

Intratumoral immune hotspots beyond tertiary lymphoid structures reflect an immunosuppressive microenvironment, different from peritumoral immune hotspots, warranting further study in the context of immunotherapies.
Abstract
Beyond tertiary lymphoid structures, a significant number of immune-rich areas without germinal center-like structures are observed in non–small cell lung cancer. Here, we integrated transcriptomic data and digital pathology images to study the prognostic implications, spatial locations, and constitution of immune rich areas (immune hotspots) in a cohort of 935 patients with lung cancer from The Cancer Genome Atlas. A high intratumoral immune hotspot score, which measures the proportion of immune hotspots interfacing with tumor islands, was correlated with poor overall survival in lung squamous cell carcinoma but not in lung adenocarcinoma. Lung squamous cell carcinomas with high intratumoral immune hotspot scores were characterized by consistent upregulation of B-cell signatures. Spatial statistical analyses conducted on serial multiplex IHC slides further revealed that only 4.87% of peritumoral immune hotspots and 0.26% of intratumoral immune hotspots were tertiary lymphoid structures. Significantly lower densities of CD20+CXCR5+ and CD79b+ B cells and less diverse immune cell interactions were found in intratumoral immune hotspots compared with peritumoral immune hotspots. Furthermore, there was a negative correlation between the percentages of CD8+ T cells and T regulatory cells in intratumoral but not in peritumoral immune hotspots, with tertiary lymphoid structures excluded. These findings suggest that the intratumoral immune hotspots reflect an immunosuppressive niche compared with peritumoral immune hotspots, independent of the distribution of tertiary lymphoid structures. A balance toward increased intratumoral immune hotspots is indicative of a compromised antitumor immune response and poor outcome in lung squamous cell carcinoma.
Significance:
Intratumoral immune hotspots beyond tertiary lymphoid structures reflect an immunosuppressive microenvironment, different from peritumoral immune hotspots, warranting further study in the context of immunotherapies.
Introduction
Despite a plethora of research dedicated to the study of non–small cell lung cancer (NSCLC) intratumor heterogeneity, deficiencies in our knowledge of the fundamental changes that occur within the tumor microenvironment have impeded progress in this area. Diverse immune responses have been proposed for different NSCLC subtypes, lung adenocarcinoma (LUAD), and lung squamous cell carcinoma (LUSC) based on the analysis of genome and transcriptome (1). Our recent findings resolve the link between spatial variability of lymphocyte infiltration with tumor immune evasion in LUAD (2). However, it is unclear how diverse immune cell subsets cooperate in a compact niche to drive pro- or antitumor responses, or whether they can be effectively measured in histology samples as part of the clinical routine (3).
We have previously demonstrated the importance of studying tumor spatial architecture for defining the clinical relevance of the immune response in estrogen receptor–positive and -negative breast cancer (4, 5). Spatial aggregation, rather than the sheer abundance of lymphocytes, is associated with recurrence-free survival in both breast cancer subtypes (4, 5). While this highlights the importance of examining not just cell abundance but also spatial patterns that can inform disease prognosis, important questions remain: (i) whether our observation extends to other cancer types beyond breast cancer, such as the highly immunogenic NSCLC; (ii) how these spatial patterns translate into immune cell functions and functional phenotypes; and (iii) how they collectively drive a microenvironment that promotes or suppresses cancer progression. In this article, we used transcriptomic data and deep learning–based digital pathology image analysis to: (i) discover the clinical relevance of spatial organization of lymphocytes in NSCLC; (ii) identify the transcriptomics underpinning of immune spatial heterogeneity; and (iii) uncover the interplay of immune cell subsets underlying distinct spatial organization patterns.
Materials and Methods
Patients and cohorts
All NSCLC whole-tumor hematoxylin and eosin (H&E) sections from The Cancer Genome Atlas (TCGA) analyzed in this study were downloaded from the TCGA data portal (n = 743 LUAD and 712 LUSC). These sections correspond to the total of 473 patients in TCGA LUAD and 466 in TCGA LUSC. Overall survival data (i.e., times to death or follow-up and vital status) was available for 935 patients with NSCLC, whereas additional clinical data (stage, age, smoking pack-years) were only available for 723 patients with NSCLC. The multivariate Cox validation took into account equally distributed discovery (n = 237 LUAD and 231 LUSC) and validation (n = 236 LUAD and 231 LUSC) cohorts.
The 10 external validation samples with multiplex IHC staining were obtained from the Leicester Archival Thoracic Tumor Investigatory Cohort (LATTICe) study, which consists of 1700 University Hospitals of Leicester Trust patients who underwent surgical treatment with curative intent for primary lung tumors. A consultant thoracic malignancy pathologist visually examined 20 H&E slides corresponding to 20 patients with high immune clustering. All 20 cases were analyzed using the immune hotspot pipeline to rank the top 10 cases. These 10 cases were then dearchived, and three serial sections were prepared for T-cell, H&E, and B-cell staining. The validation cohort was ethically approved by an NHS research ethics committee (ref. 14/EM/1159).
Automated H&E image analysis mapped heterogeneous cell populations
We collected four orthogonal data types to test the accuracy of automated H&E image analysis for single-cell classification in whole-section NSCLC images. CRImage (6) identified a total of 206,272,040 cancer cells, 35,733,365 stromal cells, and 49,953,400 lymphocytes in 1,455 H&E sections from frozen samples of 939 TCGA NSCLC patients. The balanced accuracy as an average of specification and sensitivity of the classifier was 82.13% for cancer cells, 89.16% for stromal cells and 87.81% for lymphocytes, comparing against 4,169 cancer, 1,533 stromal cells and 1,999 lymphocytes annotations by a thoracic malignancy pathologist in randomly sampled images (Fig. 1A). Automated cell percentage was more strongly correlated with molecular gene signatures than the independent scoring provided by TCGA pathologists (Supplementary Fig. S1A and S1C). This included a comparison between cancer cell percentages given by automated image analysis and pathologic scores of H&E, and tumor purity measures from the gene expression-based method ESTIMATE (7) and copy number–based ABSOLUTE (8). For stromal cell estimates, we compared H&E-derived scores with stromal scores from gene expression–based MCP-counter (9) and ESTIMATE (7). There was no pathologic score of lymphocytes available from the TCGA dataset, instead the lymphocyte percentage estimated by automated image analysis was evaluated against immune signatures inferred by TIMER (10). Automatic quantification of lymphocyte percentages significantly correlated with CD4, CD8 T-cell, and B-cell scores (R = 0.31, 0.17, and 0.24, respectively; P < 0.01; Supplementary Fig. S1B)
Figure 1.

Immune hotspots in LUSC. A, Illustrative examples of automatic cell classification results and hotspot distribution with zoomed-in regions showing areas enriched with immune cells and cancer–immune coclustering. Cancer hotspots (red) and IHs were identified based on the local clustering of cancer and lymphocytes. Intratumoral IH (green) was determined as regions enriched with both immune and cancer cells, while peritumoral IH (blue) were regions enriched with only immune cells. B, Schematic representations of tissue compartments including cancer hotspots, peritumoral IH, intratumoral IH based on automated spatial analysis, and pathologically defined TLSs and lymphoid aggregates without clear separation of T- and B-cell zones (LAG). The three spatial scores Sintra/immune, Sintra/cancer, and Sintra/tissue were determined as the proportion of intratumoral IH in the immune-rich area (intratumoral IH and peritumoral IH), cancer-rich area (intratumoral IH and cancer hotspots), and the entire tissue, respectively. C, Kaplan–Meier curves to illustrate the difference in OS in LUSC patients stratified by Sintra/immune, with the dichotomization optimized for the discovery cohort (n = 231) and assessed in the validation cohort (n = 231). D, Forest plots to show the prognostic value of Sintra/immune in multivariate models adjusted for stage, age, and smoking pack years in the validation cohort. E, Spearman correlation between lymphocyte percentage and Sintra/immune in the TCGA LUSC cohort (n = 462). F, Kaplan–Meier curve to illustrate the difference in OS in LUSC patients stratified by both Sintra/immune and lymphocyte percentage, with the stratification of the entire cohort shown (n = 462).
Bioinformatic analysis
The gene enrichment analysis for the Sintra/immune in LUSC was performed using the “TCGAbiolinks” package in R. The RNA-sequencing data (all Level 3 raw read counts, available from the TCGA data portal) were used as input to the transcriptome differential gene expression analysis. The outputs of highly enriched genes clustered under the Gene Ontology biological processes for LUSC and LUAD are shown in Fig. 2A and Supplementary Fig. S2A, respectively. The highly enriched immune genes relevant to the immune response/activation of immune response processes in LUSC included DEFB4A, LTF, FCN3, C7, MS4A1, PAX5, PLA2G1B, PLA2G2A, CHGA/CGA, FGB, BPIL1, DEFA5, CR2, PLUNC, C8B, DMBT1, KRT1. The differential gene expression analysis was performed using the ‘DESeq2’ R package to rank differentially expressed genes with logFC larger than 1 in high versus low groups for the Sintra/immune in LUSC and LUAD, with the optimal cut-off point determined on the entire cohort (Supplementary Table S1; Fig. 2B; Supplementary Fig. S2B). The B-cell signatures were estimated by seven algorithms Danaher (11), TIMER (10), CIBERSORT Absolute mode (12), MCP-counter (9), quanTIseq (13), EPIC (14), and xCell (15). The patient-level estimates were calculated as the average across slide-level scores of the patient. Comparisons between Sintra/immune high and low patients stratified by the median revealed a consistent upregulation of B-cell infiltrates in the high group (Fig. 2C). The tumor mutation burden (TMB) was computed as somatic mutations per Mb using R library “maftools” (16) for 444 TCGA LUSC tumors with mutation annotation format files available. The clonal neoantigen load was derived from a previous work (1).
Figure 2.

B-cell dominance of high-Sintra/immune group in LUSC. A, Gene Ontology (GO) biological processes found to be enriched in the DEGs according to patient groups stratified by Sintra/immune. The red line denotes the ratio of the number of DEGs in a pathway to the total number of genes in that pathway. FDR, false discovery rate. B, Volcano plot showing DEGs in the high-Sintra/immune group compared with the low group. Genes with log-fold change (logFC) smaller than 1 were excluded. Genes involved in immune responses are highlighted in yellow. C, Consistently upregulated B-cell signatures in high-Sintra/immune group across seven bioinformatics methods using the TCGA LUSC cohort, n = 459. Statistical significance was evaluated by the Wilcoxon rank sum test and adjusted by the Benjamini–Hochberg method.
Survival analysis of TCGA datasets
The investigated scores (Sintra/immune, Sintra/cancer, Sintra/tissue) were first dichotomized using Kaplan–Meier curves (R “survminer” package) to find an optimal cut-point on the discovery cohort (n = 237 LUAD and 231 LUSC). The optimal cut-off point was computed with maximally selected log-rank statistics. Next, the dichotomized cut-off point found in discovery was used in a multivariate Cox proportional hazards model (R “survival” package) adjusted for stage, age, and smoking pack-years to further substantiate the survival prognosis. The Kaplan–Meier curve and the Cox model with the same cut-off point were then tested in the validation cohort (n = 236 LUAD and 231 LUSC), which revealed that the Sintra/immune is a LUSC-specific prognosticator. The survival analysis was repeatedly performed for four patient groups determined by their smoking history (current reformed smokers for less than 15 years, current reformed smokers for more than 15 years, current smokers and lifelong nonsmokers; n = 457 LUAD and 446 LUSC in total) with the cut-off point of Sintra/immune optimized for each category. Lymphocyte percentage and TMB were dichotomized using a cut-off point optimized for the discovery cohort. To evaluate the contingency of survival analysis on the split of cohorts, we randomly split the TCGA LUSC cohort into discovery and validation by a 1:1 ratio for 100 times. In each iteration, the numerical parameters (Sintra/immune, age, pack years, and TMB) were dichotomized by the threshold optimized for the discovery cohort, followed by a log-rank test conducted in the validation cohort to determine the statistical difference in the OS of patients stratified by the dichotomized parameters. The stage was assessed as a categorical parameter in the validation cohort.
Statistical analysis
Automated cell percentages and immune scores were correlated using Pearson method with the TCGA pathologic scores, molecular gene signatures, and gene expression–based scores (Supplementary Fig. S1B). Cell percentage in IHC slides was computed as the cell counts of each immune subset divided by the total number of six immune cell subsets identified on IHC sections. For statistical comparisons among groups, Wilcoxon test was used. All statistical tests were two-sided, a P value of less than 0.05 was considered statistically significant. All statistical analyses were conducted in R version 3.5.1.
Identification of hotspots
Hotspot analysis was conducted on H&E slides using the method and parameters as previously proposed (5). Getis–Ord spatial analysis was carried out over 50×50 μm2 grids using the following equations:
 |
 |
 |
where n is the number of grids; Cj is the count of cancer cells or lymphocytes in grid j; Wi,j is 1 if grid i and j are neighbors, otherwise 0. Grids sharing a vertex or edge with the analyzed grid j were defined as first-order neighbors. Second-order neighbors included grids sharing a vertex or edge with the first order neighbors and so on. Grids within the fourth order neighborhood were considered neighbors to grid j in the equation (5). P value was calculated on the basis of Z score thresholds (17). Regions with P < 0.05 regarding cancer cells and P ≥ 0.05 regarding lymphocytes were categorized as cancer hotspots (CH). Regions with P < 0.05 regarding lymphocytes and P ≥ 0.05 regarding cancer cells were categorized as peritumoral immune hotspot (IHperi). And intratumoral immune hotspot (IHintra) was defined as regions with P < 0.05 for both cancer cells and lymphocytes. The definitions of IHperi and IHintra were described by the following equations.
 |
 |
where 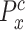 and
and 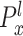 represent the P value quantifying the significance level of enrichment of cancer cells and lymphocytes in grid x, respectively.
represent the P value quantifying the significance level of enrichment of cancer cells and lymphocytes in grid x, respectively.
To quantify the spatial relationship between immune and cancer aggregates for each tumor, we derived three scores based on different normalization terms for the IHintra (5).
-
1. To quantify the proportion of IHs interfacing with tumor, the intratumoral immune hotspot score Sintra/immune was computed as the proportion of IHintra in the immune cell rich area.

-
2. To measure the amount of intratumoral immune aggregates in relation to the cancer area, Sintra/cancer was computed as the ratio of IHintra to the sum of CH and IHintra.

-
3. To quantify the colocalization of immune and cancer aggregates in the context of the entire tissue, Sintra/tissue was computed as the fraction of IHintra in the whole tissue including both hotspot area and non-hotspot area.

Multiplex IHC
Diagnostic blocks were obtained for 10 patients with LUSC in the validation cohort. Tissue blocks were fixed with formalin, embedded in paraffin, then serially sectioned into sections of 4-μm in thickness. Serial sections from the validation cohort were subjected to H&E staining and multiplex IHC using the T-cell panel: anti-CD8 (type: mouse monoclonal, clone: 4B11, source: Leica Microsystems Ltd., used at 1:100 dilution); anti-CD4 (type: mouse monoclonal, clone name: 4B12, source: Leica Microsystems Ltd., used at 1:50 dilution); anti-FOXP3 [type: mouse monoclonal, clone: 236A/E3, source: kindly gifted by Dr. G. Roncador (CNIO, Madrid, Spain), used at 1:2 dilution] and the B-cell panel: anti-CXCR5 (type: rabbit polyclonal, catalog no. ABN200, source: Merck Life Science UK Limited, used at 1:1,000 dilution); anti-CD79b (type: mouse monoclonal, clone: B29/123, source: LRF Immunodiagnostics Unit, John Radcliffe Hospital, used at 1:25 dilution); anti-CD20 (type: mouse monoclonal, clone: L26, source: Agilent Technologies LDA UK Ltd., used at 1:100 dilution); anti-P40 (type: mouse monoclonal, clone: BC28, source: Biocare Medical, used at 1:100 dilution). All slides were scanned using the NanoZoomer S210 digital slide scanner (C13239–01) and the NanoZoomer digital pathology system v.3.1.7 (Hamamatsu) at ×40 (228 nm/pixel resolution).
Deep learning pipeline to map cell populations on H&E and IHC images of the external validation samples
We performed cell detection and classification on H&E slides and the IHC T-cell panel of the validation cohort using the SCCNN pipeline (2, 18) pretrained on H&E and IHC images, respectively. Because of variations in the staining protocol, we trained an additional SCCNN-IHC classifier to discriminate CD4+FOXP3− and CD4+FOXP3+ cells from this cohort. The SCCNN pipeline consists of two models to perform cell detection and classification sequentially. The cell detection was performed on 31×31 pixels image patches to predict pixels with high probabilities of being the cell center. The output is a table of predicted cell coordinates without class label. The table is then used to retrieve single cell images of 51×51 pixels centering at the predicted cell location. The second step involves classifying nine neighboring locations near the center of the cell. The most frequent class predicted among the nine neighborhoods will be determined as the class of the cell (2, 18).
Prior to the model implementation, whole slide images of H&E and IHC samples were rescaled to 0.45-μm/pixel and divided into tiles of 2000×2000 pixels to match the resolution of training images of the SCCNN. A color normalization step was performed for H&E tiles using an example H&E tile from the training set as reference. The H&E tiles were first converted from RGB to Lab color space. Then a Reinhard stain normalization from the Image processing toolbox in Matlab R2019b (RRID:SCR_001622) was used to transform all the H&E tiles based on the reference image. We did not normalize the color for the IHC slides. Instead, the classifier was trained on single cell patches augmented by contrast and brightness adjustment, which encouraged the model to capture stain variability in the dataset.
For the B-cell panel, we trained a distance regularized dense inception network (DRDIN; ref. 19) for cell detection using masks of annotated cell locations, and an SCCNN-IHC classifier (18) to differentiate between three dominant B-cell subsets (CD20+CXCR5−, CD20+CXCR5+, and CD79b+), and the P40+ neoplastic cells. Similar to SCCNN detector, the DRDIN model was trained to predict pixels of the cell center (19). However, the input image size was of 224×224 pixels, allowing the integration of features in a larger spatial context and therefore a better capability to capture densely located cells compared with SCCNN. Models for the B-cell panel were developed on a total of 13,545 single-cell annotations collected from 5 of 10 IHC slides and randomly split into training (64%), validation (16%), and testing (20%). Cell annotations of the T-cell and B-cell panels were both generated by A. Akarca and H. Zhang, and reviewed by T. Marafioti. To ensure the quality of cell annotations, we excluded labels disagreed by the two annotators from the dataset.
On average, we identified 69,146 ± 22908 CD8+, 109,764 ± 39,028 CD4+FOXP3− and 34,861 ± 28,737 CD4+FOXP3+ in the T-cell panel, achieving an average accuracy of 0.96 on 596 cells. For the B-cell panel, we identified 13,858 ± 13,950 CD20+CXCR5−, 10,399 ± 8,510 CD20+CXCR5+, and 7,281 ± 4,336 CD79b+ cells with an average accuracy of 0.89 on 2,800 cells. To evaluate the impact of annotation budget on model performance, we retrained the classifier for the B-cell panel with different size of randomly sampled training set, and evaluated the performance on the same hold-out testing set. The model achieved an accuracy of 0.867, 0.879, 0.884, and 0.894 with 25%, 50%, 75%, and 100% of the annotations, indicating that the current amount of annotations was needed to obtain a reliable supervised classifier.
Serial section alignment
T- and B-cell sections in the validation cohort were aligned to the corresponding H&E slide using manually chosen benchmarks. The affine transformation was performed using the “fitgeotrans” function in MATLAB R2019b (RRID:SCR_001622). The quality of registration was evaluated by (i) the Target Registration Error, which represents the distance between original and transformed points normalized by the diagonal length of an image (20); (ii) the concordance between densities of cancer cells and lymphocytes detected on H&E and IHC slides, respectively.
The averaged target registration error of aligned sections was 0.13% and the densities of cancer and lymphocytes detected on IHC significantly correlated to that on H&E sections (cancer: R = 0.69, P = 0.00079; lymphocyte: R = 0.76, P = 8.7 × 10−5; Supplementary Fig. S3A and S3B).
Annotation of tertiary lymphoid structures and lymphoid aggregates
Lymphocyte clusters forming a structure were delineated by a senior pathologist on 10 digitized H&E slides from the LATTICe cohort. This manual identification was done while looking at both T-cell and B-cell serial IHC sections (Fig. 4A). Tertiary lymphoid structures (TLS) were marked as the aggregation of lymphocytes in which there was a clear zonality of T- and B-cell components, resulting in a structure. LAGs were marked as lymphocyte clusters without a distinguishable germinal center or admixed with stromal cells. Individual TLS and LAG were assigned to intratumoral, peritumoral, cancer hotspots and non-hotspot regions based on the location of the majority of its region. In total 52 TLSs were identified across 10 slides, with 49 in peritumoral IH, 2 in intratumoral IH, and 1 in non-hotspot regions. 1 out of 52 TLSs had the same area spanning intratumoral and peritumoral IH and was assigned the intratumoral IH. On the other hand, 98 LAGs were identified, with 66 in peritumoral IH, 17 in intratumoral IH, 9 in cancer hotspots and 6 in non-hotspot regions.
Figure 4.

Lymphocyte composition and the presence of TLS at intratumoral and peritumoral IH. A, Example regions showing annotations of TLS at the IH and pathologically defined lymphoid aggregates without a prominent germinal center (LAG) at the intratumoral IH. B, Lymphocyte subset densities at intratumoral (n = 10) and peritumoral IH (n = 10). Cell densities were measured as the cell counts divided by the total area of IHs. C, Pie chart displaying the average percentages of peritumoral IH and intratumoral IH identified as TLS, LAG, and non-TLS/LAG IH. D, Densities of TLS and LAG at intratumoral (n = 10) and peritumoral IH (n = 10). E, Densities of B- and T-cell subsets in TLS (n = 10), LAG (n = 10), and non-TLS/LAG IH (n = 10). Statistical significance was evaluated by the Wilcoxon signed-rank test, followed by the Benjamini–Hochberg method for P value adjustment. *, P < 0.05; **, P < 0.01. The median is marked by the horizontal line; the first and third quartiles are shown as box edges; whiskers indicate the range of error.
Identification of individual immune hotspot
Disconnected IH was identified as cluster of 50×50 μm2 grids on the 10 H&E slides of the validation cohort. The spatial connectivity was assessed using the “clump” function in the R package “raster” with directions set as 8. Each individual IH was first segregated and indexed regardless of its distribution, then partially assigned to the peritumoral or intratumoral category based on labels of grids. Grids identified as part of TLS or LAG were excluded from the analysis. This resulted in 26 ± 11 individual intratumoral IH and 39 ± 12 individual peritumoral IH per slide.
Cellular interaction analysis
We calculate the cellular interaction for each individual IH using the coordinates of immune cells within an individual IH as input. The spatially neighboring cells were connected using the Delaunay triangle graph, with the connections exceeding 250 μm excluded. The interactions between each pair of immune cell subsets were defined as the fractions of links between them among all the connections (21).
Data availability
The transcriptomic data and the digital pathology images from the TCGA cohort can be accessed on the GDC portal (portal.gdc.cancer.gov, cohorts TCGA-LUSC and TCGA-LUAD). IHC data related to the validation cohort are available upon reasonable request.
Code availability
Code and data used to generate figures and statistical information can be obtained at https://github.com/yuerua/t_b_spatial_IH. Scripts for implementing the SCCNN pipeline used for cell identification on the H&E and T-cell panel of the validation cohort are available at https://github.com/qalid7/compath. Scripts for the DRDIN pipeline used for cell identification on the B-cell panel of the validation cohort are available at https://github.com/pathdata/UNMaSk/tree/master/HE_cell_detection.
Results
High intratumoral immune hotspot score correlates with poor survival in LUSC
Image analysis and spatial statistics were used to identify spatially and histologically distinct immune-rich and cancer cell–rich areas in fresh frozen H&E slides (4) from 462 LUSC and 473 LUAD tumors from TCGA. We classified tissue neighborhoods into three compartments based on the densities of cancer or immune cells compared to the rest of the tissue: (i) Cancer hotspots: tissue regions enriched with cancer cells but not immune cells. (ii) Peritumoral IH: tissue regions enriched with immune cells but not cancer cells. (iii) Intratumoral IH, tissue regions enriched with both immune and cancer cells (Fig. 1A and B, see Materials and Methods; ref. 4).
Quantitative spatial scores Sintra/immune, Sintra/cancer, and Sintra/tissue were used to summarize the amount of intratumoral IH in relation to immune-rich regions, cancer-rich regions, and the entire tissue, respectively (Fig. 1B, see Materials and Methods; ref. 4). We observed a higher Sintra/immune in LUAD than LUSC (P = 0.011), and in females than males (P = 0.03). The Sintra/cancer was higher in females than males (P = 0.016). No statistically significant difference in the three spatial scores was found for groups categorized by other clinicopathologic characteristics, including age, smoking history, pack years, TMB, and stage (P > 0.05, Table 1).
Table 1.
. Characteristics of the included patients and their association with the Sintra/immune, Sintra/cancer, and Sintra/tissue.
| Characteristics | Categories | Total N (%) | S intra/immune P value | S intra/cancer P value | S intra/tissue P value |
|---|---|---|---|---|---|
| Histology (N = 935) | LUSC | 462 (49.41%) | 0.011 | 0.057 | 0.49 |
| LUAD | 473 (50.59%) | ||||
| Age (N = 934) | ≥65 | 566 (60.6%) | 0.61 | 0.38 | 0.38 |
| <65 | 368 (39.4%) | ||||
| Sex (N = 935) | female | 377 (40.32%) | 0.03 | 0.016 | 0.21 |
| male | 558 (59.68%) | ||||
| Smoking history (N = 903) | current reformed smoker for ⇐ 15 years | 394 (43.63%) | 0.36 | 0.17 | 0.32 |
| current reformed smoker for > 15 years | 199 (22.04%) | ||||
| current smoker | 230 (25.47%) | ||||
| lifelong nonsmoker | 80 (8.86%) | ||||
| Pack years (N = 723) | ≥35 | 478 (66.11%) | 0.23 | 0.12 | 0.20 |
| <35 | 245 (33.89%) | ||||
| TMB (N = 902) | <3 | 347 (38.47%) | 0.26 | 0.31 | 0.65 |
| ≥3 | 555 (61.53%) | ||||
| Stage (N = 935) | ia | 214 (22.89%) | 0.075 | 0.31 | 0.20 |
| ib | 257 (27.49%) | ||||
| iia | 114 (12.19%) | ||||
| iib | 159 (17.01%) | ||||
| iiia | 129 (13.80%) | ||||
| iv | 62 (6.63%) |
To determine the prognostic value of spatial scores, we carried out survival analysis in discovery and validation cohorts randomly split from the TCGA LUSC cohort, each containing 231 patients (Supplementary Table S2). Among the three scores, Sintra/immune was the only one significantly associated with the overall survival (OS) in both discovery (P = 0.019) and validation (P = 0.043) cohorts (Fig. 1C; Supplementary Fig. S4A and S4B), with high Sintra/immune indicative of an adverse outcome (threshold 0.326 determined in the discovery cohort). To examine the contingency of the prognostic value of Sintra/immune on the cohort split, we repeated the splitting of discovery and validation cohort for 100 times. In 36 iterations, Sintra/immune showed a significant prognostic effect (P < 0.05) in the validation cohort when dichotomized by the threshold optimized for the discovery cohort, with an averaged P value of 0.224 (SD, 0.261; 95% CI, 0.172–0.275). The percentage of significant iterations was higher than that of other well-known prognosticators including TNM stage (8%), age (4%), pack years (6%), and TMB (20%; Supplementary Table S3; refs. 22–25), signifying the discriminative ability of Sintra/immune to identify patients at high risk.
Multivariate analysis showed that Sintra/immune remained significant after accounting for age, stage, and smoking pack years in the validation cohort [P = 0.02; HR = 2.0 (1.12–3.7); Fig. 1D]. Sintra/immune was also significantly associated with poor outcome when tested separately in patients stratified on the basis of tobacco smoking history available in the TCGA clinical data, differentiating current reformed smokers for less than 15 years, current reformed smokers for more than 15 years, and current smokers (P < 0.05; Supplementary Fig. S5A). However, it was not significant in nonsmokers, potentially due to the small sample size (P = 0.18, n = 16; Supplementary Fig. S5A).
In LUAD tumors, Sintra/immune did not show significant prognostic value either in the validation cohort (P = 0.9, n = 236; Supplementary Fig. S6A) or within any of the smoking categories (P > 0.05; Supplementary Fig. S5B). The other two spatial scores Sintra/cancer and Sintra/tissuewere both not significant in the validation cohort (P = 0.094, P = 0.69, n = 236; Supplementary Fig. S6B and S6C).
This differential prognostic value of Sintra/immune between LUSC and LUAD pertaining to the spatial heterogeneity of lymphocyte distribution was not related to the overall immune infiltration in both NSCLC subtypes. Lymphocyte abundance, defined as the percentage of lymphocytes detected on the whole slide, was higher in LUAD than LUSC (P = 0.0018). A high level of lymphocyte abundance was associated with favorable OS in LUAD (P = 0.017) but not in LUSC (P = 0.08; Supplementary Fig. S7A and S7B). However, in multivariate analysis adjusted for stage, age and smoking pack years, lymphocyte abundance was not prognostic in either subtype (P > 0.05; Supplementary Fig. S7C and S7D). In addition, Sintra/immune did not correlate with lymphocyte abundance in LUSC (R = -0.081, P = 0.083; Fig. 1E). Tumors with high lymphocyte abundance and low Sintra/immune displayed accumulation of lymphocytes at the peritumoral region (Supplementary Fig. S8A), whereas in tumors with low lymphocyte abundance and high Sintra/immune, the majority of tumor infiltrating lymphocytes were found within the tumor nest (Supplementary Fig. S8B). Patients with high lymphocyte abundance but low Sintra/immune, indicating a preferential homing of lymphocytes to the peritumoral region, had significantly better prognosis than other patients [P = 0.00035, HR = 0.55 (0.4–0.76), Fig. 1F].
Consistent upregulation of B-cell signatures associated with high intratumoral immune hotspot score
To investigate molecular heterogeneity underpinning intratumoral and peritumoral IH, we leveraged RNA-seq data to evaluate differentially expressed genes (DEG) in groups of patients with LUSC stratified by Sintra/immune. Gene set enrichment analysis revealed that the Gene Ontology biological processes significantly enriched by these DEGs included immune response, inflammatory response, and activation of immune response (Fig. 2A). Immune related DEGs were exclusively B-cell markers, including CD79B, MS4A1 (encoding CD20), and CXCR5, which were all upregulated in the high Sintra/immune group (Fig. 2B). CXCR5 expression on B cells characterizes the subpopulation migrating into TLS, the organized lymphoid aggregates with morphologically distinct separation of T-cell and B-cell zones and serve as local sites of T-cell priming and B-cell maturation (26–30). The association between Sintra/immune and B-cell infiltration was cross-validated using seven established algorithms to estimate immune cell abundance, including Danaher (11), TIMER (10), CIBERSORT Absolute mode (12), MCP-counter (9), quanTIseq (13), EPIC (14), and xCell (15). Using these methods, we found a compelling pattern for an enrichment of B-cell populations in the high Sintra/immune group as compared with the low group according to the median (Fig. 2C).
In contrast with LUSC, Sintra/immune high group in LUAD showed a downregulation of B cell–related genes (CD79b, FCER2, CXCR5, MS4A1, CD19; Supplementary Fig. S2B). The enrichment analysis revealed that DEGs between high and low groups in LUAD were enriched for microtubule-based movement and cell-adhesion, unlike the predominance of immune-related pathways in LUSC (Supplementary Fig. S2A).
We next examined the cooccurrence of T- and B-cell genes and signatures in the high Sintra/immune LUSC group. B-cell memory genes such as CD27 were significantly correlated with FOXP3 expression (R = 0.8; Supplementary Fig. S9A), compared with a weaker correlation between CD27 and CD8A (R = 0.55; Supplementary Fig. S9B), in line with a previous report (31). Both CD27 and FOXP3 were overexpressed in the high Sintra/immune group (Supplementary Fig. S9A and S9C). The MCP-counter B-lineage signature correlated with the T-cell signature but not the cytotoxic lymphocyte or CD8 T-cell signature, indicating that T-cell subsets more likely to coexist with infiltrating B cells were CD4+ T cells, and not effector T cells (Supplementary Fig. S9D).
We then investigated the association between Sintra/immune and potential indicators of immunotherapy response, including TMB, PD-1, PD-L1 expression, and the neoantigen load (1) in patients with LUSC. We observed a significantly lower TMB in Sintra/immune high group (P = 0.032, Supplementary Fig. S10A). On the other hand, there was no statistical difference in PD-1, PD-L1 expression, or number of clonal neoantigens between Sintra/immune high and low group (PD-1, P = 0.19; PD-L1, P = 0.12; neoantigen load, P = 0.29; Supplementary Fig. S10E, S10F and S10G). Consistent with previous results (24), low TMB was associated with adverse outcomes in LUSC (P = 0.014, Supplementary Fig. S10B). Despite the negative association between Sintra/immune and TMB, high Sintra/immune independently correlated with poor OS in the multivariate analysis including TMB, age, stage, and pack years [P = 0.022, HR = 1.6 (1.07–2.5); Supplementary Fig. S10C]. In addition, high TMB and low Sintra/immune codefined a group of patients with significantly prolonged survival (P = 0.0011; Supplementary Fig. S10D).
Abundance of T- and B-cell subpopulations in intratumoral and peritumoral immune hotspots
To further investigate the distribution of B cells in relation to intratumoral and peritumoral IH as well as the interplay between T and B cells, we selected 10 LUSC tumors from the LATTICe cohort, with 6 displaying high Sintra/immune (see Materials and Methods). Guided by the gene expression analysis (Fig. 2B), we stained three consecutive sections, respectively, with a panel for B cells (CXCR5/CD79b/MS4A1, encoding CD20) and neoplastic cells (P40), a T-cell panel (CD4/CD8/FOXP3), and with H&E.
The three sections were spatially aligned using the affine transformation based on manually selected landmarks (averaged misalignment distances: 43.33 μm; averaged target registration error: 0.13%, see Materials and Methods). Deep learning approaches were used to automate the identification of three dominant B-cell subsets (CD20+CXCR5− cells, CD20+CXCR5+ cells, and CD79b+ cells; average accuracy: 0.89) and T-cell subsets (CD4+FOXP3− T cells, CD8+ T cells, and CD4+FOXP3+ T regulatory (Treg) cells; average accuracy: 0.96, see Materials and Methods). The CD79b+ cells represented B cells with high CD79b expression and different expression levels of CD20 and CXCR5. CD20+CXCR5− and CD20+CXCR5+ cells both showed low expression of CD79b. After the identification of B and T cells, we employed spatial statistics to quantify their distribution and colocalization patterns in intratumoral and peritumoral IH inferred from the corresponding H&E sections (Fig. 3A–E, see Materials and Methods).
Figure 3.

The deep learning pipeline to map T-cell and B-cell subsets at intratumoral and peritumoral IH. A, Framework of the deep learning pipeline for spatial analysis. The pipeline comprised 3 steps: serial section alignment, deep learning–based cell classification, and hotspots mapping. IHC staining with a B-cell panel (CD20/CXCR5/CD79b/P40) and a T-cell panel (CD4/FOXP3/CD8) was performed on serial sections, which were then aligned to the corresponding H&E section. Dominant B-cell subsets (CD20+CXCR5−, CD20+CXCR5+, CD79b+) and T-cell subsets (CD4+FOXP3−, CD4+FOXP3+, CD8+) were detected and classified by deep learning models and projected to the H&E slide. The magnified region example illustrates this entire framework, which combines immune cell detection from the B-/T-cell IHC sections together with hotspot identification on the corresponding H&E slide. B–E, Representative TLS, LAG, non-TLS/LAG intratumoral IH, and non-TLS/LAG peritumoral IH. Cell classification results are shown below the original image, with color codes for each cell type shown at the bottom. Scale bar, 100 μm.
In general, there was a higher density of T cells than B cells in the whole tissue, with CD4+FOXP3− and CD8+ being the first and the second most abundant T-cell subsets (Supplementary Fig. S3C). There was no significant difference among densities of B-cell subsets (Supplementary Fig. S3C). The majority of T-cell subsets were found in the non-hotspot tissue regions, whereas the B-cell subsets displayed the highest percentages in the peritumoral IH (Supplementary Fig. S3D). We further compared immune cell compositions between intratumoral and peritumoral IH. Specific B-cell subsets, CD20+CXCR5+ cells and CD79b+ cells, displayed higher densities in peritumoral IH than in intratumoral IH (P < 0.05, Fig. 4B). There was no difference between densities of T-cell subsets in intratumoral and peritumoral IH (P > 0.05, Fig. 4B). The lower B-cell subset densities at the intratumoral IH compared with the peritumoral IH suggested that the concordant increase in B-cell signatures and proportions of intratumoral IH was due to recruitment of B cells to tumors with increased tumor–immune interface, rather than preferential homing of B cells to the intratumoral IH.
TLSs account for a minor proportion of intratumoral and peritumoral immune hotspots
CXCR5 and CD79b have been recognized as markers associated with the TLS (28–30). To investigate the overlap of TLS with intratumoral and peritumoral IH, pathologists manually identified 52 TLSs with prominent germinal centers and 98 lymphoid aggregates (LAG) without a distinguishable germinal center or admixed with stromal cells from 10 H&E slides from the LATTICe cohort (Fig. 4A, see Materials and Methods).
The majority (49 of 52) TLSs were located in peritumoral IH, occupying a total of only 4.87% of the peritumoral IH area across all slides (mean per slide = 5.30%, SD = 4.62%), which was significantly higher than the proportion in intratumoral IH (2 TLS, total = 0.26%, mean per slide = 0.38%, SD = 0.49%, P = 0.0092; Fig. 4C and D). Conversely, LAGs were widespread across both compartments, with no significant difference in their proportions between intratumoral and peritumoral IH (intratumoral: n = 17, total = 2.18%, mean per slide = 2.66%, SD = 2.68%, peritumoral: n = 66, total = 3.73%, mean per slide = 3.93%, SD = 2.54%, P = 0.11; Fig. 4C and D). The rest of LAGs were found in cancer hotspots (n = 9) and non-hotspot regions (n = 6). The predominance of TLSs in peritumoral IH was consistent with the current paradigm that the majority of TLSs are located in the peritumoral area (32).
We observed a higher density of CD20+CXCR5+ and CD79b+ cells in TLS within IH than in non-TLS/LAG IH (P < 0.05; Fig. 4E). This is consistent with previous findings of the upregulation of CXCR5 and CD79b in tumors with TLS present (28–30). TLS also contained a higher amount of CD4+ T cells, CD4+FOXP3+ Treg cells, and CD20+CXCR5− B-cell subsets compared with non-TLS/LAG IH (P < 0.01, Fig. 4E), whereas only CD20+CXCR5+ and CD79b+ B cells were significantly different between TLS and LAG. Taken together, the majority of IHs are neither TLS nor LAG, based on their different B- and T-cell composition. We henceforth refer to these as non-TLS/LAG IH.
B- and T-cell spatial interactions in non-TLS/LAG immune hotspots
To further investigate the hazard underpinning intratumoral IH while excluding the confounding effect of TLS, we sought to resolve cell spatial interactions that differentiate non-TLS/LAG intratumoral and peritumoral IH. Using the Delaunay triangle graph (21), we developed a method for identifying individual non-TLS/LAG IH based on the connectivity of adjacent regions and construct cellular networks (Fig. 5A). Lymphocyte subset interactions were defined as the proportion of edges connecting two types of lymphocytes (see Materials and Methods). Both interactions and compositions of lymphocyte subsets were significantly more diverse in the peritumoral IH than in the intratumoral IH, as indicated by the higher Shannon index (P = 0.003; P = 0.003; Fig. 5B). Compared with intratumoral IH, peritumoral IH had higher frequencies of interactions between CD20+CXCR5+ and CD4+FOXP3+, CD4+FOXP3−, but not CD8+ cells (univariate logistic regression P < 0.05; Fig. 5C). On the other hand, none of the interactions involving CD79b+ B cells were observed to be significant despite the enrichment of CD79b+ B cells in peritumoral IH (P > 0.05, Fig. 5C), suggesting an independence between cell interactions and abundance.
Figure 5.

Diversity of lymphocyte interactions in intratumoral and peritumoral IH without TLS or LAG. A, Examples of lymphocyte interactions in individual peritumoral and intratumoral IH. Lymphocytes identified on the serial IHC slides were connected by the Delaunay triangle graph. Lymphocyte subsets are represented by dots of different colors. Interactions significant in the univariate logistic regression are denoted by red, otherwise blue. B, Shannon diversity of lymphocyte densities and interactions in individual peritumoral (n = 392) and intratumoral IH (n = 262). Statistical significance was evaluated by the Wilcoxon signed-rank test. C, Univariate logistic regression predicting peritumoral and intratumoral IH with individual lymphocyte interactions as variables, n = 654. The coefficients of the significant variables were all negative, suggesting a higher frequency of the interactions in peritumoral IH compared with intratumoral IH. P values were adjusted by the Benjamini–Hochberg method. Regions identified as TLS or LAG were excluded from the above analyses.
High abundance of B cells in non-TLS/LAG intratumoral immune hotspots was linked to a decrease in the CD8+/CD4+FOXP3+ ratio
We assessed the localized T-cell regulation by the CD8+/CD4+FOXP3+ ratio within individual non-TLS/LAG IH (see Materials and Methods). There was no difference between the CD8+/CD4+FOXP3+ ratio at intratumoral and peritumoral IH (intratumoral IH: mean = 4.66, SD = 7.09, peritumoral IH: mean = 3.72, SD = 4.3, P = 0.53). To further dissect the role of B cells on T-cell regulation within non-TLS/LAG IH, each non-TLS/LAG intratumoral and peritumoral IH was classified on the basis of its CD8+/CD4+FOXP3+ ratio. IH with low CD8+/CD4+FOXP3+ ratio had significantly higher percentages of CD20+CXCR5− and CD20+CXCR5+ B cells (defined as the number of cells divided by the total count of 6 immune cell types in each IH) compared to IH with high CD8+/CD4+FOXP3+ ratio, regardless of their location (P < 0.05, Fig. 6A).
Figure 6.

The relationship among B-cell, T-cell percentages and CD8+/CD4+FOXP3+ ratio in individual intratumoral IH and peritumoral IH without TLS or LAG. A, Percentages of B-cell subsets in individual intratumoral (n = 262) and peritumoral IH (n = 392) classified by CD8+/CD4+FOXP3+ ratio. The high and low groups were determined by the cut-off of median CD8+/CD4+FOXP3+ ratio in intratumoral and peritumoral IH, respectively. B, Percentages of CD8+ T cells and CD4+FOXP3+ Treg cells in individual intratumoral (n = 262) and peritumoral IH (n = 392) classified by the percentage of B cells. This B-cell percentage was calculated as the total count of all B-cell subsets divided by the total count of all 6 immune cell types. C, Percentages of CD8+ T cells in individual intratumoral (n = 262) and peritumoral IH (n = 392) classified by CD4+FOXP3+ Treg-cell percentages. Statistical significance was evaluated by the Wilcoxon rank sum test and adjusted by the Benjamini–Hochberg method.
We then investigated whether the reduced CD8+/CD4+FOXP3+ ratio at B-cell–enriched IH was associated with the exclusion of CD8+ T cells or the recruitment of Treg cells. B-cell–enriched intratumoral and peritumoral IH had significantly lower percentages of CD8+ T cells compared with IH with low B-cell percentages (intratumoral IH: P = 0.0068, peritumoral IH: P = 8.8 × 10−7, Fig. 6B). In contrast, CD4+FOXP3+ T-cell percentage was not correlated with B-cell abundance in intratumoral IH or peritumoral IH (P > 0.5, Fig. 6B). In addition, we observed a strong negative correlation between CD8+ and CD4+FOXP3+ percentages in intratumoral (P = 2.8 × 10−5) but not in peritumoral IH (P = 0.060, Fig. 6C). Taken together, these findings suggest that while a generic pattern of decreased CD8+ cells but not Treg cells in B-cell–enriched IH is observed regardless of their location, Treg cells may have a bigger impact on the regulation of CD8+ cells in intratumoral IH compared with peritumoral IH, due to the presence of negative correlation between CD8+ T-cell and Treg abundance exclusively in intratumoral IH.
Discussion
Heterogeneous spatial organization of tumor-infiltrating lymphocytes was associated with immune evasion and tumor evolution in NSCLC (2, 33). Specifically, the presence of TLS has been shown to correlate with prolonged survival in NSCLC and a variety of cancer types (28, 33–36). However, immune rich areas beyond TLS are not well studied. Immune-rich areas differ from TLS in terms of cellular densities, organizations, and spatial distributions (37–39). Understanding the role of these immune rich areas in cancer progression and treatment response may be crucial in improving therapies involving immune checkpoint blockade and induction of TLS against cancer (40). In this study, we integrated transcriptional profiling and computational pathology to deconvolve the spatial heterogeneity of immune rich areas identified by the hotspot analysis in NSCLC. We measured the proportion of IH colocalizing with tumor as a score Sintra/immune. The score was a prognostic factor associated with unfavorable outcomes in LUSC independent of age, stage, and pack-years. Despite Sintra/immune being higher in LUAD than LUSC, the score did not show prognostic value or association with immune-related genes in LUAD. Such discrepancy adds to the current paradigm of the distinct biology and immune evasion mechanisms of the two histologic subtypes, such as the different prevalence of HLA LOH and clonal neoantigens (23). This finding highlights the importance of studying IHs beyond TLS and indicates that the prognostic value of lymphocyte clustering is nonexclusive to breast cancer.
The immune spatial score Sintra/immune may be a B-cell–enriched spatial signature, given the consistent upregulation of B-cell signatures in the TCGA cohort. It was later confirmed in the IHC experiments in the validation cohort based on B-cell accumulation at the IHs. In addition, LUSC with high Sintra/immune displayed a lower TMB, raising the possibility that increased IH located inside the tumor nest might characterize tumors undergoing clonal selection or adaptation to the immune microenvironment.
One of the potential reasons for Sintra/immune being an adverse survival indicator is that Sintra/immune-high tumors had reduced peritumoral IH and therefore low TLS abundance, which has been associated with adverse outcome in NSCLC (33). TLS are characterized as organized lymphoid aggregates with a clear separation of T- and B-cell zones (28, 29). In the validation cohort, we found TLS frequently located outside the tumor nest, and contain higher densities of B cells and CD4+ T cells than intra and peritumoral IH. The majority of peritumoral and intratumoral IH were immune-rich areas without organized lymphoid structure. Given the minor proportion of TLS within both intratumoral and peritumoral IH, it is unlikely that Sintra/immune is a mere reverse approximate of TLS abundance. Comparisons between non-TLS/LAG intratumoral and peritumoral IH showed increased interactions between CD20+CXCR5+ B and CD4+ T cells, Treg cells in the non-TLS/LAG peritumoral IH. B cells have been shown to present antigen and to activate CD4+FOXP3− T cells at the interfollicular region in lymph nodes and within TLS (41, 42). It is possible that CD20+CXCR5+ B cells in non-TLS/LAG peritumoral IH also exhibit such proinflammatory functions, contributing to the antitumor immune response in peritumoral IH. The enhanced interactions between CD20+CXCR5+ B cells and Treg cells suggested that B cells in peritumoral IH were subjected to stronger regulation, potentially due to the accumulation of tumor-specific autoantibodies in peritumoral IH (43).
Furthermore, both non-TLS/LAG peritumoral and intratumoral IH exhibit a decreased CD8+ T-cell percentage and CD8+/CD4+FOXP3+ ratio in response to the increased proportions of CD20+CXCR5− and CD20+CXCR5+ B cells. In addition, an increase in Treg percentage was associated with decreased CD8+ T-cell proportions in non-TLS/LAG intratumoral IH but not in peritumoral IH (Fig. 6C). This negative relationship in intratumoral IH may indicate an immune escape strategy of tumor cells, where some tumor islands favor the recruitment of Treg over cytotoxic T cells, leading to a low-CD8+ high-Treg intratumoral IH phenotype (44, 45). Taken together, these results suggest an immunosuppressive TME imposed by the accumulation of B cells and T-cell regulation, with cytotoxic T cells subjected to greater regulation in intratumoral IH than in peritumoral IH. Therapeutic agents targeting B cells and Treg cells, such as rituximab and daclizumab, might skew the balance between intratumoral and peritumoral IH toward an immunogenic microenvironment.
This study has several limitations. The IHC panel was designed based on the DEGs inferred from the TCGA cohort, which did not include markers for the functionality of T cell subsets such as PD-1, ICOS, and GZMB (37). It is possible that CD8 T cells residing in intratumoral IH are prone to exhaustion and are expected targets of immune checkpoint blockade therapy. Likewise, this panel was unable to reveal the potential immunosuppressive role of B cells. For example, in vitro experiments demonstrated that B regulatory cells promoted the expression of FOXP3 and CTLA4 in Treg via secretion of TGFβ and cell–cell contact (46). In addition, naïve B cells and follicular lymphoma B cells have been shown to induce the conversion of CD4+ T cells to Treg in a cell–cell contact manner (47, 48), which potentially contributed to the increased signal of Treg observed in the TCGA transcriptomic data. Also, B cells in intratumoral IH can possibly acquire an IL10-secreting phenotype that displays an immunoregulatory role (49). To fully appreciate the components, functionalities, and spatial interactions of lymphocytes and myeloid cells in intra and peritumoral IH, future studies are warranted. Also, the heterogeneous distribution of IH with respect to tumor glands might be attributed to a diverse immune evasion capability of tumor clones, as indicated by the branching evolution and tissue architecture of invasive glandular tumors (50). Another caveat is the identification of TLS, which was dependent on the variable choices of markers and morphologic criteria across studies (33). Also, the shape and structure of TLS may be varied by the sectioning angle. As a result, despite best efforts, the annotated TLS and LAG regions in the validation cohort may exclude partial TLS regions and immature TLS. However, the presence of CD20+CXCR5+ and CD79b+ was consistently observed in non-TLS/LAG IH, indicating that the two types of B cells preferentially reside in the peritumoral IH with respect to intratumoral IH regardless of the presence of TLS. In addition, further investigation on the distribution of IHs in preinvasive lesions has the potential to benefit the patient risk assessment, as immune surveillance in carcinoma in situ has been associated with lesion regression (51).
In conclusion, we propose a new spatial signature, Sintra/immune, of protumor response orchestrated by B cells and T cells, as the balance between intratumoral and peritumoral IH (Fig. 7). Our study demonstrated a negative association between Sintra/immune and OS in LUSC. Increased Sintra/immune was correlated with an upregulated B-cell signature. Although limited by sample size, our preliminary results indicated the accumulation of CD20+CXCR5−/CD20+CXCR5+/CD79b+ B cells and the minor proportion of TLSs in IH. The interplay between CD8+ T cells, Tregs, and B cells underlying the spatial clustering pattern of lymphocytes characterized a protumor microenvironment in the intratumoral IH that may fuel the progression of LUSC.
Figure 7.

Immune spatial balance in LUSC. Spatial positioning of immune rich areas can reflect an immunosuppressive or an immunostimulant microenvironment. Increased intratumoral IH compared with peritumoral IH may reflect an immunosuppressive TME, characterized by decreased TLS abundance, lower densities of CD20+CXCR5+, and CD79b+ B-cell densities, decreased interactions between CD20+CXCR5+ B cells and CD4+ T cells, and the presence of a negative correlation between CD8+ T-cell and Treg cell abundance. Also, high ratio of intratumoral IH is coupled with increased B-cell and Treg infiltration in the whole tumor. Cancer hotspots, peritumoral IHs, and intratumoral IHs are denoted by red, blue, and green colors, respectively. An IH can consist of peritumoral and intratumoral compartments, denoted as circles with a combination of green and blue.
Supplementary Material
Table S3 and Fig. S1-S10
Differentially expressed genes in TCGA LUSC and LUAD patients with high versus low S_intra/immune
Lists of TCGA LUSC and LUAD cases in the discovery and validation cohort
Acknowledgments
This study was funded by an anonymous private donation in support of H. Zhang and Cancer Research UK Career Establishment Award (CRUK C45982/A21808) to Y. Yuan. Y. Yuan acknowledges funding from Cancer Research UK Career Establishment Award (CRUK C45982/A21808), CRUK Early Detection Program Award (C9203/A28770), CRUK Sarcoma Accelerator (C56167/A29363), CRUK Brain Tumor Award (C25858/A28592), Breast Cancer Now (2015NovPR638), Rosetrees Trust (A2714), Children's Cancer and Leukaemia Group (CCLGA201906), NIH U54 CA217376, NIH R01 CA185138, CDMRP Breast Cancer Research Program Award BC132057, European Commission ITN (H2020-MSCA-ITN-2019), and The Royal Marsden/ICR National Institute of Health Research Biomedical Research Centre. C. Swanton is a Royal Society Napier Research Professor (RSRP\R\210001). His work is supported by the Francis Crick Institute that receives its core funding from Cancer Research UK (CC2041), the UK Medical Research Council (CC2041), and the Wellcome Trust (CC2041). For the purpose of Open Access, the author has applied a CC BY public copyright license to any author accepted manuscript version arising from this submission. C. Swanton is funded by Cancer Research UK (TRACERx; C11496/A17786), PEACE (C416/A21999), and CRUK Cancer Immunotherapy Catalyst Network); Cancer Research UK Lung Cancer Centre of Excellence (C11496/A30025); the Rosetrees Trust, Butterfield and Stoneygate Trusts; NovoNordisk Foundation (ID16584); Royal Society Professorship Enhancement Award (RP/EA/180007); National Institute for Health Research (NIHR) University College London Hospitals Biomedical Research Centre; the Cancer Research UK-University College London Centre; Experimental Cancer Medicine Centre; the Breast Cancer Research Foundation (US) (BCRF-22-157); Cancer Research UK Early Detection an Diagnosis Primer Award (Grant EDDPMA-Nov21/100034); and The Mark Foundation for Cancer Research Aspire Award (Grant 21-029-ASP). This work was supported by a Stand Up To Cancer-LUNGevity-American Lung Association Lung Cancer Interception Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT23-17 to S.M. Dubinett and A.E. Spira). Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. C. Swanton is in receipt of an ERC Advanced Grant (PROTEUS) from the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 835297). K.S.S. Enfield was supported by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 838540 and is supported by the Royal Society (RF\ERE\210216). K.S.S. Enfield acknowledges grant support from Bristol Myers Squibb. M. Jamal-Hanjani acknowledges grant from Cancer Research UK during the conduct of the study.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
D.A. Moore reports personal fees from AstraZeneca, Eli Lilly, Takeda, ThermoFisher, Amgen, Janssen, MIM Software, and Bristol-Myers Squibb outside the submitted work. K.S.S. Enfield reports other support from European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 838540 and grants from The Royal Society (RF\ERE\210216) during the conduct of the study, and grants from Bristol Myers Squibb outside the submitted work. M. Jamal-Hanjani reports grants from Cancer Research UK during the conduct of the study; grants from Cancer Research UK, Rosetrees Trust, NIHR, NIH National Cancer Institute, IASLC International Lung Cancer Foundation, and Lung Cancer Research Foundation; nonfinancial support from Achilles Therapeutics Scientific Advisory Board and Steering Committee; other support from Pfizer, Astex Pharmaceuticals, Oslo Cancer Cluster, and Bristol Myers Squibb outside the submitted work. R. Salgado reports other support from BMS, Exact Sciences, Roche, Merck, Puma, and BMS outside the submitted work. N. McGranahan reports a patent for PCT/EP2016/059401 issued, a patent for PCT/EP2016/071471 issued, and a patent for PCT/GB2018/052004 issued. C. Swanton reports grants and personal fees from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Invitae (previously Archer DX), Ono Pharmaceuitcal, Personalis, Medicxi, China Innovation Centre of Roche (CICoR), Sarah Cannon Research Institute, Metabomed, Amgen, Novartis, GSK, MSD, Illumina, and Roche-Ventana; other support from Bicycle Therapeutics, Apogen Biotech, and personal fees and other support from Epic Bioscience during the conduct of the study. In addition, C. Swanton has a patent for PCT/2017/028013 for methods to lung cancer; a patent for PCT/EP2016/059401 targeting neoantigens pending, a patent for identifying patent response to immune checkpoint blockade (PCT/EP2016/071471) pending, a patent for determining HLA LOH (PCT/GB2018/052004) pending, a patent for predicting survival rates of patients with cancer (PCT/GB2020/050221) pending, a patent for identifying patients who respond to cancer treatment (PCT/GB2018/051912) pending, a patent for methods for lung cancer detection (US20190106751A1) pending, and a patent for (PCT/GB2017/053289) relating to assay technology to detect tumour recurrence pending. C. Swanton is an AstraZeneca advisory board member and chief investigator for the AZ MeRmaiD 1 and 2 clinical trials and is also co-chief investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL's Scientific Advisory Board. C. Swanton is a co-founder of and has stock options in Achillles Therapeutics. Y. Yuan reports personal fees from Roche and Merck and Co Inc. during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
H. Zhang: Data curation, formal analysis, methodology, writing–original draft. K. AbdulJabbar: Data curation, formal analysis, methodology, writing–original draft. D.A. Moore: Data curation, validation, investigation, visualization, writing–review and editing. A. Akarca: Data curation. K.S.S. Enfield: Writing–review and editing. M. Jamal-Hanjani: Data curation, investigation. S. Raza: Formal analysis, methodology, writing–review and editing. S. Veeriah: Investigation. R. Salgado: Investigation. N. McGranahan: Investigation. J. Le Quesne: Data curation, investigation, writing–review and editing. C. Swanton: Conceptualization, writing–review and editing. T. Marafioti: Conceptualization, data curation, writing–review and editing. Y. Yuan: Conceptualization, supervision, funding acquisition, investigation, project administration, writing–review and editing.
References
- 1. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. AbdulJabbar K, Raza SEA, Rosenthal R, Jamal-Hanjani M, Veeriah S, Akarca A, et al. Geospatial immune variability illuminates differential evolution of lung adenocarcinoma. Nat Med. 2020;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bruno TC, Ebner PJ, Moore BL, Squalls OG, Waugh KA, Eruslanov EB, et al. Antigen-presenting intratumoral B cells affect CD4+ TIL phenotypes in non–small cell lung cancer patients. Cancer Immunol Res 2017;5:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heindl A, Sestak I, Naidoo K, Cuzick J, Dowsett M, Yuan Y. Relevance of spatial heterogeneity of immune infiltration for predicting risk of recurrence after endocrine therapy of ER+ breast cancer. JNCI J Natl Cancer Inst 2018;110:10.1093/jnci/djx137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nawaz S, Heindl A, Koelble K, Yuan Y. Beyond immune density: critical role of spatial heterogeneity in estrogen receptor-negative breast cancer. Mod Pathol 2015;28:766–77. [DOI] [PubMed] [Google Scholar]
- 6. Yuan Y, Failmezger H, Rueda OM, Ali HR, Gräf S, Chin S-F, et al. Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Sci Transl Med 2012;4:157ra143. [DOI] [PubMed] [Google Scholar]
- 7. Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012;30:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li B, Severson E, Pignon J-C, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol 2016;17:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Danaher P, Warren S, Dennis L, D'Amico L, White A, Disis ML, et al. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer 2017;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med 2019;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Valencia A, editor. eLife 2017;6:e26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mayakonda A, Lin D-C, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 2018;28:1747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Getis A, Ord JK. The analysis of spatial association by use of distance statistics. Geogr Anal 1992;24:189–206. [Google Scholar]
- 18. Sirinukunwattana K, Raza SEA, Tsang YW, Snead DRJ, Cree IA, Rajpoot NM. Locality sensitive deep learning for detection and classification of nuclei in routine colon cancer histology images. IEEE Trans Med Imaging 2016;35:1196–206. [DOI] [PubMed] [Google Scholar]
- 19. Narayanan PL, Raza SEA, Hall AH, Marks JR, King L, West RB, et al. Unmasking the immune microecology of ductal carcinoma in situ with deep learning. Npj Breast Cancer. Nature Research; 2021;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fitzpatrick JM, West JB. The distribution of target registration error in rigid-body point-based registration. IEEE Trans Med Imaging 2001;20:917–27. [DOI] [PubMed] [Google Scholar]
- 21. Cheng J, Mo X, Wang X, Parwani A, Feng Q, Huang K. Identification of topological features in renal tumor microenvironment associated with patient survival. Bioinformatics 2018;34:1024–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gu C, Shi X, Qiu W, Huang Z, Yu Y, Shen F, et al. Comprehensive analysis of the prognostic role and mutational characteristics of m6A-related genes in lung squamous cell carcinoma. Front Cell Dev Biol 2021;9:661792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen directed immune escape in lung cancer evolution. Nature 2019;567:479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu Y, Li H, Huang Z, Chen K, Yu X, Sheng J, et al. Predictive values of genomic variation, tumor mutational burden, and PD-L1 expression in advanced lung squamous cell carcinoma treated with immunotherapy. Transl Lung Cancer Res 2020;9:2367–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yan D, Chen Y. Tumor mutation burden (TMB)-associated signature constructed to predict survival of lung squamous cell carcinoma patients. Sci Rep. Nature Publishing Group 2021;11:9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meylan M, Petitprez F, Becht E, Bougoüin A, Pupier G, Calvez A, et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022;55:527–41. [DOI] [PubMed] [Google Scholar]
- 27. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-chemokine gene signature identifies lymph node-like structures in melanoma: Potential for patient selection for immunotherapy? Sci Rep 2012;2:765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020;577:561–5. [DOI] [PubMed] [Google Scholar]
- 29. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautès-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol 2014;35:571–80. [DOI] [PubMed] [Google Scholar]
- 30. Hardtke S, Ohl L, Förster R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood 2005;106:1924–31. [DOI] [PubMed] [Google Scholar]
- 31. Claus C, Riether C, Schürch C, Matter MS, Hilmenyuk T, Ochsenbein AF. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res 2012;72:3664–76. [DOI] [PubMed] [Google Scholar]
- 32. Silina K, Soltermann A, Attar FM, Casanova R, Uckeley ZM, Thut H, et al. Germinal centers determine the prognostic relevance of tertiary lymphoid structures and are impaired by corticosteroids in lung squamous cell carcinoma. Cancer Res 2018;78:1308–20. [DOI] [PubMed] [Google Scholar]
- 33. Rakaee M, Kilvaer TK, Jamaly S, Berg T, Paulsen E-E, Berglund M, et al. Tertiary lymphoid structure score: a promising approach to refine the TNM staging in resected non-small cell lung cancer. Br J Cancer 2021;1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020;577:549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Munoz-Erazo L, Rhodes JL, Marion VC, Kemp RA. Tertiary lymphoid structures in cancer – considerations for patient prognosis. Cell Mol Immunol 2020;17:570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ruffin AT, Cillo AR, Tabib T, Liu A, Onkar S, Kunning SR, et al. B cell signatures and tertiary lymphoid structures contribute to outcome in head and neck squamous cell carcinoma. Nat Commun 2021;12:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Noël G, Fontsa ML, Garaud S, De Silva P, De Wind A, Van Den Eynden GG, et al. Functional Th1-oriented T follicular helper cells that infiltrate human breast cancer promote effective adaptive immunity. J Clin Invest 2021;131:e139905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wortman JC, He TF, Solomon S, Zhang RZ, Rosario A, Wang R, et al. Spatial distribution of B cells and lymphocyte clusters as a predictor of triple-negative breast cancer outcome. NPJ Breast Cancer 2021;7:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 2018;174:1373–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lauss M, Donia M, Svane IM, Jönsson G. B Cells and tertiary lymphoid structures: friends or foes in cancer immunotherapy? Clin Cancer Res 2022;28:1751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petersone L, Edner NM, Ovcinnikovs V, Heuts F, Ross EM, Ntavli E, et al. T Cell/B cell collaboration and autoimmunity: an intimate relationship. Front Immunol 2018;9:1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kinker GS, Vitiello GAF, Ferreira WAS, Chaves AS, Cordeiro de Lima VC, Medina TS. B cell orchestration of anti-tumor immune responses: a matter of cell localization and communication. Front Cell Dev Biol 2021;9:1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zaenker P, Gray ES, Ziman MR. Autoantibody production in cancer—the humoral immune response toward autologous antigens in cancer patients. Autoimmun Rev 2016;15:477–83. [DOI] [PubMed] [Google Scholar]
- 44. Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y, et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS One 2014;9:e91551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gatenbee CD, Baker AM, Schenck RO, Strobl M, West J, Neves MP, et al. Immunosuppressive niche engineering at the onset of human colorectal cancer. Nat Commun 2022;;13:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002;16:219–30. [DOI] [PubMed] [Google Scholar]
- 47. Nussbaumer O, Koslowski M. The emerging role of γδ T cells in cancer immunotherapy. Immuno-Oncol Technol 2019;1:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schwartz M, Zhang Y, Rosenblatt JD. B cell regulation of the anti-tumor response and role in carcinogenesis. J Immunother Cancer 2016;4:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Balkwill F, Montfort A, Capasso M. B regulatory cells in cancer. Trends Immunol 2013;34:169–73. [DOI] [PubMed] [Google Scholar]
- 50. Noble R, Burri D, Le Sueur C, Lemant J, Viossat Y, Kather JN, et al. Spatial structure governs the mode of tumour evolution. Nat Ecol Evol 2022;6:207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pennycuick A, Teixeira VH, AbdulJabbar K, Raza SEA, Lund T, Akarca AU, et al. Immune surveillance in clinical regression of pre-invasive squamous cell lung cancer. Cancer Discov 2020;10:1489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3 and Fig. S1-S10
Differentially expressed genes in TCGA LUSC and LUAD patients with high versus low S_intra/immune
Lists of TCGA LUSC and LUAD cases in the discovery and validation cohort
Data Availability Statement
The transcriptomic data and the digital pathology images from the TCGA cohort can be accessed on the GDC portal (portal.gdc.cancer.gov, cohorts TCGA-LUSC and TCGA-LUAD). IHC data related to the validation cohort are available upon reasonable request.