

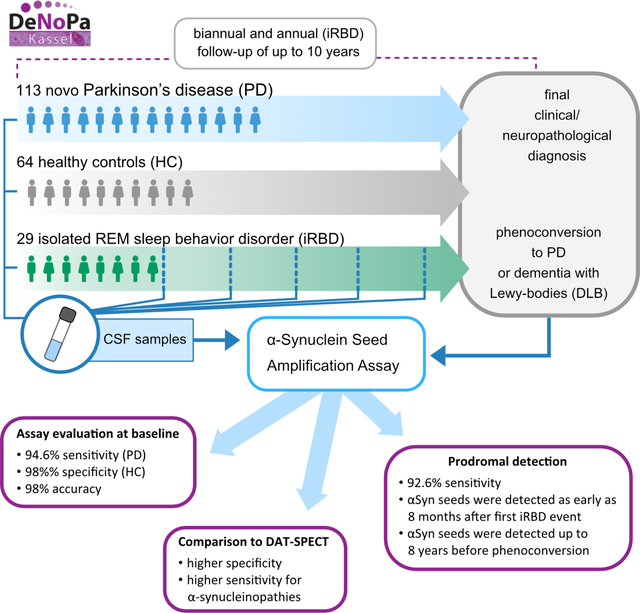
Abstract
Background.
Misfolded α-synuclein aggregates (αSyn-seeds) in cerebrospinal fluid (CSF) are biomarkers for synucleinopathies such as Parkinson’s disease (PD). αSyn-seeds have been detected in prodromal cases with isolated rapid-eye-movement (REM) sleep behavior disorder (iRBD).
Objectives.
To determine the accuracy of the αSyn seed amplification assay (αS-SAA) in a comprehensively characterized cohort with a high proportion of PD and iRBD CSF samples collected at baseline.
Methods.
We used a high-throughput αS-SAA to analyze 233 blinded CSF samples from 206 participants of the DeNovo Parkinson Cohort (DeNoPa) (113 de novo PD, 64 healthy controls, 29 iRBD confirmed by video-polysomnography). Results were compared to the final diagnosis, which was determined after up to 10 years of longitudinal clinical evaluations, including DAT-SPECT at baseline, CSF proteins, MDS-UPDRS, and various cognitive and non-motor scales.
Results.
αS-SAA detected αSyn-seeds in baseline PD-CSF with 98% accuracy. αSyn-seeds were detected in 93% of the iRBD cases. αS-SAA results showed higher agreement with the final than initial diagnosis, as 14 subjects were re-diagnosed as non-α-synuclein aggregation disorder. For synucleinopathies, αS-SAA showed higher concordance with the final diagnosis than DAT-SPECT. Statistically significant correlations were found between assay parameters and disease progression.
Conclusions.
Our results confirm αS-SAA accuracy at first clinical evaluation when a definite diagnosis is most consequential. αS-SAA conditions reported here are highly sensitive, enabling detection of αSyn-seeds in CSF from iRBD just months after first symptoms, suggesting αSyn-seeds are present in the very early prodromal phase of synucleinopathies. Therefore, αSyn-seeds are clear risk markers for synuclein-related disorders, but not for time of phenoconversion.
Keywords: α-Synuclein, seed amplification assay, early Parkinson’s disease, RBD, CSF
Graphical Abstract
Introduction
The clinical diagnosis of Parkinson’s disease (PD) relies on the presence of motor symptoms such as bradykinesia with concomitant rigidity and/or resting tremor. At the time of diagnosis, up to 70% of dopaminergic neurons are degenerated and the disease is already advanced.1 In addition, early symptoms, confounding diseases (mimics), and lack of an accessible objective diagnostic test contribute to a misdiagnosis rate as high as 42%, even for movement disorders specialists.2 Thus, an objective diagnostic test verifying different Parkinsonian syndromes would significantly reduce both the time to reach the final diagnosis and the rate of misdiagnosis.
The degeneration of dopaminergic neurons is estimated to start many years before the onset of motor symptoms.3 Several studies have shown that isolated rapid-eye-movement (REM) sleep behavior disorder (iRBD)4 is prodromal for α-synucleinopathies such as PD, dementia with Lewy bodies (DLB), and multiple system atrophy (MSA).5–8 The time from iRBD diagnosis to phenoconversion ranges from months to 29 years, but not all individuals phenoconvert within this timeframe.5–8 Therefore, there is great interest in finding tools to determine which iRBD individuals will develop a Parkinson syndrome, which type of disease, and most importantly, in which timeframe phenoconversion will occur. This would be invaluable for neuroprotective clinical trials that seek to slow or halt disease progression before the manifestation of motor symptoms.
PD etiology is unknown, but α-Synuclein (αSyn) is closely associated with the origin of the disease, if not the culprit itself.9 Misfolded αSyn aggregates (αSyn-seeds) are found as Lewy bodies in PD and DLB, or as glial cytoplasmic inclusions in MSA.10 Hence, these diseases are commonly referred to as synucleinopathies. Recently, αSyn-seed amplification assays (αS-SAA) have facilitated the validation of αSyn-seeds in cerebrospinal fluid (CSF) as biomarkers for synucleinopathies. This is now offered to patients in the U.S. under CLIA registration as a laboratory-developed test. Several independent groups have shown the accuracy of αS-SAAs in the clinical stage of the disease (under the names Real-Time Quaking-Induced Conversion [RT-QuIC], Protein Misfolding Cyclic Amplification [PMCA], or SAA), with most reporting sensitivity and specificity >90%.11–19 However, only one sub-cohort of 30 PD cases has been reported to include CSF samples collected soon after PD diagnosis (<2 years), when αS-SAA would be most relevant clinically.15,19 Remarkably, high αS-SAA positivity rates in CSF have been reported for iRBD (83%-100%) in cohorts that present 6–62% phenoconversion rates.14,20,21 Conversely, only 39% positivity was reported for a sub-cohort (18 participants) of the iRBD arm of the DeNovo Parkinson (DeNoPa) cohort, including seven phenoconverters.21 The low detection was explained by decreased specificity of clinical diagnosis at enrollment due to earlier identification of iRBD patients from community cases compared to other cohorts.21 No strong correlation between αS-SAA parameters and phenoconversion has been found to date, and it remains unknown if αSyn-seeds appear in CSF of iRBD cases at onset or later in the prodromal stage. Here, we evaluated CSF samples collected during early PD stages, and from patients with iRBD, and correlated kinetic parameters to clinical progression and neurological test results.
Materials and methods
Study participants
Study participants were part of the DeNoPa cohort [PD, healthy controls (HC), iRBD]. Inclusion criteria have been described previously22, exclusion criteria are described in Supplementary Methods. Briefly, PD patients had to be (1) newly diagnosed with PD according to UK Brain Bank Criteria (at least two of resting tremor, rigidity, bradykinesia), (2) between 40 and 85 years old, (3) not exposed to L-dopa during the four weeks prior to study enrollment and exposed for less than two weeks if previously exposed.22
RBD was diagnosed through video polysomnography (vPSG) by experienced raters (CT, FSD, LMM) on two consecutive nights according to established criteria. 23,24,25 All participants were examined by movement disorder specialists at baseline and follow-up (BM, CT, JE, SS). Follow-up visits for PD and HC were scheduled biannually. Final diagnosis in this study refers to the consensus diagnosis after up to 10 years of follow-up, including initial DAT-SPECT, biannual clinical evaluations, L-dopa challenge, lasting response to L-dopa, and the emergence of advanced PD features such as motor-fluctuations or L-dopa-induced dyskinesias. Brain autopsy was available for 9 study participants of the DeNoPa cohort (7 PD, 1 MSA and 1 HC) that died during the 10 year follow-up. In 100% of cases the neuropathological diagnosis was consistent with the clinical consensus diagnosis by movement disorder specialists (BM, CT, JE, SS). Abnormal/pathological DAT-SPECT was determined by a specialist in nuclear medicine by visual inspection or quantification. Other neurological disorders (OND) refer to definite diagnoses other than PD, non-PD refers to uncertain diagnoses only known to be inconsistent with PD. Atypical PD [progressive supranuclear palsy (PSP), DLB, MSA] was diagnosed according to established criteria.26–28
All participants were evaluated according to the revised Unified Parkinson’s Disease Rating Scale (MDS-UPDRS).29 Cognitive function was assessed using the Montreal Cognitive Assessment (MoCA)30 and the Mini-Mental State Examination (MMSE).31 Further evaluations of cognitive domains and non-motor symptoms comprised the Non-motor Symptoms Questionnaire (NMS)32 and Geriatric Depression Scale (GDS).33 Additional tests are described in the Supplementary Methods. HCs were matched using frequency matching.
CSF samples.
CSF collection has been described previously22 (Supplementary Methods). 233 CSF samples from 206 individuals were analyzed in this study. To analyze a high number of cross-sectional samples, we analyzed samples from either baseline (BL) or follow-up. In total, CSF samples were collected on average 22 months after first symptoms and 58 (51%) were collected within 12 months of first symptoms. 56 iRBD CSF samples from 29 patients were collected at enrolment (on average 5.7 years after first reported iRBD symptoms). Figure 1 describes the samples used in this study. CSF α-synuclein (α-syn), β-amyloid 1–42 (Aβ42), total and phosphorylated tau protein (t-tau, p-tau181) were quantified as previously described.34 Additional measurements included neurofilament light chain, neurogranin, and YKL-40 (Supplementary Methods).
Figure 1.
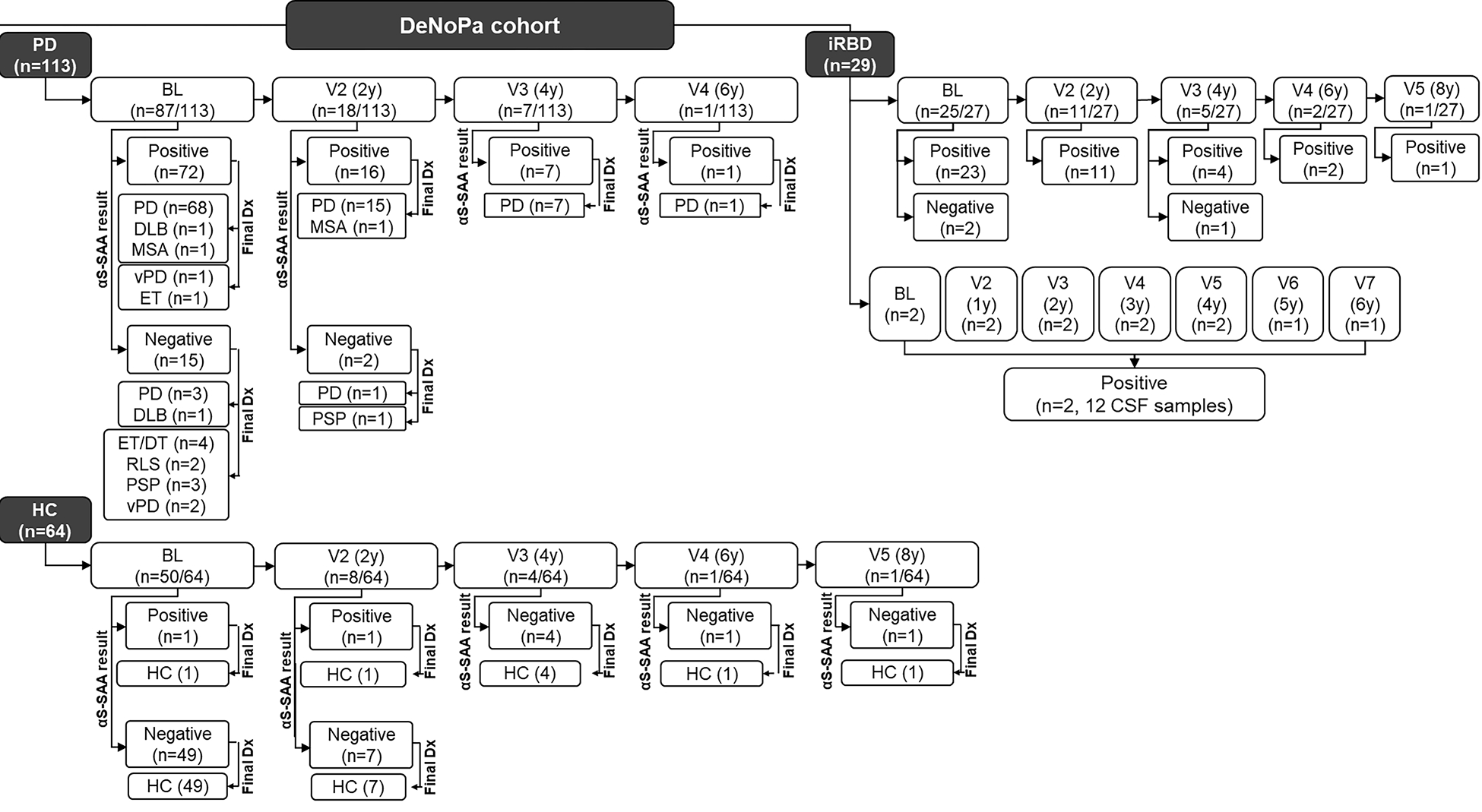
Baseline and final diagnoses and αS-SAA analysis of the DeNoPa cohort.
A total of 233 CSF samples from 206 individuals were analyzed in this study (PD, iRBD, HC). 113 PD-CSF samples were analyzed, one per PD patient, collected at BL (87), V2 (18), V3 (7), and V4 (1). 64 HC-CSF samples were analyzed, one per HC participant, collected at BL (50), V2 (8), V3 (4), V4 (1), and V5 (1).
Visits for PD and HC were scheduled biannually. 56 iRBD-CSF samples from 29 patients were analyzed and collected at BL and various follow-up visits were scheduled either annually or biannually. 96/113 PD-CSF cases were αS-SAA(+); 94/96 αS-SAA(+) PD-CSF cases had a synucleinopathy final diagnosis. 17/133 PD-CSF cases were αS-SAA(−); 12/17 αS-SAA(−) PD-CSF cases had a non-synucleinopathy final diagnosis. 62/64 HC-CSF cases were αS-SAA(−) with 64/64 cases remaining free of neurodegenerative disease until the end of the study. 53/56 iRBD-CSF samples were αS-SAA(+).
α-Synuclein seed amplification assay (αS-SAA)
The αS-SAA conditions used here have been described in detail elsewhere.19,15 Briefly, CSF samples were blindly analyzed in triplicate (40μL/well) in a reaction mixture containing 0.3mg/mL recombinant α-Synuclein (Amprion, cat# S2020), 100mM PIPES pH 6.50, 500mM NaCl, 10μM ThT, and one BSA-blocked 2.4mm Si3N4 G3 bead (Tsubaki-Nakashima). Beads were blocked in 1% BSA 100mM PIPES pH 6.50 and washed 100mM PIPES pH 6.50. αS-SAA was performed in 96-well plates (Costar, cat# 3916) using a FLUOstar Omega fluorometer. Plates were orbitally shaken at 800rpm for 1min every 29min at 37°C. Results from three replicates were considered input for a 3-output probabilistic algorithm (Supplementary material), where samples were called “positive”, “negative”, or “inconclusive”.15 Maximum fluorescence (Fmax), time to reach 50% Fmax (T50), slope, and the coefficient of determination for the fitting (R2) were calculated for each replicate using a sigmoidal equation available in Mars data analysis software (BMG). The time to reach the 5,000RFU threshold (TTT) was calculated with a user-defined equation in Mars.
Statistical analysis
Calculations were performed with R (v4.1.3). Group comparisons (PD, HC, iRBD, phenoconverter) were performed using the non-parametric Kruskal-Wallis test and sex comparison using the Chi-square test. For continuous sum scores, longitudinal modeling for all dependent variables was performed via a random intercept linear mixed model. Dependent variables were transformed to the square root scale to achieve normal distributions of the residual errors in the models, which were checked by q-q-plots of the residual errors. All models were adjusted for age, sex, and levodopa equivalent daily dose. The kinetic parameters of the αS-SAA were transformed to log10 and added to the models as a baseline covariate to assess their predictive association with the longitudinal expression of the clinical outcome. Thus, only αS-SAA kinetic parameters from samples collected at baseline (Supplementary Table I) were correlated to baseline demographic, clinical (Supplementary Table I), and biofluid parameters (Supplementary Table II). Correlations between αS-SAA parameters at BL and clinical outcomes were estimated using a nonparametric Spearman coefficient. Adjustment for multiple testing was undertaken and the false discovery rate was controlled at 5% with the Benjamini-Hochberg procedure.35
Data sharing
All relevant data are within the manuscript and its Supporting Information files. Additional data is available upon request.
Results
DeNoPA – final diagnosis of baseline PD and HC participants.
113 CSF samples (74 men, 39 women) from cases initially enrolled as PD were available (Figure 1). Of the 113 PD patients, 99 (87.6%) had a final synucleinopathy diagnosis: 95 PD (84.1%), two DLB (1.8%), two MSA (1.8%) (Figure 2A). The other 14 (12.4%) PD patients had a non-synucleinopathy as final clinical diagnosis: three (2.7%) vascular PD (vPD), five (4.4%) essential tremor (ET) and/or dystonic tremor (DT), two (1.8%) restless legs syndrome (RLS), four (3.5%) PSP. In these misdiagnosed cases, the average time from PD diagnosis to non-PD diagnosis was 3.7±2.6 years, while the average time from non-PD to final OND diagnosis was 1.9±2.5 years.
Figure 2.
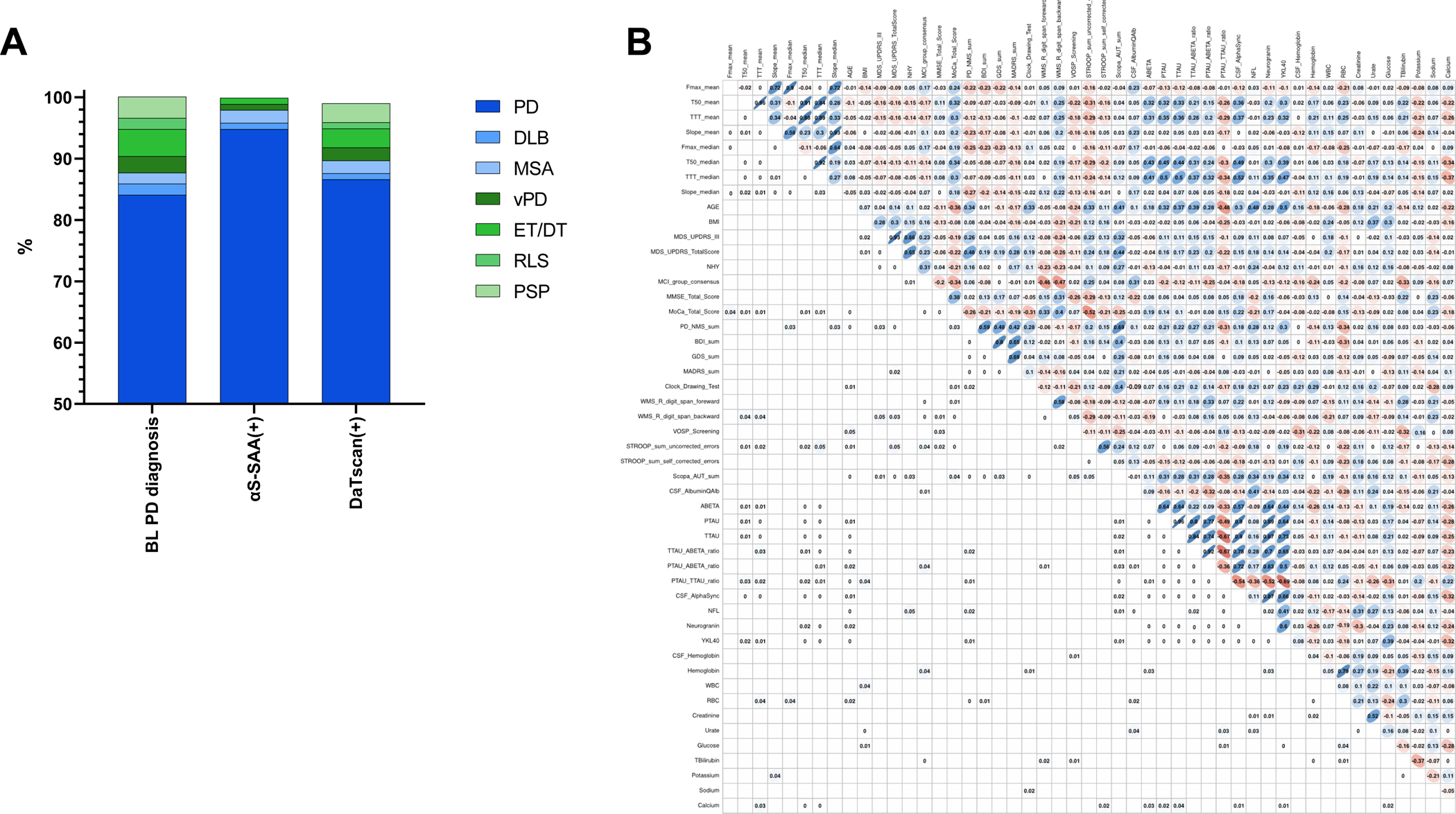
Statistical analysis of αS-SAA results from PD cases.
A) Of the 113 BL-PD cases, 87.6% had a final synucleinopathy diagnosis (PD, DLB, MSA), while 12.4% had a non-synucleinopathy final diagnosis (vPD, ET/DT, RLS< PSP). 96/113 BL-PD cases were positive in αS-SAA (αS-SAA(+)); of the 96 αS-SAA(+) cases, 97.9% had a final synucleinopathy diagnosis (PD, DLB, MSA), while only 2% had a non-synucleinopathy final diagnosis (vPD, ET/DT). 100/113 BL-PD cases had DAT-SPECT available, and 97/100 patients had abnormal DAT-SPECT (DaTscan(+)); of the 97 DaTscan(+) cases, 89.7% had a final synucleinopathy diagnosis (PD, DLB, MSA), while 10.3% had a non-synucleinopathy final diagnosis (vPD, ET/DT, RLS, PSP).
B) Spearman correlation matrix of αS-SAA kinetic parameters [maximum fluorescence (Fmax), time to reach 50% Fmax (T50), time to reach the 5,000 RFU threshold (TTT), and slope], neurological test scores, and lab-workup for αS-SAA(+) PD cases. The upper triangle of the matrix shows the nonparametric Spearman coefficient for each correlation, with blue indicating positive correlation, and red negative correlation. The intensity of color shows the value of the coefficient from 0 to 1. The lower triangle of the matrix shows the FDR adjusted significant p-values. FDR adjustments are according to Benjamini-Hochberg procedure.35 Values below 0.01 are shown as 0. Nonsignificant values (0.05) are left blank for layout convenience.
The 64 participants in the HC group remained free of neurodegenerative disease during follow-up. Supplementary Table I describes the demographics and clinical scores for those whose CSF samples were analyzed at BL.
DeNoPA – αS-SAA analysis of CSF samples from PD and HC participants.
CSF samples were blindly analyzed and assay results were compared to the final diagnosis that was considered the gold standard (Figure 1). The αS-SAA detected αSyn-seeds in CSF and was considered positive (αS-SAA(+)), in 96 (85%) of the 113 cases that were diagnosed with PD at baseline. 94 (95%) of the 99 patients with a final synucleinopathy diagnosis were αS-SAA(+), including 91 (92%) PD, one (1%) DLB, and two (2%) parkinsonian-subtype of MSA (MSA-P). One of the MSA-P samples presented the low fluorescence signature previously reported.36 αSyn-seeds were not detected (αS-SAA(−)) in the other five (5.1%) cases with synucleinopathies, which included four (4%) PD and one (1%) DLB. Thus, of the 96 αS-SAA(+) patients, there were 91 (94.8%) PD, one (1%) DLB, two (2.1%) MSA, one (1%) vascular PD (vPD), and one (1%) ET (Figure 2A). Of the 14 cases without a synucleinopathy, 12 were αS-SAA(−), and one vPD, and one ET was αS-SAA(+). Of the 64 HC participants, 62 (96.9%) were αS-SAA(−). The two positive samples did not show any symptoms consistent with prodromal disease.
αS-SAA results agreed with the final diagnosis of synucleinopathy in 95% of cases, while the agreement in the HC control group reached ~97%. Since a highly sensitive and specific assay is needed during the early-stages of synucleinopathy, we calculated sensitivity and specificity using only samples from BL. 70 of the 74 cases with a final synucleinopathy diagnosis and BL-CSF samples were αS-SAA(+), for an estimated sensitivity of 94.6% (95%CI 86.7–98.5%). 49 of the 50 HC participants with BL-CSF samples were αS-SAA(−), for an estimated specificity of 98% (95%CI 89.4–100%). Considering a synucleinopathy prevalence of 389 cases every 100,000 people,37 the assay presents a BL accuracy of 98% (95%CI 93.7–99.7%). All 39 final-PD cases with CSF collected ≤1y from first symptoms were αS-SAA(+). The high specificity of the assay was also observed when analyzing BL-CSF samples from PD cases with final non-synucleinopathy diagnoses, as 12 (85.7%) of the 14 cases were αS-SAA(−).
DeNoPA – αS-SAA results in PD patients stratified by DAT-SPECT status
DAT-SPECT was available at enrollment for 100 (88.5%) of the 113 PD patients and 97 were considered abnormal/pathological. 87 (89.7%) of the 97 PD cases with abnormal DAT-SPECT [PD-DAT(+)] had a final synucleinopathy diagnosis (84 PD, one DLB, two MSA-P) (Figure 2A). Three other subjects with neurodegenerative parkinsonian type, but non-synucleinopathy, were PSP (3.1%). The other 7 participants had a final non-synucleinopathy diagnosis: three (3.1%) ET, two (2%) RLS, two (2%) vPD. 82 (94.3%) of the 87 PD-DAT(+) were αS-SAA(+) (80 PD, two MSA-P), and four PD and one DLB were PD-DAT(+) and αS-SAA(−). Remarkably, eight of the 10 PD-DAT(+) with final non-synucleinopathy were αS-SAA(−) using CSF collected at BL in nine of the 10 cases (one PSP-CSF sample was collected at a follow-up). The three PD cases with normal DAT-SPECT were diagnosed with ET, ET/DT, and vPD, which were all αS-SAA(−). The final diagnoses for the 13 PD cases without DAT-SPECT included 11 PD, one PSP, and one DLB. The PSP case was αS-SAA(−), while the other 12 cases were αS-SAA(+).
Estimation of correlation between αS-SAA parameters and clinical parameters of PD cases
Kinetic parameters such as Fmax (RFU), T50 (h), TTT (h), and slope (RFU/h) were calculated for all PD samples. Mean and median values of these parameters were determined for each sample including the three technical replicates and their correlation with clinical test scores and lab work-up was estimated (Figure 2B). Clinical scores did not present relevant correlations with kinetic parameters, while significant but weak correlations were observed between T50 and TTT and the concentration of CSF biomarkers like amyloid-β, tau, phospho-tau, and αSyn.
DeNoPa – iRBD.
The iRBD group included 29 patients (20 men and 9 women). On average, enrolment occurred 5.35±4.0 years after first symptoms, as reported by the patients and/or respective partners (range 0.42 to 12y). Of the 29 cases, 8 (27.6%) phenoconverted after 11.4±4.7 years from the first symptoms (range 6.3–20.2y) and 3.8±3 years from enrolment (range 0.5–8.2y). The mean age at phenoconversion was 69.3±4.8 years. Of the eight phenoconverters, five (62.5%) developed PD, and three (37.5%) developed DLB. Patients have been clinically followed from enrolment for 6.6 ± 2 years on average (range 2.3 to 9.5 years).
DeNoPa – detection of αSyn-seeds by αS-SAA in CSF from iRBD
Including samples from enrolment and follow-up, 56 CSF samples from 29 iRBD patients were analyzed. 53 (94.6%) of the iRBD CSF samples were αS-SAA(+) and 27 (93.1%) of 29 had an αS-SAA(+) sample at enrolment or follow-up (Figure 1). Moreover, 25 (92.6%) of the 27 CSF samples collected at enrolment were αS-SAA(+). This level of detection at BL is 64.8% higher than previously reported for a sub-cohort of these samples and a different assay.21 All eight phenoconverters were αS-SAA(+) at enrolment. Interestingly, two iRBD patients with CSF samples collected only 8 and 9 months after the onset of first symptoms were αS-SAA(+) (Figure 3). Two iRBD cases had three αS-SAA(−) CSF samples and did not phenoconvert to manifest disease during the duration of the available follow-up (8.2 and 6.8 years after their enrollment).
Figure 3.
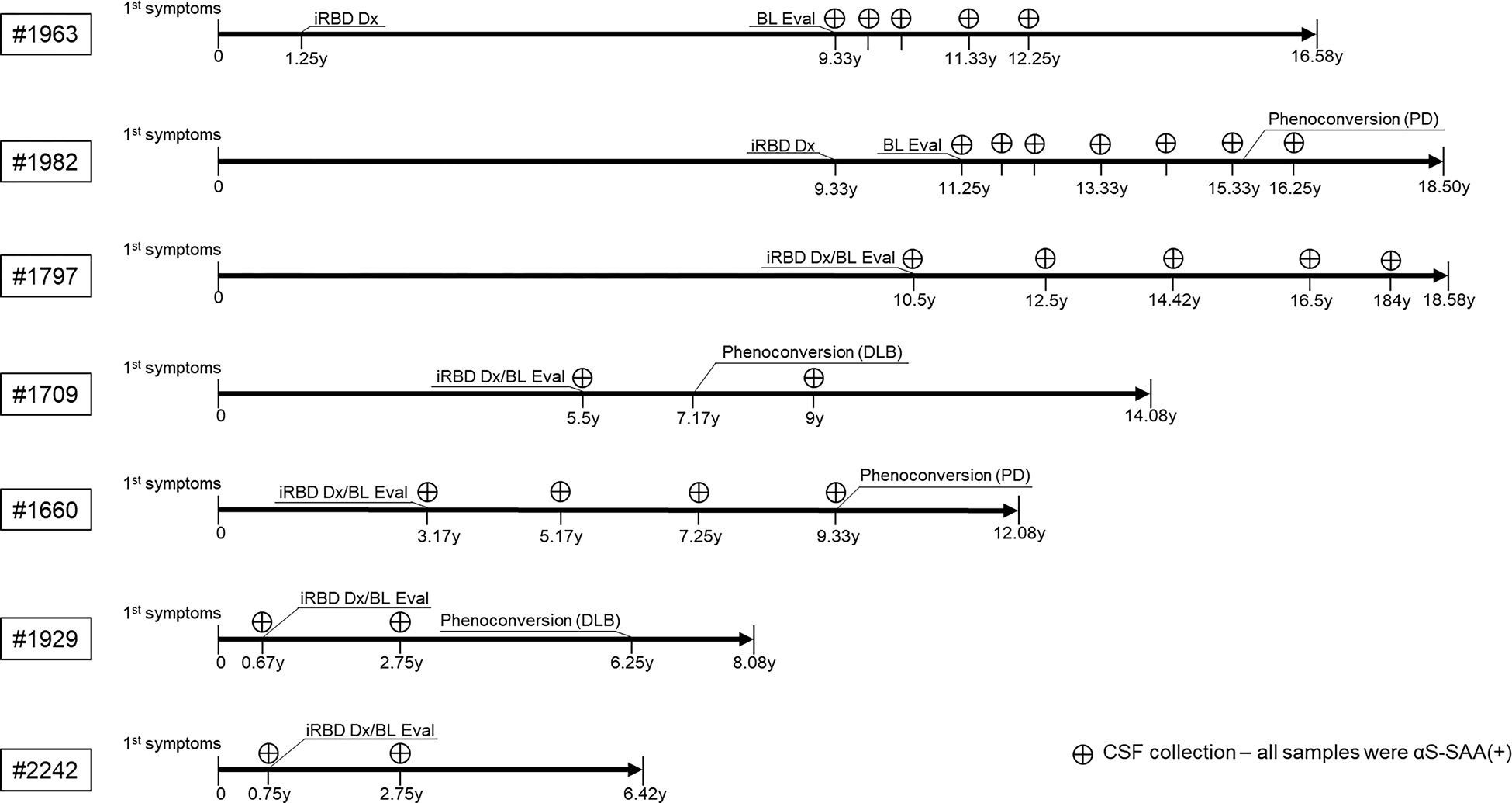
Depiction of the timeline of seven longitudinal iRBD cases from the onset of first symptoms through the duration of the study. The timeline is measured in years, with the patient ID noted on the far left. Occurrence of iRBD diagnosis (iRBD Dx) and baseline evaluation (BL Eval) is noted, along with various CSF collections and the associated αS-SAA(+) result. Phenoconversion is included for the four iRBD cases who phenoconverted from iRBD to synucleinopathy, with the final diagnosis (PD, DLB) specified in parentheses.
DeNoPa – Longitudinal detection of αSyn-seeds by αS-SAA in CSF from iRBD
Seven iRBD cases had longitudinal CSF samples available. Some samples were collected very close to the onset of first symptoms, and some before and after phenoconversion (Figure 3).
Patient #1963 was diagnosed with iRBD 1.25 years after the onset of first symptoms. CSF collection at enrolment was 8 years after diagnosis, five samples were collected over 2.92 years, all αS-SAA(+). By the end of this study, 15.3 years after iRBD diagnosis, this patient has not phenoconverted. Patient #1982 was diagnosed with iRBD 9.33 years after first symptoms and CSF collection performed 1.92 years later. Seven CSF samples were collected over five years and the final sample was collected 0.67 years after PD phenoconversion. All CSF samples were αS-SAA(+) and there were no differences in kinetic parameters (T50, TTT, Fmax, and slope) between samples collected before or after phenoconversion.
Three other patients had follow-ups biannually from enrolment and two phenoconverted. Patient #1797 was diagnosed with iRBD at enrolment, 10.5 years after first symptoms. Follow-up was ~8 years and five samples were collected, all αS-SAA(+). Patient #1709 was diagnosed with iRBD at enrolment, 5.5 years after initial symptoms, and phenoconverted to DLB 1.67 years later. CSF was collected at enrolment and 1.83 years after phenoconversion, all samples were αS-SAA(+) and showed similar aggregation profiles. Patient #1660 was diagnosed with iRBD at enrolment, 3.17 years after first symptoms. Four CSF samples were collected over the next 6.16 years when the case phenoconverted to PD. All four CSF samples were positive without evident change in the aggregation profile before and after phenoconversion. Finally, patients #1929 (DLB phenoconverter) and #2242 were evaluated at enrolment very soon after the onset of first symptoms, 0.67 and 0.75 years, respectively. In addition to the initial CSF sample at enrolment, both patients had CSF collected 2 years afterwards. All four samples were αS-SAA(+).
Correlation between clinical and αS-SAA parameters in the iRBD sub-cohort.
Kinetic αS-SAA parameters from the iRBD group were correlated to neurological test scores and biofluid work-up. Several significant correlations were found (Figure 4). The mean and median Fmax negatively correlated to the total MDS-UPDRS (−0.54, −0.57) and the p-tau/t-tau ratio (−0.73, −0,82), while T50 and TTT (both mean and median) positively correlated with the CSF/serum albumin ratio (0.69, 0.67, 0.68, 0.69). Interestingly, there were significant correlations between αS-SAA kinetic parameters and phenoconversion despite the low number of phenoconverters. Longitudinal changes in the MDS-UPDRS-III score showed a negative association with the log10(mean-Fmax) measured at enrolment after adjusting for sex, age, and the diagnosis group (p=0.047). MDS-UPDRS-III, MDS-UPDRS total, NMS sum, and GDS sum scores, showed a negative association with the log10(median Fmax) measured at BL after adjusting for sex, age, and the diagnosis group (p=0.045, p=0.028, p=0.012, and p=0.043, respectively). Because of the negative values on the log scale, this means the greater the Fmax the greater the clinical scores are, and the worse the disease status is. In reference to the HC group as a base category, the PD group led the contribution to the progression of the Total MDS-UPDRS, followed by the iRBD-phenoconverters, and lastly the non-phenoconverter iRBD group. This is consistent with the temporal progression from healthy, to prodromal, to motor disease. The model for the MoCA total score showed significant positive associations with the log10 (mean-slope) (p=0.007) and log10 (median-slope) (p=0.011) at BL, meaning the slower the amplification in the elongation phase, the more pronounced cognitive decline.
Figure 4.
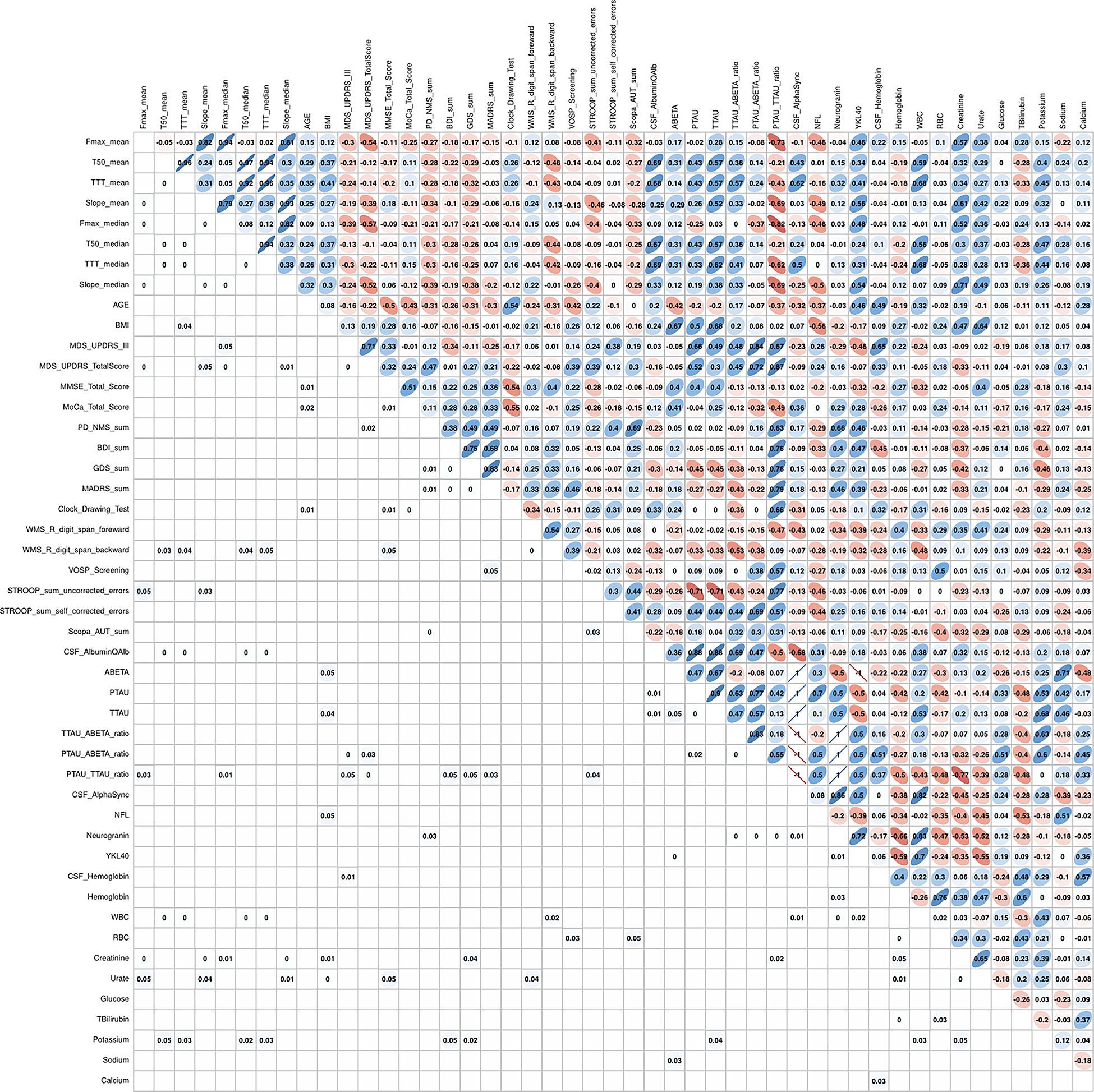
Spearman correlation matrix of αS-SAA kinetic parameters [maximum fluorescence (Fmax), time to reach 50% Fmax (T50), time to reach the 5,000 RFU threshold (TTT), and slope], neurological test scores, and biofluid workup for αS-SAA(+) iRBD cases. The upper triangle of the matrix shows the nonparametric Spearman coefficient for each correlation, with blue indicating positive correlation, and red negative correlation. The intensity of color shows the value of the coefficient from 0 to 1. The lower triangle of the matrix shows the FDR adjusted significant p-values. FDR adjustments are according to Benjamini-Hochberg procedure.35 Values below 0.01 are shown as 0. Nonsignificant values (0.05) are left blank for layout convenience.
The matrix shows the nonparametric Spearman coefficient for each correlation, with red indicating positive correlation, and blue negative correlation. The intensity of color shows the value of the coefficient from 0 to 1.
Discussion
Performing early and reliable diagnoses of synucleinopathies is a longstanding clinical challenge. In 2016, Rizzo et al. reported that there had been a complete lack of improvement in PD diagnosis for a quarter of a century.38 In the last few years, αS-SAAs have demonstrated high accuracy in detecting αSyn-seeds in CSF.11–19 Nevertheless, most reports used CSF collected from patients in the moderate to late stages of the disease when the clinical diagnosis is reinforced by typical motor- and non-motor features of PD. We have previously evaluated our αS-SAA conditions using de-novo PD samples from PPMI, but our study was limited by the low number of cases (n=30).15 Hence, in the current study we evaluated αS-SAA accuracy at the time of the initial clinical assessment against the final diagnosis, which we considered to be the “true diagnosis”, acknowledging that pathology is the current gold standard. αS-SAA performed with 94.6% sensitivity and 97% specificity, giving an overall 98% accuracy for synucleinopathies. These results match our previous report (96% sensitivity and 97% specificity)15 and strongly suggest that αS-SAA can significantly improve the diagnosis of synucleinopathies at first assessment, when accurate diagnosis would be most impactful in the clinical setting. In this cohort, differential diagnosis of non-PD in absence of αS-SAA took 3.7 years. Thus, the use of αS-SAA by general and specialized neurologists would significantly shorten the time to final diagnosis from years to days. With emerging data from other cohorts with more MSA patients, it looks like the kinetic of αS-SAA can even distinguish between PD and MSA.36,39
DAT-SPECT detects dopaminergic degeneration using a radiopharmaceutical probe against dopamine transporter40 and is used to distinguish between PD and PD-mimics such as ET or psychogenic PD, but it is not specific for synucleinopathies. Only 89.7% of cases with pathologic DAT-SPECT had a final synucleinopathy diagnosis, while αS-SAA reached 95% agreement with the final diagnosis in the same sub-cohort. Higher disagreement between DAT-SPECT and final PD diagnosis was expected since PD-mimics like PSP also present dopaminergic degeneration with pathogenic DAT-SPECT. In this regard, 80% of non-synucleinopathy cases with abnormal DAT-SPECT (including all PSP cases) were αS-SAA(−). Our study is one of the first to compare these two methods and provide evidence that αS-SAA is more accurate for synucleinopathy diagnosis. DAT-SPECT inaccuracy cannot be explained by pharmacologic interference, since all patients were monitored for drugs known to hamper the results.41
Most PD cases do not present prodromal iRBD symptoms,42 but iRBD cases are of great interest because these patients will be subject to future prevention trials (https://www.ppmi-info.org/study-design/path-to-prevention-platform-trial). 93.1% of iRBD cases were αS-SAA(+), regardless of the sampling time, as well as 92.6% of BL samples. Remarkably, αSyn-seeds were readily detected in iRBD CSF samples collected just months after the first iRBD symptoms were disclosed by the patients. The high sensitivity is comparable to αS-SAA conditions reported by others,14,20 including the Oxford and Italian cohorts reported by Poggiolini et al.21 However, a sub-cohort of the DeNoPa iRBD arm (n=18) analyzed by Poggiolini on different assay conditions reported only 39% sensitivity, which was explained by lower diagnostic specificity due to earlier enrollment from community cases. However, our results suggest that most DeNoPa iRBD cases are in fact prodromal synucleinopathy cases and they were probably found positive because of the higher sensitivity of the assay used here. These results suggest preclinical detection of αSyn-seeds is feasible since αS-SAA readily amplified αSyn-seeds from very early iRBD samples. It remains to be seen if very early and perhaps pre-clinical detection of αSyn-seeds is possible in incidental Lewy bodies and also better accessible tissues like the olfactory mucosa or other biological fluids.
Lastly, we evaluated if any kinetic parameter of aggregation correlated with a relevant clinical progression score or biofluid analyte for both PD and iRBD cohorts. Only weak correlations with clinical parameters were observed, in agreement with previous studies.18,19,43 However, correlations between assay parameters at BL with MDS-UPDRS, tau-protein, and the CSF/blood albumin ratio warrant further investigation. The low rate of phenoconversion precluded us from conclusively comparing phenoconverters and iRBD, as the small sample size of the phenoconverters may exacerbate the significance of our findings. A model in which αSyn-seed levels increase preclinically and plateau at the prodromal stage of the disease is consistent with our results. Similar behaviors are well established in amyloid-β and PrPSc in Alzheimer’s disease and prion diseases, respectively. However, kinetic parameters could be affected by CSF analytes that act as amplification accelerators or inhibitors in a patient-dependent manner.12,13,16 CSF components modulating kinetic parameters have not been identified, but there are reports in literature showing in vitro interactions between αSyn and human serum albumin, lipids, tau-protein, and other proteins that may influence amplification. 44–47 In this cohort, slower amplification correlated with a higher CSF/serum albumin ratio.
In summary, our results show that αS-SAA represents a remarkable tool for the early and even prodromal detection of synucleinopathies and suggest that αSyn-seeds are present in CSF before phenoconversion. Thus, αS-SAA in its current form represents an excellent marker of state and trait. To be used as a screening tool for preventive trials in prodromal/symptom-free individuals, αS-SAA must be further developed to determine if preclinical amplification of αSyn-seeds is possible in noninvasive samples, such as saliva.48,49 Ideally, such a tool will allow the exclusion of subjects with negative αS-SAA in future preventive trials with prodromal individuals.
Supplementary Material
Table I.
Baseline demographic and clinical data of participants with baseline CSF samples from the DeNoPa cohort
| HC (N=50) | PD (N=71) | iRBD (N=19) | iRBD converter (N=8) | p value | |
|---|---|---|---|---|---|
| Gender | 0.418 | ||||
| Male | 35 (70.0%) | 52 (73.2%) | 11 (57.9%) | 7 (87.5%) | |
| Female | 15 (30.0%) | 19 (26.8%) | 8 (42.1%) | 1 (12.5%) | |
| Age | 0.046 | ||||
| Mean (SD) | 66 (6.8) | 63.070 (9.466) | 68.597 (8.480) | 65.177 (5.973) | |
| Median (Q1, Q3) | 67 (62.250, 69) | 64 (57, 70) | 72 (62.5, 75) | 66.210 (64.75, 67.25) | |
| Min - Max | 44 – 84 | 41 – 82 | 51 – 77 | 52 – 73 | |
| BMI | 0.269 | ||||
| Mean (SD) | 26.859 (4.514) | 27.981 (4.271) | 27.084 (4.154) | 26.159 (1.543) | |
| Median (Q1, Q3) | 25.838 (24.226, 29.173) | 27.616 (25.048, 29.619) | 26.493 (24.103, 29.561) | 26.662 (25.092, 27.288) | |
| Min - Max | 19.271 – 42.253 | 20.089 – 44.789 | 20.797 – 34.136 | 23.504 – 27.757 | |
| MDS-UPDRS III | < 0.001 | ||||
| Mean (SD) | 0.680 (1.477) | 21.803 (10.861) | 2.158 (2.267) | 4.750 (3.694) | |
| Median (Q1, Q3) | 0 (0, 0) | 21.0 (13.0, 28.0) | 1.000 (0.500, 3.000) | 4.500 (1.750, 7.500) | |
| Min - Max | 0 – 6 | 3 – 54 | 0 – 7 | 0 – 10 | |
| MDS-UPDRS Total | < 0.001 | ||||
| Mean (SD) | 3.720 (3.902) | 35.345 (16.324) | 16.368 (8.187) | 18.250 (10.025) | |
| Median (Q1, Q3) | 2.000 (1.000, 5.000) | 34.000 (24.000, 46.500) | 16.000 (12.000, 18.500) | 19.500 (12.500, 24.750) | |
| Min - Max | 0.000 – 15.000 | 5.000 – 84.000 | 2.000 – 35.000 | 4.000 – 32.000 | |
| HY | < 0.001 | ||||
| Mean (SD) | 0.000 (0.000) | 1.972 (0.736) | 0.053 (0.229) | 0.000 (0.000) | |
| Median (Q1, Q3) | 0.000 (0.000, 0.000) | 2.000 (1.000, 2.500) | 0.000 (0.000, 0.000) | 0.000 (0.000, 0.000) | |
| Min - Max | 0.000 – 0.000 | 1.000 – 3.000 | 0.000 – 1.000 | 0.000 – 0.000 | |
| MMSE Total | 0.526 | ||||
| Mean (SD) | 28.571 (1.190) | 28.357 (1.263) | 28.222 (1.629) | 29.000 (0.577) | |
| Median (Q1, Q3) | 29.000 (28.000, 29.000) | 29.000 (28.000, 29.000) | 28.500 (28.000, 29.000) | 29.000 (29.000, 29.000) | |
| Min - Max | 26.000 – 30.000 | 25.000 – 30.000 | 24.000 – 30.000 | 28.000 – 30.000 | |
| Missing | 1 | 1 | 1 | 1 | |
| MoCa Total | 0.415 | ||||
| Mean (SD) | 25.700 (2.410) | 24.718 (2.987) | 24.947 (3.188) | 25.250 (3.059) | |
| Median (Q1, Q3) | 25.000 (24.250, 27.000) | 25.000 (23.000, 27.000) | 26.000 (24.000, 27.000) | 26.000 (22.750, 26.500) | |
| Min - Max | 19.000 – 30.000 | 16.000 – 30.000 | 17.000 – 29.000 | 21.000 – 30.000 |
BMI: Body Mass Index, MDS-UPDRS: Movement Disorder Society – Unified Parkinson’s Disease Rate Score, HY: Hoehn and Yahr scale, MMSE: Mental Minimal State Examination, MoCa: Montreal Cognitive Assessment test.
Acknowledgment
We thank the sample donors and their families for supporting this research by participating in the DeNoPa study. This work was partially supported by the Michael J. Fox Foundation (grant 21233 to LC).
Role of the funding source
The study sponsors provided support through an unrestricted grant and did not influence the study design, collection, and analysis of data, the writing of the paper, or the decision to submit the paper. The sponsors have been informed about the final manuscript and the submission for publication.
Dr. Sixel-Döring has received honoraria for lectures and consultancy from Ever Pharma, AbbVie, and BIAL. She served on an Advisory Board for Zambon.
Dr. Schade received institutional salaries supported by the EU Horizon 2020 research and innovation program under grant agreement No. 863664 and by the Michael J. Fox Foundation for Parkinson’s Research under grant agreement No. MJFF-021923. He is supported by a PPMI Early-Stage Investigators Funding Program fellowship of the Michael J. Fox Foundation for Parkinson’s Research under grant agreement No. MJFF-022656.
BM has received honoraria for consultancy from Roche, Biogen, AbbVie, Servier, 4D Pharma PLC, and Amprion. BM is a member of the executive steering committee of the Parkinson Progression Marker Initiative and PI of the Systemic Synuclein Sampling Study of the Michael J. Fox Foundation for Parkinson’s Research and has received research funding from the Deutsche Forschungsgemeinschaft (DFG), EU (Horizon2020), Parkinson Fonds Deutschland, Deutsche Parkinson Vereinigung, Parkinson’s Foundation, Hilde-Ulrichs-Stiftung für Parkinsonforschung, and the Michael J. Fox Foundation for Parkinson’s Research.
Funding sources:
Michael J. Fox Foundation grant to Luis Concha (MJFF-21233) and grants from NIH to CS (RF1AG055053 and RF1AG059321). This study was supported by unrestricted grants from the University Medical Centre Goettingen, the Paracelsus-Elena-Klinik, Kassel, Germany, the Michael J Fox Foundation for Parkinson’s Research, ParkinsonFonds Deutschland, and TEVA Pharma.
Footnotes
Financial disclosure/conflict of interest concerning the research related to the manuscript: Dr. Soto, Dr. Concha, Ms. Farris, and Mr. Ma are inventors of several patents related to the SAA (PMCA) technology and are affiliated with Amprion Inc., a biotech company focusing on the commercial utilization of SAA (PMCA/RT-QuIC) for biomedical diagnosis of neurodegenerative diseases. Dr. Mollenhauer is on the advisory board of Amprion.
Dr. Weber, Dr. Dakna, Ms. Lang, Mrs. Wicke, Mrs. Starke, Dr. Ebentheuer, Dr. Muntean, Dr. Sixel-Döring, and Dr. Schade have no conflicts of interest to declare.
Dr. Trenkwalder has no conflict of interest related to the research of this manuscript, she is an advisor of UCB, Ono, Abbvie, Bial and has received honoraries for lectures from Abbvie, Alexion, and Bial.
Supplementary material: Supplementary methods, Supplementary Table I, Supplementary Table II.
References
- 1.Fearnley JM & Lees AJ Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain: a journal of neurology 114 ( Pt 5, 2283–301 (1991). [DOI] [PubMed] [Google Scholar]
- 2.Beach TG & Adler CH Importance of low diagnostic Accuracy for early Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society 33, 1551–1554 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kordower JH et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain: a journal of neurology 136, 2419–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Högl B, Stefani A & Videnovic A Idiopathic REM sleep behaviour disorder and neurodegeneration - an update. Nature reviews. Neurology 14, 40–55 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Iranzo A et al. Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PloS one 9, e89741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iranzo A et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. The Lancet. Neurology 12, 443–53 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Postuma RB et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain: a journal of neurology 142, 744–759 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schenck CH, Boeve BF & Mahowald MW Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep medicine 14, 744–8 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Steiner JA, Quansah E & Brundin P The concept of alpha-synuclein as a prion-like protein: ten years after. Cell and tissue research 373, 161–173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehra S, Sahay S & Maji SK α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochimica et biophysica acta. Proteins and proteomics 1867, 890–908 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Fairfoul G et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Annals of clinical and translational neurology 3, 812–818 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shahnawaz M et al. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of α-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurology 74, 163 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Groveman BR et al. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta neuropathologica communications 6, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossi M et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta neuropathologica 140, 49–62 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Concha-Marambio L et al. Seed Amplification Assay to Diagnose Early Parkinson’s and Predict Dopaminergic Deficit Progression. Movement disorders: official journal of the Movement Disorder Society 36, 2444–2446 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bargar C et al. Streamlined alpha-synuclein RT-QuIC assay for various biospecimens in Parkinson’s disease and dementia with Lewy bodies. Acta neuropathologica communications 9, 62 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manne S et al. Ultrasensitive Detection of Aggregated α-Synuclein in Glial Cells, Human Cerebrospinal Fluid, and Brain Tissue Using the RT-QuIC Assay: New High-Throughput Neuroimmune Biomarker Assay for Parkinsonian Disorders. Journal of Neuroimmune Pharmacology (2019) doi: 10.1007/s11481-019-09835-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang UJ et al. Comparative study of cerebrospinal fluid α-synuclein seeding aggregation assays for diagnosis of Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society 34, 536–544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russo MJ et al. High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta neuropathologica communications 9, 179 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iranzo A et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. The Lancet. Neurology 20, 203–212 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Poggiolini I et al. Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain: a journal of neurology (2021) doi: 10.1093/brain/awab431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mollenhauer B et al. Nonmotor and diagnostic findings in subjects with de novo Parkinson disease of the DeNoPa cohort. Neurology 81, 1226–34 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Schenck CH, Bundlie SR, Ettinger MG & Mahowald MW Chronic behavioral disorders of human REM sleep: a new category of parasomnia. Sleep 9, 293–308 (1986). [DOI] [PubMed] [Google Scholar]
- 24.American Academy of Sleep Medicine. International Classification of Sleep Disorders Diagnostic & Coding Manual 2nd Edition. (Amer Academy of Sleep Medicine, 2005). [Google Scholar]
- 25.American Academy of Sleep Science. International Classification of Sleep Disorders. (2014).
- 26.Höglinger GU et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord 32, 853–864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wenning GK et al. The Movement Disorder Society Criteria for the Diagnosis of Multiple System Atrophy. Movement Disorders 37, 1131–1148 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKeith IG et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89, 88–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goetz CG et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Movement disorders: official journal of the Movement Disorder Society 23, 2129–70 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Nasreddine ZS et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. Journal of the American Geriatrics Society 53, 695–9 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Folstein MF, Folstein SE & McHugh PR ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12, 189–198 (1975). [DOI] [PubMed] [Google Scholar]
- 32.Chaudhuri KR et al. International multicenter pilot study of the first comprehensive self-completed nonmotor symptoms questionnaire for Parkinson’s disease: the NMSQuest study. Mov Disord 21, 916–923 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Yesavage JA et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res 17, 37–49 (1982). [DOI] [PubMed] [Google Scholar]
- 34.Mollenhauer B et al. Monitoring of 30 marker candidates in early Parkinson disease as progression markers. Neurology 87, 168–177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289–300 (1995). [Google Scholar]
- 36.Shahnawaz M et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 578, 273–277 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savica R, Grossardt BR, Bower JH, Ahlskog JE & Rocca WA Incidence and pathology of synucleinopathies and tauopathies related to parkinsonism. JAMA neurology 70, 859–66 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rizzo G et al. Accuracy of clinical diagnosis of Parkinson disease: A systematic review and meta-analysis. Neurology 86, 566–76 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Singer W et al. Alpha-Synuclein Oligomers and Neurofilament Light Chain Predict Phenoconversion of Pure Autonomic Failure. Ann Neurol 89, 1212–1220 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoessl AJ, Lehericy S & Strafella AP Imaging insights into basal ganglia function, Parkinson’s disease, and dystonia. Lancet (London, England) 384, 532–44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morbelli S et al. EANM practice guideline/SNMMI procedure standard for dopaminergic imaging in Parkinsonian syndromes 1.0. European journal of nuclear medicine and molecular imaging 47, 1885–1912 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schrag A, Horsfall L, Walters K, Noyce A & Petersen I Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. The Lancet. Neurology 14, 57–64 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Orrù CD et al. A rapid α-synuclein seed assay of Parkinson’s disease CSF panel shows high diagnostic accuracy. Annals of clinical and translational neurology 8, 374–384 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siegert A et al. Interplay between tau and α-synuclein liquid-liquid phase separation. Protein Sci 30, 1326–1336 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kachappilly N, Srivastava J, Swain BP & Thakur P Interaction of alpha-synuclein with lipids. Methods Cell Biol 169, 43–66 (2022). [DOI] [PubMed] [Google Scholar]
- 46.Mohammad-Beigi H et al. Strong interactions with polyethylenimine-coated human serum albumin nanoparticles (PEI-HSA NPs) alter α-synuclein conformation and aggregation kinetics. Nanoscale 7, 19627–19640 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Paleologou KE & El-Agnaf OMA α-Synuclein aggregation and modulating factors. Subcell Biochem 65, 109–164 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Luan M et al. Diagnostic Value of Salivary Real-Time Quaking-Induced Conversion in Parkinson’s Disease and Multiple System Atrophy. Mov Disord 37, 1059–1063 (2022). [DOI] [PubMed] [Google Scholar]
- 49.Stefani A et al. Alpha-synuclein seeds in olfactory mucosa of patients with isolated REM sleep behaviour disorder. Brain 144, 1118–1126 (2021). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.