013; Repasy et al, 2015). The induction of host‐cell death by intracellular Mtb has been extensively investigated (Mohareer et al, 2018). Here, we extend these observations using live‐cell time‐lapse microscopy of Mtb‐infected macrophages and chemical inhibition of phagocytosis to demonstrate that Mtb can also induce macrophage death “from the outside” of the cell in a contact‐dependent but uptake‐independent manner. This experimental approach allowed us to focus only on death events induced by extracellular bacteria and to characterize this process, which may allow Mtb to evade stresses associated with phagocytic uptake by macrophages.
Mtb was previously shown to induce contact‐dependent hemolysis of erythrocytes (King et al, 1993). However, the unusual structure and membrane composition of these anucleate non‐phagocytic cells makes it difficult to extrapolate these results to phagocytic immune cells such as macrophages. Other cytotoxicity assays commonly used to quantify the viability of host cells interacting with Mtb do not make a clear distinction between killing by extracellular versus intracellular bacteria (Takii et al, 2002; Hsu et al, 2003; Chen et al, 2013a, 2013b; Rybniker et al, 2014). In macrophages treated with cytochalasin D to inhibit phagocytosis, contact with extracellular Mtb was shown to be sufficient to induce inflammasome activation but cell death was not reported (Beckwith et al, 2020). Here, we show that aggregates of extracellular Mtb are much more efficient in killing cytochalasin D‐treated macrophages compared to similar numbers of single (non‐aggregated) bacteria. This may explain why the uptake‐independent killing of macrophages was not reported in a previous study where cytochalasin D‐treated macrophages were exposed to non‐aggregated bacterial suspensions (Lee et al, 2006).
We observed that the plasma membrane of cytochalasin D‐treated macrophages extends around Mtb aggregates and establishes a stable interaction with them. At this interface, the host‐cell plasma membrane stains with Annexin V, a marker of membrane perturbation. This staining is not an artifact of cytochalasin D treatment, because local Annexin V staining is also observed in the small number of untreated macrophages that establish long‐term contact with extracellular aggregates without internalizing them. We speculate that bacterial factors involved in triggering host‐cell death may be concentrated over a smaller area of the host‐cell membrane when the bacteria are aggregated rather than dispersed. This may explain why aggregates are more efficient at inducing contact‐dependent macrophage death compared to similar numbers of individual (non‐aggregated) bacteria. Alternatively, aggregated bacteria might produce higher amounts of bacterial factors required to induce host‐cell death or they might retain them more easily on their surface.
Previous studies have reported that the Mtb ESX‐1 type VII secretion system and the ESX‐1‐secreted proteins EsxA/EsxB are required for escape from the phagosome and induction of host‐cell death by intracellular bacteria (Hsu et al, 2003; van der Wel et al, 2007). Here, we show that EsxA/EsxB expression is also required to induce uptake‐independent killing of macrophages upon contact with extracellular bacterial aggregates. Unexpectedly, however, we found that elimination of EsxA/EsxB secretion by deletion of espA (Fortune et al, 2005; Chen et al, 2013a, 2013b) has little effect on macrophage killing unless the ESX‐1‐secreted EspB protein is absent. We also found that EsxA/EsxB expression is required for secretion of the cleaved 50‐kDa isoform but not the full‐length 60‐kDa isoform of EspB, in agreement with a recent study (Lim et al, 2022). Cleavage of EspB by MycP1 is required for full Mtb virulence (Ohol et al, 2010), the interaction of EspB with phospholipids (Chen et al, 2013a, 2013b), and oligomerization of EspB into channel‐shaped heptamers (Solomonson et al, 2015; Piton et al, 2020). These observations suggest that EspB can induce uptake‐independent killing of macrophages in the absence of EsxA/EsxB secretion but not in the absence of EsxA/EsxB expression, which appears to be required for MycP1‐dependent cleavage of EspB.
EspB secretion has been linked to Mtb virulence in a previous study comparing Mtb ESX‐1 mutants with different secretion patterns, although the behavior of an isogenic espB deletion strain was not reported (Ohol et al, 2010; Chen et al, 2013a, 2013b). It has been suggested that EspB heptamers mediate the secretion of other proteins (Korotkova et al, 2015; Solomonson et al, 2015; Piton et al, 2020; Gijsbers et al, 2021); however, this hypothesis is inconsistent with our proteomic analysis of the espB mutant's secretome showing that deletion of espB does not affect the secretion of any other protein. Alternative models suggest that EspB heptamers may form membrane‐spanning pores after contacting host‐cell membranes, suggesting a possible mechanism for EspB‐mediated cell death (Gijsbers et al, 2021). However, the addition of purified EspB to macrophages does not cause cell death (Chen et al, 2013a, 2013b), consistent with our observation that direct contact between Mtb aggregates and macrophages is required for killing and bystander macrophages are not affected even when they are in close proximity to Mtb aggregates. Similarly, while it is still debated whether purified EsxA alone has pore‐forming activity on membranes (de Jonge Marien et al, 2007; Conrad et al, 2017; Augenstreich et al, 2020), direct physical interaction between EsxA‐producing strains and macrophages is also required to induce uptake‐independent killing of macrophages, suggesting that secreted factors per se may not be sufficient to cause cell death.
A possible explanation for the contact‐dependency of killing may be provided by our observation that PDIM, a complex lipid‐associated with the outer membrane of Mtb (Cox et al, 1999), is also required for the killing of macrophages by Mtb aggregates, although it is not sufficient in the absence of ESX‐1. Importantly, we found that the essential role of PDIM in contact‐dependent killing is not linked to any detectable change in the Mtb secretome in the absence of PDIM. Previous reports have shown that PDIM can insert into host‐cell membranes and modify their biophysical properties (Astarie‐Dequeker et al, 2009; Augenstreich et al, 2019; Cambier et al, 2020). PDIM has also been shown to increase the membranolytic activity of EsxA against liposomes (Augenstreich et al, 2017, 2020), but its interaction with EspB has not been investigated to the best of our knowledge. The association of PDIM with the bacterial surface may explain why direct physical contact between bacteria and host cells is required to induce macrophage death.
Contact with extracellular Mtb aggregates triggers local plasma membrane perturbation and cytosolic calcium accumulation in macrophages followed by cell death. We observed that cells that display perturbed membrane foci at the site of contact with Mtb aggregate progressively accumulate cytosolic calcium before death. PDIM‐deficient bacteria cause local plasma membrane perturbation without causing cell death, suggesting that membrane perturbation per se is not sufficient to induce cell death. Membrane perturbation in macrophages requires secretion of EsxA/EsxB or EspB and may be required for cytosolic calcium accumulation because mutations that eliminate membrane perturbation (esxA and espA espB mutants) also eliminate calcium accumulation. Conversely, calcium accumulation seems not to be required for membrane perturbation because the loss of PDIM eliminates calcium accumulation without affecting membrane perturbation. Taken together, these results suggest that although PDIM and secreted EsxA/EsxB and EspB all participate in the uptake‐independent killing of macrophages after contact with extracellular Mtb aggregates, their roles are somewhat different.
We propose two alternative models that could explain these observations. EsxA/EsxB‐ or EspB‐dependent plasma membrane perturbation may facilitate PDIM insertion into the membrane, which could potentially affect the membrane permeability to ions such as calcium. Alternatively, EsxA (Augenstreich et al, 2017) or EspB may require PDIM to potentiate their membranolytic activities and to alter membrane permeability to ions. Both PDIM and a fraction of secreted EsxA and EspB are physically associated with the bacterial cell surface (Kinhikar et al, 2010; Sani et al, 2010; Raffetseder et al, 2019; Lim et al, 2022), which may explain why direct contact of Mtb with macrophages is required for induction of host‐cell death while bystander macrophages are not affected.
A general caveat of models based on analysis of esxA or espA mutants is that EsxA and EspA are mutually dependent for their secretion and it is thus unclear whether some of the phenotypes observed may depend on one or the other protein. EspA has been shown to have an essential role in EsxA secretion (Fortune et al, 2005). However, in contrast to EsxA (Hsu et al, 2003), we are not aware of any published evidence that EspA interacts with host‐cell membranes and exerts membranolytic activity. For these reasons, it is generally assumed that EsxA is the secreted bacterial effector most responsible for damaging host‐cell membranes (van der Wel et al, 2007; Welin et al, 2011; Dallenga et al, 2017).
Uptake‐independent death of macrophages by extracellular Mtb aggregates can be partially suppressed by treating the infected cells with pyroptosis inhibitors targeting NLRP3 or Caspase 1. Inhibition of this pathway reduces cell death without affecting either formation of Annexin V‐positive membrane domains or intracellular calcium accumulation. These results suggest that extracellular Mtb aggregates induce an imbalance of cytosolic calcium or other ions in contacted macrophages, leading to inflammasome activation and pyroptotic cell death. Intracellular ion imbalances and pyroptosis have similarly been observed in macrophages after plasma membrane damage caused by intracellular Mtb (Beckwith et al, 2020).
Our work highlights the importance of Mtb aggregation during host‐cell infection and demonstrates how Mtb aggregates can evade phagocytosis by inducing contact‐dependent but uptake‐independent death in macrophages. Mtb aggregation has been shown to play an important role in pathogenesis in vivo (Glickman et al, 2000; Kolloli et al, 2021), and several groups have shown that macrophages that internalize single bacteria are able to survive for days whereas uptake of Mtb aggregates typically results in rapid death (Dallenga et al, 2017; Mahamed et al, 2017). Although Mtb infections can start with very few individual bacteria (Cohen et al, 2018), these bacteria can form aggregates when they grow inside host cells (Hoff et al, 2011; Lerner et al, 2020) or on the debris of dead host cells (Lerner et al, 2017). The presence of intracellular and extracellular Mtb aggregates has been documented in live and necrotic cells in the lungs of mouse, rabbit, and guinea pig models of infection already within the first‐month post‐infection (Hoff et al, 2011; Repasy et al, 2013; Irwin et al, 2015; Kolloli et al, 2021). Moreover, aggregates have also been observed in human lung lesions and sputum samples (Kaplan et al, 2003; Timm et al, 2006; Hunter, 2011; Lerner et al, 2020; Dinkele et al, 2021; Rodel et al, 2021; Wells et al, 2021), confirming their potential relevance in human tuberculosis infection.
We propose that uptake‐independent induction of macrophage death by Mtb aggregates may promote the propagation of Mtb at different stages of infection. At early stages, the release of Mtb aggregates upon the death of infected naïve macrophages (killing “from the inside”) may allow rapid replication of extracellular bacteria on the host‐cell debris. Subsequently, uptake‐independent killing “from the outside” may allow these extracellular aggregates to evade phagocytosis by less‐permissive macrophages as host immunity begins to develop (Huang et al, 2018; Ramakrishnan, 2020). Even when Mtb aggregates are successfully internalized by macrophages, they may still evade intracellular host defenses by inducing rapid host‐cell death “from the inside” (Mahamed et al, 2017). At later stages, the toxicity of extracellular Mtb aggregates may also participate in the necrotic processes required for the expansion of lesions, formation of a large extracellular bacterial pellicle in open necrotic cavities, and bacterial spillover into the airways (Kaplan et al, 2003; Hoff et al, 2011; Hunter, 2011). Mtb aggregates that spill into the airways may induce the death of newly recruited macrophages, thereby contributing to the evasion of host immunity, exhalation of live bacteria, and transmission of the infection (Orme, 2014). Based on our results with BTP15, a small‐molecule inhibitor of ESX‐1 (Rybniker et al, 2014), we propose that bacterial spreading within the lung could be suppressed by novel therapies targeting virulence factors, such as ESX‐1 or PDIM, that are required for uptake‐independent killing of host macrophages by extracellular Mtb aggregates. By reducing host‐cell death, such therapies could also suppress the formation of necrotic lesions, where large numbers of extracellular bacteria may grow rapidly on the debris of dead host cells (Lerner et al, 2017) within an environment that is poorly penetrated by antibiotics (Hoff et al, 2011; Dartois, 2014; Strydom et al, 2019).
Materials and Methods
Bacterial strains and growth conditions
Strains used in this study include WT Mtb H37Rv Pasteur, WT Mtb Erdman, various mutants, complemented, and fluorescent strains listed in Table 1. All strains were cultured in liquid Middlebrook 7H9 liquid medium (Difco) supplemented with 10% ADC (Difco), 0.5% glycerol, and 0.02% Tyloxapol at 37°C with shaking. As a solid medium Middlebrook 7H10 (Difco) supplemented with 10% OADC enrichment (Becton Dickinson) and 0.5% glycerol was used. When required, kanamycin, hygromycin, and zeocin were added to the medium at a final concentration of 25, 50, and 25 μg/ml, respectively.
Table 1.
List of Mtb strains used in this study.
| Strain | Description | Source | Ref. |
|---|---|---|---|
| Erd | M. tuberculosis, Erdman, wild‐type | Lab collection | — |
| Erd‐tdTomato | M. tuberculosis, Erdman, wild‐type, expressing tdTomato (pND257), KanR | This study | — |
| Rv | M. tuberculosis, H37Rv, wild‐type | C. Astarie‐Dequeker | Augenstreich et al (2017) |
| Rv‐GFP | M. tuberculosis, H37Rv, wild‐type, expressing GFP (pCG211), KanR | C. Astarie‐Dequeker | Augenstreich et al (2017) |
| esxA | M. tuberculosis, H37Rv, ΔesxA | C. Astarie‐Dequeker | Augenstreich et al (2017) |
| esxA‐GFP | M. tuberculosis, H37Rv, ΔesxA, expressing GFP (pCG211), KanR | C. Astarie‐Dequeker | Augenstreich et al (2017) |
| esxA::esxBA | M. tuberculosis, H37Rv, ΔesxA, complemented with esxBA (pMVesxBA3), HygR | C. Astarie‐Dequeker | Augenstreich et al (2017) |
| esxA::esxBA‐GFP | M. tuberculosis, H37Rv, ΔesxA, complemented with esxBA (pMVesxBA3), expressing GFP (pCG211), HygR, KanR | C. Astarie‐Dequeker | Augenstreich et al (2017) |
| ΔRD1 | M. tuberculosis, H37Rv, ΔRD1 | S. Cole | Hsu et al (2003) |
| ΔRD1::2F9 | M. tuberculosis, H37Rv, ΔRD1, complemented with 2F9 cosmid, HygR | S. Cole | Hsu et al (2003) |
| espI‐eccD 1 | M. tuberculosis, Erdman, transposon insertion in espI (1,039 bp within Rv3876), HygR | Lab collection | Dhar and McKinney (2010), Zhang et al (2014) |
| espA | M. tuberculosis, Erdman, transposon insertion in espA (100 bp within Rv3616c), HygR | Lab collection | Dhar and McKinney (2010), Chen et al (2013a, 2013b) |
| espA‐tdTomato | M. tuberculosis, Erdman, transposon insertion in espA, expressing tdTomato (pND257), Kan R , HygR | This study | — |
| espA::espA | M. tuberculosis, Erdman, transposon insertion in espA, complemented with espACD (pMDespACD), HygR, KanR | Lab collection | Chen et al (2013a, 2013b) |
| espK | M. tuberculosis, Erdman, transposon insertion in espK (260 bp within the gene Rv3879c), HygR | Lab collection | Dhar and McKinney (2010) |
| espB | M. tuberculosis, Erdman, pKM464 insertion in espB, HygR | This study | — |
| espB‐tdTomato | M. tuberculosis, Erdman, pKM464 insertion in espB, expressing tdTomato (pND257), HygR, KanR | This study | — |
| espB::espB | M. tuberculosis, Erdman, pKM464 insertion in espB, complemented with espB (pMK261_espB), HygR, KanR | This study | — |
| espA espB | M. tuberculosis, Erdman, transposon insertion in espA, pKM496 insertion in espB, HygR, ZeoR | This study | — |
| espA espB‐tdTomato | M. tuberculosis, Erdman, transposon insertion in espA, pKM496 insertion in espB, expressing tdTomato (pND257), HygR, ZeoR, KanR | This study | — |
| espA espB::espB | M. tuberculosis, Erdman, transposon insertion in espA, pKM496 insertion in espB, complemented with espB (pMK261_espB) HygR, ZeoR, KanR | This study | — |
| fadD26 | M. tuberculosis, Erdman, transposon insertion in the promoter of fadD26 (112 bp upstream of Rv2930), HygR | Lab collection | Dhar and McKinney (2010) |
| fadD26‐tdTomato | M. tuberculosis, Erdman, transposon insertion in the promoter of fadD26, expressing tdTomato (pND257), HygR, KanR | This study | — |
Hyg, hygromycin; Kan, kanamycin; Zeo, zeocin.
Generation of fluorescent, mutant, and complemented strains
All the tdTomato fluorescent strains were developed by electroporating the parental strains with the integrative pND257 plasmid. pND257 is a mycobacterial L5‐based integrative plasmid in which the gene encoding TdTomato was cloned downstream of a strong constitutive promoter and confers resistance to kanamycin. Transformants were selected plating on 7H10 plates + kanamycin and verified by fluorescence microscopy.
The espB mutants were constructed using the ORBIT strategy (Murphy et al, 2018). The parental strains Mtb Erdman wild‐type and espA‐tn were electroporated with the plasmid pKM461 (Addgene, #108320) expressing tetracycline‐inducible Che9c phage RecT annealase and Bxb1 phage integrase (Murphy et al, 2018). Transformants were selected plating on 7H10 plates + kanamycin. Cells containing pKM461 were grown in liquid medium + kanamycin and supplemented with 500 ng/ml anhydrotetracycline when the culture reached an OD600 of 0.3. After 24 h induction, bacteria were electroporated with the payload plasmids pKM464 (espB mutant, Addgene, #108322) or pKM496 (espA espB mutant, Addgene, #109301) (Murphy et al, 2018) and 1 μg of the Ultramer DNA oligonucleotide (IDT) targeting espB (agcgtcaacgcccgggcgacatgcgggtccaattcgtccatgctcacttcgactccttactgtcctggcgGGTTTGTACCGTACACCACTGAGACCGCGGTGGTTGACCAGACAAACCctgcgactgcgtcatatcggatcatcctccttagtgctatagccattatcgtcgctaaactgaaaggttc). Recombinants were selected plating on 7H10 plates + hygromycin (espB mutant) or + zeocin (espA espB mutant). The recombination was verified by PCR analysis. For the selected clones the plasmid pKM461 was cured by successive growth in a kanamycin‐free medium. Loss of plasmid was verified by inoculating bacteria in liquid medium + kanamycin.
For complementation of the espB mutant strains the pMV261_espB plasmid was developed. A DNA region covering the espB gene plus 250 bp upstream of the start codon was amplified by PCR from Mtb Erdman genomic DNA using the primers espBcompF and espBcompR (Table 2). The resulting PCR product was integrated into the pMV261 (Stover et al, 1991) plasmid digested with AatII and EcoRI using the Gibson Assembly strategy according to the manufacturer protocol (NEB).
Table 2.
List of primers used in this study.
| Primer name | Sequence 5′–3′ |
|---|---|
| espBcompF | CCGCTTTGATCGGGGACGTCGCCGAATGCGACGACCTATCTCG |
| espBcompR | ACATCGATAAGCTTCGAATTGGCGACATGCGGGTCCAATTCG |
| qRT_sigA_F | AAACAGATCGGCAAGGTAGC |
| qRT_sigA_R | CTGGATCAGGTCGAGAAACG |
| qRT_espA_F | TACGACCTTCTGGGGATTGG |
| qRT_espA_R | ACGATCGAGGTCTGCCAGTT |
| qRT_espC_F | CGAATCTGTGGCGATCACTC |
| qRT_espC_R | ACAACCCGTCGATAGCCTTG |
| qRT_PPE68_F | GAAGCCTGGACTGGAGGTGG |
| qRT_PPE68_R | CGGTCAACGCGATCGGGATC |
| qRT_esxB_F | CAGATCGACCAGGTGGAGTCGA |
| qRT_esxB_R | TCGTCGGCCCTCGAGTATTG |
| qRT_esxA_F | GGGAAGCAGTCCCTGACCAAG |
| qRT_esxA_R | CCCAGTGACGTTGCCTTCGG |
| qRT_espI_F | CGACTACGACAAGCTCTTCCG |
| qRT_espI_R | GACACGAACCGCTCCGACAG |
| qRT_eccD1_F | CCGGGTGACGATCCTGACCG |
| qRT_eccD1_R | GTCGAAGCCGCCGAGTACATC |
| qRT_espB_F | GACCCACCGACTGATGTCCC |
| qRT_espB_R | CTGCCTGCACAGTTCCTTCG |
Cell cultures
BMDMs were differentiated from cryopreserved bone marrow stocks extracted from femurs of 6‐ to 8‐week‐old C57BL/6 (RRID:IMSR_JAX:000664, Jackson Laboratory) and mTmG mice (RRID:IMSR_JAX:007676, Jackson Laboratory). Mice were housed in a specific pathogen‐free facility. Animal protocols were reviewed and approved by EPFL's Chief Veterinarian, by the Service de la Consommation et des Affaires Vétérinaires of the Canton of Vaud, and by the Swiss Office Vétérinaire Fédéral. The bone marrow was cultured in Petri dishes in BMDM differentiation medium (DMEM with 10% FBS, 1% sodium‐pyruvate, 1% GlutaMax, and 20% L929‐cell‐conditioned medium as a source of granulocyte/macrophage colony‐stimulating factor). After 7 days, adherent cells were gently lifted from the plate using a cell scraper, resuspended in BMDM culture medium (DMEM with 5% FBS, 1% sodium‐pyruvate, 1% GlutaMax and 5% L929‐cell‐conditioned medium) and seeded in a 35 mm 4‐compartment Ibidi μ‐dishes or in Ibidi 24 well‐plates and incubated at 37°C, 5% CO2 before further experiments.
Macrophages infection
For infection with single non‐aggregated bacteria, 1 ml of Mtb culture at OD600 0.4–0.8 was pelleted, resuspended in 200 μl of macrophage medium, and passed through a 5 μm filter to eliminate bacterial aggregates. The resulting single‐cell suspension was used to infect BMDMs macrophages at an MOI of 1:1. After 4 h of infection, macrophages were washed extensively with macrophage medium to remove extracellular bacteria.
For infection with bacterial aggregates, 1 ml of Mtb culture at OD600 0.4–0.8 was pelleted and resuspended in 1 ml of macrophage medium. A 1:100 dilution of this solution was used to infect macrophages.
For spinfection experiments, a single‐cell suspension of Mtb or a solution of bacterial aggregates was added to BMDMs seeded in 24 well‐plates. After centrifugation at 500 g for 5 min, the medium was removed and replaced with fresh medium.
When required, 4 μM cytochalasin D (Invitrogen), 1:1,000 diluted Annexin V‐FITC (BioLegend) or Annexin V‐647 (BioLegend), 1:1,000 Draq7 (BioLegend), 50 μM BTP15 (gift from Prof. S. Cole), 50 μM Z‐IETD‐FMK (Selleck), 5 μM GSK872 (Abcam), 10 μM MCC950 (Selleck), 50 μM VX765 (Abcam), 10 μM dantrolene (Abcam), 10 mM CaCl2, 5 μM BAPTA‐AM (Selleckchem), 20 ng/ml TNFα (R&D systems), 10 μg/ml cycloheximide (Sigma–Aldrich), 50 ng/ml LPS (Sigma–Aldrich), 0.5 mM ATP (Sigma–Aldrich), 5 μM z‐VAD‐FMK (InvivoGen), and 50 nM SM164 (Selleckchem) were added at the time of infection and kept in the medium of the macrophages during the course of the experiment. When macrophages were stained with Oregon Green 488 Bapta‐1 AM (ThermoFisher Scientific), the staining was performed by adding 5 μM dye in PBS to the cells and incubating for 30 min at 37°C, 5% CO2. Macrophages were then washed three times in PBS to remove any residual extracellular dye before infection.
Immunofluorescence on macrophages infected with Mtb
Macrophages were fixed for 2 h with 4% paraformaldehyde in PBS at 24 h post‐infection. Cells were then washed three times with PBS and permeabilized for 15 min with 2% BSA, 2% saponine, 0.1% Triton x‐100 in PBS at RT. After three washes with PBS, samples were blocked for 1 h with 2% BSA in PBS at RT. Rabbit primary antibodies anti‐cleaved Caspase‐8 (Cell Signaling), ASC/TMS1 (Cell Signaling), cleaved Caspase‐1 (Cell Signaling), phospho‐RIP3 (Cell Signaling), phospho‐MLKL (Cell Signaling), mouse CD45.1‐AlexaFluor647 (BioLegend) were diluted 1:300 in 2% BSA in PBS and used for primary staining of the cells overnight at 4°C. After three washes in 2% BSA in PBS, samples (excluding those stained with anti‐mouse CD45.1‐AlexaFluor647) were incubated with a secondary goat anti‐rabbit antibody conjugated to Alexa Fluor 647 (ThermoFisher Scientific) diluted 1:300 in 2% BSA in PBS for 1 h at RT. Samples were stained with 1:1,000 Hoechst (ThermoFisher Scientific) and stored in PBS at 4°C before imaging by fluorescence microscopy.
Microscopy on macrophages infected with Mtb
For experiments quantifying cell viability, bacterial growth, and Annexin V staining, infected BMDMs were imaged by time‐lapse microscopy for up to 132 h at 1‐ or 2‐h intervals with a 20× air objective on a DeltaVision microscope or a 40× air objective on a Nikon Ti2 microscope. 3 × 1.5 μm z‐stacks were acquired on multiple XY fields.
For experiments monitoring the fluorescence of the infected cells (Annexin V and Draq7 staining, Oregon Green 488 Bapta‐1 AM fluorescence, mTmG BMDMs' membrane fluorescence), infected BMDMs were imaged by time‐lapse microscopy for up to 24 h at 20‐min intervals with a 63x oil‐objective on a DeltaVision microscope. 5 × 1 μm z‐stacks were acquired on multiple XY fields. Our DeltaVision microscope is equipped with FITC (Ex 490/20, Em 525/36), TRITC (Ex 555/25, Em 605/52), Cy‐5 (Ex 555/25, Em 605/52), and DAPI (Ex 438/24, Em 475/24) dichroic filters. On the Nikon Ti2 microscope GFP (Em 480/30, Ex 535/45) and mCherry (Em 560/40, Ex 635/60) dichroic filters were used. A stage‐top incubator (Okolab) was used to maintain the cells at 37°C in a humidified environment. Air mixed to 5% CO2 was supplied using an Okolab gas mixer. One‐half of the macrophage medium was refreshed every 3 days through custom tubing connected to the lid of the Ibidi μ‐dish.
Immunostained samples were imaged with a 63x oil‐objective on a DeltaVision microscope. The 5 × 1 μm z‐stacks were acquired on multiple XY fields.
Confocal images on fixed samples were acquired using a Leica SP8 microscope. Top and bottom z‐coordinates for all the selected cells were assigned manually and z‐stacks were acquired every 0.29 μm.
Image analysis
The microscopy images and time series were analyzed using the FIJI software from the ImageJ package (Schindelin et al, 2012). The z‐stacks acquired for the individual fluorescence channels were projected into one image using a maximum‐intensity projection. A manual threshold was set to segment the bacteria. The area above the threshold was measured and used as a proxy for the number of bacteria for each time point. To quantify the bacterial growth rate, an exponential curve was fitted to the data. In experiments without cytochalasin D treatment, bacteria were considered intracellular when they overlap with a macrophage in the bright‐field channel, otherwise they were annotated as extracellular. Macrophage infection status, Annexin V staining, and viability were monitored by visual analysis of the time‐lapse image series. Regions of interest corresponding to individual macrophages were manually drawn onto the bright‐field images and transferred to fluorescence images. Macrophages were considered infected when the region of interest overlapped with a fluorescent region in the channel used to image bacteria. Similarly, macrophages were considered Annexin V‐positive upon the appearance of positive regions in the corresponding fluorescence channel. The time of death corresponds to the frame where a macrophage stops moving and lyses. The time interval between initial infection and the appearance of the Annexin V‐positive domain or the cell death was calculated as the difference between the first frame where a macrophage was infected and the frame where the host cell became Annexin V‐positive or died. For experiments with Oregon Green 488 Bapta‐1 AM, the z‐stacks acquired in the green channel were projected into one image using a “sum slices” projection. Background subtraction was performed by subtracting from the fluorescence images a copy of the same images on which a Gaussian blur of 100 μm radius had been applied. Regions of interest (ROIs) corresponding to individual macrophages were manually drawn onto the phase images and transferred to the fluorescence images. The Oregon Green 488 Bapta‐1 AM fluorescence of each macrophage was quantified for each frame as average fluorescence intensity and normalized to the first frame imaged (for bystander cells) or to the frame of infection (for infected cells).
For images acquired from cells stained with anti‐ASC or anti‐cleaved Caspase8 (cCasp1) antibodies, ASC‐positive, or cCasp8‐positive cells were counted through manual analysis of the fluorescence images. For images acquired from cells stained with anti‐cleaved Caspase1, anti‐phosphorylated RIP3, or anti‐phosphorylated MLKL antibodies, the z‐stacks acquired in the Cy5 channel were projected into one image using a “sum slices” projection. ROIs corresponding to individual macrophages were drawn onto the phase images and transferred to the fluorescence channel to measure the background subtracted median fluorescence for each cell. Cells whose ROIs overlap with bacteria in the bacterial fluorescence channel were annotated as infected.
Confocal images were processed with ImageJ for 3D reconstruction. A Gaussian Blur filter with a 2‐pixel radius was applied to the channel corresponding to the macrophage membrane. The TransformJ:Rotate plugin was used to resample isotropically all the channels using a Quantic B‐Spline interpolation. The 3D visualization was obtained using the 3D Script—Interactive Animation tool and a combined transparency rendering.
Scanning electron microscopy
Cells were fixed for 2 h with 1.25% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, and then washed in cacodylate buffer before a secondary fix for 30 min in 0.2% osmium tetroxide in the same buffer. The cells were then dehydrated in a graded alcohol series and dried by passing them through the supercritical point of carbon dioxide in a critical point dryer (Leica Microsystems CPD300). They were then coated with a 2 nm layer of osmium metal using an osmium plasma coater (Filgen OPC60). Images of the cells were taken with a field emission scanning electron microscope (Merlin, Zeiss NTS).
CFUs counting
For colony forming units (CFUs) enumeration over time, macrophages in individual wells were infected at time point zero. At different time points, the medium of the infected cells was collected for plating. The macrophages were lysed with 0.5% TritonX‐100 in PBS for 5 min at room temperature, and the lysate was collected and mixed with the medium. Serial dilutions of this mixture were plated on a solid medium and incubated at 37°C. After 3 weeks CFUs were counted.
RNA extraction and quantitative Real‐Time PCR (qRT‐PCR)
A 10 ml Mtb cultures in the mid‐logarithmic phase were pelleted by centrifugation, resuspended in 800 μl of TRIzol Reagent (ThermoFisher), and added to a 2‐ml screw‐cap tube containing zirconia beads (BioSpec Products). Cells were disrupted by bead‐beating three times for 30 s. The cell lysate was transferred to a new tube, mixed with 200 μl of chloroform, and spun down for 15 min at 12,000 g. The top aqueous phase was collected and mixed with an iso‐volume of isopropanol. The RNA was precipitated by centrifugation for 10 min at 15,000 g, washed with 75% ethanol, air‐dried, and resuspended in DEPC‐treated water. DNase treatment was carried out using a TURBO DNA‐free kit (Invitrogen) according to the manufacturer's protocol. The RNA was transcribed to cDNA using the SuperScript IV First‐strand Synthesis System (ThermoFisher) according to the manufacturer's recommendations. qRT‐PCR were performed on an ABI 7900HT instrument, using SYBRGreen PCR Master Mix (Applied Biosystems), according to the manufacturer's instructions. Relative mRNA levels were calculated using the ΔΔCt method, normalizing transcripts levels to sigA signals. The sequences of the primers used are listed in Table 2.
Protein extracts and immunoblotting
Mtb was inoculated in 10 ml cultures (or 30 ml cultures for proteomics samples) in Sauton's medium supplemented with 0.05% Tween 80 at a starting OD600 of 0.1 and incubated at 37°C with shaking. After 4 days the cells were harvested by centrifugation, washed with PBS, and resuspended in 10 ml (or 30 ml cultures for proteomics samples) of Sauton's medium without Tween 80 and incubated at 37°C with agitation for 4 days. Cultures were pelleted by centrifugation and the collected culture media were filtered through a 0.2‐μm‐pore‐size filter. Culture filtrates (secretomes) were concentrated 50x using Amicon Ultra centrifugal filters with 3‐kDa cut‐off (Millipore), respectively. To prepare total “bacterial lysates,” the bacterial pellets were resuspended in TBS (20 mM Tris + 150 mM NaCl) plus cOmplete mini EDTA‐free protease inhibitors (Roche) and disrupted by bead beating four times for 30 s with zirconia beads (BioSpec Products). After clarification by centrifugation, the samples were filtered with a 0.2‐μm‐pore‐size filter. Total protein concentration in all the samples was determined using a Pierce BCA assay (ThermoFisher) with bovine serum albumin as the standard. Protein samples were resolved in NuPAGE 4–12% bis‐tris gels (Invitrogen) under reducing conditions. Proteins were transferred from the gel to nitrocellulose membranes using the iBlot Dry Blotting System (Invitrogen) according to the manufacturers instructions. Membranes were blocked for 1 h at room temperature with TBS + 3% BSA fraction V and then incubated overnight with the primary antibody diluted in TNT (TBS + 0.1% Tween‐20) + 2% BSA fraction V at 4°C. Membranes were washed with TNT, incubated with the secondary antibody in TNT+ 2% BSA for 30 min at room temperature, washed with TNT, and developed.
Mass spectrometry on protein samples
Mass spectrometry‐based proteomics‐related experiments were performed by the Proteomics Core Facility at EPFL. Each sample was digested by filter‐aided sample preparation (FASP) (Wiśniewski et al, 2009) with minor modifications. Proteins (40 μg) were reduced with 10 mM TCEP in 8 M urea, 0.1 M Tris–HCl pH 8.0 at 37°C for 60 min, and further alkylated in 40 mM iodoacetamide at 37°C for 45 min in the dark. Proteins were digested overnight at 37°C using a 1/50 w/w enzyme‐to‐protein ratio of mass spectrometry grade trypsin gold and LysC. Generated peptides were desalted in StageTips using 6 disks from an Empore C18 (3 M) filter based on the standard protocol (Rappsilber et al, 2007). Purified peptides were dried down by vacuum centrifugation. Samples were resuspended in 2% acetonitrile (Biosolve), 0.1% FA, and nano‐flow separations were performed on a Dionex Ultimate 3000 RSLC nano UPLC system (Thermo Fischer Scientific) on‐line connected with an Orbitrap Exploris Mass Spectrometer (Thermo Fischer Scientific). A capillary precolumn (Acclaim Pepmap C18, 3 μm‐100 Å, 2 cm × 75 μm ID) was used for sample trapping and cleaning. A 50 cm long capillary column (75 μm ID; in‐house packed using ReproSil‐Pur C18‐AQ 1.9 μm silica beads; Dr. Maisch) was then used for analytical separations at 250 nl/min over 150 min biphasic gradients. Acquisitions were performed through Top Speed Data‐Dependent acquisition mode using a cycle time of 2 s. First MS scans were acquired with a resolution of 60,000 (at 200 m/z) and the most intense parent ions were selected and fragmented by high energy collision dissociation (HCD) with a normalized collision energy (NCE) of 30% using an isolation window of 2 m/z. Fragmented ions were acquired with a resolution 15,000 (at 200 m/z) and selected ions were then excluded for the following 20 s.
Raw data were processed using MaxQuant 1.6.10.43 (Cox & Mann, 2008) against the Uniprot Reference Proteome Mycobacterium Tuberculosis Erdman strain (4222 sequences, LM201030). Carbamidomethylation was set as a fixed modification, whereas oxidation (M), phosphorylation (S, T, Y), acetylation (Protein N‐term), and glutamine to pyroglutamate were considered variable modifications. A maximum of two missed cleavages were allowed and “Match between runs” option was enabled. A minimum of 2 peptides was required for protein identification and the false discovery rate (FDR) cutoff was set to 0.01 for both peptides and proteins. Label‐free quantification and normalization were performed by Maxquant using the MaxLFQ algorithm, with the standard settings (Cox et al, 2014). The statistical analysis was performed using Perseus version 1.6.12.0 (Tyanova et al, 2016) from the MaxQuant tool suite. Reverse proteins, potential contaminants, and proteins only identified by sites were filtered out. Protein groups containing at least 2 valid values in at least one condition were conserved for further analysis. Missing values were imputed with random numbers from a normal distribution (Width: 0.3 and Down shift: 1.8 sd). A two‐sample t‐test with permutation‐based FDR statistics (250 permutations, FDR = 0.05, S0 = 1) was performed to determine significant differentially abundant candidates.
PDIM extraction
Phthiocerol dimycocerosates (PDIM) synthesis was evaluated by thin‐layer chromatography (TLC) of bacterial lipids (Kirksey et al, 2011). 10 ml of exponential‐phase bacteria were labeled with 2 μCi of [14C]‐propionate (Campro Scientific) for 48 h. Lipids were extracted from the pellets of the radiolabelled culture using 5 ml of methanol: 0.3% NaCl (10:1) and 5 ml of petroleum ether. The mixture was vortexed for 3 min, centrifuged at 930 g for 10 min, and the upper layer was collected. A second extraction on the lower phase was carried out with 5 ml of petroleum ether. The combined extracts were inactivated with an iso‐volume of chloroform for 1 h and concentrated through evaporation. The samples were spotted on a 5 × 10 cm TLC silica gel 60 F254 (Merck) and developed in petroleum ether:diethyl ether (9:1). The developed TLC plate was exposed to an Amersham Hyperfilm ECL (GE Healthcare) for chemioluminescence analysis and visualized with a Typhoon Scanner (GE Lifesciences).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.
Author contributions
Chiara Toniolo: Conceptualization; data curation; formal analysis; investigation; visualization; methodology; writing – original draft; writing – review and editing. Neeraj Dhar: Conceptualization; resources; supervision; funding acquisition; writing – review and editing. John D Mckinney: Conceptualization; resources; supervision; funding acquisition; writing – original draft.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by grants to J.D.M. from the Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (SNF) (310030B_176397). This work has received support from the Innovative Medicines Initiatives 2 Joint Undertaking grant No 853989 “ERA4TB” (https://era4tb.org/). C.T. was supported by funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement No. 665667. VIDO receives operational funding from the Government of Saskatchewan through Innovation Saskatchewan and the Ministry of Agriculture and from the Canada Foundation for Innovation through the Major Science Initiatives for its CL3 facility. The synopsis image was created using BioRender. We would like to thank Dr. Maria Pavlou, Dr. Florence Armand, and Jonathan Pittet from the Proteomics Core Facilities at the School of Life Sciences of EPFL for their help in processing and analyzing the Mtb secretome samples. We acknowledge Anaëlle Dubois, Stéphanie Rosset, Marie Croisier, and Prof. Graham W. Knott from the EPFL Biological Electron Microscopy Facility for their help in preparing and imaging samples by SEM. We acknowledge Dr. Nicholas Chiaruttini, José Artacho, and Dr. Arne Seitz from the EPFL Bioimaging and Optics Platform for their help with confocal microscopy and image analysis. We thank Dr. Daniel Sage for help with image analysis; Dr. Catherine Astarie‐Dequeker for sharing the H37Rv ∆esxA strains; Prof. Stewart Cole and Prof. Jeffrey Chen for providing antibodies, Mtb strains, and BTP15. Finally, we thank all the members of the McKinney group for fruitful discussions and feedback on the manuscript. Open access funding provided by Ecole Polytechnique Federale de Lausanne.
The EMBO Journal (2023) 42: e113490
Data availability
All strains and plasmids are available by request to the corresponding author. The mass spectrometry proteomics data generated for this study have been deposited to the ProteomeXchange Consortium (Deutsch et al, 2020) via the PRIDE (Perez‐Riverol et al, 2022) partner repository (http://proteomecentral.proteomexchange.org/cgi/GetDataset). Data relative to the Mtb Erdman strains have the dataset identifier PXD035080. Data relative to the Mtb H37Rv strains have the dataset identifier PXD035082.
References
- Aguilo JI, Alonso H, Uranga S, Marinova D, Arbués A, de Martino A, Anel A, Monzon M, Badiola J, Pardo J et al (2013) ESX‐1‐induced apoptosis is involved in cell‐to‐cell spread of Mycobacterium tuberculosis . Cell Microbiol 15: 1994–2005 [DOI] [PubMed] [Google Scholar]
- Aguilo N, Gonzalo‐Asensio J, Alvarez‐Arguedas S, Marinova D, Gomez AB, Uranga S, Spallek R, Singh M, Audran R, Spertini F et al (2017) Reactogenicity to major tuberculosis antigens absent in BCG is linked to improved protection against mycobacterium tuberculosis . Nat Commun 8: 16085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astarie‐Dequeker C, Le Guyader L, Malaga W, Seaphanh F‐K, Chalut C, Lopez A, Guilhot C (2009) Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog 5: e1000289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augenstreich J, Arbues A, Simeone R, Haanappel E, Wegener A, Sayes F, Le Chevalier F, Chalut C, Malaga W, Guilhot C et al (2017) ESX‐1 and phthiocerol dimycocerosates of mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell Microbiol 19: e12726 [DOI] [PubMed] [Google Scholar]
- Augenstreich J, Haanappel E, Sayes F, Simeone R, Guillet V, Mazeres S, Chalut C, Mourey L, Brosch R, Guilhot C et al (2020) Phthiocerol dimycocerosates from mycobacterium tuberculosis increase the membrane activity of bacterial effectors and host receptors. Front Cell Infect Microbiol 10: 420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barczak AK, Avraham R, Singh S, Luo SS, Zhang WR, Bray M‐A, Hinman AE, Thompson M, Nietupski RM, Golas A et al (2017) Systematic, multiparametric analysis of mycobacterium tuberculosis intracellular infection offers insight into coordinated virulence. PLoS Pathog 13: e1006363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, Åsberg SE, Strand TA, Haug M, Niederweis M et al (2020) Plasma membrane damage causes NLRP3 activation and pyroptosis during mycobacterium tuberculosis infection. Nat Commun 11: 2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, Hinds J, Neyrolles O, Butcher PD, Leclerc C et al (2006) Dissection of ESAT‐6 system 1 of mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect Immun 74: 88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier C, Banik SM, Buonomo JA, Bertozzi CR (2020) Spreading of a mycobacterial cell‐surface lipid into host epithelial membranes promotes infectivity. Elife 9: e60648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Zhang M, Rybniker J, Basterra L, Dhar N, Tischler AD, Pojer F, Cole ST (2013a) Phenotypic profiling of mycobacterium tuberculosis EspA point mutants reveals that blockage of ESAT‐6 and CFP‐10 secretion in vitro does not always correlate with attenuation of virulence. J Bacteriol 195: 5421–5430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Zhang M, Rybniker J, Boy‐Röttger S, Dhar N, Pojer F, Cole ST (2013b) Mycobacterium tuberculosis EspB binds phospholipids and mediates EsxA‐independent virulence. Mol Microbiol 89: 1154–1166 [DOI] [PubMed] [Google Scholar]
- Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, Sherman DR, Gerner MY, Urdahl KB (2018) Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe 24: 439–446.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad WH, Osman MM, Shanahan JK, Chu F, Takaki KK, Cameron J, Hopkinson‐Woolley D, Brosch R, Ramakrishnan L (2017) Mycobacterial ESX‐1 secretion system mediates host cell lysis through bacterium contact‐dependent gross membrane disruptions. Proc Natl Acad Sci U S A 114: 1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox JS, Chen B, McNeil M, Jacobs WR (1999) Complex lipid determines tissue‐specific replication of mycobacterium tuberculosis in mice. Nature 402: 79–83 [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ*. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallenga T, Repnik U, Corleis B, Eich J, Reimer R, Griffiths GW, Schaible UE (2017) Mycobacterium tuberculosis‐induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe 22: 519–530.e3 [DOI] [PubMed] [Google Scholar]
- Dartois V (2014) The path of anti‐tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat Rev Microbiol 12: 159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JM, Ramakrishnan L (2009) The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136: 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Bandeira N, Sharma V, Perez‐Riverol Y, Carver JJ, Kundu DJ, García‐Seisdedos D, Jarnuczak AF, Hewapathirana S, Pullman BS et al (2020) The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res 48: D1145–D1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar N, McKinney JD (2010) Mycobacterium tuberculosis persistence mutants identified by screening in isoniazid‐treated mice. Proc Natl Acad Sci U S A 107: 12275–12280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkele R, Gessner S, McKerry A, Leonard B, Seldon R, Koch AS, Morrow C, Gqada M, Kamariza M, Bertozzi CR et al (2021) Capture and visualization of live mycobacterium tuberculosis bacilli from tuberculosis patient bioaerosols. PLoS Pathog 17: e1009262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, Fortune S, Behar SM, Remold HG (2009) Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol 10: 899–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers S, Schaible U (2013) The granuloma in tuberculosis: dynamics of a host–pathogen collusion. Front Immunol 3: 411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, Bloom BR, Rubin EJ (2005) Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci U S A 102: 10676–10681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW et al (2018) Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 25: 486–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijsbers A, Vinciauskaite V, Siroy A, Gao Y, Tria G, Mathew A, Sánchez‐Puig N, López‐Iglesias C, Peters PJ, Ravelli RBG (2021) Priming mycobacterial ESX‐secreted protein B to form a channel‐like structure. Curr Res Struct Biol 3: 153–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MS, Cox JS, Jacobs WR (2000) A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of mycobacterium tuberculosis . Mol Cell 5: 717–727 [DOI] [PubMed] [Google Scholar]
- Gröschel MI, Sayes F, Simeone R, Majlessi L, Brosch R (2016) ESX secretion systems: mycobacterial evolution to counter host immunity. Nat Rev Microbiol 14: 677–691 [DOI] [PubMed] [Google Scholar]
- Hoff DR, Ryan GJ, Driver ER, Ssemakulu CC, De Groote MA, Basaraba RJ, Lenaerts AJ (2011) Location of intra‐ and extracellular M. tuberculosis populations in lungs of mice and Guinea pigs during disease progression and after drug treatment. PLoS One 6: e17550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu T, Hingley‐Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M et al (2003) The primary mechanism of attenuation of bacillus Calmette–Guérin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A 100: 12420–12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Nazarova EV, Tan S, Liu Y, Russell DG (2018) Growth of mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med 215: 1135–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RL (2011) Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis 91: 497–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SM, Driver E, Lyon E, Schrupp C, Ryan G, Gonzalez‐Juarrero M, Basaraba RJ, Nuermberger EL, Lenaerts AJ (2015) Presence of multiple lesion types with vastly different microenvironments in C3HeB/FeJ mice following aerosol infection with mycobacterium tuberculosis . Dis Model Mech 8: 591–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augenstreich J, Haanappel E, Ferré G, Czaplicki G, Jolibois F, Destainville N, Guilhot C, Milon A, Astarie‐Dequeker C, Chavent M (2019) The conical shape of DIM lipids promotes mycobacterium tuberculosis infection of macrophages. Proc Natl Acad Sci U S A 116: 25649–25658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez AJ, Maiuri P, Lafaurie‐Janvore J, Divoux S, Piel M, Perez F (2014) ESCRT machinery is required for plasma membrane repair. Science 343: 1247136 [DOI] [PubMed] [Google Scholar]
- de Jonge Marien I, Gérard P‐A, Fretz MM, Felix R, Daria B, Priscille B, Nadine H, Gilles M, Wim J, Patrick E et al (2007) ESAT‐6 from mycobacterium tuberculosis dissociates from its putative chaperone CFP‐10 under acidic conditions and exhibits membrane‐lysing activity. J Bacteriol 189: 6028–6034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan G, Post FA, Moreira AL, Wainwright H, Kreiswirth BN, Tanverdi M, Mathema B, Ramaswamy SV, Walther G, Steyn LM et al (2003) Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect Immun 71: 7099–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CH, Mundayoor S, Crawford JT, Shinnick TM (1993) Expression of contact‐dependent cytolytic activity by mycobacterium tuberculosis and isolation of the genomic locus that encodes the activity. Infect Immun 61: 2708–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinhikar AG, Verma I, Chandra D, Singh KK, Weldingh K, Andersen P, Hsu T, Jacobs WR Jr, Laal S (2010) Potential role for ESAT6 in dissemination of M. tuberculosis via human lung epithelial cells. Mol Microbiol 75: 92–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirksey MA, Tischler AD, Siméone R, Hisert KB, Uplekar S, Guilhot C, McKinney JD (2011) Spontaneous phthiocerol dimycocerosate‐deficient variants of mycobacterium tuberculosis are susceptible to gamma interferon‐mediated immunity. Infect Immun 79: 2829–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolloli A, Kumar R, Singh P, Narang A, Kaplan G, Sigal A, Subbian S (2021) Aggregation state of mycobacterium tuberculosis impacts host immunity and augments pulmonary disease pathology. Commun Biol 4: 1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova N, Piton J, Wagner JM, Boy‐Röttger S, Japaridze A, Evans TJ, Cole ST, Pojer F, Korotkov KV (2015) Structure of EspB, a secreted substrate of the ESX‐1 secretion system of mycobacterium tuberculosis . J Struct Biol 191: 236–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Remold HG, Ieong MH, Kornfeld H (2006) Macrophage apoptosis in response to high intracellular burden of mycobacterium tuberculosis is mediated by a novel caspase‐independent pathway. J Immunol 176: 4267–4274 [DOI] [PubMed] [Google Scholar]
- Lerner TR, Borel S, Greenwood DJ, Repnik U, Russell MRG, Herbst S, Jones ML, Collinson LM, Griffiths G, Gutierrez MG (2017) Mycobacterium tuberculosis replicates within necrotic human macrophages. J Cell Biol 216: 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner TR, Queval CJ, Lai RP, Russell MRG, Fearns A, Greenwood DJ, Collinson L, Wilkinson RJ, Gutierrez MG (2020) Mycobacterium tuberculosis cords within lymphatic endothelial cells to evade host immunity. JCI Insight 5: e136937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ZL, Drever K, Dhar N, Cole ST, Chen JM (2022) Mycobacterium tuberculosis EspK has active but distinct roles in the secretion of EsxA and EspB. J Bacteriol 204: e00060‐22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD (2014) Cell‐autonomous effector mechanisms against mycobacterium tuberculosis . Cold Spring Harb Perspect Med 4: a018507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed D, Boulle M, Ganga Y, Mc Arthur C, Skroch S, Oom L, Catinas O, Pillay K, Naicker M, Rampersad S et al (2017) Intracellular growth of mycobacterium tuberculosis after macrophage cell death leads to serial killing of host cells. Elife 6: e22028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohareer K, Asalla S, Banerjee S (2018) Cell death at the cross roads of host‐pathogen interaction in mycobacterium tuberculosis infection. Tuberculosis 113: 99–121 [DOI] [PubMed] [Google Scholar]
- Murphy KC, Nelson SJ, Nambi S, Papavinasasundaram K, Baer CE, Sassetti CM, Nacy CA (2018) ORBIT: a new paradigm for genetic engineering of mycobacterial chromosomes. MBio 9: e01467‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Ueno A, Saga K, Fukuzawa M, Kaneda Y (2013) Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res 74: 1056–1066 [DOI] [PubMed] [Google Scholar]
- Ohol YM, Goetz DH, Chan K, Shiloh MU, Craik CS, Cox JS (2010) Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX‐1 secretion and virulence. Cell Host Microbe 7: 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme IM (2014) A new unifying theory of the pathogenesis of tuberculosis. Tuberculosis 94: 8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik S, Kim JK, Silwal P, Sasakawa C, Jo E‐K (2021) An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol 18: 1141–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajuelo D, Gonzalez‐Juarbe N, Tak U, Sun J, Orihuela CJ, Niederweis M (2018) NAD+ depletion triggers macrophage necroptosis, a cell death pathway exploited by mycobacterium tuberculosis . Cell Rep 24: 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang X, Samten B, Cao G, Wang X, Tvinnereim AR, Chen XL, Howard ST (2013) MprAB regulates the espA operon in mycobacterium tuberculosis and modulates ESX‐1 function and host cytokine response. J Bacteriol 195: 66–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Tamayo MH, Gonzalez‐Juarrero M, Orme IM, Ordway DJ (2006) Virulent clinical isolates of mycobacterium tuberculosis grow rapidly and induce cellular necrosis but minimal apoptosis in murine macrophages. J Leukoc Biol 79: 80–86 [DOI] [PubMed] [Google Scholar]
- Passemar C, Arbués A, Malaga W, Mercier I, Moreau F, Lepourry L, Neyrolles O, Guilhot C, Astarie‐Dequeker C (2014) Multiple deletions in the polyketide synthase gene repertoire of mycobacterium tuberculosis reveal functional overlap of cell envelope lipids in host–pathogen interactions. Cell Microbiol 16: 195–213 [DOI] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Bai J, Bandla C, García‐Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks‐Zipper A, Eisenacher M et al (2022) The PRIDE database resources in 2022: a hub for mass spectrometry‐based proteomics evidences. Nucleic Acids Res 50: D543–D552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piton J, Pojer F, Wakatsuki S, Gati C, Cole ST (2020) High resolution CryoEM structure of the ring‐shaped virulence factor EspB from mycobacterium tuberculosis . J Struct Biol X 4: 100029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffetseder J, Iakobachvili N, Loitto V, Peters PJ, Lerm M (2019) Retention of EsxA in the capsule‐like layer of mycobacterium tuberculosis is associated with cytotoxicity and is counteracted by lung surfactant. Infect Immun 87: e00803‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan L (2020) Mycobacterium tuberculosis pathogenicity viewed through the lens of molecular Koch's postulates. Curr Opin Microbiol 54: 103–110 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y (2007) Protocol for micro‐purification, enrichment, pre‐fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2: 1896–1906 [DOI] [PubMed] [Google Scholar]
- Renshaw PS, Panagiotidou P, Whelan A, Gordon SV, Hewinson RG, Williamson RA, Carr MD (2002) Conclusive evidence that the major T‐cell antigens of the mycobacterium tuberculosis complex ESAT‐6 and CFP‐10 form a tight, 1:1 complex and characterization of the structural properties of ESAT‐6, CFP‐10, and the ESAT‐6·CFP‐10 complex. Implications for pathogenesis and virulence. J Biol Chem 277: 21598–21603 [DOI] [PubMed] [Google Scholar]
- Repasy T, Lee J, Marino S, Martinez N, Kirschner DE, Hendricks G, Baker S, Wilson AA, Kotton DN, Kornfeld H (2013) Intracellular bacillary burden reflects a burst size for mycobacterium tuberculosis in vivo . PLoS Pathog 9: e1003190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repasy T, Martinez N, Lee J, West K, Li W, Kornfeld H (2015) Bacillary replication and macrophage necrosis are determinants of neutrophil recruitment in tuberculosis. Microbes Infect 17: 564–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley RL, Mills CC, Nyka W, Weinstock N, Storey PB, Sultan LU, Riley MC, Wells WF (1995) Aerial dissemination of pulmonary tuberculosis. A two‐year study of contagion in a tuberculosis ward. Am J Epidemiol 142: 3–14 [DOI] [PubMed] [Google Scholar]
- Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L (2019) TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial‐lysosomal‐endoplasmic reticulum circuit. Cell 178: 1344–1361.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodel HE, Ferreira IATM, Ziegler CGK, Ganga Y, Bernstein M, Hwa S‐H, Nargan K, Lustig G, Kaplan G, Noursadeghi M et al (2021) Aggregated mycobacterium tuberculosis enhances the inflammatory response. Front Microbiol 12: 757134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybniker J, Chen JM, Sala C, Hartkoorn RC, Vocat A, Benjak A, Boy‐Röttger S, Zhang M, Székely R, Greff Z et al (2014) Anticytolytic screen identifies inhibitors of mycobacterial virulence protein secretion. Cell Host Microbe 16: 538–548 [DOI] [PubMed] [Google Scholar]
- Sani M, Houben ENG, Geurtsen J, Pierson J, de Punder K, van Zon M, Wever B, Piersma SR, Jiménez CR, Daffé M et al (2010) Direct visualization by cryo‐EM of the mycobacterial capsular layer: a labile structure containing ESX‐1‐secreted proteins. PLoS Pathog 6: e1000794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, Enninga J (2012) Phagosomal rupture by mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog 8: e1002507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomonson M, Setiaputra D, Makepeace KAT, Lameignere E, Petrotchenko EV, Conrady DG, Bergeron JR, Vuckovic M, DiMaio F, Borchers CH et al (2015) Structure of EspB from the ESX‐1 type VII secretion system and insights into its export mechanism. Structure 23: 571–583 [DOI] [PubMed] [Google Scholar]
- Stanley SA, Raghavan S, Hwang WW, Cox JS (2003) Acute infection and macrophage subversion by mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci U S A 100: 13001–13006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF et al (1991) New use of BCG for recombinant vaccines. Nature 351: 456–460 [DOI] [PubMed] [Google Scholar]
- Strydom N, Gupta SV, Fox WS, Via LE, Bang H, Lee M, Eum S, Shim T, Barry CE III, Zimmerman M et al (2019) Tuberculosis drugs' distribution and emergence of resistance in patient's lung lesions: a mechanistic model and tool for regimen and dose optimization. PLoS Med 16: e1002773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill‐Koszycki S, Schlesinger P, Chakraborty P, Haddix P, Collins H, Fok A, Allen R, Gluck S, Heuser J, Russell D (1994) Lack of acidification in mycobacterium phagosomes produced by exclusion of the vesicular proton‐ATPase. Science 263: 678–681 [DOI] [PubMed] [Google Scholar]
- Swanson KV, Deng M, Ting JP‐Y (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19: 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takii T, Yamamoto Y, Chiba T, Abe C, Belisle JT, Brennan PJ, Onozaki K (2002) Simple fibroblast‐based assay for screening of new antimicrobial drugs against mycobacterium tuberculosis . Antimicrob Agents Chemother 46: 2533–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timm J, Kurepina N, Kreiswirth BN, Post FA, Walther GB, Wainwright HC, Bekker L‐G, Kaplan G, McKinney JD (2006) A multidrug‐resistant, acr1‐deficient clinical isolate of mycobacterium tuberculosis is unimpaired for replication in macrophages. J Infect Dis 193: 1703–1710 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Warner DF (2014) Mycobacterium tuberculosis metabolism. Cold Spring Harb Perspect Med 5: a021121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid‐Burgk JL, Schmidt T, Hornung V, Cole ST et al (2015) Mycobacterium tuberculosis differentially activates cGAS‐ and inflammasome‐dependent intracellular immune responses through ESX‐1. Cell Host Microbe 17: 799–810 [DOI] [PubMed] [Google Scholar]
- van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ (2007) M. Tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129: 1287–1298 [DOI] [PubMed] [Google Scholar]
- Welin A, Eklund D, Stendahl O, Lerm M (2011) Human macrophages infected with a high burden of ESAT‐6‐expressing M. tuberculosis undergo Caspase‐1‐ and Cathepsin B‐independent necrosis. PLoS One 6: e20302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells G, Glasgow JN, Nargan K, Lumamba K, Madansein R, Maharaj K, Hunter RL, Naidoo T, Coetzer L, le Roux S et al (2021) Micro–computed tomography analysis of the human tuberculous lung reveals remarkable heterogeneity in three‐dimensional granuloma morphology. Am J Respir Crit Care Med 204: 583–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- Zhang M, Chen JM, Sala C, Rybniker J, Dhar N, Cole ST (2014) EspI regulates the ESX‐1 secretion system in response to ATP levels in mycobacterium tuberculosis . Mol Microbiol 93: 1057–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jiang X, Pfau D, Ling Y, Nathan CF (2020) Type I interferon signaling mediates mycobacterium tuberculosis–induced macrophage death. J Exp Med 218: e20200887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Khan N, Gan H, Tzelepis F, Nishimura T, Park S‐Y, Divangahi M, Remold HG (2017) Bcl‐xL mediates RIPK3‐dependent necrosis in M. tuberculosis‐infected macrophages. Mucosal Immunol 10: 1553–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]