

SUMMARY
Copper deficiency has emerged to be associated with various lipid metabolism diseases, including non-alcoholic fatty liver disease (NAFLD). However, the mechanisms that dictate the association between copper deficiency and metabolic diseases remain obscure. Here, we reveal that copper restoration caused by hepatic ceruloplasmin (Cp) ablation enhances lipid catabolism by promoting the assembly of copper-load SCO1-LKB1-AMPK complex. Overnutrition-mediated Cp elevation results in hepatic copper loss, whereas Cp ablation restores copper content to the normal level without eliciting detectable hepatotoxicity and ameliorates NAFLD in mice. Mechanistically, SCO1 constitutively interacts with LKB1 even in the absence of copper, and copper-loaded SCO1 directly tethers LKB1 to AMPK, thereby activating AMPK and consequently promoting mitochondrial biogenesis and fatty acid oxidation. Therefore, this study reveals a mechanism by which copper, as a signaling molecule, improves hepatic lipid catabolism, and it indicates that targeting copper-SCO1-AMPK signaling pathway ameliorates NAFLD development by modulating AMPK activity.
Graphical abstract
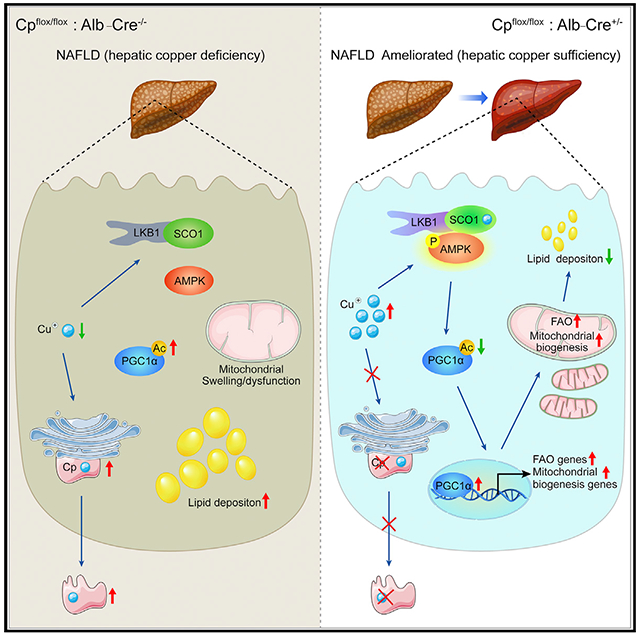
In brief
Xie et al. reveal that ceruloplasmin deletion restores hepatic copper in DIO mice and ameliorates NAFLD development via enhanced mitochondrial biogenesis and fatty acid oxidation. Mechanistically, they find that the chaperone SCO1 senses copper and activates AMPK by forming the SCO1-LKB1-AMPK complex.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is characterized by abnormal lipid metabolism, which results in lipid accumulation in hepatocytes and can lead to non-alcoholic steatohepatitis and hepatocellular carcinoma (Younossi et al., 2016). Despite that the prevalence of NAFLD worldwide is estimated at 25% (Younossi et al., 2016), approved therapies that induce durable remission of NAFLD are scarce, thereby underscoring the urgent need to identify pivotal drivers of NAFLD and therapeutic targets.
Copper is an essential dietary trace element that serves as a structural and enzymatic cofactor for proteins involved in critical cellular processes, including redox balance, mitochondrial function, and iron metabolism (Gulec and Collins, 2014; Kim et al., 2008). Liver is the central organ for copper homeostasis, and hepatic copper levels need to be maintained within a narrow range (Linder, 2020; Xu et al., 2013). Ceruloplasmin (Cp), a principal copper-binding protein secreted from the liver, plays a critical role in maintaining hepatic copper homeostasis by distributing copper from the liver to extrahepatic tissues (Hellman and Gitlin, 2002).
Circulating Cp is elevated in patients with diabetes, obesity, and NAFLD (Cunningham et al., 1995; Daimon et al., 1998; Engstrom et al., 2003; Kim et al., 2002); in parallel, hepatic copper deficiency is strongly associated with metabolic diseases, including diabetes mellitus, metabolic syndrome, and NAFLD (Aigner et al., 2008,2010; Carr and Lei, 1990; Wapnir and Devas, 1995). However, the underlying mechanisms of these observations still remain elusive.
Here, we reveal that overnutrition-caused Cp upregulation leads to a reduction in hepatic copper content along with NAFLD, while restoration of hepatic copper levels by Cp deletion, ameliorates high-fat diet (HFD)-induced NAFLD development. We identify that the SCO1-LKB1-AMPK complex perceives the elevation of copper concentration, and hepatic copper is an intriguing modulator of lipid catabolism via AMPK activation through a signaling pathway in which copper-binding protein SCO1 actively engages.
RESULTS
Copper concentrations are lower and Cp expression is higher in fatty liver and diabetes models than in controls
To investigate the roles of copper and Cp in lipid metabolism, we then measured the expression levels of Cp and copper content in human liver biopsies and mouse models of metabolic diseases. Notably, copper levels in human liver biopsies were significantly lower in NAFLD patients than healthy controls (p < 0.05) (Figure 1A). The hepatic copper content in ob/ob and HFD-induced obese (DIO) mice was also lower than that in control mice (Figures 1B and S1A). These results suggest that a reduction in hepatic copper content might be associated with NAFLD in humans and mice.
Figure 1. Cp knockdown attenuates HFD-induced hepatic steatosis.
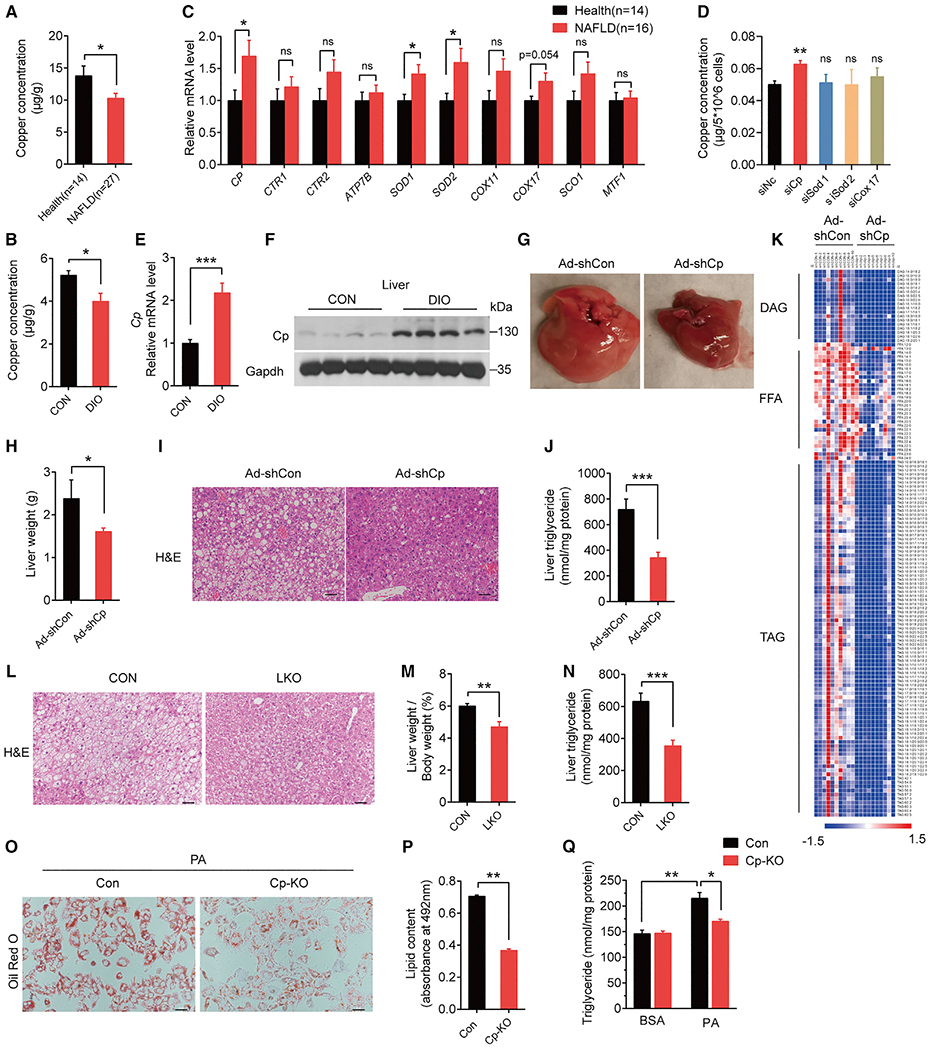
(A) ICP-MS detected hepatic copper concentrations of individuals with NAFLD (n = 27) and healthy controls (n = 14).
(B) Hepatic copper concentrations in DIO and control mice (n = 6 in each group).
(C) qPCR analysis of copper-binding proteins in livers of individuals with NAFLD (n = 16) and healthy controls (n = 14).
(D) Copper concentrations in primary hepatocytes treated with siRNAs.
(E and F) Cp mRNA (E) and protein (F) levels in livers of DIO and control mice.
(G–K) DIO mice (fed an HFD for 13 weeks) were injected with Ad-shCp or Ad-shCon (n = 10 in each group).
(G) Representative images of liver morphology.
(H) Liver weight (g) of mice.
(I) Representative images of hematoxylin and eosin (H&E)-stained liver sections (scale bar: 50 μm).
(J) Hepatic triglyceride concentrations.
(K) Heatmap of hepatic TAG, DAG, and FFA content by LC-MS analysis; each column represents data from a single mouse, and each row represents a lipid species.
(L–N) Liver-specific Cp-knockout mice (LKO) and control mice (Cpflox/flox, CON) fed an HFD for 20 weeks. n = 10 in each group (L) Representative images of H&E-stained liver sections (scale bar: 50 μm).
(M) Liver weight normalized to body weight.
(N) Hepatic triglyceride concentrations.
(O–Q) Primary hepatocytes isolated from LKO and CON mice were treated with PA.
(O) Representative images of oil red O staining.
(P) Lipid content.
(Q) TG concentrations. Data shown in (E) and (O)–(P) are from one representative experiment of at least three independent experiments. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control group. See also Figures S1 and S2, Tables S1, S2, S3, and S4.
Next, we analyzed the expressions of genes involved in copper metabolism and transportation in human liver biopsies, including CP, CTR1, and SOD1, and we found that the expression of CP, SOD1, SOD2, and COX17 was higher in liver biopsies of NAFLD patients than controls (Figure 1C). To verify which gene regulates hepatic copper abundance, we knocked down these four genes respectively in primary hepatocytes. Only knocking down of Cp substantially increased the intracellular copper concentration (Figure 1D), which is consistent with the essential role of Cp in hepatic copper homeostasis (Kim et al., 2008). In addition, we found that expression levels of hepatic Cp were significantly increased in all three NAFLD mouse models (Figures 1E, 1F, S1B–S1F).
In view of the close link between NAFLD and type 2 diabetes, Cp expression was measured in diabetes models, including streptozotocin (STZ)-injected mice (Figures S1G–S1I) and mice with genetic inactivation of hepatic insulin receptor substrate Irs1 and Irs2 double knockout (LDKO) (Figures S1J–S1L). Similar to the obese NAFLD mouse models, Cp was highly expressed in diabetic mice.
These results suggest that excess hepatic Cp might lead to reductions in hepatic copper level and subsequently result in the development of NAFLD.
Acute Cp knockdown attenuates HFD-induced hepatic steatosis
To determine the function of Cp on hepatic glucose and lipid metabolism and NAFLD development, we knocked down hepatic Cp in DIO mice (Figure S2A). Relative to controls, knockdown of hepatic Cp significantly reduced liver weight (Figures 1G and 1H), ectopic lipid accumulation in the liver (Figures 1I and 1J), and plasma free fatty acids (FFAs) (Figure S2B). Lipidomic analysis showed that the levels of diacylglycerides (DAGs), FFAs, and triglycerides (TGs) were substantially reduced in Ad-shCp-treated mouse livers (Figure 1K and Table S2). Moreover, glucose clearance and insulin sensitivity were substantially improved in Cp-knockdown mice (Figures S2C–S2F), but no differences in body weight or food intake were observed (Figures S2G and S2H).
Then, we used AAV (which is less immunogenic and widely used in clinical trials) to express Cre in mouse liver by transduction of Cpflox/flox mice with CreAAV-TBG viruses (Cpflox/flox-CreAAV-TBG). Consistent with Ad-shCp injected mice, Cpflox/flox-CreAAV-TBG mice displayed lower ectopic hepatic lipid accumulation (Figure S2I) and substantially less insulin resistance (Figure S2J). However, there were no differences in glucose tolerance between Cpflox/flox-CreAAV-TBG and Cpflox/flox-GFPAAV-TBG mice (Figure S2K).
Liver-specific Cp deletion protects DIO mice from HFD-induced hepatic steatosis
To confirm the results observed in mice with virus-mediated Cp-knockdown, we generated liver-specific Cp-knockout (LKO) mice (Figure S2L). LKO mice exhibited less lipid accumulation in the liver and less liver weight and hepatic triglyceride (Figures 1L–1N). Glucose tolerance and insulin sensitivity were improved as well in LKO mice (Figures S2M and S2N). Besides, Cp deletion attenuated inflammation in the livers (Figure S2O) and ameliorated HFD feeding-induced hepatic damage (Figures S2P and S2Q). In contrast, hepatic Cp ablation had no obvious effects on fat mass (Figures S2R and S2S), body weight (Figure S2T), and food intake (Figure S2U).
We also investigated the effect of Cp knockout (Cp-KO) on lipid accumulation in primary hepatocytes, and the result showed that Cp-KO resulted in less palmitic acid (PA)-induced lipid droplet and TG accumulation in primary hepatocytes than controls (Figures 1O–1Q).
Taken together, these data indicate that deletion of Cp ameliorates HFD-induced hepatic steatosis and improves insulin sensitivity and glucose homeostasis.
Cp knockdown increases mitochondrial biogenesis and fatty acid oxidation (FAO)
Given that mitochondria play a key role in lipid metabolism, we examined whether Cp ablation affected mitochondrial function. Electron microscopy showed that Cp deletion in mouse liver ameliorated HFD-induced mitochondria swelling and deteriorations of mitochondrial morphology (Figure 2A). In line with this, Cp ablation substantially increased mitochondrial number (Figure 2B) and the copy number of mitochondrial DNA in primary hepatocytes (Figure 2C). Moreover, expressions of genes encoding mitochondrial respiratory chain complex was upregulated, including Nd1, Nd2, Co1, and Atp6 (Figure 2D). Consistent with this, immunoblotting and immunohistochemical staining confirmed that Cp deletion efficiently upregulated protein expression of the subunits of mitochondria respiratory chain complexes, such as ATP5A, UQCRC2, MTCO1, SDHB, and NDUFB8 (Figures 2E and S2V). These results suggest that Cp deletion rescues mitochondrial morphology under HFD feeding and increases the number of healthy mitochondria.
Figure 2. Cp knockdown increases mitochondrial biogenesis and FAO.
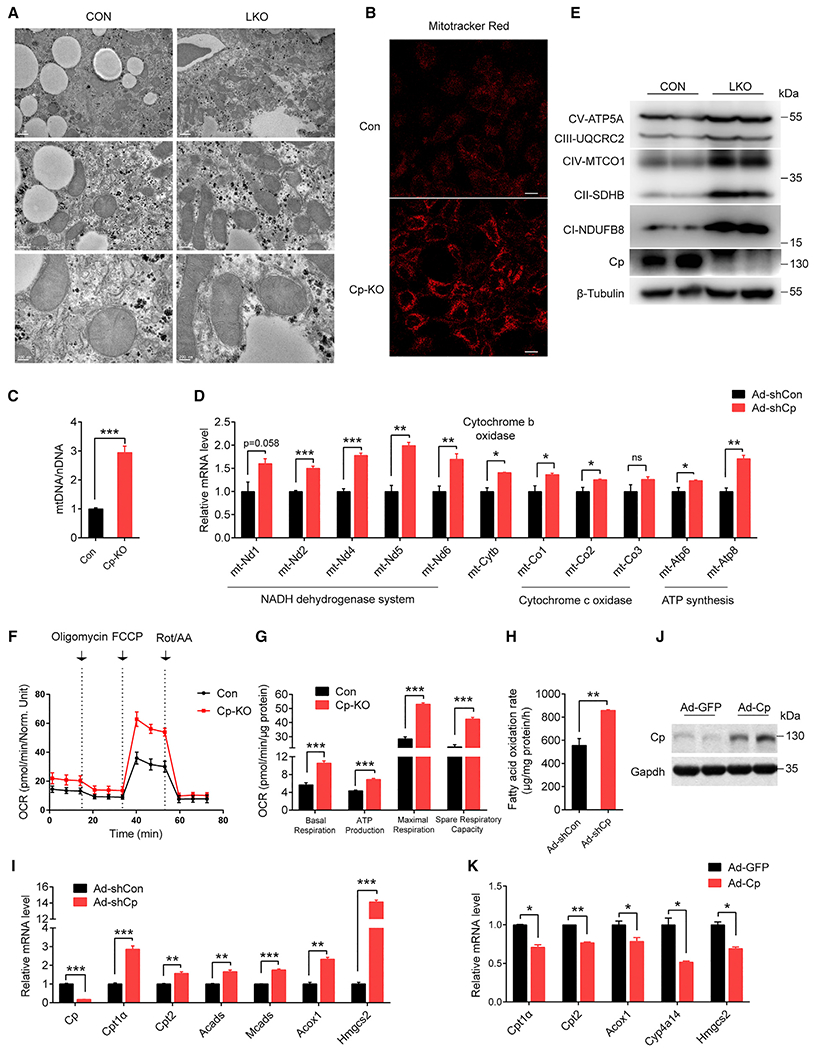
(A) Transmission electron microscopy of liver mitochondria in LKO and CON mice fed an HFD for 16 weeks; scale bar: 1 μm (line 1), 0.5 μm (line 2), 0.2 μm (line 3).
(B, C, F, G) Primary hepatocytes from CON and LKO mice termed as CON and Cp-KO.
(D, H–K) Primary hepatocytes from C57BL/6J were treated with adenovirus as indicated.
(B) Representative images of Mitotracker staining of CON and Cp-KO primary hepatocytes (scale bar: 10 μm).
(C) The ratio of mitochondrial DNA amount to nuclear DNA total amount in primary hepatocytes.
(D) mRNA expression of mitochondrial DNA in primary hepatocytes treated with Ad-shCon and Ad-shCp.
(E) Immunoblot of subunit proteins of mitochondria respiratory chain complexes in CON and LKO mice liver.
(F) Oxygen consumption rate (OCR) in CON and LKO primary hepatocytes.
(G) Average OCR analyzed from (F).
(H) Fatty acid oxidation rate of primary hepatocytes treated with Ad-shCon and Ad-shCp.
(I) mRNA expression levels of FAO genes in Ad-shCon and Ad-shCp infected primary hepatocytes.
(J and K) Cp protein abundance (J) and mRNA levels of FAO genes (K) in primary hepatocytes treated with Ad-Cp and Ad-GFP. Data shown in (C), (D), and (I)–(K) are from one representative experiment of at least three independent experiments. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control group. See also Figure S2 and Table S3.
Next, we explored the effects of Cp ablation on mitochondrial function. Notably, Cp-KO cells exhibited a significantly higher capacity for basal respiration, maximal respiration, spare respiration, and ATP production (Figures 2F and 2G), indicating that Cp-KO resulted in higher mitochondrial activity. In addition, mitochondrial FAO rate was also increased (Figure 2H). Consistent with this, Cp-KO hepatocytes showed significantly higher expressions of genes involved in mitochondrial FAO, including Cpt1α, Cpt2, and Hmgcs2 (p < 0.01) (Figure 2I), but without significant changes in the levels of lipogenic genes, including Srebp1c, Acc1, Fasn, Chrebpα, Chrebpβ, and Acly (Figure S2W). Immunohistochemical staining further confirmed that Cp deletion remarkedly upregulated Hmgcs2 expression in livers (Figure S2X). Furthermore, we found that overexpressing Cp in primary hepatocytes substantially suppressed the expressions of genes related to FAO (Figures 2J and 2K). These results indicate that Cp depletion substantially enhances mitochondrial FAO activity.
AMPK is required for amelioration of ectopic lipid accumulation caused by hepatic Cp-KO
To elucidate the molecular mechanisms by which Cp deletion reduces hepatic steatosis, we performed RNA-seq analysis of mouse livers treated with Ad-shCp or Ad-shCon. Ingenuity Pathway Analysis showed that the AMPK pathway was one of the most enriched biological process terms (Figure 3A). Incubating Cp-KO primary hepatocytes with inhibitors against various pathways reveals that only inhibitors of AMPK and SIRT1 (a direct downstream target of AMPK) blocked Cp deletion-mediated upregulation of Hmgcs2 and Creb3l3 (Figures 3B, S3A, and S3B). Moreover, AMPK and ACC were both activated in Cp-deleted hepatocytes (Figures 3C and S3C) and LKO mice liver (Figure 3D). In contrast, inhibition AMPK with Compound C (CC) impaired the Cp deletion-induced upregulation of mitochondrial respiration (Figures 3E and S3D). Consistent with this data, genetic ablation of AMPK attenuated the Cp-knockdown-mediated reduction of lipid accumulation in hepatocytes (Figures 3F, S3E, and S3F), and it also suppressed the Cp-knockdown-caused increases in mitochondria mass (Figure 3G) and the expressions of FAO genes (Figure 3H) and other mitochondrial genes (Figure 3I).
Figure 3. Cp regulates fatty acid oxidation genes via the AMPK-PGC1α axis.
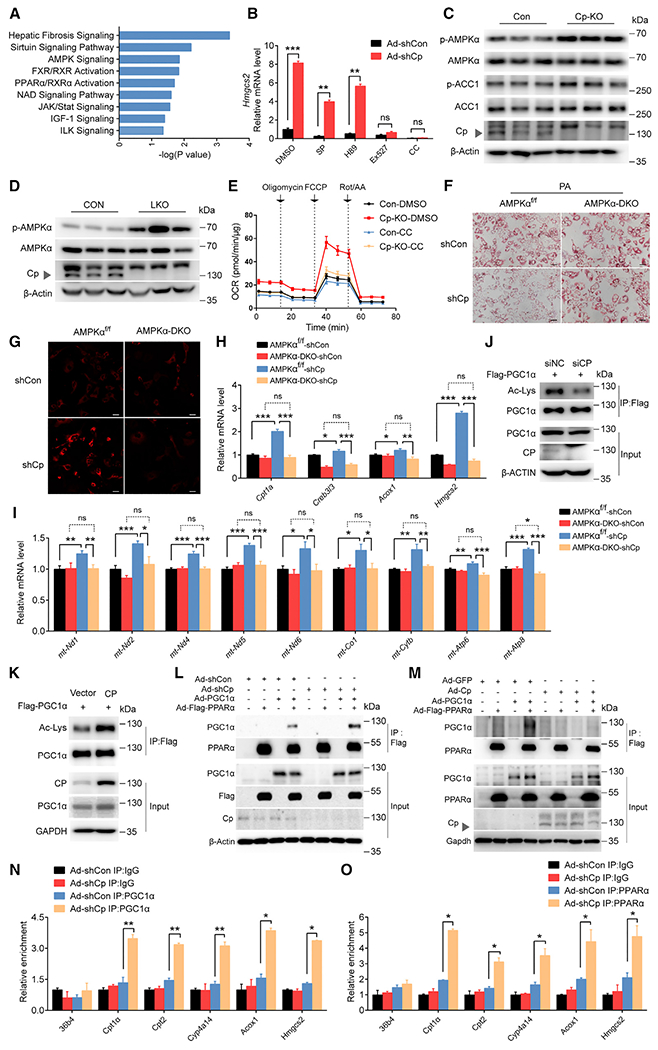
(A) Ingenuity Pathway Analysis of RNA-seq data obtained from livers of Ad-shCp- or Ad-shCon-injected DIO mice.
(B) Hmgcs2 mRNA levels in Ad-shCp- or Ad-shCon-infected and inhibitors-treated primary hepatocytes, JNK inhibitor (SP), PKA inhibitor (H89), SIRT1 inhibitor (Ex527), and AMPK inhibitor (Compound C, CC).
(C) Immunoblots (protein indicated) in Cp-KO primary hepatocytes.
(D) Immunoblots (protein indicated) in livers of LKO mice.
(E) OCR of CON and Cp-KO primary hepatocytes treated with CC.
(F–L) Primary hepatocytes were isolated from AMPKα1/α2 DKO (AMPKα-DKO) or AMPKα1flox/flox and α2flox/flox (AMPKαf/f) mice, followed by the infection with Ad-shCp or Ad-shCon.
(F) Representative images of oil red O staining of primary hepatocytes (scale bar: 50 μm).
(G) Representative images of Mitotracker staining of primary hepatocytes (scale bar: 20 μm).
(H) mRNA levels of FAO genes in primary hepatocytes.
(I) mRNA expression levels of mitochondrial DNA.
(J and K) Acetylation of PGC1α in L02 cells treated as indicated.
(L and M) Immunoblotting for PGC1α and Flag-tagged PPARα proteins after immunoprecipitation of Flag-PPARα from AML12 cells treated as indicated.
(N and O) Chromatin immunoprecipitation (ChIP) assay-qPCR analysis of the promoter regions of FAO genes in with Ad-shCp-treated primary hepatocytes, with an anti-PGC1α (N) or an anti-PPARα (O) antibody. IgG, immunoglobulin. Data shown in (B) and (H)–(I) are from one representative experiment of at least three independent experiments. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control group. See also Figures S3 and S4 and Table S3.
Collectively, these results indicate that AMPK is a critical downstream target of Cp that regulates mitochondrial function and lipid metabolism in the liver.
Cp-deletion-induced upregulation of FAO is dependent upon the AMPK-PGC1α-PPARα axis
Previous reports show that AMPK increases mitochondrial FAO by enhancing transcriptional activity of PGC1α and PPARα (Baur et al., 2006). Notably, we found that Cp ablation decreased the abundance of acetylated PGC1α, an inactivated form of PGC1α (Figure 3J); by contrast, overexpressing Cp elevated acetylated PGC1α levels (Figure 3K). Activated PGC1α promotes its interaction with PPARα, thereby increasing its co-activator ability (Piccinin et al., 2019). Here, we found that Cp deletion efficiently enhanced the interaction between PGC1α and PPARα (Figure 3L). By contrast, overexpression of Cp substantially decreased the interaction of PGC1α and PPARα (Figure 3M). We further examined PGC1α-PPARα-mediated transcription of FAO genes and revealed that knocking down Cp increased the activity of PPARE-Luc, CPT1α-Luc, and HMGCS2-Luc reporters (Figure S4A), indicating that Cp deletion enhances PPARα-transactivation. By contrast, overexpressing Cp suppressed the GW9578 (PPARα agonist)-induced increase in promoter activities of these luciferase reporters (Figure S4B). In line with this, chromatin immunoprecipitation assays confirmed that Cp deletion enhanced recruitment of PGC1α (Figure 3N) and PPARα (Figure 3O) to the promoters of PPARα target genes, whereas knocking down PGC1α (Figure S4C) or PPARα (Figure S4D) completely blocked the elevated transcription of FAO genes caused by Cp deletion. In line with this, inactivation of PPARα via GW6471, an antagonist of PPARα, abolished the effects of Cp knockdown on the reduction of lipid accumulation in hepatocytes (Figures S4E and S4F).
Restoration of hepatic copper content reduces excess lipid accumulation in liver through AMPK activation
Next, we explored how Cp deletion activated AMPK. Unexpectedly, we detected no significant alterations in AMP/ATP or ADP/ATP ratio in the livers of LKO mice (Figures S4G and S4H), suggesting that Cp deletion activates AMPK via an AMP/ADP-independent mechanism. Notably, Cp deletion efficiently prevents the decrease in hepatic copper levels caused by HFD feeding (Figures 4A, 4B, and S5A). Interestingly, treating primary hepatocytes with a copper solution increased the phosphorylation levels of AMPK and ACC in a dose-dependent manner (Figure 4C), and consistently, it activates AMPK via an AMP/ADP-independent manner (Figures S5B and S5C). Furthermore, depleting intracellular copper with copper cation-specific chelator bathocuproinedisulfonic acid disodium salt (BCS) attenuated the effect of Cp deletion on upregulation of Hmgcs2 (Figures 4D and S5D) and Cpt1α (Figures 4E and S5D). By contrast, an iron cation-specific chelator, deferasirox (DFX), had little effect on Hmgcs2 (Figure 4D) or Cpt1α (Figure 4E) mRNA levels. In addition, we performed similar experiments with additional copper chelator tetrathiomolybdate (TM) and iron chelators pyridoxal isonicotinoyl hydrazine (PIH) and deferoxamine (DFO) in primary hepatocytes, and we obtained the consistent conclusion that chelating copper, but not iron, blocked Cp-deletion-upregulated FAO gene expressions (Figures 4F and S5E–S5G). Moreover, BCS and TM attenuated increases in phosphorylated AMPK by Cp deletion in primary hepatocytes (Figure 4G). Chelating intracellular copper with BCS also prevented Cp-ablation-induced reductions in lipid accumulation, whereas the iron chelator DFX failed to do so (Figures 4H, 4I, S5H, and S5I).
Figure 4. Cp regulates AMPK signaling by changing intracellular copper concentrations and the Cu-binding protein SCO1.
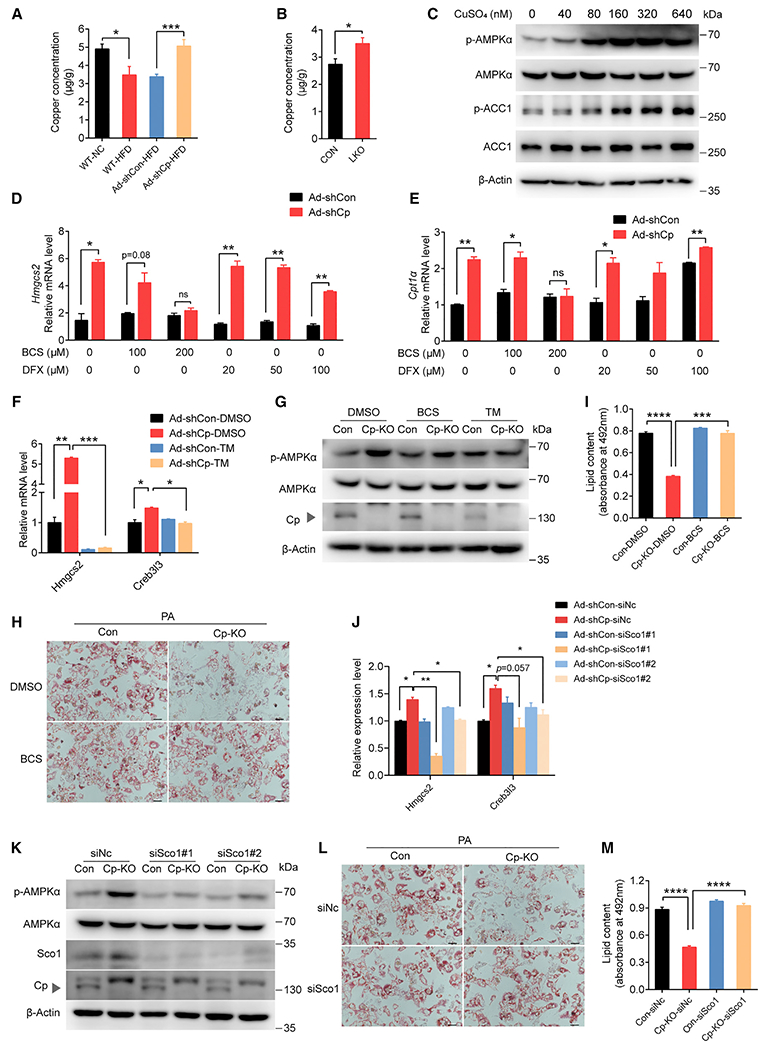
(A) Hepatic copper concentrations of wild-type (WT) mice fed chow diet (NC) (n = 6) or an HFD (n = 6) and of the mice injected with indicated adenovirus (n = 10).
(B) Copper concentrations in livers of LKO (n = 12) and CON mice (n = 10).
(C) p-AMPKα, p-ACC, ACC, and AMPKα in primary hepatocytes treated with CuSO4 for 4 h.
(D and E) mRNA levels of Hmgcs2 (D) and Cpt1α (E) in Cp-knockdown (Ad-shCp) and control (Ad-shCon) primary hepatocytes treated with a copper chelator (BCS) or an iron chelator (DFX).
(F) mRNA levels of Hmgcs2 and Creb3l3 in Cp-knockdown (Ad-shCp) and control (Ad-shCon) primary hepatocytes treated with a copper chelator (TM).
(G) Protein levels of p-AMPKα and Cp in BCS- or TM-treated hepatocytes isolated from CON and LKO mice.
(H and I) BCS-treated primary hepatocytes isolated from CON and LKO mice after PA treatment for 24 h.
(H) Representative images of oil red O staining (scale bar: 50 μm).
(I) Lipid contents.
(J) mRNA levels of Hmgcs2 in Ad-shCp-infected primary hepatocytes after knockdown of copper-binding proteins with siRNA as indicated.
(K) Protein expression of p-AMPKα, Sco1, and Cp in LKO primary hepatocytes (Cp-KO) treated with siRNA targeting Sco1 (siSco1#1, siSco1#2).
(L and M) Primary hepatocytes isolated from CON and LKO mice treated with siSco1 and PA or vehicle.
(L) Representative images of oil red O staining (scale bar: 50 μm).
(M) Lipid contents. Data shown in (D)–(F), (H)–(I), (L)–(M) are from one representative experiment of at least three independent experiments. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control group. See also Figures S5 and S6, Tables S3 and S4.
The global Cp-KO (WKO) mouse is an animal model of aceruloplasminemia that exhibits excess iron accumulation in the liver (Yamamoto et al., 2002). We also observed an increased level of hepatic iron in the WKO mouse (Figure S5J). By contrast, Prussian blue staining of the LKO mouse liver did not reveal obvious iron accumulation (Figure S5J). Although hepatic iron levels of LKO mice (measured by inductive coupled mass spectrometry [ICP-MS]) were modestly increased compared with controls (Figure S5K), they are comparable to hepatic iron levels in normal mice as previously reported (Church et al., 2015; Song et al., 2012; Wang et al., 2017).
Taken together, these results indicate that the effect of Cp deletion on the activation of AMPK and suppression of fatty liver is dependent on upregulation of hepatic copper content.
Copper upregulates hepatic FAO via SCO1
Given that copper activates AMPK and that AMPK is not reported as a copper-binding protein (González et al., 2020), we hypothesized that a copper-binding protein mediates AMPK activation. To test this hypothesis, we used a collection of siRNAs targeting key copper-binding proteins in primary hepatocytes to identify candidate protein(s) mediating the effect of Cp deletion on upregulation in FAO genes, including Hmgcs2 and Creb3l3 (Figures 4J and S6A–S6L).
We found that only knockdown of Sco1 consistently blunted the upregulation of FAO gene expression caused by Cp deletion (Figures 4J and S6L); by contrast, silencing Sco2, a paralogous gene of Sco1, has no detectable effect on the FAO gene expression and AMPK activation in present of Cp deletion (Figures S6K and S6M).
Moreover, we found that knocking down Sco1 entirely abrogated the effect of Cp ablation on increasing phosphorylated AMPK levels in hepatocytes (Figure 4K). Finally, Sco1 deficiency blocked Cp-KO-dependent suppression of lipid accumulation in hepatocytes (Figures 4L and 4M).
Taken together, these results suggest that SCO1 plays a critical role in copper-dependent AMPK activation and lipid catabolism.
SCO1 promotes AMPK activation by directly interacting with AMPK and LKB1
Next, we explored the mechanism by which SCO1 regulates AMPK activity. SCO1 was reported as a mitochondrial protein (Leary et al., 2007). However, we detected that some SCO1 signals overlapped well with that of β-actin (Figure 5A), suggesting that a number of SCO1 proteins localized in cytoplasm. Moreover, co-immunoprecipitated assay showed that exogenous SCO1 interacted with exogenous AMPKα1 and endogenous LKB1 (Figure 5B). Reverse co-immunoprecipitation further confirmed the interaction among AMPK, SCO1, and LKB1 (Figure 5C). Besides, the endogenous interaction between SCO1 and AMPK was also detected in L02 cells (Figure 5D). In addition, SCO1 could interact with the AMPK α, β, and γ subunits (Figure 5E). Furthermore, pull-down assays showed a direct interaction between SCO1 and AMPK (Figures 5F and 5G) as well as between SCO1 and LKB1 (Figures 5H and 5I). In addition, confocal images revealed the colocalization of AMPK and Sco1 in hepatocytes (Figure 5J). To explore whether LKB1 is required for the activation of AMPK by copper, we treated Lkb1-silenced hepatocytes with CuSO4 solution. The results showed that knockdown of Lkb1 substantially suppressed the activation of AMPK induced by copper (Figure 5K), which indicated that LKB1 is an essential component for copper-induced AMPK activation. Next, we performed similar co-immunoprecipitated assay in subcellular fractionation. The cytoplasm and mitochondria were respectively purified from primary hepatocytes (Figure 5L), and we only detected the interaction between Sco1 and AMPK in cytosol but not in the lysate of mitochondria (Figures 5M and 5N). The co-immunoprecipitated assay in AMPKα-DKO primary hepatocytes showed that SCO1 interacted with LKB1 even in the absence of AMPK, suggesting that Sco1 interacts with LKB1 independent of AMPK (Figure 5O). Taken together, these results suggest that SCO1 acts as a scaffold protein to tether the activating kinase LKB1 to AMPK, resulting in AMPK phosphorylation.
Figure 5. SCO1 tethers LKB1 to AMPK and activates AMPK.
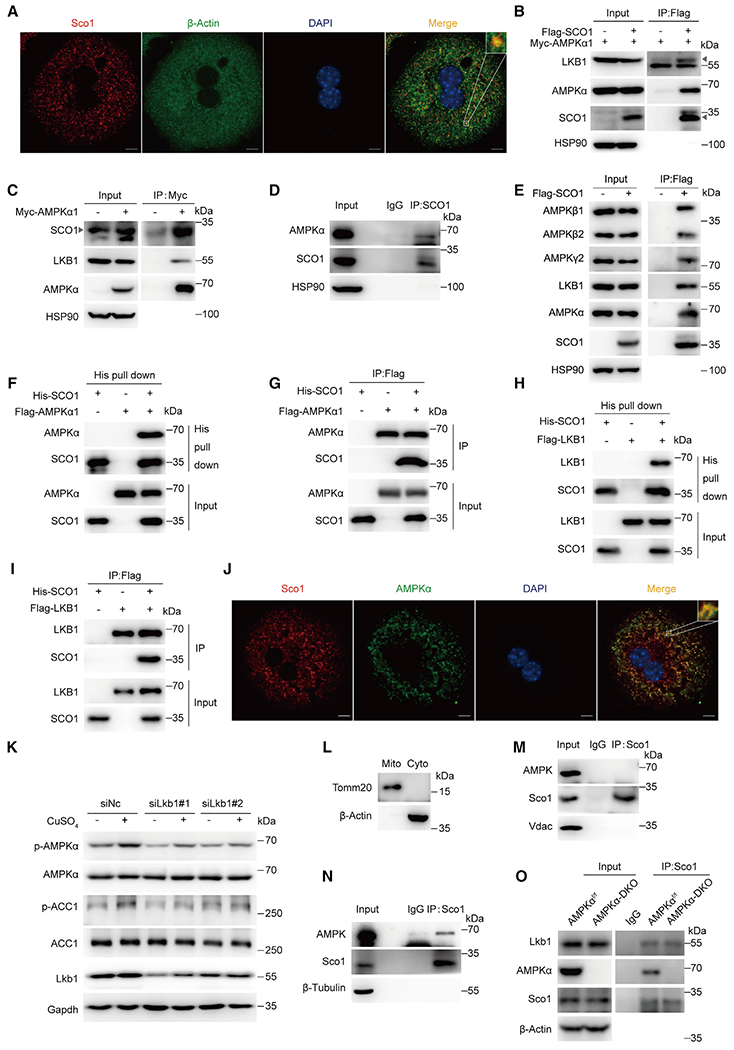
(A) Immunofluorescence of SCO1 and β-actin. Cells were reacted with indicated antibodies. Nuclei were counterstained with DAPI.
(B) Immunoblotting for LKB1, AMPKα, and Flag-tagged SCO1 proteins after immunoprecipitation of SCO1 from HEK293T cells. Triangle indicates LKB1 (up) and SCO1 (down).
(C) Immunoblotting for SCO1, LKB1, and AMPKα proteins after immunoprecipitation of AMPKα1 from HEK293T cells.
(D) AMPKα and SCO1 protein levels after immunoprecipitation of SCO1 from L02 cells.
(E) Immunoblotting for AMPKα, AMPKβ1/2, AMPKγ2, LKB1, and Flag-tagged SCO1 proteins after immunoprecipitation of SCO1 from HEK293T cells.
(F–I) Direct interaction between SCO1 and AMPK complex. Pull-down assays were performed with Ni-NTA beads against His-SCO1 (F, G) or an anti-Flag antibody against Flag-tagged AMPKα (H, I), followed by immunoblotting with the antibodies as indicated.
(J) Immunofluorescence of Sco1 and AMPK. Cells were reacted with indicated antibodies. Nuclei were counterstained with DAPI.
(K) Immunoblotting for p-AMPKα, ACC1, and LKB1 in primary hepatocytes treated with siRNA targeting Lkb1 (siLkb1#1, siLkb1#2) combined with CuSO4 (320 nM) treatment for 4 h.
(L–N) Immunoblotting for endogenous Sco1 and AMPKα after immunoprecipitation of Sco1 in different subcellular fraction.
(O) Protein levels of Lkb1, AMPKα, and Sco1 after immunoprecipitation of Sco1 from primary hepatocytes isolated from AMPKαf/f and AMPKα-DKO mice.
Copper-loaded SCO1 is essential for AMPK activation by forming SCO1-LKB1-AMPK complex
Next, we determined whether copper or Cp regulates the assembly of the SCO1-LKB1-AMPK complex. A co-immunoprecipitation assay showed that overexpression of CP reduced the interaction between SCO1 and AMPK, but it had little effect on SCO1-LKB1 interaction (Figure 6A). Interestingly, we failed to detect CP protein in SCO1-LKB1-AMPK precipitation (Figure 6A), which further confirmed that CP modulation of AMPK activity is not through its direct physical interaction with AMPK. Given that overexpression of CP might lead to a reduction in intracellular copper via copper excretion, we hypothesized that copper affects the assembly of the SCO1-LKB1-AMPK complex. To confirm this hypothesis, we depleted intracellular copper with BCS, and the co-immunoprecipitation result showed that BCS treatment significantly reduced the interaction between SCO1 and AMPK (Figure 6B), indicating the important role of copper in the SCO1-AMPK interaction. In another experiment, we constructed an SCO1 copper-binding deficient mutant (C152/156A) by substituting CyS152 and CyS156 with alanine, as reported (Balatri et al., 2003; Nittis et al., 2001). An obvious shift of the SCO1 (C152/156A) band (Figure 6C, line 7), similar with BCS treatment (Figure 6C, line 6), revealed that its copper-binding capacity was abolished. Notably, a co-immunoprecipitation assay showed that the C152/156A mutation in SCO1 impaired its interaction with AMPK, but it had no effects on its interaction with LKB1 (Figure 6D). Moreover, the pull-down assay demonstrated that the interaction between truncated SCO1 (aa 119-284, His-Truncated) and AMPK is comparable to that between full-length SCO1 and AMPK (Figure 6E); by contrast, we only detected feeble interaction between truncated SCO1 (aa 119–284) and LKB1 (Figures 6F and 6G). Meanwhile, overexpression of the truncated SCO1 abolished the Cp-silencing-caused activation of AMPK (Figure 6H). We next performed a similar pull-down assay with copper-loaded His-Truncated (Cu-Truncated) or copper-deficient His-Truncated protein (apo-Truncated). An obvious upshift of apo-Truncated band in non-reducing gel electrophoresis confirmed its copper-deficient status, and the pull-down result demonstrated that Cu-Truncated had a stronger binding affinity to AMPK than apo-Truncated (Figure 6I).
Figure 6. Copper-deficient SCO1 is unable to form SCO1-LKB1-AMPK complex and dampens AMPK activation.
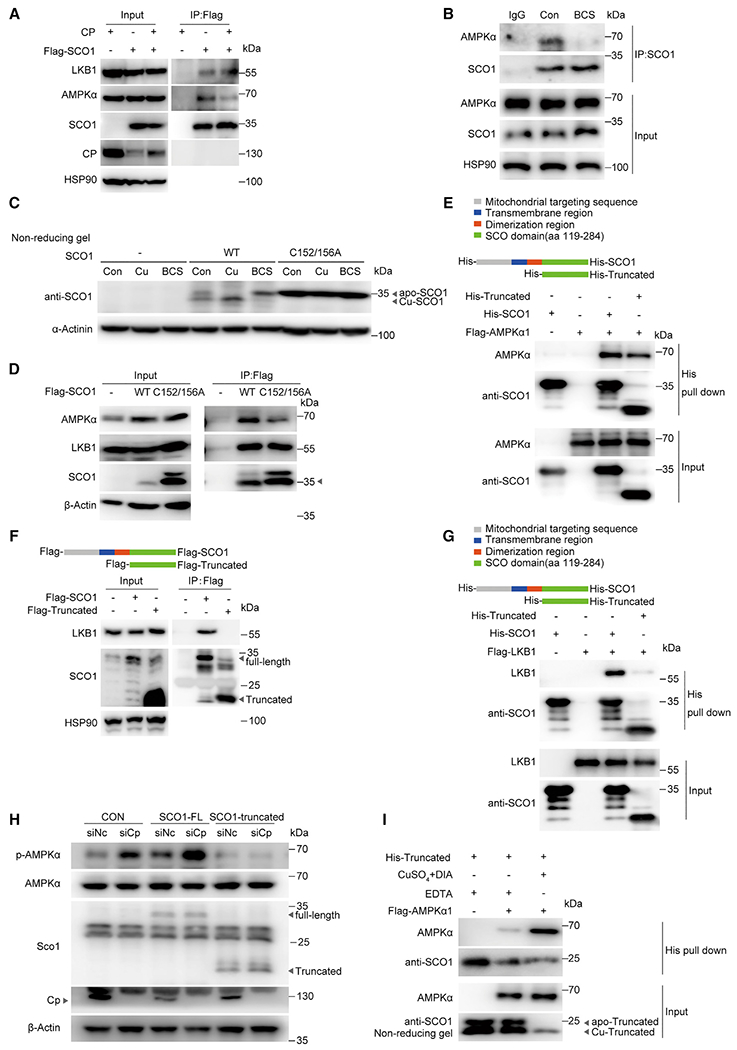
(A) Immunoblotting for LKB1, AMPKα, and Flag-tagged SCO1 proteins after immunoprecipitation of SCO1 from HEK293T cells overexpressing Cp and Flag-tagged SCO1.
(B) Immunoblotting for endogenous SCO1, AMPKα, and LKB1 after immunoprecipitation of SCO1 from L02 cells treated with BCS for 24 h.
(C) Immunoblotting in non-reducing SDS-PAGE for the protein levels of copper-loaded SCO1 (Cu-SCO1) and copper-deficient SCO1 (apo-SCO1) in HEK293T cells transfected with WT or mutant (C152/156A) SCO1, followed by treatment with CuSO4 (50 mM) or BCS (200 mM) for 24 h.
(D) Immunoblotting for AMPKα, LKB1, and Flag-tagged SCO1 proteins after immunoprecipitation of SCO1 from HEK293T cells transfected with indicated vectors; arrow indicates SCO1.
(E) Direct interaction between AMPKα and full-length SCO1 (His-SCO1) or truncated SCO1 (His-Truncated). Pull-down assays were performed with Ni-NTA beads against SCO1, followed by immunoblotting with the antibodies indicated.
(F and G) Immunoprecipitation of FLAG M2 beads against SCO1 (F) or pull-down assays with Ni-NTA beads against SCO1 (G) were performed between LKB1 and full-length SCO1 or truncated SCO1, followed by immunoblotting with the antibodies indicated.
(H) Immunoblotting for p-AMPKα, Sco1, and Cp in primary hepatocytes treated with siRNA targeting Cp combined with overexpression of full-length SCO1 (SCO1-FL) or truncated SCO1 (SCO1-Truncated).
(I) Pull-down assays were performed between AMPKα with truncated SCO1 (His-Truncated) after depleting copper binding to His-truncated with EDTA, or loading copper to His-Truncated with CuSO4. D-isoascorbic acid (DIA) was used to reduce Cu2+ to Cu1+. Immunoblotting in non-reducing SDS-PAGE for the protein levels of copper-loaded His-Truncated (Cu-Truncated) and copper-deficient His-Truncated (apo-Truncated).
Taken together, these results reveal that intracellular copper increase activates AMPK through promoting the assembly of the SCO1-LKB1-AMPK complex.
Adequate hepatic copper levels are necessary for Cp-deletion-induced amelioration of hepatic steatosis
Given that SCO1 deficiency blocked Cp-KO-dependent suppression of lipid accumulation in hepatocytes (Figures 4L and 4M) and depleting intracellular copper by BCS produced copper-deficient Sco1 (Figure 6C, line 6), we thus deprived the copper of Sco1 by treating mice with BCS to explore its effect on lipid metabolism in vivo (Figures 7A and S7A). ICP-MS analysis showed that BCS treatment substantially blunted Cp-LKO-caused elevation of hepatic copper content (Figure 7B). Consistently, BCS administration reduced AMPK phosphorylation levels upregulated by Cp-LKO (Figures 7C and S7B–S7D), presumably due to depletion copper from SCO1. In line with this, BCS treatment also abolished the effects of Cp-LKO on the amelioration of hepatic lipid accumulations (Figures 7D–7F). Moreover, BCS treatment impaired the effects of Cp-LKO on the upregulation of FAO (Figure 7G) and other mitochondrial genes (Figure 7H), as well as maintaining healthy mitochondria morphology in the liver (Figure 7I). There were no significant differences in body weight and food intake between the two groups of mice (Figures S7E and S7F).
Figure 7. Copper chelation aggravates hepatic steatosis.
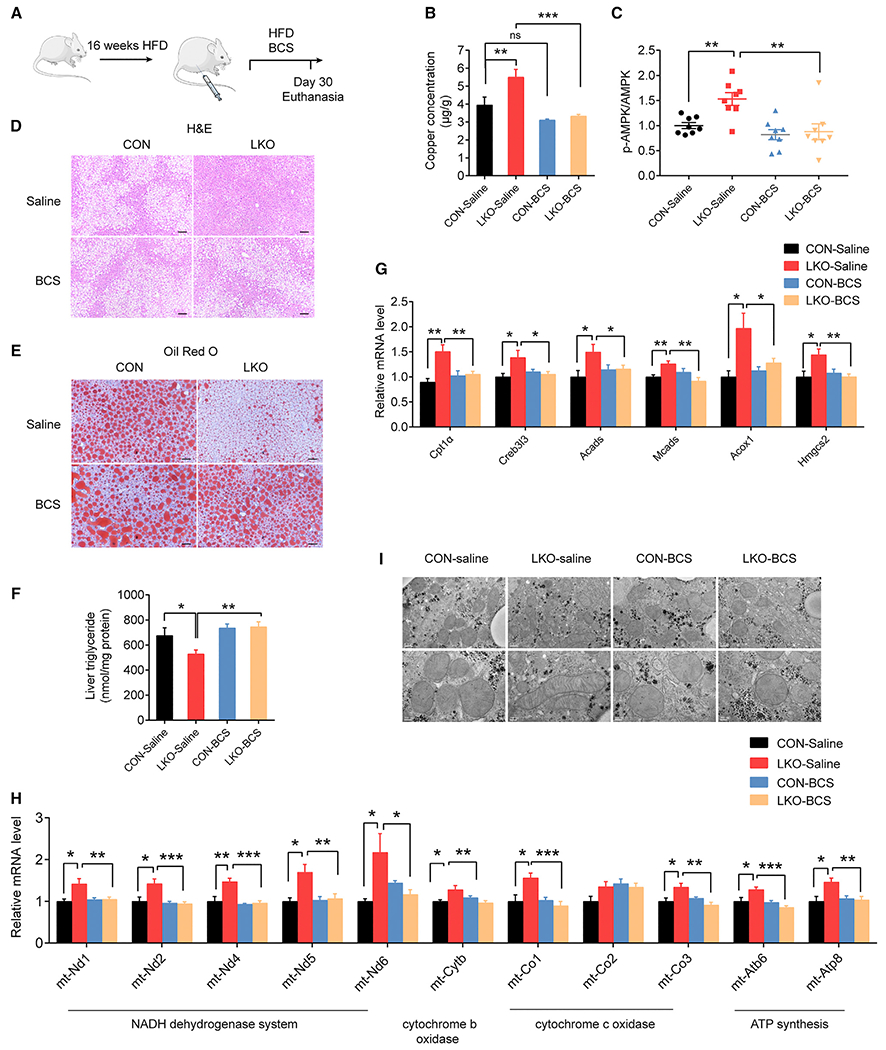
(A) Workflow for administration of BCS in liver-specific Cp knockout and control mice (n = 10 in each group).
(B) Copper concentrations in the livers of LKO and CON mice.
(C) The quantified ratio of signals of p-AMPKα to total AMPKα in mouse livers using western blotting analysis in Figures S7B–S7D.
(D and E) Representative images of H&E (D) and oil red O staining (E) of mouse liver sections (H&E staining scale bar: 100 μm; oil red O staining: 50 μm).
(F) Hepatic TG concentrations.
(G) mRNA levels of FAO genes in mice livers.
(H) mRNA levels of mitochondrial DNA in mouse livers.
(I) Transmission electron microscopy analysis of mitochondria in mice livers; scale bar: 0.5 μm (line 1), 0.2 μm (line 2). Data shown in (G)–(H) are from one representative experiment of at least three independent experiments. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control group. ns, not significant. See also Figure S7 and Table S3.
Moreover, the activity of key regulators in unfold protein response was detected by immunoblot, and it revealed the upregulation of the phosphorylation levels of Ire1α, Eif2α, and Perk and protein levels of Atf4 in the livers of BCS-treated mice (Figures S7G and S7H). These results suggest that BCS treatment induced hepatic ER stress. Notably, BCS treatment had no detectable effect on proliferation of hepatocytes (Figure S7I), nor of causing obvious liver injury (Figure S7J) in the murine models, as evidenced by the Ki67 staining (Figure S7I) and TUNEL staining (Figure S7J), respectively.
Taken together, these findings suggest that copper is required to increase AMPK activation and hepatic lipid catabolism in mouse liver with Cp deletion.
DISCUSSION
Copper is a unique transition metal and is required in a variety of essential biological processes. Conventionally, copper is regarded as a static cofactor within enzyme active sites; however, emerging evidence suggests non-canonical roles for copper. For example, copper serves as a modulator in autophagy signaling pathway (Paris et al., 2009; Tsang et al., 2020) and as an essential cofactor for the copper-dependent transcriptional regulation (Itoh et al., 2008). The total copper content in the adult body is 50–120 mg, and maintaining the amount of copper in the liver and other tissues is key for the homeostasis of biological processes (Ross et al., 2012). Therefore, it is reasonable to postulate that at least one system exists to sense fluctuations in the amount of copper in the liver; however, this system and its downstream biological impact have not been defined.
Here, we demonstrate that AMPK is activated by copper in a dose-dependent manner, and hepatic Cp deletion restores hepatic copper levels and consequently activates AMPK via Sco1. Knockdown of Sco1 entirely blocked the activation of AMPK by Cp deletion. Interestingly, we detected SCO1, but not CP, in AMPK-LKB1 complex, suggesting that CP regulation of AMPK activation is directly mediated through SCO1. SCO1 is known as a copper chaperone for maturation of mitochondrial complex IV, the cytochrome c oxidase complex (Leary et al., 2007). However, knockdown of other copper-binding proteins that are essential for transferring copper to this complex had no effect on the AMPK activation by silencing hepatic Cp, demonstrating a specific effect of SCO1 on AMPK. Impairment of the copper-binding function of SCO1, either by mutation of the copper-binding motif of SCO1 or BCS-mediated intracellular copper depletion, blunted the interaction between SCO1 and AMPK, but it had little effect on SCO1 binding to LKB1. Previous reports show that oxidation of SCO1 on cysteine residues induced by copper loading results in a disulfide band formation that changes the structure and confirmation of SCO1 (Abajian and Rosenzweig, 2006). In addition, the interaction between the truncated SCO1 (only with copper binding domain) and AMPK was comparable to that between full-length SCO1 and AMPK. By contrast, this truncated SCO1 was unable to interact with LKB1. Therefore, copper-mediated conformational changes in SCO1 likely promote the interaction between AMPK and SCO1. LKB1 binds to the region in SCO1 that is distal from the copper-loading domain of SCO1, so the copper-caused conformational alteration in SCO1 has negligible effect on the interaction between SCO1 and LKB1. Thus, these findings show that the SCO1-LKB1-AMPK complex is a copper sensor.
In the canonical mechanism of AMPK activation, AMP binds three of the four cystathionine β-synthetase (CBS) sites of the γ subunit of AMPK trimeric complex and stimulates its kinase activity (Lin and Hardie, 2018). However, we didn’t detected significant alterations in AMP/ATP or ADP/ATP ratio of the copper-treated hepatocytes, although copper treatment substantially activated AMPK. Recent studies revealed that AMPK also responds to glucose starvation via a non-canonical, AXIN-dependent mechanism, in which AXIN acts as an adaptor for LKB1 to interact with and phosphorylate AMPK (Li et al., 2019; Zhang et al., 2013, 2014). Here, we report that SCO1 is a “LKB1 adaptor,” which also promotes the interaction between LKB1 and AMPK, thereby responding to alteration in hepatic copper levels.
The role of copper and copper-containing proteins in regulating lipid metabolism by activating AMPK is an unusual finding. This mechanism may exist because adequate hepatic copper is indispensable in assembling functioning mitochondrial enzymes, such as COX, which support normal mitochondrial function. When the hepatic copper concentration is adequate, the SCO1-LKB1-AMPK complex may be activated, consequently promoting mitochondrial biogenesis and lipid catabolism. In parallel, copper also acts as a signaling modulator and triggers mitochondria biogenesis and lipid utilization via the SCO1-LKB1-AMPK complex. Moreover, our study reveals that 4-h fasting does not change the hepatic copper level, probably because the hepatic copper concentration is strictly controlled within a narrow range, although the precise mechanisms maintaining physiological hepatic copper content remain unclear. Thus, our results imply a role for low hepatic copper content led by high hepatic Cp levels, to be concurrent with the high energy status in NAFLD, and that activation of AMPK by copper activates catabolic processes, including FAO and mitochondrial biogenesis and oxidative phosphorylation. We found that HFD administration substantially increases hepatic Cp expression in mice, which exported copper out of liver and caused a reduction in hepatic copper content in these mice. Consistent with this, it is well documented that overnutrition results in low hepatic copper levels (Aigner et al., 2010; Church et al., 2015; Kennedy et al., 1986; Song et al., 2012; Tallino et al., 2015). Notably, the restoration of copper by Cp ablation restored copper content within the normal range (as Figure 4A shows), since once the level of copper reaches over a certain range in hepatocytes, ATP7B traffics to the canalicular and mediates copper efflux for biliary excretion (Gulec and Collins, 2014). Therefore, different from marked copper accumulation-caused tissue damage like ATP7B and ATP7A deficiency (Członkowska et al., 2018; Kaler, 2011), Cp ablation restored the hepatic copper level without the cytotoxic effects induced by copper overload, suggesting Cp might be a safe target for NAFLD treatment, compared with other copper transporters.
As a ferroxidase, intracellular iron export is impaired in mice and humans with aceruloplasminemia (loss of function of Cp) (Yamamoto et al., 2002). Although circulating Cp is mainly produced and secreted by liver, extrahepatic Cp expression has been verified in a broad arrange of tissues including spleen, lung, testis, and brain (Aldred et al., 1987; Yang et al., 1996), as well as in many cell types, such as macrophages, glial cells, neurons, and Sertoli cells (Fleming and Gitlin, 1990; Klomp et al., 1996; Wright et al., 1981). Consistent with this, we found that global Cp knockout (WKO) leads to excess iron accumulation in the liver, whereas hepatocyte-specific Cp deletion (LKO) only results in a moderate increase in hepatic iron content, which is within the normal physiological range (Church et al., 2015; Song et al., 2012; Wang et al., 2017). Taken together, our data suggest that LKO does not phenocopy mouse models of aceruloplasminemia and Wilson’s disease (Wooton-Kee et al., 2020; Yamamoto et al., 2002).
In conclusion, the SCO1-LKB1-AMPK complex serves as a sensor for alterations in hepatic copper levels. Once copper binds to SCO1 and causes allosteric transformation, copper-SCO1-LKB1 anchors to AMPK and thus activates AMPK, stimulating mitochondrial biogenesis and lipid catabolism. Importantly, our findings reveal that restoration of hepatic copper content and targeting hepatic copper-binding proteins may present promising therapeutic approaches for the treatment of NAFLD.
Limitations of the study
We have reported a function of AMPK in sensing hepatic copper content, but whether AMPK regulates copper homeostasis deserves further investigation. Moreover, our results demonstrate that restoration of hepatic copper triggers the assemble of SCO1-LKB1-AMPK complex that consequently results in enhanced mitochondrial biogenesis and FAO; however, the impact of SCO1-LKB1-AMPK complex on other AMPK regulated processes warrants future investigation. Furthermore, although the effect of copper-SCO1-LKB1-AMPK complex on lipid catabolism is validated in hepatocytes and in vivo, the generality of the findings in other types of cells and extrahepatic tissues needs to be verified in the future. In addition, we reveal that high-fat feeding elevates hepatic Cp levels, but the underlying mechanism remains to be elucidated.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Junli Liu (liujunli@sjtu.edu.cn).
Materials availability
All unique materials and reagents generated in this study are available from the lead contact upon request without restriction.
Data and code availability
The datasets generated by RNA sequencing are available from Mendeley data (https://doi.org/10.17632/g5pxd2dcvm.1). The published paper includes the datasets generated by LC-MS during this study.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
The animal experiments were performed under the guidelines of Provision and General Recommendation of Chinese Experimental Animals Administration Legislation and approved by the Science and Technology Department and with the approval of the Institutional Animal Care and Research Advisory Committee at the Boston Children’s Hospital. The mice were maintained under specific pathogen-free (SPF) conditions in a temperature-controlled environment (25 ± 1°C), on a 12h light/dark cycle, and free access to a standard chow diet and water or special diet as described in the main text and figure legends. Male C57BL/6J mice (5–6 weeks old) and male ob/ob mice (8 weeks old) (SPF grade) were purchased from GemPharmatech Company (Nanjing, China) or the Jackson Laboratory (Bar Harbor, USA).
Cell lines
HEK293T, HEK293A, AML12 cells were purchased from ATCC. L02 cells were purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd. (Shanghai, China). Primary hepatocytes were isolated from mice liver.
All cells were cultured at 37°C with 5% CO2. HEK 293T and 293A cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA), and the L02 cells were maintained in RPMI 1640 medium (Gibco, USA). The AML12 cells were maintained in DMEM/F12 (Gibco, USA) supplemented with 40 ng/mL of dexamethasone (DEX), 5 μg/mL of insulin, 5 μg/mL of transferrin, and 5 ng/mL of selenium (ITS, Sigma-Aldrich, USA; 41400045). The primary hepatocytes were plated on collagen-coated culture dishes with Medium 199 (Sigma) containing 0.3 g/L glutamine for 3–4 h until cells were attached. All media were supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin/streptomycin (Gibco, USA;15140–122).
Human samples
Human liver biopsies were collected from 14 healthy individuals, and 27 NAFLD patients recruited by the Shanghai Jiao Tong University Affiliated Sixth People’s Hospital from 2016 to 2020 (Approval no: ChiCTR2100048122). Informed consent was obtained from each patient, and the clinical data were extracted from the medical records. The gender, age, and steatosis level (%) of each individual are provided in Table S1.
METHOD DETAILS
Mouse models
Cpflox/flox mice and Cp-WKO mice were generated by the Institute of Laboratory Animals Science (CAMS & PUMC) using the CRISPR–Cas9 system. For the Cpflox/flox mice, Cas9 mRNA, single guide RNAs (sgRNAs: forward sequence: 5′-TAGGAGTCTAACAGACCACGGG-3′; reverse sequence: 5′-AAACCCCGTGGTCTGTTAGACT-3′), and donor were co-injected into the zygotes. The 2nd exon of Cp was flanked with the loxP sequence. Cp-WKO mice were generated by deletion of a specific sequence from the 2nd exon of the Cp gene. Liver-specific Cp knockout (Cp-LKO) mice were generated by crossing the Cpflox/flox mice with albumin-Cre mice (The Jackson Laboratory). The five-week-old male C57BL/6J mice, Cp LKO mice, and their control littermates were fed a high-fat diet (HFD) (60% fat from calories, Research Diet) for 12–16 weeks to induce NAFLD before subsequent treatment (indicated in figure legends). Livers were collected after mice were euthanized and frozen in liquid nitrogen for analysis. DIO mice were administered AdshCp and control Ad-sh Lacz via tail vein injection at a dose of 1 × 109 plaque-forming unit (pfu) per mouse for three weeks for adenovirus-mediated silencing of the target genes. The primer sequences for the Ad-shCp were as follows: forward primer sequence: 5′-CACCGCAACAGGATGTACTCTATAACGAATTATAGAGTACATCCTGTTGC-3′; reverse primer sequence: 5′AAAAGCAACAGGATGTACTCTATAATTCGTTATAGAGTACATCCTGTTGC-3′). For AAV-mediated liver-specific silencing of the target genes, Cpflox/flox mice were fed an HFD for eight weeks and then administered AAV2/9-TBG-GFP and AAV2/9-TBG-Cre-GFP via tail vein injection at a dose of 1 × 1012 vector genomes (vg) per mouse for additional 12 weeks. The protocol for STZ induction of type 2 diabetes mice is as follows: C57BL/6J mice were fed an HFD diet for 12 weeks. Streptozotocin 40 mg/kg STZ (Sigma-Aldrich, 18883–66–4) was injected intraperitoneally for three days daily. Hepatic AMPKα1 and β2 double knockout mice (AMPKα-DKO) and AMPKα1flox/flox and α2flox/flox (AMPKαf/f) mice were kindly provided by Dr. Jingya Li (Chinese Academy of Sciences). To deplete copper from the mice, the Cpflox/flox (CON, control) mice and Cp-LKO mice were intraperitoneally injected with bathocuproinedisulfonic acid, BCS (20 mg/kg) (Sigma-Aldrich, B1125) daily for one month as previously described (Ruiz et al., 2014). Primary hepatocytes were isolated from 6-to 10-week-old male Cp-LKO mice and control littermates (CON). The mice were housed individually one day before sacrifice for accurate and consistent measurement of the p-AMPKα levels from the liver. After anesthesia, the liver lobes were clamped tightly using a liquid nitrogen-cooled clamp and were flash-frozen immediately in liquid nitrogen (Zhang et al., 2018).
Cell treatment
The primary hepatocytes were isolated as described previously (Sun et al., 2020). In brief, an infusion needle was inserted into the vena cava after the mice were anesthetized and then the liver was infused continuously with solutions containing collagenase Type I (0.02 mg/mouse). After the perfusion, the digested liver was disaggregated with forceps and passed through a 100-μm filter and centrifuged at 400 × g for 3 min. After removal of the supernatant fraction, the hepatocytes were centrifuged with 45% percoll (Biodee, DE-17-0891-02G) at 700 × g for 10 min at 4°C. Next, the hepatocytes were resuspended and plated on collagen-coated culture dishes with Medium 199 (Sigma) containing 0.3 g/L glutamine for 3–4 h until cells were attached. All media were supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin/streptomycin (Gibco, USA;15140–122). For the compound treatment experiment, the hepatocytes were treated with DMSO or SP (5 μM, Cayman Chemical Company, USA; 10010466), H-89 dihydrochloride (10 μM, MedChemExpress Life Science Reagents, USA; HY-15979), Ex527 (5 μM, Cayman Chemical Company, USA; 10009798), CC (10 μM, MedChemExpress Life Science Reagents, USA; HY-13418A), GW9578 (10 μM, Cayman Chemical Company, USA; 10011211), GW6471 (10 μM, Cayman Chemical Company, USA; 11697), BCS (100 μM, 200 μM, Sigma-Aldrich, USA; B1125), TM (toluene, 10 μM, Sigma-Aldrich, USA; 323446), DFX (Deferasirox, 20 μM, 50 μM, 100 μM, Selleck Chemicals, USA; S1712), PIH (Pyridoxal isonicotinoyl hydrazone, 50 μμ, Cayman Chemical Company, USA; 19,357), DFO (100 μM, Selleck Chemicals, USA; S5742) for 24–36 h. The BSA-conjugated PA (250 μM, Sigma-Aldrich, USA; P9767) was incubated for 24 h. For the CuSO4 treatment, the hepatocytes were treated with the vehicle or CuSO4 (40 nM, 80 nM, 160 nM, 320 nM, and 640 nM) for 4 h in a serum-free medium.
Liver tissue lipidomic profiling by LC-MS
The LC-MS analysis of the hepatic lipids was conducted at the Dalian Institute of Chemical Physics, Chinese Academy of Sciences. The extraction solvents for weighed liver tissues from Ad-shCon and Ad-shCp were prepared by supplementing HPLC-grade methanol with various lipids and free fatty acid (FFA) standards. These included ceramide CER (d18:1/17:0), lysophosphatidylcholine LPC (19:0), phosphatidylcholine, PC (19:0/19:0), phosphatidylethanolamine, PE (17:0/17:0), sphingomyelin, SM (d18:1/12:0), triacylglycerol, TG (15:0/15:0/15:0), d3-hexadecanoic acid (C16:0), d3-octadecanoic acid (C18:0). Liquid-liquid MTBE (Sigma) extraction was conducted as described previously with slight modifications (Chen et al., 2013; Matyash et al., 2008). Global lipidomic profiling was performed on the ACQUITY™ Ultra Performance Liquid Chromatography (UPLC) system (Waters, Milford, MA, USA) coupled with the AB SCIEX triple TOF 5600 plus mass spectrometer (Applied Biosystems SCIEX, Foster City, CA, USA) as described before (Xuan et al., 2018). Parameters for MS analysis were established as follows: ion spray voltage, 5500 V(+) and 4500 V(−); declustering potential, 100 V (+) and – 100 V(−); collision energy, 10 V(+) and – 10 V (−); and interface heater temperature of 550°C. The pressure of curtain gas, GS1, and GS2 was maintained at 35, 55, and 55 psi, respectively. After data processing, the lipids were assigned based on their distinct patterns of MS/MS fragmentation, accurate molecular weight, and retention times. The number of lipids was normalized by the peak area of the internal standard and the weight of the tissue samples. The fold change and statistical differences (p-value) between the two groups are depicted in Table S2.
RNA sequencing
Total RNA was extracted from the livers of mice fed the HFD and injected with Ad-shCon or Ad-shCp (n = 3). The quality and purity were examined by NanoDrop analysis (OD260/280 = 1.8–2.0, OD230/260 = 0.4–0.5). RNA sequencing libraries were prepared from 4 mg of total RNA using the mRNA-seq Library Prep Kit v2 from Illumina Vazyme Biotech, China) according to the manufacturer’s instructions. The quality of the sequencing libraries was controlled by the Agilent 2100 Bioanalyzer Instrument (Agilent Technologies, CA, USA) and sequenced in a 350 bp paired-end run on an Illumina HiSeq PE150 Sequencing System. The PhiX (1%) control library (Illumina) was spiked into each lane as the sequencing control.
Inductive coupled mass spectrometry (ICP-MS)
Tissues from humans or mice were excised and dissected (30–150 mg) in liquid nitrogen to avoid freeze-thawing. Tissues and cells were digested in a mixture of nitric acid and hydrogen peroxide. All samples were incubated for 15 min at 25°C and then heated at 120°C for a few hours for complete dissolution. The samples were further diluted using ultrapure water (18.2 MΩ cm−1). The amount of iron and copper was analyzed using an iCAP Q ICP-MS instrument (Thermo Fisher Scientific, Bremen, Germany). The quantity was normalized to μg/g after dividing by the corresponding liver weight.
Mitotracker staining
The culture media of the primary hepatocytes were removed from the dish, and a prewarmed (37°C) staining solution containing 100 nM of the MitoTracker® probe (Thermo Fisher, M7512) was added to the cells. After incubation for 15–45 min undergrowth conditions and washing twice with PBS, the fluorescence from the mitochondria was observed with a confocal laser scanning microscopy (ZEISS) at the excitation wavelength of 599 nm.
Seahorse experiment
Cellular OCR (Oxygen consumption rate) was measured with an Agilent Seahorse XF96 Analyzer (Seahorse Bioscience). Primary hepatocytes were isolated from 6-to 10-week-old male Cp-LKO mice and their control littermates. The hepatocytes were seeded in 80 μL medium in 96-well XF Cell Culture Microplates (Agilent) at a cell count of 7000 cells/well for 24 h. They were incubated with 200 μL/well of the assay medium (XF base medium with 25 mM glucose, 1 mM sodium pyruvate, and 2 mM L-glutamine) in a non-CO2 incubator for 60 min at 37°C. The Sensor Cartridge (Agilent) was loaded sequentially with the following compounds for the mitochondrial stress test: the ATP synthase inhibitor oligomycin (3 mM), mitochondrial uncoupler carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 2 mM), and electron transport inhibitors rotenone and antimycin A (1 mM each). The hepatocytes were pre-incubated with the AMPK inhibitor compound C (10 μM) for 24 h (Figures 3 and S3). Basal respiration, ATP production, maximal respiration, and the spare respiratory capacity were measured. The maximal respiratory rate was calculated as the highest OCR after adding FCCP, and the spare respiratory capacity was defined as the difference between the maximal and basal respiration.
Triglyceride assay
Triglyceride levels in the livers or in cultured hepatocytes were measured with a commercially available kit (Applygene, E1013–105) following the manufacturer’s instructions. Briefly, 50 mg of liver tissues or cultured hepatocytes were homogenized in 1 mL of the lysis buffer and incubated on ice for 10 min. Subsequently, the supernatant was heated at 70°C for 10 minutes and centrifuged at 2,000 rpm for 5 min at room temperature. The triglyceride levels in the supernatant were detected with the SpectraMax i3 Multi-Mode Detection Platform (Molecular Devices, USA) at 550 nm. The results were normalized to the protein concentrations measured by the BCA reagent (Thermo Fisher, USA; 23225).
Luciferase reporter assay
Luciferase reporter assays were performed as previously reported (Xu et al., 2020). AML12 cells were transfected with 50–200 ng/well of the plasmids encoding the firefly luciferase reporter (PPARE-Luc, HMGCS2-Luc, CPT1α-Luc) with Lipofectamine 2000. The Renilla plasmid (50–200 ng/well) was the internal control for evaluating the transfection efficiency. After 24 h of transfection, the cells were transduced with adenoviruses (Ad-shCon, Ad-shCp, Ad-GFP, and Ad-Cp), with or without GW9578 (10 μM, Cayman Chemical Company, USA; 10011211) for additional 24 h. The cellular luciferase activity was measured using a commercial Dual-Luciferase Assay Kit (Promega, E1960). The firefly luciferase activity was normalized to the Renilla activity.
Plasmid construction
FLAG-AMPKα1 and FLAG-LKB1 plasmids were kindly provided by Prof. Shengcai Lin (Xiamen University). The cDNA sequence of AMPKα1 was cloned into pcDNA.3.1/myc-His A vector. The full-length coding regions of the murine Sco1 and SCO domain (Truncated SCO1, aa 119–284) were subcloned into the pET28a vector, and the pHAGE-fEF1a-IRES-ZsGreen vector. Meanwhile, the SCO1 copper-binding-inactive mutant (C152/156A) was also constructed by mutating Cys152 and Cys156 to alanine using the KOD-Plus-Mutagenesis Kit (Toyobo Co., Ltd., Japan; SMK-101). The PPARE-Luc construct was created by annealing two complementary DNAs containing three repeats of the canonical PPAR responsive elements followed by ligation into a pGL3-Basic vector (Agilent, USA). The CPT1α and HMGCS2-Luc constructs were produced by inserting ~1 kb promoter regions of human CPT1α or HMGCS2-containing PPAR responsive elements into the pGL3-Basic vector.
Purification of the SCO1 and truncated SCO1 proteins
BL21 bacteria were transformed with pET28a-SCO1 or pET28a truncated-SCO1 vectors and were shaken at 37°C at 220 rpm until the OD600 value reached 0.6–0.8. Then, 0.3 mM of IPTG (Yeasen Biotechnology Co., Ltd., China; 10902ES08) was added, and the bacteria were allowed to grow for an additional 4 h at 37°C. Full-length SCO1 was present in the inclusion bodies and was purified as following. (1) The cells were resuspended with lysis buffer (50 mM Tris, 5 mM Imidazole, 500 mM NaCl, pH = 8.0) and were sonicated for 30 min (10 sON, 10 s OFF) with 300 W. The supernatant was removed by centrifugation. The precipitate was sequentially washed in turn with buffer A (50 mM KH2PO4, 5 mM EDTA, 300 mM KCl, pH = 8.0), buffer B (50 mM KH2PO4,5 mM EDTA,300 mM KCl,2 M Urea, pH = 8.0), and buffer C (50 mM KH2PO4,5 mM EDTA,300 mM KCl,0.1% DOC). Each step was repeated thrice. (2) The precipitated pellets were resuspended in the binding buffer (20 mM Tris, 8 M Urea, 150 mM NaCl, 10 mM Imidazole, pH = 8.0) and centrifuged at 1,2000 rpm. Further, the supernatant was incubated overnight with 1 mL of Ni-NTA Agarose beads (Qiagen, Germany; 30210) at 4°C. The beads were pelleted down and washed cautiously. The binding proteins were eluted using the elution buffer (50 mM Tris, 500 mM NaCl, 250 mM Imidazole, pH = 8.0).
The truncated SCO1 was soluble, so it was purified as follows: The cells were sonicated for 30 min (10 s ON, 10 s OFF) with 300 W. The supernatant was incubated overnight with 1 mL of Ni-NTA Agarose beads (Qiagen, Germany; #30210) at 4°C. The remaining steps were the same as that used for full-length SCO1. The copper-loaded truncated SCO1 (Cu-Truncated SCO1) was purified by adding 1 mM CuSO4 and 3 mM D-isoascorbic acid (Sigma-Aldrich, USA; 856061) to the LB medium of the bacteria transformed with the pET28a-truncated-SCO1 vector. For purification of the apo-Truncated SCO1 protein, the LB medium was supplemented with 10 mM EDTA.
Purification of the AMPK and LKB1 proteins
HEK 293T cells were transfected with the FLAG-AMPKα1 and FLAG-LKB1 vectors and lysed using the IP lysis buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, pH = 7.4). After centrifugation for 30 min with 15,000 rpm at 4°C, the supernatant was transferred to a new 1.5-mL centrifuge tube and incubated with the anti-FLAG affinity gel (Abmart, China, 324085) at 4°C for 6 h or overnight. The affinity gel was pelleted down and then washed thrice with the IP lysis buffer. The protein was eluted with 200 ng/mL of the 3X FLAG peptide solution. The protein concentration in the elution buffer was measured with the BCA kit.
In vitro pull-down assay
For this assay, 2 μg of 6X His-SCO1 or truncated SCO1 was incubated with 1 μg of FLAG-AMPKα1 in the binding buffer (20 mM Tris, 1 mM EDTA, 100 mM NaCl, 1 mM DTT, 10 mM MgCl2, and 1% Triton X-100, pH = 7.4) at 4°C for 4 h. Next, 50 μL of the Ni-NTA Agarose beads or the anti-FLAG affinity gel was added to the mixture and incubated at 4°C for 3 h. The beads were pelleted down, washed thrice with the IP lysis buffer, and boiled with 2X SDS-PAGE loading buffer. The bands were resolved by SDS-PAGE. For the pull-down assay of the full-length SCO1, 15–25 μL of SCO1 in Ni-NTA Agarose beads or AMPKα1 in anti-FLAG affinity gel suspension solution were mixed with the other protein in the binding buffer and incubated overnight at 4°C (Figures 5F–5I, 6E, and 6G). The beads were pelleted down, washed thrice with the IP lysis buffer, and boiled with 2X SDS-PAGE loading buffer. The bands were resolved by SDS-PAGE.
Immunoprecipitation (IP)
The cells were lysed with the IP lysis buffer. The supernatant was incubated overnight with the primary antibodies at 4°C, followed by continuous incubation with protein A/G Sepharose beads (Beyotime Biotechnology, China; P2055) for 3 h. Next, the beads were pelleted down, washed thrice with the IP lysis buffer, and boiled in 2 X SDS-PAGE loading buffer. The supernatant was harvested for Western blotting.
For the FLAG-tagged protein IP assay, the supernatant was incubated overnight with the anti-FLAG affinity gel at 4°C.
Immunofluorescent staining
The primary hepatocytes were fixed with 10% formalin. After washing with PBS and perforating with 0.5% Triton in PBS for 15 min, the cells were blocked with 5% BSA/PBS and then incubated with anti-SCO1 (Proteintech, USA; 12614–1-AP; 1:200 dilution) and anti-β-Actin (Earthox, China; E021090-02; 1:200 dilution) antibodies for 12 h at 4°C. After washing with PBS, the cells were incubated with Alexa 488 and Cy3-conjugated secondary antibodies (1:200 dilution) for 1 h. The nuclei were stained with DAPI (4’,6-diamidino-2-phenylindole, 300 nM) for 5 min. After washing with PBS, the cells were mounted and observed by a confocal laser scanning microscope (ZEISS, Germany).
Subcellular fractionation and co-immunoprecipitation
Mitochondria were isolated from primary hepatocytes with slight modifications (Yoo et al., 2020). Primary hepatocytes were rapidly washed twice with KPBS (136 mM KCl, 10 mM KH2PO4, pH = 7.2) and then gently scraped with 1 mL KPBS plus protease inhibitors. Cells were carefully homogenized with 50 strokes of a 2 mL Dounce tissue homogenizerto prevent air bubble formation. The homogenate was centrifuged at 700 g for 7 min, and the pellet was discarded. The supernatant was centrifuged at 8,000 g for 10 min at 4°C, and the supernatant was discarded. Next, the pellet was washed with KPBS for five times and then lysed with the IP lysis buffer. Finally, the supernatant was co-immunoprecipitated with the SCO1 antibody.
Isolation of cytosolic proteins from the primary hepatocytes was performed using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific, USA; 78833) according to the manufacturer’s protocol. This was followed by co-immunoprecipitation with the SCO1 antibody.
Chromatin immunoprecipitation (ChIP)
The ChIP assays were performed according to previous studies with minor modifications (Hu et al., 2017; Miao et al., 2006). Briefly, the primary hepatocytes transduced with Ad-shCp or Ad-shCon were cross-linked with 1% formaldehyde for 15 min, and 125 mM glycine was added for 5 min to quench the reaction. The cells were lysed with the lysis buffer (1% SDS, 10 mM EDTA, and 50 mM Tris-HCl, pH = 8.0) and sonicated eight times on ice (15 s ON, 90 s OFF). The supernatant was incubated overnight with the PGC1α (Abcam, UK: ab54481) or PPARα (Abcam, UK; ab24509) antibodies. After precipitation with protein A/G Sepharose beads, the fragmented DNA was pulled down and subjected to qPCR analysis. The sequences of the primers are listed in Table S3.
Transmission electron microscopy
The electron microscopy was conducted at the Shanghai Jiao Tong University Instrumental Analysis Center. Briefly, the liver tissues from Cp-LKO and littermate controls were excised into pieces of 1 mm3 and fixed overnight in 2.5% glutaraldehyde at room temperature. The samples were post-fixed in potassium ferrocyanide-reduced osmium tetroxide (Electron Microscopy Sciences, USA) for 1.5 h and dehydrated in a bath with gradually increasing concentrations of ethanol (50%, 70%, 90%). The ethanol was then eliminated with gradually increasing acetone concentrations (90% ethanol: 90% acetone = 1:1, 90% acetone, 100% acetone). Further, the tissues were incubated with a mixture of acetone: EPON (liquid epoxy resin) (1:1,1:2, and 1:3) and ultimately embedded with pure EPON (Electron Microscopy Sciences, USA). The picture was captured using a biological transmission electron microscope (Tecnai G2 Spirit BioTWIN, Thermo Fisher Scientific, USA). The liver tissues from the BCS-treated Cp-LKO mice and the control group were analyzed following the same procedures.
Fatty acid oxidation assay
The complete fatty acid oxidation rate was measured as previously reported (Haas et al., 2012). Briefly, freshly separated primary hepatocytes were cultured in DMEM-high glucose (25 mM) + 10% FBS, transduced with adenovirus for 24 h, and then incubated overnight in DMEM-low glucose (5 mM) media. Cells were then incubated with 0.1 mM palmitate (Sigma, USA; P9767) conjugated with 0.3% (w/v) fatty acid-free bovine serum albumin (BSA, Sigma, USA; A8806), and 0.4 μCi/mL [1–14C]-palmitate (Perkin Elmer,). After 4 h, the medium was transferred to a CO2 trap, and CO2 was absorbed in 1 M NaOH in the adjacent wells. The captured CO2 (complete oxidation) was subjected to scintillation counting.
Blood chemistry
Free fatty acids were detected with the LabAssay NEFA kit (FUJIFILM Wako Chemicals USA Corp., USA; 294–63601), following the manufacturer’s instructions. The serum levels of ALT and AST were detected following the manufacturer’s instructions (NJJCBIO: C009–2–1 for ALT, C010–2–1 for AST).
Histological staining
Liver tissues were fixed in 4% paraformaldehyde (PFA), embedded in paraffin (SAV LP), and cut into 5 mm slices for hematoxylin and eosin (H&E) staining, Prussian blue staining, TUNEL assay, or immunohistochemical staining of Ndufb8, Hmgcs2 and Ki67.
The terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining assay was performed using the DeadEnd™ Fluorometric TUNEL System (Promega Corporation, USA; G3250) following the manufacturer’s instructions.
For Ki67, Ndufb8 and Hmgcs2 staining, the sections were deparaffinized, and antigen retrieval was performed; that is, they were treated with 10 mmol/L citrate buffer (pH = 6.0) with microwave heating (2 min at 700 W, repeated thrice). Next, the sections were blocked with 5% BSA/PBS for 30 min at room temperature and incubated overnight with antibodies against Ki67 (Abcam, UK; ab16667; 1:100 dilution), NDUFB8 (Abcam, UK; ab192878; 1:500 dilution), or HMGCS2 (Abcam, UK; ab137043; 1:500 dilution) in 1% BSA/PBS at 4°C. The next day, the sections were incubated with a biotinylated anti-rabbit IgG secondary antibody (1:200 dilution) for 1 h at room temperature, followed by treatment with the VECTASTAIN Elite ABC HRP Kit reagents (Vector Laboratories Inc., USA; PK-6100) for 1 h. Finally, the sections were processed with a DAB peroxidase substrate kit (Vector Laboratories Inc., USA; SK-4100), followed by hematoxylin staining.
For Oil Red O staining, the tissues were placed in a 15% sucrose solution at 4°C for dehydration and then transferred to a 30% sucrose solution for dewatering at 4°C. Finally, the tissues were embedded in OCT (optimal cutting temperature) compound and stored at −20°C. Subsequently, the embedded tissues were excised into 8 μm slices, and Oil Red O staining was performed according to the manufacturer’s instructions. For hepatocyte Oil Red O staining, the primary hepatocytes were fixed in 4% paraformaldehyde for 30 min, and 0.5% of the Oil Red O stock was mixed with water (v/v = 3:2) as the working solution. The fixed cells were washed with PBS, incubated with the working solution for 1 h at room temperature, and washed thrice with PBS. The picture was captured by an inverted microscope (Olympus LS, Tokyo, Japan).
Quantitative PCR (qPCR)
Total RNA was extracted from hepatocytes or liver tissues using the TRIzol reagent (Life Technologies, USA; 15596). RNA was reverse-transcribed to cDNA using the Hiscript II Q RT SuperMix (Nanjing Vazyme Biotech Co, China; R222–01). qPCR was then performed with the AceQ Universal SYBR qPCR Master Mix (Nanjing Vazyme Biotech Co, China; Q511–02) on a Roche qPCR instrument (Roche Life Science, Switzerland). To measure mtDNA/nDNA, DNA was extracted from hepatocytes using the QIAquick PCR Purification kit according to the manufacturer’s instructions (Qiagen, USA; 28106). The expression of mRNA was normalized to that of 36B4 or 18S reference genes. The sequences of the primers are listed in Table S3.
Glucose tolerance and insulin tolerance tests
Glucose tolerance and insulin tolerance tests were performed as described previously (Sun et al., 2020). For glucose tolerance tests, the mice were intraperitoneally (i.p.) injected with glucose (1.0 g/kg for DIO mice) after 16 h of fasting. Glucose concentrations in the blood were monitored at the indicated times using a glucometer (Roche Life Science, Switzerland; Accu-chek). For insulin tolerance tests, insulin (Eli Lilly and Company, USA; 1.3 U/kg for DIO mice) was intraperitoneally injected into the mice after 6 h of fasting. Glucose concentrations in the blood were measured at the indicated times after the injection.
Micro-computed tomography (micro-CT) analysis
CT imaging was performed on an Inveon MM Platform (Siemens Preclinical Solutions, Knoxville, USA) with a computer-controlled bed and 8.5 cm transaxial and 5.7 cm axial fields of view (FOV). During the scanning process, the mice were anesthetized with isoflurane and placed prone on the CT scanner bed near the central field of view. The Inveon Acquisition Workplace (IAW) 1.5 was used, and thirty min 2-bed CT was scanned with a power of 80 kV and 500 μA and an exposure time of 1100 ms. The CT data were reconstructed in real-time by the COBRA_Exxim Cone Beam Reconstruction Package. The 3-D regions of interest (ROIs) were drawn guided by CT thresholds and technician experience, and the image HU (Hounsfield unit) values and volumes were measured using the Inveon Research Workplace (IRW) 4.2 software. Individual quantification of the ROI in each of them was calculated. The density value of the ROI was determined by a standard curve of the phantom.
Small interfering RNA transfection
Scrambled siRNA control or gene-targeting siRNAs (GenePharma, China) were transfected into the cells with Lipofectamine RNAiMAX Transfection reagent (Invitrogen, USA; 13778075) following the manufacturer’s instructions. The sequences of siRNAs are listed in Table S4.
Immunoblotting
Liver tissue or the hepatocytes lysates were generated as described previously (Zhang et al., 2018). After separating the proteins from the SDS-PAGE gel, they were transferred onto a polyvinylidene fluoride (PVDF) membrane (pore size: 0.22 mm, Millipore, USA). The membranes were incubated overnight with primary antibodies at 4°C, followed by incubation with the secondary antibodies for 1 h at room temperature. The antibody of SCO1 (Santa Cruz Biotech., USA; sc-398001) detects the specific epitope located in the SCO domain of SCO1. For the non-reducing SDS-PAGE, the cell lysates were boiled with a non-reduced loading buffer (Cwbio IT Group, China; CW0028S), and the immunoblots were performed as described above (Brady et al., 2010). The ImageJ software (NIH) was used to quantify the relative intensities of the bands.
ADP, AMP, and ATP analysis
ATP, ADP and AMP were analyzed by CE (G7100A, Agilent) couple to time of flight (TOF) mass spectrometry (G6224A, Agilent) as previously described (Yan et al., 2019). Briefly, a 10-cm dish of primary hepatocytes (80–90% confluence) was prepared for each treatment. The cells were rinsed with 25 mL mannitol solution (5% dissolved in water, Sigma, USA; M4125) and instantly frozen in liquid nitrogen. Then, 1 mL of methanol coupled with the Standard 1 (IS1, Human Metabolome Technologies, HMT, H3304-1002, 50 μM, 1:200), was added to the dish, followed by a quick scraping of the cells from the dish. The tissue was excised by freeze-clamping, then ground in 1 mL of methanol with 50 μM IS1. The lysate from cells or tissues were then mixed with 1 mL of chloroform and 400 μL of water by 20 sec of vortexing. After centrifugation at 15,000 g for 15 min at 4°C, 420 μL of the aqueous phase was collected. Aqueous phases from cells or tissues were then ultra-filtrated with a 5-kDa cut-off filter (Millipore, USA; UFC3LCCNB-HMT) by centrifuging at 12,000 g for 3 h at 4°C. The filtered samples were then freeze-dried in a vacuum concentrator (labconco, CentriVap Bench-top centrifugal vacuum concentrator, free zone-2.5L) at −50°C and dissolved in 100 μL of water coupled with Internal Standard 2 (IS2, Human Metabolome Technologies, Japan; H3304–1104; 1:200 dilution). A total of 20 μL of redissolved solution was then loaded into an injection vial with a conical insert for CE-QTOF MS analysis. The fused silica capillary (50 μm i.d. × 80 cm, HMT, Tsuruoka, Japan) was used for sample separation. Detailed CE-MS methods were described before (Ma et al., 2022). The qualitative analysis was proceeded based on the pre-analyzed metabolite standard library (HMT), and identification for AMP, ADP and ATP at m/z 346.0558, m/z 426.0221, and m/z 505.9885, respectively. Peak extraction and identification were carried out with Quantitative Analysis Software (Agilent). Note that a portion of ADP and ATP could lose one phosphate group during in-source fragmentation, thus leaving the same m/z ratios as AMP and ADP, and should be corrected according to their different retention times in the capillary. Therefore, the total amount of ADP is the sum of the latter peak of the m/z 346.0558 spectrogramme and the former peak of the m/z 426.0221 spectrogramme, and the same is applied for ATP.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were presented as the mean ± SEM. The Student’s t-test, Wilcoxon signed-rank test, and one-way or two-way analysis of variance (ANOVA) were employed to compare the means of the continuous variables. Statistical analyses were conducted with SPSS version 25.0 (IBM, Armonk, New York, USA) or GraphPad Prism version 9.0 (GraphPad Software Inc., San Diego, CA, USA). Before analysis of the significance, the Sharpiro-Wilk test and Kolmogorov-Smirnov test were applied to analyze the normality of the data. Two-tailed, unpaired Student’s t-test was employed to analyze data that conformed to the normal distribution. At the same time, the Wilcoxon Rank-Sum test was used to analyze data with the abnormal distribution. A single symbol represents a p-value <0.05, two symbols represent a p-value <0.01, and three symbols denote the p-value of <0.001. Experiments determining the blood glucose, food intake, and body weight of mice were performed blind. Histological analysis was also performed and analyzed in a blinded manner. The sample size in each experiment was determined based on previous studies and preliminary results (Chen et al., 2021; Gao et al., 2019). No data points were excluded from the study.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-PGC1α antibody, WB, dil:1/1,000 | Abcam | Cat#ab54481; RRID: AB_881987 |
| Rabbit monoclonal anti-Phospho-AMPKα (Thr172) (40H9) antibody, WB, dil:1/1,000 | Cell Signaling Technology | Cat#2535; RRID: AB_331250 |
| Rabbit polyclonal anti- PPARα antibody, WB, dil:1/1,000 | Abcam | Cat#ab24509; RRID: AB_448110 |
| Rabbit monoclonal anti-AMPKα (D5A2) antibody, WB, dil:1/1,000 | Cell Signaling Technology | Cat#5831; RRID: AB_10622186 |
| Rabbit monoclonal anti-Acetyl-CoA Carboxylase (C83B10) antibody, WB, dil:1/1,000 | Cell Signaling Technology | Cat#3676; RRID: AB_2219397 |
| Rabbit monoclonal anti-Phospho-Acetyl-CoA Carboxylase (Ser79) (D7D11) antibody, WB, dil:1/1,000 | Cell Signaling Technology | Cat#11818; RRID: AB_2687505 |
| Rabbit monoclonal anti-Total OXPHOS antibody, WB, dil:1/500 | Abcam | Cat#ab110413; RRID: AB_2629281 |
| Rabbit monoclonal anti-β-Actin (D6A8) antibody, WB, dil:1/3,000 | Cell Signaling Technology | Cat#8457; RRID: AB_10950489 |
| Rabbit monoclonal anti-GAPDH (D16H11) antibody, WB, dil:1/3,000 | Cell Signaling Technology | Cat#5174; RRID: AB_10622025 |
| Rabbit polyclonal anti-Hsp90 antibody, WB, dil:1/2,000 | Cell Signaling Technology | Cat#4874; RRID: AB_2121214 |
| Rabbit monoclonal anti-α-Actinin (D6F6) antibody, WB, dil:1/2,000 | Cell Signaling Technology | Cat# 6487; RRID: AB_11179206 |
| Rabbit monoclonal anti-β-Tubulin (9F3) antibody, WB, dil:1/2,000 | Cell Signaling Technology | Cat#2128; RRID: AB_823664 |
| Rabbit polyclonal anti-Ceruloplasmin antibody, WB, dil:1/1,000 | Thermo Fisher Scientific | Cat#PA5-14225; RRID: AB_2085718 |
| Mouse monoclonal anti-SCO1 (D-1) antibody, WB, dil:1/500; IP, dil:1/100 | Santa Cruz Biotechnology | Cat#sc-398001; |
| Rabbit polyclonal anti-SCO1 antibody, WB, dil:1/1,000; IF, dil:1/200 | Proteintech | Cat#12614-1-AP; RRID: AB_2184641 |
| Mouse anti-β-Actin antibody, IF, dil:1/200 | Earthox | Cat#E021090-02; |
| Alexa Fluor 488 Secondary Antibody, IF, dil:1/200 | Thermo Fisher Scientific | Cat#A32731; RRID: AB_2633280 |
| CoraLite®488-Conjugated AMPK Alpha Monoclonal Antibody | Proteintech | Cat# CL488-66536; RRID: AB_2883352 |
| Rabbit monoclonal anti-LKB1(D60C5) antibody, WB, dil:1/1,000 | Cell Signaling Technology | Cat#3047; RRID: AB_2198327 |
| Rabbit polyclonal anti-acetylated-Lysine antibody, WB, dil: 1/1,000 | Cell Signaling Technology | Cat#9441; RRID: AB_331805 |
| Mouse monoclonal anti-Myc-Tag antibody, WB, dil:1/1,000; IP, dil: 1/250 | OriGene | Cat#TA150121; RRID: AB_2622266 |
| Mouse monoclonal anti-DYKDDDDK tag antibody, WB, dil:1/5,000; IP, dil: 1/500 | Proteintech | Cat#66008-3-Ig; RRID: AB_2749837 |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H+L), WB, dil:1/50,000 | Jackson ImmunoResearch | Cat#Fs111-035-003; RRID: AB_2313567 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L), WB, dil:1/50,000 | Jackson ImmunoResearch | Cat#Fs115-035-003; RRID: AB_10015289 |
| Protein A/G PLUS-Agarose | Santa Cruz Biotechnology | Cat#sc-2003; RRID: AB_10201400 |
| Protein A+G Agarose | Beyotime | Cat#P2055 |
| Anti-Flag Affinity Gel | Bimake | Cat#B23101 |
| Pierce™ Protein A/G Magnetic Beads | Thermo Fisher Scientific | Cat#88803 |
| Anti-DYKDDDDK-Tag Mouse mAb (Agarose Conjugated) | Abmart | Cat#M20018 |
| Ni-NTA Agarose | Qiagen | Cat#30210 |
| Phospho-IRE1-S724 Rabbit mAb, B, dil:1/1,000 | Abclonal | Cat#AP0878; RRID: AB_2771207 |
| IRE1α (14C10) Rabbit mAb, WB, dil:1/1,000 | Cell Signaling Technology | Cat#3294S; RRID: AB_823545 |
| Phospho-eIF2α-S51 Rabbit mAb, WB, dil:1/1,000 | Abclonal | Cat#AP0692; RRID: AB_2863809 |
| eIF2α Antibody, WB, dil:1/1,000 | Cell Signaling Technology | Cat#9722S; RRID: AB_2230924 |
| Phospho-PERK (Thr980) (16F8) Rabbit mAb, WB, dil:1/1,000 | Cell Signaling Technology | Cat#3179S; RRID: AB_2095853 |
| PEK/PERK (Ab-981) Antibody, WB, dil:1/1,000 | Biorbyt | Cat#Ab-981; RRID: AB_10685785 |
| ATF4 (D4B8) Rabbit mAb, WB, dil:1/1,000 | Cell Signaling Technology | Cat#11815S; RRID: AB_2095853 |
| Anti-Ki67 antibody, IF, dil:1/100 | Abcam | Cat#ab16667; RRID: AB_302459 |
| Anti-NDUFB8 antibody, IF, dil:1/500 | Abcam | Cat#ab192878; RRID: AB_2847808 |
| Anti-HMGCS2 antibody, IF, dil:1/500 | Abcam | Cat#ab137043; RRID: AB_2749817 |
| Bacterial and virus strains | ||
| Adenoviral ceruplasmin shRNA | This paper | N/A |
| Adenoviral shLacz | This paper | N/A |
| HBAAV2/9-TBG-EGFP | Hanbio Biotechnology (Shanghai) | HH20190118GKL-AVP01 |
| HBAAV2/9-TBG-Cre-EGFP | Hanbio Biotechnology (Shanghai) | HH20190118GKL-AVP01 |
| Adenoviral PPARE-Luc | This paper | N/A |
| Adenoviral HMGCS2-Luc | This paper | N/A |
| Adenoviral CPT1α-Luc | This paper | N/A |
| Biological samples | ||
| Human liver tissue sample | Shanghai Jiao Tong University Affiliated Sixth People’s Hospital |
N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Oil Red O | Sigma-Aldrich | Cat#O0625 |
| Dexamethasone | Sigma-Aldrich | Cat#D4902 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat#A8806 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#S8830 |
| PhosSTOP™ | Sigma-Aldrich | Cat#4906837001 |
| Sodium palmitate | Sigma-Aldrich | Cat#P9767 |
| Insulin | Roche | Cat#11376497001 |
| D-(+)-Glucose | Sigma-Aldrich | Cat#G7021 |
| Bathocuproinedisulfonic acid disodium salt (BCS) | Sigma-Aldrich | Cat#B1125 |
| Compound C (CC) | MedChemExpress | Cat#HY-13418A |
| Deferasirox (DFX) | Selleck | Cat#S1712 |
| Ammonium tetrathiomolybdate (TM) | Sigma-Aldrich | Cat#323446 |
| SP 600125 | Cayman Chemical | Cat#10010466 |
| Ex527 | Cayman Chemical | Cat#10009798 |
| GW 6471 | Cayman Chemical | Cat#11697 |
| GW 9578 | Cayman Chemical | Cat#10011211 |
| H89 | MedChemExpress | Cat#HY-15979 |
| Deferoxamine mesylate | Selleck | Cat#S5742 |
| Pyridoxal isonicotinoyl hydrazone | Cayman Chemical | Cat#19357 |
| Copper (II) sulfate | SHANGHAI LINFENG CHEMICAL | Cat#20160728 |
| SDS-PAGE Loading Buffer (non-Reducing, 5x) | Cwbiotech | Cat#CW0028S |
| Tetrakis(acetonitrile)copper(I) Hexafluorophosphate | Tcichemical | Cat#T2665 |
| D-isoascorbic acid | Sigma-Aldrich | Cat#856061 |
| IPTG | Yeasen | Cat#10902ES08 |
| Proteinase K Solution | ThermoFisher Scientific | Cat#AM2548 |
| Passive Lysis Buffer, 5X | Promega | Cat#E1941 |
| Penicillin/Streptomycin | ThermoFisher Scientific | Cat#15140-122 |
| Dulbecco’s modified Eagle’s medium (DMEM) | ThermoFisher Scientific | Cat#C11995500BT |
| ITS Liquid Media Supplement (100x) | Sigma-Aldrich | Cat# 41400045 |
| Medium 199 | Sigma-Aldrich | Cat#M2154 |
| L-Glutamine | Sigma-Aldrich | Cat#G3126 |
| Collagenase type I | Worthington biochemical corporation | Cat#4196 |
| Rat Collagen I | R&D Systems | Cat#3440-005-01 |
| PERCOLL | BIODEE | Cat#DE-17-0891-02G |
| RPMI 1640 medium | ThermoFisher Scientific | Cat#A10491-01 |
| DMEM/F12 | ThermoFisher Scientific | Cat#21331020 |
| TRIzol reagent | ThermoFisher Scientific | Cat#15596 |
| Lipofectamine RNAiMAX Transfection Reagent | ThermoFisher Scientific | Cat#13778075 |
| Fetal Bovine Serum | ThermoFisher Scientific | Cat#16000-044 |
| STZ | Sigma-Aldrich | Cat#18883-66-4 |
| Critical commercial assays | ||
| BCA assay kit | Thermo Fisher Scientific | Cat#23225 |
| Hiscript II Q RT SuperMix for qPCR | Vazyme, China | Cat#R222-01 |
| AceQ Universal SYBR qPCR Master Mix | Vazyme, China | Cat#Q511-02 |
| VAHTS mRNA-seq V2 Library Prep Kit for Illumina | Vazyme, China | Cat#NR601 |
| MitoTracker Red probe | ThermoFisher Scientific | Cat#M7512 |
| XF Cell Mito Stress Test Kit | Agilent Technologies | Cat#103015-100 |
| TG Assay kit | Applygene | Cat#E1013-105 |
| KOD -Plus Mutagenesis Kit | TOYOBO | Cat#SMK-101 |
| QIAquick PCR Purification Kit | Qiagen | Cat#28106 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat#E1960 |
| DeadEnd™ Fluorometric TUNEL System | Promega | Cat#G3250 |
| NE-PER kit | Thermo Fisher | 78833 |
| AST kit | NJJCBIO | C010-2-1 |
| ALT kit | NJJCBIO | C009-2-1 |
| Deposited data | ||
| RNA sequencing data | This paper | Mendeley data (https://doi.org/10.17632/g5pxd2dcvm.1) |
| Experimental models: Cell lines | ||
| Mouse: AML12 cells | ATCC | Cat#CRL-2254 |
| Mouse: Primary hepatocytes | This paper | N/A |
| Human: L02 cells | Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd. | N/A |
| Human: 293A | Invitrogen | Cat#R70507 |
| Human: HEK293T | ATCC | Cat#CRL-1573 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | GemPharmatech | Cat#N000013 |
| Mouse: ob/ob (B6/JGpt-Lepem1Cd25/Gpt) | GemPharmatech | Cat#T001461 |
| LDKO mice | GemPharmatech | N/A |
| Mouse: Cpflox/flox | Institute of Laboratory Animals Science | N/A |
| Mouse: global Cp KO | Institute of Laboratory Animals Science | N/A |
| Mouse: Albumin-Cre transgenic mice | The Jackson Laboratory | N/A |
| Mouse: AMPKα1flox/flox and α2flox/flox (AMPKαf/f) | Gift from Jingya Li (Chinese Academy of Sciences) | N/A |
| Mouse: AMPKα1 and α2 double liver KO (AMPKα-DKO) | Gift from Jingya Li (Chinese Academy of Sciences) | N/A |
| Oligonucleotides | ||
| Primers for qPCR, see Table S3 | This paper | N/A |
| Primers for plasmid construction, see Table S3 | This paper | N/A |
| siRNAs, see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| pET28a-SCO1-HIS | This paper | N/A |
| SCO1-Flag | This paper | N/A |
| SCO1-His-Trunated | This paper | N/A |
| SCO1-C152/156A | This paper | N/A |
| pcDNA-3.0-PGC1α-Flag | This paper | N/A |
| CP-pLVX-IRES-Puro | This paper | N/A |
| pcDNA-3.1-AMPKα1-Myc | This paper | N/A |
| pcDNA-3.1-AMPKα1-Flag | Gift from Shengcai Lin (Xiamen University) | N/A |
| LKB1-Flag | Gift from Shengcai Lin (Xiamen University) | N/A |
| Software and algorithms | ||
| GraphPad Prism version 9.0 | GraphPad Software | https://www.graphpad.com/ |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| SPSS version 25.0 | IBM | https://www.ibm.com/analytics/spss-statistics-software |
| Other | ||
| High-fat diet | Research diet | Cat#D12492 |
| Chow diet | SLACOM | Cat#P1101F-25 |
Highlights.
Hepatic copper deficiency mediated by ceruloplasmin upregulation is a cause of NAFLD
Ceruloplasmin deletion restores copper levels and alleviates hepatic steatosis via AMPK
The copper chaperone SCO1 promotes assembly of SCO1-LKB1-AMPK complex for AMPK activation
ACKNOWLEDGMENTS
We thank Dr. Shengcai Lin from Xiamen University for providing AMPKα1 and LKB1 plasmids. We thank Dr. Jingya Li from the Chinese Academy of Sciences for providing AMPKα1 and α2 double liver KO mice (AMPKα-DKO) and AMPKα1flox/flox and α2flox/flox (AMPKαf/f) mice. We are also grateful to the “Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education” and “Shanghai Frontiers Science Center of Cellular Homeostasis and Human Diseases” for supporting the research platforms. This work was supported by the following grants to J.L.: National Key R&D Program ofChina (2021YFA0804800); Training Program of the Major Research Plan of the National Natural Science Foundation of China (no. 91857111), China; National Key R&D Program of China (2018YFA0800600); Innovative research team of high-level local universities in Shanghai (SHSMU-ZDCX20212501); Shanghai Municipal Commission of Science and Technology (no. 20410713200; no. 21S11909000); National Facility for Translational Medicine (Shanghai) (TMSK-2020-102); “Shuguang Program” supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (20SG10); and Innovative research team of high-level local universities in Shanghai (SHSMU-ZDCX20212501). This work was also supported by the following grants to S.Z.C.: National Natural Science Foundation of China (no. 82170863), Shanghai Rising-Star Program (no. 21QA1407000), and Lingang Laboratory (no. LG-QS-202205-06). This work was also supported by the following grants to Y.M.Y.: National Natural Science Foundation of China (no. 82103389) and China Postdoctoral Science Foundation (no. 2021M692125). This work was also supported by the following grants to L.H.: China National Postdoctoral Program for Innovative Talents (BX2021191), China Postdoctoral Science Foundation (no. 2021M702168), and Shanghai Sixth People’s Hospital (ynqn202104). This work was also supported by National Natural Science Foundation of China (no. 81702792 to S.M.X.) and (no. 82130020, 82072646, 8213000134 to J.Y.G.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111498.
DECLARATION OF INTERESTS
The authors declare no conflict of interest associated with this study.
REFERENCES
- Abajian C, and Rosenzweig AC (2006). Crystal structure of yeast Sco1. J. Biol. Inorg. Chem 11, 459–466. 10.1007/s00775-006-0096-7. [DOI] [PubMed] [Google Scholar]
- Aigner E, Strasser M, Haufe H, Sonnweber T, Hohla F, Stadlmayr A, Solioz M, Tilg H, Patsch W, Weiss G, et al. (2010). A role for low hepatic copper concentrations in nonalcoholic Fatty liver disease. Am. J. Gastroenterol 105, 1978–1985. 10.1038/ajg.2010.170. [DOI] [PubMed] [Google Scholar]
- Aigner E, Theurl I, Haufe H, Seifert M, Hohla F, Scharinger L, Stickel F, Mourlane F, Weiss G, and Datz C (2008). Copper availability contributes to iron perturbations in human nonalcoholic fatty liver disease. Gastroenterology 135, 680–688. 10.1053/j.gastro.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Aldred AR, Grimes A, Schreiber G, and Mercer JF (1987). Rat ceruloplasmin. Molecular cloning and gene expression in liver, choroid plexus, yolk sac, placenta, and testis. J. Biol. Chem 262, 2875–2878. [PubMed] [Google Scholar]
- Balatri E, Banci L, Bertini I, Cantini F, and Ciofi-Baffoni S (2003). Solution structure of Sco1: a thioredoxin-like protein Involved in cytochrome c oxidase assembly. Structure 11, 1431–1443. 10.1016/j.str.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. (2006). Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342. 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady GF, Galbán S, Liu X, Basrur V, Gitlin JD, Elenitoba-Johnson KSJ, Wilson TE, and Duckett CS (2010). Regulation of the copper chaperone CCS by XIAP-mediated ubiquitination. Mol. Cell Biol 30, 1923–1936. 10.1128/mcb.00900-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr TP, and Lei KY (1990). High-density lipoprotein cholesteryl ester and protein catabolism in hypercholesterolemic rats induced by copper deficiency. Metabolism 39, 518–524. 10.1016/0026-0495(90)90011-z. [DOI] [PubMed] [Google Scholar]
- Chen S, Hoene M, Li J, Li Y, Zhao X, Häring HU, Schleicher ED, Weigert C, Xu G, and Lehmann R (2013). Simultaneous extraction of metabolome and lipidome with methyl tert-butyl ether from a single small tissue sample for ultra-high performance liquid chromatography/mass spectrometry. J. Chromatogr. A 1298, 9–16. 10.1016/j.chroma.2013.05.019. [DOI] [PubMed] [Google Scholar]
- Chen S, Liu X, Peng C, Tan C, Sun H, Liu H, Zhang Y, Wu P, Cui C, Liu C, et al. (2021). The phytochemical hyperforin triggers thermogenesis in adipose tissue via a Dlat-AMPK signaling axis to curb obesity. Cell Metab. 33, 565–580.e7. 10.1016/j.cmet.2021.02.007. [DOI] [PubMed] [Google Scholar]
- Church SJ, Begley P, Kureishy N, McHarg S, Bishop PN, Bechtold DA, Unwin RD, and Cooper GJS (2015). Deficient copper concentrations in dried-defatted hepatic tissue from ob/ob mice: a potential model for study of defective copper regulation in metabolic liver disease. Biochem. Biophys. Res. Commun 460, 549–554. 10.1016/j.bbrc.2015.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham J, Leffell M, Mearkle P, and Harmatz P (1995). Elevated plasma ceruloplasmin in insulin-dependent diabetes mellitus: evidence for increased oxidative stress as a variable complication. Metabolism 44, 996–999. 10.1016/0026-0495(95)90095-0. [DOI] [PubMed] [Google Scholar]
- Cz1onkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, Rybakowski JK, Weiss KH, and Schilsky ML (2018). Wilson disease. Nat. Rev. Dis. Primers 4, 21. 10.1038/s41572-018-0018-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daimon M, Susa S, Yamatani K, Manaka H, Hama K, Kimura M, Ohnuma H, and Kato T (1998). Hyperglycemia is a factor for an increase in serum ceruloplasmin in type 2 diabetes. Diabetes Care 21, 1525–1528. 10.2337/diacare.21.9.1525. [DOI] [PubMed] [Google Scholar]
- Engström G, Stavenow L, Hedblad B, Lind P, Eriksson KF, Janzon L, and Lindgärde F (2003). Inflammation-sensitive plasma proteins, diabetes, and mortality and incidence of myocardial infarction and stroke: a population-based study. Diabetes 52, 442–447. 10.2337/diabetes.52.2.442. [DOI] [PubMed] [Google Scholar]
- Fleming RE, and Gitlin JD (1990). Primary structure of rat ceruloplasmin and analysis of tissue-specific gene expression during development. J. Biol. Chem 265, 7701–7707. [PubMed] [Google Scholar]
- Gao P, Li L, Yang L, Gui D, Zhang J, Han J, Wang J, Wang N, Lu J, Chen S, et al. (2019). Yin Yang 1 protein ameliorates diabetic nephropathy pathology through transcriptional repression of TGFbeta1. Sci. Transl. Med 11, eaaw2050. 10.1126/scitranslmed.aaw2050. [DOI] [PubMed] [Google Scholar]
- González A, Hall MN, Lin SC, and Hardie DG (2020). AMPK and TOR: the Yin and Yang of cellular nutrient sensing and growth control. Cell Metab 31, 472–492. 10.1016/j.cmet.2020.01.015. [DOI] [PubMed] [Google Scholar]
- Gulec S, and Collins JF (2014). Molecular mediators governing iron-copper interactions. Annu. Rev. Nutr 34, 95–116. 10.1146/annurevnutr-071812-161215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas JT, Miao J, Chanda D, Wang Y, Zhao E, Haas ME, Hirschey M, Vaitheesvaran B, Farese RV Jr., Kurland IJ, et al. (2012). Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 15, 873–884. 10.1016/j.cmet.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman NE, and Gitlin JD (2002). Ceruloplasmin metabolism and function. Annu. Rev. Nutr 22, 439–458. 10.1146/annurev.nutr.22.012502.114457. [DOI] [PubMed] [Google Scholar]
- Hu Y, Shin DJ, Pan H, Lin Z, Dreyfuss JM, Camargo FD, Miao J, and Biddinger SB (2017). YAP suppresses gluconeogenic gene expression through PGC1alpha. Hepatology 66, 2029–2041. 10.1002/hep.29373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, Akram K, McKinney RD, Ushio-Fukai M, and Fukai T (2008). Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J. Biol. Chem 283, 9157–9167. 10.1074/jbc.M709463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler SG (2011). ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol 7, 15–29. 10.1038/nrneurol.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy ML, Failla ML, and Smith JC Jr. (1986). Influence of genetic obesity on tissue concentrations of zinc, copper, manganese and iron in mice. J. Nutr 116, 1432–1441. 10.1093/jn/116.8.1432. [DOI] [PubMed] [Google Scholar]
- Kim BE, Nevitt T, and Thiele DJ (2008). Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol 4, 176–185. 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- Kim CH, Park JY, Kim JY, Choi CS, Kim YI, Chung YE, Lee MS, Hong SK, and Lee KU (2002). Elevated serum ceruloplasmin levels in subjects with metabolic syndrome: a population-based study. Metabolism 51, 838–842. 10.1053/meta.2002.33348. [DOI] [PubMed] [Google Scholar]
- Klomp LW, Farhangrazi ZS, Dugan LL, and Gitlin JD (1996). Ceruloplasmin gene expression in the murine central nervous system. J. Clin. Invest 98, 207–215. 10.1172/jci118768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary SC, Cobine PA, Kaufman BA, Guercin GH, Mattman A, Palaty J, Lockitch G, Winge DR, Rustin P, Horvath R, and Shoubridge EA (2007). The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab. 5, 9–20. 10.1016/j.cmet.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Li M, Zhang CS, Zong Y, Feng JW, Ma T, Hu M, Lin Z, Li X, Xie C, Wu Y, et al. (2019). Transient receptor potential V channels are essential for glucose sensing by aldolase and AMPK. Cell Metab. 30, 508–524.e12. 10.1016/j.cmet.2019.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, and Hardie DG (2018). AMPK: sensing glucose as well as cellular energy status. Cell Metab. 27, 299–313. 10.1016/j.cmet.2017.10.009. [DOI] [PubMed] [Google Scholar]
- Linder MC (2020). Copper homeostasis in mammals, with emphasis on secretion and excretion. A review. Int. J. Mol. Sci 21, E4932. 10.3390/ijms21144932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Tian X, Zhang B, Li M, Wang Y, Yang C, Wu J, Wei X, Qu Q, Yu Y, et al. (2022). Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature 603, 159–165. 10.1038/s41586-022-04431-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, and Schwudke D (2008). Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res 49, 1137–1146. 10.1194/jlr.D700041-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Fang S, Bae Y, and Kemper JK (2006). Functional inhibitory cross-talk between constitutive androstane receptor and hepatic nuclear factor-4 in hepatic lipid/glucose metabolism is mediated by competition for binding to the DR1 motif and to the common coactivators, GRIP-1 and PGC-1alpha. J. Biol. Chem 281, 14537–14546. 10.1074/jbc.M510713200. [DOI] [PubMed] [Google Scholar]
- Nittis T, George GN, and Winge DR (2001). Yeast Sco1, a protein essential for cytochrome c oxidase function is a Cu(I)-binding protein. J. Biol. Chem 276, 42520–42526. 10.1074/jbc.M107077200. [DOI] [PubMed] [Google Scholar]
- Paris I, Perez-Pastene C, Couve E, Caviedes P, LeDoux S, and Segura-Aguilar J (2009). Copper dopamine complex induces mitochondrial autophagy preceding caspase-independent apoptotic cell death. J. Biol. Chem 284, 13306–13315. 10.1074/jbc.M900323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinin E, Villani G, and Moschetta A (2019). Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: the role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol 16, 160–174. 10.1038/s41575-018-0089-3. [DOI] [PubMed] [Google Scholar]
- Ross AC, Caballero BH, Cousins RJ, Tucker KL, and Ziegler TR (2012). Modern Nutrition in Health and Disease, Eleventh edition (Wolters Kluwer Health Adis (ESP)). [Google Scholar]
- Ruiz LM, Jensen EL, Bustos RI, Arg€uelloa G, Gutierrez-Garcia R, González M, Hernández C, Paredes R, Simon F, Riedel C, et al. (2014). Adaptive responses of mitochondria to mild copper deprivation involve changes in morphology, OXPHOS remodeling and bioenergetics. J. Cell. Physiol 229, 607–619. 10.1002/jcp.24484. [DOI] [PubMed] [Google Scholar]
- Song M, Schuschke DA, Zhou Z, Chen T, Pierce WM Jr., Wang R, Johnson WT, and McClain CJ (2012). High fructose feeding induces copper deficiency in Sprague-Dawley rats: a novel mechanism for obesity related fatty liver. J. Hepatol 56, 433–440. 10.1016/j.jhep.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Wei G, Liu H, Xiao D, Huang J, Lu J, Miao J, Liu J, and Chen S (2020). Inhibition of XBP1s ubiquitination enhances its protein stability and improves glucose homeostasis. Metabolism 105, 154046. 10.1016/j.metabol.2019.154046. [DOI] [PubMed] [Google Scholar]
- Tallino S, Duffy M, Ralle M, Corté s MP, Latorre M, and Burkhead JL (2015). Nutrigenomics analysis reveals that copper deficiency and dietary sucrose up-regulate inflammation, fibrosis and lipogenic pathways in a mature rat model of nonalcoholic fatty liver disease. J. Nutr. Biochem 26, 996–1006. 10.1016/j.jnutbio.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang T, Posimo JM, Gudiel AA, Cicchini M, Feldser DM, and Brady DC (2020). Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma. Nat. Cell Biol 22, 412–424. 10.1038/s41556-020-0481-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, An P, Xie E, Wu Q, Fang X, Gao H, Zhang Z, Li Y, Wang X, Zhang J, et al. (2017). Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66, 449–465. 10.1002/hep.29117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapnir RA, and Devas G (1995). Copper deficiency: interaction with high-fructose and high-fat diets in rats. Am. J. Clin. Nutr 61, 105–110. 10.1093/ajcn/61.1.105. [DOI] [PubMed] [Google Scholar]
- Wooton-Kee CR, Robertson M, Zhou Y, Dong B, Sun Z, Kim KH, Liu H, Xu Y, Putluri N, Saha P, et al. (2020). Metabolic dysregulation in the Atp7b (−/−) Wilson’s disease mouse model. Proc. Natl. Acad. Sci. USA 117, 2076–2083. 10.1073/pnas.1914267117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WW, Musto NA, Mather JP, and Bardin CW (1981). Sertoli cells secrete both testis-specific and serum proteins. Proc. Natl. Acad. Sci. USA 78, 7565–7569. 10.1073/pnas.78.12.7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Liu Y, Hu R, Wang M, Stohr O, Xiong Y, Chen L, Kang H, Zheng L, Cai S, et al. (2020). TAZ inhibits GR and coordinates hepatic glucose homeostasis in normal physiologic states. Preprint at bioRxiv 10.1101/2020.05.12.091264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Barrientos T, and Andrews NC (2013). Iron and copper in mitochondrial diseases. Cell Metab. 17, 319–328. 10.1016/j.cmet.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Q, Hu C, Yu D, Wang L, Zhou Y, Zhao X, Li Q, Hou X, and Xu G (2018). Development of a high coverage pseudotargeted lipidomics method based on ultra-high performance liquid chromatography–mass spectrometry. Anal. Chem 90, 7608–7616. 10.1021/acs.analchem.8b01331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Yoshida K, Miyagoe Y, Ishikawa A, Hanaoka K, Nomoto S, Kaneko K, Ikeda S.i., and Takeda S (2002). Quantitative evaluation of expression of iron-metabolism genes in ceruloplasmin-deficient mice. Biochim. Biophys. Acta 1588, 195–202. 10.1016/s0925-4439(02)00165-5. [DOI] [PubMed] [Google Scholar]
- Yan M, Qi H, Xia T, Zhao X, Wang W, Wang Z, Lu C, Ning Z, Chen H, Li T, et al. (2019). Metabolomics profiling of metformin-mediated metabolic reprogramming bypassing AMPKα. Metabolism 91, 18–29. 10.1016/j.metabol.2018.11.010. [DOI] [PubMed] [Google Scholar]
- Yang F, Friedrichs WE, deGraffenried L, Herbert DC, Weaker FJ, Bowman BH, and Coalson JJ (1996). Cellular expression of ceruloplasmin in baboon and mouse lung during development and inflammation. Am. J. Respir. Cell Mol. Biol 14, 161–169. 10.1165/ajrcmb.14.2.8630266. [DOI] [PubMed] [Google Scholar]
- Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, Kim JK, Heo Y, Lee HS, Lee MY, et al. (2020). A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 31, 267–283.e12. 10.1016/j.cmet.2019.11.020. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, and Wymer M (2016). Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL, Wu YQ, Li TY, Liang Y, Lu Z, et al. (2014). The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 20, 526–540. 10.1016/j.cmet.2014.06.014. [DOI] [PubMed] [Google Scholar]
- Zhang CS, Li M, Zong Y, and Lin SC (2018). Determining AMPK activation via the lysosomal v-ATPase-ragulator-AXIN/LKB1 Axis. Methods Mol. Biol 1732, 393–411. 10.1007/978-1-4939-7598-3_25. [DOI] [PubMed] [Google Scholar]
- Zhang YL, Guo H, Zhang CS, Lin SY, Yin Z, Peng Y, Luo H, Shi Y, Lian G, Zhang C, et al. (2013). AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab 18, 546–555. 10.1016/j.cmet.2013.09.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated by RNA sequencing are available from Mendeley data (https://doi.org/10.17632/g5pxd2dcvm.1). The published paper includes the datasets generated by LC-MS during this study.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.