

Abstract
Cyclic(alkyl)(amino)carbene (CAAC) ligands are found to perturb regioselectivity of the copper-catalyzed carboboration of terminal alkynes, favoring the less commonly observed internal alkenylboron regiosomer through an α-selective borylcupration step. A variety of carbon electrophiles participate in the reaction, including allyl alcohols derivatives and alkyl halides. The method provides a straightforward and selective route to versatile tri-substituted alkenylboron compounds that are otherwise challenging to access.
Keywords: organoboron, copper, carboboration, CAAC ligand, regioselectivity
Graphical Abstract
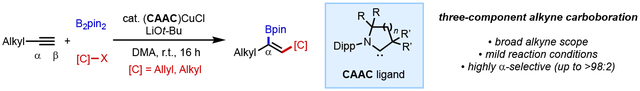
Organoboron compounds play a unique role in the chemical sciences. Carbon–boron bonds can readily be converted into a diverse array of carbon–carbon and carbon–heteroatom linkages via an ever-expanding battery of methods,[1–4] and organoboron molecules themselves possess myriad of functions in the context of biology[5–6] and materials science.[7–9] The invention of new methods to assemble organoboron compounds from simple chemical inputs streamlines access to important families of molecules. Multi-component catalytic couplings, in which three or more building blocks are united in a single reaction, hold tremendous promise in enabling direct synthesis of densely functionalized organoboron compounds. In this context, copper-catalyzed borylative 1,2-difunctionalization of alkynes is an established means of preparing tri- and tetrasubstitued alkenylboron targets via a mechanism involving migratory insertion of an alkyne into a Ln•CuI–boryl intermediate followed by coupling of the resulting Ln•CuI(alkenyl) species with an electrophile.[10–13] Controlling the regioselectivity of these processes in a way that grants access to either regioisomer in a predictable manner remains challenging (Scheme 1). With terminal alkynes, the vast majority of catalytic systems deliver the boryl group to the terminal (β) position, restricting access to the opposite alkenylboron regioisomers (Scheme 1A). Here, we demonstrate that appropriately tuned cyclic(alkyl)(amino)carbene (CAAC)-ligated copper catalysts enables regioselective carboboration to give internal (α) alkenylboron compounds with a broad collection of carbon electrophiles (Scheme 1C).
Scheme 1.
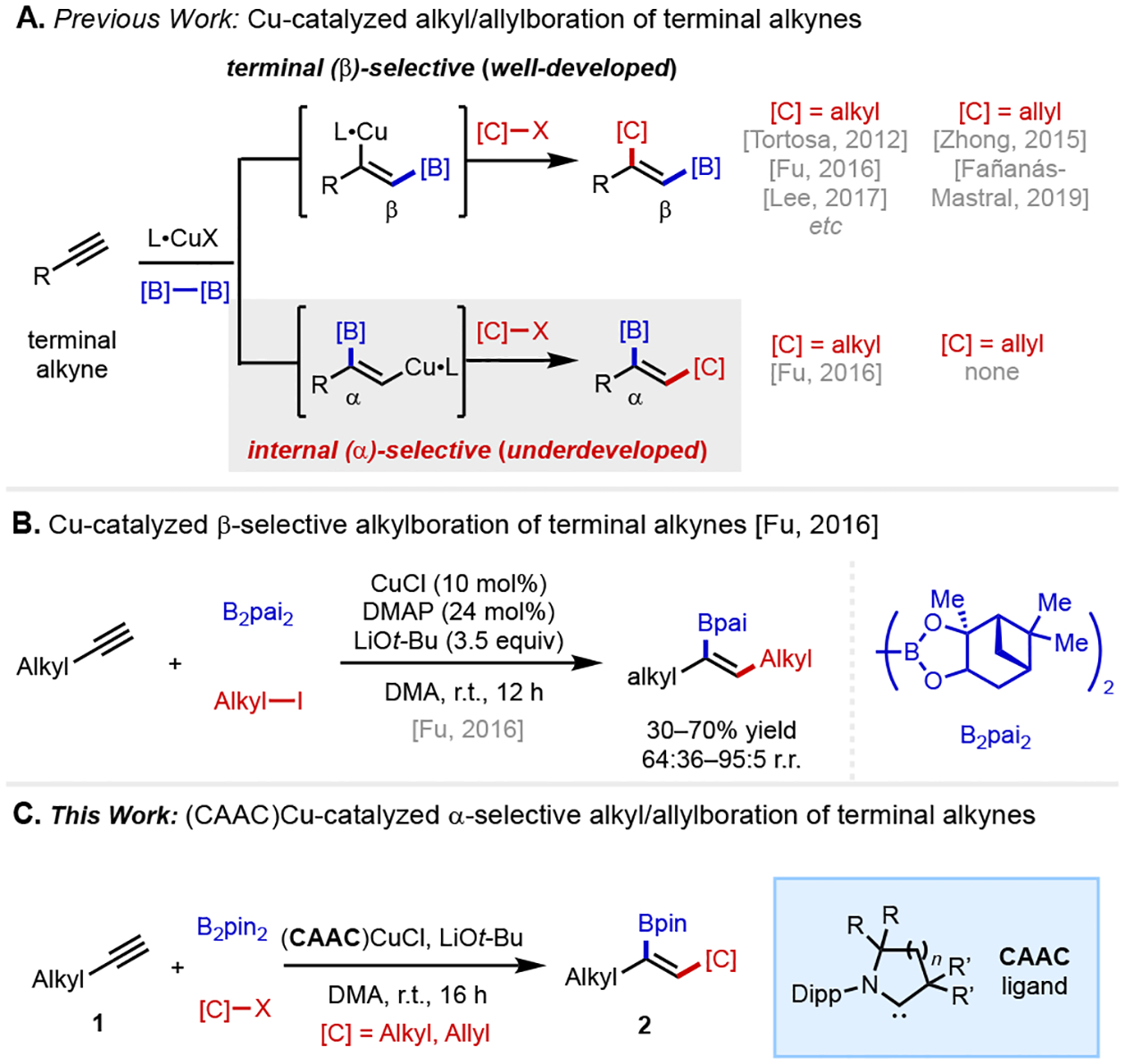
Overview of Cu-catalyzed regioselective carboboration of terminal alkynes.
Regioselectivity trends in Ln•Cu–boryl alkyne addition processes are complex and reflect an interplay between the steric and electronic properties of the ligand, the identity of the boryl group, and the substituent(s) on the alkyne substrate.[14–16] N-Heterocyclic carbene (NHC) ligands[17–18] have been widely used in catalytic Ln•Cu–boryl catalysis and generally favor boryl transfer to the terminal position of terminal alkynes with Bpin and related boryl groups, though either position can predominate depending on the nature of substrate and the ligand environment around boron. We recently demonstrated that strongly σ-donating CAAC ligands[19–21] override substituent effects of the boryl group and the alkyne, allowing for reliably Markovnikov (α-selective) protoboration of diverse terminal alkynes with a variety of bis-boron nucleophiles.[22] Based on this result, we questioned whether it would be possible to employ C(sp3)-based electrophiles in lieu of a proton to develop a three-component carboboration, with regioselectivity and product substitution patterns that would complement existing methodology.[23–32] Of relevance to this proposal, Xiao and Fu disclosed an important study in which the combination of CuCl (10 mol%) as the precatalyst, DMAP (24 mol%) as the ligand, and B2pai2 (pai = (+)-pinanediolato) as the bis-boron reagent leads to branched-selective carboboration, though in this case yields and regioselectivities were variable (30–70% yield, 64:36–95:5 r.r.) (Scheme 1B). The less common and more expensive B2pai2 nucleophile was employed to maximize regioselectivity, and some synthetically useful carbogenic groups were incompatible with this protocol (e.g., allyl electrophiles).[33]
To reduce this idea to practice, we examined carboboration of model terminal alkyne 1a with two representative carbon electrophiles, allyl diethyl phosphate and methyl iodide. The former was selected because allyl electrophiles have not been previously employed in α-selective carboboration of alkynes, despite being used in several reports of linear selectivity.[30–32,34] The latter was selected because it was found to be low-yielding under previously published conditions (one example, 87:13 r.r., 32% yield).[33]
A library of CAAC•CuCl precatalysts with different steric and electronic properties was tested, and a summary of the data is shown in Table 1. To our delight, EtCAAC5-ligated Cu complex (L1CuCl) promoted both transformations with high conversion and high α-selectivity. Replacement of the ethyl groups on the α-carbon of L1[35] with either an electron-withdrawing group (L2) [36] or more sterically bulky groups (L3, L4) [35,37] led to decreased yield and α:β ratio. EtCAAC6 ligand (L5), [38] a much stronger electron-donor than L1, gave poor yields in both transformations, though high α:β ratio (84:16) was observed in the methylboration reaction. Interestingly, BiCAAC ligands,[39] i-PrBiCAAC (L6) and PhEtBiCAAC (L7), which are also strong electron-donors, furnished the desired product methylborylated product 2ad with high α-selectivity (97 % and 92 %, respectively). But neither of them could deliver any desired allylborylated product 2aa. Moreover, further exploration of substrate scope for methylboration using L6 suggested that this ligand could not tolerate the presence of Lewis basic functional groups. For example, when an ether-containing substrate was attempted (see 2kd below), only 23% yield and 47% α-selectivity were observed. A control experiment with IPr (L8), a representative N-heterocyclic carbene ligand commonly used in copper–boryl chemistry,[10–13] led to low yield with both electrophiles.
Table 1.
Optimization of reaction conditions.a
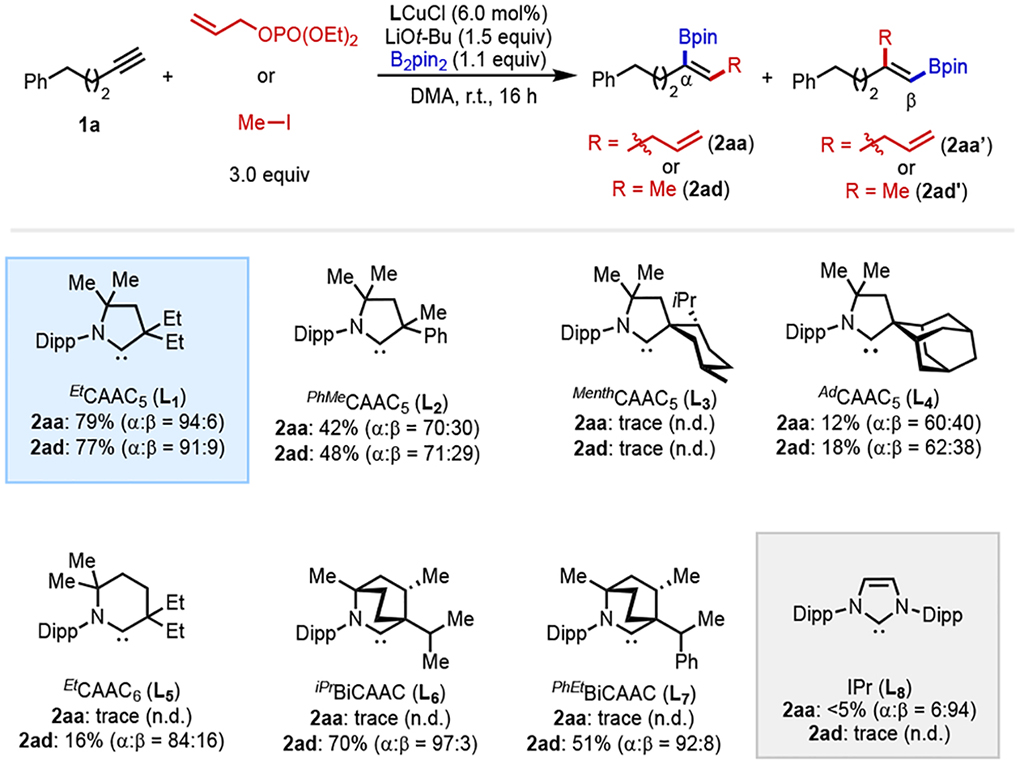
|
Yields of products (2aa or 3aa) and regioselectivity (±2%) were determined by 1H NMR spectroscopy (600 MHz) using CH2Br2 as the internal standard. n.d. = not determined.
With the optimized conditions in hand, we examined the scope of the allylboration reaction (Table 2). Terminal alkynes bearing primary alkyl groups provided the corresponding products in excellent yields with high levels of regioselectivity (2aa and 2ba). In addition, functional groups such as ether (2ca), cyano (2da), halogen (2ea), protected amines (2fa and 2ga) and pendant piperidine (2ha) and azetidine (2ia) were well tolerated, furnishing desired products in good yields and high α-selectivity, except in the case of 2ga and 2ia, where moderate α-selectivity (65% and 70%, respectively) was observed. Notably, when phenylacetylene was subjected to the optimal reaction conditions, the desired product 2ja was generated with 75% α-selectivity. Allyl electrophiles with phenyl and n-propyl groups substituted at γ-position were also compatible under the reaction conditions, furnishing desired products in high yields (60–71%) and excellent regioselectivity (>90% α-borylation, 93–97% SN2’ allylation) (2ab and 2ac).
Table 2.
Scope of α-selective allylboration of terminal alkynes.a
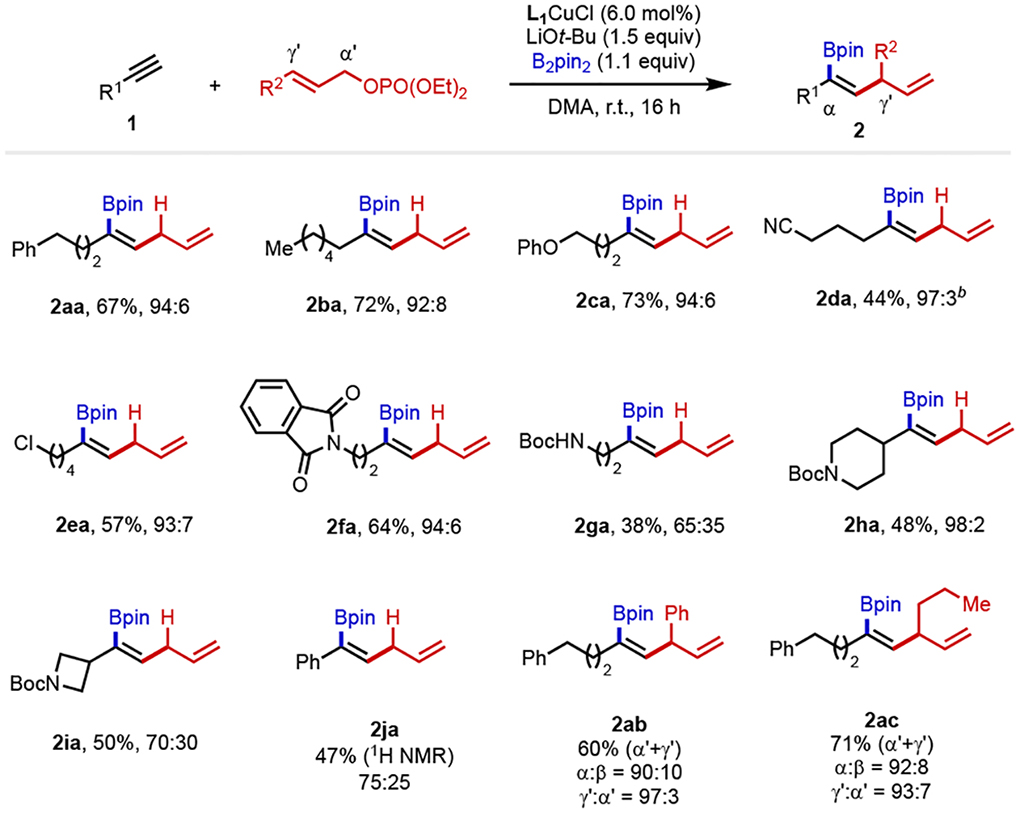
|
Conditions: 1 (0.10 mmol), B2pin2 (0.11 mmol), allyl electrophile (0.30 mmol), L1CuCl (0.006 mmol), LiOt-Bu (0.15 mmol) and DMA (0.60 mL), r.t. Ratios of α:β (±2) were determined via 1H NMR spectroscopy (600 MHz) of the crude reaction mixtures. Percentages represent isolated yields of the α-borylated product.
The corresponding protoboration side product (23%) was observed by 1H NMR analysis of the crude reaction mixture.
We next explored the scope of terminal alkynes for alkylboration. Alkynes containing different primary alkyl chains readily underwent efficient methylboration with high α-selectivity (2ad and 2bd). In addition, a range of functional groups, including ether (2kd), chloro (2ed, 2md), cyano (2cd), amide (2od) and protected amino group (2ld), were tolerated, furnishing the desired products in good yields and high regioselectivity. The reactions of alkynes bearing secondary alkyl groups at the α-position (2qd and 2rd) gave high α-selectivity as well. However, similarly to the previous reported (CAAC)Cu-catalyzed protoboration reactions, tert-butyl acetylene (2td) has very low reactivity under the optimal conditions. Alkynes with medicinally relevant functional groups such as pendant piperidine and azetidine were both competent coupling partners (2hd and 2id). Unfortunately, poor α-selectivity was observed when benzyl protected propargyl alcohol (2sd) or phenylacetylene (2jd) were used as substrates.
We next explored the scope of the alkyl electrophile. Deuterated methyl iodide works well, showing the ability of this method to assemble specifically labelled compounds efficiently. The reactions of primary alkyl electrophiles with 1a afforded the α-selective alkylboration products with high yield, though relatively lower α:β ratios were observed compared to the reaction using methyl iodide (2af, 2ag). Notably, the alkyl electrophiles with functional groups, such as terminal alkene, silyl ether and ester, were compatible under our reaction conditions, giving 60%–66% yield and 73%–75% α-selectivity (2ah–2aj). When benzyl bromide was used as electrophile, the desired product (2ak) was generated in excellent yield and high α-selectivity. Similar α:β ratio was observed when an alkyne bearing secondary alkyl groups at the α-position was applied (2hf).
A plausible catalytic cycle for this reaction is depicted in Scheme 2A. One possible explanation for the observation that regioselectivity varies across the different C(sp3) electrophiles tested in Table 3 is that the borylcupration step could be reversible. Under such a scenario, the nature of the C(sp3) electrophile and the rate of C–C bond formation may influence regioselectivity. To test this hypothesis, we performed a crossover experiment between alkyne 1a and 1j. In this experiment, L1•CuCl was reacted with LiOt-Bu, then B2pin2, followed by alkyne 1j in a J-Young tube and monitored by NMR to show a 70% yield of the in situ-generated complex III (Scheme 2B). Subsequent addition of alkyne 1a affords a mixture of borylcuprated species III and III’ observed by 1H- and 13C-NMR. These results support a reversible borylcupration in which complex III reverts to boryl complex II (Sheme 2A) followed by reinsertion with alkyne 1a to afford the observed mixture. In parallel, we also performed kinetic studies of the reaction of alkyne 1a and BnBr and found a first order rate dependence in catalyst [L1•CuCl] and electrophile [BnBr]. Meanwhile, a zeroth order rate dependence was observed from the alkyne [1a] and the borane/base combination [B2pin2•LiOt-Bu] (Scheme 2C). The resulting rate law is consistent with the mechanism proposed in Scheme 2A wherein a reversible borylcupration precedes the regio- and rate-determining electrophile substitution. These studies taken together support the proposed mechanism wherein a reversible borylcupration would account for the change in regioselectivity as a function of electrophile identity. To investigate the mechanism of the alkylation step, we performed a radical clock experiment with cyclopropylmethyl iodide (Scheme 2D). The results of this study show exclusive formation of the ring-intact product (2al) with no detectable quantity of ring-opened product (2am) observed. This indicates that the alkylation step likely does not proceed through a radical-based mechanism.
Scheme 2.
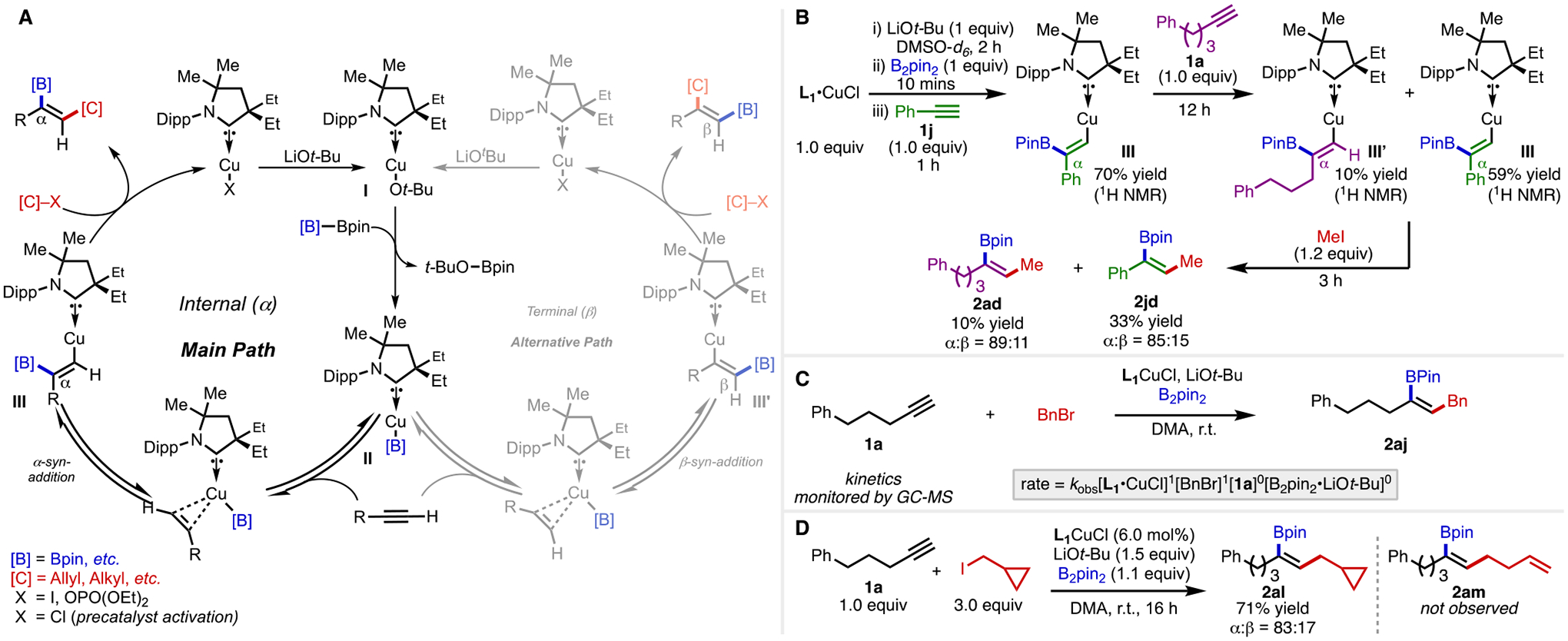
Catalytic cycle and mechanistic studies.
Table 3.
Scope of α-selective alkylboration of terminal alkynes.a
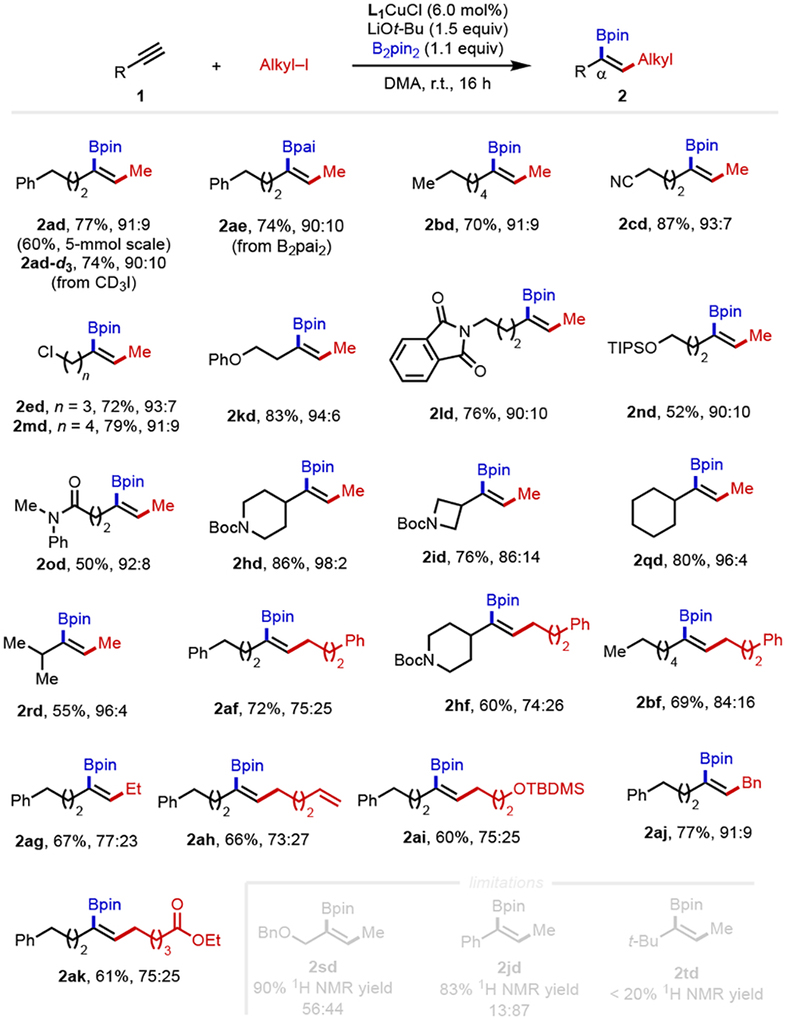
|
Conditions: 1 (0.10 mmol), B2pin2 (0.11 mmol), alkyl iodide (0.30 mmol), L1CuCl (0.006 mmol), LiOt-Bu (0.15 mmol) and DMA (0.60 mL), r.t. Ratios of α:β (±2) were determined via 1H NMR spectroscopy (600 MHz) of the crude reaction mixtures. Percentages represent isolated yields of the α-borylated products.
In conclusion, we have extended our investigations of (CAAC)Cu–boryl catalysis to the three-component carboboration of terminal alkynes and have found that high levels of α-selectivity are maintained across different carbon electrophiles, including allyl electrophiles, which have not been previously employed in an α-selective reaction system. The generality of the method across different alkyne substrates offers a convenient means of preparing tri-substituted alkenylboron compounds with established utility in organic synthesis.
Supplementary Material
ACKNOWLEDGMENT
This work was financially supported by the National Institutes of Health (5R35GM125052-04 and diversity supplement, 5R35GM125052-04S1) and an ACS PRF Doctoral New Investigator Grant (K.M.E.). This work was also supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Catalysis Science Program, under Award No. DE-SC0009376 (G.B.), National Science Foundation Award No. CHE-1800598 (D.B.G.), and the Agence Nationale de la Recherche Award No. ANR-19-276CE07-0017 (R.J). Individual fellowships were provided by National Science Foundation Award No. CHE-2213302 (S.D.M.) and an SDSU University Graduate Fellowship (S.Y.).
Footnotes
Supporting Information
This material is available free of charge via the Internet at http://pubs.acs.org.”
Experimental procedures and spectral data (PDF)
NMR data (MNova format) (ZIP)
The authors declare no competing financial interest.
REFERENCES
- (1).Miyaura N; Suzuki A Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev 1995, 95, 2457–2483. [Google Scholar]
- (2).Scott HK; Aggarwal VK Highly Enantioselective Synthesis of Tertiary Boronic Esters and their Stereospecific Conversion to other Functional Groups and Quaternary Stereocentres. Chem. Eur. J 2011, 17, 13124–13132. [DOI] [PubMed] [Google Scholar]
- (3).Lennox AJJ; Lloyd-Jones GC Selection of Boron Reagents for Suzuki–Miyaura Coupling. Chem. Soc. Rev 2014, 43, 412–443. [DOI] [PubMed] [Google Scholar]
- (4).West MJ; Fyfe JWB; Vantourout JC; Watson AJB Mechanistic Development and Recent Applications of the Chan–Lam Amination. Chem. Rev 2019, 119, 12491–12523. [DOI] [PubMed] [Google Scholar]
- (5).António JPM; Russo R; Carvalho CP; Cal PMSD; Gois PMP Boronic Acids as Building Blocks for the Construction of Therapeutically Useful Bioconjugates. Chem. Soc. Rev 2019, 48, 3513–3536. [DOI] [PubMed] [Google Scholar]
- (6).Baker SJ; Ding CZ; Akama T; Zhang Y-K; Hernandez V; Xia Y Therapeutic Potential of Boron-Containing Compounds. Future Med. Chem 2009, 1, 1275–1288. [DOI] [PubMed] [Google Scholar]
- (7).Jäkle F Lewis Acidic Organoboron Polymers. Coord. Chem. Rev 2006, 250, 1107–1121. [Google Scholar]
- (8).Mellerup SK; Wang S Boron-Based Stimuli Responsive Materials. Chem. Soc. Rev 2019, 48, 3537–3549. [DOI] [PubMed] [Google Scholar]
- (9).Miao J; Wang Y; Liu J; Wang L Organoboron Molecules and Polymers for Organic Solar Cell Applications. Chem. Soc. Rev 2022, 51, 153–187. [DOI] [PubMed] [Google Scholar]
- (10).Yoshida H Borylation of Alkynes under Base/Coinage Metal Catalysis: Some Recent Developments. ACS Catal 2016, 6, 1799–1811. [Google Scholar]
- (11).Hemming D; Fritzemeier R; Westcott SA; Santos WL; Steel PG Copper-Boryl Mediated Organic Synthesis. Chem. Soc. Rev 2018, 47, 7477–7494. [DOI] [PubMed] [Google Scholar]
- (12).Carreras J; Caballero A; Pérez PJ Alkenyl Boronates: Synthesis and Applications. Chem. Asian J 2019, 14, 329–343. [DOI] [PubMed] [Google Scholar]
- (13).Whyte A; Torelli A; Mirabi B; Zhang A; Lautens M Copper-Catalyzed Borylative Difunctionalization of π-Systems. ACS Catal 2020, 10, 11578–11622. [Google Scholar]
- (14).Tsushima T; Tanaka H; Nakanishi K; Nakamoto M; Yoshida H Origins of Internal Regioselectivity in Copper-Catalyzed Borylation of Terminal Alkynes. ACS Catal 2021, 11, 14381–14387. [Google Scholar]
- (15).Jang H; Zhugralin AR; Lee Y; Hoveyda AH Highly Selective Methods for Synthesis of Internal (α-) Vinylboronates through Efficient NHC–Cu-Catalyzed Hydroboration of Terminal Alkynes. Utility in Chemical Synthesis and Mechanistic Basis for Selectivity. J. Am. Chem. Soc 2011, 133, 7859–7871. [DOI] [PubMed] [Google Scholar]
- (16).Moure AL; Mauleón P; Gómez Arrayás R; Carretero JC Formal Regiocontrolled Hydroboration of Unbiased Internal Alkynes via Borylation/Allylic Alkylation of Terminal Alkynes. Org. Lett 2013, 15, 2054–2057. [DOI] [PubMed] [Google Scholar]
- (17).Hopkinson MN; Richter C; Schedler M; Glorius F An Overview of N-Heterocyclic Carbenes. Nature 2014, 510, 485–496. [DOI] [PubMed] [Google Scholar]
- (18).Díez-González S; Marion N; Nolan SP N-Heterocyclic Carbenes in Late Transition Metal Catalysis. Chem. Rev 2009, 109, 3612–3676. [DOI] [PubMed] [Google Scholar]
- (19).Jazzar R; Soleilhavoup M; Bertrand G Cyclic (Alkyl)- and (Aryl)-(amino)carbene Coinage Metal Complexes and Their Applications. Chem. Rev 2020, 120, 4141–4168. [DOI] [PubMed] [Google Scholar]
- (20).Morvan J; Mauduit M; Bertrand G; Jazzar R Cyclic (Alkyl)(amino)carbenes (CAACs) in Ruthenium Olefin Metathesis. ACS Catal 2021, 11, 1714–1748. [Google Scholar]
- (21).Melaimi M; Jazzar R; Soleilhavoup M; Bertrand G Cyclic (Alkyl)(amino)carbenes (CAACs): Recent Developments. Angew. Chem. Int. Ed 2017, 56, 10046–10068. [DOI] [PubMed] [Google Scholar]
- (22).Gao Y; Yazdani S; Kendrick IV A; Junor GP; Kang T; Grotjahn DB; Bertrand G; Jazzar R; Engle KM Cyclic (Alkyl)(amino)carbene Ligands Enable Cu-Catalyzed Markovnikov Protoboration and Protosilylation of Terminal Alkynes: A Versatile Portal to Functionalized Alkenes. Angew. Chem. Int. Ed 2021, 60, 19871–19878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Alfaro R; Parra A; Alemán J; García Ruano JL; Tortosa M Copper(I)-Catalyzed Formal Carboboration of Alkynes: Synthesis of Tri- and Tetrasubstituted Vinylboronates. J. Am. Chem. Soc 2012, 134, 15165–15168. [DOI] [PubMed] [Google Scholar]
- (24).Zhang L; Cheng J; Carry B; Hou Z Catalytic Boracarboxylation of Alkynes with Diborane and Carbon Dioxide by an N-Heterocyclic Carbene Copper Catalyst. J. Am. Chem. Soc 2012, 134, 14314–14317. [DOI] [PubMed] [Google Scholar]
- (25).Yoshida H; Kageyuki I; Takaki K Copper-Catalyzed Three-Component Carboboration of Alkynes and Alkenes. Org. Lett 2013, 15, 952–955 [DOI] [PubMed] [Google Scholar]
- (26).Itoh T; Shimizu Y; Kanai M Ligand-Enabled, Copper-Catalyzed Regio- and Stereoselective Synthesis of Trialkylsubstituted Alkenylboronates from Unactivated Internal Alkynes. J. Am. Chem. Soc 2016, 138, 7528–7531. [DOI] [PubMed] [Google Scholar]
- (27).Zhou Y; You W; Smith KB; Brown MK Copper-Catalyzed Cross-Coupling of Boronic Esters with Aryl Iodides and Application to the Carboboration of Alkynes and Allenes. Angew. Chem. Int. Ed 2014, 53, 3475–3479. [DOI] [PubMed] [Google Scholar]
- (28).Cheng L-J; Mankad NP Copper-Catalyzed Borocarbonylative Coupling of Internal Alkynes with Unactivated Alkyl Halides: Modular Synthesis of Tetrasubstituted β-Borylenones. Angew. Chem. Int. Ed 2018, 57, 10328–10332. [DOI] [PubMed] [Google Scholar]
- (29).Zhurakovskyi O; Dias RMP; Noble A; Aggarwal VK Stereo- and Regiocontrolled Methylboration of Terminal Alkynes. Org. Lett 2018, 20, 3136–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bin H-Y; Wei X; Zi J; Zuo Y-J; Wang T-C; Zhong C-M Substrate-Controlled Regio- and Stereoselective Synthesis of Boron-Substituted 1,4-Dienes via Copper-Catalyzed Boryl–Allylation of Alkynes with Allyl Phosphates and Bis(pinacolato)diboron. ACS Catal 2015, 5, 6670–6679. [Google Scholar]
- (31).Mateos J; Rivera-Chao E; Fañanás-Mastral M Synergistic Copper/Palladium Catalysis for the Regio- and Stereoselective Synthesis of Borylated Skipped Dienes. ACS Catal 2017, 7, 5340–5344. [Google Scholar]
- (32).Rivera-Chao E; Mitxelena M; Varela JA; Fañanás-Mastral M Copper-Catalyzed Enantioselective Allylboration of Alkynes: Synthesis of Highly Versatile Multifunctional Building Blocks. Angew. Chem. Int. Ed 2019, 58, 18230–18234. [DOI] [PubMed] [Google Scholar]
- (33).Su W; Gong T-J; Zhang Q; Zhang Q; Xiao B; Fu Y Ligand-Controlled Regiodivergent Copper-Catalyzed Alkylboration of Unactivated Terminal Alkynes. ACS Catal 2016, 6, 6417–6421. [Google Scholar]
- (34). Control experiments revealed that the α-selective reaction conditions from Ref. 33 did not lead to appreciable product formation (<5%) when allyl electrophiles were employed.
- (35).Lavallo V; Canac Y; Prasang C; Donnadieu B; Bertrand G Stable Cyclic (Alkyl)(Amino)Carbenes as Rigid or Flexible, Bulky, Electron-Rich Ligands for Transition-Metal Catalysts: A Quaternary Carbon Atom Makes the Difference. Angew. Chem. Int. Ed 2005, 44, 5705–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Morvan J; Vermersch F; Zhang Z; Falivene L; Vives T; Dorcet V; Roisnel T; Crévisy C; Cavallo L; Vanthuyne N; Bertrand G; Jazzar R; Mauduit M Optically Pure C1-Symmetric Cyclic(alkyl)(amino)carbene Ruthenium Complexes for Asymmetric Olefin Metathesis. J. Am. Chem. Soc 2020, 142, 19895–19901. [DOI] [PubMed] [Google Scholar]
- (37).Lavallo V; Frey GD; Kousar S; Donnadieu B; Bertrand G Allene Formation by Gold Catalyzed Cross-Coupling of Masked Carbenes and Vinylidenes. Proc. Natl. Acad. Sci. USA 2007, 104, 13569–13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Weinstein CM; Junor GP; Tolentino DR; Jazzar R; Melaimi M; Bertrand G Highly Ambiphilic Room Temperature Stable Six-Membered Cyclic (Alkyl)(amino)carbenes. J. Am. Chem. Soc 2018, 140, 9255–9260. [DOI] [PubMed] [Google Scholar]
- (39).Tomás-Mendivil E; Hansmann MM; Weinstein CM; Jazzar R; Melaimi M; Bertrand G Bicyclic (Alkyl)(amino)carbenes (BICAACs): Stable Carbenes More Ambiphilic than CAACs. J. Am. Chem. Soc 2017, 139, 7753–7756. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.