

Abstract
The dihydropyrimidinase-like (DPYSL) proteins, also designated as the collapsin response mediators (CRMP) proteins, constitute a family of five cytosolic phosphoproteins abundantly expressed in the developing nervous system but down-regulated in the adult mouse brain. The DPYSL proteins were initially identified as effectors of semaphorin 3A (Sema3A) signaling and consequently involved in regulation of growth cone collapse in young developing neurons. To date, it has been established that DPYSL proteins mediate signals for numerous intracellular/extracellular pathways and play major roles in variety of cellular process including cell migration, neurite extension, axonal guidance, dendritic spine development and synaptic plasticity through their phosphorylation status. The roles of DPYSL proteins at early stages of brain development have been described in the past years, particularly for DPYSL2 and DPYSL5 proteins. The recent characterization of pathogenic genetic variants in DPYSL2 and in DPYSL5 human genes associated with intellectual disability and brain malformations, such as agenesis of the corpus callosum and cerebellar dysplasia, highlighted the pivotal role of these actors in the fundamental processes of brain formation and organization. In this review, we sought to establish a detailed update on the knowledge regarding the functions of DPYSL genes and proteins in brain and to highlight their involvement in synaptic processing in later stages of neurodevelopment, as well as their particular contribution in human neurodevelopmental disorders (NDDs), such as autism spectrum disorders (ASD) and intellectual disability (ID).
Keywords: dihydropyrimidinase-like proteins, collapsin response mediator proteins, neurodevelopmental disorders (NDDs), human genetics research, neuronal development, synaptic physiopathology, missense variants, animal model
1. Introduction
The Dihydropyrimidinase-like (DPYSL) proteins, also designated as the Collapsin response mediators (CRMP) proteins, constitute a family of five cytosolic phosphoproteins (Quinn et al., 1999), abundantly expressed in the developing nervous system but down-regulated in the adult mouse brain (Minturn et al., 1995; Byk et al., 1996; Wang and Strittmatter, 1996; Fukada et al., 2000; Yuasa-Kawada et al., 2003). The DPYSL proteins were initially identified as effectors of semaphorin 3A (Sema3A) signaling and consequently involved in regulation of growth cone collapse (Goshima et al., 1995).
To date, it has been established that DPYSL proteins mediate signals for numerous intracellular/extracellular pathways and play major roles in variety of cellular process including cell migration (Yamashita et al., 2006), neurite extension (Brot et al., 2010), axonal guidance (Goshima et al., 1995; Arimura et al., 2005; Uchida and Goshima, 2005; Yoshimura et al., 2005), dendritic spine development (Yamashita et al., 2007) and synaptic plasticity (Yamashita et al., 2011) through their phosphorylation status.
The roles of DPYSL at early stages of brain development have been described in the past years, particularly for DPYSL2 and DPYSL5. In this review, we sought to establish a detailed synthesis on the functions of DPYSL genes and proteins in brain and to highlight their involvement in synaptic processing in later stages of neurodevelopment, as well as their contribution in human neurodevelopmental disorders (NDDs), such as autism spectrum disorders (ASD) and intellectual disability (ID). This synthesis will likely provide a new perspective regarding the specific function of DPYSL genes and proteins in developing and functioning brain, as well as their respective role in the fine regulation of brain developmental stages.
2. The DPYSL proteins family
In mammals, five DPYSL (or CRMP) proteins have been identified and are encoded by their respective coding-genes (CRMP1 or DPYSL1, DPYSL2, DPYSL3, DPYSL4, and DPYSL5) (Table 1). The predicted secondary structure of human DPYSL proteins family is well described and allowed to determine that DPYSL1-4 display around 75% sequence homology with each other, whereas DPYSL5 is more distant phylogenetically with only 50% of sequence homology (Schmidt and Strittmatter, 2007; Tang et al., 2015). Alignment and comparison of amino acid sequence of mouse DPYSL5 with other DPYSL proteins show that the conservation level is lower at the C-terminal region (Fukada et al., 2000; Tang et al., 2015). The DPYSL associate generally in homo-tetramer or hetero-tetramer complexes with single or multiple isoforms.
Table 1.
List of known human DPYSL/CRMP genes and corresponding proteins.
| Human gene (HGNC) | Locus | Transcript RefSeq | Human protein | Alias for protein name | Protein RefSeq |
|---|---|---|---|---|---|
| CRMP1 | 4p16.2 | NM_001014809.3 | DPYSL1 | CRMP1 | NP_001014809.1 |
| DPYSL2 | 8p21.2 | NM_001197293.3 | DPYSL2 | CRMP2 | NP_001184222.1 |
| DPYSL3 | 5q32 | NM_001197294.2 | DPYSL3 | CRMP4 | NP_001184223.1 |
| DPYSL4 | 10q26.3 | NM_006426.3 | DPYSL4 | CRMP3 | NP_006417.2 |
| DPYSL5 | 2p23.3 | NM_001253723.2 | DPYSL5 | CRMP5 | NP_001240652.1 |
The official name of the human genes is indicated according to the Hugo Gene Nomenclature Committee (HGNC) nomenclature. The chromosomal locus of each gene corresponds to the Genome Reference Consortium Human Build 38 patch release 14 (GRCh38.p14). The reference sequence (RefSeq) for genes and protein was extracted from the National Center for Biotechnology Information (NCBI) database.
3. Physiological pathways involving DPYSL proteins
The DPYSL proteins participate in several major physiological pathways, from cellular migration, neurite growth and guidance to synapse maturation, particularly through their C-terminal domain, which includes the last 50 amino-acids, and which is the target of numerous post-translational modifications sites that regulate the interaction between DPYSL and various types of proteins, including receptors, ion channels, cytoskeletal and motor proteins.
As an example, numerous kinases such as glycogen kinase 3β (GSK3β), cyclin-dependent kinase 5 (Cdk5), dual specificity tyrosine phosphorylation-regulated kinase 2 (DYRK2) and Rho-associated kinase 2 (ROCK2) target the C-terminal regions of DPYSL proteins (Uchida and Goshima, 2005; Yoshimura et al., 2005; Cole et al., 2006; Arimura and Kaibuchi, 2007; Uchida et al., 2009; Yamashita and Goshima, 2012; Nakamura et al., 2014). Despite differences in the C-terminal part of DPYSL5 compared to other DPYSL proteins, it is noted that many consensus sequences for DPYSL phosphorylation sites are retrieved for DPYSL5 (Fukada et al., 2000).
The C-terminal domain is extensively conserved among DPYSL isoforms and across species, and is sufficient to associate with assembled microtubules in vivo (Soutar et al., 2009). Precisely, phosphorylation/dephosphorylation status of DPYSL proteins is essential to control their spatiotemporal functions, by modulating their binding to cytoskeleton and signaling proteins (Yamashita and Goshima, 2012).
Thus, DPYSL proteins can coordinate cytoskeleton dynamic regulating filopodia formation, axonal guidance, neurite outgrowth and establishment of neuronal polarity by interacting with tubulin and actin in brain (Fukata et al., 2002; Arimura et al., 2005; Hotta et al., 2005; Rosslenbroich et al., 2005; Brot et al., 2010; Higurashi et al., 2012; Ji et al., 2014; Khazaei et al., 2014; Tan et al., 2015; Gong et al., 2016; Yu-Kemp and Brieher, 2016). Non-phosphorylated DPYSL2 promotes axonal elongation and branching by binding to tubulin heterodimer (Schmidt and Strittmatter, 2007) whereas its phosphorylation by GSK3β, ROCK2 and Cdk5 lowers binding affinity of DPYSL2 to tubulin leading to growth cone collapse and arrest of axonal outgrowth (Fukata et al., 2002; Schmidt and Strittmatter, 2007; Khanna et al., 2012). The binding of DPYSL1-3 to tubulin allows polymerization and stabilization of microtubules (Fukata et al., 2002; Lin et al., 2011; Khazaei et al., 2014) while the tubulin-DPYSL4 or tubulin-DPYSL5 complex interaction causes inhibition of microtubule polymerization (Aylsworth et al., 2009; Brot et al., 2010).
In addition to phosphorylation, the functions of DPYSL proteins are also regulated by other post-translational modifications including acylation, SUMOylation and O-GlcNAcylation (Leney et al., 2017; Myllykoski et al., 2017; Chew and Khanna, 2018). For instance, DPYSL2 phosphorylation at Serine 522 by Cdk5 promotes association between DPYSL2 and cytoplasmic loops of CaV2.2 (Brittain et al., 2012; Chew and Khanna, 2018), leading to an increase of Ca2+ influx through the Cav2.2 channel and the release of neurotransmitters (Brittain et al., 2009, 2012). Similarly, SUMOylation of DPYSL2 alters calcium influx (Ju et al., 2013) and increases cell surface expression of NaV1.7 channel (Dustrude et al., 2016). Dephosphorylation of DPYSL2 at Thr514 and deSUMOylation at Lys374 sites promote the formation and maturation of dendritic spines, however, no interference is found between these two post-translational modifications in the regulation of dendritic spine morphology (Zhang et al., 2018).
4. Neuronal expression of DPYSL genes and proteins
Based on in situ hybridization (Wang and Strittmatter, 1996) and immunostaining analyses (Bretin et al., 2005), the DPYSL proteins are detected at a higher level in post-mitotic neural cells during the embryonic stage than in adult mouse brain stage (Wang and Strittmatter, 1997; Ricard et al., 2001). The mRNA expression level of Dpysl genes is intense during the neonatal period (Embryonic day 18 – Postnatal day 5) in the central nervous system of mice (Charrier et al., 2003). At Postnatal day 1, all DPYSL except DPYSL4 are strongly expressed in cortex and hippocampus (Wang and Strittmatter, 1996; Ricard et al., 2001) essential for social communication and cognitive functions. In addition, a peak of the expression level of DPYSL proteins is observed during the first postnatal week corresponding to a period of neuronal maturation and synaptogenesis (Byk et al., 1996; Wang and Strittmatter, 1996; Bretin et al., 2005; Schmidt and Strittmatter, 2007).
The BrainSpan transcriptome of the developing human brain shows a similar kinetics of DPYSL genes expression level, indicating their preponderant role in prenatal and perinatal periods when neurogenesis, dendritic development and synaptogenesis stages occur (Figures 1A–E) (Sunkin et al., 2013). Interestingly, DPYSL2 displays similar expression levels from prenatal to adult stages, suggesting that it may also have a role in later stages of development such as in myelination or synaptic pruning (Figures 1A–E). Indeed, DPYSL2 mediates Semaphorin 3F dependent synapse pruning (Ziak et al., 2020).
Figure 1.

Dynamic expression profile of DPYSL genes in the human brain. (A–E) Spatio-temporal profile of expression of DPYSL genes. The database used to perform the gene expression heatmap is BrainSpan. The level expression is expressed in RPKM (reads per kilobase of transcript per million reads mapped) and these data are obtained from RNA sequencing and exon microarray. Pcw, post-conceptual week. (F) DPYSL expression in different cell populations of the humain cerebrum. Data are from BBI allen single cell atlases. The single-cell atlases is realized from human fetal samples (72–129 days post-conceptual age).
A transcriptomic study of human fetal brain development (BBI Allen single cell atlases) using single-cell RNA sequencing indicated that around 25–30% of cells display DPYSL expression in cerebrum, except DPYSL4 which is very weakly expressed (only 2% of cells) (Cao et al., 2020). DPYSL mRNAs are mainly found in neurons (excitatory/inhibitory) but also in glial cells (Figure 1F), which suggest a contribution in neuronal degeneration/regeneration, as well as in inflammatory pathways in the context of neurological diseases inflammation and neurodegeneration pathways (Nagai et al., 2017).
5. Antagonistic/synergistic roles of DPYSL proteins
Each DPYSL protein displays a distinct subcellular neuronal localization both in time and space demonstrating their divergent functions during development (Goshima et al., 1995; Minturn et al., 1995; Byk et al., 1996; Wang and Strittmatter, 1996; Byk et al., 1998; Kamata et al., 1998; Bretin et al., 2005).
In primary hippocampal mouse neurons, during the axonogenesis, DPYSL2 is specifically enriched in neurite which is the future axon while DPYSL5 is strongly retrieved in dendrites maintaining dendrites at a quiescent state (Brot et al., 2010, 2014). At Days in vitro (DIV) 4–5, a switch is observed. DPYSL5 is detected at a very low level in dendrites and DPYSL2 level remains constant in same area allowing dendritic outgrowth (Brot et al., 2010). The transient DPYSL5 expression in different neuronal compartments regulates the establishment of neuronal polarity (Bretin et al., 2005; Brot et al., 2010, 2014). DPYSL5 forms a ternary complex with tubulin and microtubule associated protein 2 (MAP2) and inhibits the neurites outgrowth by reducing DPYSL2-tubulin interaction complex (Brot et al., 2010; Brot, 2014). It is not yet excluded that the inhibition of DPYSL2 activity by DPYSL5 may occur through their hetero-oligomerization as DPYSL2 and DPYSL5 have a very similar structure and form hetero-tetramer in vivo (Fukada et al., 2000; Brot et al., 2010; Petratos and Lee, 2013; Ponnusamy and Lohkamp, 2013).
Despite that DPYSL5 does not inhibit axonal growth (Brot et al., 2010), its deficiency results in an increase on DPYSL2-induced axon elongation and on multiple axon formation (Inagaki et al., 2001). These results suggest that in vivo DPYSL5 also modulates DPYSL2 activity on axonal growth and formation.
Although the biological functions associated with each homo- or hetero-tetramer of DPYSL proteins are still poorly known, it has been demonstrated that DPYSL2 and DPYSL3 are complexing and work together to regulate growth cone development and axonal elongation in vivo (Tan et al., 2015). For instance, overexpression of DPYSL2 and DPYSL3 in hippocampus stimulate axonal growth and this effect is abolished when DPYSL2 is co-transfected with the truncated construct DPYSL3ΔC471 (unable to bind actin) or when DPYSL3 is co-transfected with DPYSL2ΔC322 (unable to bind tubulin). These findings suggest that DPYSL2/DPYSL3 hetero-tetramer complex creates a link between microtubules and actin, aiming to coordinate cytoskeleton dynamics, and axonal development regulation in hippocampal neurons (Tan et al., 2015). These findings illustrate the various actions of DPYSL proteins and highlight their ability to form homo- or hetero-tetramer complex, in order to modulate and regulate the function of other DPYSL proteins during neural network formation.
DPYSL proteins appear to play antagonistic but also complementary roles during neurodevelopment (Byk et al., 1998; Yuasa-Kawada et al., 2003; Brot, 2014; Makihara et al., 2016). In vivo studies demonstrate that DPYSL1 and DPYSL2 have synergistic but distinct roles in mediating Sema3A signaling in order to regulate dendritic development and spine maturation (Hamajima et al., 1996; Sasaki et al., 2002; Morita et al., 2006; Yamashita et al., 2007, 2012).
In fact, abnormalities were observed in dendritic patterning (branching and length dendritic) of cortical (layer V) neurons from distinctly Dpysl1−/− and Sema3A−/− mouse model, compared to their littermate neurons. These defects in dendritic morphology are not retrieved in KO Dpysl2−/− and double-heterozygous KO Dpysl1+/–Dpysl2+/− mouse models. Moreover, the level of DPYSL1 increases in Dpysl2−/− compared to wild-type cortical brain lysates, highlighting a DPYSL1 compensatory mechanism for DPYSL2 deficiency (Diss et al., 2014; Makihara et al., 2016). A proteomic analysis in cortex of Dpysl2ki/ki mice (where serine 522 is mutated to alanine preventing its potential phosphorylation) demonstrated an increase of DPYSL3, DPYSL4 and DPYSL5 (Nakamura et al., 2018) as well as in Dpysl2−/− (Nakamura et al., 2016), thereby suggesting that the phosphorylation or loss of functions of DPYSL2 have an impact on other DPYSL proteins.
A study from Yamashita and colleagues showed that both DPYSL1 and DPYSL2 are required for regulating dendritic branch trajectory in cerebral cortical neurons reinforcing their synergistic role in dendritic organization (Yamashita et al., 2012). In addition, DPYSL1-4 may have a redundant role in dendritic growth and maturation in neurons (Quach et al., 2008; Khazaei et al., 2014; Cha et al., 2016; Makihara et al., 2016; Takaya et al., 2017; Kawashima et al., 2021).
6. Synaptic functions of DPYSL proteins
6.1. Role in the formation and maturation of dendritic spines
When maturation of neurons and synaptic connections is strongly active (around first postnatal week in rodents), DPYSL expression is the highest (Charrier et al., 2003). All five DPYSL proteins are expressed in synaptosomes from rat brain at neonatal postnatal day 1 (P1) (Charrier et al., 2006; Brittain et al., 2009; Yamashita et al., 2012) and are postsynaptic density (PSD) proteins (Collins et al., 2006; Laumonnier et al., 2007), suggesting a role in synaptogenesis and neurotransmission. Dendrites are the first site of synapse formation (Purves and Hume, 1981) and synaptogenesis represents an essential process for the establishment of cognitive and communication function as well as for learning and memory (Elston, 2000).
Studies on genetic deletion of Dpysl members in mice establish a direct link between loss of DPYSL and impairment of dendritic patterning and spine development (Table 2; Charrier et al., 2006; Quach et al., 2008; Yamashita et al., 2012). The synaptic density is reduced in Dpysl1−/−, Dpysl2−/− mutant mice (Yamashita et al., 2007; Makihara et al., 2016).
Table 2.
Neuronal and behavioral phenotypes observed in mouse models invalidated for the Dpysl/Crmp genes.
| Mouse genotype | Neuroanatomical and cellular phenotype | Molecular defect | Electrophysiology | Behavioral phenotype | References |
|---|---|---|---|---|---|
| Dpysl1−/− or Crmp1−/− | - Abnormal dendritic development of CA1 pyramidal neurons - Reduction of synapse density in CA1 hippocampus - Reduced number of mature dendritic spines in cortical neurons |
- Decreased PDS95 and GAP-43 protein levels in CA1 hippocampus | - Decreased LTP in CA1 hippocampus | - Hyperactivity, impaired emotional behavior - Decreased pre-pulse inhibition - Impaired learning and memory |
Su et al. (2007) and Yamashita et al. (2007, 2013) |
| Dpysl2−/− or Crmp2−/− | - Altered dendritic morphology (number spine and dendritic branching) of CA1 pyramidal neurons and cortical neurons (layer V) - Altered dendritic spine pruning in dentate gyrus - Abnormal axon pruning arising from hippocampus and visual cortex - Dysgenesis of corpus callosum |
- Abnormal NMDA receptor composition - Decreased level of synaptic proteins NSF, PRKACB, GNAI1, GRIA2, SNAP25 - Increased level of synaptic proteins SHANK3, SHANK2, GRIA1 | - Reduced LTP induction in hippocampus | - Decreased anxiety - Hyperactivity - Impaired social behavior, learning and memory - Defects in locomotor activity |
Nakamura et al. (2016), Zhang et al. (2018), and Ziak et al. (2020) |
| Dpysl3−/− or Crmp4−/− | - Increased dendritic total length and branching in primary hippocampal neurons - Defective infrapyramidal bundle of mossy fibers of the dentate gyrus (DG) pruning in the hippocampus |
- Altered mRNA expression levels of genes related to neurotransmission and cell adhesion in hippocampus, cortex and olfactory bulb |
- Decreased social interaction - Alterations of sensory responses (temperature and olfactory) |
Niisato et al. (2012), Tsutiya et al. (2016), Takaya et al., 2017, and Tsutiya et al. (2017) | |
| Dpysl4−/− or Crmp3−/− | - Abnormal dendrite and spine morphogenesis in hippocampus - Defect in infrapyramidal bundle of mossy fibers pruning in hippocampus |
- Impairment of LTP induction in CA1 hippocampus | Quach et al. (2018) | ||
| Dpysl5−/− or Crmp5−/− | - Aberrant Purkinje cell morphology in cerebellum | - Impaired LTD induction between parallel fibers and Purkinje cells | - Abnormal limb-clasping reflexes | Yamashita et al. (2011) |
The SEMA3A protein is essential for induction of mature spines formation through the Fyn-Cdk5 cascade in cultured cortical neurons (Sasaki et al., 2002; Li et al., 2004; Cole et al., 2006; Morita et al., 2006; Figure 2). Nevertheless, Sema3A is not able to induce an increase in functional synapses density in cortical neurons from Dpysl1−/− and Cdk5−/− mice (Yamashita et al., 2007). Several studies revealed the importance of CDK5 phosphorylation of DPYSL1 at Thr509 and Ser522 sites and of DPYSL2 at Ser522 site for SEMA3A-induced spine development and maturation (Yamashita et al., 2007; Jin et al., 2016; Makihara et al., 2016). Conversely, DPYSL2 dephosphorylated forms increase the number of dendritic spines and the amplitude of miniature excitatory postsynaptic currents (mEPSCs) (Zhang et al., 2018). This suggest that dephosphorylated forms of DPYSL2 promotes polymerization of tubulin (Fukata et al., 2002; Uchida and Goshima, 2005) and thus, spinogenesis. A recent study demonstrated that DPYSL2 is not only a mediator of Sema3A-signaling regulating spine development but also plays a key role in synaptic refinement through Semaphorin 3F (Ziak et al., 2020). Loss of Dpysl2 causes axonal pruning defects and inadequate elimination of dendritic spines in multiples areas of the brain and in cultures of hippocampal neurons (Table 2). This defect is accompanied by social behavior abnormalities (see section “DPYSL genes and neurodevelopmental disorders”).
Figure 2.

Regulation of dendritic spine maturation in cortical neurons by DPYSL proteins through Sema3A-cdk5 signaling pathway. We propose a model involving the Sema3A-DPYSL pathway in the formation and maturation of dendritic spines. Fyn phosphorylates semaphorin receptor (Plexin A2) and facilitates PlexinA2 interaction to Sema3A. Fyn also promotes the phosphorylation of kinases like cyclin dependent kinase-5 (Cdk5) which in turn phosphorylates DPYSL proteins. We suggest that DPYSL proteins will thus play a role in the genesis and maturation of dendritic spines. AS an example, DPYSL1 can regulate spine development through Sema3A–Cdk5 signaling. Sema3A binds a receptor complex of the transmembrane proteins Neuropilin-1 (NP-1) and Plexin A1 (Plex-A1). After interacting with plexin A1, Fyn stimulates kinase activity of Cdk5 via Tyr15 phosphorylation of Tyr15. Cdk5 Tyr15 phosphorylation will then phosphorylates DPYSL1 tetramer at Thr509 and Ser522 locations. Sema3A through phosphorylation of DPYSL1 by Cdk5 will increase the density of PSD-95 and synapsin I clusters at dendrites and thus, promote formation of mature spines in cultured cortical primary neurons.
DPYSL3 is also critical for spine formation and maturation in cultured hippocampal neurons via the interaction with actin cytoskeleton by its C-terminal region (Rosslenbroich et al., 2005; Cha et al., 2016). Overexpression of DPYSL3 wild-type or DPYSL3 with actin-binding domain constructs increase frequency of mEPSCs in comparison with control GFP or with form of DPYSL3ΔC471 (lacking the domain of interaction with actin) transfected neurons. These results indicate that DPYSL3-actin interaction increases number of functional synapses and thus, influences synaptic transmission (Cha et al., 2016). Similarly, DPYSL5 deficiency in cerebellum induces an aberrant Purkinje cell morphology. In Dpysl5+/− mice, Brain-derived neurotrophic factor (BDNF) increased the number of primary dendrites per neurons in the hippocampus while this effect is lost in neurons from complete KO Dpysl5−/− brains (Table 2). Consequently, they demonstrate that DPYSL5 phosphorylation by TrkB is involved in BDNF–TrkB signaling to regulate dendritic morphology and synaptic plasticity in Purkinje cells (Yamashita et al., 2011).
The phosphorylated/dephosphorylated state of DPYSL proteins seems to be crucial for the regulation of their interaction with cytoskeleton proteins and for the control of dendritic architecture (Arimura et al., 2005; Yamashita and Goshima, 2012; Makihara et al., 2016; Zhang et al., 2018). Several post-translational modifications of DPYSL2 allow modulation of membrane addressing of the CaV2.2 and NaV1.7 ion channels, as well as the formation and maturation of dendricic spines (see section Physiological pathways involving DPYSL proteins). These data highlight the importance of future research on their post-translational modifications and associated signaling pathways to clarify their function in synapse formation and in neurotransmission.
6.2. Role in physiology and synaptic plasticity
In addition to synapse formation process, DPYSL proteins interact with presynaptic and postsynaptic machinery and may also have a role in synaptic plasticity. For instance, loss of Dpysl1-4 in murine models cause dysregulation of genes expression related to excitatory and/or inhibitory synaptic transmission explaining synaptic plasticity dysfunction (Yamashita et al., 2011; Tsutiya et al., 2015; Zhang et al., 2016; Tsutiya et al., 2017). In fact, abnormal NMDA receptor composition, including GluN2B and GluN1, is observed in hippocampus of Dpysl2 knock-out (KO) mice resulting in a reduction of long-term potentiation (LTP) induction and in defects in learning function (Zhang et al., 2016). Dpysl2−/− mice also showed altered expression of proteins involved in GABAergic synapse (NSF, PRKACB, GNAI1), glutamatergic synapse (GRIA2, PRKACB, GNAI1, SHANK3, SHANK2, GRIA1) and neurotrophin signaling pathways (Table 2). These alterations of both inhibitory and excitatory synapse related proteins may contribute to the behavioral phenoytpe of these mice (Nakamura et al., 2016).
An altered LTP is found in CA1 hippocampi neurons of Dpysl1−/− and Dpysl4−/− mice models (Table 2; Su et al., 2007; Quach et al., 2008). Deletion of Dpysl1 leads to a decrease in the expression of GAP43 and PSD95 proteins (Su et al., 2007) and inactivation of Dpysl3 also disturbs the mRNA expression levels of genes encoding GluR1, GluR2, VgluT1, VgluT2, GABAɑ1, GABAAγ2, GABAB receptor 1 and vGAT, in a region-dependent manner (Tsutiya et al., 2017). To date, no reports have shown that DPYSL5 is required for LTP formation, but in cerebellum of Dpysl5−/− mice the induction of long-term depression (LTD) is deficient between parallel fibers and Purkinje cells (Table 2; Yamashita et al., 2011). Consequently, involvement of DPYSL in LTP and in LTD is a critical mechanism for memory and learning processes (Malenka and Bear, 2004; Stacho and Manahan-Vaughan, 2022).
In parallel, three studies showed the involvement of DPYSL proteins in the dynamic trafficking of AMPA receptors (AMPARs) (Khazaei et al., 2014; Lin et al., 2019) and NMDA receptors (NMDARs) (Bretin et al., 2006). It is well-known that the trafficking of glutamatergic receptor, which enables the endocytosis, recycling and exocytosis of receptors is crucial for synaptic strength and plasticity. Moreover, the interaction between dephosphorylated DPYSL2 and endophilin2 promotes insertion of the GluA1 subunit of AMPARs to the post-synaptic membrane and increases amplitude and frequency of mEPSCs in cultured hippocampal neurons (Figure 3; Zhang et al., 2018). In contrast, DPYSL2 downregulates the amount of the NR2B subunit of the NMDARs on the surface of cortical neurons (Bretin et al., 2006). DPYSL5 protein also regulate the endocytosis of GluA1 subunit of the AMPARs via phosphorylation of GluA2 at Serine 880, illustrating a specific function of DPYSL5 at glutamatergic synapses (Lin et al., 2019; Figure 3). On the other hand, it is also shown that DPYSL5 could modulate GluA2 endocytosis via GluA2 phosphorylation site on Serine 880 (S880), triggering social deficit (Lin et al., 2019).
Figure 3.

Representation of the contribution of DPYSL2 and DPYSL5 proteins in the control of synaptic plasticity. DPYSL2 binds and regulates the trafficking of both voltage gated Na2+ (NaV1.7) and Ca2+ (CaV2.2) channels at presynaptic terminal. DPYSL2 phosphorylation at Ser 522 by Cdk5 promotes its binding to Cav2.2. This interaction causes an increased number of CaV2.2 at cell surface leading to an increase in Ca2+ influx and glutamate release. SUMOylation is enhanced by phosphorylation of DPYSL2 through CDK5 action. This SUMOylation induces an increase of NaV1.7 channel at surface and in neuronal excitability. At postsynaptic level, DPYSL2 phosphorylation by GSK3β inhibits interaction with endophilin2 and reduces the number of GluA1 subunits of AMPARs at membrane. Similarly, DPYSL5 via GluA2 S880 phosphorylation can modulate traffic at the surface of the GluA2 subunit of AMPA receptors. (Adapted from Lin et al., 2019; Stratton et al., 2020; Henley et al., 2021).
Complementary to their role at the postsynaptic level, DPYSL are also expressed at presynaptic terminal. DPYSL2 and DPYSL4 have been identified as main regulators of ion currents voltage dependent (Brittain et al., 2009; Quach et al., 2011, 2013). Alike, DPYSL4 facilitates the depolarization-evoked Ca2+ response of L- and N-type Ca2+ channels to promote dendrite morphogenesis of hippocampal neurons (Quach et al., 2013; Figure 3). DPYSL2 binds and regulates the trafficking to membrane of both presynaptic voltage-gated Na2+channels (NaV1.7) (Dustrude et al., 2013, 2016, 2017) and N-type voltage-gated Ca2+ channel (CaV2.2) (Brittain et al., 2009, 2011; Moutal et al., 2016).
Several post-translational modifications of DPYSL2 allow modulation of membrane addressing. For instance, DPYSL2 phosphorylation at Serine 522 by Cdk5 promotes association between DPYSL2 and cytoplasmic loops of CaV2.2 (Brittain et al., 2012; Chew and Khanna, 2018), leading to an increase of Ca2+ influx through the Cav2.2 channel and the release of neurotransmitters (Brittain et al., 2009, 2012). Similarly, SUMOylation of DPYSL2 alters calcium influx (Ju et al., 2013) and increases cell surface expression of NaV1.7 channel (Dustrude et al., 2016). These findings suggest that DPYSL2 can regulate synaptic activity and plasticity by modifying the membrane localization of ion channels and thus controlling associated currents (Figure 3). Moutal et al. identified syntaxin1 as a novel DPYSL2 protein partner (Moutal et al., 2017), and this protein is involved in synaptic vesicle endocytosis neurotransmitter release (Rizo, 2022).
Interestingly, DPYSL3 interacts with proteins involved in synaptic vesicle recycling (Quinn et al., 2003) and electrophysiological experiments demonstrated that DPYSL3 enhances Ca2+ current density in hippocampal neurons (Wang et al., 2010).
Together, the DPYSL proteins act as neuromodulators of Ca2+ channel function and seem to play a major role in synaptic vesicle exocytosis and transmitter releasing in synaptic cleft (Figure 3).
Thus, the combination of these findings converged on the fact that DPYSL proteins might be key regulators of synapses architecture and activity via an interaction with cytoskeletal proteins but also with synaptic scaffolding proteins. Future protein interaction studies shall further clarify DPYSL protein interactome at the synapses.
7. DPYSL genes and neurodevelopmental disorders
Consistently with the major role of DPYSL proteins in dendritic organization and in formation and maturation of synapse, various studies suggested that they would contribute in the pathophysiology of psychiatric diseases such as schizophrenia and NDDs (Table 3; Edgar et al., 2000; Charrier et al., 2003; Hong et al., 2005; Beasley et al., 2006; Bader et al., 2012; Braunschweig et al., 2013; Yamashita et al., 2013; Lee et al., 2015; Quach et al., 2015; Tsutiya et al., 2017; Quach et al., 2021; Murtaza et al., 2022) (database SFARI, denovo-db). Interestingly, dendritic and spine dysfunctions are described in NDDs including schizophrenia, Down’s syndrome, Fragile X syndrome, Rett syndrome and ASD (Huttenlocher, 1991; Kaufmann and Moser, 2000; Martínez-Cerdeño, 2017; Nelson and Bender, 2021; Quach et al., 2021). Dpysl KO mouse models displayed morphological abnormalities in neurons as well as behavioral defects similar to those found in schizophrenia (hyperactivity, learning and memory deficits...) or in ASD (Yamashita et al., 2013; Nakamura et al., 2016; Tsutiya et al., 2017; Ohtani-Kaneko, 2019).
Table 3.
Summary table of de novo heterozygous missense variants in DPYSL genes and their contribution in neurodevelopmental diseases.
| Genes | Allele change | Residue change | Behavioral phenotype | References |
|---|---|---|---|---|
| DPYSL1 | c.1052T > C | p.Phe351Ser | ID, behavioral problems | Ravindran et al. (2022) |
| c.1280C > T | p.Thr427Met | ASD, no ID, delayed motor development | Ravindran et al. (2022) | |
| c.1766C > T | p.Pro589Leu | ID, ASD, delayed motor development | Ravindran et al. (2022) | |
| DPYSL2 | c.42C > A | p.Ser14Arg | ID | Suzuki et al. (2022) |
| c.1028G > A | p.Arg343His | ASD | Satterstrom et al. (2020); Database: SFARI | |
| c.1312C > A | p.His438Asn | ASD | De Rubeis et al. (2014); Database: SFARI, de novo-db | |
| c.1693C > T | p.Arg565Cys | ID | Suzuki et al. (2022) | |
| c.1801C > T | p.Arg601Cys | ASD | Iossifov et al. (2014); Database: SFARI, de novo-db | |
| DPYSL3 | c.415G > A | p.Val139Ile | ASD | Iossifov et al. (2014); Database: SFARI, de novo-db |
| c.1622C > A | p.Ser541Tyr | ASD | Tsutiya et al. (2017) | |
| DPYSL5 | c.121G > A | p.Glu41Lys | Severe ID Behavioral problems | Jeanne et al. (2021); Database: de novo-db |
| c.139G > A | p.Gly47Arg | ID Ritscher-Schinzel syndrome | Jeanne et al. (2021) | |
| c.241G > A | p.Asp81Asn | Developmental disorder | Database: de novo-db | |
| c.1090G > A | p.Val364Ile | ASD | Database: de novo-db |
Fragile X mental retardation protein (FRMP) encoded by the FMR1 gene, is an RNA binding protein involved in fragile X syndrome, and regulates the function of many neuronal mRNAs crucial for neuronal development, synaptic plasticity and dendritic spine architecture (Banerjee et al., 2018). Interestingly, a proteomic analysis on extracts of nucleus laminaris from chicken identified CRMP1 and DPYSL2 as candidate substrates for FMRP (Sakano et al., 2017). Post-transcriptional modifications of DPYSL proteins, such as SUMOylation, impact their function in synapse, which is of particular interest for Fragile X syndrome since the activation of mGluR5 receptors promotes the SUMOylation of FRMP, leads to the dissociation of FRMP from mRNA granules to regulate spine elimination and maturation (Khayachi et al., 2018). Moreover, DPYSL2 protein expression can be controlled by the mTOR signaling pathway that is dysregulated in fragile X syndrome (Sharma et al., 2010). Both DPYSL2 and mTOR are associated with common physiological functions such as neuronal polarity, axonal outgrowth and synaptic strength as well as brain disorders including schizophrenia (Pham et al., 2016; Na et al., 2017; Izumi et al., 2022). Taken together, these data suggest that deregulation of the mTor-DPYSL2 molecular pathway may be involved in NDDs such as schizophrenia or ID.
For instance, genetic variants of DPYSL1 or DPYSL2 genes and alteration of DPYSL1 and DPYSL2 proteins levels have been reported in post-mortem brains of schizophrenic patients (Beasley et al., 2006; Martin-de-souza et al., 2010; Nomoto et al., 2021). Additionally, a link between DPYSL1 and DPYSL2 and the maternal antibody-related ASD subtype (MAR ASD) has been established (Braunschweig et al., 2013; Ramirez-Celis et al., 2021). Maternal antibodies in the placenta target fetal proteins and would cause alterations in neurodevelopment leading to behaviors associated with autism. A recent study highlighted that maternal IgG reactivity during pregnancy to both DPYSL1 + DPYSL2 increased at 16-fold the odds of an ASD diagnosis compared to the control group and over 6-fold relative to the ID group (Ramirez-Celis et al., 2022). This pattern DPYSL1 + DPYSL2 of MAR ASD is associated with ASD + ID diagnosis and ASD no-ID (Ramirez-Celis et al., 2022).
In addition to DPYSL1-2, DPYSL3, and DPYSL5 are also involved in psychiatric disorders with the description of de novo missense mutations in DPYSL2, 3, and 5 in individuals with NDDs (Table 3 and Figure 4).
Figure 4.
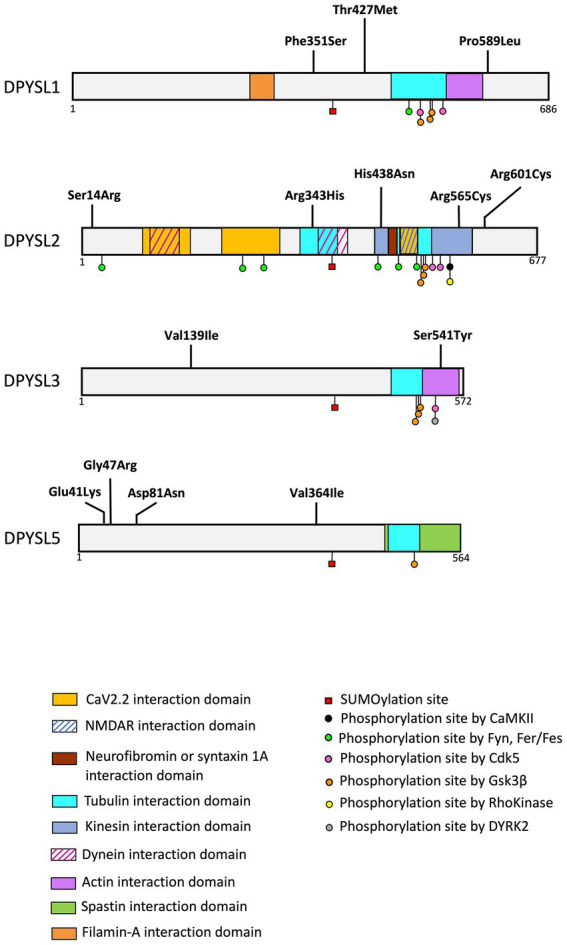
Schematic representation of protein interaction domains, post-translational. The interaction between DPYSL proteins and the represented proteins was identified in animal models (mouse, rat). Alignment of the amino acid sequences corresponding to these domains demonstrates sequence conservation in humans. The list of binding sites and post translational modifications provided in the figure is non-exhaustive. (Adapted from Nakamura et al., 2020).
7.1. Genetic variants in DPYSL1
A recent study reported heterozygous de novo variants in the DPYSL1 gene in three unrelated individuals with muscular hypotonia, ID and/or ASD (Table 3, Figure 4) (Ravindran et al., 2022). Whole exome sequencing identified two variants associated with ID (p.Pro589Leu for the long isoform of DPYSL1 or p.Pro475Leu for the short isoform; p.Phe351Ser for the long isoform or p.Phe237Ser for the short isoform), and one variant in an individual with ASD (p.Thr427Met for the long isoform or p.Thr313Met for the short isoform). These variants are predicted to affect the ternary structure of DPYSL1 and to impact the oligomerization of DPYSL1 proteins. When using the short isoform of DPYLS1 protein, the p.Thr313Met and p.Pro475Leu variants are positioned next to the dimer/tetramer interface of CRMP1B, and they impair the homo-oligomerization of DPYSL1. The overexpression of variants p.Thr313Met and p.Pro475Leu in mouse cortical neurons caused a decrease in neuritic outgrowth (Ravindran et al., 2022), which is a morphological phenotype associated with many neurodevelopmental disorders (Quach et al., 2015; Prem et al., 2020). Interestingly, Dpysl1−/− mice have defects in dendritic spines (Yamashita et al., 2007; Makihara et al., 2016) and an inability to induce LTP (Su et al., 2007). In addition, these mice show schizophrenia like behaviors (Yamashita et al., 2013).
7.2. Genetic variants in DPYSL2
Three de novo missense variants in DPYSL2, predicted deleterious in silico, were described in individuals with ASD from the Autism Sequencing consortium (variant p.His438Asn) (Veron et al., 2018) and in two individuals of the Simons Simplex Collection (p.Arg343His, p.Arg601Cys) (Iossifov et al., 2014; Satterstrom et al., 2020). Of interest, a recent study described two unrelated patients with ID and hypoplasia of the corpus callosum associated with a de novo missense variant (p.Ser14Arg or p.Arg565Cys) of DPYSL2 (Table 3 and Figure 4; Suzuki et al., 2022).
Functional assays in zebrafish model showed that p.Ser15Arg and p.Arg566Cys variants (corresponding to codons Ser14 and Arg 565 of human DPYSL2) led to the loss of function of DPYSL2 protein. Cell transfection experiments of DPYSL2 protein variants demonstrated that both mutations caused a decrease in DPYSL2 protein levels, probably due to increased degradation by the proteasome. Moreover, both variants impaired DPYSL2 interaction with tubulin. These results collectively support the pathogenity impact of p.Ser14Arg and p.Arg565Cys variants causing intellectual disability in humans (Suzuki et al., 2022). It is interesting to note that the patients described by Suzuki et al., have dysplasia of the corpus callosum which has also been found in Dpysl2−/− mouse model that display a dysgenesis of corpus callosum and defects in callosal axon guidance (Ziak et al., 2020).
In mice, Dpysl2 deficiency induces a reduction of spine density and dendritic branching in CA1 hippocampal neurons and in layer V of cortical neurons of mice (Makihara et al., 2016; Zhang et al., 2016). Moreover, brain-specific Dpysl2-KO mice display hyperactivity and social, cognitive and affective behavioral impairments, reminiscent of deficits associated with schizophrenia (Zhang et al., 2016). On the other hand, total deletion of Dpysl2 in mice leads to histological and behavioral alterations similarly to “ASD-related phenotype” such as axonal pruning defects and inadequate elimination of dendritic spines in dentate gyrus of hippocampi (Ziak et al., 2020). Very interestingly, a defect in synaptic pruning in layer V pyramidal neurons has been reported in temporal lobe of postmortem ASD patients (Tang et al., 2014). Furthermore, Dpysl2−/− mice exhibit ASD-related social behavior changes such as ultrasonic vocalization deficits in the early postnatal period (P8, P12) and social behavioral deficits in adult (Ziak et al., 2020).
7.3. Genetic variants in DPYSL3
The genetic analysis of the Simon Simplex Collection reported two de novo missense variants (p.Ser541Tyr and p.Val139Ile) of the DPYSL3 gene associated with ASD (Iossifov et al., 2014; Tsutiya et al., 2017; Table 3 and Figure 4).
The study of Dpysl3-KO cultured hippocampal neurons showed that DPYSL3 deficiency was associated with longer dendrites with more branching (Tsutiya et al., 2017). The Dpysl3-KO neurons transfected with pEGFP-DPYSL3S540Y exhibited an increasing in dendritic branching compared to control Dpysl3-KO neurons transfected with pEGFP-DPYSL3WT (the human DPYSL3 Serine 541 corresponds to mouse DPYSL3 codon Serine 540). To conclude, the p.Ser541Tyr mutation alters dendritic morphology and impairs the function of DPYSL3 (Tsutiya et al., 2017).
In addition, Dpysl3-KO mice exhibit several ASD-like phenotypes, including deficits in social interaction (determined by the three-chambers test) and alterations of sensory response measured by the emission of ultrasonic vocalization of mouse pups after different sensory stimuli. Interestingly, the serine 541, which is mutated into Tyrosine in an ASD patient (Tsutiya et al., 2017), is a phosphorylation site of DPYSL3 (Figure 4; Mertins et al., 2016) (database PhosphositePlus). As phosphorylation is essential for DPYSL cellular functions, it is plausible that DPYSL3 p.Ser541Tyr mutation may cause a loss-of-function of DPYSL3 leading to defects in dendritic arborization associated with behavioral deficits. Furthermore, Tsutiya and colleagues highlighted that Dpysl3 deficiency altered mRNA expression of Gria1 and Gria2 (encoding GLUR1 and GLUR2 subunits of the AMPA receptor), essential for dendritic development and maturation (Chen, 2009). In addition, it remains essential to highlight that previous studies also revealed contribution of these two AMPA receptors subunits in mice with social deficits and in patients with ASD or other NDDs (Purcell et al., 2001; Ramanathan et al., 2004; Essa et al., 2013; Erickson et al., 2014; Uzunova et al., 2014; Kim et al., 2019).
7.4. Genetic variants in DPYSL5
The DPYSL5 gene (and its respective protein DPYSL5) is the latest discovered member of DPYSL family (Fukada et al., 2000; Inatome et al., 2000; Ricard et al., 2001), and has been recently described as a novel candidate gene for NDDs (Table 3 and Figure 4). An international collaboration allowed to identify nine families including patients with ID associated with cerebral malformations, and carriers of de novo heterozygous missense variants in DPYSL5. A recurrent de novo variant p.Glu41Lys has been identified in eight unrelated subjects with ID, corpus callosum agenesis and posterior fossa abnormalities. Furthermore, a p.Gly47Arg variant was found in two sisters with Ritscher-Schinzel syndrome (Jeanne et al., 2021). It is critical to note that all individuals with p.Glu41Lys and p.Gly47Arg mutations in DPYSL5 display an agenesis of corpus callosum which is a neuroanatomical malformation already associated with ASD and ID (Halgren et al., 2012; Wegiel et al., 2018; Li et al., 2019; Mimura et al., 2019; Nabais Sá et al., 2020; Qi et al., 2022).
Very interestingly, a dysgenesis of the corpus callosum has also been described for the two patients with ID and carrying the mutations p.Ser14Arg and p.Arg565Cys in DPYSL2 gene. Another similarity worth mentioning is a hypoplasia of the cerebellum in patients carrying both variants of DPYSL5 which is also found in the patient with mutation p.Ser14Arg of DPYSL2 gene (Jeanne et al., 2021; Suzuki et al., 2022). DPYSL5 protein may form a homo- or hetero-tetramer with DPYSL2-4 (Wang and Strittmatter, 1997; Stenmark et al., 2007). As previously described in Jeanne et al. publication, DPYSL5 p.Glu41Lys and p.Gly47Arg variants do not affect oligomeric assembly but by adding a positive charge to the electrostatic surface of the protein, which may alter the interaction between DPYSL5 and its partners (Jeanne et al., 2021). It is well-characterized that primary neuronal cultures, overexpressing DPYSL5 inhibits tubulin polymerization and neurite growth (Brot et al., 2014). However, overexpression of missense variants of DPYSL5 results in the loss of the inhibitory regulation of DPYSL5 on dendritic growth. Both mutations altered the function of DPYSL5 by preventing the formation of the complex with MAP2 and βIII-Tubulin. In addition, p.Gly47Arg substitution increased the binding of DPYSL5 to DPYSL2 (Jeanne et al., 2021). Thus, it has been hypothesized that p.Gly47Arg modulates the neuronal function of DPYSL2 by increasing the formation of DPYSL2/DPYSL5 complex. This study highlighted the importance of DPYSL5 in neuronal development and put forward that defect in these regulatory mechanisms is responsible for a syndromic form of NDD with brain anomalies.
No studies have reported behavioral defects related to NDDs in Dpysl5 mutant animal models but Lin et al., demonstrated that the hippocampal overexpression of DPYSL5 triggers social interaction deficits in both control mice and in 3xTg-Ad mice, a classical mouse model of Alzheimer’s disease (Lin et al., 2019) suggesting that DPYSL5 closely controls social behavior. Overall results reveal that impairments in DPYSL2, DPYSL3 and DPYSL5 functions can lead to ID, ASD or schizophrenia. Although genetic causes of ASD and ID include mutations in genes coding for proteins involved in various pathways, such as chromatin remodeling, transcriptional regulation or the dynamics and reorganization of the cytoskeleton, a majority of genes/proteins mutated in NDDs contribute to the architecture and activity of the synapses (Guilmatre et al., 2009; Pavlowsky et al., 2012). Thus, in this review we have provided compelling evidence that dysregulation of DPYSL expression may also impair synaptic function and consequently lead to early-onset cognitive disorders, demonstrating that DPYSL genes and proteins defects may also contribute to “synaptopathies.”
8. Discussion
The DPYSL proteins family appear to be involved in various biological events during the development including differentiation, axon guidance, neurites extension, dendritic branching and axonal regeneration (Ip et al., 2014). Here, we gathered various evidence from an extensive review of the literature that DPYSL genes and proteins are necessary for regulating the formation and the maturation of synapses, the neurotransmission and synaptic plasticity, mainly due to their synaptic localization at both pre and post synaptic terminals (Collins et al., 2006; Laumonnier et al., 2007; Brittain et al., 2009, 2011).
Genetic deletion of Dpysl in mice leads to synaptic impairment as well as cognitive and behavioral disorders, which are common defects associated with NDDs. As summarized in Table 3, genetic studies uncovered the contribution of de novo missense mutations in the DPYSL genes in NDDs (Iossifov et al., 2014; Veron et al., 2018; Satterstrom et al., 2020; Jeanne et al., 2021; Suzuki et al., 2022) (database: SFARI; denovo-db), suggesting their central role in the brain formation and functioning and the pathogenesis of NDDs. This review also highlights that DPYSL may have antagonistic or complementary activity and that their predisposition to homo- and hetero-oligomerization may have a direct impact on their physiological role. It is likely that the localization of variants in specific interaction and/or functional domains necessary for oligomerization of DPYSL proteins may have a consequence on their synaptic functions and thus lead to NDD.
Further understanding of signaling pathways located upstream and downstream of DPYSL for each homo or hetero-tetramer assembly will likely help to elucidate the physiological contribution of DPYSL proteins during brain formation and maturation and the pathogenic mechanisms leading to neurodevelopmental disorders.
Online database
BBI-Allen single cell atlases, https://descartes.brotmanbaty.org/ (accessed January 19, 2023).
National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/ (accessed January 19, 2023).
SFARI gene, https://gene.sfari.org/ (accessed January 19, 2023).
Denovo-DB, https://denovo-db.gs.washington.edu/denovo-db/ (accessed January 19, 2023).
PhosphoSitePlus, https://www.phosphosite.org/ (accessed January 19, 2023).
Author contributions
FD and FL wrote the first draft of the manuscript. DU, PV, and MJ contributed in the revision of the initial version. All authors revised and approved the final version of the manuscript submitted for publication.
Funding
FD was supported by a PhD fellowship from the Région Centre Val de Loire. Work in the lab of FL devoted to DPYSL is supported by the Inserm (GOLD cross-cutting program on genomic variability), the Association pour le Développement de la Neurogénétique.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- Arimura N., Kaibuchi K. (2007). Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 8, 194–205. doi: 10.1038/nrn2056 [DOI] [PubMed] [Google Scholar]
- Arimura N., Ménager C.´, Kawano Y., Yoshimura T., Kawabata S., Hattori A., et al. (2005). Phosphorylation by rho kinase regulates CRMP-2 activity in growth cones. Mol. Cell. Biol. 25, 9973–9984. doi: 10.1128/MCB.25.22.9973-9984.2005, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylsworth A., Jiang S. X., Desbois A., Hou S. T. (2009). Characterization of the role of full-length CRMP3 and its calpain-cleaved product in inhibiting microtubule polymerization and neurite outgrowth. Exp. Cell Res. 315, 2856–2868. doi: 10.1016/j.yexcr.2009.06.014, PMID: [DOI] [PubMed] [Google Scholar]
- Bader V., Tomppo L., Trossbach S. V., Bradshaw N. J., Prikulis I., Leliveld S. R., et al. (2012). Proteomic, genomic and translational approaches identify CRMP1 for a role in schizophrenia and its underlying traits. Hum. Mol. Genet. 21, 4406–4418. doi: 10.1093/hmg/dds273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A., Ifrim M. F., Valdez A. N., Raj N., Bassell G. J. (2018). Aberrant RNA translation in fragile X syndrome: from FMRP mechanisms to emerging therapeutic strategies. Brain Res. 1693, 24–36. doi: 10.1016/j.brainres.2018.04.008, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley C. L., Pennington K., Behan A., Wait R., Dunn M. J., Cotter D. (2006). Proteomic analysis of the anterior cingulate cortex in the major psychiatric disorders: evidence for disease-associated changes. Proteomics 6, 3414–3425. doi: 10.1002/pmic.200500069 [DOI] [PubMed] [Google Scholar]
- Braunschweig D., Krakowiak P., Duncanson P., Boyce R., Hansen R. L., Ashwood P., et al. (2013). Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl. Psychiatry 3:e277. doi: 10.1038/tp.2013.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretin S., Reibel S., Charrier E., Maus-Moatti M., Auvergnon N., Thevenoux A., et al. (2005). Differential expression of CRMP1, CRMP2A, CRMP2B, and CRMP5 in axons or dendrites of distinct neurons in the mouse brain. J. Comp. Neurol. 486, 1–17. doi: 10.1002/cne.20465 [DOI] [PubMed] [Google Scholar]
- Bretin S., Rogemond V., Marin P., Maus M., Torrens Y., Honnorat J., et al. (2006). Calpain product of WT-CRMP2 reduces the amount of surface NR2B NMDA receptor subunit. J. Neurochem. 98, 1252–1265. [DOI] [PubMed] [Google Scholar]
- Brittain J. M., Duarte D. B., Wilson S. M., Zhu W., Ballard C., Johnson P. L., et al. (2011). Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat. Med. 17, 822–829. doi: 10.1038/nm.2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain J. M., Piekarz A. D., Wang Y., Kondo T., Cummins T. R., Khanna R. (2009). An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 284, 31375–31390. doi: 10.1074/jbc.M109.009951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain J. M., Wang Y., Eruvwetere O., Khanna R. (2012). Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. Febs Letter 586, 3813–3818. doi: 10.1016/j.febslet.2012.09.022, PMID: [DOI] [PubMed] [Google Scholar]
- Brot S. (2014). Collapsin response-mediator protein 5 (CRMP5) phosphorylation at threonine 516 regulates neurite outgrowth inhibition. Eur. J. Neurosci. 40, 3010–3020. doi: 10.1111/ejn.12674 [DOI] [PubMed] [Google Scholar]
- Brot S., Auger C., Bentata R., Rogemond V., Ménigoz S., Chounlamountri N., et al. (2014). Collapsin response mediator protein 5 (CRMP5) induces mitophagy, thereby regulating mitochondrion numbers in dendrites. J. Biol. Chem. 289, 2261–2276. doi: 10.1074/jbc.M113.490862, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brot S., Rogemond V., Perrot V., Chounlamountri N., Auger C., Honnorat J., et al. (2010). CRMP5 interacts with tubulin to inhibit neurite outgrowth, thereby modulating the function of CRMP2. J. Neurosci. 30, 10639–10654. doi: 10.1523/JNEUROSCI.0059-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byk T., Dobransky T., Cifuentes-Diaz C., Sobel A. (1996). Identification and molecular characterization of Unc-33-like phosphoprotein (Ulip), a putative mammalian homolog of the axonal guidance-associated unc-33 gene product. J. Neurosci. 16, 688–701. doi: 10.1523/JNEUROSCI.16-02-00688.1996, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byk T., Ozon S., Sobel A. (1998). The Ulip family phosphoproteins--common and specific properties. Eur. J. Biochem. 254, 14–24. doi: 10.1046/j.1432-1327.1998.2540014.x [DOI] [PubMed] [Google Scholar]
- Cao J., O’Day D. R., Pliner H. A., Kingsley P. D., Deng M., Daza R. M., et al. (2020). A human cell atlas of fetal gene expression. Science 370:eaba7721. doi: 10.1126/science.aba7721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha C., Zhang J., Ji Z., Tan M., Li S., Wu F., et al. (2016). CRMP4 regulates dendritic growth and maturation via the interaction with actin cytoskeleton in cultured hippocampal neurons. Brain Res. Bull. 124, 286–294. doi: 10.1016/j.brainresbull.2016.06.008, PMID: [DOI] [PubMed] [Google Scholar]
- Charrier E., Mosinger B., Meissirel C., Aguera M., Rogemond V., Reibel S., et al. (2006). Transient alterations in granule cell proliferation, apoptosis and migration in postnatal developing cerebellum of CRMP1−/− mice. Genes Cells 11, 1337–1352. doi: 10.1111/j.1365-2443.2006.01024.x [DOI] [PubMed] [Google Scholar]
- Charrier E., Reibel S., Rogemond V., Aguera M., Thomasset N., Honnorat J. (2003). Collapsin response mediator proteins (CRMPs): involvement in nervous system development and adult neurodegenerative disorders. Mol. Neurobiol. 28, 51–64. doi: 10.1385/MN:28:1:51 [DOI] [PubMed] [Google Scholar]
- Chen W. (2009). AMPA GluR1 and GluR2 receptor subunits regulate dendrite complexity and spine motility in neurons of the developing neocortex. Neuroscience 159, 172–182. doi: 10.1016/j.neuroscience.2008.11.038, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew L. A., Khanna R. (2018). CRMP2 and voltage-gated ion channels: potential roles in neuropathic pain. Neuronal. Signals 2:NS20170220. doi: 10.1042/NS20170220, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole A. R., Causeret F., Yadirgi G., Hastie J. C., McLauchlan H., McManus E. J., et al. (2006). Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J. Biol. Chem. 281, 16591–16598. doi: 10.1074/jbc.M513344200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M. O., Husi H., Yu L., Brandon J. M., Anderson C. N. G., Blackstock W. P., et al. (2006). Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 97, 16–23. doi: 10.1111/j.1471-4159.2005.03507.x, PMID: [DOI] [PubMed] [Google Scholar]
- De Rubeis S., He X., Goldberg A. P., Poultney C. S., Samocha K., Cicek A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diss G., Ascencio D., DeLuna A., Landry C. R. (2014). Molecular mechanisms of paralogous compensation and the robustness of cellular networks. J. Exp. Zool. B Mol. Dev. Evol. 322, 488–499. doi: 10.1002/jez.b.22555 [DOI] [PubMed] [Google Scholar]
- Dustrude E. T., Moutal A., Yang X., Wang Y., Khanna M., Khanna R. (2016). Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proc. Natl. Acad. Sci. U. S. A. 113, E8443–E8452. doi: 10.1073/pnas.1610531113, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustrude E. T., Perez-Miller S., François Moutal L., Moutal A., Khanna M., Khanna R. (2017). A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels 11, 316–328. doi: 10.1080/19336950.2017.1299838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustrude E. T., Wilson S. M., Ju W., Xiao Y., Khanna R. (2013). CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J. Biol. Chem. 288, 24316–24331. doi: 10.1074/jbc.M113.474924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar P. F., Douglas J. E., Cooper G. J., Dean B., Kydd R., Faull R. L. (2000). Comparative proteome analysis of the hippocampus implicates chromosome 6q in schizophrenia. Mol. Psychiatry 5, 85–90. doi: 10.1038/sj.mp.4000580, PMID: [DOI] [PubMed] [Google Scholar]
- Elston G. N. (2000). Pyramidal cells of the frontal lobe: all the more spinous to think with. J. Neurosci. 20:RC95. doi: 10.1523/JNEUROSCI.20-18-j0002.2000, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson C. S., Lee S. J., Barlow-Anacker A. J., Druckenbrod N. R., Epstein M. L., Gosain A. (2014). Appearance of cholinergic myenteric neurons during enteric nervous system development: comparison of different ChAT fluorescent mouse reporter lines. Neurogastroenterol. Motil. 26, 874–884. doi: 10.1111/nmo.12343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essa M. M., Braidy N., Vijayan K. R., Subash S., Guillemin G. J. (2013). Excitotoxicity in the pathogenesis of autism. Neurotox. Res. 23, 393–400. doi: 10.1007/s12640-012-9354-3 [DOI] [PubMed] [Google Scholar]
- Fukada M., Watakabe I., Yuasa-Kawada J., Kawachi H., Kuroiwa A., Matsuda Y., et al. (2000). Molecular characterization of CRMP5, a novel member of the collapsin response mediator protein family. J. Biol. Chem. 275, 37957–37965. doi: 10.1074/jbc.M003277200 [DOI] [PubMed] [Google Scholar]
- Fukata Y., Itoh T. J., Kimura T., Ménager C., Nishimura T., Shiromizu T., et al. (2002). CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4, 583–591. doi: 10.1038/ncb825 [DOI] [PubMed] [Google Scholar]
- Gong X., Tan M., Gao Y., Chen K., Guo G. (2016). CRMP-5 interacts with actin to regulate neurite outgrowth. Mol. Med. Rep. 13, 1179–1185. doi: 10.3892/mmr.2015.4662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima Y., Nakamura F., Strittmatter P., Strittmatter S. M. (1995). Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 376, 509–514. doi: 10.1038/376509a0 [DOI] [PubMed] [Google Scholar]
- Guilmatre A., Dubourg C., Mosca A.-L., Legallic S., Goldenberg A., Drouin-Garraud V., et al. (2009). Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry 66, 947–956. doi: 10.1001/archgenpsychiatry.2009.80, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren C., Kjaergaard S., Bak M., Hansen C., El-Schich Z., Anderson C. M., et al. (2012). Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin. Genet. 82, 248–255. doi: 10.1111/j.1399-0004.2011.01755.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamajima N., Matsuda K., Sakata S., Tamaki N., Sasaki M., Nonaka M. (1996). A novel gene family defined by human dihydropyrimidinase and three related proteins with differential tissue distribution. Gene 180, 157–163. doi: 10.1016/s0378-1119(96)00445-3 [DOI] [PubMed] [Google Scholar]
- Henley J. M., Seager R., Nakamura Y., Talandyte K., Nair J., Wilkinson K. A. (2021). SUMOylation of synaptic and synapse-associated proteins: An update. J. Neurochem. 156, 145–161. doi: 10.1111/jnc.15103, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higurashi M., Iketani M., Takei K., Yamashita N., Aoki R., Kawahara N., et al. (2012). Localized role of CRMP1 and CRMP2 in neurite outgrowth and growth cone steering. Dev. Neurobiol. 72, 1528–1540. doi: 10.1002/dneu.22017 [DOI] [PubMed] [Google Scholar]
- Hong L. E., Wonodi I., Avila M. T., Buchanan R. W., McMahon R. P., Mitchell B. D., et al. (2005). Dihydropyrimidinase-related protein 2 (DRP-2) gene and association to deficit and nondeficit schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 136B, 8–11. doi: 10.1002/ajmg.b.30181, PMID: [DOI] [PubMed] [Google Scholar]
- Hotta A., Inatome R., Yuasa-Kawada J., Qin Q., Yamamura H., Yanagi S. (2005). Critical role of Collapsin response mediator protein-associated molecule CRAM for Filopodia and growth cone development in neurons. Mol. Biol. Cell 16, 32–39. doi: 10.1091/mbc.E04-08-0679, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher P. R. (1991). Dendritic and synaptic pathology in mental retardation. Pediatr. Neurol. 7, 79–85. doi: 10.1016/0887-8994(91)90001-2 [DOI] [PubMed] [Google Scholar]
- Inagaki N., Chihara K., Arimura N., Ménager C., Kawano Y., Matsuo N., et al. (2001). CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 4, 781–782. doi: 10.1038/90476, PMID: [DOI] [PubMed] [Google Scholar]
- Inatome R., Tsujimura T., Hitomi T., Mitsui N., Kuroda S., Yamamura H., et al. (2000). Identification of CRAM, a novel Unc-33 gene family protein that associates with CRMP3 and protein-tyrosine kinase(s) in the developing rat brain. J. Biol. Chem. 275, 27291–27302. doi: 10.1074/jbc.M910126199 [DOI] [PubMed] [Google Scholar]
- Iossifov I., O’Roak B., Sanders S. J., Ronemus M., Krumm N., Levy D. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip J. P. K., Fu A. K. Y., Ip N. Y. (2014). CRMP2: functional roles in neural development and therapeutic potential in neurological diseases. Neuroscientist 20, 589–598. doi: 10.1177/1073858413514278 [DOI] [PubMed] [Google Scholar]
- Izumi R., Hino M., Nagaoka A., Shishido R., Kakita A., Hoshino M., et al. (2022). Dysregulation of DPYSL2 expression by mTOR signaling in schizophrenia: multi-level study of postmortem brain. Neurosci. Res. 175, 73–81. doi: 10.1016/j.neures.2021.09.004 [DOI] [PubMed] [Google Scholar]
- Jeanne M., Demory H., Moutal A., Vuillaume M.-L., Blesson S., Thépault R.-A., et al. (2021). Missense variants in DPYSL5 cause a neurodevelopmental disorder with corpus callosum agenesis and cerebellar abnormalities. Am. J. Hum. Genet. 108, 951–961. doi: 10.1016/j.ajhg.2021.04.004, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z., Tan M., Gao Y., Zhang J., Gong X., Guo G., et al. (2014). CRMP-5 interacts with tubulin to promote growth cone development in neurons. Int. J. Clin. Exp. Med. 7, 67–75. [PMC free article] [PubMed] [Google Scholar]
- Jin X., Sasamoto K., Nagai J., Yamazaki Y., Saito K., Goshima Y., et al. (2016). Phosphorylation of CRMP2 by Cdk5 regulates dendritic spine development of cortical neuron in the mouse hippocampus. Neural Plast. 2016:6790743. doi: 10.1155/2016/6790743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W., Li Q., Wilson S. M., Brittain J. M., Meroueh L., Khanna R. (2013). SUMOylation alters CRMP2 regulation of calcium influx in sensory neurons. Channels (Austin) 7, 153–159. doi: 10.4161/chan.24224, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata T., Daar I. O., Subleski M., Copeland T., Kung H. F., Xu R. H. (1998). Xenopus CRMP-2 is an early response gene to neural induction. Brain Res. Mol. Brain Res. 57, 201–210. doi: 10.1016/s0169-328x(98)00082-5 [DOI] [PubMed] [Google Scholar]
- Kaufmann W. E., Moser H. W. (2000). Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex 10, 981–991. doi: 10.1093/cercor/10.10.981 [DOI] [PubMed] [Google Scholar]
- Kawashima T., Jitsuki-Takahashi A., Takizawa K., Jitsuki S., Takahashi T., Ohshima T., et al. (2021). Phosphorylation of collapsin response mediator protein 1 (CRMP1) at tyrosine 504 residue regulates semaphorin 3A-induced cortical dendritic growth. J. Neurochem. 157, 1207–1221. doi: 10.1111/jnc.15304 [DOI] [PubMed] [Google Scholar]
- Khanna R., Wilson S. M., Brittain J. M., Weimer J., Sultana R., Butterfield A., et al. (2012). Opening Pandora’s jar: a primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol. 7, 749–771. doi: 10.2217/FNL.12.68, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayachi A., Gwizdek C., Poupon G., Alcor D., Chafai M., Cassé F., et al. (2018). Sumoylation regulates FMRP-mediated dendritic spine elimination and maturation. Nat. Commun. 9:757. doi: 10.1038/s41467-018-03222-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazaei M. R., Girouard M.-P., Alchini R., Ong Tone S., Shimada T., Bechstedt S., et al. (2014). Collapsin response mediator protein 4 regulates growth cone dynamics through the actin and microtubule cytoskeleton. J. Biol. Chem. 289, 30133–30143. doi: 10.1074/jbc.M114.570440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-W., Park K., Kang R. J., Gonzales E. L. T., Kim D. G., Oh H. A., et al. (2019). Pharmacological modulation of AMPA receptor rescues social impairments in animal models of autism. Neuropsychopharmacology 44, 314–323. doi: 10.1038/s41386-018-0098-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F., Cuthbert P. C., Grant S. G. N. (2007). The role of neuronal complexes in human X-linked brain diseases. Am. J. Hum. Genet. 80, 205–220. doi: 10.1086/511441, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Joo J., Nah S.-S., Kim J. W., Kim H.-K., Kwon J.-T., et al. (2015). Changes in Dpysl2 expression are associated with prenatally stressed rat offspring and susceptibility to schizophrenia in humans. Int. J. Mol. Med. 35, 1574–1586. doi: 10.3892/ijmm.2015.2161, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leney A. C., El Atmioui D., Wu W., Ovaa H., Heck A. J. R. (2017). Elucidating crosstalk mechanisms between phosphorylation and O-GlcNAcylation. Proc. Natl. Acad. Sci. U. S. A. 114, E7255–E7261. doi: 10.1073/pnas.1620529114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Becker B., Jiang X., Zhao Z., Zhang Q., Yao S., et al. (2019). Decreased interhemispheric functional connectivity rather than corpus callosum volume as a potential biomarker for autism spectrum disorder. Cortex 119, 258–266. doi: 10.1016/j.cortex.2019.05.003 [DOI] [PubMed] [Google Scholar]
- Li C., Sasaki Y., Takei K., Yamamoto H., Shouji M., Sugiyama Y., et al. (2004). Correlation between semaphorin3a-induced facilitation of axonal transport and local activation of a translation initiation factor eukaryotic translation initiation factor 4E. J. Neurosci. 24, 6161–6170. doi: 10.1523/JNEUROSCI.1476-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P.-C., Chan P. M., Hall C., Manser E. (2011). Collapsin response mediator proteins (CRMPs) are a new class of microtubule-associated protein (MAP) that selectively interacts with assembled microtubules via a taxol-sensitive binding interaction. J. Biol. Chem. 286, 41466–41478. doi: 10.1074/jbc.M111.283580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-S., Lin Y.-F., Chen K. C., Yang Y. K., Hsiao Y.-H. (2019). Collapsin response mediator protein 5 (CRMP5) causes social deficits and accelerates memory loss in an animal model of Alzheimer’s disease. Neuropharmacology 157:107673. doi: 10.1016/j.neuropharm.2019.107673, PMID: [DOI] [PubMed] [Google Scholar]
- Makihara H., Nakai S., Ohkubo W., Yamashita N., Nakamura F., Kiyonari H., et al. (2016). CRMP1 and CRMP2 have synergistic but distinct roles in dendritic development. Genes Cells 21, 994–1005. doi: 10.1111/gtc.12399, PMID: [DOI] [PubMed] [Google Scholar]
- Malenka R. C., Bear M. F. (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. doi: 10.1016/j.neuron.2004.09.012 [DOI] [PubMed] [Google Scholar]
- Martin-de-souza D., Schmitt A., Röder R., Lebar M., Schneider Axmann T., Falkai P., et al. (2010). Sex-specific proteome differences in the anterior cingulate cortex of schizophrenia. J. Psychiatr. Res. 44, 9–991. doi: 10.1016/j.jpsychires.2010.03.003 [DOI] [PubMed] [Google Scholar]
- Martínez-Cerdeño V. (2017). Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol. 77, 393–404. doi: 10.1002/dneu.22417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P., Mani D. R., Ruggles K. V., Gillette M. A., Clauser K. R., Wang P., et al. (2016). Proteogenomics connects somatic mutations to signaling in breast cancer. Nature 534, 55–62. doi: 10.1038/nature18003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura K., Oga T., Sasaki T., Nakagaki K., Sato C., Sumida K., et al. (2019). Abnormal axon guidance signals and reduced interhemispheric connection via anterior commissure in neonates of marmoset ASD model. NeuroImage 195, 243–251. doi: 10.1016/j.neuroimage.2019.04.006 [DOI] [PubMed] [Google Scholar]
- Minturn J. E., Fryer H. J., Geschwind D. H., Hockfield S. (1995). TOAD-64, a gene expressed early in neuronal differentiation in the rat, is related to unc-33, a C. elegans gene involved in axon outgrowth. J. Neurosci. 15, 6757–6766. doi: 10.1523/JNEUROSCI.15-10-06757.1995, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita A., Yamashita N., Sasaki Y., Uchida Y., Nakajima O., Nakamura F., et al. (2006). Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J. Neurosci. 26, 2971–2980. doi: 10.1523/JNEUROSCI.5453-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutal A., François-Moutal L., Perez-Miller S., Cottier K., Chew L. A., Yeon S. K., et al. (2016). (S)-Lacosamide binding to collapsin response mediator protein 2 (CRMP2) regulates CaV2.2 activity by subverting its phosphorylation by Cdk5. Mol. Neurobiol. 53, 1959–1976. doi: 10.1007/s12035-015-9141-2 [DOI] [PubMed] [Google Scholar]
- Moutal A., Wang Y., Yang X., Ji Y., Luo S., Dorame A., et al. (2017). Dissecting the role of the CRMP2–neurofibromin complex on pain behaviors. Pain 158, 2203–2221. doi: 10.1097/j.pain.0000000000001026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaza N., Cheng A. A., Brown C. O., Meka D. P., Hong S., Uy J. A., et al. (2022). Neuron-specific protein network mapping of autism risk genes identifies shared biological mechanisms and disease-relevant pathologies. Cell Rep. 41:111678. doi: 10.1016/j.celrep.2022.111678, PMID: [DOI] [PubMed] [Google Scholar]
- Myllykoski M., Baumann A., Hensley K., Kursula P. (2017). Collapsin response mediator protein 2: high-resolution crystal structure sheds light on small-molecule binding, post-translational modifications, and conformational flexibility. Amino Acids 49, 747–759. doi: 10.1007/s00726-016-2376-z [DOI] [PubMed] [Google Scholar]
- Na E. J., Nam H. Y., Park J., Chung M. A., Woo H. A., Kim H.-J. (2017). PI3K-mTOR-S6K signaling mediates neuronal viability via collapsin response mediator Protein-2 expression. Front. Mol. Neurosci. 10:288. doi: 10.3389/fnmol.2017.00288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabais Sá M. J., Venselaar H., Wiel L., Trimouille A., Lasseaux E., Naudion S., et al. (2020). De novo CLTC variants are associated with a variable phenotype from mild to severe intellectual disability, microcephaly, hypoplasia of the corpus callosum, and epilepsy. Genet. Med. 22, 797–802. doi: 10.1038/s41436-019-0703-y [DOI] [PubMed] [Google Scholar]
- Nagai J., Baba R., Ohshima T. (2017). CRMPs function in neurons and glial cells: potential therapeutic targets for neurodegenerative diseases and CNS injury. Mol. Neurobiol. 54, 4243–4256. doi: 10.1007/s12035-016-0005-1 [DOI] [PubMed] [Google Scholar]
- Nakamura F., Kumeta K., Hida T., Isono T., Nakayama Y., Kuramata-Matsuoka E., et al. (2014). Amino- and carboxyl-terminal domains of filamin-a interact with CRMP1 to mediate Sema3A signalling. Nat. Commun. 5:5325. doi: 10.1038/ncomms6325 [DOI] [PubMed] [Google Scholar]
- Nakamura F., Ohshima T., Goshima Y. (2020). Collapsin response mediator proteins: their biological functions and pathophysiology in neuronal development and regeneration. Front. Cell. Neurosci. 14:188. doi: 10.3389/fncel.2020.00188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H., Takahashi-Jitsuki A., Makihara H., Asano T., Kimura Y., Nakabayashi J., et al. (2018). Proteome and behavioral alterations in phosphorylation-deficient mutant collapsin response mediator Protein2 knock-in mice. Neurochem. Int. 119, 207–217. doi: 10.1016/j.neuint.2018.04.009 [DOI] [PubMed] [Google Scholar]
- Nakamura H., Yamashita N., Kimura A., Kimura Y., Hirano H., Makihara H., et al. (2016). Comprehensive behavioral study and proteomic analyses of CRMP2-deficient mice. Genes Cells 21, 1059–1079. doi: 10.1111/gtc.12403 [DOI] [PubMed] [Google Scholar]
- Nelson A. D., Bender K. J. (2021). Dendritic integration dysfunction in neurodevelopmental disorders. Dev. Neurosci. 43, 201–221. doi: 10.1159/000516657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niisato E., Nagai J., Yamashita N., Abe T., Kiyonari H., Goshima Y., et al. (2012). CRMP4 suppresses apical dendrite bifurcation of CA1 pyramidal neurons in the mouse hippocampus. Dev. Neurobiol. 72, 1447–1457. doi: 10.1002/dneu.22007 [DOI] [PubMed] [Google Scholar]
- Nomoto M., Konopaske G. T., Yamashita N., Aoki R., Jitsuki-Takahashi A., Nakamura H., et al. (2021). Clinical evidence that a dysregulated master neural network modulator may aid in diagnosing schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 118:e2100032118. doi: 10.1073/pnas.2100032118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani-Kaneko R. (2019). Crmp4-KO mice as an animal model for investigating certain phenotypes of autism spectrum disorders. Int. J. Mol. Sci. 20:2485. doi: 10.3390/ijms20102485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlowsky A., Chelly J., Billuart P. (2012). Emerging major synaptic signaling pathways involved in intellectual disability. Mol. Psychiatry 17, 682–693. doi: 10.1038/mp.2011.139, PMID: [DOI] [PubMed] [Google Scholar]
- Petratos S., Lee J. Y. (2013). Stop CRMPing my style: a new competitive model of CRMP oligomerization. J. Neurochem. 125, 800–802. doi: 10.1111/jnc.12224, PMID: [DOI] [PubMed] [Google Scholar]
- Pham X., Song G., Lao S., Goff L., Zhu H., Valle D., et al. (2016). The DPYSL2 gene connects mTOR and schizophrenia. Transl. Psychiatry 6:e933. doi: 10.1038/tp.2016.204, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnusamy R., Lohkamp B. (2013). Insights into the oligomerization of CRMPs: crystal structure of human collapsin response mediator protein 5 - PubMed. J. Neurochem. 125, 855–868. doi: 10.1111/jnc.12188, PMID: [DOI] [PubMed] [Google Scholar]
- Prem S., Millonig J. H., DiCicco-Bloom E. (2020). Dysregulation of Neurite Outgrowth and Cell Migration in Autism and Other Neurodevelopmental Disorders. Adv. Neurobiol. 25, 109–153. doi: 10.1007/978-3-030-45493-7_5 [DOI] [PubMed] [Google Scholar]
- Purcell A. E., Jeon O. H., Zimmerman A. W., Blue M. E., Pevsner J. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57, 1618–1628. doi: 10.1212/wnl.57.9.1618, PMID: [DOI] [PubMed] [Google Scholar]
- Purves H., Hume R. I. (1981). The relation of postsynaptic geometry to the number of presynaptic axons that innervate autonomic ganglion cells. J. Neurosci. 1, 441–452. doi: 10.1523/JNEUROSCI.01-05-00441.1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C., Feng I., Costa A. R., Pinto-Costa R., Neil J. E., Caluseriu O., et al. (2022). Variants in ADD1 cause intellectual disability, corpus callosum dysgenesis, and ventriculomegaly in humans. Genet. Med. 24, 319–331. doi: 10.1016/j.gim.2021.09.014, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach T. T., Auvergnon N., Khanna R., Belin M. F., Kolattukudy P. E., Honnorat J., et al. (2018). Opposing Morphogenetic Defects on Dendrites and Mossy Fibers of Dentate Granular Neurons in CRMP3-Deficient Mice. Brain Sci. 8:196. doi: 10.3390/brainsci8110196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach T. T., Honnorat J., Kolattukudy P. E., Khanna R., Duchemin A.-M. (2015). CRMPs: critical molecules for neurite morphogenesis and neuropsychiatric diseases | molecular psychiatry. Mol. Psychiatry 20, 1037–1045. doi: 10.1038/mp.2015.77 [DOI] [PubMed] [Google Scholar]
- Quach T. T., Massicotte G., Belin M.-F., Honnorat J., Glasper E. R., Devries A. C., et al. (2008). CRMP3 is required for hippocampal CA1 dendritic organization and plasticity. FASEB J. 22, 401–409. doi: 10.1096/fj.07-9012com, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach T. T., Stratton H. J., Khanna R., Kolattukudy P. E., Honnorat J., Meyer K., et al. (2021). Intellectual disability: dendritic anomalies and emerging genetic perspectives. Acta Neuropathol. 141, 139–158. doi: 10.1007/s00401-020-02244-5, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach T. T., Wang K. R., Chounlamountri N., Auvergnon N., Honnorat J., Duchemin A.-M. (2011). Effect of CRMP3 expression on dystrophic dendrites of hippocampal neurons. Mol. Psychiatry 16, 689–691. doi: 10.1038/mp.2011.6 [DOI] [PubMed] [Google Scholar]
- Quach T. T., Wilson S. M., Rogemond V., Chounlamountri N., Kolattukudy P. E., Martinez S., et al. (2013). Mapping CRMP3 domains involved in dendrite morphogenesis and voltage-gated calcium channel regulation. J. Cell Sci. 126, 4262–4273. doi: 10.1242/jcs.131409 [DOI] [PubMed] [Google Scholar]
- Quinn C. C., Chen E., Kinjo Ashi G., Gali K., Bell A. W., Elliott R. C., et al. (2003). TUC-4b, a novel TUC family variant, regulates neurite outgrowth and associates with vesicles in the growth cone. J. Neurosci. 23, 2815–2823. doi: 10.1523/JNEUROSCI.23-07-02815.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn C. C., Gray G. E., Hockfield S. (1999). A family of proteins implicated in axon guidance and outgrowth. J. Neurobiol. 41, 158–164. [PubMed] [Google Scholar]
- Ramanathan S., Woodroffe A., Flodman P. L., Mays L. Z., Hanouni M., Modahl C. B., et al. (2004). A case of autism with an interstitial deletion on 4q leading to hemizygosity for genes encoding for glutamine and glycine neurotransmitter receptor sub-units (AMPA 2, GLRA3, GLRB) and neuropeptide receptors NPY1R, NPY5R. BMC Med. Genet. 5:10. doi: 10.1186/1471-2350-5-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Celis A., Becker M., Nuño M., Schauer J., Aghaeepour N., Van de Water J. (2021). Risk assessment analysis for maternal autoantibody-related autism (MAR-ASD): a subtype of autism. Mol. Psychiatry 26, 1551–1560. doi: 10.1038/s41380-020-00998-8, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Celis A., Croen L. A., Yoshida C. K., Alexeeff S. E., Schauer J., Yolken R. H., et al. (2022). Maternal autoantibody profiles as biomarkers for ASD and ASD with co-occurring intellectual disability. Mol. Psychiatry, 27, 3760–3767. doi: 10.1038/s41380-022-01633-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran E., Arashiki N., Becker L. L., Takizawa K., Levy J., Rambaud T., et al. (2022). Monoallelic CRMP1 gene variants cause neurodevelopmental disorder. Elife 11:e80793. doi: 10.7554/eLife.80793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard R. V., Charrier E., Aguera B., Belin M.-F., Thomasset N., Honnorat J. (2001). Isolation and expression pattern of human Unc-33-like phosphoprotein 6/collapsin response mediator protein 5 (Ulip6/CRMP5): coexistence with Ulip2/CRMP2 in Sema3a- sensitive oligodendrocytes. J. Neurosci. 21, 7203–7214. doi: 10.1523/JNEUROSCI.21-18-07203.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J. (2022). Molecular Mechanisms Underlying Neurotransmitter Release. Annu. Rev. Biophys. 51, 377–408. doi: 10.1146/annurev-biophys-111821-104732, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosslenbroich V., Dai L., Baader S. L., Noegel A. A., Gieselmann V., Kappler J. (2005). Collapsin response mediator protein-4 regulates F-actin bundling. Exp. Cell Res. 310, 434–444. doi: 10.1016/j.yexcr.2005.08.005 [DOI] [PubMed] [Google Scholar]
- Sakano H., Zorio D. A. R., Wang X., Ting Y. S., Noble W. S., MacCoss M. J., et al. (2017). Proteomic analyses of nucleus laminaris identified candidate targets of the fragile X mental retardation protein. J. Comp. Neurol. 525, 3341–3359. doi: 10.1002/cne.24281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y., Cheng C., Uchida Y., Nakajima O., Ohshima T., Yagi T., et al. (2002). Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron 35, 907–920. doi: 10.1016/s0896-6273(02)00857-7 [DOI] [PubMed] [Google Scholar]
- Satterstrom F. K., Kosmicki J. A., Wang J., Breen M. S., De Rubeis S., An J.-Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cells 180, 568–584.e23. doi: 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt E. F., Strittmatter S. M. (2007). The CRMP family of proteins and their role in Sema3A signaling. Adv. Exp. Med. Biol. 600, 1–11. doi: 10.1007/978-0-387-70956-7_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A., Hoeffer C. A., Takayasu Y., Miyawaki T., McBride S. M., Klann E., et al. (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702. doi: 10.1523/JNEUROSCI.3696-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutar M. P. M., Thornhill P., Cole A. R., Sutherland C. (2009). Increased CRMP2 phosphorylation is observed in Alzheimer’s disease; does this tell us anything about disease development? Curr. Alzheimer Res. 6, 269–278. doi: 10.2174/156720509788486572 [DOI] [PubMed] [Google Scholar]
- Stacho M., Manahan-Vaughan D. (2022). The intriguing contribution of hippocampal long-term depression to spatial learning and long-term memory. Front. Behav. Neurosci. 16:806356. doi: 10.3389/fnbeh.2022.806356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark P., Ogg D., Flodin S., Flores A., Kotenyova T., Nyman T., et al. (2007). The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J. Neurochem. 101, 906–917. doi: 10.1111/j.1471-4159.2006.04401.x, PMID: [DOI] [PubMed] [Google Scholar]
- Stratton H., Boinon L., Moutal A., Khanna R. (2020). Coordinating synaptic signaling with CRMP2. Int. J. Biochem. Cell Biol. 124:105759. doi: 10.1016/j.biocel.2020.105759 [DOI] [PubMed] [Google Scholar]
- Su K.-Y., Chien W.-L., Fu W.-M., Yu I.-S., Huang H.-P., Huang P.-H., et al. (2007). Mice deficient in collapsin response mediator protein-1 exhibit impaired long-term potentiation and impaired spatial learning and memory. J. Neurosci. 27, 2513–2524. doi: 10.1523/JNEUROSCI.4497-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkin S. M., Ng L., Lau C., Dolbeare T., Gilbert T L., Thompson C. L., et al. (2013). Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 41, D996–D1008. doi: 10.1093/nar/gks1042, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Li S., Tokutomi T., Takeuchi C., Takahashi M., Yamada M., et al. (2022). De novo non-synonymous DPYSL2 (CRMP2) variants in two patients with intellectual disabilities and documentation of functional relevance through zebrafish rescue and cellular transfection experiments. Hum. Mol. Genet. 31:ddac166. doi: 10.1093/hmg/ddac166 [DOI] [PubMed] [Google Scholar]
- Takaya R., Nagai J., Piao W., Niisato E., Nakabayashi T., Yamazaki Y., et al. (2017). CRMP1 and CRMP4 are required for proper orientation of dendrites of cerebral pyramidal neurons in the developing mouse brain. Brain Res. 1655, 161–167. doi: 10.1016/j.brainres.2016.11.003 [DOI] [PubMed] [Google Scholar]
- Tan M., Cha C., Ye Y., Zhang J., Li S., Wu F., et al. (2015). CRMP4 and CRMP2 interact to coordinate cytoskeleton dynamics, regulating growth cone development and axon elongation. Neural Plast. 2015:947423. doi: 10.1155/2015/947423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y., Ye Z., Wei Y., Lin C., Wang Y., Qin C., et al. (2015). Vertebrate paralogous CRMPs in nervous system: evolutionary, structural, and functional interplay. J. Mol. Neurosci. 55, 324–334. doi: 10.1007/s12031-014-0327-2 [DOI] [PubMed] [Google Scholar]
- Tang G., Gudsnuk K., Kuo S.-H., Cotrina M. L., Rosoklija G., Sosunov A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutiya A., Nakano Y., Hansen-Kiss E., Kelly B., Nishihara M., Goshima Y., et al. (2017). Human CRMP4 mutation and disrupted Crmp4 expression in mice are associated with ASD characteristics and sexual dimorphism. Sci. Rep. 7:16812. doi: 10.1038/s41598-017-16782-8, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutiya A., Watanabe H., Nakano Y., Nishihara M., Goshima Y., Ohtani-Kaneko R., et al. (2016). Deletion of collapsin response mediator protein 4 results in abnormal layer thickness and elongation of mitral cell apical dendrites in the neonatal olfactory bulb. J. Anat. 228, 792–804. doi: 10.1111/joa.12434, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutiya A., Nishihara M., Goshima Y., Ohtani-Kaneko R. (2015). Mouse pups lacking collapsin response mediator protein 4 manifest impaired olfactory function and hyperactivity in the olfactory bulb - PubMed. Eur. J. Neurosci. 42, 2335–2345. doi: 10.1111/ejn.12999 [DOI] [PubMed] [Google Scholar]
- Uchida Y., Goshima Y. (2005). Molecular mechanism of axon guidance mediated by phosphorylation of CRMP2. Seikagaku 77, 1424–1427. doi: 10.3389/fnbeh.2022.806356 [DOI] [PubMed] [Google Scholar]
- Uchida Y., Ohshima T., Yamashita N., Ogawara M., Sasaki Y., Nakamura F., et al. (2009). Semaphorin3A signaling mediated by Fyn-dependent tyrosine phosphorylation of collapsin response mediator protein 2 at tyrosine 32. J. Biol. Chem. 284, 27393–27401. doi: 10.1074/jbc.M109.000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzunova G., Hollander E., Shepherd J. (2014). The role of ionotropic glutamate receptors in childhood neurodevelopmental disorders: autism spectrum disorders and fragile x syndrome. Curr. Neuropharmacol. 12, 71–98. doi: 10.2174/1570159X113116660046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veron A. D., Bienboire-Frosini C., Girard S. D., Sadelli K., Stamegna J.-C., Khrestchatisky M., et al. (2018). Syngeneic transplantation of olfactory ectomesenchymal stem cells restores learning and memory abilities in a rat model of global cerebral ischemia. Stem Cells Int. 2018:2683969. doi: 10.1155/2018/2683969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Brittain J. M., Wilson S. M., Khanna R. (2010). Emerging roles of collapsin response mediator proteins (CRMPs) as regulators of voltage-gated calcium channels and synaptic transmission. Commun Integr Biol 3, 172–175. doi: 10.4161/cib.3.2.10620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.-H., Strittmatter S. M. (1996). A family of rat CRMP genes is differentially expressed in the nervous system. J. Neurosci. 16, 6197–6207. doi: 10.1523/JNEUROSCI.16-19-06197.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. H., Strittmatter S. M. (1997). Brain CRMP forms heterotetramers similar to liver dihydropyrimidinase. J. Neurochem. 69, 2261–2269. doi: 10.1046/j.1471-4159.1997.69062261.x, PMID: [DOI] [PubMed] [Google Scholar]
- Wegiel J., Kaczmarski W., Flory M., Martinez-Cerdeno V., Wisniewski T., Nowicki K., et al. (2018). Deficit of corpus callosum axons, reduced axon diameter and decreased area are markers of abnormal development of interhemispheric connections in autistic subjects. Acta Neuropathol. Commun. 6:143. doi: 10.1186/s40478-018-0645-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N., Goshima Y. (2012). Collapsin response mediator proteins regulate neuronal development and plasticity by switching their phosphorylation status. Mol. Neurobiol. 45, 234–246. doi: 10.1007/s12035-012-8242-4, PMID: [DOI] [PubMed] [Google Scholar]
- Yamashita N., Morita A., Uchida Y., Nakamura F., Usui H., Ohshima T., et al. (2007). Regulation of spine development by semaphorin3A through cyclin-dependent kinase 5 phosphorylation of collapsin response mediator protein 1. J. Neurosci. 27, 12546–12554. doi: 10.1523/JNEUROSCI.3463-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N., Mosinger B., Roy A., Miyazaki M., Ugajin K., Nakamura F., et al. (2011). CRMP5 (collapsin response mediator protein 5) regulates dendritic development and synaptic plasticity in the cerebellar Purkinje cells. J. Neurosci. 31, 1773–1779. doi: 10.1523/JNEUROSCI.5337-10.2011, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N., Ohshima T., Nakamura F., Kolattukudy P., Honnorat J., Mikoshiba K., et al. (2012). Phosphorylation of CRMP2 (collapsin response mediator protein 2) is involved in proper dendritic field organization. J. Neurosci. 32, 1360–1365. doi: 10.1523/JNEUROSCI.5563-11.2012, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N., Takahashi A., Takao K., Yamamoto T., Kolattukudy P., Miyakawa T., et al. (2013). Mice lacking collapsin response mediator protein 1 manifest hyperactivity, impaired learning and memory, and impaired prepulse inhibition. Front. Behav. Neurosci. 7:216. doi: 10.3389/fnbeh.2013.00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N., Uchida Y., Ohshima T., Hirai S., Nakamura F., Taniguchi M., et al. (2006). Collapsin response mediator protein 1 mediates reelin signaling in cortical neuronal migration. J. Neurosci. 26, 13357–13362. doi: 10.1523/JNEUROSCI.4276-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T., Kawano Y., Arimura N., Kawabata S., Kikuchi A., Kaibuchi K. (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cells 120, 137–149. doi: 10.1016/j.cell.2004.11.012, PMID: [DOI] [PubMed] [Google Scholar]
- Yuasa-Kawada J., Suzuki R., Kano F., Ohkawara T., Murata M., Noda M. (2003). Axonal morphogenesis controlled by antagonistic roles of two CRMP subtypes in microtubule organization. Eur. J. Neurosci. 17, 2329–2343. doi: 10.1046/j.1460-9568.2003.02664.x, PMID: [DOI] [PubMed] [Google Scholar]
- Yu-Kemp H.-C., Brieher W. M. (2016). Collapsin response mediator Protein-1 regulates Arp2/3-dependent actin assembly. J. Biol. Chem. 291, 658–664. doi: 10.1074/jbc.C115.689265, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Kang E., Wang Y., Yang C., Yu H., Wang Q., et al. (2016). Brain-specific Crmp2 deletion leads to neuronal development deficits and behavioural impairments in mice. Nat. Commun. 7:11773. doi: 10.1038/ncomms11773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Zhao B., Zhu X., Li J., Wu F., Li S., et al. (2018). Phosphorylation and SUMOylation of CRMP2 regulate the formation and maturation of dendritic spines. Brain Res. Bull. 139, 21–30. doi: 10.1016/j.brainresbull.2018.02.004, PMID: [DOI] [PubMed] [Google Scholar]
- Ziak J., Weissova R., Jeřábková K., Janikova M., Maimon R., Petrasek T., et al. (2020). CRMP2 mediates Sema3F-dependent axon pruning and dendritic spine remodeling. EMBO Rep. 21:e48512. doi: 10.15252/embr.201948512 [DOI] [PMC free article] [PubMed] [Google Scholar]