

Summary
Purpose.
Emerging evidence suggests that 5’ Adenosine Monophosphate-Activated Protein Kinase (AMPK), a key regulator of cellular bioenergetics, is a novel target for the treatment of glioblastoma (GBM), a lethal brain tumor. SBI-0206965, an aminopyrimidine derivative, is a potent AMPK inhibitor being investigated for the treatment of GBM. Here we characterized the systemic and brain pharmacokinetics (PK) and hepatic metabolism of SBI-0206965.
Methods.
We performed intracerebral microdialysis to determine brain partitioning of SBI-0206965 in jugular vein cannulated rats. We assessed systemic PK of SBI-0206965 in rats and C57BL/6 mice following oral administration. Employing human, mouse, and rat liver microsomes we characterized the metabolism of SBI-0206965.
Results.
SBI-0206965 is quickly absorbed, achieving plasma and brain extracellular fluid (ECF) peak levels within 0.25 – 0.65 h. Based on the ratio of Cmax and AUC in brain ECF to plasma (corrected for protein binding), brain partitioning is ~ 0.6—0.9 in rats. However, the compound has a short elimination half-life (1–2 h) and low relative oral bioavailability (~ 0.15). The estimated in-vitro hepatic intrinsic clearance of SBI-0206965 in mouse, rat and human was 325, 76 and 68 mL/min/kg, respectively. SBI-0206965 metabolites included desmethylated products, and the metabolism was strongly inhibited by ketoconazole, a CYP3A inhibitor.
Conclusion.
SBI-0206965 has adequate brain permeability but low relative oral bioavailability which may be due to rapid hepatic metabolism, likely catalyzed by CYP3A enzymes. Our observations will facilitate further development of SBI-0206965, and/or other structurally related molecules, for the treatment of GBM and other brain tumors.
Keywords: Glioblastoma, 5’ Adenosine Monophosphate-Activated Protein Kinase, Brain partitioning, In-vitro metabolism
Introduction
Glioblastoma (GBM), a grade IV astrocytoma, is one of the most aggressive and infiltrative brain tumors, with a dismal prognosis. The five-year survival rate for GBM patients is only 6–7%, and the median survival is only 14.6 months [1, 2]. According to the National Brain Tumor Society an estimated 12500 patients will be newly diagnosed with malignant brain tumor in 2022 [3]. The current standard of care for GBM includes maximally safe tumor resection surgery followed by concurrent adjuvant radiation treatment and chemotherapy with the DNA alkylating agent temozolomide. Unfortunately, despite this aggressive treatment the tumors recur in virtually all patients and there is an urgent need for the development of novel therapies for the treatment of GBM. Some of the formidable barriers to the development of effective new therapeutic agents for GBM include: (i) lack of novel validated GBM-specific targets that may facilitate selective drug therapy and, (ii) failure of the anti-glioma agents to cross blood–brain barrier (BBB).
5’ Adenosine Monophosphate-Activated Protein Kinase (AMPK) appears to be an attractive and “druggable” novel target for the treatment of GBM. GBMs remodel metabolism to fuel survival, proliferation, and invasion like most of the cancers. AMPK is a major energetic biosensor and metabolic switch that controls a broad array of cellular biosynthetic and catabolic pathways [4]. The role of AMPK in cancer is debatable as AMPK can be referred as both a “conditional” tumor suppressor and “contextual” oncogene [5, 6]. This is because although, on one hand, active AMPK inhibits mammalian target of rapamycin (mTOR) and lipogenesis which are two crucial arms of cancer growth, AMPK also ensures viability by metabolic reprogramming in cancer cells [7]. Recently, Chhipa et al. (2018) provided evidence strongly suggesting that AMPK is an attractive novel target for GBM therapy [8]. By applying a multifaceted approach, they showed: i) AMPK subunits were highly expressed in GBM tumor tissue and numerous primary GBM cell lines, ii) AMPK depletion in vitro and in orthotopic GBM xenografts significantly reduced cell viability and suppressed tumor growth and iii) systemic AMPK knockout was well tolerated in adult mice with no discernible phenotype suggestive of minimal systemic adverse effects.
However, AMPK-inhibitory compounds are largely limited to Compound C, which has a poor target-selectivity profile [9]. SBI-0206965, an aminopyrimidine derivative, was originally identified through screening a library of pyrimidine analogues for ATP-competitive inhibitors of the autophagy initiator Unc-51 Like Autophagy Activating Kinase 1 (ULK1) with the ability to inhibit ULK signaling. Egan et al. (2015) first reported that SBI-0206965 inhibits autophagy and enhances apoptosis in human U87MG GBM and lung cancer cells coupled with nutrient stress or mTOR inhibition [10]. It was recently evaluated that SBI-0206965 directly inhibits AMPK with 40-fold greater potency and markedly lower kinase promiscuity than Compound C and a co-crystal structure of the AMPK kinase domain/SBI-0206965 complex shows that the drug occupies a pocket that partially overlaps the ATP active site in a type IIb inhibitor manner [9]. Recently, Deng et al. (2019) also provided a potential therapeutic strategy to treat neuroblastoma by targeting the β2-adrenergic receptor (β2-AR) pathway in combination with SBI-0206965 [11].
Little is known about the pharmacokinetics (PK) of SBI-0206965, and its BBB penetration, which are important determinants of the clinical viability of a therapeutic agent for the treatment of CNS malignancies. Therefore, in this study, we conducted a comprehensive evaluation of the plasma PK and relative oral bioavailability of SBI-0206965 in animal models and the time-course and brain partitioning of the compound. Also, employing liver microsomes we studied the metabolism of SBI-0206965 to gain insights into the structure of the metabolites formed, potential involvement of hepatic metabolism and implications for the PK of the compound in humans.
Materials and methods
Materials
SBI-0206965 was purchased from Cayman Chemical Company (Ann Arbor, MI). Concentric-style microdialysis probes were constructed as described elsewhere [12]. Dialysis membrane (MW cutoff, 13,000 Da and outside diameter, 210 μm) was purchased from Spectrum Laboratories, CA. Cannulation materials for the implantation of venous catheters were purchased from Plastics One Inc. (Roanoke, VA) and VWR scientific (Batavia, IL). BD Microtainer® K2EDTA coated tubes were purchased from VWR Scientific (Philadelphia, PA). Rapid Equilibrium Dialysis Device Single-Use Protein binding kit and phosphate-buffered saline were purchased from Thermo Scientific™. C57BL/6 mouse, male IGS – Sprague Dawley rat and human liver microsomes and rapidSTART™ NADPH-regenerating system (2 mM NADP+, 10 mM glucose-6 phosphate, 0.4 U/ml glucose-6-phosphate dehydrogenase) were purchased from Sekisui XenoTech, LLC (Kansas city, KS). Quinidine and quercetin were purchased from Sigma Chemical Co. (St. Louis, MO) and ketoconazole was purchased from Janssen Biotech N.V. (distributed by Research Diagnostics, INC. Flanders, NJ).
Animals
Male Sprague–Dawley (SD) rats (200–250 g, 6–7 weeks of age), purchased from Charles River Laboratories (Wilmington, MA), were housed individually in a room maintained at 22°C, 55% relative humidity with a 12/12 h light/dark cycle and provided access to water and standard laboratory chow ad libitum. The experimental protocol was approved by the University of Cincinnati Institutional Animal Care and Use Committee (IACUC, Protocol No. 20–01–24–01). C57BL/6 mice were generated by in-house breeding at the Cincinnati Children’s Hospital Medical Center (CCHMC; Cincinnati, OH). Housing and experimental protocols employing C57BL/6 mice were approved by the CCHMC IACUC (Protocol No. IACUC2021–0031).
Methods
Microdialysis surgery in rats
Microdialysis studies in rats were performed as described in our previous publications [13–15]. Briefly, rats (N = 5) were implanted with stainless steel guide cannula under ketamine/xylazine (70/6 mg/kg i.p.) anesthesia 48 to 72 h before the microdialysis experiment. On the day of the experiment, a concentric style dialysis probe was inserted through the guide cannula into the striatum. The coordinates for the guide cannula were 1.2 mm anteroposterior and 3.1 mm lateral from bregma, according to the stereotaxic atlas of Paxinos et al. [16]. The active portion of the membrane for the striatum was 4.5 mm. The probes were connected to an infusion pump set to deliver modified Dulbecco’s phosphate buffered saline at a rate of 2.0 μL/min.
Drug administration and sampling protocol
After an equilibration period of 30 min, SBI-0206965 (25 mg/kg) was administered via intraperitoneal (i.p.) route. Dialysate (via probe outlet tubing) and blood (via jugular catheter) samples were collected at 0, 0.5, 1, 2, 4, 6, 8 and 10 h. Both dialysate and plasma samples were stored at −80 °C until analyzed.
Retrodialysis for assessing in vivo recovery of SBI-0206965 from dialysate samples
Buffer containing SBI-0206965 (25 μM) was perfused through the probe in rats. Following an equilibration for 1 h, dialysis samples were collected every 30 min for 3 h. The % loss of SBI-0206965 was estimated as an estimate of in vivo recovery.
Systemic pharmacokinetics of SBI-0206965 in C57BL/6 mice
SBI-0206965 (25 mg/kg) was administered via oral gavage (SBI-0206965 was formulated in 10% DMSO, 40% polyethylene glycol 300, 5% Tween 80 and 45% saline) to C57BL/6 mice (N = 3/time point). The blood samples were collected at 0, 0.5, 1, 2, 4, 6, 12 and 24 h, immediately centrifuged to collect plasma fraction, which were then stored at −80 °C until analyzed.
In vitro metabolism in liver microsomes
SBI-0206965 (1 μM) was incubated with mouse, rat, or human liver microsomal fractions (protein concentration 1 mg/mL, 3 mM MgCl2, 1 mM EDTA and the rapid-START™ NADPH-regenerating system) in potassium phosphate buffer (50 mM, pH 7.4). After incubation at 37 °C for various time periods, the metabolic reaction was stopped by cooling on ice with simultaneous addition of 900 μL ethyl acetate and the reaction mixtures were vortexed for 30 s and centrifuged at 11,000 g for 15 min. Supernatant (850 μL) was transferred in a glass tube and organic fraction was evaporated using a rotary evaporator. The residue was reconstituted in acetonitrile and transferred into glass vials for LC/MS analysis. The degradation rate constant and in vitro hepatic intrinsic clearance of SBI-0206695 was determined as described previously [17, 18].
Metabolic inhibition study
Rat liver microsomal metabolism of SBI-0206965 was performed as described above in the presence of cytochrome P450 (CYP) inhibitors ketoconazole (1 and 5 μM), quinine (1 and 5 μM) and quercetin (6 and 12 μM) which are FDA-recommended prototypical (“indexed”) inhibitors of CYP3A, CYP2C and CYP2D enzymes, respectively. The reaction velocity (reduction in SBI-0206965 concentration over a 15 min period) vs SBI-0206965 concentrations in the presence and the absence of ketoconazole was fitted to Michaelis–Menten kinetics as described before [19].
Bioanalysis
All samples were extracted using protein precipitation method. Chromatographic separations were achieved on a Waters XBridge™ C18 column (3.5 μm, 2.1 × 100 mm). Elution was performed using an isocratic condition (50:50 water: acetonitrile 0.1% formic acid) for 7 min at flow rate of 200 μL/min with an injection volume of 5 μL. Retention time of SBI-0206965 was 2.85 min. The analysis was performed using a Thermo Scientific LTQ-FT™ hybrid mass spectrometer operated in positive-ion electrospray mode. The source voltage was held at 5 kV, with a capillary temperature of 275 °C. The product ion scans were acquired in profile mode using an isolation width of 1.6 and a normalized collision energy of 25 for SBI-0206965.
For identification of SBI-0206965 metabolites, chromatographic separations were achieved on a Symmetry C18 column (5 μm; 2.1 × 150 mm) using a gradient method (Solvent A = 95:5 water: acetonitrile 0.1% formic acid and Solvent B = 95:5 acetonitrile: water 0.1% formic acid) at a flow rate of 200 μL/min for 20 min. The product ion scans were acquired in profile mode using an isolation width of 1.6 – 2 (1.6 for the MS2 stage and broadened to 2.0 for the subsequent MS3 stage) and a normalized collision energy of 25 for SBI-0206965.
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Non-compartmental analysis was performed using Phoenix® WinNonlin version 8.3 (Certara) to estimate the PK parameters. GraphPad Prism version 8.0.2 was used to perform non-linear regression, one-way ANOVA and Michaelis–Menten enzyme kinetics.
Results
BBB partitioning and systemic pharmacokinetics of SBI-0206965
The plasma and brain PK profiles of SBI-0206965 are shown in Fig. 1 and PK data are provided in Table 1. First, to address the brain penetration of the compound, we employed microdialysis which allows collecting serial brain ECF samples simultaneously with blood samples thus facilitating an insight into the time course and the rate and extent of drug penetration into the brain. Following SBI-0206965 administration as intraperitoneal injection, the peak plasma concentration of 3502 ng/mL was attained in 15 min post administration. The peak brain ECF concentration of 358 ng/mL was also attained relatively quickly with a Tmax of about 30 min (Fig. 1a). The half-life of the drug in ECF was similar to that in plasma (about 1–2 h). The ratio of brain ECF Cmax concentrations and the AUC0–5 to the unbound plasma Cmax and AUC0–5 are 0.6 and 0.9, respectively, which is suggestive of relatively facile drug penetration across the BBB (Table 1). Next, we also compared the PK profile of the compound following oral administration. While we did not employ the i.v. route for the determination of absolute bioavailability, a comparison of the plasma AUC following oral route vs i.p. route provides an estimate of the relative oral bioavailability (Fig. 1b). As shown the elimination profile following oral administration was similar to that of the i.p. administration and the relative bioavailability was 0.15 (Table 1). We also conducted an oral PK study of the compound in C57BL/6 mice, since mice are typically used in xenograft studies for determining anti-tumor efficacy. The elimination half-life in mice was also fairly short (~ 1 h) suggesting that SBI-0206965 is also rapidly eliminated in mice and is it likely to have low oral bioavailability.
Fig. 1.
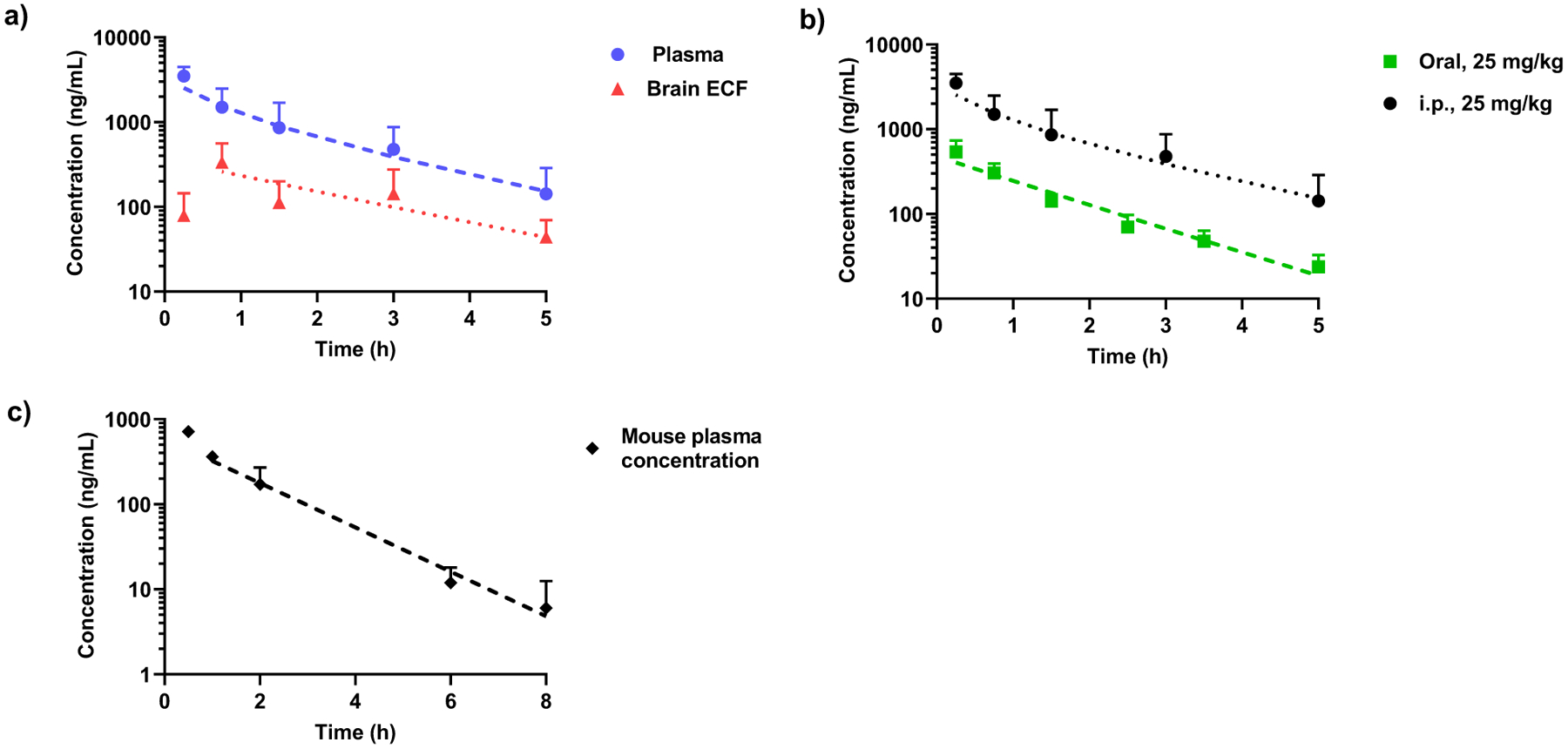
Brain and Systemic Pharmacokinetics of SBI-0206965. a) Plasma and brain extracellular fluid (ECF) levels of SBI-0206965 in male SD rats (N = 5) at single intra-peritoneal dose of 25 mg/kg. b) Plasma concentration–time profile of SBI-0206965 in SD rats after single intra-peritoneal (N = 5) or oral (N = 4) dose. c) Plasma concentration–time profile of SBI-0206965 in C57BL/6 mice (N = 3) at single oral dose of 25 mg/kg. Data are presented as mean ± SD. Dotted lines represent the best-bit regression curves plotted using Phoenix WinNonLin.® (Certara)
Table 1.
Pharmacokinetic parameters for oral and i.p. administration of SBI-0206965 (25 mg/kg) in male SD rats obtained using non-compartmental analysis. Data are presented as mean ± SD
| PK parameters | Plasma (Oral) | Plasma (i.p.) | Plasma unbound (i.p.) | Brain ECF (i.p.) | Ratio (brain ECF/plasmaub) |
|---|---|---|---|---|---|
| Tmax (h) | 0.25 | 0.25 | 0.65 ± 0.20 | - | |
| T1/2 (h) | 1.08 ± 0.16 | 1.06 ± 0.70 | 2.02 ± 0.73 | - | |
| Cmax (ng/mL) | 539.06 ± 219.94 | 3502.42 ± 988.90 | 615.03 ± 173.65 | 358.33 ± 205.39 | 0.58 |
| AUC0–5 (h*ng/mL) | 666.71 ± 215.16 | 4194.92 ± 2040.20 | 736.63 ± 358.26 | 660.81 ± 464.32 | 0.90 |
| AUC0-inf (h*ng/mL) | 703.74 ± 222.82 | 4549.50 ± 2208.84 | 798.89 ± 387.87 | 783.67 ± 494.92 | - |
| Relative oral bioavailability (F) | 0.15 | - | - |
Liver microsomal metabolism, structural elucidation of metabolites and estimation of hepatic intrinsic clearance
To better understand the factors contributing to the observed rapid elimination and low relative oral bioavailability of SBI-0206965, we conducted an in-vitro metabolism study using mouse, rat, and human liver microsomes. After optimizing the initial reaction conditions (microsomal protein and substrate concentrations), we evaluated the rate of SBI-0206965 metabolism in these microsomal fractions over a 30 min incubation period. As shown in Fig. 2a, the compound was rapidly metabolized in all three incubations and the degradation constant (Kdeg) ranged from 0.04 to 0.11 min−1. Based on these calculations the estimated intrinsic hepatic clearance in humans is projected to be similar to that in rats (Table 2).
Fig. 2.
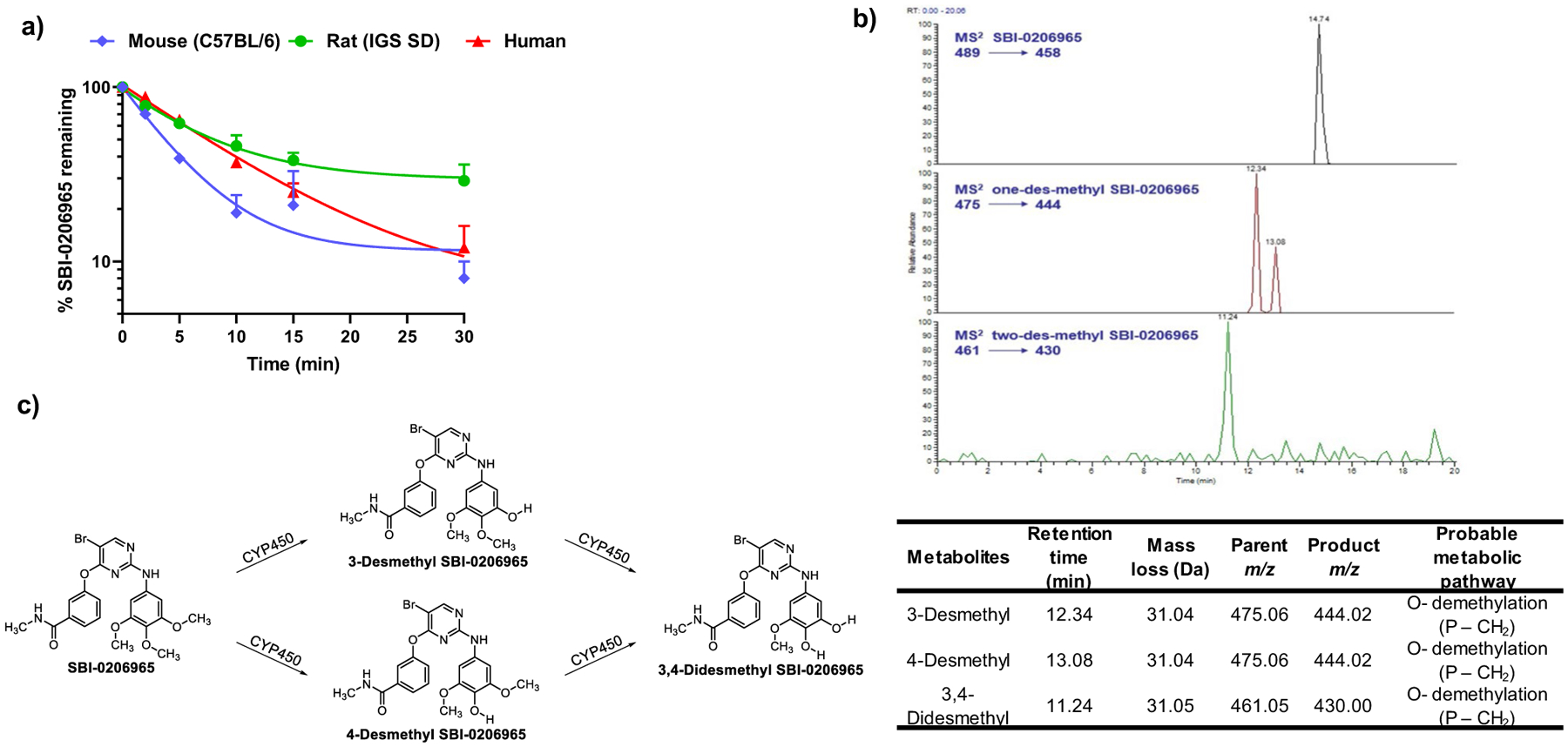
Microsomal Metabolism of SBI-0206965. a) Time course of SBI-0206965 metabolism in mouse (C57BL/6), rat (IGS SD) and human liver microsomes (N = 2). b) Extracted ion chromatograms of SBI-0206965 metabolites and in-vitro characterization of SBI-0206965 metabolites using Thermo Scientific LTQ-FT™ hybrid mass spectrometer. P, parent (SBI-0206965); m/z, mass charge ratio. c) Probable metabolic pathway for the formation of SBI-0206965 metabolites
Table 2.
Kinetics of SBI-0206965 microsomal degradation and estimated intrinsic and total hepatic clearance C57BL/6 mice, IGS SD rats and humans
| PK parameters | Mouse (C57BL/6) | Rat (IGS SD) | Human |
|---|---|---|---|
| Kdeg (min−1) | 0.11 | 0.04 | 0.07 |
| T1/2 (min) | 6.24 | 16.5 | 9.63 |
| CLint, app (μL/min/mg) | 111.06 | 42 | 71.96 |
| CLint, scaled (mL/min/kg body weight) | 324.84 | 75.6 | 68 |
The extracts from microsomal incubations at the end of the treatment period were analyzed using LC/MS. Precursor ions specifically found in SBI-0206965-containing incubation samples were viewed as potential metabolites and were thus conducted for MS2 analysis. During this preliminary experiment, mainly two oxidative metabolites were detected in rat liver microsomes. The extracted ion chromatograms of these metabolites are shown in Fig. 2b. The retention times and measured masses of the proposed metabolites are tabulated below the figure. The following ion chromatograms were acquired and quantified: for SBI-0206965, the m/z 489.08 parent ion producing the m/z 458.03 product ion; for 3- and 4-desmethyl SBI-0206965, 475.06 parent ion producing the m/z 444.02 product ion; for 3, 4-didesmethyl SBI-0206965, the m/z 461.05 parent ion producing the m/z 430.00 product ion. Thus, based on their accurate masses, fragment ions, and retention times, it appears that SBI-0206965 is primarily metabolized initially to two desmethylated products which are further oxidized to form didesmethylated products as shown in Fig. 2c.
Inhibition of SBI-0206965 metabolism by cytochrome P450 (CYP)-specific inhibitors
Given that oxidative metabolism shown above is primarily catalyzed by CYP enzymes, we conducted rat liver microsomal metabolism of SBI-0206965 in the presence and the absence of ketoconazole (1 and 5 μM), quinine (1 and 5 μM) and quercetin (6 and 12 μM) that are FDA-recommended CYP-specific inhibitors of CYP3A, CYP2C and CYP2D enzymes, respectively. Only ketoconazole (KTZ) reduced the metabolic loss of SBI-0206965 suggesting that CYP3A enzymes are primarily involved in the metabolism of this compound (Fig. 3a). Furthermore, the metabolism of SBI-0206965 ± KTZ was analyzed using Michaelis–Menten kinetics. The maximum velocity (Vmax) and Michaelis–Menten constant (Km) values in the absence of KTZ were 1.92 nmol/mg of protein/min and 11.19 μM and in the presence of KTZ were 1.22 nmol/mg of protein/min and 14.45 μM, respectively (Fig. 3b). Based on the Vmax and Km values, it appears that KTZ is mixed-type inhibitor of SBI-0206965 metabolism.
Fig. 3.
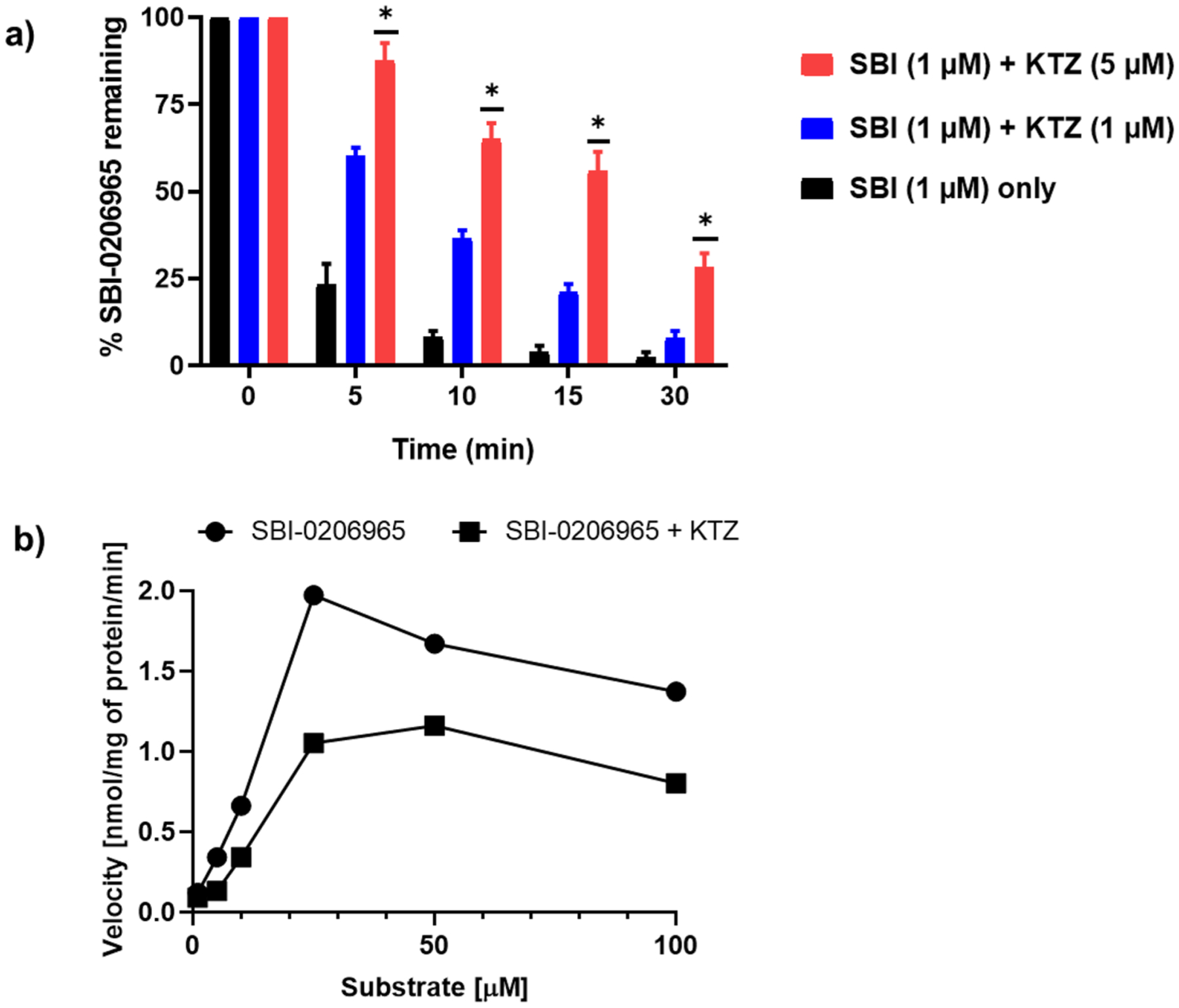
Inhibition of SBI-0206965 metabolism by ketoconazole. a) Inhibitory effect of KTZ (1 and 5 μM) on SBI-0206965 metabolism by rat liver microsomes (N = 3) measured at the indicated incubation time period. b) Michaelis–Menten plots of SBI-020695 metabolism ± ketoconazole (1 μM). Data represented as Mean ± SD. The data were analyzed for statistical significance using One-way ANOVA using GraphPad Prism 8.0.2, * = p < 0.05
Discussion
SBI-0206965, originally identified as an inhibitor of the autophagy initiator kinase ULK1, has recently been reported as a more potent and selective AMPK inhibitor relative to the widely used, but promiscuous inhibitor, Compound C/Dorsomorphin [20]. Given the emerging evidence of these regulators in various malignancies including CNS tumors, SBI-0206965 and its derivatives are being investigated for the treatment of brain cancers such as GBM and neuroblastoma [21, 22]. SBI-0206965 (compound 1, Fig. 2c), is a first-generation inhibitor in its class that was discovered from a targeted screening approach and medicinal chemistry efforts to identify ULK-1 inhibitors with “druglike” physicochemical properties. SBI-0206965 has a molecular weight of 489 g/mol, logP of 3.8, and hydrogen bonding counts which comply with the Lipinski’s Rules [9, 23]. However, the systemic pharmacokinetics of this molecule were not characterized and given its emerging significance for the treatment of brain tumors, its CNS pharmacokinetic profile needs to be investigated.
Our study shows that SBI-0206965 easily penetrates the BBB with brain ECF Cmax in rats of 360 ng/mL (0.75 μM) within 40–60 min following i.p. administration of 25 mg/kg. SBI-0206965 inhibits ULK1 in kinase inhibition assays with IC50 of 108 nM, while the cytotoxicity IC50 values range from 5–10 μM. The blood supply to the GBM is somewhat disrupted resulting in higher tumoral concentrations relative to normal brain [24]. Accordingly, in our previous study we observed that the tumoral uptake of letrozole in the orthotopically implanted C6 glioma is two-fold higher than that in normal brain in rats [14]. Collectively, our results should guide the dosing strategy for further animal experiments.
We assessed the PK of SBI-0206965 after oral administration in rats and mice. In both species, the half-life was around 1–2 h suggesting rapid drug elimination. Further, comparing the resulting plasma AUC from oral vs i.p. administration suggest a relatively low oral bioavailability of 0.15 in rats. For a mechanistic understanding of low oral availability and short elimination half-life, we assessed the metabolism of SBI-0206965 in mouse, rat, and human liver microsomes. We then determined intrinsic hepatic clearance based on published values for protein content/mg liver tissue and average total liver tissue weight in different species [18]. Further, incorporating the hepatic blood flow the total hepatic clearance was estimated, as described previously [25]. The rapid metabolic loss observed in the hepatic microsomal incubations is reflected in the estimated high hepatic clearance in each species.
The LC/MS analytical approach used here permitted structural elucidation of SBI-0206965 metabolites. The metabolites include desmethyl and didesmethylated products. In liver microsomal incubations these products are formed as a result of metabolic reactions catalyzed by CYP monooxygenases. An important point to note here is that these metabolites are also observed in rat plasma but not in the brain ECF. As a fraction of the concentrations of the parent SBI-0206965 molecule, higher metabolite levels are seen following oral administration relative to i.p. administration which suggests that this compound undergoes significant first-pass metabolism. Due to the unavailability of the metabolites from commercial sources, we were not able to evaluate if these metabolites retain the pharmacological activity of SBI-0206965.
Further experiments are required to characterize SBI-0206965 metabolism and confirm the structure of the metabolites. Additional structural elucidation and quantitative studies are warranted if SBI-0206965 is further developed for human trials. In our initial attempts for reaction phenotyping, we employed FDA recommended CYP-specific inhibitors for major CYP enzymes and noted that only ketoconazole inhibited SBI-0206965 metabolism [26, 27]. Ketoconazole is an imidazole anti-fungal agent, which is used as probe for CYP3A involvement in the metabolism of a compound [28, 29]. Given that other CYP-specific inhibitors did not alter SBI-0206965 metabolism it appears that CYP3A may be primary enzyme involved in the observed microsomal metabolism of this compound. Further, since CYP3A are the most abundant hepatic CYP enzymes accounting for nearly 40% of the total liver CYP content, it is likely that the SBI-0206965 metabolism in vivo is primarily mediated by CYP3A enzymes [30].
The observed low oral bioavailability of SBI-0206965 relative to that with intraperitoneal administration may be due poor absorption and/or hepatic first pass metabolism catalyzed by CYP3A. Further experiments are required to delineate the factors impacting low oral bioavailability, but our data suggest it may be largely due to first pass metabolism. The extent of hepatic first-pass metabolism following intraperitoneal administration is markedly lower relative to oral administration. The disappearance of drugs from the peritoneal cavity following an intraperitoneal injection is due to transport by capillary blood or lymph, enzymatic metabolism and biding to tissue proteins. The transport may occur by the mesenterial-portal vasculature, the mesenterial-extra portal and the extra-mesenterial vasculature, and the lymph vessels. Accordingly, absorption occurs rather rapidly but compounds are only partially subjected to hepatic first-pass elimination [31]. In contrast, following per oral administration, the fraction absorbed is sequentially and completely carried to the liver via the portal vein where the compound is subjected to hepatic first-pass. The distinct difference in the oral vs intraperitoneal bioavailability has indeed been observed for many CYP3A4 substrates such as midazolam and docetaxel [32–34]. The primary CYP3A enzyme in adult human liver, CYP3A4 participates in metabolism of over 60% of all marketed drugs. CYP3A4 is also susceptible to induction/inhibition by many other drugs and xenobiotics and as such CYP3A4 substrates are predisposed to elevated risk of drug-drug interactions [35].
In summary, the brain penetration of SBI-0206965 in rats was in the range of 0.6—0.9 based on comparing the Cmax and AUC of brain ECF to the unbound plasma levels of the compound. While inter-species differences in BBB penetration are well documented, our data suggests that SBI-0206965 is likely to have adequate BBB penetration in humans. However, SBI-0206965 has a short half-life and low oral bioavailability in rodents. The hepatic elimination of the compound is rapid primarily entailing demethylation catalyzed by CYP3A enzyme. Therefore, this agent is likely to have short elimination half-life and low oral bioavailability in humans as well. The oral route of administration resulted in markedly reduced Cmax and AUC relative to the i.p administration. While we did not conduct microdialysis studies following oral administration, based on the assumption of linear pharmacokinetics and unchanged blood–brain partitioning, it is unlikely that therapeutically relevant brain concentrations of SBI-0206965 will be achieved using the oral route. In the event that SBI-0206965 exhibits promising anti-GBM efficacy, efforts to develop sustained release parenteral formulations may be warranted. Another approach may entail localized delivery of compound to the brain. For instance, such methods are under investigation for temozolomide, the only drug specifically approved for the treatment of GBM, that has a short elimination half-life of approximately 2 h [36]. Alternatively, as we have suggested newer SBI-0206965 analogs with improved pharmacokinetic properties and BBB penetration may be needed for maximal exploitation of AMPK/ULK1 as therapeutic targets.
Acknowledgements
University of Cincinnati Brain Tumor Center (Pankaj B. Desai) and National Institutes of Health (NIH) partially funded this project grants R01NS099162 and R01NS114074 (Biplab DasGupta).
Funding
This project was partially funded by University of Cincinnati Brain Tumor Center (Pankaj B. Desai) and National Institutes of Health (NIH) grants R01NS099162 and R01NS114074 (Biplab DasGupta).
Footnotes
Ethics approval The experimental protocol for rat study was approved by the University of Cincinnati Institutional Animal Care and Use Committee (IACUC, Protocol No. 20–01–24–01). C57BL/6 mice were generated by in-house breeding at the Cincinnati Children’s Hospital Medical Center (CCHMC; Cincinnati, OH). Housing and experimental protocols employing C57BL/6 mice were approved by the CCHMC IACUC (Protocol No. IACUC2021–0031).
Competing interests The authors have no relevant financial or nonfinancial interests to disclose.
Data availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS (2020) CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol 22(12 Suppl 2):iv1–iv96. 10.1093/neuonc/noaa200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson DR, O’Neill BP (2012) Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol 107(2):359–364. 10.1007/s11060-011-0749-4 [DOI] [PubMed] [Google Scholar]
- 3.National Brain Tumor Society website (2022) Quick Brain Tumor Facts. https://braintumor.org/brain-tumor-information/brain-tumor-facts/. Accessed 18 Mar 2022
- 4.Bonini MG, Gantner BN (2013) The multifaceted activities of AMPK in tumor progression–why the “one size fits all” definition does not fit at all? IUBMB Life 65(11):889–896. 10.1002/iub.1213 [DOI] [PubMed] [Google Scholar]
- 5.Dasgupta B, Chhipa RR (2016) Evolving Lessons on the Complex Role of AMPK in Normal Physiology and Cancer. Trends Pharmacol Sci 37(3):192–206. 10.1016/j.tips.2015.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zadra G, Batista JL, Loda M (2015) Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol Cancer Res 13(7):1059–1072. 10.1158/1541-7786.MCR-15-0068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, Kumar A, Zhou X, Sun Y, Quinn B, McPherson C, Warnick RE, Kendler A, Giri S, Poels J, Norga K, Viollet B, Grabowski GA, Dasgupta B (2014) Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci USA 111(4):E435–E444. 10.1073/pnas.1311121111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chhipa RR, Fan Q, Anderson J, Muraleedharan R, Huang Y, Ciraolo G, Chen X, Waclaw R, Chow LM, Khuchua Z, Kofron M, Weirauch MT, Kendler A, McPherson C, Ratner N, Nakano I, Dasgupta N, Komurov K, Dasgupta B (2018) AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat Cell Biol 20(7):823–835. 10.1038/s41556-018-0126-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dite TA, Langendorf CG, Hoque A, Galic S, Rebello RJ, Ovens AJ, Lindqvist LM, Ngoei KRW, Ling NXY, Furic L, Kemp BE, Scott JW, Oakhill JS (2018) AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J Biol Chem 293(23):8874–8885. 10.1074/jbc.RA118.003547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, Teriete P, Asara JM, Turk BE, Cosford ND, Shaw RJ (2015) Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell 59(2):285–297. 10.1016/j.molcel.2015.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng J, Jiang P, Yang T, Huang M, Xie J, Luo C, Qi W, Zhou T, Yang Z, Zou Y, Gao G, Yang X (2019) β2-adrenergic receptor signaling promotes neuroblastoma cell proliferation by activating autophagy. Oncol Rep 42(4):1295–1306. 10.3892/or.2019.7266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto BK, Pehek EA (1990) A neurochemical heterogeneity of the rat striatum as measured by in vivo electrochemistry and microdialysis. Brain Res 506(2):236–242. 10.1016/0006-8993(90)91256-g [DOI] [PubMed] [Google Scholar]
- 13.Apparaju SK, Gudelsky GA, Desai PB (2008) Pharmacokinetics of gemcitabine in tumor and non-tumor extracellular fluid of brain: an in vivo assessment in rats employing intracerebral micro-dialysis. Cancer Chemother Pharmacol 61(2):223–229. 10.1007/s00280-007-0464-1 [DOI] [PubMed] [Google Scholar]
- 14.Dave N, Gudelsky GA, Desai PB (2013) The pharmacokinetics of letrozole in brain and brain tumor in rats with orthotopically implanted C6 glioma, assessed using intracerebral microdialysis. Cancer Chemother Pharmacol 72(2):349–357. 10.1007/s00280-013-2205-y [DOI] [PubMed] [Google Scholar]
- 15.Arora P, Gudelsky G, Desai PB (2021) Gender-based differences in brain and plasma pharmacokinetics of letrozole in sprague-dawley rats: Application of physiologically-based pharmacokinetic modeling to gain quantitative insights. PLoS ONE 16(4):e0248579. 10.1371/journal.pone.0248579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paxinos G, Watson CR, Emson PC (1980) AChE-stained horizontal sections of the rat brain in stereotaxic coordinates. J Neurosci Methods 3(2):129–149. 10.1016/0165-0270(80)90021-7 [DOI] [PubMed] [Google Scholar]
- 17.Naritomi Y, Terashita S, Kimura S, Suzuki A, Kagayama A, Sugiyama Y (2001) Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab Dispos 29(10):1316–24. https://dmd.aspetjournals.org/content/29/10/1316 [PubMed] [Google Scholar]
- 18.Hosea NA, Collard WT, Cole S, Maurer TS, Fang RX, Jones H, Kakar SM, Nakai Y, Smith BJ, Webster R, Beaumont K (2009) Prediction of human pharmacokinetics from preclinical information: comparative accuracy of quantitative prediction approaches. J Clin Pharmacol 49(5):513–533. 10.1177/0091270009333209 [DOI] [PubMed] [Google Scholar]
- 19.Chen A, Zhou X, Tang S, Liu M, Wang X (2016) Evaluation of the inhibition potential of plumbagin against cytochrome P450 using LC-MS/MS and cocktail approach. Sci Rep 6:28482. 10.1038/srep28482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahwazi D, Neopane K, Markby GR, Kopietz F, Ovens AJ, Dall M, Hassing AS, Gräsle P, Alshuweishi Y, Treebak JT, Salt IP, Göransson O, Zeqiraj E, Scott JW, Sakamoto K (2021) Investigation of the specificity and mechanism of action of the ULK1/AMPK inhibitor SBI-0206965. Biochem J 478(15):2977–2997. 10.1042/BCJ20210284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y, Liu L, Wang Y, Xiao S, Mai R, Zhu Z, Cao Y (2021) Glioblastoma stem cell (GSC)-derived PD-L1-containing exosomes activates AMPK/ULK1 pathway mediated autophagy to increase temozolomide-resistance in glioblastoma. Cell Biosci 11(1):63. 10.1186/s13578-021-00575-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dower CM, Bhat N, Gebru MT, Chen L, Wills CA, Miller BA, Wang HG (2018) Targeted Inhibition of ULK1 Promotes Apoptosis and Suppresses Tumor Growth and Metastasis in Neuroblastoma. Mol Cancer Ther 17(11):2365–2376. 10.1158/1535-7163.MCT-18-0176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.National Center for Biotechnology Information (2022) PubChem Compound Summary for CID 92044402. https://pubchem.ncbi.nlm.nih.gov/compound/sbi-0206965. Accessed 18 Mar 2022
- 24.Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, Giannini C, Burns TC, Kizilbash SH, Laramy JK, Swanson KR, Kaufmann TJ, Brown PD, Agar N, Galanis E, Buckner JC, Elmquist WF (2018) Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol 20(2):184–191. 10.1093/neuonc/nox175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi GW, Lee YB, Cho HY (2019) Interpretation of Non-Clinical Data for Prediction of Human Pharmacokinetic Parameters: In Vitro-In Vivo Extrapolation and Allometric Scaling. Pharmaceutics 11(4):168. 10.3390/pharmaceutics11040168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.FDA/CEDR resources page. Food and Drug Administration Web site. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#table1-2. Accessed 18 Mar 2022
- 27.FDA/CEDR resources page. Food and Drug Administration Web site. In Vitro Drug Interaction Studies - Cytochrome P450 Enzyme and Transporter-Mediated Drug Interactions Guidance for Industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Accessed 18 Mar 2022
- 28.Eagling VA, Tjia JF, Back DJ (1998) Differential selectivity of cytochrome P450 inhibitors against probe substrates in human and rat liver microsomes. Br J Clin Pharmacol 45(2):107–114. 10.1046/j.1365-2125.1998.00679.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahatthanatrakul W, Sriwiriyajan S, Ridtitid W, Boonleang J, Wongnawa M, Rujimamahasan N, Pipatrattanaseree W (2021) Effect of cytochrome P450 3A4 inhibitor ketoconazole on risperi-done pharmacokinetics in healthy volunteers. J Clin Pharm Ther 37(2):221–225. 10.1111/j.1365-2710.2011.01271.x [DOI] [PubMed] [Google Scholar]
- 30.de Wildt SN, Kearns GL, Leeder JS, van den Anker JN (1999) Cytochrome P450 3A: ontogeny and drug disposition. Clin Pharmacokinet 37(6):485–505. 10.2165/00003088-199937060-00004 [DOI] [PubMed] [Google Scholar]
- 31.Claassen V (1994) Intraperitoneal Drug Administration. In: Techniques in the behavioral and neural sciences, Vol. 12C, Elsevier, pp 46–58 [Google Scholar]
- 32.Lau CE, Ma F, Wang Y, Smith C (1996) Pharmacokinetics and bioavailability of midazolam after intravenous, subcutaneous, intraperitoneal and oral administration under a chronic food-limited regimen: relating DRL performance to pharmacokinetics. Psychopharmacology 26(3):241–248. 10.1007/BF02246454 [DOI] [PubMed] [Google Scholar]
- 33.Kruijtzer CM, Beijnen JH, Schellens JH (2002) Improvement of oral drug treatment by temporary inhibition of drug transporters and/or cytochrome P450 in the gastrointestinal tract and liver: an overview. Oncologist 7(6):516–530. 10.1634/theoncologist.7-6-516 [DOI] [PubMed] [Google Scholar]
- 34.Hendrikx JJ, Lagas JS, Rosing H, Schellens JH, Beijnen JH, Schinkel AH (2013) P-glycoprotein and cytochrome P450 3A act together in restricting the oral bioavailability of paclitaxel. Int J Cancer 132(10):2439–2447. 10.1002/ijc.27912 [DOI] [PubMed] [Google Scholar]
- 35.Sato S, Matsumiya K, Tohyama K, Kosugi Y (2021) Translational CNS Steady-State Drug Disposition Model in Rats, Monkeys, and Humans for Quantitative Prediction of Brain-to-Plasma and Cerebrospinal Fluid-to-Plasma Unbound Concentration Ratios. AAPS J 23(4):81. 10.1208/s12248-021-00609-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adhikari B, Li J, Brandel MG, Futalan D, Akers J, Deming T, Chen CC, Carter BS (2017) The use of TMZ embedded hydrogels for the treatment of orthotopic human glioma xenografts. J Clin Neurosci 45:288–292. 10.1016/j.jocn.2017.07.027 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.