

Abstract
Ferroptosis is a unique iron-dependent form of regulated cell death. Recently, researchers found that ferroptosis was sensitive to cell density, regulated by Hippo signaling. This article summarizes the roles of the Hippo pathway effectors YAP and TAZ in ferroptosis and the therapeutic potential of activating ferroptosis in cancer.
Keywords: Ferroptosis, Hippo pathway, YAP, TAZ
Cell death occurs in every multicellular organism. The two primary cell death mechanisms are apoptosis (programmed cell death) and necrosis (pathological pre-mature cell death). Recently, a new type of iron-dependent, regulated cell death mechanism called ferroptosis was discovered [1]. Ferroptosis is highly dependent on reactive oxygen species (ROS) availability. Cancer cells have high levels of intracellular ROS, making them especially susceptible to ferroptosis. Since its discovery, ferroptosis has been the focus of many laboratories, as it represents a novel method for treating cancer, especially the cancers resistant to conventional therapies.
Ferroptosis happens when polyunsaturated fatty acid (PUFA)-containing phospholipids (PUFA-PLs) interact with intracellular ROS, leading to lipid peroxidation. To generate PUFA-PLs, cells use acyl-coenzyme A (CoA) synthetase long chain family member 4 (ACSL4) to ligate free PUFAs with CoA to generate PUFA-CoAs. Lysophosphatidylcholine acyltransferase 3 (LPCAT3) subsequently re-esterify and incorporate PUFA-CoAs into PLs, forming PUFA-PLs. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) oxidizes NADPH, giving rise to NADP+ and hydrogen peroxide (H2O2). H2O2 then reacts with the free iron in the cytoplasm through Fenton reaction, producing ROS. ROS will in turn peroxidize PUFA-PLs on the cell membrane, leading to membrane rupture and cell death [2] (Figure 1).
Figure 1. Mechanism of Ferroptosis.
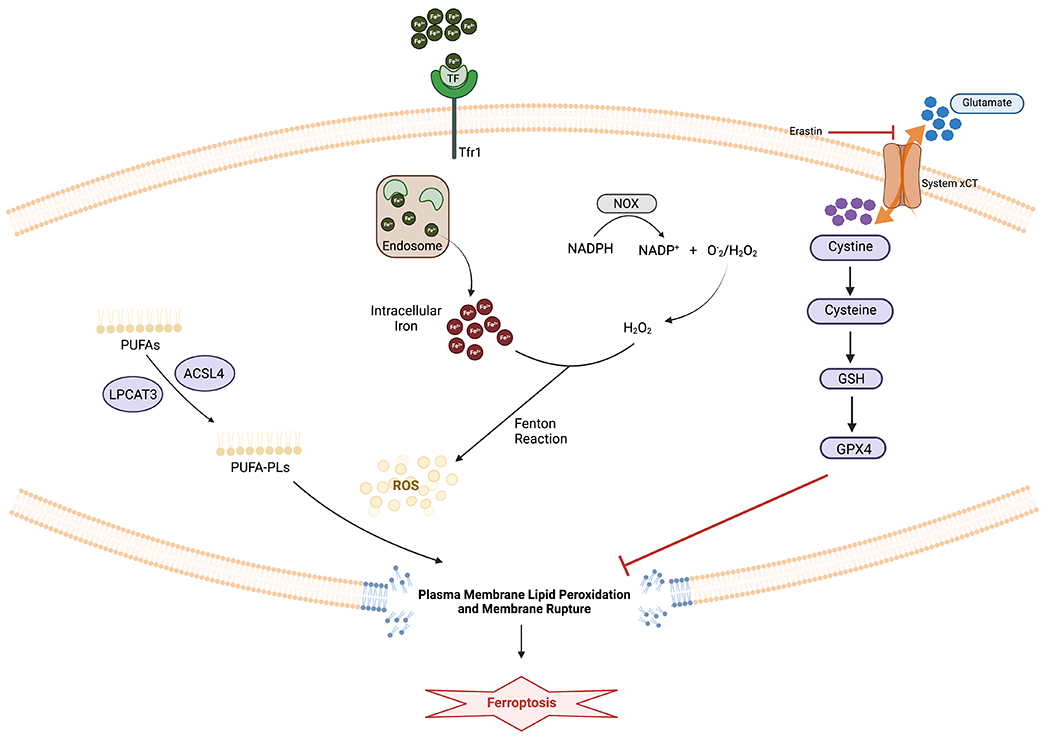
Ferroptosis happens when polyunsaturated fatty acid (PUFA)-containing phospholipids (PUFA-PLs) interact with intracellular ROS, leading to lipid peroxidation. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) oxidizes NADPH, giving rise to NADP+ and hydrogen peroxide (H2O2). H2O2 then reacts with the free iron in the cytoplasm through Fenton reaction, producing ROS. ROS will peroxidize PUFA-PLs on the cell membrane, leading to membrane rupture and cell death. The glutathione peroxidase 4 (GPX4) - glutathione (GSH) system neutralizes lipid peroxidation and prevents ferroptosis.
To neutralize lipid peroxidation and to prevent ferroptosis, cancer cells have developed many defense mechanisms. Among them the most well-known is the glutathione peroxidase 4 (GPX4) - glutathione (GSH) system. Cancer cells use the glutamate-cysteine antiporter to import cysteine, an important precursor of GSH. GPX4, bound to its cofactor GSH, can reduce peroxidized lipids to their corresponding alcohols [3] (Figure 1).
Genetic and non-genetic factors can both modulate ferroptosis. Exciting recent discoveries showed that cell density, a non-genetic factor, regulate cellular sensitivity to ferroptosis: cancer cells are vulnerable to ferroptosis when cultured at low density but are resistant to it when confluent [3,4]. For example, in primary mammary epithelial and breast cancer cells, GPX4 inhibition induced ferroptosis only in low cell density conditions [3]. These discoveries raised the question of how changes in cell density led to ferroptosis, and how we can utilize this information to induce ferroptosis in cancer for cancer elimination.
Hippo pathway is an evolutionarily conserved pathway that relays cell confluence signals to downstream gene expression [5]. This forum article presents the emerging concepts that Hippo pathway effectors Yes-Association Protein (YAP) and Transcriptional coactivator with PDZ-binding motif (TAZ) play essential roles in regulating ferroptosis. YAP/TAZ are highly expressed in most solid tumors and are notorious for contributing to therapy-resistance [6]. Thus, linking YAP/TAZ to ferroptosis presents unique opportunities for targeting these cancers. We will then review and propose novel methods of activating ferroptosis and modulating YAP/TAZ for cancer treatments.
HIPPO PATHWAY IN FERROPTOSIS
Hippo pathway controls tissue growth, organ size, and development by regulating cell proliferation and self-renewal. The Hippo pathway is composed of core kinases: mammalian Ste20-like kinases 1/2 (MST1/2), large tumor suppressor 1/2 (LATS1/2), and the two major downstream effectors which are paralogous transcription coactivators: YAP and TAZ. In high cell density condition, the Hippo pathway is turned on: E-cadherin, a membrane adhesion molecule enriches at cell-cell contact sites, where it recruits Neurofibromatosis 2 (NF2). NF2 subsequently recruits MST1/2 and LATS1/2 to the cell membrane, where MST1/2 form complexes with Salvador Family WW Domain Containing Protein 1 (SAV1) to phosphorylate and activate LATS1/2. LATS1/2 binds to adaptor protein monopolar spindle-one-binder 1 and phosphorylates YAP/TAZ, rendering YAP/TAZ inactive and sequestered in the cytoplasm, bound to adaptor proteins such as 14-3-3. In low cell density condition, the Hippo pathway is turned off. Unphosphorylated YAP/TAZ translocate to the nucleus to interact with transcription factors, such as TEA-domain transcription factor 1-4 (TEAD1-4), to transcribe target genes [5]. The target genes of YAP/TAZ regulate ferroptosis in various ways, including regulating PUFA-PL synthesis (ACSL4), intracellular iron availability (TFRC: transferrin receptor), and ROS production (NOX2/NOX4) [7]. In the following paragraphs, we describe in detail how YAP/TAZ regulate ferroptosis in specific cancer settings.
TAZ induces ferroptosis in various cancers. When renal cell carcinoma cell lines (RCC4, 786O) and patient-derived xenograft cell line (13-789) were plated at low density, Hippo pathway was inactivated, leading to TAZ nuclear localization. Inside the nucleus, TAZ transcriptionally activated epithelial membrane protein 1 (EMP1), which stabilized NOX4 and induced ferroptosis. TAZ knockout rendered these cells insensitive to ferroptosis [6]. In a panel of eight ovarian cancer cell lines (TOV-21G, ES-2, RMG-2, RMG-V, OVCA432, OVCA429, OVCA420, 41M) overexpression of TAZ using TAZS89A (an active form of TAZ) activated NOX2 through Angiopoietin-Like 4 (ANGPTL4), a direct target gene of TAZ. This sensitized cells to ferroptosis. siRNA knockdown of TAZ in chemo-resistant recurrent ovarian cancer cell line (CAOV2R) decreased susceptibility to ferroptosis [6].
The most prominent example of YAP activating ferroptosis happens in mesothelioma, an aggressive tumor forming in lung, heart, or abdomen. NF2 or Hippo pathway inactivation is found in 50% of malignant mesothelioma patients. Experimental disruptions of the NF2-Hippo axis in mesothelioma cell lines activated YAP, which enhanced cellular sensitivity to ferroptosis. As mesothelioma cell density increased, E-cadherin enriches at cell-cell contact sites, activating the NF2-Hippo axis which suppressed YAP and ferroptosis [4]. These results indicate that mesothelioma may be especially sensitive to ferroptosis due to YAP activation.
Interestingly, YAP/TAZ do not always induce ferroptosis: their roles in ferroptosis are cell context specific. In sorafenib (a chemotherapeutic drug) resistant cells, YAP/TAZ forms complexes with Activating Transcription Factor 4 (ATF4) and induces its nuclear localization. ATF4 transcriptionally activates Solute Carrier Family 7 Member 11 (SLC7A11), a glutamate-cysteine antiporter component, which increases GSH levels and prevents ferroptosis. The binding of YAP/TAZ with ATF4 was also observed in HT1080 fibrosarcoma, MDA-MB-231 breast cancer cells and heptacellular carcinoma [8]. These examples indicate that the transcription factors to which YAP/TAZ bind actively regulate ferroptosis. Therefore, it is essential to view the relationship between YAP/TAZ and ferroptosis in a specific cellular and molecular context.
THERAPEUTIC POTENTIAL
While there are numerous methods to treat cancer, the development of resistance to cancer therapy is detrimental. Inducing ferroptosis is an ideal method to tackle therapy resistance. Some existing ferroptosis-inducing drugs include erastin, RSL3, sulfasalazine, and sorafenib. These drugs inhibit xCT-mediated cystine uptake, resulting in ferroptosis [9–11]. Since YAP/TAZ drive therapy resistance and modulate ferroptosis, there is great potential to develop novel ferroptosis-inducing cancer therapies by modulating YAP/TAZ. This can be done either by regulating YAP/TEAD interactions or upstream YAP/TAZ regulators (Figure 2).
Figure 2. Diagram showing different ways of targeting ferroptosis.
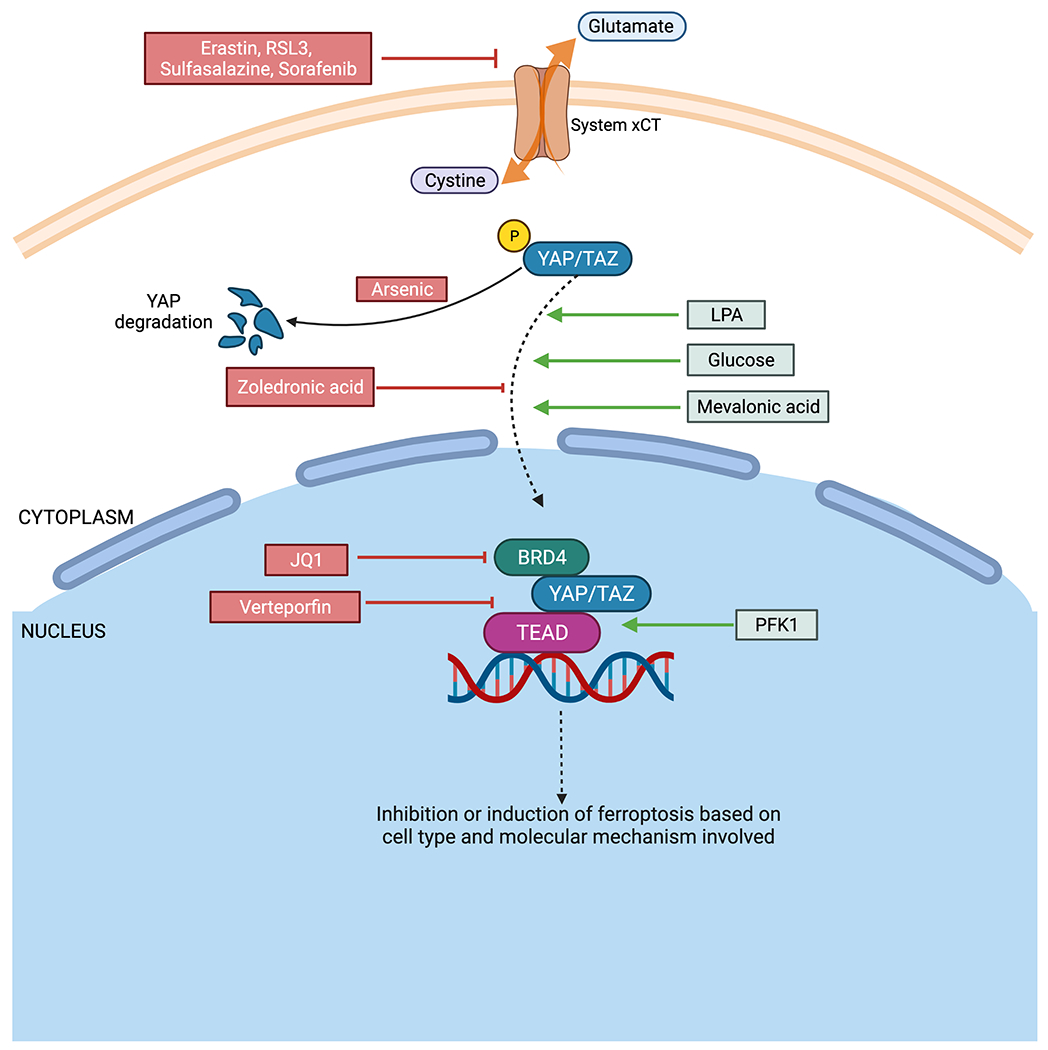
Drugs such as Erastin, RSL3, Sulfasalazine, Lanperisone and Sorafenib can inhibit system xCT and thereby induce ferroptosis. Arsenic treatment causes YAP ubiquitination and degradation.
Ferroptosis can be induced by activating YAP/TAZ. Lysophosphatidic acid (LPA) and mevalonic acid aid in nuclear localization of YAP/TAZ while glucose stabilizes the activity of YAP/TAZ through inactivation of AMP-activated protein kinase (AMPK) and the Hippo pathway. Phosphofructokinase 1 (PFK1) promotes YAP/TAZ binding with transcription factor, TEAD. Inhibition of YAP/TAZ can be mediated by maintaining dephosphorylated state using mevalonate pathway inhibitors such as zoledronic acid. Verteporfin inhibits interaction between YAP/TAZ with TEAD, thereby preventing binding with DNA for transcription. JQ1 inhibits BRD4, which forms a part of the YAP/TAZ-TEAD complex.
Images constructed with Biorender.
In cancers where YAP/TAZ induce ferroptosis, such as renal cell carcinoma and ovarian cancer (where TAZ is the dominant Hippo pathway effector) [6] and malignant mesothelioma (where YAP is the dominant Hippo pathway effector) [4], activators of YAP/TAZ signaling can be used to induce ferroptosis and treat cancer. Lysophosphatidic acid (LPA), a serum phospholipid, can dephosphorylate YAP/TAZ and enhance the interaction between YAP and TEAD. Glucose can sustain the activities of YAP/TAZ through inactivation of AMP-activated protein kinase (AMPK) and the Hippo pathway. Phosphofructokinase 1 (PFK1) promotes YAP/TAZ interaction with TEAD. Mevalonic acid activates YAP/TAZ and aids in their nuclear localization [12]. Therefore, LPA, glucose, mevalonic acid are viable options to upregulate YAP/TAZ and TEAD signaling to activate ferroptosis. However, caution is needed to ensure that ferroptosis overrides the cancer-promoting effects of YAP/TAZ and TEAD activation, such as enhanced cell proliferation, anti-apoptosis, activation of nutrient transporters, therapy resistance, and increased metastasis [5]. Success requires careful modulation of different YAP/TAZ-activated genes. This can be achieved by modulating the transcription factor partners (such as TEAD or ATF4) of YAP/TAZ which determine activation of specific genes [8].
In cancers where YAP/TAZ inhibit ferroptosis, such as sorafenib resistant hepatocellular carcinoma [8], YAP/TAZ inhibitors can be exploited to induce ferroptosis. Mevalonate pathway inhibitors like zoledronic acid inhibit the entry of YAP/TAZ into the nucleus by maintaining them in a phosphorylated state [5]. Verteporfin (VP) inhibits binding between TEAD and YAP/TAZ to limit their transcriptional activity. The Bromodomain and Extra-Terminal (BET) motif inhibitor JQ-1 inhibits YAP-mediated transcription by inhibiting Bromodomain-containing protein 4 (BRD4), an essential component of the YAP/TAZ-TEAD complex [13]. In esophageal squamous cell carcinoma (ESCC) cells, arsenic-ferrosoferric oxide conjugated nano complex can degrade YAP, increasing ESCC sensitivity to radiation and chemotherapy in vivo [14]. These YAP/TAZ inhibitors are all potential candidates for inducing ferroptosis in these specific cancers. However, there are certain challenges to using YAP/TAZ inhibitors. For example, VP is phototoxic and proteotoxic, used at high concentration, and is sometimes not specific: some of the tumor-inhibitory effects of VP are independent of YAP [15]. Therefore, special considerations are needed to ensure the optimal YAP/TAZ inhibitor type and dosage is used to induce ferroptosis.
The complex nature of YAP/TAZ signaling and its role in ferroptosis can be attributed to cell-type specificity and the various mechanisms involved. The molecules associated with YAP/TAZ play central roles in driving or inhibiting ferroptosis. YAP/TAZ targeted therapy is an attractive option that could improve the clinical outcomes of cancer patients.
CONCLUDING REMARKS
The field of ferroptosis is relatively new but has revealed exciting possibilities in disease pathology and treatments. A notable observation is how cell density alters cellular sensitivity to ferroptosis. Hippo pathway, as a major pathway responding to cell density, has a significant role in regulating ferroptosis [3,4,6,8]. Modulating YAP/TAZ, the essential effectors of the Hippo pathway to induce ferroptosis is a novel method to overcome therapy resistance. While ferroptosis is specific to different cell types and dependent on the binding partners of YAP/TAZ, this field of research holds great potential for treating therapy-resistant cancers.
Acknowledgements
This work is supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM142837. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also acknowledge the help from the Editorial Assistance Services Initiative from Johns Hopkins University.
Footnotes
Declaration of interests
D.C. is a consultant of Faze Medicines Inc.
REFERENCES:
- 1.Dixon SJ et al. (2012) Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 149, 1060–1072. 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lei G et al. (2022) Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 10.1038/s41568-022-00459-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vucetic M et al. (2020) Together we stand, apart we fall: how cell-to-cell contact/interplay provides resistance to ferroptosis. Cell Death Dis 11. 10.1038/s41419-020-02994-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu J et al. (2019) Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402-+. 10.1038/s41586-019-1426-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng YG and Pan DJ (2019) The Hippo Signaling Pathway in Development and Disease. Dev Cell 50, 264–282. 10.1016/j.devcel.2019.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun T and Chi JT (2021) Regulation of ferroptosis in cancer cells by YAP/TAZ and Hippo pathways: The therapeutic implications. Genes Dis 8, 241–249. 10.1016/j.gendis.2020.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang WH and Chi JT (2020) Hippo pathway effectors YAP/TAZ as novel determinants of ferroptosis. Mol Cell Oncol 7. 10.1080/23723556.2019.1699375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao RZ et al. (2021) YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. Embo Mol Med 13. 10.15252/emmm.202114351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su YW et al. (2020) Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett 483, 127–136. 10.1016/j.canlet.2020.02.015 [DOI] [PubMed] [Google Scholar]
- 10.Wu YN et al. (2020) Ferroptosis in Cancer Treatment: Another Way to Rome. Front Oncol 10. 10.3389/fonc.2020.571127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin XY et al. (2020) The Mechanism of Ferroptosis and Applications in Tumor Treatment. Frontiers in Pharmacology 11. 10.3389/fphar.2020.01061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Totaro A et al. (2018) YAP/TAZ upstream signals and downstream responses. Nat Cell Biol 20, 888–899. 10.1038/s41556-018-0142-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanconato F et al. (2018) Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med 24, 1599-+. 10.1038/s41591-018-0158-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou W et al. (2020) Arsenic nano complex induced degradation of YAP sensitized ESCC cancer cells to radiation and chemotherapy. Cell Biosci 10. 10.1186/s13578-020-00508-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pobbati AV and Hong WJ (2020) A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 10, 3622–3635. 10.7150/thno.40889 [DOI] [PMC free article] [PubMed] [Google Scholar]