

In this study, Zacarías-Fluck et al. report the use of Omomyc, a MYC inhibitor, in targeting melanoma development, metastasis, and recurrence in human cell lines and CDX mouse models. By blocking MYC's transcriptional influence, Omomyc elicited gene expression signatures associated with good patient prognoses, pointing to the therapeutic potential of MYC inhibition in melanoma.
Keywords: MYC, Omomyc, melanoma, signature
Abstract
MYC's key role in oncogenesis and tumor progression has long been established for most human cancers. In melanoma, its deregulated activity by amplification of 8q24 chromosome or by upstream signaling coming from activating mutations in the RAS/RAF/MAPK pathway—the most predominantly mutated pathway in this disease—turns MYC into not only a driver but also a facilitator of melanoma progression, with documented effects leading to an aggressive clinical course and resistance to targeted therapy. Here, by making use of Omomyc, the most characterized MYC inhibitor to date that has just successfully completed a phase I clinical trial, we show for the first time that MYC inhibition in melanoma induces remarkable transcriptional modulation, resulting in severely compromised tumor growth and a clear abrogation of metastatic capacity independently of the driver mutation. By reducing MYC's transcriptional footprint in melanoma, Omomyc elicits gene expression profiles remarkably similar to those of patients with good prognosis, underlining the therapeutic potential that such an approach could eventually have in the clinic in this dismal disease.
Cutaneous melanoma, the most aggressive and deadly form of skin cancer, is the fifth most diagnosed cancer in Europe and one of the most frequent causes of cancer death. Its incidence and mortality trends are both increasing. In 2020, >105,000 people were diagnosed and 16,000 have died of this disease in the EU (https://ecis.jrc.ec.europa.eu/factsheets.php, accessed August 8, 2022). In the US alone, almost 100,000 new melanoma cases (representing 5% of all new cancers) are expected to be diagnosed in 2022, resulting in >7000 deaths (https://seer.cancer.gov/statfacts/html/melan.html, accessed August 8, 2022).
Melanomas arise from the malignant transformation of melanocytes as a result of a combination of exogenous and endogenous triggers as well as tumor-intrinsic and immune-related factors (Schadendorf et al. 2018). A seminal study by TCGA Network established a genomic classification according to driving mutations BRAF, NRAS, NF1, and triple wild type (WT), with potential consequent clinical implications (The Cancer Genome Atlas Network 2015). Intratumor heterogeneity studies performed in the last years have shown that cutaneous melanoma, along with lung cancer, harbors a significantly larger mutational burden than other tumor types. This could potentially be explained by years of exposure of the skin to UV light, a natural exogenous mutagen, leading to malignant transformation (McGranahan and Swanton 2017).
One main reason for the high death rate associated with melanoma is its highly aggressive and metastatic nature (Shain and Bastian 2016). Current therapeutic options for stage III and IV metastatic melanoma consist of immunotherapy (anti-PD-1 alone or in combination with anti-CTLA-4) or, for BRAF mutated melanomas, BRAF inhibitors (BRAFis) combined with MEK inhibitors (MEKis) (Álamo et al. 2021; Garbe et al. 2022). Although the 5-yr overall survival rate has improved thanks to these therapies, a significant number of patients do not benefit from them due to innate or acquired resistance (Czarnecka et al. 2020), generating a dire need for new treatments.
MYC is a transcription factor acting as a hub that integrates intracellular and extracellular cues leading to cell growth and proliferation. In order to function, MYC dimerizes with Max to then bind DNA and activate the transcription of genes that allow cells to divide in an orderly manner. However, its tightly controlled activity is lost in most human cancers, where MYC drives uncontrolled proliferation (Whitfield and Soucek 2021).
Melanomas with increased copy number gains in 8q24, harboring the MYC locus, occur in nonchronically sun-damaged skin, are amelanotic, and show an aggressive clinical course (Pouryazdanparast et al. 2012) with visceral metastases (Kraehn et al. 2001). MYC overexpression (independently of copy number gains) is found in 34% of melanomas (Bennett 2016), where it has been shown to increase aggressiveness (Schlagbauer-Wadl et al. 1999). In contrast, MYC down-regulation sensitizes melanomas to chemotherapy and radiotherapy (Biroccio et al. 2001), significantly decreasing their proliferation (Mannava et al. 2008). Finally, MYC deregulation is associated with several pathways of treatment resistance (Singleton et al. 2017). Hence, MYC inhibition seems to hold promise for an important therapeutic impact in all of these contexts.
Our laboratory has developed and validated Omomyc (Soucek et al. 1998; Massó-Vallés and Soucek 2020), the best-characterized direct MYC inhibitor to date, which functions as a dominant negative for MYC transcriptional activity. In a nutshell, Omomyc sequesters MYC away from DNA and occupies target gene promoters in the form of inactive homodimers or heterodimers with Max, shutting down their transcription. The different proportions of Omomyc dimers depend on the cellular context, as the relative amounts of MYC and Max will determine stoichiometrically the likelihood of formation of the different complexes. Omomyc has been used as a transgene in a wide range of murine cancer models, showing a remarkable effect on tumor growth, with only minor, reversible side effects in normal tissues (Massó-Vallés and Soucek 2020). Among these different models, we demonstrated that transgenic expression of Omomyc in suprabasal skin of a model of MYC-induced papillomatosis induces apoptosis and reduces proliferation of MYC-overexpressing cells in the absence of side effects in normal tissue (Soucek et al. 2004). However, the therapeutic impact of Omomyc in melanoma has not been studied yet. Hence, here we modeled MYC inhibition in melanoma using a panel of cell lines of human origin with different driver mutations and assessed both in vitro proliferation and in vivo tumor growth in the presence and absence of Omomyc. We also investigated the effect of MYC inhibition on melanoma metastatic capacity and transcriptional profile, relating Omomyc-induced changes to transcriptional data from melanoma patients. Our results indicate that specific gene sets reflecting decreased MYC transcriptional activity are associated with reduced metastatic capacity and increased immune response, and their expression can also be used to identify patients with good prognosis, highlighting the therapeutic relevance of MYC inhibition in the context of melanoma.
Results
Expression of Omomyc-RFP in human melanoma cells impairs their proliferation in vitro
In order to analyze the effect of Omomyc expression in melanoma cells, we transfected a panel of nine human melanoma cell lines comprising both BRAF mutated and wild-type cell lines, with a plasmid coding for the Omomyc-RFP fusion protein under the control of doxycycline. We then evaluated Omomyc's impact on proliferation by Crystal Violet staining in 96-well plates. Despite the fact that RFP levels showed high variability of Omomyc expression both at the single-cell level and among the different cell lines (Supplemental Fig. 1A), at 10 d, Omomyc induced a significant decrease in proliferation in all of them with respect to the untreated control (Fig. 1A). Importantly, this striking effect was already evident at 3 d in highly proliferating cell lines, (Fig. 1B) and was independent of their mutational profile, as we observed no differences related to the presence of BRAFV600E, BRAFwt, or even NRASQ61L/R/K or mutated NF1 (Fig. 1A). Interestingly, when cells were plated at very low density for colony formation studies, this antiproliferative effect was even more noticeable, with very few colonies arising in Omomyc-expressing cells compared with their control counterparts (Fig. 1C). In a subset of cell lines (namely, A375, SkMel37, and SkMel103), Omomyc also triggered cell death at 7 d (Supplemental Fig. S1B). With the aim of better characterizing the effect of Omomyc over a longer period, we analyzed proliferation and cell death up to 4 wk in both A375 (Fig. 1D, top panels) and SkMel147 (Fig. 1D, bottom panels). In both cell lines, we saw sustained growth arrest. In the case of A375, this was accompanied by a constant and significant increase in cell death with respect to the control from day 7 onward. However, in the case of SkMel147, the increase in cell death was only evident at day 21 and became significant at day 28 (Fig. 1D; Supplemental Fig. S1C). Of note, during this long-term experiment, RFP levels decreased in both cell lines (Supplemental Fig. S1D). This could be explained by either the death of cells expressing higher levels of Omomyc, counterselection of the Omomyc transgene, or a combination of both.
Figure 1.

Omomyc induces growth arrest in melanoma human cell lines in vitro. (A) Growth inhibition by transgenic Omomyc at 10 d by Crystal Violet staining. (B) Early effect of Omomyc on proliferation by cell count. (C) Colony formation assay. (D) Proliferation and cell death following Omomyc expression as a late event in A375 (top panels) and SkMel147 (bottom panels). (E) Cell cycle profiles during the first 4 d of Omomyc expression of A375 and SkMel147. Student's t-test of Omomyc-expressing samples to their relative controls (n = 3). (E) Relation between Omomyc expression and inhibition of proliferation. (F) MYC and Max expression at 7 d of Omomyc expression (representative image from triplicates). (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001.
Western blot (WB) analysis with an anti-Omomyc antibody revealed that Omomyc expression levels are highly correlated with cell number reduction (r2 = 0.278; P = 0.0056) (Supplemental Fig. S1E). Thus, as previously shown in triple-negative breast cancer (TNBC) (Massó-Vallés et al. 2022), Omomyc's ability to reduce proliferation depends on its expression levels and appears to be independent of the mutational profile of the cell lines.
To further characterize the antiproliferative effect caused by MYC inhibition, we performed a cell cycle analysis at 1, 2, 3, and 4 d of expression of Omomyc in A375 and SkMel147, two cell lines with different oncogenic drivers: BRAFV600E and NRASQ61K, respectively. Despite these differences, we observed a very similar change in their cell cycle profile as a consequence of Omomyc expression already since day 1: a significant increase in G0–G1 populations that peaked above 80% on day 4, with a concomitant and steady decrease in the proportion of cells in S phase, down to a marginal 5%, and G2–M phases to even lower levels, with almost no changes in sub-G1 populations (Fig. 1E; Supplemental Fig. S1F).
Taken together, these results show that Omomyc induces an increase in the percentage of cells in G1 already at day 1 that results in evident in vitro growth arrest at day 3, which is maintained for at least 4 wk.
Omomyc expression reduces MYC levels in different melanoma cell lines
To determine whether Omomyc had any effect on MYC and Max protein levels in the different cellular contexts, we performed WB analysis after 1 wk of induction. Interestingly, while we found that Omomyc caused a decrease in MYC levels in nearly all cell lines, with the exception of A375 (lower expression but statistically nonsignificant), SkMel37 (higher, though not significant), and SkMel173 (no change), the modulation of Max was less consistent: Lower levels were observed in A375 and 501Mel, and higher levels were observed in SkMel173 (Fig. 1F; quantified in Supplemental Fig. S1G). Additionally, we found a significant correlation between relative MYC levels and proliferation (r2= 0.535, P = 0.0002) (Supplemental Fig. S1H).
Omomyc expression induces extensive transcriptional modulation in melanoma cells
Given the role of MYC as a master regulator of gene expression, we analyzed the transcriptional effect of MYC inhibition by Omomyc. In order to do so, we plated BRAFV600E mutated A375 cells in the presence or absence of doxycycline and performed a microarray analysis at 4 d, when Omomyc expression levels had reached their steady-state levels (Fig. 2A), to look at transcriptional changes that coincided with the antiproliferative effect of Omomyc. As expected, principal component analysis showed that Omomyc-expressing samples clustered together and had a gene expression pattern distinct from that of the vehicle counterparts (Fig. 2B). Specifically, 3203 genes were down-regulated by Omomyc expression (17.25%), while 3875 were up-regulated (20.87%) (Fig. 2C; Supplemental Table S1A), with an overall modulation of expression of ∼40% of the genes (Fig. 2D, heat map for the top 152 genes with absolute log FC > 2.25). To delve into Omomyc's mechanism of transcriptional modulation, we performed qPCR analysis of five genes as early as 6 h, 12 h, 24 h, and 4 d after Omomyc expression, which is the same time point of the microarray. The transcriptional changes were only significant at 4 d, with the exception of HSPD1, which already showed a small but significant decrease at 24 h (Fig. 2E). Of note, we believe that the variability of the results at 6 and 12 h reflects the low and incomplete expression of Omomyc in the cells, which instead reaches much higher levels at 24 h, when the gene expression levels are much less variable.
Figure 2.

Omomyc induces transcriptional modulation in the A375 melanoma human cell line in vitro. (A) Omomyc-RFP levels determined by cytometry, with percentage of positive cells and mean intensity. (B) Principal component analysis showing the separation of the five Omomyc-expressing (Dox) replicates from the control ones. (C) Heat map of A375 cells expressing Omomyc or not at day 4 (absolute log FC > 2.25). (D) Volcano plot of A375 cells expressing Omomyc. (E) qPCR validation of microarray results of selected genes at 6, 12, 24, and 96 h of Omomyc expression. (F) Heat maps of the top 10 regulated genes in selected gene sets. Representative MYC-related (G) and melanoma and other skin cancer (H) gene sets modulated by Omomyc. (I) Immune-related gene set enrichment induced by Omomyc expression.
In line with this massive change in gene expression, gene set enrichment analysis revealed a plethora of different transcriptional programs affected by Omomyc (Fig. 2F–I; Supplemental Table S1B,C). In fact, and intimately related to MYC's role in tumor progression (Kress et al. 2015), MYC and E2F targets as well as cell cycle checkpoint gene sets were significantly down-regulated (Fig. 2F,G). Interestingly, MYC inhibition by Omomyc expression also led to a metabolic reprogramming, with a significant decrease in the oxidative phosphorylation gene set, a pathway notoriously associated with invasion of primary melanoma (Fig. 2F; Salhi et al. 2020).
Also directly related to melanoma, Omomyc impaired the expression of several gene sets relevant to its progression and up-regulated genes normally found underrepresented in melanoma metastasis (i.e., Alonso metastasis down and Schlingemann skin carcinogenesis TPA down) (Fig. 2H).
Finally, in line with MYC's role in immune suppression, MYC inhibition led to a proinflammatory expression profile (Fig. 2I).
Hence, in vitro expression of Omomyc induces broad transcriptional reprogramming, not only affecting MYC targets and MYC-related transcriptional programs but also down-regulating key genes involved in melanoma metastases, invasion, and relapse.
MYC inhibition by Omomyc leads to cell cycle arrest through down-regulation of multiple targets
In line with the results of the cell cycle analyses, gene expression analysis of A375 showed that most of the changes in cell cycle-related genes are present at 4 d of Omomyc expression (Fig. 3A). Intriguingly, at this time point, Omomyc induced down-regulation of TP53 mRNA levels, yet its downstream target CDKN1A (Jalili et al. 2012), a key cell cycle inhibitor, was up-regulated.
Figure 3.
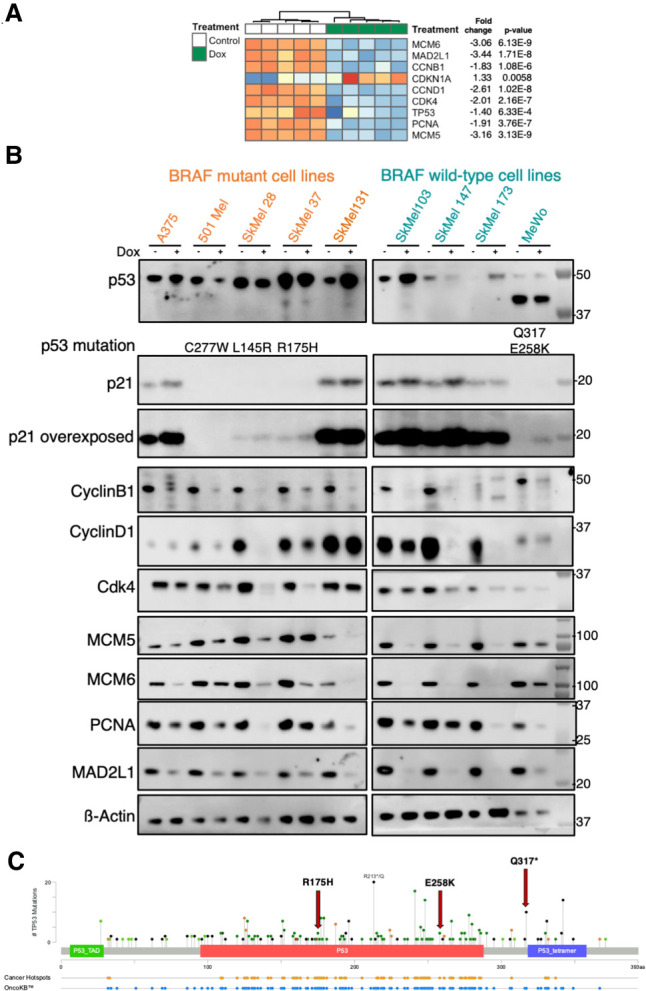
Omomyc induces a dramatic growth arrest in melanoma human cell lines in vitro. Changes in the expression of genes related to cell cycle and division in A375 cells at the mRNA (A) and protein (B) levels after 7 d of OmomycRFP expression. (C) Mutations in TP53 gene (cBioPortal).
We then extended this cell cycle-related analysis to the remaining human melanoma cell lines in our panel, performing WB at 7 d of Omomyc expression (Fig. 3B). At this later time point, Omomyc led to a clear and significant stabilization of p53 in most wild-type p53 cell lines with a concomitant and significant increase in the levels of p21 protein, including in A375, where TP53 mRNA levels were decreased, and with the exception of SkMel173, in which p21 levels were lower in Omomyc-expressing cells than in control cells. Four of the cell lines included in the panel bear a total of five mutations in the TP53 gene; of these four are in the DNA binding domain (L145R, R175H, E258K, and C277W) and one is a truncating mutation before the tetramerization domain (Q317) (Fig 3C; Supplemental Table S2). Interestingly, we found a marked and significant down-regulation of the p53 mutant forms in three out of four p53 mutant cell lines. Moreover, p21 levels increased in two of these cell lines (SkMel37 and MeWo), pointing to a p53-independent increase, maybe through inhibition of MYC-Miz-1-induced repression (Wu et al. 2003) or Akt phosphorylation (Shamloo and Usluer 2019). Thus, Omomyc induces cell cycle arrest with concomitant stabilization of wild-type p53 and down-regulation of mutant p53 that leads to increased levels of cyclin-dependent kinase inhibitor p21.
We then analyzed CyclinB1, which is necessary for the control of G2/M transition (Wang et al. 2014), and Cyclin D1 and Cdk4, both of which are involved in G1/S transition (Diehl 2002). We found a general down-regulation of all three proteins, although, intriguingly, the extent of down-regulation is very variable across the cell lines.
Finally, we analyzed MCM5 and MCM6, two components of the minichromosome maintenance complex (MCM), the replicative helicase essential for the DNA replication during cell cycle and DNA repair (Costa et al. 2011), and again found a very clear decrease in their levels in all of the cell lines. Similarly, we found decreased levels of PCNA, a cofactor of DNA polymerase δ (Moldovan et al. 2007), and saw remarkable down-regulation of MAD2L1, a mitotic spindle assembly checkpoint component and MYC target (see Supplemental Fig. S2 for all of the quantifications; Menssen et al. 2007).
In conclusion, Omomyc induces cell cycle arrest through down-regulation of several key genes—and their respective protein products—controlling cell cycle and DNA replication, irrespective of the melanoma driving mutation or p53 status.
Omomyc induces tumor regression in human melanoma CDXs
Encouraged by these in vitro results, we decided to assess the effect of Omomyc expression in vivo, making use of the two melanoma cell lines that we picked as representative of BRAF mutated and NRAS mutated melanoma: A375 and SkMel147, respectively. We inoculated these cells subcutaneously into immunocompromised mice, and when the mean tumor volume reached 100 mm3, mice were randomized into vehicle and doxycycline groups (Supplemental Fig. S3A). Systemic administration of doxycycline in the drinking water did not affect mouse weight, although we observed an initial slight decrease in both models, most probably due to animal adjustment to the change in taste of the drinking water (Supplemental Fig. S3B). Strikingly, Omomyc caused dramatic regression of A375 tumors (Fig. 4A, left panel; Supplemental Fig. S3C) and significantly prolonged mouse survival (Fig 4A, right panel) until the end of the experiment (4 wk), when most of the control mice had to be euthanized due to maximum tumor volume (P < 0.0001) (Fig. 4A; Supplemental Fig. S3C). A similar striking effect was observed in SkMel147 tumors: Here, there was a first phase of tumor stabilization followed by tumor regression. Again, also in this case, there was a significant increase in mouse survival, with all of the vehicle-treated mice needing euthanasia by day 15, whereas all of the mice bearing Omomyc-expressing tumors survived at least to the end point, established at 26 d, corresponding to double the survival time of control mice (P < 0.0001) (Fig. 4B; Supplemental Fig. S3D). It is noteworthy that tumors of mice drinking doxycycline did eventually start to grow later in the experiment. This is due to outgrowth of cells lacking expression of Omomyc, as shown in Supplemental Figure S3E. This could be a consequence of negative selection, leading to the complete loss of the transgene (which seems to be the case in A375 cells), or the selective proliferative advantage of cells expressing the lowest—or null—levels of Omomyc (like in SkMel147, where very faint RFP levels can be detected in some cells that, importantly, still lack Ki67 expression).
Figure 4.

Omomyc induces tumor regression and inhibition of metastases in human melanoma cell lines in vivo. Omomyc significantly impairs the growth of A375 (A) and SkMel147 (B), increasing the survival of tumor-bearing mice. Ki67 staining after 3 d of Omomyc expression in the tumor margin of A375 (C) and SkMel147 (D). Percentage of mice with spontaneous lung, liver, and lymph node metastases in A375 (E) and SkMel147 (F) models. Recurrence and lung metastases after tumor resection in A375 (G) and SkMel147 (H). (Veh) Vehicle, (Dox) doxyclycline.
Notably, after only 3 d of Omomyc expression, the percentage of Ki67+ cells in the invasive front of the tumor had significantly decreased in A375 (P = 0.0123) (Fig. 4C), as did the tumor volume (Supplemental Fig. S3F, left panel). In the case of SkMel147, due to their high growth rate and the slow kinetics of Omomyc expression triggered by doxycycline, by day 3 the median volume of the treated group was already 264.6 mm3 (IQR: 192.4–517.4 mm3, similar to the vehicle group) (Supplemental Fig. S3F, right panel). Nonetheless, despite its delay, the percentage of Ki67+ cells was at that point lower in mice drinking doxycycline (P = 0.0176) (Fig 4D), anticipating the final result: a durable tumor regression induced by Omomyc expression.
A similar effect of inhibition of growth and regression is achieved by Omomyc also in a p53 mutated context of human melanoma cell lines (SkMel28 and SkMel37, bearing mutations in both p53 and BRAFV600E) (Supplemental Fig. S3G,H).
Hence, Omomyc expression in melanoma cells in vivo is sufficient to induce a rapid and durable tumor regression even in advanced tumors, independently of their TP53 status.
Omomyc curbs metastases in human melanoma CDXs and prevents postsurgery tumor recurrence
Melanomas are intrinsically metastatic, and this is the main cause of death among patients. Thus, after confirming the outstanding effect of Omomyc on primary tumor growth in vivo, we looked for any effect on metastases by ex vivo imaging of specific organs. Not surprisingly, we found metastatic colonization of the lungs, liver, and lymph nodes of the control mice (Fig. 4E). Encouragingly, instead, Omomyc significantly impaired the metastatic spread of BRAF mutant A375 cells in vivo, decreasing the percentage of mice with metastases (P < 0.001 in all cases) (Fig. 4E). Notably, we observed an even more dramatic result in NRAS mutant SkMel147, where Omomyc completely inhibited the metastatic spread (P < 0.001 in all cases) (Fig. 4F). In this case, when we found residual metastases under doxycycline treatment, these did not express Omomyc anymore, meaning that they were actual escapees that had lost the expression of the transgene (Supplemental Fig. S3I) and were not considered in the analysis.
Finally, to test the effect of Omomyc in the adjuvant setting, we inoculated A375 and SkMel147 cells into nude mice and let the tumors grow until 500 mm3, at which point the primary lesions were surgically removed and mice were randomized to receive either doxycycline or vehicle in the drinking water with periodical IVIS scanning to detect metastases (Supplemental Fig. S3J). Although A375-bearing mice showed a very low percentage of tumor recurrence and lung metastases after tumor resection, tumors recurred only in control mice but not in those under doxycycline treatment, with a concomitant nonsignificant reduction of lung metastases in treated mice (P = 0.068) (Fig. 4G). This was also confirmed in the SkMel147 model with even more striking results, as tumors recurred in >50% of control mice but not in Omomyc-expressing ones, which survived to the end point with significantly fewer metastases (P < 0.001) (Fig. 4H).
Taken together, these data show that Omomyc reduces melanoma cell proliferation, causes tumor regression in vivo, reduces the metastatic capacity of melanoma cells, and prevents tumor recurrence when expressed after surgical removal of the primary tumor.
Omomyc-induced tumor regression in vivo is accompanied by transcriptional modulation
In order to gain more insight into the mechanism of action leading to therapeutic impact of MYC inhibition by Omomyc in melanoma in vivo, we took tumor samples after 1 wk of Omomyc expression in A375 and SkMel147 subcutaneous tumors and subjected them to microarray analysis (Supplemental Fig. S4A). For this study, we specifically selected tumors of similar sizes in order to avoid any gene expression change due to this variable (Supplemental Fig. S4B, bottom panels). Principal component analysis showed that Omomyc-expressing tumors from both cell lines have a transcriptional profile that is distinct from their vehicle counterparts (Supplemental Fig. S4C). Despite some variability in gene expression among tumors (PC2 axis in Supplemental Fig. S4C), we were able to identify a large cadre of genes significantly regulated by Omomyc in both cell line-derived tumors (Supplemental Fig. S4D; Supplemental Table S3A,B). Supplemental Figure S4E shows regulation of the top 200 genes, from which we selected and validated some by qPCR (Supplemental Fig. S4F).
Despite the fact that A375 and SkMel147 are driven by different oncogenic mutations (BRAFV600E and NRASQ61R, respectively), we found considerable overlap in transcriptional modulation by Omomyc: 51% of down-regulated genes (DN) in A375 were also down-regulated in SkMel147, whereas 26% of the up-regulated genes (UP) in A375 showed the same modulation in SkMel147 (Fig. 5A; Supplemental Table S4). We found a clear link between the 436 DN genes and gene categories relevant for both MYC and melanoma biology: genes involved in melanoma progression and metastases, MYC targets, cell cycle and division, DNA structure and replication, RNA processing and transcription, and DNA repair and metabolism. In fact, the majority (75.7%) of the regulated genes belong to at least one of these categories (Fig. 5B; Supplemental Table S5A,B).
Figure 5.

Omomyc-induced transcriptional modulation independent of melanoma driver mutation targets key melanoma genes. (A) Overlaps of modulated genes by Omomyc in vivo. (B) Chord diagram of common down-regulated genes linked to categories of down-regulated gene sets. (C) Selected common down-regulated gene sets. (D) Supervised K-means clustering string analysis of down-regulated genes. (E) Transcription factor binding site profiles in the ENCODE and ChEA libraries.
Confirming the specificity and therapeutic relevance of MYC inhibition in vivo, gene set enrichment analysis revealed a conserved collection of gene sets comprising both pathways related to melanoma and MYC that were significantly down-regulated by Omomyc (summarized in Fig. 5C; Supplemental Table S6). Notably, the heat maps from the melanoma metastasis and relapse and the MYC target gene sets show that the vast majority of the genes in these categories are down-regulated by Omomyc (Supplemental Fig. S4G).
In addition, unsupervised string analysis of common DN genes groups them into four different clusters (Supplemental Fig. S5A; Supplemental Table S7). When we analyzed these clusters, we found that they encompassed genes involved in (1) chromatin modification, organization, and remodeling, as well as transcription; (2) cell metabolism; (3) cell cycle; and (4) RNA processing and metabolism (Fig. 5D; Supplemental Table S8A–D). Analogously, common UP genes show two clusters after unsupervised string analysis (Supplemental Fig. S5B; Supplemental Table S8E,F), including genes involved in (1) metabolism, lysosomes/autophagy, vesicle-mediated transport, subcellular organization, and immune response, and (2) immune response (Supplemental Fig. S5C).
In order to gain more insight into the regulation of the DN genes, we subjected them to Enrichr analysis (Chen et al. 2013; Kuleshov et al. 2016; Xie et al. 2021). Specifically, we queried which transcription factor (TF) binding sites were present in the promoter region of these genes in the ENCODE and ChEA libraries. As expected, we found that the TFs most significantly represented in MYC-induced DN genes in vivo presented MYC, Max, and E2F TF binding sites (Fig. 5E; Supplemental Table S9A). Interestingly, when these genes were compared with kinase and GPCR perturbation libraries or Gene Expression Omnibus (GEO) drug perturbation pathways, we found that the most significantly similar profiles were those associated with knockdown of tyrosine and serine/threonine kinases involved in cell cycle, proliferation, and transcriptional regulation of genes—among others—in melanoma (Supplemental Fig. S5D; Supplemental Table S9B), or inhibition of RTK–AKT–MAPK pathway components and cell cycle regulators in cancers of different origins (Supplemental Fig. S5E; Supplemental Table S9C). These analyses clearly show that MYC, Max, and E2F are the most prominent TFs regulating the expression of DN genes and that Omomyc-expressing tumors phenocopy melanomas with compromised key signaling pathways.
Overall, these results obtained through different approaches show that Omomyc induces a severe transcriptional modulation in vivo—conserved between melanomas with different driver mutations—that affects not only single genes but entire transcriptional programs involved in melanoma progression and metastasis, as well as chromatin modification, metabolism, and, as expected, a variety of MYC-related programs relevant for tumorigenesis.
Finally, to take a closer look at the metastatic process and early transcriptional changes that could account for the outstanding effect of Omomyc, we performed a microarray analysis of lymph node metastases after only 2 d of doxycycline treatment (before their quick disappearance caused by Omomyc), which is equivalent to ∼1 d of suboptimal Omomyc expression. We inoculated SkMel147 cells subcutaneously and scanned the mice for spontaneous metastases on day 18. Mice were randomized, and doxycycline was administered. After 2 d, mice were scanned again, and metastases were confirmed in eight of the 10 initial mice (Supplemental Fig. S6A). Despite the extreme transcriptional variability observed among the metastases from vehicle-treated mice (Supplemental Fig. S6B), we found several gene sets modulated by Omomyc, both down-regulated (DN) and up-regulated (UP) (Supplemental Fig. S6C; Supplemental Table S10). Although the transcriptional analysis of primary tumors was performed at 7 d while that of lymph node metastases was only at 2 d, almost 50% of the DN gene sets were common to the two settings (Supplemental Fig. S6C). Among the DN gene sets, we found several that could at least partially explain the remarkable effect of Omomyc on metastases (Supplemental Fig. S6D): Besides general transcriptional programs such as translation and DNA replication, we found the expected MYC target and melanoma metastasis gene sets. Interestingly, EZH2 targets were also down-regulated. The enhancer of zeste homolog 2 (EZH2) is a histone-lysine N-methyltransferase whose inhibition has been shown to impact the invasive capacity of NRAS mutant cells, like the SkMel147 used in this experiment (Terranova 2021). Notably, some of the gene sets whose expression was exclusively abrogated in metastases are intimately related to melanoma metastases. The most prominent ones are lamellipodium membrane, notch signaling, and the ILK and ATF2 pathways. Indeed, lamellipodia, together with filopodia, contributes to invasion and metastases of tumor cells (Machesky 2008), while melanoma metastases are promoted by Notch signaling through its interaction with β-catenin (Bedogni 2014) and by the transcription factor ATF2 by the suppression of protein fucosylation (Lau et al. 2015). Finally, integrin-linked kinase (ILK) is a serine/threonine kinase important for melanoma cell migration through the formation of actin cytoskeleton and motility structures and in the signal transduction upon engagement to ECM attachment (Wong et al. 2007).
Among the few up-regulated gene sets found in LN metastases from mice under doxycycline treatment, we found one reinforcing the inhibition of ATF2 activity (ATF2 UP.V1 DN; genes down-regulated by ATF2 when hyperactive, also present in the primary tumor after 7 d) and three gene sets consisting of genes down-regulated during skin cancer progression, specifically found in metastases: hummerich skin cancer progression DN, benign skin tumor DN, and malignant skin tumor DN.
Taken together, these results confirm that Omomyc targets not only the proliferation and survival of primary and, by extension, metastatic tumor cells but also different mechanisms directly involved in the metastatic spread.
Melanoma patients classified by an Omomyc-derived six-gene signature show improved OS, decreased MYC activity and metastatic capacity, and increased immune response
The ultimate goal of our MYC inhibition study is to predict how such a therapeutic approach could potentially impact patient treatment and/or survival and how Omomyc-induced transcriptional reprogramming relates to melanoma patient prognosis. To this end, we accessed TCGA data of melanomas with RNA-seq and clinical information (Hoadley et al. 2018) and analyzed how differential expression of every single DN or UP gene in both BRAFV600E- and NRASQ61L-driven melanomas impacts patient survival. Out of 436 commonly DN genes, lower than the median expression of 143 of them (32.8%) is significantly associated with increased overall survival (OS) in melanoma patients. Similarly, high expression of 70 out of 156 UP genes (44.9%) in Omomyc-expressing tumors is associated with increased OS in patients (Supplemental Table S11). Next, in an attempt to see whether Omomyc's transcriptional signature could be beneficial to patients, we focused on genes associated with better prognosis. With this filter, we generated a hybrid six-gene signature encompassing BIRC5, CCNB2, and ORC1 from the DN genes and DDX60, PARP9, and DTX3L from the UP genes. As expected, patients with this signature have significantly increased OS (205 mo [162–NA mo] vs. 42 mo [28–63 mo], P = 7 × 10−7), a lower hypoxia score (P < 10−10), and a lower fraction of altered genomes (P = 3.884 × 10−4) (Fig. 6A,B).
Figure 6.

Melanoma patients stratified by a six-gene signature show improved OS with lower metastatic capacity and increased immune response. (A) Survival of melanoma patients discriminated by a six-gene signature. (B) Patients with good prognosis have a lower hypoxia index and a lower fraction of the genome altered. (C) Volcano plot depicting gene expression of good prognosis versus poor prognosis patients. (D) Overlap between Omomyc-modulated genes in preclinical settings (percentage shown) and genes significantly enriched in good and poor prognosis patients. Preranked GSEA shows decreased MYC transcriptional activity, metastatic capacity, and melanoma relapse (E) and decreased expression of EMT genes, E2F targets, and cell cycle genes (F). (G) Tumors from patients with good prognosis show enrichment of genes related to immune response. (H) There was no evidence of differential MYC transcriptional repression activity in either group of patients.
This six-gene signature discriminated patients with differential gene expression, with a plethora of genes down-regulated and up-regulated in patients with good prognosis with respect to bad prognosis (summarized in Fig. 6C).
In more detail, 63% of genes up-regulated by Omomyc in vivo are enriched in patients with good prognosis, whereas 56% of genes down-regulated by Omomyc are enriched in poor prognosis patients (Fig. 6D), pointing to a reasonable predictive value of the results obtained with Omomyc in a clinical setting.
In order to analyze how these data related specifically to MYC biology and melanoma, we performed preranked GSEA in patients and found that good prognosis patients have lower expression of MYC target genes as well as of genes involved in melanoma metastases and relapse (Fig. 6E). Consistently, these patients also showed significantly lower expression of genes involved in epithelial-to-mesenchymal transition, E2F targets, and cell cycle (Fig. 6F), as well as increased expression of adaptive and inflammatory immune response genes (Fig. 6G). Finally, in line with what we had seen in vivo, we found no differences in the expression of genes repressed by MYC (Fig. 6H), which may reflect the fact that these genes do not have a clear impact on melanoma patient survival.
Discussion
MYC's role in tumor progression and maintenance has long been established for most human cancers. In melanoma, it has been shown to be deregulated by amplification of the 8q24 chromosome or by upstream activating mutations in the RAS/RAF/MAPK pathway, which results in an aggressive clinical course and resistance to targeted therapy.
To date and to our knowledge, direct inhibition of MYC in melanoma has only been tested using small interfering RNA (siRNA) (Hong et al. 2007) or antisense oligonucleotides directed against MYC (Dinçer et al. 2010; Massó-Vallés and Soucek 2020), where it induced inhibition of proliferation through cell cycle arrest, senescence, and apoptosis, or using nanoparticles (NPs) containing the MYC inhibitor 10058-F4 in a 72-h proliferation assay, where Alamar Blue metabolic testing showed a fourfold decrease in the proliferation of C32 BRAF mutant human melanoma cells (Pan et al. 2015).
Here we made use of Omomyc, the most-characterized MYC inhibitor to date, which has just successfully completed a phase I clinical trial, to assess the therapeutic impact of MYC inhibition in melanoma both in vitro and in vivo in the presence of different mutational profiles. Omomyc had previously shown an outstanding antitumorigenic effect in a wide range of murine preclinical models of cancer, without any severe side effects in normal tissues (Massó-Vallés and Soucek 2020). Most recently, it has also been shown to target and curb the metastatic spread in TNBC models (Massó-Vallés et al. 2022). For these reasons and for the undisputed role of MYC in melanoma, we sought to model MYC inhibition in this disease and investigate its effects on progression and metastases, the main causes of death for melanoma patients. Using a panel of human cell lines with different driving mutations, we were able to show that Omomyc expression in vitro has a significant impact on proliferation and colony formation of the different lines, and this effect correlates with Omomyc expression levels. Although the induction of apoptosis after 1 wk of Omomyc expression is only evident in one-third of the cell lines, prolonged analysis up to 4 wk showed that, after a period of stable growth arrest, cell death increases significantly in different cellular contexts.
In regard to cell cycle, MYC overexpression has been reported to enable cancer cells to enter it by inducing the expression of cell cycle regulators and inhibiting cell cycle checkpoints (Dhanasekaran et al. 2022). When we performed cell cycle analysis in BRAF mutant A375 and NRAS mutant SkMel147 melanoma cells expressing Omomyc, we found consistent results: accumulation of cells in G1 phase with a concomitant remarkable decrease of cells in S and G2/M phases. We also found a significant decrease in the expression levels of the cell cycle regulators CyclinB1, CyclinD1, and MYC target Cdk4 (Hermeking et al. 2000). However, progression through the cell cycle is not only regulated by cyclins and cyclin-dependent kinases but also involves, among other processes, the replication of DNA and cell division. For example, minichromosome maintenance proteins (MCMs) are part of DNA helicase complexes that help unwind DNA during its replication, recombination, and repair (Seo and Kang 2018). In addition, proliferating cell nuclear antigen (PCNA) is a cofactor of DNA polymerase δ and, as a tetramer, helps increase the processivity of leading strand synthesis during DNA replication (Moldovan et al. 2007). Finally, the mitotic spindle assembly checkpoint maintains genome stability during chromosome segregation before cell division (Lara-Gonzalez et al. 2012), and one of its key components is MAD2L1, which is a direct MYC target (Menssen et al. 2007). Remarkably, Omomyc expression induced a striking down-regulation of all of them (MCM5, MCM6, MAD2L1, and PCNA).
p53 is probably the best-studied tumor suppressor and is key in preventing malignancy. It is capable of controlling the expression of a large number of genes, including those involved in the cell cycle by p21-dependent indirect transcriptional repression (Engeland 2022). cBioPortal data of 1847 melanoma patients from eight different studies show that p53 or its inhibitors MDM2 and MDM4 are altered in 27% of samples analyzed in most of cases corresponding to putative drivers (cBioPortal, https://bit.ly/3K2Hra9). The panel of cell lines used in this study contained five cell lines with wild-type and four cell lines with mutated p53. Interestingly, while in p53 wild-type cells Omomyc induced stabilization of the protein, in mutant cells it caused a significant reduction in the expression of their mutated isoforms. In most of the cases, this modulation was accompanied by an increase in cyclin-dependent kinase inhibitor p21. Hence, by decreasing the level of multiple proteins involved in the progression of the cell cycle and increasing at least one cell cycle inhibitor, Omomyc counteracts one of MYC's fundamental roles in tumor progression.
To gain further insight into the mechanism of action of Omomyc in melanoma, we performed a microarray analysis of BRAF mutant A375 melanoma cells in vitro. As previously found in fibroblasts, non-small cell lung cancer, and TNBC (Savino et al. 2011), Omomyc expression abrogated several transcriptional programs, including MYC and E2F targets, RNA metabolism, cell cycle, ribosome biogenesis, and RNA polymerase II-mediated transcription. Furthermore, it up-regulated genes that are normally repressed by MYC. Thus, Omomyc is able to counteract MYC's protumorigenic transcriptional activity by reverting its ability to activate and, at least in vitro, repress gene expression.
To complement these studies with an in vivo analysis, we tested Omomyc's ability to affect tumor growth in two different xenograft models, driven by BRAFV600E and NRASQ61R, respectively. In line with the in vitro results, there was a profound inhibition of tumor growth and even regression in both cases. Most remarkably, Omomyc also curbed the metastatic spread, as analyzed in the lymph nodes, lungs, and liver. Furthermore, it completely prevented tumor recurrence after surgery and, again, abrogated the appearance of late metastases. Importantly, in many of the residual metastases found in mice drinking doxycycline, we saw no sign of Omomyc expression, confirming that at least a proportion of these metastases was the result of Omomyc transgene loss, a phenomenon already observed before in transgenic mouse models of Omomyc (Soucek et al. 2013). These results were also confirmed in BRAF and p53 double-mutant cell lines.
Thus, Omomyc not only is able to halt primary tumor growth but, importantly, also targets the metastatic spread both in the presence of the primary tumor and as adjuvant after tumor resection.
It should be noted that our experiments have been conducted in immune-deficient models. Given the clear role of MYC in immune suppression (Dhanasekaran et al. 2022) and the proinflammatory reprogramming that we observed in A375 cells, our prediction is that Omomyc's effect could be even more pronounced in an immunocompetent context.
Despite their different driver mutations, microarray analyses of in vivo tumors showed a significant overlap of modulated genes. Up to half of the down-regulated genes in one cell line were also down-regulated in the other, while for up-regulated genes this overlap was ∼25%. In order to determine the relevance of this in vivo transcriptional reprogramming of melanoma cells, we curated and analyzed gene categories relevant to MYC and melanoma biology. Strikingly, up to 75% of the genes down-regulated by Omomyc belong to one of these categories, confirming the on-target effect and the biological relevance of Omomyc in melanoma. Different analytical approaches (namely, string and gene set enrichment analyses) confirmed these results.
In addition, Enrichr analysis showed that the most prevalent transcription factor binding sites present in these common down-regulated genes are those for MYC and Max, along with E2F6.
We also show that Omomyc's effect in melanoma phenocopies the knockdown of components of the cell cycle or RAS–RAF–MAPK pathway as well as inhibition of different kinases, including mutant BRAF. Consequently, targeting MYC renders tumor cells incapable of proliferation and survival in a manner similar to that attained with tyrosine kinases inhibitors but without causing their undesired toxic effects and in the absence of potential signal rewiring leading to resistance. Moreover, given that tumors expressing Omomyc acquired a proinflammatory profile, it would be reasonable to think that combination of Omomyc with IO would be beneficial for melanoma patients. Finally, further transcriptional analysis of spontaneous metastases showed that Omomyc targets multiple mechanisms involved in the metastatic spread and aggressiveness of melanoma.
The ultimate goal of our work for a very long time has been to bring Omomyc to the clinical setting, a landmark that was achieved in 2021, when it entered a phase I clinical trial that was successfully completed in 2022. Now, our current results suggest that its clinical applicability could extend to melanoma. Hence, in this work, we also analyzed whether Omomyc-induced transcriptional modulation in our preclinical models could relate to patient outcome. First, we assessed whether the expression of Omomyc-modulated genes could be related to survival and found that approximately half of them confer survival advantage when their expression in patients is lower than the median. In contrast, half of the UP genes confer survival advantage when their expression is higher than the median.
With the aim of extrapolating a useful predictive signature from our preclinical data to be used in melanoma patients, we generated a hybrid gene signature that would be able to discriminate patients with good and poor prognoses. This signature includes low expression of three genes with a known role in cancer progression (although only one of them has been thoroughly studied in melanoma): Origin recognition complex subunit 1 (ORC1) is essential for the initiation of DNA replication and is tightly regulated during the cell cycle (https://www.ncbi.nlm.nih.gov/gene/4998), BIRC5/Survivin prevents apoptotic cell death, and CCNB2 is a key component of the cell cycle regulatory machinery and a potential prognostic predictor for melanoma (Chen et al. 2020). The signature also includes three genes with high expression, with limited study in cancer to date and even scarcer in melanoma: DDX60 is a putative RNA helicase implicated in cellular processes involving RNA binding and alteration of RNA secondary structure (https://www.ncbi.nlm.nih.gov/gene/55601), DTX3L is an E3 ubiquitin–protein ligase, and the ADP-ribosyltransferase PARP9, together with DTX3L, plays a role in DNA damage repair (Takeyama et al. 2003). Although DTX3L has been proposed to stimulate melanoma metastases (Thang et al. 2015), patient data from cBioPortal (Liu et al. 2018) and Human Protein Atlas (https://www.proteinatlas.org/ENSG00000137628-DDX60) show that its expression is directly correlated with increased patient survival.
The two cohorts of patients classified by the signature present radically different survival, with a fivefold increase for the good prognosis patients, who also show a lower hypoxic index and fraction of the genome altered. Notably, in melanoma, as in most solid tumors, hypoxia is associated with aggressiveness and metastases (Kalaora et al. 2022). The fraction of genome altered is the percentage of genome that has been affected by copy number gains or losses, and it has recently been shown that, in melanoma, it increases in visceral metastases compared with their respective primary tumors (Papp et al. 2021).
Furthermore, transcriptomic analysis also shows that ∼60% of the genes up-regulated by Omomyc are enriched in good prognosis patients, while genes down-regulated by Omomyc are enriched in poor prognosis patients, meaning that the transcriptional reprogramming observed in mice upon Omomyc expression in melanoma cells resembles that of patients with good prognosis. Accordingly, gene set enrichment analysis showed a remarkable overlap of our preclinical data with data from patients classified according to the Omomyc-derived gene signature (low ORC1-–BIRC5–CCNB2 and high PARP9–DDX60–DTX3L): Expression of MYC and E2F targets, melanoma metastases and relapse, G2/M checkpoints, and epithelial-to-mesenchymal transition gene sets is significantly decreased, while adaptive and inflammatory immune response gene sets are enriched. In fact, the most biologically relevant transcriptional programs are equally modulated in Omomyc-expressing melanoma cells and in patients with good prognosis.
In summary, the data presented here demonstrate that MYC inhibition is an effective therapeutic approach against melanoma for both primary tumors and metastases, able to reshape the tumor transcriptional landscape toward an immunogenic and less proliferative profile, erasing MYC's transcriptional footprint independently of the melanoma driver mutation. Additionally, by relating our preclinical models to patient data, we unveil a gene signature that discriminates patient prognosis and underlines the therapeutic potential that such an approach could eventually have in the clinic in this dismal disease.
Materials and methods
Cell lines and viral infections
A375, 501Mel, SkMel28, SkMel103, and SkMel147 human melanoma cell lines were a kind gift from Dr Aroa Soriano (Vall d'Hebron Institute of Research [VHIR], Barcelona) while SkMel37, SkMel131 (clone 1.36–1.5), SkMel173, and MeWo human melanoma cell lines were a kind gift of Cristina Ruiz Herguido (Institut Hospital del Mar d'Investigacions Mèdiques [IMIM], Barcelona). All cell lines were routinely tested for mycoplasma contamination. Cells were grown in DMEM supplemented with 10% fetal bovine serum and 1% L-glutamine (all from Life Technologies). A375 and SkMel147 were validated at High Technology Unit (VHIR).
Omomyc was cloned into the pTRIPZ lentiviral vector (Open Biosystems and Thermo Scientific) to generate a doxycycline-inducible Omomyc-RFP expression cassette, and the shRNA sequences were removed (Annibali et al. 2014). For infections, 293T cells were seeded at 70% confluence, and the following morning, 25 mM chloroquine was added. Two hours later, 293T cells were transfected with pTRIPZ-Omomyc plus the lentiviral vectors pMD2G and psPAX2 by the CaPO4 method. The medium was changed the following day, and sodium butyrate was added at 5 mM. Viral supernatants were harvested on the subsequent 2 d, filtered, and added to target cells with 0.8 µg/mL polybrene. Retroviral infection using 293 cells transduced with pQCXIH-firefly luciferase vector was used to modify cells for in vivo experiments.
Western blots
Western blots were performed as previously published (Massó-Vallés et al. 2022), with modifications. Briefly, cells were plated with or without 1 µg/mL doxycycline and grown for 4 or 7 d. Media was aspirated, cells were washed with PBS, and dry plates were stored at −80°C until processing. Frozen cells were scraped with RIPA buffer supplemented with protease and phosphatase inhibitors (Roche), incubated for 30 min, and centrifuged at maximum speed for another 30 min. Supernatants were collected, total protein concentration was quantified by DC protein assay (Bio-Rad), and absorbance at 560 nm was measured with a TECAN Spark spectrophotometer (Life Sciences). Protein extracts were run on 10% or 12% precast gels (Life Technologies), transferred to nitrocellulose membranes (Millipore), and incubated with antibodies against c-Myc (Y69; Abcam ab32072), Cdk4 (D9G3E; Cell Signaling Technology 12790), CyclinD1 (92G2; Cell Signaling Technology 2978), Cyclin B1 (Cell Signaling Technology 4138), p53 (Dako M7001), p21 (clone CP74; Thermo Ab-11 MS-891-P0), Max (C-17; Santa Cruz Biotechnology sc-197 and R&D AF4304), MCM5 (EP2683Y; Abcam ab75975), MCM6 (H-8; Santa Cruz Biotechnology sc-393618), MAD2L1 (Sigma-Aldrich HPA003348), PCNA (BioLegend 307904), β-Actin (Sigma-Aldrich A-5441), or Omomyc (noncommercial primary rabbit polyclonal anti-Omomyc antibody affinity-purified and selected against the MYC b-HLH-LZ epitope). ECL images were acquired using an Amersham Imager 600 and iBright CL1500 imaging system (Thermo Fisher). Protein expression was quantified using the Fiji image processing package containing ImageJ (Schindelin et al. 2012).
Propidium iodide (PI) and cell cycle analysis
Cell cycle was done as previously published (Massó-Vallés et al. 2022). Briefly, cells were treated with 1 µg/mL doxycycline upon plating or untreated and grown for 4 d, followed by trypsinization, two washes in PBS, and fixation in ethanol. Next, 5 × 105 cells were treated for 20 min with 25 mg/L PI, 10 µg/mL RNase, and 0.1% Triton X-100 in the dark. Navios (Beckman Coulter Life Sciences) and FACSCanto II (BD Biosciences) flow cytometers were used for data acquisition, and FCS Express 4 software was used for data analysis.
Proliferation assay
For Crystal Violet staining, cells were plated at low density (between 100 and 2000 cells, depending on the cell line) in 96-well plates in the presence or absence of 1 µg/mL doxycycline and incubated for 10 d. Next, media was removed from the wells, and cells were fixed with 4% PFA for 12 min, washed twice, stained with Crystal Violet for 20 min, and washed twice. For quantification, 10% acetic acid was added to the wells, and absorbance at 580 nm was read after 10 min.
For cell count, A375 and SkMel147 were plated in p60 plates in the presence or or absence of 1 µg/mL doxycycline and incubated for 1, 2, 3, and 4 d in one experiment and for 1, 2, 3, and 4 wk in another experiment. Media and PBS from the washing were recovered and mixed with the cells after trypsinization. Live and dead cells were counted with Vi-Cell XR cell viability analyzer (Trypan Blue dye exclusion method).
All experiments were repeated in at least three independent experiments, and samples were prepared at least in triplicates.
Cell death assay
To analyze the viability of the cells at 7 d of Omomyc expression, 15,000–130,000 cells, depending on the cell line, were plated in p100 plates and grown for 1 wk in the presence or absence of doxycycline. After trypsinization and recovery of cells, live and dead cells were counted as above.
Colony formation assay
Cells were plated at low density (250–1000 cells, depending on the cell line) in six-well plates in the presence or absence of doxycycline and incubated until the colonies in the control wells started showing signs of cell death due to local overconfluence. At the end point, media was removed from the wells, and cells were fixed with 4% PFA for 12 min, washed twice, stained with Crystal Violet for 20 min, and washed twice.
qRT-PCR
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed as previously described (Massó-Vallés et al. 2022). Briefly, to mimic the microarrays conditions, A375 and SkMel147 Omomyc-RFP cells were treated with 1 μg/mL doxycycline for 6, 12, and 24 h and for 4 d or left untreated. RNA was then extracted with TRIzol and quantified using NanoDrop. Equal amounts of RNA were reverse-transcribed to generate cDNA using iScript reverse transcription Supermix for qRT-PCR (Bio-Rad). SYBR Green qRT-PCR analysis was then performed on these cDNA samples with PerfeCTa SYBR Green FastMix and Low Rox (Quantabio) using the QuantStudio 6 FLEX system (Applied Biosystems). The data thus obtained were analyzed following the comparative (ΔΔCt) method described by Livak and Schmittgen (2001). Analysis of relative gene expression data was performed using real-time quantitative PCR and the 2−ΔΔC(T) method (Livak and Schmittgen 2001). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and β-Tubulin were used as housekeeping genes. Sequences of primers used are listed in Supplemental Table S12.
Animal studies
All of the animal studies were performed in accordance with ARRIVE guidelines and following the three-R rule of replacement, reduction, and refinement principles. Mice were housed and treated in accordance with protocols approved by the CEEA (Ethical Committee for the Use of Experimental Animals) at the Vall d'Hebron Institute of Oncology (VHIO), Barcelona.
For the in vivo experiments, 12 mice were used per treatment group, and they were randomized by tumor size and weight. The experimental end point was established a priori when tumor volume reached 1750 mm3, when metastases showed clear signs of growth, or when mice showed any sign of discomfort. Experiments were not performed in a blinded fashion.
All of the schematics were created with Biorender.com.
Cell line-derived subcutaneous model
A375 (2 × 106), SkMel147 (2 × 106), SkMel37 (4 × 106), or SkMel28 (10 × 106) human melanoma cells were inoculated subcutaneously into the dorsal flanks of 6-wk-old athymic BALB/cAnN-Foxn1nu/nu/Rj mice (Janvier Laboratories). Once the tumors reached 100 mm3, mice were randomized for treatment. One group was given 2 g/L doxycycline in 5% sucrose in the drinking water to induce expression of the Omomyc-RFP transgene in the melanoma cells, while the control group was given 5% sucrose. At the end point, mice were euthanized by CO2 inhalation. Tumors were then excised, fixed for 48 h in buffered 4% formaldehyde, transferred to 70% ethanol, and embedded in paraffin.
Tumor excision model
To analyze the effect of Omomyc in the metastatic capacity of the melanoma cell lines after the primary tumor was excised, cells were inoculated as above, and tumors were excised when they reached 500 mm3. Mice were subjected to surgery and allowed to recover. Two days after, they were scanned to detect luciferase-expressing tumor cells that could have been left behind during surgery in order to discard including mice with incomplete tumor resection. Once the success of the surgery was confirmed, mice were randomized into two groups as above; i.e., 5% sucrose in drinking water with or without 2 g/L doxycycline. Mice were routinely checked for tumor recurrence and scanned in IVIS to detect metastases.
Transcriptional analysis of lymph node metastases
SkMel147 cells were inoculated as above, and mice were scanned for metastases when they reached ∼750 mm3. At this point, 10 mice were randomized in vehicle and doxycycline groups as above. Two days after treatment onset, mice were euthanized by CO2 inhalation, and lymph nodes, lungs, and brains were excised and scanned to confirm the presence of metastases. Finally, eight lymph node metastases, four per group, were snap-frozen and stored until RNA was extracted.
In vivo imaging
Metastatic colonization was monitored by in vivo bioluminescence imaging using the IVIS-200 imaging system from Xenogen (Perkin Elmer) as previously described (Massó-Vallés et al. 2022). Briefly, mice were injected intraperitoneally with a d-luciferin solution (150 mg/kg in PBS) for 5–10 min prior to acquisition. Mice were anesthetized with isoflurane (5% for induction and 2% during acquisition), and air flow was set at 0.8 L/min. IVIS data were analyzed with Living Image software (Perkin Elmer). Study analysis consisted of a light radiance quantification. Signals from the light sources were detected and characterized. Working units were photons/second/square centimeter/steradian, which allowed comparison between images obtained by different acquisition parameters. Acquisition and analysis were carried out by the Preclinical Imaging Platform staff at VHIR.
Immunohistochemistry and confocal microscopy
Paraffin-embedded tumor sections were cut at 4 µm thick and stained with H&E for pathological examination. For Ki-67 immunofluorescence, antigen retrieval was performed by heating for 20 min at 400 W in a microwave in 0.01 M citrate buffer (pH 6.0). After blocking for 45 min in 3% BSA and washing in PBS, slides were incubated overnight at 4°C with anti-Ki-67 (Thermo Scientific RM-9106-S0) and diluted 1:100 in Dako ready-to-use diluent (Dako S2022). After a PBS wash, slides were incubated with goat antirabbit IgG (H+L)-Alexa Fluor 488 conjugate (Thermo Fisher Scientific A-11008) diluted 1:200, stained with DAPI (Life Technologies D1306) diluted 1:10,000, washed once with water, and mounted with fluorescence mounting medium (Dako S3023). For anti-Omomyc immunohistochemistry, the same procedure was performed, but a primary rabbit polyclonal anti-Omomyc antibody (affinity-purified and to remove MYC cross-reactive antibodies) was used at a 0.02 mg/mL final concentration. Confocal microscopy images were captured using a Nikon C2+ confocal microscope and NIS-Elements software. Images were acquired and analyzed in a blind fashion.
Microarray analysis
For RNA extraction from A375 cells, cells were seeded in the presence or absence of doxycycline and incubated for 3 d (n = 5). Plates were washed twice with PBS and frozen at −80°C until processing. For tumors, a viable section of tumors from control or treated mice (n = 5 for A375 and n = 4 for SkMel147) were snap-frozen until processed. For lymph node metastases, ipsilateral axillary lymph nodes with confirmed metastases (n = 4 per group) were snap-frozen until processing. In all of the samples, RNA was extracted with TRIzol reagent according to the manufacturer's instructions (Invitrogen). The quality of RNA was confirmed with an Agilent 2100 Bioanalyzer. Clariom S human HT microarray plates (Applied Biosystems) were processed at VHIR's High Technology Unit. Gene expression was analyzed with Partek Genomic suite (Partek Incorporated) and R 4.1.0 (https://www.R-project.org) under R Studio desktop version 1.4.1717. Raw expression values were obtained directly from .CEL files and preprocessed using the robust multiarray average method (Irizarry et al. 2003). Differentially expressed genes between groups were obtained after applying a linear model analysis with empirical Bayes moderation of the variance estimates by using the limma package from Bioconductor. To deal with multiple testing issues, P-values were adjusted to obtain strong control over the false discovery rate (FDR) using the Benjamini and Hochberg method (Benjamini and Hochberg 1995).
The following R packages were used: Tidyverse (ggplot2, tidyr, dplyr, and forcats), ggforce, ggrepel, pheatmap, survminer, ggvenn, ggridges, circlize, cowplot, and Bioconductor (oligo, clariomshumantranscriptcluster.db, sva, and limma).
Gene set enrichment analysis (GSEA) was performed using publicly available software provided by the Broad Institute (version 3.0) with the hallmark, curated, motif, GO, and oncogenic signature gene sets from the MSigDB (http://www.broadinstitute.org/gsea/msigdb). The number of permutations was set to 1000, and the genes were ranked according to Signal2Noise (cell lines) or log2 ratio of classes (mouse tumors).
Gene categories based on melanoma- and MYC-related gene sets
Gene categories depicted in Figure 4 were manually curated based on gene sets relevant to melanoma and MYC biology. These categories consisted of up to 2000 genes. The complete list of gene sets and their corresponding genes is in Supplemental Table S5. To link common down-regulated genes by Omomyc in vivo with the genes in these categories, we generated a binary matrix where 1 denotes connection and 0 indicates the absence of it. This matrix then was used to create the chord diagram with the Circlize package.
String analysis
The lists of common DN and UP genes were subjected to unsupervised string analysis at https://string-db.org. The settings were as follows: network type: full STRING network; meaning of network edges: confidence; active interaction sources: all; minimum required interaction score: medium confidence (0.400); maximum number of interactions to show: first shell: none/query proteins only, second shell: none; and network clustering: no clustering.
Subsequent supervised analysis was performed with the same settings except for the network clustering, where the number of clusters was set to three (DN genes) or two (UP genes) (string analysis of DN genes: https://version-11-5.string-db.org/cgi/network?networkId=bzTgvKhrb4qU; string analysis of UP genes: https://version-11-5.string-db.org/cgi/network?networkId=bPIrLa5TIVBo).
The list of genes belonging to every cluster was downloaded and subjected to the “investigate gene sets” tool from Molecular Signatures Database (MSigDB) (Subramanian et al. 2005), from which we obtained the lists of gene sets to which these genes belonged. Finally, based on these lists, we assigned the gene categories to which each cluster belonged. The full list of genes and gene sets is in Supplemental Table 8, A–F.
Enrichr analysis
The list of common DN genes was subjected to the Ma'ayan laboratory's Enrichr analysis (https://maayanlab.cloud/Enrichr). Specifically, we used the following libraries: ENCODE_and_ChEA_Consensus_TFs_from_ChIP-X, Kinase_Perturbations_from_GEO_down, Kinase_Perturbations_from_GEO_up, L1000_Kinase_and_GPCR_Perturbations_down, and L1000_Kinase_and_GPCR_Perturbations_up.
Cbioportal for cancer genomics data
For cancer genomics data, we accessed the skin cutaneous melanoma (TCGA and PanCancer Atlas) data set (https://www.cbioportal.org/study/summary?id=skcm_tcga_pan_can_atlas_2018).
Omomyc-modulated genes and patient survival relation study
We analyzed melanoma patient survival according to the expression of each of the common Omomyc-induced DEGs (UP + DN genes). Lower than the median expression of DN genes or higher than the median expression of UP genes that associated with significantly longer OS or PFS were considered. We also included genes whose expression were in quartiles 1 (highest) or 4 (lowest) and associated with significantly longer OS or PFS. The full list of genes is in Supplemental Table S10.
Generation of gene signatures based on Omomyc-induced down-regulated (DN) and up-regulated (UP) genes
From the list of common DN and UP genes associated with improved survival, we tested which combination of three DN and three UP genes would discriminate patients with good or poor survival and represent a significant proportion of the patients in the study. The BIRC5–CCNB2–ORC1–DDX60–DTX3L–PARP9 signature discriminated 45 patients with good prognosis and 63 patients with poor prognosis. The analysis in cBioPortal is available at https://bit.ly/3xApUj7.
Skin cutaneous melanoma (TCGA and PanCancer Atlas) patient information
Once we classified patients according to the gene signatures, we looked at clinical attributes and mRNA under “comparison” to look for patient characteristics that could explain or be related to their differences in survival.
Preranked GSEA
Gene set enrichment analysis of patients with good or bad prognosis was performed as preranked. To do so, we downloaded mRNA expression data from patients belonging to both groups and ordered the genes according to their fold change. GSEA v4.1.0 with the hallmark, curated, motif, GO, and oncogenic signature gene sets from the MSigDB was used. Among the parameters, we used “classic” enrichment statistic and “meandiv” normalization score.
Code availability
All of the code for the bioinformatic analysis will be shared on request.
Data availability statement
The data generated in this study are available here and in the Supplemental Data files. Microarray data are publicly available in Gene Expression Omnibus (GEO) as follows: A375 in vitro microarray: GSE227840, A375 in vivo microarray: GSE227841, SkMel147 in vivo microarray: GSE227842, and SkMel147 lymph node metastases microarray: GSE227843.
Statistical analysis
All analyses, except the bioinformatic ones, were done using GraphPad Prism 6 software. Normal distribution of the data was assessed for each group using D'Agostino–Pearson test. Differences in samples’ mean values were analyzed using Student's t-test or ANOVA (parametric) for normally distributed data or Mann–Whitney or Kruskal–Wallis test when otherwise. F-test was used to calculate the difference in the variances of the groups. Differences in the quantifications of the Western blots were analyzed using one-sample t-tests to a mean = 100. We did not use statistical methods to predetermine sample size in animal studies but did make efforts to achieve scientific goals using the minimum number of animals.
Supplementary Material
Acknowledgments
M.F.Z.-F. was supported by the Juan de la Cierva Programme of the Spanish Ministry of Economy and Competitiveness (IJCI-2014-22403) and Fundació La Marató de TV3 (grant 474/C/2019); F.G. was supported by Spanish Ministry of Science and Innovation Contratos Predoctorales de Formación en Investigación en Salud (PFIS; FI20/00274); I.G.-L. was supported by a grant from the University Teacher Training Program (FPU), Ministry of Universities (FPU20/04812); and S.M.-M. was supported by the Generalitat de Catalunya “Contractació de Personal Investigador Novell (FI-DGR)” 2016 fellowship (2016FI_B 00592). This project was funded by grants from the Spanish Ministry of Science and Innovation (Fondo de Inversión en Salud [FIS] PI19/01277, which also supported I.G.-L. and S.M.-M, and Retos-Colaboración 2019 RTC2019-007067-1), La Marató TV3, the Generalitat de Catalunya AGAUR 2017 grant SGR-3193, and the European Research Council (ERC-PoC II/3079/SYST-iMYC [813132]). We thank the rest of the Soucek laboratory for critical reading of the manuscript, and the personnel at Vall d'Hebron Research Institute (VHIR) High Technology Unit. We acknowledge Vall d'Hebron Institute of Oncology and the Cellex Foundation for providing research facilities and equipment.
Author contributions: M.F.Z.-F. conceived the study, curated the data, performed the formal analysis of the data, acquired the funding, performed the investigation, was responsible for the methodology, administered and supervised the project administration, performed the visualization, wrote the original draft of the manuscript, and reviewed and edited the manuscript. D.M.-V. performed the formal analysis and was responsible for the methodology. F.G., I.G.L., and J.K. performed the formal analysis and the investigation. S.C.-S. performed the formal analysis. T.J., S.M.-M., and M.-E.B. performed the methodology. G.M.-F., E.S.d.P., L.F., and J.G. performed the investigation. L.N. curated the data and performed the formal analysis of the data. J.R.W. acquired the funding, performed the methodology, and reviewed and edited the manuscript. L.S. conceived the study, acquired the funding, adminstered and supervised the project, and reviewed and edited the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.350078.122.
Freely available online through the Genes & Development Open Access option.
Competing interest statement
M.F.Z.-F., S.C.-S., L.F., and J.R.W. are shareholders of Peptomyc, a company focused on developing Myc inhibitors for cancer treatment. D.M.-V., S.C.-S., T.J., S.M.-M., G.M.-F., L.F., J.G., and M.-E.B. report personal fees from Peptomyc during the completion of this project. L.S. and M.B. are cofounders and shareholders of Peptomyc.
References
- Álamo MDC, Ochenduszko S, Crespo G, Corral M, Oramas J, Sancho P, Medina J, Garicano F, Lopez P, Campos Balea B, et al. 2021. Durable response to vemurafenib and cobimetinib for the treatment of BRAF-mutated metastatic melanoma in routine clinical practice. Onco Targets Ther 14: 5345–5352. 10.2147/OTT.S325208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annibali D, Whitfield JR, Favuzzi E, Jauset T, Serrano E, Cuartas I, Redondo-Campos S, Folch G, Gonzàlez-Juncà A, Sodir NM, et al. 2014. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat Commun 5: 4632. 10.1038/ncomms5632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedogni B. 2014. Notch signaling in melanoma: interacting pathways and stromal influences that enhance notch targeting. Pigment Cell Melanoma Res 27: 162–168. 10.1111/pcmr.12194 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Bennett DC. 2016. Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res 29: 122–140. 10.1111/pcmr.12422 [DOI] [PubMed] [Google Scholar]
- Biroccio A, Benassi B, Amodei S, Gabellini C, Del Bufalo D, Zupi G. 2001. c-Myc down-regulation increases susceptibility to cisplatin through reactive oxygen species-mediated apoptosis in M14 human melanoma cells. Mol Pharmacol 60: 174–182. 10.1124/mol.60.1.174 [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. 2015. Genomic classification of cutaneous melanoma. Cell 161: 1681–1696. 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128. 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Liu Z, Li M, Huang Y, Wang M, Zeng W, Wei W, Zhang C, Gong Y, Guo L. 2020. Potential prognostic predictors and molecular targets for skin melanoma screened by weighted gene co-expression network analysis. Curr Gene Ther 20: 5–14. 10.2174/1566523220666200516170832 [DOI] [PubMed] [Google Scholar]
- Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, Berger JM. 2011. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat Struct Mol Biol 18: 471–477. 10.1038/nsmb.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnecka AM, Bartnik E, Fiedorowicz M, Rutkowski P. 2020. Targeted therapy in melanoma and mechanisms of resistance. Int J Mol Sci 21: 4576. 10.3390/ijms21134576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran R, Deutzmann A, Mahauad-Fernandez WD, Hansen AS, Gouw AM, Felsher DW. 2022. The MYC oncogene—the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol 19: 23–36. 10.1038/s41571-021-00549-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA. 2002. Cycling to cancer with cyclin D1. Cancer Biol Ther 1: 226–231. 10.4161/cbt.72 [DOI] [PubMed] [Google Scholar]
- Dinçer S, Oskay EK, Piskin AK, Zeybek ND, Pişkin E. 2010. Growth inhibition of SK-MEL-30 human melanoma cells by antisense c-myc oligonucleotides delivered by poly(N-isopropylacrylamide)/poly(ethyleneimine) copolymer. J Tissue Eng Regen Med 4: 284–290. 10.1002/term.239 [DOI] [PubMed] [Google Scholar]
- Engeland K. 2022. Cell cycle regulation: p53–p21–RB signaling. Cell Death Differ 29: 946–960. 10.1038/s41418-022-00988-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe C, Amaral T, Peris K, Hauschild A, Arenberger P, Basset-Seguin N, Bastholt L, Bataille V, Del Marmol V, Dréno B, et al. 2022. European consensus-based interdisciplinary guideline for melanoma. Part 2: treatment - update 2022. Eur J Cancer 170: 256–284. 10.1016/j.ejca.2022.04.018 [DOI] [PubMed] [Google Scholar]
- Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O'Connell BC, Mateyak MK, Tam W, Kohlhuber F, et al. 2000. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci 97: 2229–2234. 10.1073/pnas.050586197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et al. 2018. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173: 291–304.e6. 10.1016/j.cell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Zhao Y, Li Z, Huang W. 2007. esiRNA to eri-1 and adar-1 genes improving high doses of c-myc-directed esiRNA effect on mouse melanoma growth inhibition. Biochem Biophys Res Commun 361: 373–378. 10.1016/j.bbrc.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264. 10.1093/biostatistics/4.2.249 [DOI] [PubMed] [Google Scholar]
- Jalili A, Wagner C, Pashenkov M, Pathria G, Mertz KD, Widlund HR, Lupien M, Brunet JP, Golub TR, Stingl G, et al. 2012. Dual suppression of the cyclin-dependent kinase inhibitors CDKN2C and CDKN1A in human melanoma. J Natl Cancer Inst 104: 1673–1679. 10.1093/jnci/djs373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaora S, Nagler A, Wargo JA, Samuels Y. 2022. Mechanisms of immune activation and regulation: lessons from melanoma. Nat Rev Cancer 22: 195–207. 10.1038/s41568-022-00442-9 [DOI] [PubMed] [Google Scholar]
- Kraehn GM, Utikal J, Udart M, Greulich KM, Bezold G, Kaskel P, Leiter U, Peter RU. 2001. Extra c-myc oncogene copies in high risk cutaneous malignant melanoma and melanoma metastases. Br J Cancer 84: 72–79. 10.1054/bjoc.2000.1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress TR, Sabò A, Amati B. 2015. MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15: 593–607. 10.1038/nrc3984 [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. 2012. The spindle assembly checkpoint. Curr Biol 22: R966–R980. 10.1016/j.cub.2012.10.006 [DOI] [PubMed] [Google Scholar]
- Lau E, Feng Y, Claps G, Fukuda MN, Perlina A, Donn D, Jilaveanu L, Kluger H, Freeze HH, Ronai ZA. 2015. The transcription factor ATF2 promotes melanoma metastasis by suppressing protein fucosylation. Sci Signal 8: ra124. 10.1126/scisignal.aac6479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. 2018. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173: 400–416.e11. 10.1016/j.cell.2018.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Machesky LM. 2008. Lamellipodia and filopodia in metastasis and invasion. FEBS Lett 582: 2102–2111. 10.1016/j.febslet.2008.03.039 [DOI] [PubMed] [Google Scholar]
- Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, Mathews CK, Shewach DS, Nikiforov MA. 2008. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 7: 2392–2400. 10.4161/cc.6390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massó-Vallés D, Soucek L. 2020. Blocking Myc to treat cancer: reflecting on two decades of Omomyc. Cells 9: 883. 10.3390/cells9040883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massó-Vallés D, Beaulieu M-E, Jauset T, Giuntini F, Zacarías-Fluck MF, Foradada L, Martínez-Martín S, Serrano E, Martín-Fernández G, Casacuberta-Serra S, et al. 2022. MYC inhibition halts metastatic breast cancer progression by blocking growth, Invasion, and Seeding. Cancer Res Commun 2: 110–130. 10.1158/2767-9764.CRC-21-0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Swanton C. 2017. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168: 613–628. 10.1016/j.cell.2017.01.018 [DOI] [PubMed] [Google Scholar]
- Menssen A, Epanchintsev A, Lodygin D, Rezaei N, Jung P, Verdoodt B, Diebold J, Hermeking H. 2007. c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle 6: 339–352. 10.4161/cc.6.3.3808 [DOI] [PubMed] [Google Scholar]
- Moldovan GL, Pfander B, Jentsch S. 2007. PCNA, the maestro of the replication fork. Cell 129: 665–679. 10.1016/j.cell.2007.05.003 [DOI] [PubMed] [Google Scholar]
- Pan D, Kim B, Hu G, Gupta DS, Senpan A, Yang X, Schmieder A, Swain C, Wickline SA, Tomasson MH, et al. 2015. A strategy for combating melanoma with oncogenic c-Myc inhibitors and targeted nanotherapy. Nanomedicine 10: 241–251. 10.2217/nnm.14.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp O, Doma V, Gil J, Markó-Varga G, Kárpáti S, Tímár J, Vízkeleti L. 2021. Organ specific copy number variations in visceral metastases of human melanoma. Cancers 13: 5984. 10.3390/cancers13235984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouryazdanparast P, Brenner A, Haghighat Z, Guitart J, Rademaker A, Gerami P. 2012. The role of 8q24 copy number gains and c-MYC expression in amelanotic cutaneous melanoma. Mod Pathol 25: 1221–1226. 10.1038/modpathol.2012.75 [DOI] [PubMed] [Google Scholar]
- Salhi A, Jordan AC, Bochaca II, Izsak A, Darvishian F, Houvras Y, Giles KM, Osman I. 2020. Oxidative phosphorylation promotes primary melanoma invasion. Am J Pathol 190: 1108–1117. 10.1016/j.ajpath.2020.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savino M, Annibali D, Carucci N, Favuzzi E, Cole MD, Evan GI, Soucek L, Nasi S. 2011. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS One 6: e22284. 10.1371/journal.pone.0022284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, Stang A, Roesch A, Ugurel S. 2018. Melanoma. Lancet 392: 971–984. 10.1016/S0140-6736(18)31559-9 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlagbauer-Wadl H, Griffioen M, van Elsas A, Schrier PI, Pustelnik T, Eichler HG, Wolff K, Pehamberger H, Jansen B. 1999. Influence of increased c-Myc expression on the growth characteristics of human melanoma. J Invest Dermatol 112: 332–336. 10.1046/j.1523-1747.1999.00506.x [DOI] [PubMed] [Google Scholar]
- Seo YS, Kang YH. 2018. The human replicative helicase, the CMG complex, as a target for anti-cancer therapy. Front Mol Biosci 5: 26. 10.3389/fmolb.2018.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, Bastian BC. 2016. From melanocytes to melanomas. Nat Rev Cancer 16: 345–358. 10.1038/nrc.2016.37 [DOI] [PubMed] [Google Scholar]
- Shamloo B, Usluer S. 2019. p21 in cancer research. Cancers 11: 1178. 10.3390/cancers11081178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton KR, Crawford L, Tsui E, Manchester HE, Maertens O, Liu X, Liberti MV, Magpusao AN, Stein EM, Tingley JP, et al. 2017. Melanoma therapeutic strategies that select against resistance by exploiting MYC-driven evolutionary convergence. Cell Rep 21: 2796–2812. 10.1016/j.celrep.2017.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. 1998. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 17: 2463–2472. 10.1038/sj.onc.1202199 [DOI] [PubMed] [Google Scholar]
- Soucek L, Nasi S, Evan GI. 2004. Omomyc expression in skin prevents Myc-induced papillomatosis. Cell Death Differ 11: 1038–1045. 10.1038/sj.cdd.4401443 [DOI] [PubMed] [Google Scholar]
- Soucek L, Whitfield JR, Sodir NM, Massó-Vallés D, Serrano E, Karnezis AN, Swigart LB, Evan GI. 2013. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev 27: 504–513. 10.1101/gad.205542.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeyama K, Aguiar RC, Gu L, He C, Freeman GJ, Kutok JL, Aster JC, Shipp MA. 2003. The BAL-binding protein BBAP and related Deltex family members exhibit ubiquitin–protein isopeptide ligase activity. J Biol Chem 278: 21930–21937. 10.1074/jbc.M301157200 [DOI] [PubMed] [Google Scholar]
- Terranova CJ. 2021. Chromatin state profiling reveals PRC2 inhibition as a therapeutic target in NRAS-mutant melanoma. Mol Cell Oncol 8: 1986350. 10.1080/23723556.2021.1986350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thang ND, Yajima I, Kumasaka MY, Iida M, Suzuki T, Kato M. 2015. Deltex-3-like (DTX3L) stimulates metastasis of melanoma through FAK/PI3K/AKT but not MEK/ERK pathway. Oncotarget 6: 14290–14299. 10.18632/oncotarget.3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fan M, Candas D, Zhang TQ, Qin L, Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, et al. 2014. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell 29: 217–232. 10.1016/j.devcel.2014.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield JR, Soucek L. 2021. The long journey to bring a Myc inhibitor to the clinic. J Cell Biol 220: e202103090. 10.1083/jcb.202103090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RP, Ng P, Dedhar S, Li G. 2007. The role of integrin-linked kinase in melanoma cell migration, invasion, and tumor growth. Mol Cancer Ther 6: 1692–1700. 10.1158/1535-7163.MCT-07-0134 [DOI] [PubMed] [Google Scholar]
- Wu S, Cetinkaya C, Munoz-Alonso MJ, von der Lehr N, Bahram F, Beuger V, Eilers M, Leon J, Larsson LG. 2003. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 22: 351–360. 10.1038/sj.onc.1206145 [DOI] [PubMed] [Google Scholar]
- Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. 2021. Gene set knowledge discovery with Enrichr. Curr Protoc 1: e90. 10.1002/cpz1.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available here and in the Supplemental Data files. Microarray data are publicly available in Gene Expression Omnibus (GEO) as follows: A375 in vitro microarray: GSE227840, A375 in vivo microarray: GSE227841, SkMel147 in vivo microarray: GSE227842, and SkMel147 lymph node metastases microarray: GSE227843.