

Abstract
During development of flowering plants, some MIKC-type MADS-domain transcription factors (MTFs) exert their regulatory function as heterotetrameric complexes bound to two sites on the DNA of target genes. This way they constitute “floral quartets” or related “floral quartet-like complexes” (FQCs), involving a unique multimeric system of paralogous protein interactions. Tetramerization of MTFs is brought about mainly by interactions of keratin-like (K) domains. The K-domain associated with the more ancient DNA-binding MADS-domain during evolution in the stem group of extant streptophytes (charophyte green algae + land plants). However, whether this was sufficient for MTF tetramerization and FQC formation to occur, remains unknown. Here, we provide biophysical and bioinformatic data indicating that FQC formation likely originated in the stem group of land plants in a sublineage of MIKC-type genes termed MIKCC-type genes. In the stem group of this gene lineage, the duplication of the most downstream exon encoding the K-domain led to a C-terminal elongation of the second K-domain helix, thus, generating the tetramerization interface found in extant MIKCC-type proteins. In the stem group of the sister lineage of the MIKCC-type genes, termed MIKC*-type genes, the duplication of two other K-domain exons occurred, extending the K-domain at its N-terminal end. Our data indicate that this structural change prevents heterodimerization between MIKCC-type and MIKC*-type proteins. This way, two largely independent gene regulatory networks could be established, featuring MIKCC-type or MIKC*-type proteins, respectively, that control different aspects of plant development.
Keywords: MADS-box gene, MIKC-type MADS-domain transcription factor, keratin-like domain, protein–protein interaction, floral quartet, cooperative DNA binding
Introduction
MADS-box genes, encoding MADS-domain transcription factors (MADS-TFs), constitute a conserved family of developmental control genes that are found in almost all eukaryotic organisms and that fulfil important functions in animals, plants, and fungi. MADS-TFs of animals, fungi, and plants only share the conserved DNA-binding MADS-domain that specifically binds to cis-regulatory DNA elements termed CArG-box (for “C-A-rich-G”). MIKC-type MADS-domain transcription factors (termed MTFs henceforth for simplicity) of plants possess a more complex domain structure. In addition to the MADS-domain, they carry a conserved keratin-like protein–protein interaction domain giving rise to the eponymous “MIKC” domain architecture comprising an N-terminal MADS-domain (M) followed by an intervening− (I), keratin-like (K), and C-terminal (C) domain (Kaufmann et al. 2005; Theißen and Gramzow 2016). Exactly when and how the K-domain originated is not known yet. However, as (almost) all streptophytes (i.e. charophyte green algae + land plants) carry MIKC-type genes, whereas chlorophytes (another group of green algae and sister group of streptophytes) do not, it appears likely that the K-domain evolved in the stem group of extant streptophytes (Tanabe et al. 2005; Derelle et al. 2006). In land plants, MTFs subdivide into the two subfamilies of MIKCC- and MIKC*-type proteins that display structural differences within the I- and K-domain (Henschel et al. 2002; Kaufmann et al. 2005) and that have been shown to preferentially bind to different types of CArG-boxes, termed serum response factor (SRF)-type (consensus sequence 5′-CC(A/T)6GG-3′) and N10-type (5′-C(A/T)8G-3′), respectively (Verelst et al. 2007; Zobell et al. 2010; Wu et al. 2011). Both land plant–specific subfamilies most likely originate from the duplication of an ancestral MIKC-type MADS-box gene prior to the transition of plants to land (Gramzow and Theißen 2010). However, the exact phylogenetic relationship of charophyte MIKC-type genes and MIKCC- and MIKC*-type genes as well as the structural features that distinguish both land plant subfamilies have been discussed controversially in the literature (Henschel et al. 2002; Tanabe et al. 2005; Kwantes et al. 2012; Nishiyama et al. 2018; Thangavel and Nayar 2018). Whereas charophytes usually carry only one MIKC-type gene, their number heavily increased during land plant evolution giving rise to 39 MIKCC- and 6 MIKC*-type genes in Arabidopsis thaliana, for example (Gramzow and Theißen 2010).
In seed plants, MTFs are involved in the control of a plethora of developmental processes, ranging from root development to floral induction, flower and fruit development of angiosperms (Smaczniak, Immink, Angenent, et al. 2012). In order to exert their regulatory function, the transcription factors bind to DNA as homo- or heteromeric complexes of two or four proteins. According to large-scale interaction data, MIKCC- and MIKC*-type proteins form two mainly independent protein-protein interaction networks that predominantly control aspects of sporophyte (MIKCC) and gametophyte (MIKC*) development, respectively (de Folter et al. 2005; Immink et al. 2009; Smaczniak, Immink, Angenent, et al. 2012). The best studied complexes of MTFs are part of so called “floral quartets” that are presumed to define the identities of the different floral organs during flower development of angiosperms (Theißen 2001; Theißen and Saedler 2001; Theißen et al. 2016). A floral quartet consists of an MTF tetramer that simultaneously binds to two separated DNA-binding sites via looping the DNA in between both binding sites. The simultaneous binding to both binding sites and the DNA-loop formation thereby presumably activates target gene expression by yet largely hypothetical epigenetic mechanisms (Mendes et al. 2013; Theißen et al. 2016). The existence of MTF tetramers has been shown not to be limited to floral homeotic proteins controlling flower development (Wang et al. 2010; Ruelens et al. 2017). Rather it appears likely that at least most MIKCC-type proteins of seed plants can be incorporated into floral quartet-like complexes (FQCs; Espinosa-Soto et al. 2014; Puranik et al. 2014; Rümpler et al. 2018) and it has been hypothesized that the ability of MIKCC-type proteins to tetramerize was probably an important precondition for establishing and diversifying sharp developmental switches (Theißen et al. 2016).
The protein–protein interactions that facilitate MIKCC-type protein dimerization and tetramerization are mainly mediated by the K-domain (Yang and Jack 2004; Melzer et al. 2009; Puranik et al. 2014; Rümpler et al. 2018). According to structural data of the floral homeotic MIKCC-type protein SEPALLATA 3 (SEP3) from A. thaliana, the K-domain folds into two amphipathic α-helices that constitute coiled coils (Puranik et al. 2014), a common and well-studied class of protein–protein interaction domains (Lupas and Gruber 2005). The first helix and the N-terminal part of helix 2 strengthen the dimeric interaction between two SEP3 monomers bound to one DNA-binding site. The C-terminal part of helix 2 allows for the interaction of two DNA-bound dimers, thus facilitating FQC formation (Puranik et al. 2014; Rümpler et al. 2018). Due to its high sequence conservation, it is presumed that the K-domains of most MIKCC-type proteins follow a structure that is very similar to that determined for SEP3 (Rümpler et al. 2018).
There is growing evidence that FQC formation is widespread among seed plant MIKCC-type proteins (Espinosa-Soto et al. 2014; Rümpler et al. 2018) and that tetramerization is of high functional relevance (Mendes et al. 2013; Hugouvieux et al. 2018). However, neither is anything known about FQC formation capabilities of MIKC*-type proteins nor when during evolution MTFs “learned” to constitute quartets and which evolutionary changes facilitated this important ability. Here, we present bioinformatical and molecular biophysical data which demonstrate that FQC origin most likely coincides with the transition of plants to land more than 500 million years ago. FQC formation was likely facilitated by the duplication of a K-domain exon of an ancestral MIKCC-type gene. In contrast, two other K-domain exons very likely got duplicated during early MIKC*-type gene evolution, resulting in structural differences in the protein–protein interaction interfaces of MIKCC- and MIKC*-type proteins, eventually preventing heteromeric interactions between members of both subfamilies. We hypothesize that the ancient exon duplications created the molecular prerequisites for the evolution of effective and diverse developmental switches and the establishment of two independent interaction networks controlling sporophyte and gametophyte development of land plants, respectively.
Results
MIKCC-type Protein Representatives From Ferns, Lycophytes and Mosses Form FQCs
The ability of MTFs to constitute FQCs has so far only been tested for a limited set of MIKCC-type proteins from seed plants (Melzer and Theißen 2009; Melzer et al. 2009; Wang et al. 2010; Smaczniak, Immink, Muino, et al. 2012; Jetha et al. 2014; Ruelens et al. 2017; Rümpler et al. 2018). To investigate the tetramerization capabilities and DNA binding of representative MIKCC-type proteins from ferns, lycophytes, and bryophytes, we used a well-established electrophoretic mobility shift assay (EMSA; Melzer and Theißen 2009; Melzer et al. 2009; Rümpler et al. 2018). The rationale of our approach is described in detail in Materials and Methods. We synthesized and cloned the coding sequences of PHYSCOMITRELLA PATENS MADS 1 (PPM1) from the moss Physcomitrium patens (formerly Physcomitrella patens), SELAGINELLA MOELLENDORFFII MADS 3 (SmMADS3) from the lycophyte Selaginella moellendorffii, and CERATOPTERIS RICHARDII MADS 3 (CRM3) from the fern Ceratopteris richardii. The encoded proteins were produced in vitro and their ability to form FQCs was analyzed via EMSA using a radioactively labeled DNA probe that carried two SRF-type CArG-boxes of the sequence 5′-CCAAATAAGG-3′ in a distance of 63 bp (about six helical turns; probe 1; Melzer and Theißen 2009; Melzer et al. 2009; Rümpler et al. 2018). A distance of six helical turns was chosen, because this was determined to be the optimal distance for FQC formation for a number of MIKCC-type proteins from A. thaliana (Jetha et al. 2014). When increasing amounts of in vitro translated protein were coincubated with a constant amount of DNA, a single retarded fraction of reduced electrophoretic mobility was observed for all of the investigated MIKCC-type proteins (fig. 1a and c, e and supplementary fig. S1a and c, e, Supplementary Material online). This is in contrast to most seed plant MIKCC-type proteins investigated so far, which usually produce two retarded fractions (at least for low amounts of applied protein), which constitute complexes of two and four proteins bound to DNA, respectively (Melzer and Theißen 2009; Melzer et al. 2009; Wang et al. 2010; Jetha et al. 2014; Rümpler et al. 2018).
Fig. 1.

FQC formation capabilities of MIKCC- and MIKC*-type MADS-TFs from Physcomitrium patens, Selaginella moellendorffii, Ceratopteris richardii, and Arabidopsis thaliana. (a, c, e) Increasing amounts of in vitro transcribed/translated (a) PPM1, (c) SmMADS3, and (e) CRM3 protein, respectively, were coincubated together with constant amounts of radioactively labeled DNA probe 1. Two fractions of different electrophoretic mobility occur—a fraction of high electrophoretic mobility, constituting unbound DNA probe (labeled with “0” on the left of the gel picture), and a retarded fraction constituting a DNA probe bound by four proteins (“4”). (b, d, f) To determine the stoichiometry of the protein-DNA complexes observed in a, c, and e, (b) PPM1, (d) SmMADS3, and (f) CRM3 wild-type proteins were coexpressed at different ratios with PPM1ΔC, SmMADS3-GFP, and CRM3-GFP, respectively, and coincubated together with constant amounts of DNA probe 1. An overlay of measured signal intensities of the individual lanes is shown on the right. Each peak of the graph is labeled according to the ratio of wild-type and truncated/elongated protein of the corresponding fraction. (g-l) Increasing amounts of in vitro transcribed/translated (g) PPM4, (h) SmMADS2, (i) CRM14 + CRM16, (j) CRM13 + CRM15, (k) AGL66, and (l) AGL104 was coincubated together with constant amounts of radioactively labeled DNA probe 2. Three fractions of different electrophoretic mobility occur—a fraction of high electrophoretic mobility constituting unbound DNA probe (labeled with “0”), a fraction of intermediate electrophoretic mobility constituting a DNA probe bound by a single protein dimer (“2”) and a fraction of low electrophoretic mobility constituting a DNA probe bound by four proteins (“4”). Signal intensities of different fractions were measured and plotted against the amount of applied protein (triangles, free DNA; squares, DNA probe bound by two proteins; circles, DNA probe bound by four proteins). Graphs were fitted according to equations (1)–(3) described in Materials and Methods to eventually quantify and express FQC formation capabilities by Kd1/Kd2. In case of (g) PPM4, a double band was observed for the fraction of intermediate electrophoretic mobility, likely caused by different conformations of the DNA-bound protein dimer. As negative control, 2 µl of in vitro transcription/translation mixture loaded with the empty pTNT plasmid were added to the binding reaction (“Δ”). (a, c, e, g–l) Applied amounts of in vitro transcription/translation products were (lanes 1–8) 0, 0.05, 0.1, 0.2, 0.4, 0.6, 1, and 2 µl, whereby CRM3, PPM4, SmMADS2, CRM14 + CRM16, and CRM13 + CRM15 were prediluted 1:10 and PPM1 and SmMADS3 were prediluted 1:20 with BSA (10 mg/ml). (b, d, f) 3 µl of in vitro transcription/translation product were applied to each lane. Ratios of both template plasmids used for in vitro transcription/translation were (lanes 1–11): 0:1, 1:9, 1:7, 1:5, 1:3, 1:1, 3:1, 5:1, 7:1, 9:1, 1:0.
The electrophoretic mobility of the single retarded fraction observed for the fern, lycophyte, and moss MIKCC-type proteins suggests a complex of four DNA-bound proteins. However, to determine the stoichiometry of the observed complexes more accurately, as had been previously established (Melzer et al. 2009), we generated a truncated version of PPM1 (PPM1ΔC) that only comprises MADS-, I-, and K-domain of PPM1 but lacks most parts of the C-terminal domain (amino acids 175–283) and coincubated variable amounts of wild-type and C-terminally truncated protein together with labeled DNA probe. Following the assumption that all PPM1 full-length proteins within the DNA-bound complex can be substituted by a truncated PPM1ΔC protein, the number of fractions with different electrophoretic mobility reveals the stoichiometry of the DNA-bound complex. Similar to PPM1 full length protein, PPM1ΔC alone produced a single retarded fraction but with a higher electrophoretic mobility due to the reduced protein size (fig. 1b, lane 1 and supplementary fig. S1b, Supplementary Material online). If PPM1 and PPM1ΔC were mixed at different ratios, in total, five retarded fractions of different electrophoretic mobility occurred (fig. 1b, lanes 2–10 and supplementary fig. S1b, Supplementary Material online), representing DNA-bound complexes of PPM1/PPM1ΔC in protein ratios of 0:4, 1:3, 2:2, 3:1, and 4:0, respectively. This indicates that the single retarded fraction observed in figure 1a constitutes a DNA probe bound by four PPM1 proteins. Since in figure 1a no fraction of intermediate electrophoretic mobility (i.e., a single protein dimer bound to DNA) was observed even at low amounts of applied protein, PPM1 seems to form FQCs with very high affinity under our experimental conditions (i.e., it almost immediately occupies both DNA-binding sites), either because dimerization of DNA-bound MTF dimers occurs in a highly cooperative way, or even MTF tetramers are formed in free solution before binding to DNA.
Because SmMADS3 and CRM3 have considerably shorter C-terminal domains than PPM1, we conducted the stoichiometry tests with elongated instead of shortened protein versions. We generated elongated versions of SmMADS3 and CRM3 via fusion to the green fluorescent protein (SmMADS3-GFP and CRM3-GFP) and coincubated variable amounts of wild-type and GFP-fused proteins together with labeled DNA probe. Similar to PPM1, we observed in total five retarded fractions of different electrophoretic mobility for SmMADS3/SmMADS3-GFP and for CRM3/CRM3-GFP (fig. 1d and f and supplementary fig. S1d and f, Supplementary material online), demonstrating that also SmMADS3 and CRM3 bind to DNA immediately as tetramers and thus form FQCs with very high affinity under the applied conditions.
Previous studies have shown, that FQC formation of MTFs highly depends on the spacing of the two CArG-boxes (Melzer and Theißen 2009; Melzer et al. 2009; Jetha et al. 2014). If both CArG-boxes are not spaced by an integral number of helical turns, the binding sites are not directed to the same side of the DNA helix. Consequently, DNA-loop formation would require an energetically costly twist of the DNA making FQC formation less favorable (Melzer et al. 2009). Therefore, we tested PPM1, SmMADS3, and CRM3 against a DNA probe where both CArG-boxes are spaced by 7.5 helical turns (79 bp) instead of the previously used 6 helical turns. Even to a DNA probe with an unfavorable spacing of the CArG-boxes, PPM1, SmMADS3, and CRM3 almost exclusively bound as tetramers (supplementary fig. S2, Supplementary Material online). In addition, we tested binding of the three MIKCC-type proteins against a DNA-probe where one of the two CArG-boxes was mutated. Surprisingly, even to a single CArG-box SmMADS3 bound as a tetramer, although binding strength was considerably weaker compared with a DNA probe containing two binding sites (supplementary fig. S2b, Supplementary Material online). PPM1 and CRM3 showed barely any binding to a DNA probe carrying a single binding site, probably because binding affinity to a single CArG-box was too low to show proper binding under our experimental conditions (supplementary fig. S2a and c, Supplementary Material online). Taken together, these results suggest that PPM1, SmMADS3 and CRM3 probably already form tetramers in solution.
All Investigated MIKC*-type Proteins are Unable to Form FQCs
To examine the FQC formation capabilities of MIKC*-type proteins, we cloned the coding sequences of PPM4 from P. patens, SmMADS2 from S. moellendorffii, CRM13, CRM14, CRM15, and CRM16 from C. richardii, and AGAMOUS-LIKE 66 (AGL66) and AGL104 from A. thaliana and expressed the proteins in vitro. Because MIKC*-type proteins are known to preferentially bind to N10-type CArG-box sequences (Verelst et al. 2007; Zobell et al. 2010; Wu et al. 2011), we tested all MIKC*-type proteins for their FQC formation capabilities against a radioactively labeled DNA-probe that carried two N10-type CArG boxes of the sequence 5′-CTATATATAG-3′ in a distance of 63 bp (about six helical turns; probe 2). At low amounts of applied protein, all of the tested MIKC*-type proteins produced a fraction of intermediate electrophoretic mobility, and with increasing protein amounts an additional fraction of low electrophoretic mobility occurred (fig. 1g–l and supplementary fig. S3, Supplementary Material online). A similar DNA-binding behavior is typically observed for seed plant MIKCC-type MADS-TFs, where the fraction of intermediate electrophoretic mobility corresponds to a DNA probe bound by two proteins (i.e., binding of a single dimer) and the fraction of low electrophoretic mobility constitutes a DNA probe bound by four proteins (i.e., binding of two dimers or one tetramer) (Melzer and Theißen 2009; Melzer et al. 2009; Rümpler et al. 2018). By measuring the signal intensities of the three different fractions (free DNA, DNA probe bound by two proteins, and DNA probe bound by four proteins) the cooperative DNA binding and thus FQC formation capabilities of the examined MIKC*-type proteins can be quantified and expressed as ratio of the dissociation constants for the binding reaction of the first and the second dimer Kd1/Kd2 (for details, see Materials and Methods), as described previously (Melzer et al. 2009; Jetha et al. 2014; Rümpler et al. 2018). All investigated MIKC*-type proteins produced very low Kd1/Kd2 values ranging from 1 to 7, indicating no or very weak positive interaction of two DNA-bound dimers and thus no ability to cooperatively form FQCs under our experimental conditions. For comparison, similar tests with seed plant MIKCC-type proteins capable of forming FQCs, such as the SEP proteins (SEP1, SEP2, SEP3, SEP4) from A. thaliana or GNETUM GNEMON MADS 3 (GGM3), GGM9, GGM11 from Gnetum gnemon, resulted in cooperativity values of 100 or higher at similar experimental conditions indicating highly cooperative DNA binding (Melzer et al. 2009; Wang et al. 2010; Jetha et al. 2014).
PPM4 and SmMADS2, that turned out to also bind SRF-type CArG-boxes with moderate affinity, showed similar binding behavior to a DNA probe carrying SRF-type and N10-type CArG-boxes, respectively (supplementary fig. S4, Supplementary Material online). This suggests that the different CArG-boxes that were used to test MIKCC- and MIKC*-type proteins do not interfere with cooperative DNA binding and FQC formation of the investigated protein.
Duplications of Distinct K-domain Exons Differentiate MIKCC- and MIKC*-type Genes
MIKCC- and MIKC*-type genes are known to differ with respect to their exon–intron structure, although there is no consensus in the literature as to whether these differences pertain to the I-domain and/or the K-domain encoding part (Henschel et al. 2002; Tanabe et al. 2005; Kwantes et al. 2012; Nishiyama et al. 2018; Thangavel and Nayar 2018). To investigate which sequence determinants might account for the observed differences in FQC formation capabilities, we conducted a large-scale exon homology analysis based on a multiple sequence alignment of all MIKCC- and MIKC*-type genes from P. patens, S. moellendorffii, C. richardii, and A. thaliana. With a few exceptions, all analyzed MIKCC-type proteins are encoded by one MADS-domain exon, one I-domain exon, four K-domain exons and a variable number of C-terminal domain exons (fig. 2). K-domain exons were defined based on whether they were homologous to exons encoding for the K-domain of SEP3 from A. thaliana for which the crystal structure has been determined (Puranik et al. 2014). In contrast to MIKCC-type proteins, the analyzed MIKC*-type proteins are usually encoded by six instead of four K-domain exons. Exon homology analyses demonstrate that only three K-domain exons are shared by MIKCC- and MIKC*-type genes (highlighted by black arrows in fig. 2). The first three (most upstream) K-domain exons of MIKC*-type genes are not found among MIKCC-type genes and the last (most downstream) K-domain exon of MIKCC-type genes is not found among MIKC*-type genes (highlighted by red arrowheads in fig. 2).
Fig. 2.
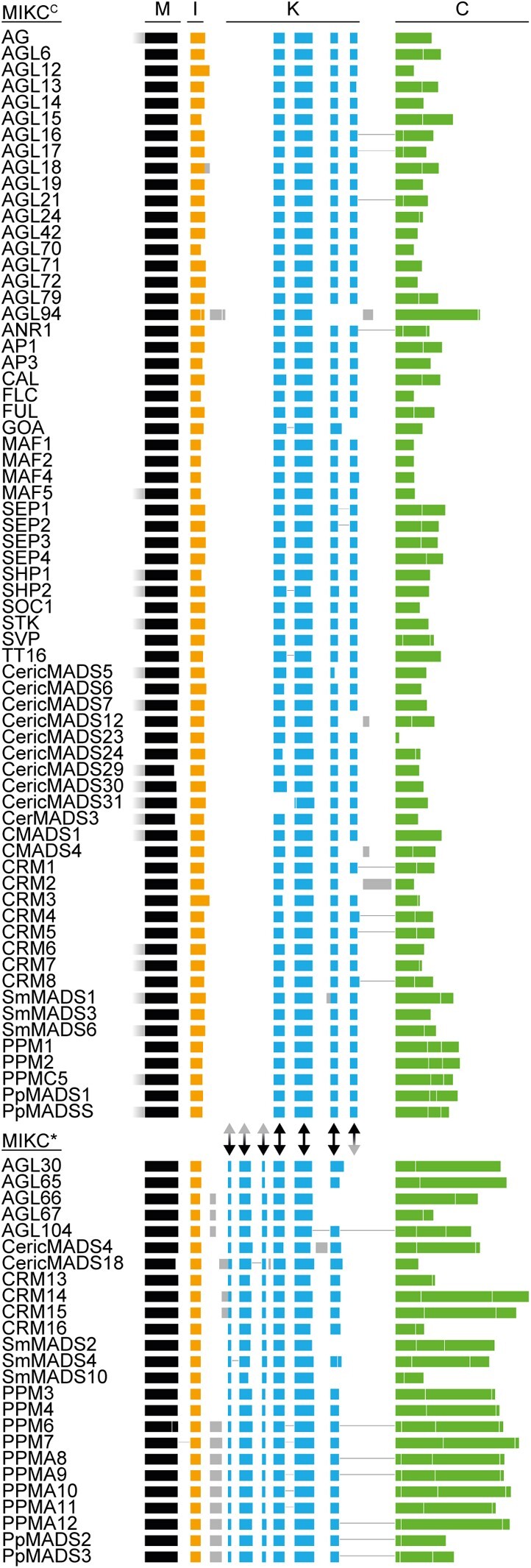
Exon homology of MIKCC- and MIKC*-type genes from Arabidopsis thaliana, Ceratopteris richardii, Selaginella moellendorffii, and Physcomitrium patens. Colored boxes represent coding exons of all MIKCC- and MIKC*-type genes from A. thaliana, C. richardii, S. moellendorffii, and P. patens. Introns and noncoding exons are not shown. Exons encoding for the MADS-domain, the intervening domain (I-domain), the keratin-like domain (K-domain), and the C-terminal domain (C-domain) are labeled on top and are additionally color coded in black, yellow, blue, and green, respectively. Exons with uncertain homology assignment are color-coded in gray. Exons encoding for MADS-, I-, and K-domain were aligned according their homology based on a multiple sequence alignment of the encoded proteins back translated into a codon alignment (for details, see Materials and Methods). Fused exons are connected by horizontal black lines. Two-headed arrows between MIKCC- and MIKC*-type genes illustrate presence (black) or absence (gray arrowhead) of homologous exons in either of the two subfamilies.
The K-domain exons that differentiate MIKCC- and MIKC*-type genes show some remarkable peculiarities. The last two K-domain exons of MIKCC-type genes are of similar size, almost invariably encoding for 14 amino acids each. Analysis of the amino acid sequence encoded by these exons revealed that they also show similarity on the sequence level (fig. 3a). Likewise, the first four K-domain exons of MIKC*-type genes show similarities in size and sequence. The first and the third K-domain exons often encode for 7 amino acids each. The second and the fourth K-domain exons usually encode for 21 amino acids each (fig. 3b). Due to the similar size and the high level of sequence similarity, it appears very likely that the last two K-domain exons of MIKCC-type genes as well as the first four K-domain exons of MIKC*-type genes evolved by exon duplications of an ancestral and a pair of ancestral exons, respectively. As the hypothetically duplicated exons are present in almost all analyzed MIKCC- and MIKC*-type genes, it appears likely that the exon duplication events took place in the stem lineage of MIKCC- and MIKC*-type genes, respectively and thus in the stem group of extant land plants.
Fig. 3.

Similarity of K-domain exons hypothesized to be duplicated. (a, b) Multiple sequence alignment of the amino acids encoded by (a) exons 5 and 6 of the MIKCC-type genes SEP3, CRM3, SmMADS3, and PPM1 and (b) exons 4–7 of the MIKC*-type gene AGL66 and exons 3–6 of CRM13, CRM15, SmMADS2, and PPM4, respectively. (c) Exon–intron structure of the MIKCC- and MIKC*-type genes shown in a and b, respectively, together with the exon–intron structure of the charophyte MIKC-type gene KnMADS1. Homologous exons were aligned and identical nucleotide positions of neighboring sequences are connected with solid gray lines to illustrate homology. K-domain exons shared by MIKCC-, MIKC*-, and charophyte MIKC-type genes are color-coded in green, hypothetically duplicated K-domain exons of MIKCC- and MIKC*-type genes are color-coded in different shades of blue and yellow/orange, respectively. (d) X-ray crystal structure of the MIKCC-type protein SEP3 (PDB-ID: 4OX0). Subdomains encoded by exons 3, 4, 5, and 6, as indicated, are color-coded in yellow, green, light blue, and dark blue, respectively. (e) Tetramer of four SEP3 K-domains following the same color-coding as in d. Protein structure images were generated with Swiss-PdbViewer (Guex and Peitsch 1997).
Charophyte MIKC-type Genes Show no Duplications of K-domain Exons
Land plant MIKCC- and MIKC*-type genes very likely evolved by a gene duplication of an ancestral MIKC-type gene in the stem lineage of extant land plants (Henschel et al. 2002; Kaufmann et al. 2005; Gramzow and Theißen 2010). Consequently, MIKC-type genes from charophyte green algae, a grade of freshwater algae that represent land plants’ closest extant relatives, phylogenetically belong to neither of the two land-plant specific subfamilies (Nishiyama et al. 2018). Instead charophyte MIKC-type genes likely constitute direct descendants of an ancestral MIKC-type gene, prior to the split into MIKCC- and MIKC*-type. We analyzed the exon–intron structures of charophyte MIKC-type genes, for which genomic information is available, and indeed found the hypothetical exon–intron structure of an ancestral MIKC-type gene. The charophyte MIKC-type protein KnMADS1 from Klebsormidium nitens (Klebsormidiophyceae) is encoded by one MADS-domain exon, one I-domain exon, four K-domain exons and five C-terminal domain exons (fig. 3c). The first two K-domain exons of KnMADS1 show high similarity in length and sequence to the first and second, as well as to the third and fourth K-domain exon of MIKC*-type genes, whereas the last K-domain exon of KnMADS1 shows high similarity to the last two K-domain exons of MIKCC-type genes (fig. 3c). This observation corroborates the hypothesis that ancestral MIKCC- and MIKC*-type genes underwent exon duplications causing structural divergence of their K-domains.
To better comprehend which structural changes may have been brought about by the different exon duplications, we plotted the protein regions encoded by the different K-domain exons on the known crystal structure of the K-domain of the MIKCC-type protein SEP3 from A. thaliana (Puranik et al. 2014). SEP3 exon 3 (i.e., the first K-domain exon) encodes for the first (N-terminal) α-helix harboring dimerization interface 1. Exon 4 encodes for the kink region separating both K-domain helices and for the N-terminal half of the second α-helix harboring dimerization interface 2. Exons 5 and 6 encode for the C-terminal half of the second α-helix comprising the tetramerization interface (fig. 3d and e). Consequently, the duplication of the last K-domain exon of an ancestral MIKCC-type gene likely elongated the second (C-terminal) K-domain helix and thereby gave rise to the full-length tetramerization interface found in extant MIKCC-type proteins. In contrast, the duplication of the first two K-domain exons of an ancestral MIKC*-type protein probably elongated the dimerization interface of the first (N-terminal) helix.
Artificial Exon Deletion Impedes FQC Formation of MIKCC-type Proteins
To investigate the functional importance of the last two K-domain exons for FQC formation of MIKCC-type proteins, we generated mutant versions of PPM1, SmMADS3, CRM3, and SEP3, lacking the sequence of either of the two hypothetically duplicated K-domain exons (exons 5 and 6, respectively), by mutagenesis PCR. The resulting mutant proteins PPM1ΔE5, PPM1ΔE6, SmMADS3ΔE5, SmMADS3ΔE6, CRM3ΔE5, CRM3ΔE6, SEP3ΔE5, and SEP3ΔE6 were expressed in vitro and tested for their ability to form FQCs in EMSA. In contrast to the wild-type proteins, all exon deletion mutants, except for CRM3ΔE6, produced a fraction of intermediate electrophoretic mobility (DNA probe bound by two proteins) at low amounts of applied protein. With increasing protein amounts an additional fraction of low electrophoretic mobility (DNA probe bound by four proteins) occurred (fig. 4 and supplementary fig. S5, Supplementary Material online). By quantification of the signal intensities of the different fractions, FQC formation capabilities were estimated and expressed as Kd1/Kd2. Except for CRM3ΔE6, all mutated MIKCC-type proteins produced low cooperativity values, comparable to those determined for MIKC*-type proteins (fig. 4). This indicates no or very weak positive interaction between the two DNA-bound dimers and thus no ability for cooperative DNA binding and FQC formation. CRM3ΔE6 showed an uncommon DNA-binding behavior as it produced two fractions of low electrophoretic mobility. Furthermore, the bound and unbound fractions did not follow the usually observed sigmoidal increase and decrease, respectively (fig. 4f). However, as no signal of a DNA probe bound by only two proteins was observed, it appears likely that CRM3ΔE6 is still able to form FQCs.
Fig. 4.

FQC formation capabilities of exon deletion mutants of the MIKCC-type proteins PPM1, SmMADS3, CRM3, and SEP3. Increasing amounts of in vitro transcribed/translated (a) PPM1ΔE5, (b) PPM1ΔE6, (c) SmMADS3ΔE5, (d) SmMADS3ΔE6, (e) CRM3ΔE5, (f) CRM3ΔE6, (g) SEP3ΔE5, and (h) SEP3ΔE6 were coincubated together with constant amounts of DNA probe 1. For details, see legend of figure 1. (f) Because CRM3ΔE6 produced no signal of intermediate electrophoretic mobility constituting a DNA probe bound by a single protein dimer, Kd1/Kd2 cannot be determined. Applied amounts of in vitro transcription/translation products were (lanes 1–10) 0, 0.0125, 0.025, 0.05, 0.1, 0.2, 0.4, 0.5, 1, and 2 µl, whereby PPM1ΔE5, PPM1ΔE6, SmMADS3ΔE5, SmMADS3ΔE6, CRM3ΔE5, and CRM3ΔE6 were prediluted 1:5 with BSA (10 mg/ml).
Artificial Duplication of Exon 6 Enables KnMADS1 to Form FQCs
To further study the functional implications of the exon duplications during early evolution of MIKCC- and MIKC*-type genes, we used the charophyte MIKC-type gene KnMADS1 as a proxy for the ancestral state of land plant MIKCC- and MIKC*-type genes immediately after the split into both gene families. We generated three mutated versions of KnMADS1 (fig. 5a). To mimic an early MIKCC-type gene, we duplicated exon 6 (KnMADS1duplE6) and in addition deleted exon 3 (KnMADS1Δ3duplE6), as exon 3 of KnMADS1 is not found among extant MIKCC-type genes (figs. 2 and 3c). To mimic an early MIKC*-type gene, we duplicated KnMADS1 exons 3 and 4 (KnMADS1duplE3E4). KnMADS1 wild-type and mutated proteins were produced in vitro and tested for their ability to form FQCs in EMSA. In accordance to our hypothesis that the duplication of the last K-domain exon facilitated FQC formation, KnMADS1 wild-type protein produced very low cooperativity values and thus was unable to form FQCs under our experimental conditions (fig. 5b and supplementary fig. S6a, Supplementary Material online). In contrast, the two MIKCC-type mimicking mutants KnMADS1duplE6 and KnMADS1ΔE3duplE6 showed no signal of intermediate electrophoretic mobility (DNA probe bound by only two proteins), but instead bound to DNA immediately as tetramer, similar to the tested MIKCC-type proteins from Physcomitrium, Selaginella, and Ceratopteris (fig. 5c and d and supplementary fig. S6b and c, Supplementary material online). The MIKC*-type mimicking mutant KnMADS1duplE3E4 showed a DNA-binding behavior similar to that of the KnMADS1 wild-type protein, although the signal of intermediate electrophoretic mobility appeared weaker, resulting in slightly higher cooperativity values (fig. 5e and supplementary fig. S6d, Supplementary Material online).
Fig. 5.

FQC formation capabilities of exon duplication and deletion mutants of the charophyte MIKC-type protein KnMADS1. (a) Exon–intron structure of KnMADS1 wild type and the mutated version KnMADS1duplE6, KnMADS1ΔE3duplE6, and KnMADS1duplE3E4. K-domain exons are color coded according to figure 3c. Black triangle highlights the position at which the coding sequence was terminated to generate C-terminally truncated versions of KnMADS1 and KnMADS1duplE3E4. (b-e) Increasing amounts of in vitro transcribed/translated (b) KnMADS1, (c) KnMADS1duplE6, (d) KnMADS1ΔE3duplE6, and (e) KnMADS1duplE3E4 was coincubated together with constant amounts of DNA probe 1. For details, see legend of figure 1. Because (c) KnMADS1duplE6 and (d) KnMADS1ΔE3duplE6 produced no signals of intermediate electrophoretic mobility constituting a DNA probe bound by a single protein dimer, Kd1/Kd2 cannot be determined. (f) KnMADS1 wild-type protein was coexpressed at different ratios with KnMADS1ΔC and coincubated together with constant amounts of DNA probe 1. An overlay of measured signal intensities of the individual lanes is shown on the right. Each peak of the graph is labeled according to the ratio of full-length and truncated protein of the corresponding fraction. The cartoons below the gel illustrate the composition of the different fractions with full-length and truncated proteins shown in yellow and green, respectively. (g, h) To test for heteromeric interaction capabilities between KnMADS1duplE6, KnMADS1ΔE3duplE6, and KnMADS1duplE3E4, the same assay as in f was conducted using (g) KnMADS1duplE6 together with KnMADS1duplE3E4ΔC and (h) KnMADS1ΔE3duplE6 together with KnMADS1duplE3E4ΔC, respectively. (b–e) Applied amounts of in vitro transcription/translation products were (lanes 1–8) 0, 0.05, 0.1, 0.2, 0.4, 0.6, 1, and 2 µl. (f–h) 3 µl of in vitro transcription/translation product were applied to each lane. Ratios of both template plasmids used for in vitro transcription/translation were (lanes 1–11): 0:1, 1:9, 1:7, 1:5, 1:3, 1:1, 3:1, 5:1, 7:1, 9:1, 1:0.
Exon Duplication Mutants of KnMADS1 are Unable to Form Heterodimers
Based on large-scale interaction data of MIKCC- and MIKC*-type proteins from seed plants, both subfamilies form two mainly independent protein–protein interaction networks (de Folter et al. 2005; Immink et al. 2009; Smaczniak, Immink, Angenent, et al. 2012). We aimed to investigate, whether the structural divergence of the K-domains of MIKCC- and MIKC*-type proteins, that was brought about by the duplication of different K-domain exons, may impede interactions between members of both subfamilies. We generated C-terminal truncation mutants of KnMADS1 wild type (KnMADS1ΔC) and KnMADS1duplE3E4 (KnMADS1duplE3E4ΔC) in order to be able to differentiate heteromeric complexes of different composition in EMSA due to their different size. When we coincubated variable amounts of KnMADS1 and KnMADS1ΔC together with labeled DNA probe, five retarded fractions of different electrophoretic mobility occurred, demonstrating that each DNA-bound KnMADS1 wild-type protein can be substituted by a C-terminally truncated version (fig. 5f and supplementary fig. S6e, Supplementary Material online). When we conducted the same experiment using variable amounts of KnMADS1duplE6 and KnMADS1duplE3E4ΔC, only four retarded fractions of different electrophoretic mobility occurred (fig. 5g and supplementary fig. S6f, Supplementary Material online). The fraction representing a DNA probe bound by three KnMADS1duplE3E4ΔC proteins and one KnMADS1duplE6 protein (fraction labeled with “3:1” in fig. 5f) was completely absent. When we coincubated variable amounts of KnMADS1ΔE3duplE6 and KnMADS1duplE3E4ΔC, only three retarded fractions occurred, likely representing a DNA probe bound by four KnMADS1ΔE3duplE6 proteins (labeled with “0:4”), by two KnMADS1ΔE3duplE6 proteins and two KnMADS1duplE3E4ΔC proteins (“2:2”) and by four KnMADS1duplE3E4ΔC proteins (“4:0”), respectively (fig. 5h and supplementary fig. S6g, Supplementary Material online). No DNA probe bound by three KnMADS1duplE3E4ΔC proteins and one KnMADS1ΔE3duplE6 protein or by one KnMADS1duplE3E4ΔC protein and three KnMADS1ΔE3duplE6 proteins was observed. Consequently, the duplication of exons 3 and 4 of one partner and the duplication of exon 6 (and deletion of exon 3) of the other partner seems to impede heterodimer formation of KnMADS1. Therefore, it appears plausible that the structural divergence of the K-domains of MIKCC- and MIKC*-type proteins, that was caused by the different exon duplications, prevents heteromeric interactions between members of both subfamilies.
Some Charophyte MIKC-type Proteins Form FQCs
Besides KnMADS1, we cloned and tested a number of other charophyte MIKC-type proteins for their ability to form FQCs. Similar to KnMADS1, also CaMADS1 from Chlorokybus atmophyticus (Chlorokybophyceae) and CglMADS1 from Chaetosphaeridium globosum (Coleochaetophyceae) produced clear signals of intermediate electrophoretic mobility (i.e., binding of a single protein dimer) resulting in low cooperativity values indicating no FQC formation (fig. 6a and b and supplementary fig. S7a and b, Supplementary material online). In contrast, ZspMADS1 from Zygnema sp. (Zygnematophyceae) produced only very weak signals of intermediate electrophoretic mobility resulting in high cooperativity values denoting strong FQC formation capabilities (fig. 6c and supplementary fig. S7c, Supplementary Material online). All three tested proteins from the genus Coleochaete (Coleochaetophyceae), CiMADS1 from Coleochaete irregularis, CoMADS1 from Coleochaete orbicularis, and CsMADS1 from Coleochaete scutata, showed no signals of intermediate electrophoretic mobility at all, indicating no binding of single dimers and thus very strong FQC formation (fig. 6d–f, supplementary fig. S7d–f, Supplementary material online). All experimentally investigated charophyte MIKC-type genes show no duplication of the last K-domain exon. However, the last K-domain exon of charophyte MIKC-type genes already encodes for the N-terminal half of the tetramerization interface found in land plant MIKCC-type proteins. Therefore, it appears plausible, that to a certain extend already a shorter C-terminal K-domain helix is sufficient to mediate MTF tetramerization or that the helix was extended by another mechanism. In case of CsMADS1, coiled-coil predictions (Gruber et al. 2006) indeed suggest that the C-terminal K-domain helix extends beyond the border of the K-domain as the pattern of regularly spaced hydrophobic residues, that is characteristic for amphipathic α-helices, extends into the C-terminal domain (supplementary fig. S8a, Supplementary Material online). In accordance to this observation, truncation of the complete C-terminal domain of CsMADS1 prevented FQC formation (supplementary fig. S8a and c, Supplementary material online).
Fig. 6.

FQC formation capabilities of the charophyte MIKC-type proteins CaMADS1, CglMADS1, ZspMADS1, CiMADS1, CoMADS1, and CsMADS1. Increasing amounts of in vitro transcribed/translated (a) CaMADS1ΔC, (b) CglMADS1ΔC, (c) ZspMADS1ΔC, (d) CiMADS1, (e) CoMADS1, and (f) CsMADS1, were coincubated together with constant amounts of DNA probe 1. For details see legend of figure 1. Because (d) CiMADS1, (e) CoMADS1, and (f) CsMADS1 produced no signal of intermediate electrophoretic mobility constituting a DNA probe bound by a single protein dimer, Kd1/Kd2 cannot be determined. In case of (a) CaMADS1, a double band was observed for the fraction of intermediate electrophoretic mobility, likely caused by different conformations of the DNA-bound protein dimer. Because CaMADS1, CglMADS1, and ZspMADS1 full-length proteins comprise very long C-terminal domains, resulting in low DNA-binding affinities and blurry bands, C-terminally truncated mutants were used instead. Applied amounts of in vitro transcription/translation products were (a, c–e) (lanes 1–9) 0, 0.025, 0.05, 0.1, 0.2, 0.4, 0.8, 1, and 2 µl, whereby CaMADS1ΔC and ZspMADS1ΔC were prediluted 1:10 with BSA (10 mg/ml), (b) (lanes 1–9) 0.02, 0.04, 0.1, 0.2, 0.4, 0.8, 1, 2, and 3 µl, (f) (lanes 1–8) 0, 0.005, 0.01, 0.02, 0.04, 0.06, 0.1, and 0.2 µl.
Discussion
Floral-quartet-like complexes represent a unique system of gene regulation involving heterotetramers of MIKCC-type MTFs. FQCs control important developmental processes in plants, but how they originated remained unknown for decades. Here we present data clarifying the evolutionary origin of FQCs.
Like many other proteins, MTFs bind with sufficient strength to specific DNA sequences only as dimers, a feature that they share also with all other MADS-domain proteins (Shore and Sharrocks 1995; Amoutzias et al. 2008). In addition, however, many MTFs constitute also tetrameric complexes. These tetrameric MTFs recognize their target genes by binding to two CArG-boxes, involving DNA-loop formation between the binding sites (Egea-Cortines et al. 1999; Theißen 2001; Theißen and Saedler 2001; Theißen et al. 2016). Tetramerization of regulatory proteins, and DNA-binding on two cis-regulatory elements involving DNA looping, is well known from bacterial repressors and activators, such as the lac repressor and lambda repressor/activator (Hochschild and Ptashne 1986; Oehler et al. 1990; Lewis 2005). In these cases, protein–protein interactions between protein dimers provide Gibbs free energy (ΔG°) in addition to that available by protein–DNA interactions alone, and hence lead to a cooperative formation of tetramers bound to DNA. Consequently, a switch-like on-off interaction of the regulatory proteins with their target genes occurs (Hochschild and Ptashne 1986).
Cooperative binding has also been demonstrated for MTFs in FQCs (Melzer et al. 2009; Jetha et al. 2014; Rümpler et al. 2018). The proximate (molecular) and ultimate (evolutionary) mechanisms that may have led to the origin and maintenance of MTF tetramerization and FQC formation have already been discussed elsewhere (Theißen et al. 2016). Briefly, like in case of viral and bacterial activators and repressors, cooperative formation of MTFs could lead to a sharp transcriptional response of target genes. Even small increases of MTF concentrations may lead to drastic changes in the regulatory response of target genes, allowing for a switch-like, all-or-nothing kind of regulation of target genes. Since MTFs act as genetic switches that control discrete developmental processes—the establishment of different kinds of tissue or organ identities is a good case in point—cooperativity could well be one crucial mechanism that transforms the quantitative nature of molecular interactions between MTFs and DNA into discrete phenotypic outputs (Theißen and Melzer 2007; Kaufmann et al. 2010). In case of for example organ identity genes the biological relevance of this behavior is quite plausible, since it leads to the development of organs with unambiguous identities (such as stamens and carpels) and avoids the formation of chimeric organs. Accordingly, it has been hypothesized that the ability of MIKCC-type proteins to establish and diversify sharp developmental switches may have facilitated the evolution of different tissues and organs as required during evolution of the increasingly complex body plans of plants on land (Theißen et al. 2016; Nishiyama et al. 2018).
We consider tetramerization of MTFs and FQC formation an important evolutionary novelty in gene regulation. It has long been known that MADS-domain proteins act in multimeric (often called “ternary”) complexes. In cases other than MIKCC-type proteins, however, dimers of MADS-domain proteins interact with non-MADS-domain proteins. Well-characterized examples are the different complexes of the general yeast transcription factor MINICHROMOSOME MAINTENANCE 1 (MCM1), some of which involve, for example, the homeodomain protein α2 and the HMG-domain protein a2, and the animal SRF that interacts among others with members of the myocardin family of transcription factors and with ternary complex factors (Shore and Sharrocks 1995; Messenguy and Dubois 2003; Tsong et al. 2006; Onuh and Qiu 2021). However, tetrameric complexes composed exclusively of MADS-domain proteins encoded by the same or paralogous genes appear to be unique to MTFs in plants.
Heterotetramerization of paralogous MTFs represents not only a regulatory system with important functions in flowering plants, but, by inference, very likely also in all other land plants (embryophytes). Hence, tetramerization of MTFs and cooperative formation of FQCs is of considerable biological and even agronomic interest. However, surprisingly little has been known about its evolutionary origin. It has been shown that in addition to MIKCC-type proteins of flowering plants also some MIKCC-type proteins of extant gymnosperms are able to form FQCs (Wang et al. 2010; Ruelens et al. 2017). Since the lineages that led to extant gymnosperms and angiosperms separated roughly about 300 million years ago, this pushes the origin of FQC formation accordingly. Corresponding information about nonseed plants, and about MIKC*-type proteins from any plants, was lacking so far. It remains unclear, therefore, when (in time) and where (in which lineage) tetramerization of MTFs actually originated. Here we demonstrate that FQC formation has much deeper roots than seed plants, down to streptophyte green algae, which are close relatives of land plants. FQC formation had therefore been established at least 450–500 million years ago already.
Almost 20 years ago two hypothetical scenarios have been proposed to explain the causal link between the K-domain and MTF tetramerization (Kaufmann et al. 2005). One model has it that the K-domain was initially just involved in protein dimerization and is hence more ancient than tetramerization. Only later tetramerization may have been acquired as a second function of the K-domain. Alternatively, the K-domain might have had a primary role in tetramerization right from the beginning, and no or only a minor role in dimerization. Under this assumption, the MTFs and tetramerization would have occurred simultaneously.
Based on the data presented here, we now can draw a clear picture of the molecular changes that facilitated MTF tetramerization (fig. 7). In the stem group of extant streptophytes, a K-domain encoding sequence originated downstream of the MADS-box, by a yet unknown mechanism. In its ancestral state, the K-domain was likely encoded by four exons and probably already constituted a fold similar to that determined for the MIKCC-type protein SEP3 (Puranik et al. 2014), comprising helix 1, a rigid kink and helix 2. This ancestral type of K-domain (as still retained in some charophyte green algae) may had already been able to facilitate tetramerization to a certain extent, but does not do so in all cases (fig. 6), so that it may have primarily originated as an additional dimerization domain. In the stem group of extant land plants, a gene duplication of an ancestral MIKC-type gene occurred (fig. 7b). In the lineage of one of the copies, the first two K-domain exons got duplicated, generating MIKC*-type genes as has already been hypothesized before (fig. 3b and c; Kwantes et al. 2012). In the lineage of the second copy, the first K-domain exon likely got lost, whereas the last K-domain exon was duplicated, generating MIKCC-type genes (fig. 3a and c). By the duplication of the last K-domain exon, helix 2 of the K-domain got elongated, now constituting a strong tetramerization interface (fig. 3d and e). Since no MIKC-type genes encoding ancestral K-domains have been found in land plant genomes yet, it appears very likely that such genes have been lost in the stem group of extant land plants, even though other scenarios currently cannot be completely ruled out. Ancestral MTFs may have negatively interfered with the assembly or function of transcription factor complexes once more “advanced” MIKCC-type and MIKC*-type proteins had originated.
Fig. 7.

Hypothesized mode of MTF evolution. (a) Simplified phylogenetic tree of green plants with highlighted major evolutionary changes of Type II MADS-box genes (branching according to Wickett et al. [2014]). In the stem group of extant streptophytes (charophytes + land plants), an ancestral type II MADS-box gene (illustrated by gray branch color) acquired the K-domain, giving rise to the eponymous “MIKC” domain architecture of the encoded MTFs. In the stem group of extant land plants, a gene duplication of an ancestral MIKC-type gene (illustrated by green branch color) gave rise to the two, land plant–specific, MTF subfamilies MIKCC (blue branch color) and MIKC* (orange branch color). Gray arrow depicts the time point, when the evolutionary changes shown in b occurred. Cartoons of FQCs and DNA-bound MTF dimers indicate presence or absence, respectively, of FQC formation capabilities of MTFs in the respective plant lineages. (b) Following the gene duplication of an ancestral MIKC-type gene (upper part, green background), the first two K-domain exons got duplicated in the gene lineage of MIKC*-type genes (left part, orange background), whereas the first K-domain exon got lost and the last K-domain exon got duplicated in the gene lineage of MIKCC-type genes (right part, blue background).
During the diversification of land plants into bryophytes (hornworts, liverworts, mosses), lycophytes, monilophytes (ferns and their allies), and spermatophytes (gymnosperms and angiosperms), gene duplications and sequence diversifications led to a moderate increase in the number of MIKC*-type genes and a strong increase in the number of MIKCC-type genes (Gramzow and Theißen 2010; Thangavel and Nayar 2018). In seed plants, MIKCC-type genes are almost exclusively involved in the control of sporophyte developmental processes (Smaczniak, Immink, Angenent, et al. 2012). Given that the sporophyte became the dominant life phase during land plant evolution, it is tempting to speculate that the gain of tetramerization in the MIKCC-type lineage, the strong increase in the number of MIKCC-type genes during land plant evolution, and the gain in importance of the sporophyte life phase were causally linked. MIKCC-type genes might have been coopted to control developmental processes of the increasingly complex sporophyte, because they were able to cooperatively constitute FQCs in different combinations and therefore bore a higher combinatorial potential than MIKC*-type genes. The ability of MIKCC-type proteins to constitute FQCs may have enabled the control of numerous different tissues and organs and hence may have facilitated the origin of land plant diversity from the species to the body plan level.
Large scale interaction data of MIKCC- and MIKC*-type proteins from flowering plants have shown, that both protein subfamilies form two largely independent interaction networks (de Folter et al. 2005; Immink et al. 2009; Gramzow and Theißen 2010). Based on our data, it appears likely that the structural changes within the K-domain caused by duplications of different exons, laid the foundation for this functional separation and the subsequent evolution of independent gene regulatory networks.
Materials and Methods
Cloning Procedures and Site-directed Mutagenesis
The plasmid for in vitro transcription/translation of A. thaliana SEP3 has been generated previously (Melzer et al. 2009). Coding sequences of Zygnema sp. ZspMADS1 (1kP: STKJ-2019062), C. globosum CglMADS1 (1kP: DRGY-2036441), Co. scutata CsMADS1 (GenBank: AB035568.1), Co. irregularis CiMADS1 (1kP: QPDY-2028842), Co. orbicularis CoMADS1 (GenBank: GBSL01006298.1), K. nitens KnMADS1 (GenBank: DF237509; locus tag KFL_005600030), Ch. atmophyticus CaMADS1 (1kP: AZZW-2004234), P. patens PPM1 (GenBank: AF150931_1) and PPM4 (GenBank: AY509529.1), S. moellendorffii SmMADS2 (NCBI Ref: XM_002974738.1) and SmMADS3 (NCBI Ref: XM_002984875.1), C. richardii CRM3 (GenBank: Y08239_1), CRM13 (GenBank: FM995267.1), CRM14 (GenBank: FM995269.1), CRM15 (GenBank: FM995271.1), and CRM16 (GenBank: FM995273.1), as well as A. thaliana AGL66 (NCBI Ref: NM_106447.4) and AGL104 (NCBI Ref: NM_102063.3) were codon optimized for Oryctolagus cuniculus (European rabbit), synthesized via the GeneArt gene synthesis service (Thermo Fisher Scientific) and cloned into pTNT (Promega) using EcoRI and SalI restriction sites. Plasmids for in vitro transcription/translation of C-terminally truncated proteins, exon duplication and exon deletion mutants were generated by site-directed mutagenesis PCR following the manufacturer's instructions of the Q5 Site-Directed Mutagenesis Kit (New England Biolabs). To generate the plasmids for in vitro transcription/translation of the fusion proteins SmMADS3-GFP and CRM3-GFP, a HindIII restriction site was introduced into plasmids pTNT-SmMADS3 and pTNT-CRM3 by site-directed mutagenesis substituting the stop codons of SmMADS3 and CRM3. Subsequently, the coding sequence of the enhanced GFP gene mGFP6 was PCR amplified from pGreenII-35S::mGFP6 (Hellens et al. 2000) while adding HindIII restriction sites at the 3′ and 5′ end. The purified PCR product was cloned into pTNT-SmMADS3 and pTNT-CRM3 using the introduced HindIII restriction site.
Generation of DNA Probes
The DNA probes for EMSA have been generated essentially as described by Melzer et al. (2009). In total four different DNA probes were used in this study (supplementary table S1, Supplementary Material online). Probe 1: a 151-bp DNA probe carrying two SRF-type CArG-boxes of the sequence 5′-CCAAATAAGG-3′ in a distance of 63 bp, about 6 helical turns; probe 2: same as probe 1 except that the SRF-type CArG-boxes were substituted by N10-type CArG-boxes of the sequence 5′-CTATATATAG-3′; probe 3: a 167-bp DNA probe carrying two SRF-type CArG-boxes of the sequence 5′-CCAAATAAGG-3′ in a distance of 79 bp, about 7.5 helical turns; probe 4: same as probe 1 but with a randomized sequence instead of the second (right) CArG-box and its flanking 15 bp. The DNA probes were radioactively labeled with [α-P32] dATP by a Klenow fill-in reaction of 5′-overhangs.
In Vitro Transcription/Translation and Electrophoretic Mobility Shift Assay
Proteins were produced in vitro via the rabbit reticulocyte based TNT SP6 Quick Coupled Transcription/Translation System (Promega). If two proteins were to be tested together in a single binding reaction, the proteins were coexpressed by loading the transcription/translation system with template plasmids of both proteins at the desired ratio. EMSA was performed essentially as described by Melzer et al. (2009). The composition of the protein-DNA binding reaction buffer was essentially as described by Egea-Cortines et al. (1999) with final concentrations of 1.6 mM EDTA, 10.3 mM HEPES, 1 mM DTT, 1.3 mM Spermidine hydrochloride, 33.3 ng/µl Poly dI/dC, 2.5% CHAPS, 4,3% glycerol, and 5.2–7.7 ng/µl BSA. To test for FQC formation capabilities, 0.1 ng of radioactively labeled DNA probe was coincubated with variable amounts of in vitro transcribed/translated protein ranging from 0.0025 to 2 µl. To test for complex stoichiometry and heteromeric interactions, 0.1 ng of radioactively labeled DNA probe was coincubated with 3 µl of in vitro transcription/translation reaction product that had been loaded with both template plasmids at the indicated ratios: 0:1, 1:9, 1:7, 1:5, 1:3, 1:1, 3:1, 5:1, 7:1, 9:1, 1:0.
Rationale of our FQC Formation Assay and Quantification of FQC-formation Capabilities
In this study, we analyze FQC formation of MTFs via a previously established EMSA. As has been explained in detail elsewhere, our assay allows us to discriminate between two MTF dimers that individually bind to two adjacent CArG-boxes, and a single MTF tetramer that simultaneously binds to both CArG-boxes while looping the DNA in between and thus forming an FQC (Melzer and Theißen 2009; Melzer et al. 2009). FQC formation is indicated by a high cooperativity of four MTFs binding to specific DNA sequences.
In brief, we incubate increasing amounts of the MTF of interest together with a constant amount of a radioactively labeled DNA probe carrying two identical CArG-boxes. In case of an MTF incapable of forming FQCs, dimers of MTFs will independently bind with the same affinity to both CArG-boxes. In other words, the affinity of the first dimer, binding to an unbound DNA probe, is equal to the affinity of the second dimer, binding to a DNA probe at which one binding site is already occupied. In contrast, in case of an MTF capable of forming FQCs, the two DNA-binding dimers positively interact with each other by forming a tetramer. Due to this positive interaction between both dimers, the affinity of the second dimer, that binds to a DNA probe at which one binding site is already occupied, is increased, compared with the DNA-binding affinity of the first dimer. This increased DNA-binding affinity of the second dimer, shifts the equilibrium of the binding reaction towards a DNA probe with two occupied binding sites. As a consequence, MTFs that are capable of forming FQCs produce weaker or no signals of a DNA probe bound by only one dimer than MTFs incapable of forming FQCs.
The signal intensities of the three fractions of different electrophoretic mobility (unbound DNA probe, DNA probe bound by two proteins, and DNA probe bound by four proteins) were quantified using Multi Gauge 3.1 (Fujifilm). Quantification of FQC-formation capabilities was performed by nonlinear regression to equations described by Melzer et al. (2009) and Senear and Brenowitz (1991). In brief, if the relative concentration of unbound DNA probe [Y0], DNA probe bound by two proteins [Y2], and DNA probe bound by four proteins [Y4] are described as a function of the amount of applied protein [P2]:
| (1) |
| (2) |
| (3) |
Kd1 and Kd2 are the dissociation constants for the binding reaction of a protein dimer to an unbound DNA probe and the binding reaction of a second protein dimer to a DNA probe where one binding site is already occupied, respectively. Kd1 and Kd2 were estimated by nonlinear regression to equations (1)–(3) using GraphPad Prism 9 (GraphPad Software). The ability to form FQCs can subsequently be expressed by dividing Kd1 by Kd2.
Exon Homology Analysis
Coding sequences of all MIKCC- and MIKC*-type genes from A. thaliana, C. richardii, S. moellendorffii, and P. patens were translated into protein sequences, aligned with Mafft applying default settings (Katoh and Standley 2013), and subsequently back-translated into a codon alignment using RevTrans (Wernersson and Pedersen 2003). Exon borders were determined by aligning coding sequences to the corresponding genomic sequences using Splign (Kapustin et al. 2008). Information about sequence similarity and exon borders were combined into a graphical presentation utilizing a customized perl script previously described by Hoffmeier et al. (2018). This way homologous exons were identified and manually aligned. Exons were color coded, depending on the protein domain they encode for. Exons were defined as K-domain exons if they were homologous to exons encoding for the K-domain of SEP3 from A. thaliana, for which the crystal structure has been determined (Puranik et al. 2014). The codon alignments (including accession numbers for all genes) used for generation of figures 2 and 3 are given in supplementary Supplementary Data 1 and 2, Supplementary Material online, respectively.
Supplementary Material
Acknowledgments
Part of this work was funded by grant TH417/12-1 from the German Research Foundation (DFG) to GT and FR in the framework of the Priority Program “MAdLand—Molecular Adaptation to Land: Plant Evolution to Change” (SPP 2237). The authors thank Felix Althoff and Sabine Zachgo (Osnabrück) for fruitful discussions about MADS-box genes on MAdLand, and to Stefan Rensing (Marburg/Freiburg) and Jan de Vries (Göttingen) for helpful information and stimulating discussions about diverse Charophyte genomes. Many thanks also to Philipp Gehlhaar for some experiments during initial stages of the project.
Contributor Information
Florian Rümpler, Matthias Schleiden Institute/Genetics, Friedrich Schiller University Jena, Jena, Germany.
Chiara Tessari, Matthias Schleiden Institute/Genetics, Friedrich Schiller University Jena, Jena, Germany.
Lydia Gramzow, Matthias Schleiden Institute/Genetics, Friedrich Schiller University Jena, Jena, Germany.
Christian Gafert, Matthias Schleiden Institute/Genetics, Friedrich Schiller University Jena, Jena, Germany.
Marcus Blohs, Matthias Schleiden Institute/Genetics, Friedrich Schiller University Jena, Jena, Germany.
Günter Theißen, Matthias Schleiden Institute/Genetics, Friedrich Schiller University Jena, Jena, Germany.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Author Contributions
G.T. and F.R. initiated, conceived and supervized the project; F.R., C.T., C.G., and M.B. generated the constructs for in vitro transcription/translation and conducted the EMSA experiments; C.T. and L.G. compiled the sequence collection and L.G. conducted the exon homology analysis; F.R. prepared figures; F.R. and G.T. wrote the manuscript; all authors read and approved the submitted version of the manuscript.
Data Availability
Alignments used for the exon homology analysis of figure 2 and the exon duplication analysis of figure 3 are available through supplementary Data 1 and 2, Supplementary Material online, respectively.
References
- Amoutzias GD, Robertson DL, Van de Peer Y, Oliver SG. 2008. Choose your partners: dimerization in eukaryotic transcription factors. Trends Biochem Sci. 33:220–229. [DOI] [PubMed] [Google Scholar]
- de Folter S, Immink RGH, Kieffer M, Parenicova L, Henz SR, Weigel D, Busscher M, Kooiker M, Colombo L, Kater MM, et al. . 2005. Comprehensive interaction map of the Arabidopsis MADS box transcription factors. Plant Cell. 17:1424–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derelle E, Ferraz C, Rombauts S, Rouze P, Worden AZ, Robbens S, Partensky F, Degroeve S, Echeynie S, Cooke R, et al. . 2006. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci U S A. 103:11647–11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea-Cortines M, Saedler H, Sommer H. 1999. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. EMBO J. 18:5370–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Soto C, Immink RGH, Angenent GC, Alvarez-Buylla ER, de Folter S. 2014. Tetramer formation in Arabidopsis MADS domain proteins: analysis of a protein-protein interaction network. BMC Syst Biol. 8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramzow L, Theißen G. 2010. A Hitchhiker’s Guide to the MADS world of plants. Genome Biol. 11:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber M, Soding J, Lupas AN. 2006. Comparative analysis of coiled-coil prediction methods. J Struct Biol. 155:140–145. [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- Hellens RP, Edwards EA, Leyland NR, Bean S, Mullineaux PM. 2000. Pgreen: a versatile and flexible binary Ti vector for Agrobacterium-mediated plant transformation. Plant Mol Biol. 42:819–832. [DOI] [PubMed] [Google Scholar]
- Henschel K, Kofuji R, Hasebe M, Saedler H, Münster T, Theißen G. 2002. Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol Biol Evol. 19:801–814. [DOI] [PubMed] [Google Scholar]
- Hochschild A, Ptashne M. 1986. Cooperative binding of lambda-repressors to sites separated by integral turns of the DNA Helix. Cell 44:681–687. [DOI] [PubMed] [Google Scholar]
- Hoffmeier A, Gramzow L, Bhide AS, Kottenhagen N, Greifenstein A, Schubert O, Mummenhoff K, Becker A, Theissen G. 2018. A dead gene walking: convergent degeneration of a clade of MADS-box genes in crucifers. Mol Biol Evol. 35:2618–2638. [DOI] [PubMed] [Google Scholar]
- Hugouvieux V, Silva CS, Jourdain A, Stigliani A, Charras Q, Conn V, Conn SJ, Carles CC, Parcy F, Zubieta C. 2018. Tetramerization of MADS family transcription factors SEPALLATA3 and AGAMOUS is required for floral meristem determinacy in Arabidopsis. Nucleic Acids Res. 46:4966–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immink RGH, Tonaco IA, de Folter S, Shchennikova A, van Dijk AD, Busscher-Lange J, Borst JW, Angenent GC. 2009. SEPALLATA3: the ‘glue’ for MADS box transcription factor complex formation. Genome Biol. 10:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetha K, Theißen G, Melzer R. 2014. Arabidopsis SEPALLATA proteins differ in cooperative DNA-binding during the formation of floral quartet-like complexes. Nucleic Acids Res. 42:10927–10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapustin Y, Souvorov A, Tatusova T, Lipman D. 2008. Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct. 3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K, Melzer R, Theißen G. 2005. MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene 347:183–198. [DOI] [PubMed] [Google Scholar]
- Kaufmann K, Pajoro A, Angenent GC. 2010. Regulation of transcription in plants: mechanisms controlling developmental switches. Nat Rev Genet. 11:830–842. [DOI] [PubMed] [Google Scholar]
- Kwantes M, Liebsch D, Verelst W. 2012. How MIKC* MADS-box genes originated and evidence for their conserved function throughout the evolution of vascular plant gametophytes. Mol Biol Evol. 29:293–302. [DOI] [PubMed] [Google Scholar]
- Lewis M. 2005. The lac repressor. C R Biol. 328:521–548. [DOI] [PubMed] [Google Scholar]
- Lupas AN, Gruber M. 2005. The structure of alpha-helical coiled coils. Adv Protein Chem. 70:37–38. [DOI] [PubMed] [Google Scholar]
- Melzer R, Theißen G. 2009. Reconstitution of ‘floral quartets’ in vitro involving class B and class E floral homeotic proteins. Nucleic Acids Res. 37:2723–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer R, Verelst W, Theißen G. 2009. The class E floral homeotic protein SEPALLATA3 is sufficient to loop DNA in ‘floral quartet’-like complexes in vitro. Nucleic Acids Res. 37:144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes MA, Guerra RF, Berns MC, Manzo C, Masiero S, Finzi L, Kater MM, Colombo L. 2013. MADS domain transcription factors mediate short-range DNA looping that is essential for target gene expression in Arabidopsis. Plant Cell. 25:2560–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messenguy F, Dubois E. 2003. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 316:1–21. [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Sakayama H, de Vries J, Buschmann H, Saint-Marcoux D, Ullrich KK, Haas FB, Vanderstraeten L, Becker D, Lang D, et al. . 2018. The chara genome: secondary complexity and implications for plant terrestrialization. Cell 174:448–464.e24. [DOI] [PubMed] [Google Scholar]
- Oehler S, Eismann ER, Kramer H, Müller-Hill B. 1990. The three operators of the lac operon cooperate in repression. EMBO J. 9:973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuh JO, Qiu HY. 2021. The serum response factor/cofactors interactions and their implications in diseases. FEBS J. 288:3120–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puranik S, Acajjaoui S, Conn S, Costa L, Conn V, Vial A, Marcellin R, Melzer R, Brown E, Hart D, et al. . 2014. Structural basis for the oligomerization of the MADS domain transcription factor SEPALLATA3 in Arabidopsis. Plant Cell. 26:3603–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruelens P, Zhang ZC, van Mourik H, Maere S, Kaufmann K, Geuten K. 2017. The origin of floral organ identity quartets. Plant Cell. 29:229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rümpler F, Theißen G, Melzer R. 2018. A conserved leucine zipper-like motif accounts for strong tetramerization capabilities of SEPALLATA-like MADS-domain transcription factors. J Exp Bot. 69:1943–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senear DF, Brenowitz M. 1991. Determination of binding constants for cooperative site-specific protein-DNA interactions using the gel mobility-shift assay. J Biol Chem. 266:13661–13671. [PubMed] [Google Scholar]
- Shore P, Sharrocks AD. 1995. The mads-box family of transcription factors. Eur J Biochem. 229:1–13. [DOI] [PubMed] [Google Scholar]
- Smaczniak C, Immink RG, Angenent GC, Kaufmann K. 2012. Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139:3081–3098. [DOI] [PubMed] [Google Scholar]
- Smaczniak C, Immink RG, Muino JM, Blanvillain R, Busscher M, Busscher-Lange J, Dinh QD, Liu S, Westphal AH, Boeren S, et al. . 2012. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc Natl Acad Sci U S A. 109:1560–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y, Hasebe M, Sekimoto H, Nishiyama T, Kitani M, Henschel K, Münster T, Theißen G, Nozaki H, Ito M. 2005. Characterization of MADS-box genes in charophycean green algae and its implication for the evolution of MADS-box genes. Proc Natl Acad Sci U S A. 102:2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel G, Nayar S. 2018. A survey of MIKC type MADS-box genes in non-seed plants: algae, bryophytes, lycophytes and ferns. Front Plant Sci. 9:510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theißen G. 2001. Development of floral organ identity: stories from the MADS house. Curr Opin Plant Biol. 4:75–85. [DOI] [PubMed] [Google Scholar]
- Theißen G, Gramzow L. 2016. Structure and evolution of plant MADS-domain transcription factors. In: Gonzalez DH, editor. Plant transcription factors: evolutionary, structural and functional aspects. Philadelphia (PA): Elsevier. p. 127–138. [Google Scholar]
- Theißen G, Melzer R. 2007. Molecular mechanisms underlying origin and diversification of the angiosperm flower. Ann Bot. 100:603–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theißen G, Melzer R, Rümpler F. 2016. MADS-domain transcription factors and the floral quartet model of flower development: linking plant development and evolution. Development. 143:3259–3271. [DOI] [PubMed] [Google Scholar]
- Theißen G, Saedler H. 2001. Floral quartets. Nature 409:469–471. [DOI] [PubMed] [Google Scholar]
- Tsong AE, Tuch BB, Li H, Johnson AD. 2006. Evolution of alternative transcriptional circuits with identical logic. Nature 443:415–420. [DOI] [PubMed] [Google Scholar]
- Verelst W, Saedler H, Münster T. 2007. MIKC* MADS-protein complexes bind motifs enriched in the proximal region of late pollen-specific Arabidopsis promoters. Plant Physiol. 143:447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-Q, Melzer R, Theißen G. 2010. Molecular interactions of orthologues of floral homeotic proteins from the gymnosperm Gnetum gnemon provide a clue to the evolutionary origin of ‘floral quartets’. Plant J. 64:177–190. [DOI] [PubMed] [Google Scholar]
- Wernersson R, Pedersen AG. 2003. Revtrans: multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 31:3537–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, Ayyampalayam S, Barker MS, Burleigh JG, Gitzendanner MA, et al. . 2014. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc Natl Acad Sci U S A. 111:E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WW, Huang XT, Cheng JA, Li ZG, de Folter S, Huang ZR, Jiang XQ, Pang HX, Tao SH. 2011. Conservation and evolution in and among SRF- and MEF2-type MADS domains and their binding sites. Mol Biol Evol. 28:501–511. [DOI] [PubMed] [Google Scholar]
- Yang YZ, Jack T. 2004. Defining subdomains of the K domain important for protein-protein interactions of plant MADS proteins. Plant Mol Biol. 55:45–59. [DOI] [PubMed] [Google Scholar]
- Zobell O, Faigl W, Saedler H, Münster T. 2010. MIKC* MADS-box proteins: conserved regulators of the gametophytic generation of land plants. Mol Biol Evol. 27:1201–1211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Alignments used for the exon homology analysis of figure 2 and the exon duplication analysis of figure 3 are available through supplementary Data 1 and 2, Supplementary Material online, respectively.