

Abstract
Purpose of review
To summarize recent literature relating early-life environmental exposures on DNA methylation in the placenta, to identify how variation in placental methylation is regulated in an exposure-specific manner and to encourage additional work in this area.
Recent findings
Multiple studies have evaluated associations between prenatal environmental exposures and placental methylation in both gene-specific and epigenome-wide frameworks. Specific exposures lead to unique variability in methylation, and cross-exposure assessments have uncovered certain genes that demonstrate consistency in differential placental methylation. Exposure studies that assess methylation effects in a trimester-specific approach tend to find larger effects during 1st trimester exposure. Earlier studies have more targeted gene-specific approaches to methylation, while later studies have shifted towards epigenome-wide, array-based approaches. Studies focusing on exposures such as air pollution, maternal smoking, environmental contaminants, and trace metals appear to be more abundant, while studies of socioeconomic adversity and circadian disruption are scarce but demonstrate remarkable effects.
Summary
Understanding the impacts of early-life environmental exposures on placental methylation is critical to establishing the link between the maternal environment, epigenetic variation, and long-term health. Future studies into this field should incorporate repeated measures of exposure throughout pregnancy, in order to determine the critical windows in which placental methylation is most heavily affected. Additionally, the use of methylation-based scores and sequencing technology could provide important insights into epigenetic gestational age and uncovering more genomic regions where methylation is affected. Studies examining the impact of other exposures on methylation, including pesticides, alcohol, and other chemicals are also warranted.
Keywords: environmental exposures, DNA methylation, epigenetics, placenta, developmental origins of health and disease
Introduction
DNA methylation is the best-characterized and most stable epigenetic modification, influencing gene expression through the disruption of transcription factor binding, chromatin structure, and subsequent gene silencing [1–3]. DNA methylation typically involves methylation of the 5th carbon position at a cytosine residue within a CpG dinucleotide (CpG), resulting in 5-methylcytosine (5mC). There are ~28 million CpGs in the genome, most of which are methylated [4]. However, CpGs located in “CpG islands” (regions of high CpG density and commonly found in gene promoters) tend to be unmethylated, as methylation in these regions generally represses transcription of the gene [5]. DNA methylation is primarily measured in this locus/gene-specific way, though other forms of measuring methylation do exist. One such example is building a methylation profile across several unique sites in the genome, through a genome-wide analysis [6]. Global methylation is another form of methylation profiling, and is defined as the total level of 5mC content in a sample relative to total cytosine content [6]. Though these types of methylation are generally studied in nuclear DNA, other types of DNA have proven to be useful tools for understanding methylation. Altered methylation of mitochondrial DNA (mtDNA) which is located outside the nucleus and plays an important role in cell life and death [7], has been implicated in a number of human diseases including cancer [8], and cardiovascular disease (CVD) [9].
DNA methylation can be influenced by both genetic and environmental factors, and recent studies have provided concrete evidence of a link between methylation and certain environmental exposures, including tobacco smoke [10–12], air pollution [13–15], toxic metals [16–18], and chemical compounds [19–21]. Exposure to these contaminants, particularly early in life, is associated with an increased later-life disease risk [22–24] (Figure 1). Much of this literature linking the maternal environment, epigenetic variation, and developmental programming has focused on DNA methylation in cord blood, due to its availability in birth cohort studies and its utility as a surrogate marker of target offspring tissue or as a target itself of environmental impacts on the developing immune system [25, 26].
Figure 1.
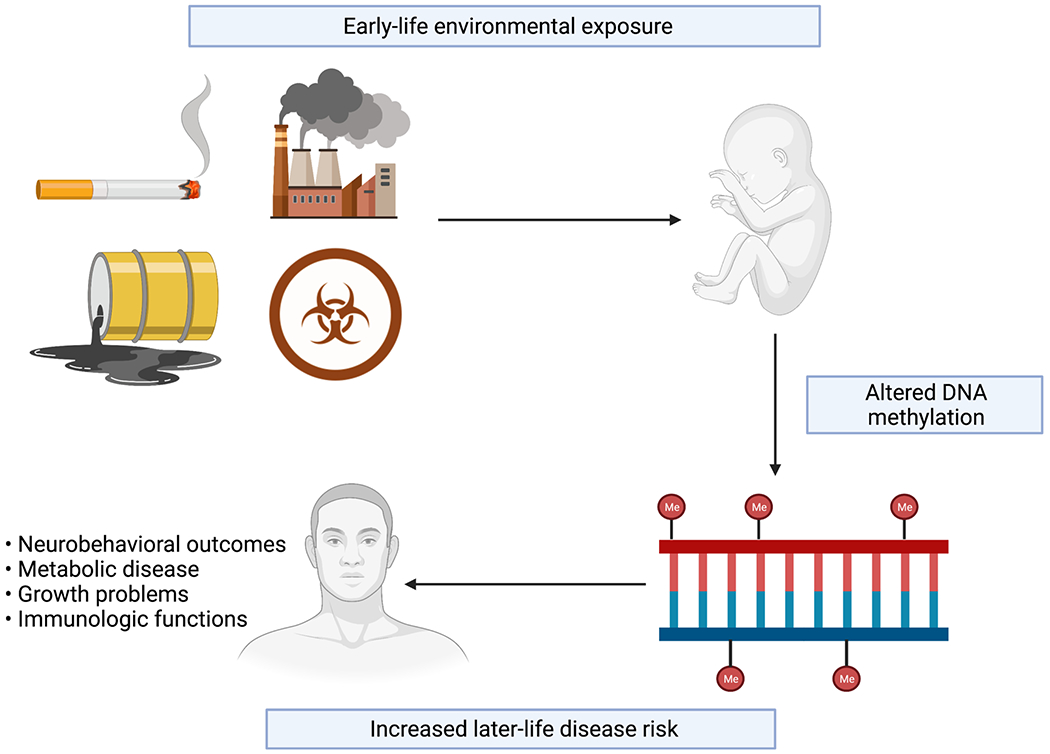
Early-life environmental exposures and their impact on genome-wide DNA methylation and disease risk.
The placenta, though, may also be a highly relevant target tissue for the environment during in utero development. This ephemeral organ acts as a regulator of the intrauterine environment and physiologic interface between the mother and developing fetus. It is the first organ to develop and plays an important role throughout pregnancy, coordinating nutrient, gas, and waste exchange, as well as acting as an immunologic and endocrine organ [27–29]. An overview of placental structure and its role in function is described in Figure 2. Placental development is essential for proper fetal development, and can be influenced by the maternal environment [30, 31]. Exogenous exposures including environmental contaminants, pharmaceuticals, and psychosocial factors, as well as endogenous characteristics including maternal metabolic state contribute to that maternal environment and subsequently influence fetal development and potentially lifelong offspring health [32–34] through interactions with or effects on the placenta [35, 36]. Those environmental impacts to the placenta can be reflected in its molecular landscape, including changes to gene and protein expression and the upstream mechanisms which control these cellular products, in particularly DNA methylation, which exhibits a unique profile in the placental genome [37].
Figure 2.
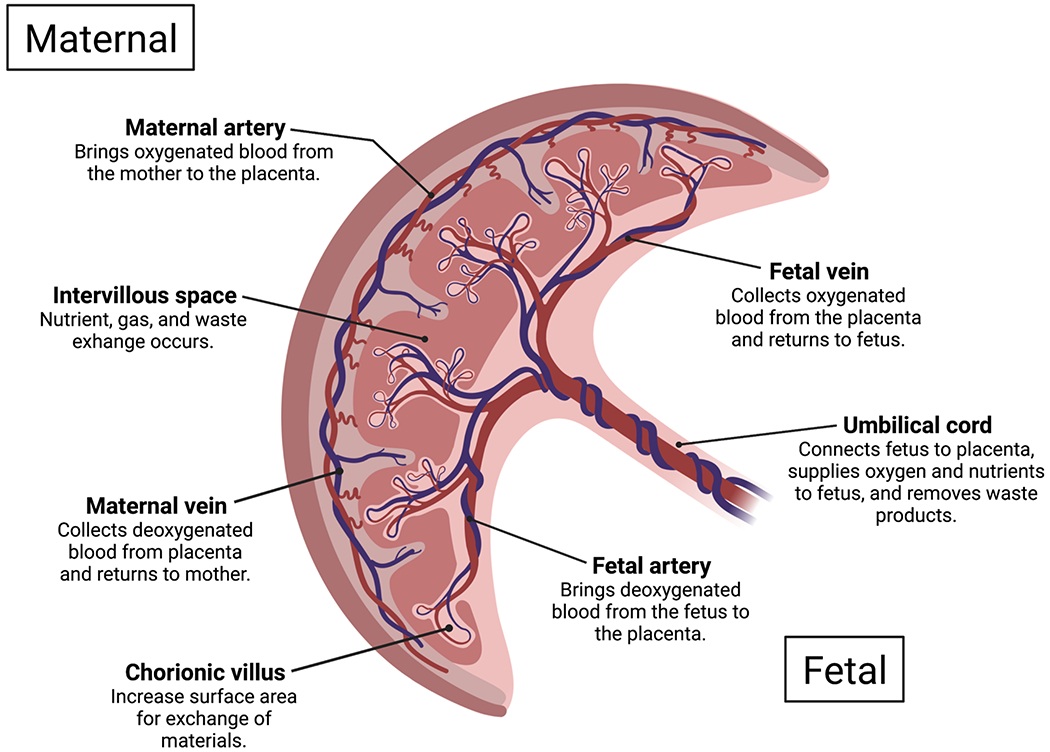
Human placental structure and function.
In this review, we summarize the existing literature relating early-life environmental exposures on DNA methylation in the placenta, to identify how variation in placental methylation is regulated in an exposure-specific manner and to encourage additional work in this area.
Methods
In December 2021, we searched PubMed for literature concerning placental DNA methylation and early-life environmental exposures. We utilized general keywords “placenta”, “methylation”, “environment”, “exposures”, as well as specific exposure keywords “smoking”, “pollution”, “metals”, “chemicals”, “circadian”, and “socioeconomic.” From our queries, we included studies published in English within the last 5 years that represented new research on placental methylation in relation to in-utero environmental exposures and were either open access or accessible through a typical University library. We excluded studies that were systematic review articles, non-human studies, and studies where environmental exposures’ impacts were assessed in-vitro. If multiple papers assessed the same environmental exposure and methylation type in nested, overlapping sample sources, only the paper with the largest sample size was retained. Papers that met inclusion criteria but were later found to have a scope not matching inclusion criteria were also excluded. See Figure 3 for flow chart of literature search strategy. For papers meeting inclusion criteria, relevant data, including paper title, authors, year and place of publication, cohort size and source, exposure and specific type of DNA methylation assessed, research question, methods, and key findings, was extracted and maintained in a local database.
Figure 3.
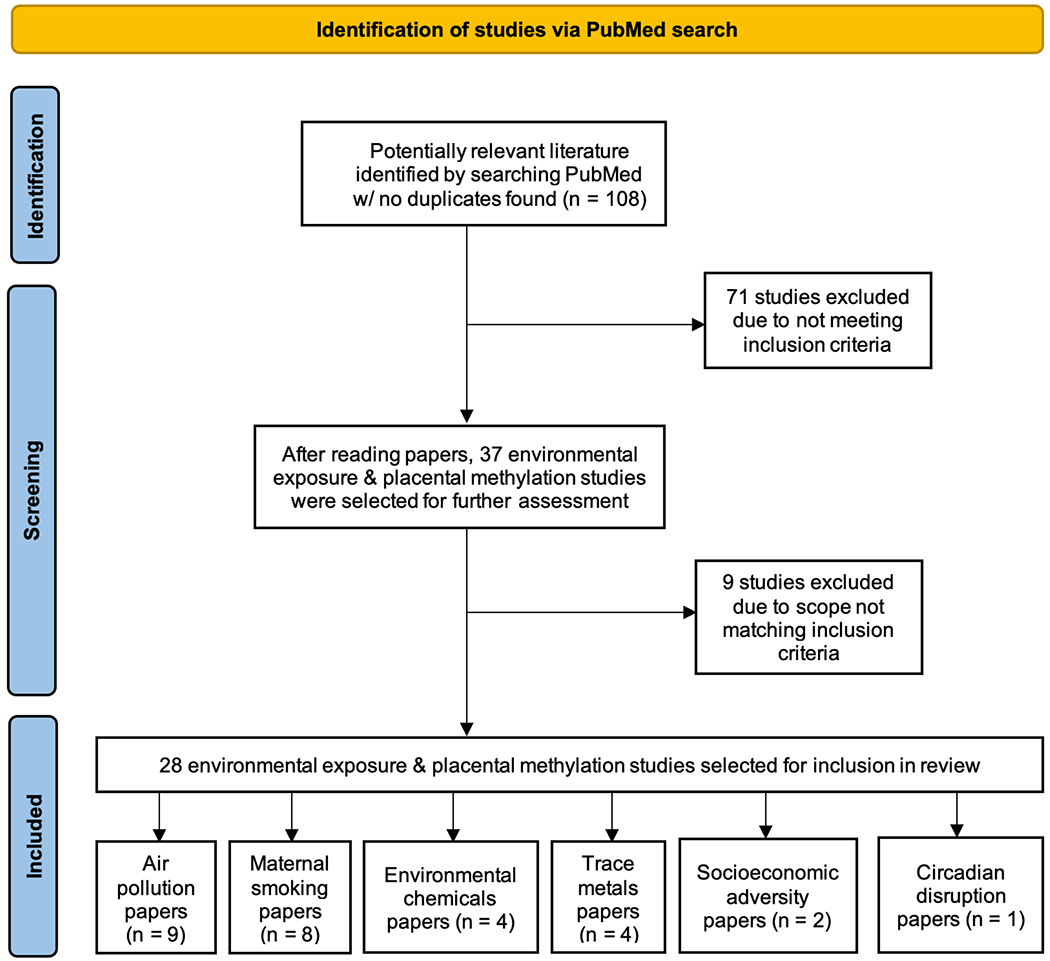
Flow chart illustrating paper selection process for conducting literature search
Results
A total of 108 studies were initially retrieved for screening and assessed for possible inclusion. After exclusion of non-pertinent articles, 28 studies met final inclusion criteria for assessing placental DNA methylation in relation to an environmental exposure; 9 (32.1%) studied air pollution, 8 (28.6%) studied maternal smoking, 4 (14.3%) studied environmental chemicals, 4 (14.3%) studied trace metals, 2 (7.1%) studied socioeconomic status (SES), and 1 (3.6%) studied circadian disruption (Fig. 3). Results from all studies are summarized in Table 1.
Table 1.
Summary of recent publications focusing on various environmental exposures in relation to placental DNA methylation.
| 1st author, year | Study cohort & size | Exposure | Assay | Direction of effect on DNAme |
|---|---|---|---|---|
| Air Pollution Studies | ||||
| Janssen et. al, 2013 [32] | ENVIRONAGE (n = 240) | PM2.5 | Bisulfite sequencing | ↓ global DNAme (1st trimester, whole pregnancy, implantation) |
| Saenen et. al, 2017 [34] | ENVIRONAGE (n = 361) | PM2.5 | Bisulfite sequencing | ↓ LEP (2nd trimester) |
| Nawrot et. al, 2018 [35] | ENVIRONAGE (n = 407) | PM2.5 | Bisulfite sequencing | ↑ NPAS2, CRY1, PER2, PER3 (3rd trimester) ↓ PER1, CLOCK (3rd trimester) |
| Neven et. al, 2018 [36] | ENVIRONAGE (n = 463) | PM2.5, black carbon, NO2 | Bisulfite sequencing | ↑ APEX1, OGG1, ERCC4, p53 (PM2.5) ↓ DAPK1 (PM2.5) ↑ APEX1, ERCC4 (black carbon) |
| Zhao et. al, 2021 [37] | Shanghai MCPC (n = 287) | PM2.5 | Infinium 450K | ↑ IGF2, ↓ BID (2nd & 3rd trimester, whole pregnancy) ↑ FOX3N (2nd trimester) |
| Maghbooli et. al, 2018 [8] | • Prenatal care clinics in Tehran, Iran (n = 92) ○ Polluted = 48 ○ Non-polluted = 44 |
PM2.5, PM10 | HPLC | ↑ global DNAme (PM2.5 & PM10, 1st trimester) ↑ global DNAme (PM2.5, 3rd trimester, polluted) |
| Yang et. al, 2020 [41] | COCOA (n = 1,180) Grouped analysis: ○ n = 6 for each group (8 groups) ■ High/low exposure groups, high/low CB VD within those, AD within those |
PM2.5 | Infinium 450K | ↓ AHRR, DPP10, HLA-DRB1 (high PM2.5, low CB VD, and AD group) |
| Abraham et. al, 2018 [43] | EDEN (n = 668) | NO2, PM10 | Infinium 450K | ↓ ADORA2B (NO2, 1st & 2nd trimester, whole pregnancy) ↑ PXT1, KCTD20 (NO2, 2nd trimester & whole pregnancy) ↓ TUBGCP, ↑ TGM6, ADCK5 (PM10, month before birth) ↓ LINE-1, Alu (PM10, 1st trimester) |
| Ladd-Acosta et. al, 2019 [48] | EARLI (n = 133) | NO2, O3 | Infinium 450K | ↓ All probes, CpG islands (NO2) ↑ shores, islands, ↓ shelves (O3) ↓ ZNF442, PTPRH, SLC25A44, F11R (NO2), STK38 (O3) |
| Maternal Smoking Studies | ||||
| Janssen et. al, 2017 [52] | • ENVIRONAGE (n = 382) ○ Smokers (n = 62) ○ Past-smokers (n = 65) ○ Non-smokers (n = 255) |
Cigarette smoke | Bisulfite sequencing | ↑ MT-RNR1 ↓ CYP1A1 |
| Fa et. al, 2017 [54] | • Women seeking legal abortions in Denmark (n = 39) ○ Smoking-exposed (n = 17) |
Cigarette smoke | Bisulfite sequencing | ↑ AHRR |
| Vos et. al, 2021 [55] | • ENVIRONAGE (n = 60) ○ Smokers (n = 20) |
Cigarette smoke | Bisulfite sequencing | ↑ D-loop |
| Cardenas et. al, 2019 [58] | • Gen3G (n = 441) ○ Smokers (n = 38) ○ Non-smokers (n = 403) |
Cigarette smoke | Infinium 450K | ↑ MDS2, PBX1, CYP1A2, VPBRP, CD28, CDK6 ↓ WBP1L |
| Shorey-Kendrick et. al, 2021 [59] | • VCSIP (n = 96) ○ Smokers (n = 72) ■ VCS (n = 37) ■ Placeb ○ (n = 35) ○ Non-smokers (n = 24) |
Cigarette smoke | Infinium 450K | ↑ DIP2C, APOH/PRKCA (VCS) |
| Van Otterdijk et. al, 2017 [60] | • HEBC (n = 120) ○ Smoked during entire pregnancy (n = 27) ○ Quit smoking while pregnant (n = 32) ○ Non-smokers (n = 61) |
Cigarette smoke | Bisulfite sequencing | ↓ AHRR, CYP1A1 (smoked throughout pregnancy) ↓ GFI1 (quit while pregnant) |
| Rousseaux et. al, 2020 [61] | • EDEN (n = 568) ○ Current smokers (n = 117) ○ Former smokers (n = 70) ○ Non-smokers (n = 381) |
Cigarette smoke | Infinium 450K | ↓ LINE-1, ↑ DMRs (current smokers) |
| Everson et. al, 2021 [63] | • PACE (n = 1,700) ○ Any MSDP (n = 344) ○ Sustained MSDP (n = 163) ○ No MSDP (n = 1,193) |
Cigarette smoke | Infinium 450K | ↓ CpG DNAme (Any/sustained MSDP) |
| Chemical Studies | ||||
| Song et. al, 2021 [64] | • S-MBCS (n = 146) ○ Low BPA (n = 108) ○ High BPA (n = 38) |
BPA | Infinium 450K | ↑ CpG DNAme, HLA-DRB6 (high BPA) |
| Jedynak et. al, 2021 [67] | EDEN (n = 202) | Phenols | Infinium 450K | ↑ DMPs, DMRs |
| Kim et. al, 2018 [70] | CHECK (n = 109) | POPs | Bisulfite sequencing | ↓ LINE-1, H19, ↑ IGF2 |
| Zhao et. al, 2019 [73] | • Wenzhou Birth Cohort (n = 249) ○ FGR cases (n = 124) ○ Controls (n = 125) |
PBDEs | Bisulfite sequencing | ↓ IGF2, ↑ HSD11B2 |
| Trace M etal Studies | ||||
| Appleton et. al, 2017 [77] | RICHS (n = 222) | As, Cd, Pb, Mn, Hg, Zn | Bisulfite sequencing | ↑ NR3C1 (higher As, Cd, Pb, Mn, Hg, lower Zn) |
| Everson et. al, 2018 [82] | • RICHS (n = 141) • NHBCS (n = 343) |
Cd | Infinium 450K | ↑ CpG DNAme |
| Tian et. al, 2020 [83] | • RICHS (n = 141) • NHBCS (n = 343) |
Se | Infinium 450K | ↑ GFI1, CAPN9, SKIDA1, ↓ ZNF496, TBC1D5 |
| Kennedy et. al, 2020 [84] | • RICHS (n = 141) • NHBCS (n = 306) |
Cu | Infinium 450K | ↓ Enhancers, active TSS |
| Socioeconomic Adversity Studies | ||||
| Santos et. al, 2019 [86] | ELGAN (n = 426) | SES | Infinium 450K | ↑ ↓ DMPs |
| Appleton et. al, 2013 [87] | RICHS (n = 444) | SES | Bisulfite sequencing | ↓ HSD11B2 |
| CD Studies | ||||
| Clarkson-Townsend et. al, 2019 [88] | • RICHS (n = 237) ○ Night shift (n = 53) ○ No night shift (n = 184) |
CD | Infinium 450K | ↓ DMPs, NAV1, MXRA8, GABRG1, PRDM16, WNT5A, FOXG1 (night shift) ↑ TDO2, ADAMTSL3, DLX2, SERPINA1 |
Abbreviations: ENVIRONAGE = Environmental Influence on Early Aging; PM2.5 = particulate matter ≤ 2.5 μm; DNAme = DNA methylation; NO2 = nitrogen dioxide; Shanghai MCPC = Shanghai Maternal-Child Pairs Cohort; PM10 = particulate matter ≤ 10 μm; HPLC = high performance liquid chromatography; COCOA = Cohort for Childhood Origin of Asthma and Allergic Diseases; CB = cord blood; VD = vitamin D; AD = atopic dermatitis; EARLI = Early Autism Risk Longitudinal Investigation; O3 = ozone; CpG shores = CpG probe ± 2 kb from CpG island; CpG shelves = CpG probe ± 2-4 kb from island; D-loop = displacement loop; Gen3G = Genetics of Glucose regulation in Gestation and Growth; VCSIP = Vitamin C to decrease the effects of smoking in pregnancy on infant lung function; VCS = vitamin C supplement; HEBC = Harvard Epigenetic Birth Cohort; DMRs = differentially methylated regions; PACE = Pregnancy and Childhood Epigenetics; MSDP = maternal smoking during pregnancy; S-MBCS = Shanghai-Minhang Birth Cohort Study; BPA = bisphenol A; DMPs = differentially methylated probes; CHECK = Children’s Health and Environmental Chemicals in Korea; POPs = persistent organic pollutants; FGR = fetal growth retardation; PBDEs = polybrominated diphenyl ethers; RICHS = Rhode Island Child Health Study; As = arsenic; Cd = cadmium; Pb = lead; Mn = manganese; Hg = mercury; Zn = zinc; NHBCS = New Hampshire Birth Cohort Study; Se = selenium; Cu = copper; TSS = transcription start site; ELGAN = Extremely Low Gestational Age Newborns; SES = socioeconomic status; CD = circadian disruption
Air pollution
From our review, exposure to air pollution was the most highly-studied environmental exposure in relation to placental DNA methylation. One study assessed global placental DNA methylation based on PM2.5 exposure during different during different time windows of pregnancy [38], observing an overall decrease in methylation as exposure to PM2.5 increased during whole pregnancy, particularly with exposure during the 1st trimester and specifically the early first trimester when implantation occurs. A separate study assessed methylation of 7 CpGs in the promoter of leptin, a hormone that plays a functional role in embryo implantation, intrauterine development, and fetal growth during pregnancy [39]. The authors observed decreased methylation across all 7 sites among mothers with increased PM2.5 exposure in the 2nd trimester [40].
A 2018 study assessed placental methylation among genes within the circadian clock pathway in response to PM2.5 exposure [41]. The authors observed that increased PM2.5 exposure during 3rd trimester led to increased DNA methylation in the promoters of NPAS2, CRY1, PER2, and PER3, while an inverse association was seen between 1st trimester exposure and CLOCK methylation. Another study examined exposure to PM2.5, black carbon, and NO2 on promoter methylation of tumor suppressor and DNA repair genes, including genes on the nucleotide excision repair (NER) and base excision repair (BER) pathways [42]. Increased PM2.5 exposure throughout pregnancy was found to be positively associated with promoter methylation of repair genes APEX1, OGG1, and ERCC4, as well as tumor-suppressor gene p53. Increased black carbon exposure was also positively associated with methylation in APEX and ERCC4. Interestingly, pollution exposure during the 1st and 2nd trimesters of pregnancy mainly affected methylation of tumor suppressor genes, whereas later pregnancy exposure affected genes of the BER pathway. These gene and trimester-specific trends were observed in additional studies, including one that studied PM2.5 exposure on promoter methylation among 5 candidate placental genes and its role as a mediator of the exposure’s impact on fetal growth [43]. The authors noted that in the case of IGF2, a growth hormone that plays a crucial role in fetal development [44], increased PM2.5 exposure during 2nd or 3rd trimester and entire pregnancy was associated with decreases in promoter methylation, while increases in exposure across the same windows resulted in increased promoter methylation of BID, an apoptosis regulator that has been shown to be susceptible to oxidative stress and immune response induced by environmental risk factors [45, 46]. A mediation analysis also showed that PM2.5 exposure might influence fetal growth through BID methylation.
Several studies used qualitative measures of air pollution exposure, grouping their participants based on exposure levels. This included a 2018 study done in Tehran, Iran, assessing impact of PM2.5 and PM10 exposure on global placental DNA methylation [14]. Positive correlations were observed between PM2.5/PM10 exposure in 1st trimester and methylation of all participants in “polluted” and “non polluted” groups. Stronger correlations were also seen in the “polluted” group compared to “non-polluted” group. Another study assessed prenatal PM2.5 exposure on placental methylation and how these changes modulate vitamin D deficiency and atopic dermatitis in offspring [47]. They observed significant hypomethylation in the promoter of the AHRR gene, as well as decreased expression of AHRR targets (AHR), in mothers with high PM2.5 exposure, low cord blood vitamin D, and offspring atopic dermatitis compared to other groups. AHR is a transcription factor that responds to chemicals regulating expression of genes with toxic or protective effects [48]. Placental hypomethylation of AHRR could suppress expression of AHR, thereby decreasing AHR signaling and disrupting immune response, which could increase risk of atopic dermatitis in offspring.
Several studies also investigated genomic regions that exhibit differential methylation patterns in response to varying levels of pollution exposure. One study assessed NO2 and PM10 exposure on global methylation in Alu and LINE-1 repetitive elements, as well as in specific CpG-sites [49]. The authors identified 27 differentially methylated regions (DMRs) associated with air pollutants, including 4 located in CD81, DAXX, NOTCH3, and P2RX4 genes, all of which have been implicated in preeclampsia phenotypes [50–53]. A similar trend was recently reported in a study looking at NO2 and O3 exposure impact on methylation across genomic locations, including CpG islands, shores, shelves, and open seas [54]. The authors noted location-specific variability in methylation, but more importantly they identified 5 hypomethylated DMRs in placenta mapped to genes ZNF442, PTPRH, SLC25A44, F11R, and STK38. Several of these genes are involved in immune and inflammatory processes, and these processes have been implicated as biological targets of air pollutant exposure [55, 56].
Studies of air pollution are often unique in that they can consider various time windows of exposure in pregnancy, something that is generally not seen in studies requiring biomarkers of exposure, due to the burden and cost of sampling multiple times throughout pregnancy [57]. Several genes exhibiting variable methylation in response to air pollution exposure have been previously implicated in environmental exposure-induced changes to immune response and oxidative stress [43, 47], making them good candidates for future studies on how changes in their methylation levels may impact these processes. Finally, a few studies assessed global methylation in the form of LINE-1 and Alu non-coding, repetitive elements, but such studies of global patterns of methylation are becoming more rare, replaced with studies of gene-specific and genome-wide methylation, made possible with array-based technologies and sequencing.
Maternal smoking
There is also an abundance of studies focusing on maternal smoking either prior to or during pregnancy, and placental methylation. In one study [58], investigators aimed to assess the effect of prenatal exposure to smoking on methylation of mtDNA and in the promoter of the CYP1A1 gene, which is involved in detoxification and may be activated by constituents of tobacco smoke [59]. The authors noted that direction of effect varies based on the type of DNA (genomic vs. mtDNA); current smokers had neonates with lower CpG-specific methylation at CYP1A1 compared to non-smokers, but higher mtDNA methylation at specific loci (specifically the MT-RNR1 gene). Another study that also assessed CYP1A1 promoter methylation found no association with prenatal smoking exposure, though it is worth nothing that this study only sampled placenta from pre-term births [60]. This may denote that early-pregnancy smoking is not sufficient to elicit CYP1A1 methylation changes. Likewise, a recent study [61] that also assessed placental mtDNA methylation found significantly higher D-loop methylation in smokers compared to non-smokers, but no difference in methylation of LDLR2 between groups. The D-loop and LDLR2 both lie on the displacement loop of mtDNA, with D-loop on the heavy chain and LDLR2 on the light chain [62, 63]. These findings suggest that prenatal exposure to smoking may have a differential impact on the displacement loop of placental mtDNA.
Recently, a large epigenome-wide association study (EWAS) aimed to understand if placental methylation would mediate the established association of prenatal smoking and lower infant birthweight [64]. Authors found 153 DMRs between smokers and non-smokers, with increases and decreases based on exposure. Interestingly, methylation of 7 CpGs had a mediating effect of lower birthweight in smokers. A separate study looking at impacts of Vitamin C supplementation on smoking-associated changes in placental methylation found differential methylation between those groups and suggested Vitamin C could be a potential intervention [65].
One study investigated maternal smoking effects on tissue-specific impacts in the placenta [66]. Authors observed significant decreases in methylation on the fetal side of the placenta among those who smoked throughout pregnancy, and, to a lesser extent, quit during pregnancy compared to non-smokers. Methylation levels on maternal sides of the placenta displayed less significant changes between groups, suggesting that smoking-induced alterations reflect smoking through pregnancy rather than long-term smoking history, under the assumption that maternal tissue would already have the smoking associated differences. along the lines of understanding the “biological memory” of smoking on methylation [67], a recent EWAS found that ~ 75% of DMRs showed “reversible” alterations of DNA methylation present in placentas of current smokers, and 26 of these DMRs were also present in placentas of former smokers (which were not exposed directly to cigarette smoke), suggesting an “epigenetic memory” of placentas exposed to smoking prior to pregnancy. These DMRs also contained “enhancer-like” epigenetic marks enriched for H3K4me1 and H3K27ac, suggesting these placenta enhancer regions are particularly sensitive to tobacco smoke [68].
A recent EWAS performed by the Pregnancy and Childhood Epigenetics (PACE) consortium [69] represents the largest study of smoking and placental methylation, and serves as a complement to their prior work linking smoking and cord blood methylation [11]. This analysis identified over 400 differentially methylated CpG sites, with very few overlapping with those identified in cord blood, and many exhibiting effect estimates substantially larger than those observed in blood. Additionally, many of these CpGs were associated with smoking-related birth outcomes, including preterm birth and small birthweight, lending support to the critical role that the placenta plays in mediating the impact of adverse environmental exposures on newborn health.
Environmental chemicals
Environmental chemicals including persistent organic pollutants (POPs), bisphenol A (BPA), and polybrominated diphenyl ethers (PBDEs), also emerged as widely-studied prenatal exposure related to placental DNA methylation. Effects on placental methylation varied in both methylation-specific and chemical-specific contexts. A recent study conducted in the Shanghai-Minhang Birth Cohort Study (S-MBCS) assessed prenatal BPA exposure on CpG-specific methylation in the placenta [70]. Authors noted a trend towards hypermethylation of CpGs in the high BPA-exposed group. This result aligns with a previous study in mouse placentas that showed increased BPA exposure led to increased DNA methylation (and subsequently reduced expression) of WNT-2, a gene involved in cell proliferation and differentiation during embryogenesis [71, 72]. A similar association was seen in an EWAS evaluating prenatal exposure to 9 synthetic phenols on methylation [73]. Researchers identified 596 phenol-associated CpG sites, with > 90% of sites showing a positive association between urinary phenol concentration and methylation. A similar association was seen in 97% of DMRs (n = 180), with many of these DMRs related to exposure to triclosan, which has been shown in mice to have adverse effects on placental development and nutrient transport [74, 75]. Triclosan-associated DMRs were also shown to overlap with imprinted genes, suggesting triclosan could impact these important drivers of fetal development.
Examining imprinted placental genes was further investigated in a couple of studies. The first [76] was a 2018 study looking at exposure to POPs on IGF2 and H19, expressed from the paternal and maternal allele, respectively, on chromosome 11. IGF2 lies upstream of H19, and when it is down-regulated, expression of H19 increases. Decreased IGF2 levels have been shown to impair fetal growth [77]. Researchers observed significant hypermethylation of CpGs in IGF2 and hypomethylation in H19 in response to higher serum POP concentrations. Increased methylation in IGF2 may lead to decreased expression, and since IGF2 is known to be a major regulator of placental and fetal growth [78], this could severely impair fetal growth. The second study assessed methylation of IGF2 and HSD11B2, a non-imprinted gene, in response to in-utero exposures to PBDEs among a cohort of children with fetal growth restriction (FGR) [79]. In contrast to the 2018 study, authors observed hypomethylation at IGF2-associated CpGs in response to increased cord blood concentrations of BDE-17-190, a PBDE congener. However, they noted a positive association between that congener and methylation of HSD11B2. HSD11B2 converts maternal cortisol to cortisone [80], and hypermethylation of its promoter has been shown to reduce placental expression [81], leading to increased cortisol being able to enter fetal circulation. High levels can affect fetal growth [82], which may explain the prevalence of FGR in these infants.
Overall, the reviewed chemical-methylation association studies showed variability in methylation in a chemical-and-gene specific framework. Specifically, BPA and phenol exposures appeared to show a more positive association with placental methylation, while POP and PBDE exposures tend to show more variability, with opposite effect directions based on specific genes. One trend to note was that we did not find any recent placental methylation studies looking at exposure to specific pesticides, although there are a number of such studies greater than five years ago.
Trace metals
Our group has been at the forefront of understanding the effects of prenatal trace metal exposures on placental methylation with a number of studies conducted by our group within the Rhode Island Child Health Study (RICHS) or New Hampshire Birth Cohort Study (NHBCS) which have extensive data on placental DNA methylation. One study [83] looked at concentrations of various neurotoxic metals and their impact on methylation of NR3C1, a glucocorticoid receptor that has been linked to neurobehavioral outcomes at birth [84, 85], and is involved in the development of a child’s hypothalamic-pituitary-adrenal (HPA) axis [86]. We found that higher levels of Arsenic (As), Cadmium (Cd), Lead (Pb), Manganese (Mn), and Mercury (Hg), and lower levels of Zinc (Zn) were associated with increased methylation of NR3C1. We noted that this metal-induced hypermethylation may reduce expression of NR3C1, thereby affect the child’s developing HPA axis, which may increase cognitive and neurodevelopmental risk later in life.
Cadmium (Cd) is a unique metal to examine in the placenta, given that it can be sequestered in the placenta and not pass into fetal circulation, and so may elicit its toxicity within the placenta itself [87]. In a study of Cd-associated placental methylation [88], we found that increased Cd concentrations in the placenta was associated with differential methylation at 17 CpGs. Additionally, these Cd-associated CpGs may play a functional role in birth outcomes as methylation at specific CpGs was associated with increased expression of genes such as TNFAIP2 and ACOT7. Higher expression of these genes is associated with lower birthweight in our cohorts. In another study [89], we identified that increasing placental selenium was associated with increased methylation of a CpG in the GFI1 gene, and that methylation of that gene was associated with greater muscle tone in the infants. In an EWAS [90] on placental copper (Cu) and DNA methylation, we identified Cu-associated differentially methylated sites and regions, including the antioxidant GSTP1 gene, and the ZNF197 transcription factor, which has as transcriptional targets a number of Cu metabolism genes [91]. These studies of metals suggest that DNA methylation likely plays a role in the impacts of these metals on various fetal and newborn health outcomes and may be involved in the regulation of those metals in the placenta.
Socioeconomic Adversity
It is also important to consider how demographic, social, and structural factors, which contribute to exposure to adverse environmental agents, also impact placental DNA methylation and may act to modify the effects of the exposures. In one study conducted in the Extremely Low Gestational Age Newborns (ELGAN) [92] cohort, an adversity risk score was developed, based on four indicators: maternal education, marital status, eligibility for public health insurance, and supplemental nutrition assistance. Higher scores indicate greater socioeconomic adversity. Investigators identified 33 CpGs with methylation associated with at least one indicator, with 19 (58%) hypermethylated and 14 (42%) hypomethylated. Additionally, 15 (45%) of these were associated with the summative risk score, with placentas from female infants showing more robust differential methylation than male placentas. Thus, epigenomic effects may be linked to embedding of adversity, potentially effecting long term health outcomes. However, these effects may be attributed to this cohort consisting only of pre-term infants, and therefore these findings may not be generalizable to placentas of term pregnancies.
Our group had previously assessed adversity on placental methylation in RICHS [93]. Similar to the ELGAN study, we developed a cumulative risk score for adversity. We tested the association of this exposure on HSD11B2 methylation, and found that infants whose mothers experienced the greatest levels of adversity during pregnancy had the lowest extent of placental HSD11B2 methylation, particularly among males. Low maternal education, prenatal tobacco use, and higher cumulative risk scores were associated with significant HSD11B2 hypomethylation, with a one-unit increase in risk score correlating with a 2.3% decrease in methylation.
In general, there is an overall lack of literature on the impact of adversity on placental methylation, and additional work in this area may provide important insights on the mechanisms underlying the impacts of adversity on health across generations. Thus, additional studies about adversity and incorporating measures of adversity with other environmental exposures is warranted.
Circadian Disruption (CD)
The last environmental exposure we sought literature on was disruptions in circadian rhythm (CD). Our team was one of the first (and to date, only few) groups to examine CD on placental methylation, which we assessed based on night-shift work in the RICHS cohort [94]. We observed differential methylation at 298 CpG sites in night shift workers, with an average methylation decrease of 1.7% compared to non-night shift workers. We hypothesized that this could be due to increased transcription factor (TF) binding to DNA, leading to chromatin changes causing hypomethylation [95]. Additionally, CLOCK, a core component of the circadian clock, acts as a histone acetyltransferase [96], and thus CD could be impacting the epigenetic activity of CLOCK, affecting chromatin state and accessibility. The 298 probes were also found to be associated with traits such as psoriasis, lupus, type-1 diabetes (T1D), and multiple sclerosis (MS). This finding is in concert with a growing literature of the impacts of CD on various health outcomes [97]. Thus, our study shows that CD is impacting placental epigenetics and may also play a role in the development of diseases, though additional studies on this exposure’s effect on placental methylation are required.
Conclusions and Future Perspectives
In this review, we have outlined the current state of evidence pertaining to early-life environmental exposures and their impact on placental DNA methylation. We have examined 6 well-studied exposure categories but recognize there were a number of other exposures, including pesticides, alcohol, and other chemicals, that we did not include in this review. From what we have summarized, we can identify a few prevailing themes: 1) Specific exposures lead to unique variability in methylation, though cross-exposure assessment shows certain genes demonstrating consistency in differential methylation across exposures; 2) Exposure studies that have looked at trimester-specific exposures’ impact on methylation patterns tend to find effects that are most striking during the 1st trimester; 3) Earlier studies have more targeted gene-specific approaches to methylation or assessed repetitive elements such as LINE-1 and Alu, while later studies are epigenome-wide, array-based.
One challenge in the study of DNA methylation in the placenta has been limitations in the ability to control for cellular heterogeneity in genome-wide studies. Until recently, many studies made use of reference-free methods to address this issue given a lack of reference data. This limitation has been recently overcome, though, with the publication of a reference panel and method, through R package planet, to estimate the cellular composition from array-based DNA methylation data [98]. Additionally, since methylation and exposure assessment are often observed coincidentally at birth, it is difficult to elucidate any form of temporality or causation. Incorporating repeated measures of exposure throughout pregnancy could improve this issue and allow for a better understanding of the critical windows during which exposure impacts placental methylation.
As for additional directions of placental epigenetic research, the use of DNA methylation-based scores is likely to become more prevalent as various risk scores are developed. For example, a placental epigenetic clock has been developed for estimating epigenetic gestational age from placental methylation levels [99]. Like other epigenetic clocks, the deviation between actual gestational age and the estimated age can be used as an outcome and future studies that aim to assess epigenetic gestational age could consider this approach. Also, as sequencing technology continues to improve, it is likely there will be more studies utilizing such an approach in the placenta, and this could provide important new insights to genomic regions that thus far are being missed using array-based and targeted approaches. Finally, as we begin to uncover specific genes that have identified epigenetic alterations related to exposures, there will be opportunities for the development of more robust biomarkers, leading to a better understanding of how environmental exposures work.
The placenta clearly plays a critical role in fetal development and newborn health, and is increasingly being recognized as a critical organ in the developmental origins of long-term health. As cohorts and studies with placental data mature, there will be incredible opportunities to look empirically at these relationships and we encourage ongoing efforts to establish the links between the developmental environment, the placenta, and long-term health.
Funding
This work was supported by the National Institutes of Health (NIH-NIGMS T32GM008490, NIH-NIEHS R24ES028507, NIH-NIEHS R01ES025145, NIH-NIEHS P30 ES019776).
Footnotes
Conflicts of Interest
Michael Mortillo and Carmen Marsit declare they have no conflicts of interest.
Human and animal rights informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Newell-Price J, Clark AJ, King P. DNA methylation and silencing of gene expression. Trends Endocrinol Metab. 2000;11(4):142–8. [DOI] [PubMed] [Google Scholar]
- 2.Hannon E, Knox O, Sugden K, Burrage J, Wong CCY, Belsky DW, et al. Characterizing genetic and environmental influences on variable DNA methylation using monozygotic and dizygotic twins. PLoS Genet. 2018;14(8):e1007544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bird A DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. [DOI] [PubMed] [Google Scholar]
- 4.Robinson WP, Price EM. The human placental methylome. Cold Spring Harb Perspect Med. 2015;5(5):a023044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39(4):457–66. [DOI] [PubMed] [Google Scholar]
- 6.Vryer R, Saffery R. What’s in a name? Context-dependent significance of ‘global’ methylation measures in human health and disease. Clin Epigenetics. 2017;9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stoccoro A, Coppede F. Mitochondrial DNA Methylation and Human Diseases. Int J Mol Sci. 2021;22(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong Z, Pu L, Cui H. Mitoepigenetics and Its Emerging Roles in Cancer. Front Cell Dev Biol. 2020;8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baccarelli AA, Byun HM. Platelet mitochondrial DNA methylation: a potential new marker of cardiovascular disease. Clin Epigenetics. 2015;7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsaprouni LG, Yang TP, Bell J, Dick KJ, Kanoni S, Nisbet J, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics. 2014;9(10):1382–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am J Hum Genet. 2016;98(4):680–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elliott HR, Tillin T, McArdle WL, Ho K, Duggirala A, Frayling TM, et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenetics. 2014;6(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gruzieva O, Xu CJ, Breton CV, Annesi-Maesano I, Anto JM, Auffray C, et al. Epigenome-Wide Meta-Analysis of Methylation in Children Related to Prenatal NO2 Air Pollution Exposure. Environ Health Perspect. 2017;125(1):104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maghbooli Z, Hossein-Nezhad A, Adabi E, Asadollah-Pour E, Sadeghi M, Mohammad-Nabi S, et al. Air pollution during pregnancy and placental adaptation in the levels of global DNA methylation. PLoS One. 2018;13(7):e0199772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mostafavi N, Vlaanderen J, Portengen L, Chadeau-Hyam M, Modig L, Palli D, et al. Associations Between Genome-wide Gene Expression and Ambient Nitrogen Oxides. Epidemiology. 2017;28(3):320–8. [DOI] [PubMed] [Google Scholar]
- 16.Goodrich JM, Basu N, Franzblau A, Dolinoy DC. Mercury biomarkers and DNA methylation among Michigan dental professionals. Environ Mol Mutagen. 2013;54(3):195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tajuddin SM, Amaral AF, Fernandez AF, Rodriguez-Rodero S, Rodriguez RM, Moore LE, et al. Genetic and non-genetic predictors of LINE-1 methylation in leukocyte DNA. Environ Health Perspect. 2013;121(6):650–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tellez-Plaza M, Tang WY, Shang Y, Umans JG, Francesconi KA, Goessler W, et al. Association of global DNA methylation and global DNA hydroxymethylation with metals and other exposures in human blood DNA samples. Environ Health Perspect. 2014;122(9):946–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanna CW, Bloom MS, Robinson WP, Kim D, Parsons PJ, vom Saal FS, et al. DNA methylation changes in whole blood is associated with exposure to the environmental contaminants, mercury, lead, cadmium and bisphenol A, in women undergoing ovarian stimulation for IVF. Hum Reprod. 2012;27(5):1401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh H, Iwasaki M, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, et al. Association between serum organochlorines and global methylation level of leukocyte DNA among Japanese women: a cross-sectional study. Sci Total Environ. 2014;490:603–9. [DOI] [PubMed] [Google Scholar]
- 21.Watkins DJ, Wellenius GA, Butler RA, Bartell SM, Fletcher T, Kelsey KT. Associations between serum perfluoroalkyl acids and LINE-1 DNA methylation. Environ Int. 2014;63:71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark NA, Demers PA, Karr CJ, Koehoorn M, Lencar C, Tamburic L, et al. Effect of early life exposure to air pollution on development of childhood asthma. Environ Health Perspect. 2010;118(2):284–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindoso L, Mondal K, Venkateswaran S, Somineni HK, Ballengee C, Walters TD, et al. The Effect of Early-Life Environmental Exposures on Disease Phenotype and Clinical Course of Crohn’s Disease in Children. Am J Gastroenterol. 2018;113(10):1524–9. [DOI] [PubMed] [Google Scholar]
- 24.Pesce G, Sese L, Calciano L, Travert B, Dessimond B, Maesano CN, et al. Foetal exposure to heavy metals and risk of atopic diseases in early childhood. Pediatr Allergy Immunol. 2021;32(2):242–50. [DOI] [PubMed] [Google Scholar]
- 25.Lin X, Teh AL, Chen L, Lim IY, Tan PF, MacIsaac JL, et al. Choice of surrogate tissue influences neonatal EWAS findings. BMC Med. 2017;15(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lura MP, Gorlanova O, Muller L, Proietti E, Vienneau D, Reppucci D, et al. Response of cord blood cells to environmental, hereditary and perinatal factors: A prospective birth cohort study. PLoS One. 2018;13(7):e0200236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher SJ. The placenta dilemma. Semin Reprod Med. 2000;18(3):321–6. [DOI] [PubMed] [Google Scholar]
- 28.Maltepe E, Fisher SJ. Placenta: the forgotten organ. Annu Rev Cell Dev Biol. 2015;31:523–52. [DOI] [PubMed] [Google Scholar]
- 29.Nugent BM, Bale TL. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol. 2015;39:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barker DJ, Thornburg KL. Placental programming of chronic diseases, cancer and lifespan: a review. Placenta. 2013;34(10):841–5. [DOI] [PubMed] [Google Scholar]
- 31.Kusuyama J, Alves-Wagner AB, Makarewicz NS, Goodyear LJ. Effects of maternal and paternal exercise on offspring metabolism. Nat Metab. 2020;2(9):858–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitro SD, Johnson T, Zota AR. Cumulative Chemical Exposures During Pregnancy and Early Development. Curr Environ Health Rep. 2015;2(4):367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson JF, Hamilton EG, Lam J, Chen H, Woodruff TJ. Differences in cytochrome p450 enzyme expression and activity in fetal and adult tissues. Placenta. 2020;100:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sachdeva P, Patel BG, Patel BK. Drug use in pregnancy; a point to ponder! Indian J Pharm Sci. 2009;71(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myatt L Placental adaptive responses and fetal programming. J Physiol. 2006;572(Pt 1):25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saenen ND, Martens DS, Neven KY, Alfano R, Bove H, Janssen BG, et al. Air pollution-induced placental alterations: an interplay of oxidative stress, epigenetics, and the aging phenotype? Clin Epigenetics. 2019;11(1):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5(8):e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janssen BG, Godderis L, Pieters N, Poels K, Kicinski M, Cuypers A, et al. Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol. 2013;10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sagawa N, Yura S, Itoh H, Kakui K, Takemura M, Nuamah MA, et al. Possible role of placental leptin in pregnancy: a review. Endocrine. 2002;19(1):65–71. [DOI] [PubMed] [Google Scholar]
- 40.Saenen ND, Vrijens K, Janssen BG, Roels HA, Neven KY, Vanden Berghe W, et al. Lower Placental Leptin Promoter Methylation in Association with Fine Particulate Matter Air Pollution during Pregnancy and Placental Nitrosative Stress at Birth in the ENVIRONAGE Cohort. Environ Health Perspect. 2017;125(2):262–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nawrot TS, Saenen ND, Schenk J, Janssen BG, Motta V, Tarantini L, et al. Placental circadian pathway methylation and in utero exposure to fine particle air pollution. Environ Int. 2018;114:231–41. [DOI] [PubMed] [Google Scholar]
- 42.Neven KY, Saenen ND, Tarantini L, Janssen BG, Lefebvre W, Vanpoucke C, et al. Placental promoter methylation of DNA repair genes and prenatal exposure to particulate air pollution: an ENVIRONAGE cohort study. Lancet Planet Health. 2018;2(4):e174–e83. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Y, Wang P, Zhou Y, Xia B, Zhu Q, Ge W, et al. Prenatal fine particulate matter exposure, placental DNA methylation changes, and fetal growth. Environ Int. 2021;147:106313. [DOI] [PubMed] [Google Scholar]
- 44.White V, Jawerbaum A, Mazzucco MB, Gauster M, Desoye G, Hiden U. IGF2 stimulates fetal growth in a sex- and organ-dependent manner. Pediatr Res. 2018;83(1-1):183–9. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-Perez C, Roy SS, Naghdi S, Lin X, Davies E, Hajnoczky G. Bid-induced mitochondrial membrane permeabilization waves propagated by local reactive oxygen species (ROS) signaling. Proc Natl Acad Sci U S A. 2012;109(12):4497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang P, Lindsay J, Owens TW, Mularczyk EJ, Warwood S, Foster F, et al. Phosphorylation of the proapoptotic BH3-only protein bid primes mitochondria for apoptosis during mitotic arrest. Cell Rep. 2014;7(3):661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang SI, Lee SH, Lee SY, Kim HC, Kim HB, Kim JH, et al. Prenatal PM2.5 exposure and vitamin D-associated early persistent atopic dermatitis via placental methylation. Ann Allergy Asthma Immunol. 2020;125(6):665–73 e1. [DOI] [PubMed] [Google Scholar]
- 48.Hao N, Whitelaw ML. The emerging roles of AhR in physiology and immunity. Biochem Pharmacol. 2013;86(5):561–70. [DOI] [PubMed] [Google Scholar]
- 49.Abraham E, Rousseaux S, Agier L, Giorgis-Allemand L, Tost J, Galineau J, et al. Pregnancy exposure to atmospheric pollution and meteorological conditions and placental DNA methylation. Environ Int. 2018;118:334–47. [DOI] [PubMed] [Google Scholar]
- 50.Ding H, Dai Y, Lei Y, Wang Z, Liu D, Li R, et al. Upregulation of CD81 in trophoblasts induces an imbalance of Treg/Th17 cells by promoting IL-6 expression in preeclampsia. Cell Mol Immunol. 2019;16(1):302–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Novakovic B, Evain-Brion D, Murthi P, Fournier T, Saffery R. Variable DAXX gene methylation is a common feature of placental trophoblast differentiation, preeclampsia, and response to hypoxia. FASEB J. 2017;31(6):2380–92. [DOI] [PubMed] [Google Scholar]
- 52.Roberts VH, Webster RP, Brockman DE, Pitzer BA, Myatt L. Post-Translational Modifications of the P2X(4) purinergic receptor subtype in the human placenta are altered in preeclampsia. Placenta. 2007;28(4):270–7. [DOI] [PubMed] [Google Scholar]
- 53.Zhao WX, Huang TT, Jiang M, Feng R, Lin JH. Expression of notch family proteins in placentas from patients with early-onset severe preeclampsia. Reprod Sci. 2014;21(6):716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ladd-Acosta C, Feinberg JI, Brown SC, Lurmann FW, Croen LA, Hertz-Picciotto I, et al. Epigenetic marks of prenatal air pollution exposure found in multiple tissues relevant for child health. Environ Int. 2019;126:363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johannesson S, Andersson EM, Stockfelt L, Barregard L, Sallsten G. Urban air pollution and effects on biomarkers of systemic inflammation and coagulation: a panel study in healthy adults. Inhal Toxicol. 2014;26(2):84–94. [DOI] [PubMed] [Google Scholar]
- 56.Ruckerl R, Hampel R, Breitner S, Cyrys J, Kraus U, Carter J, et al. Associations between ambient air pollution and blood markers of inflammation and coagulation/fibrinolysis in susceptible populations. Environ Int. 2014;70:32–49. [DOI] [PubMed] [Google Scholar]
- 57.Strauss WJ, Ryan L, Morara M, Iroz-Elardo N, Davis M, Cupp M, et al. Improving cost-effectiveness of epidemiological studies via designed missingness strategies. Stat Med. 2010;29(13):1377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Janssen BG, Gyselaers W, Byun HM, Roels HA, Cuypers A, Baccarelli AA, et al. Placental mitochondrial DNA and CYP1A1 gene methylation as molecular signatures for tobacco smoke exposure in pregnant women and the relevance for birth weight. J Transl Med. 2017;15(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ezzeldin N, El-Lebedy D, Darwish A, El-Bastawisy A, Hassan M, Abd El-Aziz S, et al. Genetic polymorphisms of human cytochrome P450 CYP1A1 in an Egyptian population and tobacco-induced lung cancer. Genes Environ. 2017;39:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fa S, Larsen TV, Bilde K, Daugaard TF, Ernst EH, Lykke-Hartmann K, et al. Changes in first trimester fetal CYP1A1 and AHRR DNA methylation and mRNA expression in response to exposure to maternal cigarette smoking. Environ Toxicol Pharmacol. 2018;57:19–27. [DOI] [PubMed] [Google Scholar]
- 61.Vos S, Nawrot TS, Martens DS, Byun HM, Janssen BG. Mitochondrial DNA methylation in placental tissue: a proof of concept study by means of prenatal environmental stressors. Epigenetics. 2021;16(2):121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma H, Singh A, Sharma C, Jain SK, Singh N. Mutations in the mitochondrial DNA D-loop region are frequent in cervical cancer. Cancer Cell Int. 2005;5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Loon NM, Lindholm D, Zelcer N. The E3 ubiquitin ligase inducible degrader of the LDL receptor/myosin light chain interacting protein in health and disease. Curr Opin Lipidol. 2019;30(3):192–7. [DOI] [PubMed] [Google Scholar]
- 64.Cardenas A, Lutz SM, Everson TM, Perron P, Bouchard L, Hivert MF. Mediation by Placental DNA Methylation of the Association of Prenatal Maternal Smoking and Birth Weight. Am J Epidemiol. 2019;188(11):1878–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shorey-Kendrick LE, McEvoy CT, O’Sullivan SM, Milner K, Vuylsteke B, Tepper RS, et al. Impact of vitamin C supplementation on placental DNA methylation changes related to maternal smoking: association with gene expression and respiratory outcomes. Clin Epigenetics. 2021;13(1):177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Otterdijk SD, Binder AM, Michels KB. Locus-specific DNA methylation in the placenta is associated with levels of pro-inflammatory proteins in cord blood and they are both independently affected by maternal smoking during pregnancy. Epigenetics. 2017;12(10):875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rousseaux S, Seyve E, Chuffart F, Bourova-Flin E, Benmerad M, Charles MA, et al. Immediate and durable effects of maternal tobacco consumption alter placental DNA methylation in enhancer and imprinted gene-containing regions. BMC Med. 2020;18(1):306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol. 2007;23(3):297–307. [DOI] [PubMed] [Google Scholar]
- 69.Everson TM, Vives-Usano M, Seyve E, Cardenas A, Lacasana M, Craig JM, et al. Placental DNA methylation signatures of maternal smoking during pregnancy and potential impacts on fetal growth. Nat Commun. 2021;12(1):5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Song X, Wang Z, Zhang Z, Miao M, Liu J, Luan M, et al. Differential methylation of genes in the human placenta associated with bisphenol A exposure. Environ Res. 2021;200:111389. [DOI] [PubMed] [Google Scholar]
- 71.Steinhart Z, Angers S. Wnt signaling in development and tissue homeostasis. Development. 2018;145(11). [DOI] [PubMed] [Google Scholar]
- 72.Ye Y, Tang Y, Xiong Y, Feng L, Li X. Bisphenol A exposure alters placentation and causes preeclampsia-like features in pregnant mice involved in reprogramming of DNA methylation of WNT2. FASEB J. 2019;33(2):2732–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jedynak P, Tost J, Calafat AM, Bourova-Flin E, Busato F, Forhan A, et al. Pregnancy exposure to synthetic phenols and placental DNA methylation - An epigenome-wide association study in male infants from the EDEN cohort. Environ Pollut. 2021;290:118024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cao X, Hua X, Wang X, Chen L. Exposure of pregnant mice to triclosan impairs placental development and nutrient transport. Sci Rep. 2017;7:44803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, Chen X, Feng X, Chang F, Chen M, Xia Y, et al. Triclosan causes spontaneous abortion accompanied by decline of estrogen sulfotransferase activity in humans and mice. Sci Rep. 2015;5:18252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim S, Cho YH, Lee I, Kim W, Won S, Ku JL, et al. Prenatal exposure to persistent organic pollutants and methylation of LINE-1 and imprinted genes in placenta: A CHECK cohort study. Environ Int. 2018;119:398–406. [DOI] [PubMed] [Google Scholar]
- 77.St-Pierre J, Hivert MF, Perron P, Poirier P, Guay SP, Brisson D, et al. IGF2 DNA methylation is a modulator of newborn’s fetal growth and development. Epigenetics. 2012;7(10):1125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417(6892):945–8. [DOI] [PubMed] [Google Scholar]
- 79.Zhao Y, Song Q, Ge W, Jin Y, Chen S, Zhao Y, et al. Associations between in utero exposure to polybrominated diphenyl ethers, pathophysiological state of fetal growth and placental DNA methylation changes. Environ Int. 2019;133(Pt B):105255. [DOI] [PubMed] [Google Scholar]
- 80.Bro-Rasmussen F, Buus O, Trolle D. Ratio cortisone/cortisol in mother and infant at birth. Acta Endocrinol (Copenh). 1962;40:579–83. [DOI] [PubMed] [Google Scholar]
- 81.Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM. Epigenetic regulation of 11 beta-hydroxysteroid dehydrogenase type 2 expression. J Clin Invest. 2004;114(8):1146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao Y, Gong X, Chen L, Li L, Liang Y, Chen S, et al. Site-specific methylation of placental HSD11B2 gene promoter is related to intrauterine growth restriction. Eur J Hum Genet. 2014;22(6):734–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Appleton AA, Jackson BP, Karagas M, Marsit CJ. Prenatal exposure to neurotoxic metals is associated with increased placental glucocorticoid receptor DNA methylation. Epigenetics. 2017;12(8):607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bromer C, Marsit CJ, Armstrong DA, Padbury JF, Lester B. Genetic and epigenetic variation of the glucocorticoid receptor (NR3C1) in placenta and infant neurobehavior. Dev Psychobiol. 2013;55(7):673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Monk C, Feng T, Lee S, Krupska I, Champagne FA, Tycko B. Distress During Pregnancy: Epigenetic Regulation of Placenta Glucocorticoid-Related Genes and Fetal Neurobehavior. Am J Psychiatry. 2016;173(7):705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seckl JR. Glucocorticoids, feto-placental 11 beta-hydroxysteroid dehydrogenase type 2, and the early life origins of adult disease. Steroids. 1997;62(1):89–94. [DOI] [PubMed] [Google Scholar]
- 87.Gundacker C, Hengstschlager M. The role of the placenta in fetal exposure to heavy metals. Wien Med Wochenschr. 2012;162(9–10):201–6. [DOI] [PubMed] [Google Scholar]
- 88.Everson TM, Punshon T, Jackson BP, Hao K, Lambertini L, Chen J, et al. Cadmium-Associated Differential Methylation throughout the Placental Genome: Epigenome-Wide Association Study of Two U.S. Birth Cohorts. Environ Health Perspect. 2018;126(1):017010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tian FY, Everson TM, Lester B, Punshon T, Jackson BP, Hao K, et al. Selenium-associated DNA methylation modifications in placenta and neurobehavioral development of newborns: An epigenome-wide study of two U.S. birth cohorts. Environ Int. 2020;137:105508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kennedy E, Everson TM, Punshon T, Jackson BP, Hao K, Lambertini L, et al. Copper associates with differential methylation in placentae from two US birth cohorts. Epigenetics. 2020;15(3):215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Le A, Shibata NM, French SW, Kim K, Kharbanda KK, Islam MS, et al. Characterization of timed changes in hepatic copper concentrations, methionine metabolism, gene expression, and global DNA methylation in the Jackson toxic milk mouse model of Wilson disease. Int J Mol Sci. 2014;15(5):8004–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Santos HP Jr., Bhattacharya A, Martin EM, Addo K, Psioda M, Smeester L, et al. Epigenome-wide DNA methylation in placentas from preterm infants: association with maternal socioeconomic status. Epigenetics. 2019;14(8):751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Appleton AA, Armstrong DA, Lesseur C, Lee J, Padbury JF, Lester BM, et al. Patterning in placental 11-B hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity. PLoS One. 2013;8(9):e74691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Clarkson-Townsend DA, Everson TM, Deyssenroth MA, Burt AA, Hermetz KE, Hao K, et al. Maternal circadian disruption is associated with variation in placental DNA methylation. PLoS One. 2019;14(4):e0215745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martin EM, Fry RC. A cross-study analysis of prenatal exposures to environmental contaminants and the epigenome: support for stress-responsive transcription factor occupancy as a mediator of gene-specific CpG methylation patterning. Environ Epigenet. 2016;2(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006;125(3):497–508. [DOI] [PubMed] [Google Scholar]
- 97.Lyons AB, Moy L, Moy R, Tung R. Circadian Rhythm and the Skin: A Review of the Literature. J Clin Aesthet Dermatol. 2019;12(9):42–5. [PMC free article] [PubMed] [Google Scholar]
- 98.Yuan V, Price EM, Del Gobbo G, Mostafavi S, Cox B, Binder AM, et al. Accurate ethnicity prediction from placental DNA methylation data. Epigenetics Chromatin. 2019;12(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee Y, Choufani S, Weksberg R, Wilson SL, Yuan V, Burt A, et al. Placental epigenetic clocks: estimating gestational age using placental DNA methylation levels. Aging (Albany NY). 2019;11(12):4238–53. [DOI] [PMC free article] [PubMed] [Google Scholar]