

Abstract
Chronic inflammation is frequently invoked as a mechanism of neurodegeneration and yet inflammatory cell infiltrates are seldom seen in brains of these disorders. Different disciplines utilize different technologies and methodologies to describe what is immunologically defined as the innate immune response (IIR). We examined murine models of the human neurodegenerative disease Aicardi‐Goutières Syndrome, where an IIR is initiated by aberrant RNA metabolism secondary to a mutation in adenosine deaminase acting on RNA gene (ADAR1). We previously showed that these mice demonstrated a deficit in RNA editing that lead to MDA‐5 mediated RNA sensing pathway activation of the IIR with massive interferon stimulated gene transcription and translation. As early as 2 weeks of age, in situ hybridization demonstrated that different central nervous system (CNS) cell lineages expressed very high levels of distinct interferon stimulated genes (ISGs) in the absence of interferon and absence of immune cell infiltrates. We have expanded these studies to more completely describe the breadth of ISG expression systemically and in CNS using double label in situ hybridization. Within the CNS aberrant ISG expression was mostly limited to neurons, microglia, ependyma, choroid plexus, and endothelial cells with little expression in oligodendroglia and astrocytes except for STAT1. Wild type controls showed a similar pattern of ISG expression but only in aged mice and at levels minimally detectable by in situ hybridization. Despite months of elevated ISG expression in mutant mice, there was essentially no inflammatory infiltrate, no interferon production and minimal glial reaction. Histomorphological neurodegenerative pathology of ventricular dilatation and deep gray matter mineralization were evident in mutant mice 8–13 months of age but this did not show a spatial relationship to ISG expression. This IIR without immune cell infiltration leads to neurodegeneration through non‐canonical pathways that may accentuate normal aging pathways.
Keywords: innate immune response, interferon stimulated genes, neurodegeneration, neuroinflammation
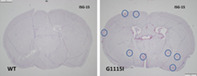
The brains of 8‐week‐old wildtype (WT) mice (left) and mice carrying mutations (D1113H) in the ADAR1 gene (right) probed for expression of Interferon Stimulated Gene 15 (ISG‐15) using In situ hybridization (red). WT mice show little to no expression of ISG‐15 while ADAR1 mutant mouse brain shows a broad and chaotic distribution of expression throughout the brain (8 of many foci circled).
1. BACKGROUND
Aicardi‐Goutières Syndrome (AGS) is an early‐onset encephalopathy characterized by intellectual and motor disability [1, 2, 3]. Disease onset begins in utero in a quarter of patients and within the first year of life for most of the remaining patients. An antecedent infection or vaccination has been noted in about three quarters of patients [3]. Initial clinical symptoms mimic congenital infection and include hepatosplenomegaly, elevated serum liver enzymes, and peripheral blood “Interferon signature” (i.e., increase interferon [IFN] stimulated gene [ISG] expression detected by quantitative polymerase chain reaction). Approximately three quarters of patients are disabled with an encephalopathy including irritability, fever, and delayed head growth. A common systemic sign of chilblain skin lesions in fingers and toes develops in 40% of patients.
Diagnosis is suggested by the above clinical signs and symptoms in conjunction with neuroimaging demonstrating deep gray and white matter calcification, dystrophy, and vasculopathy. After an initial encephalopathic stage further neurological deterioration is not prominent [1]. Increased IFN type 1 activity in cerebrospinal fluid and serum and increased ISG expression in peripheral blood characterize the disease. The diagnosis is confirmed by demonstrating biallelic mutations in any of 7 genes (ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and TREX1) or heterozygous dominant mutations in TREX1, ADAR, or IFIH1. Lethality depends upon the involved gene but in a meta‐analysis of 374 patients only 19% had died, most before the age of 15 [1].
The genetics support the hypothesis that the pathogenesis of AGS relates to deficient nucleic acid editing generating double stranded RNA sequences that activate the IIR through a MDA5 dependent RNA sensing pathway, eliciting a cascade of ISG expression and translation. Neurodegeneration occurs in the context of an “interferon signature” of gene expression but in the relative absence of IFN and inflammatory infiltrates. Detailed descriptions of the neuropathology associated with AGS are sparse [4, 5, 6, 7, 8]. While it is not clear if all genetic mutations associated with AGS have identical pathology, some degenerative features are shared. In general atrophy and gliosis are observed in the context of a possible microangiopathy with vasculocentric mineralization. Microinfarcts have been described, but primary inflammatory infiltrates are conspicuously absent.
We used two new murine models of AGS to better characterize altered gene expression within the central nervous system (CNS) and its relationship to neurodegeneration. Our initial studies of murine models of AGS with D1113H or K999N mutations in the catalytic domains of ADAR1 showed growth retardation and altered RNA editing [9]. Elevated ISG expression in absence of IFN in the brain was associated with mild astrocytosis and microgliosis at 8 weeks of age. Genetic deletion of MDA5 rescued mice from this phenotype. We have expanded these studies to assess systemic ISG expression and confirm the CNS morphological localization of ISG expression using double‐label in situ hybridization (ISH) with cell lineage markers. As seen in the CNS, ISG was also increased systemically. Organ ISG expression showed a broad and chaotic multifocal distribution in tissue histiocytes, endothelial cells and select organ specific cells (e.g., hepatocytes, myocytes). While there was no significant inflammatory infiltrate in the brain or systemic organs, with age the brain showed neurodegenerative changes (atrophy, gliosis, and vascular mineralization) spatially unrelated to ISG expression. Aged wild type (WT) control mice also began to show ISG expression in a pattern analogous to the robust expression seen in the mutant mice.
2. MATERIALS AND METHODS
Mouse genome mutagenesis was carried out as previously described through the CRISPR/Cas9 gene editing approach. Mutation in the catalytic domain of ADRA1 was confirmed by Sanger sequencing. Mice were maintained in a SPF animal facility in University of Pittsburgh School of Medicine. Studies were approved by IACUC at the University of Pittsburgh. Aberrations in RNA were confirmed as previously described. Based upon RNA and protein screening assays of brain extracts, a group of nine genes (Table S2) were selected for further analysis by in situ hybridization.
A total of 44 mice were used in these studies (see Table 1).
TABLE 1.
Mice were used in these studies
| Strain | Age | Number |
|---|---|---|
| K999N | 8 weeks | 5 |
| WT control | 8 weeks | 10 |
| K1113D | 8 weeks | 8 |
| K999N | 6 months | 4 |
| K1113D | 8 months | 3 |
| WT | 8 months | 4 |
| K1113D | 12–13 months | 3 |
| WT | 12–13 months | 4 |
| WT | 20 months | 3 |
Mice were humanely sacrificed, and brain and systemic tissues (heart, lung, liver, spleen, intestine, and kidney) were immersion fixed in 4% paraformaldehyde, before dehydration and embedding in paraffin. Five‐micron thick sections were stained with H&E, immunohistochemistry for glial fibrillary acidic protein (GFAP), and Ionized calcium binding adaptor molecule 1 (Iba1) with antibodies; mouse anti‐GFAP (catalog# 837202, BioLegend) and rabbit anti‐IBA‐1 (catalog# WDG5619, WAKO) each at a dilution of 1:1000, followed by secondary antibodies and peroxidase. ISH studies were performed on FFPE tissue sections using commercial RNAscope Target Probes (Advanced Cell Diagnostics, Hayward, CA; see Table S2). Pretreatment, hybridization, and detection techniques (RNAscope 2.5HD) were performed according to manufacturer's protocols and as previously described []. Table S2 lists the cell lineage and IIR gene probe catalog numbers and targeted sequences. For double label ISH, a cell lineage probe in channel 1 and ISG probe in channel 2 were used according to manufacturer's specifications and developed with a double chromogenic reaction (RNAscope 2.5 Duplex Reagent Kit Mouse [cat. no 322436]).
3. RESULTS
3.1. At 8 weeks of age, the mutant ADAR1 (D1113H and K999N) and WT mice showed no histologic evidence of inflammation or other significant pathology
Systemic organs including: lung, heart, kidney, liver, spleen, and intestine from WT and ADAR1 mutant mice were stained with H&E. The H&E histology of the systemic organs showed no developmental nor histopathological abnormalities. Specifically, there was no evidence of inflammation or tissue loss.
Brains of WT and ADAR1 mutant mice at 8 weeks of age were indistinguishable by H&E histology (Figure 1). There was no evidence of an inflammatory infiltrate, nor subjective loss of neuronal or glial tissue. To more sensitively assess subtle neuropathological changes, brains were immunohistochemically stained for GFAP (Figure 1B,E) and ionized calcium‐binding adapter molecule 1 (IBA1; Figure 1C,F) to discern astrocytosis or microgliosis, respectively. (Figure 1) Subjectively mild accentuation of IBA1 and GFAP stain could occasionally be appreciated but neither of these special stains demonstrated significant histopathology in WT or D1113H or K999N mice at 8 weeks of age.
FIGURE 1.
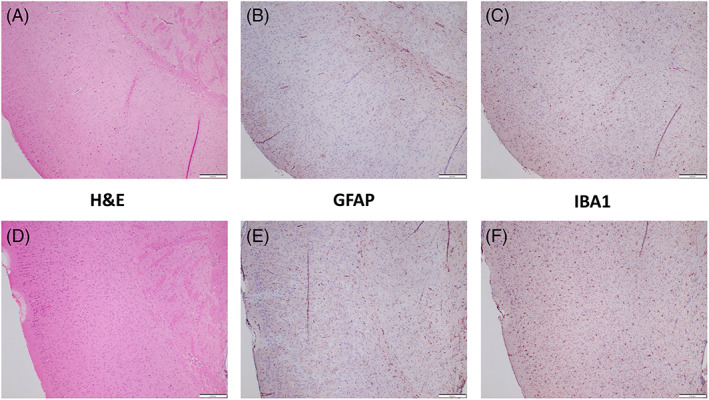
Paraffin sections of formalin fixed paraffin embedded (FFPE) brains of wild type (WT) (top) and D1113H (bottom) 8‐week‐old mice. (A,D) H&E stain of cortex demonstrated no developmental abnormalities or histopathology. Specifically, there was no evidence of immune cell infiltration. Immunohistochemical stain for GFAP (B,E) and IBA1 (C,F) showed no evidence of significant astrocytosis or microgliosis. Bar = 200 microns
3.2. Aged (8–13 months old) D1113H mice showed brain atrophy and mineralization of thalamic blood vessels in the absence of inflammatory cell infiltration
To assess pathologic changes in older D1113H mice, three 8‐month‐old and three 13‐month‐old mutant mice were sacrificed and histologically evaluated and compared to age‐matched controls. For the most part aged mutant mouse brains showed atrophy and minimal gliosis (Figure S1). D1113H brains were smaller and showed lateral and third ventricle dilation but no inflammatory cell infiltrates were noted in these older mice. The 8‐month‐old mice brains showed mild focal and symmetrical astrocytosis in the thalamus. Mild vascular mineralization was note in these regions with surrounding astrocytosis. In 12–13‐month‐old mice, the vascular mineralization and perivascular astrocytosis were similarly distributed but more pronounced. (Figure 2)
FIGURE 2.
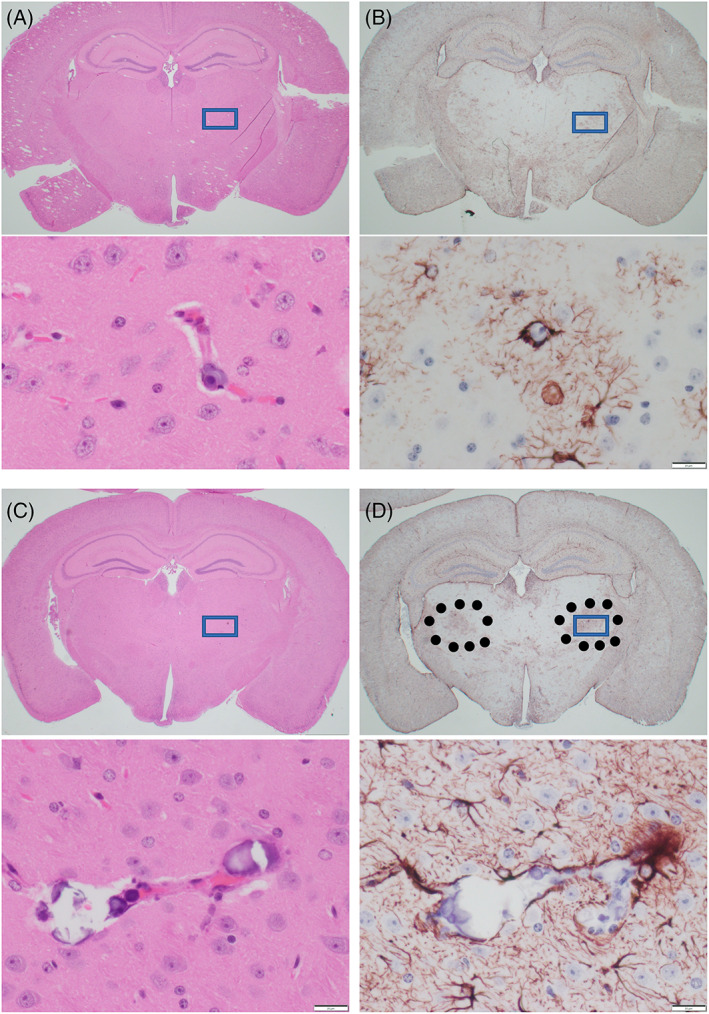
Paraffin sections of FFPE brains of 8‐month (A,B) and 13‐month‐old (C,D) D1113H mice. (A) H&E stain of coronal section of 8‐month‐old D1113H mouse brain. At low power no significant pathology is appreciated. In the inset at high power rare blood vessel in the thalamus shows focal mineralization. (B) In a successive section stained for GFAP symmetric regions of mild gliosis are noted in the thalamus. In the inset at high power these regions show reactive astrocytes surrounding mineralized blood vessels noted in the H&E. (C) H&E stain of coronal section of 13‐month‐old D1113H mouse brain. At low power no significant pathology is appreciated. In the inset at high power blood vessels in the thalamus shows focal vascular mineralization. (D) In a successive section stained for GFAP symmetric regions of gliosis are noted in the thalamus (surrounded by dots). In the inset at high power these regions show reactive astrocytes surrounding mineralized blood vessels as noted in the H&E. Bar in inset = 20 microns
3.3. By ISH mutant D1113H and K999N mouse systemic organs showed cell lineage specific expression of ISGs in a multifocal chaotic distribution
To assess tissue and cell specific expression of select ISGs (ISG‐15, CXCL10, and CCL5), chromogenic ISH was performed as previously described (Figure S2). By ISH, the liver of both mutant strains demonstrated expression of ISG15 in widely scattered foci of parenchymal hepatocytes (Figure S2A, left). Expression of CXCL10 (Figure S2A, middle) showed a similar pattern with additional intensely stained and widely distributed cells morphologically consistent with Kupffer cells. Expression of CCL5 (Figure S2A, right) was limited to discrete intensely stained cells in a histological distribution consistent with endothelia or Kupffer cells. The heart (Figure S2B) showed no expression of ISG‐15. CXCL10 expression was limited to rare foci of intensely stained myocardial cells. CCL5 showed rare staining of cells with elongated cytoplasmic processes consistent with endothelia. The lung (Figure S2C) showed expression of ISG‐15 in bronchial epithelium. CXCL10 showed expression in discrete foci of alveoli. As the foci were scattered and did not follow anatomical or discernably random distribution patterns, we use the expression “multifocal chaotic” to describe them. CCL5 showed a broad even distribution of intensely stained cells in a distribution consistent with endothelia. The intestine (Figure S2D) showed expression of ISG‐15 within epithelium, stronger in apical than basal regions. CXCL10 expression was limited to macrophages within discrete regions of lamina propria. CCL5 expression was more limited than CXCL10 expression but also limited to macrophages within lamina propria. The kidney (Figure S2E) showed background staining of proximal tubules in addition to discrete staining of occasional tubule epithelial foci. CXCL10 expression was observed in multiple foci on tubular epithelium in addition to scattered interstitial cells. CCL5 expression was widely distributed in interstitial cells morphologically and histologically consistent with macrophages. The spleen (Figure S2F) showed expression of all three cytokines predominantly within the center of follicles in cells morphologically consistent with macrophages. These results are summarized in the Table S1.
3.4. By ISH D1113H and K999N mutant mouse brains showed cell lineage specific expression of ISGs in a multifocal and chaotic pattern
The brains of mutant mice showed strong and multifocal staining for ISG15, OASL2, and STAT1 with less intensive hybridization noted for ifit1 (Figure 3A–G). Superficial cortical neurons showed higher signal than deep cortical or subcortical neurons. However, the staining pattern was not uniform and instead showed elements of a chaotic patch nature conforming neither to anatomical or vascular territories (see Section 4). Positive regions of neuronal ISG15 hybridization were in register with positive regions of other ISG expression by other cell types (e.g., CXCL10 in microglia) (especially Figure 3E,F). None of the screened ISGs showed elevated hybridization in astrocytes or oligodendrocytes except for STAT1 which showed moderate increased staining in oligodendrocytes and astrocytes compared to WT controls. Endothelium showed modest elevation of CCL5, OASL1, OASL2, and IRF7. Ependyma showed two patterns of hybridization; uniform expression of ISG15, OASL2, IRF7, and STAT1 in all epithelial cells, more limited staining of cells interdigitating in the ependyma (representing from 1:5 to 1:20 of lining cells) staining with CCL5, OASL1, and ifit1 (3C). There was modest sub‐pial and meningeal staining. Scattered cells in deep white matter also stain, however, their lineage was difficult to distinguish with certainty on morphology alone, although they were most consistent with endothelia.
FIGURE 3.
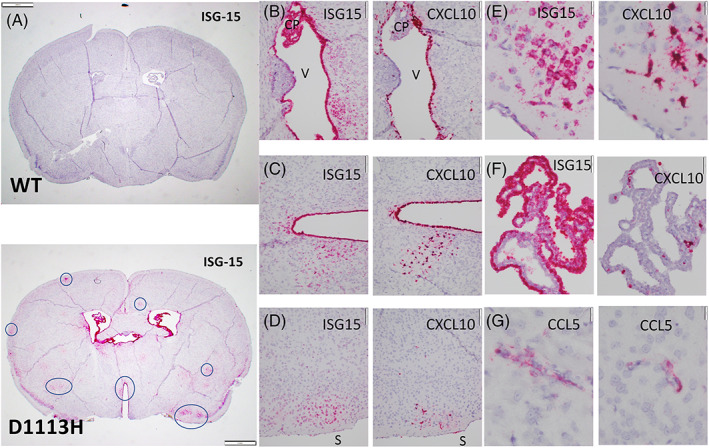
ISH on sections of FFPE paraffin embedded brains. (A) Low power complete coronal section from 8‐week‐old WT (top) and D1113H (bottom) mice were hybridized for ISG‐15. At 8 weeks WT mice showed essentially no hybridization. The brains of D1113H mice showed lateral and third ventricular dilation and multiple variably distributed foci of strong and diffuse staining for ISG15 in neurons (7 foci circled). Staining was greater in superficial neurons than deep neurons. There was intense staining of ependymal and choroid plexus epithelium with more modest sub‐pial and meningeal staining. Bar = 1 mm. (B) High power image of successive sections of periventricular region (“V” in ventricle) of D1113H mice hybridized for ISG‐15 (left) and CXCL10 (right). ISG‐15 shows intense expression in the ependyma and choroid plexus (CP) along with subependymal neurons. CXCL10 expression is also intense in some portions of the ependymal lining but there are regions without staining. CXCL10 expression in the choroid plexus is limited to individual macrophages. Bar = 100 microns. (C) High power image of successive sections of periventricular region of D1113H mice hybridized for ISG‐15 (left) and CXCL10 (right). ISG‐15 shows intense expression of the ependyma and subependymal neurons. CXCL10 expression in ependyma is more limited. Bar = 100 microns. (D) High power image of successive sections of subpial cortical region of D1113H mice (brain surface labeled “S”) hybridized for ISG‐15 (left) and CXCL10 (right). ISG‐15 shows intense expression in a focus of superficial neurons while CXCL10 expression in the successive section shows intense expression in the same focus but not in neurons but rather cells with morphology consistent with microglia. Bar = 100 microns. (E) Higher power of D demonstrating the neuronal morphology of the ISG‐15 positive cells and the microglial morphology of the CXCL10 positive cells. Bar = 20 microns. (F) High power of successive images of choroid plexus showing intense hybridization of ISG‐15 (left) in epithelia cells compared to intense hybridization for CXCL10 in macrophages of choroid plexus. Bar = 50 microns. (G) High power of select cortical regions hybridized for CCL5 showing discrete staining of cells with morphology consistent with endothelia. Bar = 20 microns
The brains of mutant mice demonstrated a different expression pattern of ISH for CXCL10 as opposed to ISG‐15. Scattered foci of microglia showed intense staining for CXCL10. There was intense staining of ependyma and choroid plexus macrophages. There was weak staining of scattered neuronal elements.
The brains of mutant mice demonstrated yet another expression pattern of ISH staining for the cytokine CCL5. Overall staining for CCL5 was less intense than that for CXCL10 and ISG‐15. Scattered microglia and endothelia showed modest staining (3G). Regions varied from 1:1 to 1:10 ependymal cells staining while choroid plexus epithelium was negative. However intense staining of macrophages in the choroid plexus was observed. The ISH staining of ISG expression in the brain is summarized in the Table 2.
TABLE 2.
Summary of in situ hybridization staining of interferon stimulated genes in the brains of ADAR1 mutant mice
| ISG15 | CXCL10 | CCL5 | OASL1 | OASL2 | IRF7 | ifih1/MDA5 | ifit1 | STAT1 | IFNb | |
|---|---|---|---|---|---|---|---|---|---|---|
| Neurons | Frequent foci | − | − | Rare foci | Occasional foci | − | Rare foci | Occasional foci | Most neurons | − |
| Astrocytes | − | − | − | − | − | − | − | − | Moderate in some cells | − |
| Oligodendroglia | Occasional cells weak | ‐ | − | − | − | − | − | − | Moderate in some cells | − |
| Ependyma epithelium | Strong most cells | ~50%–75% of cells | Interdigitating cells | Interdigitating cells | Strong most cells | Strong most cells | Moderate most cells | Moderate most cells | Strong most cells | − |
| Endothelia and perithelial cells a | + | − | + | + | ++ | ++ | + | ++ | ++ | − |
| Microglial nodules | − | +++ | + | − | − | − | − | − | − | − |
| Choroid plexus | Strong most cells | Interdigitating cells | Interdigitating cells | Interdigitating cells | strong most cells | Interdigitating cells | Moderate most cells | − | Strong most cells | − |
Given spatial proximity of perivascular microglia and endothelial cells, distinguishing these elements in capillaries of brain even with double label ISH is difficult (see Section 3).
3.5. Double label ISH staining for cell lineage markers and ISGs confirmed the expression patterns based upon cell morphology
To confirm the morphologic definition of cell lineage specific ISH, double label ISH was performed using probes for specific cell types (Synaptophysin for neurons, Olig2 for oligodendroglia, GFAP for astrocytes, and CD31 for endothelial cells) and three select ISGs (ISG‐15, CXCL10, and CCL5). To illustrate the predominant ISG expression in specific cell lineages, examples of the double label results are shown in Figure S3.
Microglia have a complex distribution in the brain. Many are tightly associated with endothelia (i.e., pericytes) while others show greater extension into the brain parenchyma. In regions of high neuronal ISG‐15 expression, microglia showed strong CXCL10 staining (Figure 3E). In other areas where ISG expression was noted in association with small vessels, the vascular anatomy of endothelial cells and microglia was so proximate that it was difficult to attribute the ISG expression to either endothelial cells versus microglia. We attempted to use double label ISH with endothelial (CD31) or microglial (Aif1) specific cell lineage probes in conjunction with ISG probes. Even the double label ISH technique did not permit unambiguous definition of the expression cell. However, within lightly stained microglial nodules CXCL10 staining clearly associated with Aif1 staining (Figure S3C) supporting the morphological attribution of CXCL10 positive cells as microglia (see Section 4).
3.6. ISG expressing regions did not specifically show elevated expression of astrocytic (GFAP) or microglial markers (IBA1)
Successive paraffin brain sections were used to compare H&E histology, immunohistochemistry for astrocytosis (GFAP) and microgliosis (IBA1) along with expression of the 3 cytokines (Figure 4). These comparisons highlight the discordance between intense cytokine expression and absence of histopathology.
FIGURE 4.
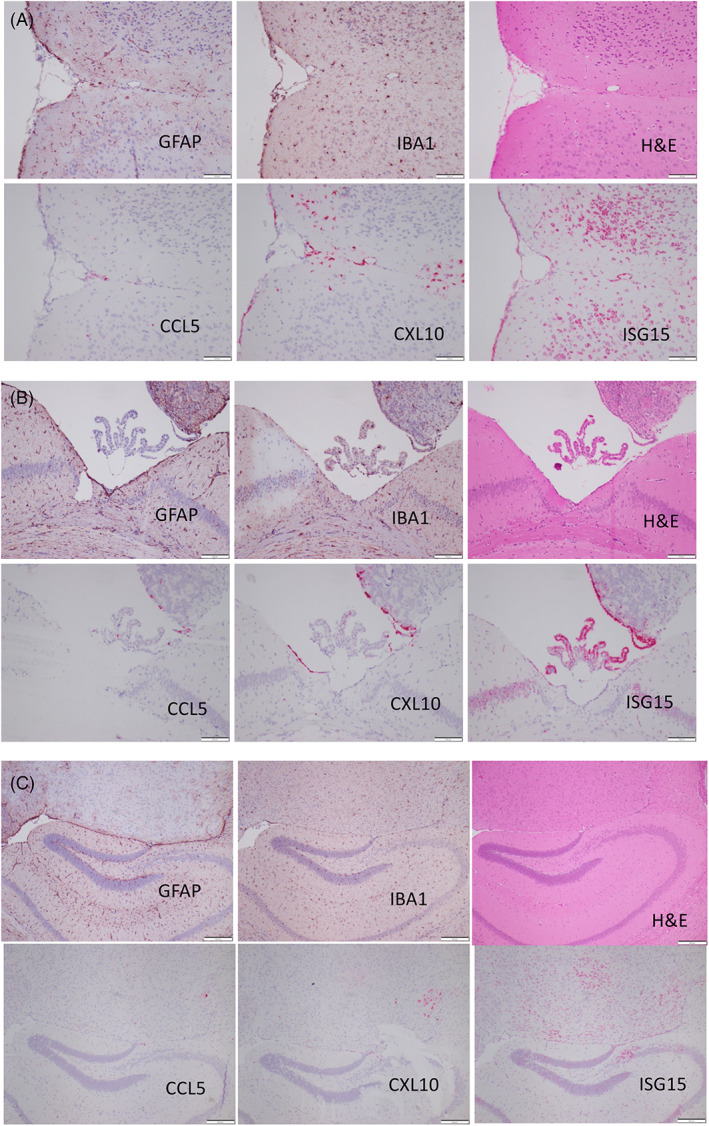
Successive paraffin sections of different brain regions demonstrating absence of astrocytosis (GFAP) and microgliosis (IBA1), in cortical (A), ventricular (B), and hippocampal (C) regions of D1113H mice with intense hybridization for ISG‐15, CXCL10 and less intense staining for CCL5. Bar = 100 microns
3.7. Aged D1113H mice showed persistent cell specific expression of ISGs while aged WT mice showed ISG staining in a similar but much less intense pattern
As seen at 8 weeks of age (Figure 3), ISH staining in 13‐month‐old D1113H mice for ISG‐15 (Figure 5B,D,F) showed intense staining in ependyma, choroid plexus, and neurons. However, unlike 8‐week‐old WT mice which showed no appreciable hybridization for ISG 15, 13‐month‐old WT mice (Figure 5A,C,E) showed weak staining in a similar distribution to D1113H mice. Higher power of choroid plexus, ependyma, and subependymal neurons showed attenuate staining in aged WT (Figure 5C,E) compared to aged mutant (Figure 5D,F) brains.
FIGURE 5.
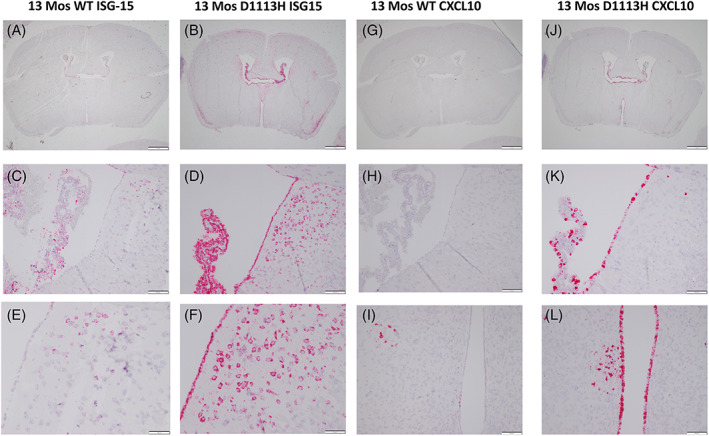
Aged (13‐month‐old) WT and D1113H mutant mice showed cell specific expression of ISG‐15 (A–E) and CXCL10 (F–L). While 8‐week‐old WT mice showed essentially no ISG expression (Figure 3A) aged WT mice began to show ISG staining in a pattern but much less intense to the in mutant mice. At low power (A,B,G,J) 13‐month D1113H mouse brain is a little smaller and shows ventricular dilatation along with persistent ISG expression. Faint hybridization is seen in the 13‐month WT mouse choroid plexus even at this low power (A). Higher power of select regions of ISH for ISG‐15 and CXCL10 permit comparison of aged WT and D1113H mutant expression. The pattern of cell staining is the same for aged WT and D1113H mice, however, signal intensity is very attenuate in the aged WT mouse brain. Bar = 1 mm in A,B,G,J, Bar = 100 microns in C,D,E,F,H,K,I,L
As seen at 8 weeks of age, ISH staining in 13‐month‐old D1113H mice for CXCL10 (Figure 5H,J,L) showed intense staining in ependyma and choroid plexus. Again while 8‐week‐old WT mice showed no ISH staining 13‐month‐old WT mice (Figure 5G,I,K) showed weak staining in a similar distribution to 8‐month‐old D1113H mice.
In summary, young adult WT mice showed little to no expression of ISGs. However aged WT mice (8–13 months old) showed attenuated ISG expression in a pattern analogous to that seen in the ADAR1 mutant mice (Figure S4).
4. DISCUSSION
4.1. Summary of findings
The principal findings from this study of a murine model of AGS are; (1) Mutations in the ADAR1 gene are associated with abundant tissue expression of ISGs in the absence of significant immune cell infiltration. (2) Nevertheless, with age, mutant animals show brain atrophy and mineralization. (3) ISH demonstrates a multifocal “chaotic” distribution of cell lineage specific ISG expression in brain and systemic organs. (4) The brains of aged wild‐type mice showed similar but less intense ISG expression as that shown in young adult ADAR1 mutant mice.
AGS is associated with mutations in seven proteins related to either RNA editing or detection of non‐edited RNA by the cellular innate immune response (IIR) [1]. While it is theoretically possible that abnormally edited specific RNAs could account for a degenerative phenotype, the fact that mutations in any of the AGS genes leads to a similar phenotype despite their acting on select RNAs, would support the hypothesis of a more general pathogenic mechanism. In its simplest rendition, one could hypothesize that detection of non‐edited RNAs by the cellular IIR initiates the general pathological process. That knockout of either MDA5 or MAVS in the RNA‐sensing pathway rescues the various mouse AGS models [10], supports the hypothesis that MDA5 detection of any of several non‐edited RNAs leads to signaling in the nucleus and induction of ISGs even in the absence of IFN. If this is the pathogenic mechanism, our findings raise several important questions.
4.2. Why do different cell lineages express different ISGs?
Despite all cells having the same genetic mutation in nucleic acid editing, different cell lineages express different cytokines. Said another way, rather than all cells expressing all of the “interferon signature” cytokines, different cell lineages express select cytokines. This would be consistent with a general IIR acting on cells with different promoter and enhancer availability [10]. It may be possible to experimentally dissect this with in vitro studies. Given the role of microglia as sentinel CNS cells in inflammation, we expected many ISGs would be expressed by these cells. However, only CXCL10 showed robust expression in microglia. It is possible that some ISG expressing interdigitating cells within choroid plexus or ependyma or cells intimately associated with endothelia are of microglia phenotype, but morphology alone did not permit that resolution and the Aif1 double labels could not resolve that unambiguously. In vitro isolation of microglia would be necessary to fully address this question. In RNA‐seq analysis that identified injury and aging‐associated microglial subsets, one cluster expressed CXCL10 [11]. Conceivably, a microglial subpopulation could account for the topographic distribution of heightened CXCL10 expression in the ADAR1 mutant mice.
A report by Inoue et al. [9] identified ISG RNAs in distinct neuronal subtypes in the K999N mutant mouse using RNA seq. Their analysis of excitatory neurons, inhibitory neurons, oligodendrocytes, and astrocytes showed variably heightened levels of their analyzed ISGs in all of these cell types. That analysis did not capture microglia, and did not report ISG15 and CXCL10 levels, so we cannot compare our topographic analysis with that RNA seq investigation of whole brain ISGs. Notably, as in our investigations, Inoue et al did not observe type 1 IFN expression in mutant ADAR1 brain. This supports the possibility that internal signaling triggers engage upstream transcription factors such as STAT1, Sting, or NF‐kB to promote the ISG response.
4.3. What determines what regions of the brain show induction of ISGs?
The multifocality of the differential cytokine expression is also enigmatic. The brain hybridizations demonstrate widespread multifocal expression that does not obey neuroanatomical or neurophysiological principles. What is driving selective regional expression? The fact that different cell types (e.g. neurons and microglia) in the same region are showing selective expression suggests a microenvironmental stimulus stochastically initiating expression. Is a “second hit” (e.g., physiological stress) involved in activating different foci? The multifocal “chaotic” regional expression pattern is similar in young and old mutant mice. Whether specific regions wax and wane over time, remains to be determined with more dynamic assays. Whether ADAR1 mutation uncovers distinct neuronal [12] or microglial subtypes [13] dispersed through the brain or triggers distinct ISG expression in a microenvironment‐dependent way also remains to be determined. The possibility of neuronal/microglial crosstalk supporting sustained expression of distinct ISGs in these regions may be presaged by effects of CXCL10‐expressing microglia on neuronal biology in other settings [14].
4.4. With such abundant ISG expression why is there so little gliotic reaction and so little inflammatory cell infiltrate?
In many ways the histopathological findings in the D1113H and K999N mouse model of AGS are particularly remarkable for their absence. Despite markedly elevated expression and translation of potent cytokines, a clear “Interferon signature” within the nervous system and body, there is no significant evidence of an immune inflammatory infiltrate. Even an innate tissue response like gliosis is missing until the mice have aged. This is unexpected and unlike several other transgenic models where artificial expression of individual cytokines has been associated with an intense immune response [15, 16, 17]. A hypothetical block in cytokine protein translation is not supported by the clear concordance of RNA abundance and Illumina protein assays. Other potential blocks in cytokine function include absence of; cytokine processing, cytokine release or functional cytokine receptors. Whatever the block, findings described here clearly show that simple activation of the cytokine induction by the IIR does not lead to inflammatory cell infiltrate and that an additional process (e.g., cell stress, infection) needs to be sought. It is important to note that the ISH observations of increased ISG expression in the AGS murine model are occurring in the context of no elevation of IFN. While this may seem paradoxical, IFN independent induction of ISGs has been observed in viral infections (e.g., HCMV [18]) and found to be IRF3 and cell type dependent [19].
4.5. Does chronic activation of IIR with transcription and translation of ISGs exact a metabolic toll on individual cells?
It is difficult to imagine that the observed robust cytokine expression does not incur a metabolic cost to the cell. While gliosis was not observed in young adult mutant mouse brains, with age, mild degenerative gliosis and vascular mineralization were seen but not in regions particularly notable for ISG expression. Does chronic IIR activation exacerbate age related neurodegenerative changes? Would sensitive behavioral or physiological markers of age‐related neurodegeneration show exacerbation or earlier onset in AGS mice? Would other cellular stresses (e.g., infection) exacerbate the chronic IIR induced pathogenic changes? Given the absence of ISH staining in 8‐week‐old WT mice, we were surprised to see cytokine expression in the aged WT mice in a pattern that mimicked mutant mice. This finding suggests that changes in RNA editing and an associated ISG burden may underly age‐related neurodegeneration.
4.6. Limitations of study
The current study is limited to analysis of mutation in only one of seven genes associated with AGS. Whether mutation in the other six genes will generate similar patterns of ISG expression remains to be shown empirically. However, all seven genes contribute to metabolism of nucleic acids that are detected by the IIR, thus a similar final common pathway to ISG expression could be conjectured. As noted above, the findings raise many unanswered questions most importantly; what explains the “chaotic” pattern of ISG expression? Future work applying technology like digital spatial profiling may permit identification of regional cell–cell signaling that could explain the chaotic expression. That similar expression changes are seen in aged wild‐type mice raises the intriguing possibility that the degenerative mechanism seen in this AGS model may be related to the general pathway of age‐related neurodegeneration.
4.7. Conclusion
Mice carrying mutation in ADAR1 gene show activation of IIR with high level expression of ISGs in brain and systemic organs in the absence of immune cell infiltration. ISG expression in mutant mice mimics that seen in aged wild‐type mice raising the possibility that activation of IIR is a general pathway to neurodegeneration.
AUTHOR'S CONTRIBUTION
Drs. Wiley, Steinman and Wang equally contributed to the experimental design, data interpretation and writing of this manuscript.
CONFLICT OF INTEREST
The authors acknowledge no commercial conflicts of interest in this work.
Supporting information
Figure S1 FFPE sections of 13‐month WT and D1113H mouse brains were stained with H&E (left), GFAP (middle) and IBA1 (right). While the mutant mouse brain (D,E,F and J,K,L) is a little smaller and has larger ventricles than the age matched WT (A,B,C and G,H,I) mouse brain, for the most part there is no significant difference in gliosis markers. A–F Bar = 1 mm, G–L Bar = 200 microns.
Figure S2 (A–F) ISH on sections of FFPE paraffin embedded systemic organs from 8‐week old D1113H mice were hybridized for ISG‐15 (left), CXCL10 (middle), or CCL5 (right). (A) The liver demonstrated expression of ISG15 in widely scattered foci of parenchymal hepatocytes. Expression of CXCL10 showed a similar pattern with additional intensely stained and widely distributed cells morphologically consistent with Kupffer cells. Expression of CCL5 was limited to discrete intensely stained cells in a histological distribution consistent with endothelia or Kupffer cells. (B) The heart showed no expression of ISG‐15. CXCL10 expression was limited to rare foci of intensely stained myocardial cells. CCL5 showed rare staining of elongated cytoplasmic processes consistent with endothelia. (C) The lung showed expression of ISG‐15 in bronchial epithelium. CXCL10 showed expression in discrete foci of alveoli. CCL5 showed a broad even distribution of intensely stained cells in a distribution consistent with endothelia. (D) The intestine showed expression of ISG‐15 within epithelium stronger in apical than basal regions. CXCL10 expression was limited to macrophages within discrete regions of lamina propria. CCL5 expression was more limited than CXCL10 expression but also limited to macrophages within lamina propria. (E) The kidney showed background staining of proximal tubules (diffuse non‐granular pink staining) in addition to discrete staining of occasional tubule epithelial foci. CXCL10 expression was observed in multiple foci on tubular epithelium in addition to scattered interstitial cells. CCL5 expression was widely distributed in interstitial cells morphologically and histologically consistent with macrophages. (F) The spleen showed expression of all three cytokines predominantly within the red pulp in cells morphologically consistent with macrophages.
Figure S3 FFPE sections of 8‐week D1113H mouse brains stained with double label ISH for interferon stimulated genes (red; A–D = ISG‐15, B,C = CXCL10) and cell lineage markers (blue). Double label staining confirms the morphological interpretation that neurons express abundant ISG‐15 (A) while oligodendroglia and astrocytes and endothelial cells express little of these gene product. Double label staining for CXCL10 confirms the morphological interpretation that neurons, oligodendroglia, astrocytes, and endothelial cells express little of this gene product (D–H). Double staining for CXCL10 and AIF1 shows frequent colocalization in microglial nodules (I–K) while choroid plexus contains macrophage elements double labeled for both CXCL10 and AIF1 (L). Bar = 50 microns.
Figure S4 FFPE sections of 13‐month WT mouse brains were hybridized for ISG‐15 (A–C) and CXCL10 (D–F). While no significant expression of ISGs was detected in 8‐week‐old WT mice brains, at 13 months regions positive in mutant mouse brains have begun to show hybridization in pia, choroid plexus, ependyma, and multifocal nodules, though at a much‐attenuated level. Bar = 100 microns.
Table S1 Summary of ISH findings for three cytokines in systemic organs and brain
Table S2 ISH probes
ACKNOWLEDGMENTS
This study was supported by National Institute of Health Grant R01AI139544 and VA grant I01RX001455. We would also like to thank and acknowledge Guoji Wang, Mark Stauffer, Kelly Puglisi, and Xinfeng Guo for their superb histology and laboratory services.
Wiley CA, Steinman RA, Wang Q. Innate immune activation without immune cell infiltration in brains of murine models of Aicardi‐Goutières Syndrome. Brain Pathology. 2023;33(3):e13118. 10.1111/bpa.13118
[Correction added on 26 October 2022, after first online publication: “Aicardi–Gutiérrez” has been changed to “Aicardi‐Goutières” throughout the article. Richard A. Steinman's affiliation has been amended.]
Contributor Information
Clayton A. Wiley, Email: wileyca@upmc.edu.
Qingde Wang, Email: wangqd@pitt.edu.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;167A(2):296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crow YJ, Manel N. Aicardi‐Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15(7):429–40. [DOI] [PubMed] [Google Scholar]
- 3. Rice GI, Kitabayashi N, Barth M, Briggs TA, Burton ACE, Carpanelli ML, et al. Genetic, phenotypic, and interferon biomarker status in ADAR1‐related neurological disease. Neuropediatrics. 2017;48(3):166–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barth PG. The neuropathology of Aicardi‐Goutieres syndrome. Eur J Paediatr Neurol. 2002;6(Suppl A):A27; discussion A7‐9, A77‐86–31. [DOI] [PubMed] [Google Scholar]
- 5. Barth PG, Walter A, van Gelderen I. Aicardi‐Goutieres syndrome: a genetic microangiopathy? Acta Neuropathol. 1999;98(2):212–6. [DOI] [PubMed] [Google Scholar]
- 6. Klok MD, Bakels HS, Postma NL, van Spaendonk RM, van der Knaap MS, Bugiani M. Interferon‐alpha and the calcifying microangiopathy in Aicardi‐Goutieres syndrome. Ann Clin Transl Neurol. 2015;2(7):774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kumar D, Rittey C, Cameron AH, Variend S. Recognizable inherited syndrome of progressive central nervous system degeneration and generalized intracranial calcification with overlapping phenotype of the syndrome of Aicardi and Goutieres. Am J Med Genet. 1998;75(5):508–15. [PubMed] [Google Scholar]
- 8. Marguet F, Laquerriere A, Goldenberg A, Guerrot AM, Quenez O, Flahaut P, et al. Clinical and pathologic features of Aicardi‐Goutieres syndrome due to an IFIH1 mutation: a pediatric case report. Am J Med Genet A. 2016;170A(5):1317–24. [DOI] [PubMed] [Google Scholar]
- 9. Guo X, Wiley CA, Steinman RA, Sheng Y, Ji B, Wang J, et al. Aicardi‐Goutieres syndrome‐associated mutation at ADAR1 gene locus activates innate immune response in mouse brain. J Neuroinflammation. 2021;18(1):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Inoue M, Nakahama T, Yamasaki R, Shibuya T, Kim JI, Todo H, et al. An Aicardi‐Goutieres syndrome‐causative point mutation in Adar1 gene invokes multiorgan inflammation and late‐onset encephalopathy in mice. J Immunol. 2021;207:3016–27. [DOI] [PubMed] [Google Scholar]
- 11. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue‐specific macrophage identities. Cell. 2014;159(6):1327–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hammond TR, Dufort C, Dissing‐Olesen L, Giera S, Young A, Wysoker A, et al. Single‐cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell‐state changes. Immunity. 2019;50(1):253–71. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cho H, Proll SC, Szretter KJ, Katze MG, Gale M Jr, Diamond MS. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive‐stranded RNA viruses. Nat Med. 2013;19(4):458–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stratoulias V, Venero JL, Tremblay ME, Joseph B. Microglial subtypes: diversity within the microglial community. EMBO J. 2019;38(17):e101997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Weering HR, Boddeke HW, Vinet J, Brouwer N, de Haas AH, van Rooijen N, et al. CXCL10/CXCR3 signaling in glia cells differentially affects NMDA‐induced cell death in CA and DG neurons of the mouse hippocampus. Hippocampus. 2011;21(2):220–32. [DOI] [PubMed] [Google Scholar]
- 16. Boztug K, Carson MJ, Pham‐Mitchell N, Asensio VC, DeMartino J, Campbell IL. Leukocyte infiltration, but not neurodegeneration, in the CNS of transgenic mice with astrocyte production of the CXC chemokine ligand 10. J Immunol. 2002;169(3):1505–15. [DOI] [PubMed] [Google Scholar]
- 17. Pagenstecher A, Lassmann S, Carson MJ, Kincaid CL, Stalder AK, Campbell IL. Astrocyte‐targeted expression of IL‐12 induces active cellular immune responses in the central nervous system and modulates experimental allergic encephalomyelitis. J Immunol. 2000;164(9):4481–92. [DOI] [PubMed] [Google Scholar]
- 18. Stalder AK, Carson MJ, Pagenstecher A, Asensio VC, Kincaid C, Benedict M, et al. Late‐onset chronic inflammatory encephalopathy in immune‐competent and severe combined immune‐deficient (SCID) mice with astrocyte‐targeted expression of tumor necrosis factor. Am J Pathol. 1998;153(3):767–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ashley CL, Abendroth A, McSharry BP, Slobedman B. Interferon‐independent upregulation of interferon‐stimulated genes during human cytomegalovirus infection is dependent on IRF3 expression. Viruses. 2019;11(3):246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang W, Xu L, Su J, Peppelenbosch MP, Pan Q. Transcriptional regulation of antiviral interferon‐stimulated genes. Trends Microbiol. 2017;25(7):573–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 FFPE sections of 13‐month WT and D1113H mouse brains were stained with H&E (left), GFAP (middle) and IBA1 (right). While the mutant mouse brain (D,E,F and J,K,L) is a little smaller and has larger ventricles than the age matched WT (A,B,C and G,H,I) mouse brain, for the most part there is no significant difference in gliosis markers. A–F Bar = 1 mm, G–L Bar = 200 microns.
Figure S2 (A–F) ISH on sections of FFPE paraffin embedded systemic organs from 8‐week old D1113H mice were hybridized for ISG‐15 (left), CXCL10 (middle), or CCL5 (right). (A) The liver demonstrated expression of ISG15 in widely scattered foci of parenchymal hepatocytes. Expression of CXCL10 showed a similar pattern with additional intensely stained and widely distributed cells morphologically consistent with Kupffer cells. Expression of CCL5 was limited to discrete intensely stained cells in a histological distribution consistent with endothelia or Kupffer cells. (B) The heart showed no expression of ISG‐15. CXCL10 expression was limited to rare foci of intensely stained myocardial cells. CCL5 showed rare staining of elongated cytoplasmic processes consistent with endothelia. (C) The lung showed expression of ISG‐15 in bronchial epithelium. CXCL10 showed expression in discrete foci of alveoli. CCL5 showed a broad even distribution of intensely stained cells in a distribution consistent with endothelia. (D) The intestine showed expression of ISG‐15 within epithelium stronger in apical than basal regions. CXCL10 expression was limited to macrophages within discrete regions of lamina propria. CCL5 expression was more limited than CXCL10 expression but also limited to macrophages within lamina propria. (E) The kidney showed background staining of proximal tubules (diffuse non‐granular pink staining) in addition to discrete staining of occasional tubule epithelial foci. CXCL10 expression was observed in multiple foci on tubular epithelium in addition to scattered interstitial cells. CCL5 expression was widely distributed in interstitial cells morphologically and histologically consistent with macrophages. (F) The spleen showed expression of all three cytokines predominantly within the red pulp in cells morphologically consistent with macrophages.
Figure S3 FFPE sections of 8‐week D1113H mouse brains stained with double label ISH for interferon stimulated genes (red; A–D = ISG‐15, B,C = CXCL10) and cell lineage markers (blue). Double label staining confirms the morphological interpretation that neurons express abundant ISG‐15 (A) while oligodendroglia and astrocytes and endothelial cells express little of these gene product. Double label staining for CXCL10 confirms the morphological interpretation that neurons, oligodendroglia, astrocytes, and endothelial cells express little of this gene product (D–H). Double staining for CXCL10 and AIF1 shows frequent colocalization in microglial nodules (I–K) while choroid plexus contains macrophage elements double labeled for both CXCL10 and AIF1 (L). Bar = 50 microns.
Figure S4 FFPE sections of 13‐month WT mouse brains were hybridized for ISG‐15 (A–C) and CXCL10 (D–F). While no significant expression of ISGs was detected in 8‐week‐old WT mice brains, at 13 months regions positive in mutant mouse brains have begun to show hybridization in pia, choroid plexus, ependyma, and multifocal nodules, though at a much‐attenuated level. Bar = 100 microns.
Table S1 Summary of ISH findings for three cytokines in systemic organs and brain
Table S2 ISH probes
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.