

Abstract
Mitochondrial translation defects are a continuously growing group of disorders showing a large variety of clinical symptoms including a wide range of neurological abnormalities. To date, mutations in PTCD3, encoding a component of the mitochondrial ribosome, have only been reported in a single individual with clinical evidence of Leigh syndrome. Here, we describe three additional PTCD3 individuals from two unrelated families, broadening the genetic and phenotypic spectrum of this disorder, and provide definitive evidence that PTCD3 deficiency is associated with Leigh syndrome. The patients presented in the first months of life with psychomotor delay, respiratory insufficiency and feeding difficulties. The neurologic phenotype included dystonia, optic atrophy, nystagmus and tonic–clonic seizures. Brain MRI showed optic nerve atrophy and thalamic changes, consistent with Leigh syndrome. WES and RNA‐seq identified compound heterozygous variants in PTCD3 in both families: c.[1453‐1G>C];[1918C>G] and c.[710del];[902C>T]. The functional consequences of the identified variants were determined by a comprehensive characterization of the mitochondrial function. PTCD3 protein levels were significantly reduced in patient fibroblasts and, consistent with a mitochondrial translation defect, a severe reduction in the steady state levels of complexes I and IV subunits was detected. Accordingly, the activity of these complexes was also low, and high‐resolution respirometry showed a significant decrease in the mitochondrial respiratory capacity. Functional complementation studies demonstrated the pathogenic effect of the identified variants since the expression of wild‐type PTCD3 in immortalized fibroblasts restored the steady‐state levels of complexes I and IV subunits as well as the mitochondrial respiratory capacity. Additionally, minigene assays demonstrated that three of the identified variants were pathogenic by altering PTCD3 mRNA processing. The fourth variant was a frameshift leading to a truncated protein. In summary, we provide evidence of PTCD3 involvement in human disease confirming that PTCD3 deficiency is definitively associated with Leigh syndrome.
Keywords: Leigh syndrome, mitochondria, mitochondrial disorder, mitochondrial translation, PTCD3
In this manuscript the authors describe three patients from two unrelated families carrying mutations in PTCD3 and provide definitive evidence that PTCD3 deficiency is associated to Leigh syndrome. Functional complementation studies performed in both, immortalized patients' fibroblasts and cell models demonstrated the pathogenicity of the identified variants and their impact on the mitochondrial function.
1. INTRODUCTION
The oxidative phosphorylation (OXPHOS) system is responsible for the production of the major bulk of cellular energy in the form of ATP. This molecular machinery is located in the inner mitochondrial membrane and is composed of five heteropolymeric protein complexes, the subunits of which are encoded by the nuclear and mitochondrial (mtDNA) genomes [1]. As a result, the proper function of the OXPHOS system relies on the coordinated expression of both genetic systems. The vast majority of the OXPHOS system components are encoded by the nuclear genome, being synthesized in the cytosol and specifically imported into the mitochondria [2]. On the other hand, the mtDNA encodes only 13 OXPHOS structural subunits which are transcribed in the mitochondrial matrix as polycistronic mRNAs and translated by the mitoribosomes before their incorporation into the OXPHOS complexes [3, 4, 5].
Aberrant mitochondrial protein translation leads to combined OXPHOS deficiency and disrupted energy metabolism [6]. In the recent years, mitochondrial translation defects have emerged as a growing group of mitochondrial diseases. This heterogeneous group of pathological entities is caused by mtDNA alterations targeting mitochondrial tRNA and rRNA as well as by mutations in nuclear genes encoding mitochondrial protein translation factors, mitochondrial aminoacyl‐tRNA synthetases and mitochondrial ribosome‐associated proteins (MRPs) [7, 8]. To date, disease‐causing mutations in 13 genes encoding MRPs have been identified [9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22]. The clinical symptoms associated with MRP deficiencies are highly heterogeneous and affect multiple systems and organs. The disease manifests during the neonatal period or in infancy, and the course of the disease is severe, frequently resulting in early death. Myopathy, cardiac abnormalities, hepatopathy, renal dysfunction, deafness and dysmorphic features are the most common clinical phenotypes associated with MRP deficiencies. Neurological findings range from deficits without apparent brain abnormalities to severe structural lesions and Leigh syndrome. Biochemically, MRP defects are characterized by lactic acidosis and combined OXPHOS deficiencies [8]. Leigh syndrome is the most prevalent manifestation of mitochondrial disease in children. This neurodegenerative disorder is genetically heterogeneous; to date, pathogenic mutations in more than 75 genes, encoded either by mitochondrial and nuclear genomes, have been identified [23, 24]. Frequently, neurodevelopmental deterioration is accompanied by brainstem dysfunction, including tone, reflex, ataxia, dysphagia abnormalities, and seizures. Lesions in the brainstem, cerebellum, basal ganglia, oculomotor and cranial nerves are typically detected by neuroimaging.
PTCD3 (MIM* 614918), also known as MRPS39, encodes a member of the pentatricopeptide repeat domain protein family, which is associated with the small subunit of the mitochondrial ribosome [25, 26]. This protein plays a crucial role in the initial stages of intramitochondrial protein translation and is, therefore, required for the proper function of the OXPHOS system [27, 28]. Until now, functional evidence of disease‐causing variants in PTCD3 has only been reported in a single individual presenting psychomotor regression, optic atrophy, and Leigh syndrome [9].
In the present study, we provide further evidence of PTCD3 involvement in human disease by reporting a detailed clinical, biochemical and functional description in three additional patients from two unrelated families. Functional complementation studies performed in immortalized patients' fibroblasts and minigene assays in cell models demonstrated disease causality for the identified variants and provided evidence that their pathogenic effect was primarily caused by defective mRNA processing. In addition, we established that PTCD3 deficiency is definitively associated to Leigh syndrome.
2. MATERIAL AND METHODS
2.1. Patients
2.1.1. Patient 1 (P1)
This patient is a Spanish boy born to healthy, non‐consanguineous parents. Family history, pregnancy, and delivery were uneventful. Neurodevelopment was considered normal until the 10th month of life, when delayed adquisicion of milestones and hypertonia were noted. At 2 years old, he had marked papillary paleness and an akinetic‐rigid syndrome characterized by generalized hypertonicity, few spontaneous movements, reduced facial expressiveness, and an intention tremor. Trunk, limb tonic extension, and tonic cephalic deviation during sleep were also observed. At 2.5 years old, he was admitted to the intensive care unit due to acute severe respiratory distress and neurological deterioration, which included rigidity and absent visual contact and speech. He was admitted a few more times due to respiratory symptoms (bronchospasm), developing chronic respiratory failure and progressive dysphagia requiring gastrostomy. At 3 years old, he presented with spastic and dystonic tetraparesis predominantly on the left side, optic atrophy, and null response to a visual evoked potential study. At 4 years old, he had a diffuse sensory and motor axonal neuropathy, and the auditory evoked potential revealed a bilateral central affection. The patient was initially treated with L‐Dopa, but no improvement was observed. Treatment with carnitine, thiamine, biotin, gabapentin, baclofen, and risperidone was also ineffective and had no effect on the disease progression. At the time of his last revision, he was 10 years old and had no eye contact, social smile, or interaction with the environment as well as a drug resistance status epilepticus, characterized by tonic–clonic seizures. He died at 11 years of age from pneumonia, which resulted in respiratory failure and acidosis.
Muscle biopsy at 3.5 years of age revealed muscle fibres without fibre size variability with occasional angulated fibres, that were hyperchromatic for NADH staining, consistent with denervated fibres. Slow and fast myosin analysis showed no fibre‐type grouping. COX staining showed a population of COX‐negative fibres in mosaic, with increased amounts of lipid material (Figure 1). These findings supported the diagnosis of mitochondrial disease. Mitochondrial respiratory chain analysis in fibroblasts revealed decreased activity of complex IV. However, when normalized to citrate synthetase, the values fall within the control range (Table S1). At 10 years of age, brain MRIs revealed a volume deficit with signal changes in both the globus pallidus and thalamus, as well as optic nerve atrophy (Figure 2), which remained stable over time. The EEGs revealed a slow and poorly structured background. On the echocardiogram, he had a slight hypertrophy of the left ventricle wall. The metabolic workup (which had been normal in previous analysis) showed only a mild increase of lactate and glutamine in plasma (Table 1). Of note, this individual has previously been included in a large cohort study of Mendelian disorders [29]. However, that manuscript did not provide details on the clinical, biochemical and functional data demonstrating the pathogenicity of the identified variants.
FIGURE 1.
Muscle biopsy findings. (A, B) Hematoxylin–eosin showed a conserved pattern with mild variability in the measurement of fibers with peripheral myonuclei. (C) Gomori's modified trichrome staining showed no increased endomysial or perimysial connective tissue and no subsarcolemmal reinforcements of mitochondria or ragged‐red fibers. (D) Sudan black revealed fibers with increased number and size of lipid droplets (arrows). (E) NADH staining showed preserved intermyofibrillar pattern. (F) COX‐negative fibers (arrows) with a mosaic distribution pattern were observed. (G) SDH staining showed no fibers with mitochondrial proliferation. (H) Blue fibers with SDH‐COX combined staining indicated absence of complex IV (arrows). (I, J) Immunohistochemical stain for slow (I) and fast myosin (J) revealed a pattern of distribution by fiber types in mosaic. No grouping was observed. (K, L) Gomori's modified trichrome (K) and NADH staining (L) showed angulated fibers (arrow) with hyperchromasia for NADH, consistent with denervated fibers.
FIGURE 2.
Brain MRI and thoracoabdominal X‐rays in PTCD3 patients. T1W axial brain MRI showing atrophy of both optic nerves (white arrows) in patient 1 (A) and patient 2.1 (B). (C) T2W axial brain MRI. Hyperintensity and volume loss of the thalami (white arrows) in patient 2.1. (D) Thoracoabdominal X‐ray demonstrating severe lumbar scoliosis in patient 2.1.
TABLE 1.
Clinical, biochemical, and molecular characteristics of PTCD3 patients
| Borna et al. (2019) | P1 (this report) | P2.1 (this report) | P2.2 (this report) | |
|---|---|---|---|---|
| Age of onset | Neonatal period | Neonatal period | 3 months | 3 months |
| Alive/dead | Dead at 1 year 4 months | Dead at 11 years | Alive at 19 years | Dead at 18 years |
| IUGR | + | − | − | − |
| Microcephaly | + | − | − | − |
| Developmental delay | + | + | + | + |
| Respiratory insufficiency | + | + | − | + |
| Dysphagia | + | + | − | + |
| Tonic clonic seizures/epileptic status | Myoclonic epilepsy | Tonic clonic seizures | Tonic clonic seizures | Tonic clonic seizures |
| Optic atrophy | + | + | − | − |
| Nystagmus | + | + | − | + |
| Hearing loss | + | + | − | − |
| Rigidity | + | + | − | − |
| Dystonia | NR | + | + | + |
| Scoliosis | NR | − | + | + |
| Brain MRI | T2W Increased signal in thalamus, caudate nucleus, and putamen. Punctate lesions in brainstem. Mild cerebral atrophy. | T2W Increased signal in both the globus pallidus and thalamus. Optic nerve hypoplasia | Optic nerve hypoplasia with thalamic volume loss. Basal ganglia volume and signal were normal. | ND |
| Lactate | ||||
| Plasma (CV: 0.55–1.77 mmol/L) | High | 2.3 | 8.6 | 7.8 |
| CSF | High | ND | ND | ND |
| Other biochemical analysis (plasma) | ||||
| Pyruvate (CV: 0.03–0.10 mmol/L) | NR | 0.09 | 0.20 | 0.20 |
| Alanine (CV: 226–416 μmol/L) | NR | 380 | 545 | 371 |
| Glutamine (CV: 330–632 μmol/L) | NR | 832 | 731 | 630 |
| Glycine (CV: 109–293 μmol/L) | NR | 237 | 385 | 337 |
| CoQ10 (CV: 0.41–1.15 μmol/L) | NR | 0.64 | 0.32 | 0.37 |
| OXPHOS system activity defects | CI, CIV | CIV | CI, CIV | ND |
| Genotype | c.[415‐2A>G]; [1747_1748insCT] | c.[1453‐1G>C]; [1918C>G] | c.[710delC]; [902C>T] | c.[710delC]; [902C>T] |
| Effect on protein | p.[Cys139GlufsTer71]; [Phe583SerfsTer3] | p.[Ala485IlefsTer12]; [Ser608_Lys660del] | p.[Thr237AsnfsTer14]; [Ala289GlufsTer18] | p.[Thr237AsnfsTer14]; [Ala289GlufsTer18] |
Note: Altered results are highlighted in bold. Other mitochondrial biomarkers such as plasma GDF‐15/GFG21 or urine organic acids disclosed normal results.
Abbreviations: IUGR, intrauterine growth retardation; ND, not done; NR, not reported.
2.1.2. Patient 2.1 (P2.1)
The proband is a 19‐year‐old Brazilian girl born by cesarean section to non‐consanguineous parents. Family history and pregnancy were uneventful. During the first year of life, abnormal eye movements and severe developmental delay were observed, with no acquisition of gross and fine motor milestones and progressive deterioration of eye contact. At 6 and 10 years of age, she had two episodes of focal unilateral tonic–clonic seizures. At 12 years of age, progressive generalized dystonia and scoliosis appeared. Her last physical examination, at 18 years old, revealed that she had a normal level of consciousness with no language development and poor eye contact, without nystagmus or other abnormal eye movements. She showed generalized dystonia and spasticity with hyperreflexia and several muscle contractures and truncal hypotonia, but was able to hold her head up. She kept her hands clenched without any manipulative intent. She also had severe thoracolumbar scoliosis and amenorrhea. She is being treated with a mitochondrial cocktail (coenzyme Q10, carnitine), calcium, vitamin D, trihexyphenidyl, baclofen, gabapentin, and diazepam. However, this treatment did not result in a perceptible improvement of the clinical condition.
At 15 years of age, brain MRI revealed significant optic nerve atrophy as well as thalamic volume loss. The volume and signal of the basal ganglia were normal (Figure 2). Visual evoked potential had a poorly structured response. The video EEG revealed no epileptiform abnormalities.
Metabolic workup revealed high blood lactate, pyruvate, alanine, glycine and glutamine with low plasma coenzyme Q10 levels (Table 1). Mitochondrial respiratory chain analysis in fibroblasts revealed decreased activity of complex I, complexes I + III and complex IV (Table S1).
2.1.3. Patient 2.2 (P2.2)
Patient 2.2 (P2.2) is the twin sister of patient 2.1 and she presented with a similar phenotype. She also had a severe early developmental delay, noticeable from 3 months of age, as well as abnormal eye movements. She was diagnosed with dystonia and scoliosis at a younger age (4–5 months) than her sister. She was treated with a mitochondrial cocktail (coenzyme Q10, vitamin C, carnitine and thiamine) as well as diazepam, baclofen, L‐dopa and botulinum toxin for dystonia. At 17 years of age, she presented with several afebrile generalized tonic–clonic seizures associated with acute respiratory insufficiency, requiring admission to the pediatric intensive care unit for noninvasive mechanical ventilation and antiepileptic treatment (midazolam, valproic acid and levetiracetam). Because of dysphagia, she was fed through a nasogastric tube. Considering her overall condition, it was decided that she should be monitored by the palliative care unit. She died at home a month later. Biochemical analysis showed similar results to those observed in her sister (Table 1). Brain MRI was not performed.
2.2. Whole exome sequencing
Whole‐exome sequencing (WES) was performed at the Centre Nacional d'Anàlisi Genòmica (CNAG‐CRG). For exome enrichment, we used the Nimblegen SeqCap EZ MedExome+mtDNA 47 Mb capture kit, followed by sequencing using the Illumina HiSeq 2000 genome analyzer platform. The primary data files (FASTQ files) were analyzed using the pipeline developed by CNAG‐CRG. Sequence reads were mapped to Human genome build hg19/GRCh37. The variant calls of P1 were analyzed using the URDCAT genome‐phenome analysis platform (GPAP), filtered by frequency (allele frequency < 1% in population databases, including 1000G and gnomAD), by functional impact on the encoded protein, as well as for the clinical and biochemical phenotype of the patients. Annotation and variant analysis of P2.1 and P2.2 were performed using VarAFT [30]. Selected candidate variants were validated and segregated by Sanger sequencing. Primers and conditions used for PCR amplification and sequencing are available on demand. Sequence alterations are reported according to the Human Genome Variation Society (HGVS) nomenclature guidelines. The gnomAD (v.2.1.1 and 3.1.3), Iraniome and Great Middle East Variome databases were accessed on November 2021. Mutations are referred to transcript ENST00000254630.7.
2.3. RNA sequencing
RNA‐seq was performed at CNAG‐CRG using total RNA extracted from P1 fibroblasts. The quality control of the total RNA was done using the Qubit® RNA HS Assay (Life Technologies) and RNA 6000 Nano Assay on a Bioanalyzer 2100 (Agilent). Libraries were prepared using the TruSeq® Stranded mRNA LT Sample Prep Kit (Illumina Inc., Rev.E, October 2013). The libraries were sequenced on a HiSeq 2500 (Illumina) in paired‐end mode (2 × 76 bp). Primary data analysis, image analysis, base calling and quality scoring of the run were processed using the manufacturer's software Real‐Time Analysis (RTA 1.18.66.3), followed by generation of FASTQ sequence files. Reads from RNA‐seq were demultiplexed and then mapped with STAR (v2.7.0a) to the hg19 genome assembly. Analysis of the aligned data was done using DROP in order to detect aberrantly expressed genes, aberrantly spliced genes and monoallelic expression of rare variants [31, 32, 33].
2.4. Cell culture
Skin‐derived fibroblasts were maintained in MEM (1 g/L glucose, 10% fetal calf serum and 1% penicillin–streptomycin) and grown to confluence in 25 cm2 flasks. When indicated, cells were treated with cycloheximide (chx) (Sigma‐Aldrich) as previously described [34].
2.5. PTCD3 cDNA analysis
Total RNA was extracted from P1 and P2.1 as well as from control fibroblasts with RNeasy kit (Qiagen). Single‐stranded complementary DNA (cDNA) was obtained using M‐MLV Reverse Transcriptase, RNase H Minus Point Mutant (Promega) according to the manufacturer's protocol. PTCD3 cDNA was analyzed by reverse transcription‐PCR (RT‐PCR) using specific primers (Table S2) and Sanger sequencing.
2.6. Protein expression analysis
Fibroblasts were homogenized using RIPA lysis buffer containing a protease inhibitor cocktail (1862209, Merck). Briefly, cells were scraped in lysis buffer, incubated on ice for 10 minutes, and centrifuged at 10,000g at 4°C for 10 min. Protein extracts were subjected to SDS‐PAGE and electroblotted. Proteins were visualized by immunostaining with specific antibodies followed by colorimetric detection using the Opti‐4CNTM Substrate Kit (Bio‐Rad). The antibodies used in this study were: anti‐PTCD3 (HPA041382, Sigma‐Aldrich), anti‐Oxphos cocktail (45‐8199, Invitrogen) and anti‐GAPDH (sc‐47724, Santa Cruz Biotechnology).
2.7. High‐resolution respirometry
High‐resolution respirometry was performed using polarographic oxygen sensors in a two‐chamber Oxygraph‐2k system at 37°C according to the manufacturer's instructions (Oroboros Instruments). Manual titration of OXPHOS inhibitors (Oligomycin, Antimycin) and uncouplers (CCCP) was performed using Hamilton syringes (Hamilton Company) as previously described [35]. Data were recorded and analyzed using the DatLab software v5.1.1.9 (Oroboros Instruments).
2.8. Functional analysis of PTCD3 variants effect on mRNA processing
To determine the effect on mRNA processing of the discovered PTCD3 variants (c.1453‐1G>C, c.1918C>G, and c.902C>T), a minigene assay was performed. Briefly, the genomic regions containing the three variants and their corresponding wild‐type alleles were cloned into the Exon Trap Cloning Vector pET01 (MoBiTec GmbH). To test the effect of these variants on mRNA processing, the wild‐type (pET01‐WT) and mutant (pET01‐MUT) plasmids were transfected into HAP1 cells using Lipofectamine3000 (Promega) according to the manufacturer's recommendations. Total RNA was extracted 48 h after transfection and subjected to RT‐PCR using primers corresponding to the 5′ and 3′ exons of the pET01 vector (Table S2). The PCR products were visualized in agarose gels and analyzed by Sanger sequencing.
2.9. Functional complementation studies
Control and patients' fibroblasts were immortalized by lentiviral transduction of the pLOX‐Ttag‐iresTK vector (Addgene, #12246). GFP‐tagged wild‐type PTCD3 cDNA vector was purchased from Origene (RC204119L2). Lentiviral particles were generated in HEK293T packaging cells, as described [36]. The recovery of the steady‐state levels of OXPHOS subunits and the respiratory capacity was analyzed as mentioned above.
2.10. Statistics
In all cases, statistical analyses were performed using the two‐tailed Student's t‐test to compare the means of two independent groups of normally distributed data. Data were reported as the mean ± SD p Values lower than 0.05 were considered statistically significant.
3. RESULTS
3.1. Identification of PTCD3 variants in two families
WES was performed in the three patients reported here: P1 (family 1); P2.1 and P2.2 (family 2). Using the filtering steps described in Section 2, we identified compound heterozygous variants in PTCD3 gene (NM_017952.5) in both families: c.1453‐1G>C and c.1918C>G in P1, and c.710del and c.902C>T in P2.1 and P2.2. In addition, RNA‐seq was also performed on P1 fibroblasts, which prioritized PTCD3 as a candidate disease gene, as an aberrant splicing was detected. Interestingly, PTCD3 expression levels detected in P1 were the lowest of the entire cohort of transcriptomes (Figure S1).
RT‐PCR and Sanger sequencing of P1 and P2.1 fibroblasts grown with or without cycloheximide (chx) were used to assess the effect of the identified variants (Figure 3); cDNA analysis of P1 revealed a skipping of exon 19 caused by the c.1453‐1G>C substitution, which altered a canonical splice acceptor site. This aberrant transcript was only detected when patient's fibroblasts were grown in chx‐containing media, suggesting that it is subjected to nonsense‐mediated mRNA decay (NMD). This finding is consistent with the decreased PTCD3 expression levels observed by RNA‐seq analysis. The skipping of exon 19 is predicted to produce the substitution of alanine 485 for isoleucine and a frameshift leading to a truncated protein (p.Ala485IlefsTer12). Furthermore, PTCD3 cDNA analysis of this patient revealed the skipping of exon 23, which contained the second WES‐identified variant (c.1918C>G). This mRNA alteration was predicted to result in an in‐frame deletion of the 28 amino acids encoded by exon 23 (p.Ser608_Lys660del).
FIGURE 3.
Genetic analyses of individuals carrying PTCD3 variants. The effect of the identified variants was evaluated by RT‐PCR and Sanger sequencing in patient fibroblasts grown with or without cycloheximide (chx). C, control; P, patient; PTC, premature termination codon.
WES analysis also identified two PTCD3 heterozygous variants (c.710del and c.902C>T) in P2.1 and P2.2. RT‐PCR studies were only performed in P2.1, as P2.2 fibroblasts were unavailable. The c.710del variant resulted in a threonine substitution for asparagine at position 237, leading to a frameshift and a premature termination codon (p.Thr237AsnfsTer14) (Figure 3). The analysis of the second variant (c.902C>T) by Human Splicing Finder (HSF) suggested an alteration of the normal splicing pattern. Indeed, the study of PTCD3 cDNA revealed the skipping of exon 12, where the c.902C>T variant is located. This alteration was predicted to result in glutamate 289 to alanine substitution, provoking a frameshift leading to a premature termination codon at position 307 of the protein (p.Ala289GlufsTer18). The identified variants segregate into separate alleles, in both families.
3.2. PTCD3 mRNA processing is altered in both families
The splicing defects observed in patients' fibroblasts prompted us to determine whether the c.1918C>G and c.1453‐1G>C variants from family 1, and the c.902C>T variant from family 2 could alter PTCD3 mRNA processing. HAP1 cells transiently transfected with minigene vectors, encompassing the genomic regions of the three variants, as well as their wild‐type alleles, were analyzed by RT‐PCR.
Results showed that HAP1 cells transfected with plasmids carrying the PTCD3 wild‐type sequences produced the expected products, indicating unaltered splicing events (Figure 4). Cells expressing the c.1918C>G, c.1453‐1G>C and c.902C>T variants had lower molecular weight bands in the agarose gel analysis, indicative of potential splicing alterations. Sanger sequencing of these products confirmed the skipping of the involved exons in all cases. It is worth noting that the c.1453‐1G>C variant, which abolished a canonical splice acceptor site, had the strongest effect, as almost all detected transcripts corresponded to mRNA with the skipped exon. On the other hand, c.902C>T and c.1918C>G variants generated a mixture of correctly spliced and exon‐skipped transcripts. Overall, these results demonstrated that the c.1918C>G, c.1453‐1G>C, and c.902C>T variants were responsible for the PTCD3 mRNA processing defects observed in patients' fibroblasts.
FIGURE 4.
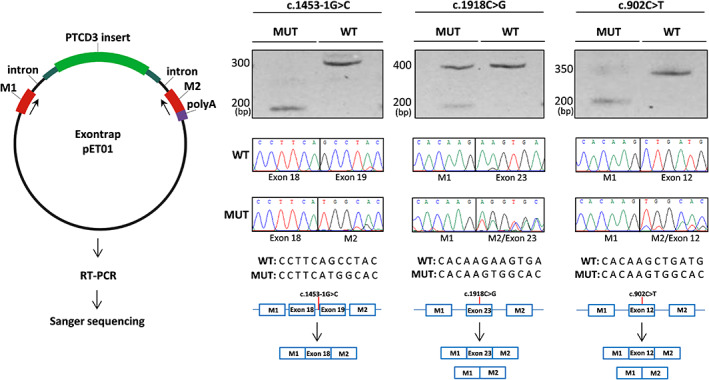
mRNA processing defects caused by PTCD3 variants. A minigene assay was performed to determine the effect of the c.1453‐1G>C, c.1918C>G, and c.902C>T variants. As indicated in the left panel, the wild‐type (pET01‐WT) and mutant (pET01‐MUT) plasmids were transfected into HAP1 cells. RT‐PCR followed by agarose gel and Sanger sequencing demonstrated altered splicing in the cells expressing mutated PTCD3 vectors, compared to those transfected with the wild‐type plasmids. M1 and M2 indicate pET01 exonic sequences. MUT, mutant; WT, wild‐type.
3.3. PTCD3 and OXPHOS subunits are reduced in patients' fibroblasts
To determine the effect of the identified variants, we first analyzed the protein levels of PTCD3 in P1 and P2.1 fibroblasts by western blot. Results demonstrated that PTCD3 protein was significantly reduced in patients' cells, with 70% (P1) and 85% (P2.1) reduction when compared to controls (p < 0.01) (Figure 5A).
FIGURE 5.
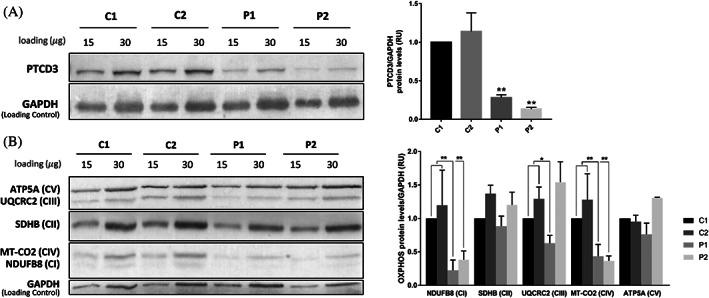
Steady‐state levels of PTCD3 and OXPHOS subunits are reduced in patients' fibroblasts. (A) Western blot showed reduced levels of PTCD3 protein in patients' fibroblasts compared with control individuals. (B) P1 and P2 showed reduced levels of NDUFB8 (complex I) and MT‐CO2 (complex IV). The levels of UQCRC2 (complex III) were also reduced in P1, but not in P2. Complexes II and V subunits remained unchanged. GAPDH was used as a loading control. Two biological replicates were performed for each sample. C, control; P, patient. *p < 0.05, **p < 0.01.
Since PTCD3 is a component of the mitochondrial ribosome, we investigated the steady‐state levels of representative subunits of the OXPHOS system (Figure 5B). A significant reduction of NDUFB8 (complex I) and MT‐CO2 (complex IV) in both patients (p < 0.001) was observed. Furthermore, steady‐state levels of UQCRC2 (complex III) were significantly reduced in P1, but not in P2.1 (p < 0.01). ATP5A1 (complex V) and SDHB (complex II) protein levels remained unaltered.
3.4. Respiratory capacity is compromised in PTCD3 patients' fibroblasts
The mitochondrial respiratory capacity in P1 and P2.1 fibroblasts was investigated by measuring the oxygen consumption rate (OCR) with high‐resolution respirometry. Results showed a slight but significant decrease in the patients' basal respiratory rate compared to control cells (p < 0.001). Treatment with the mitochondrial uncoupler CCCP resulted in a significant reduction in maximal respiratory capacity in both patients (p < 0.001) (Figure 6).
FIGURE 6.
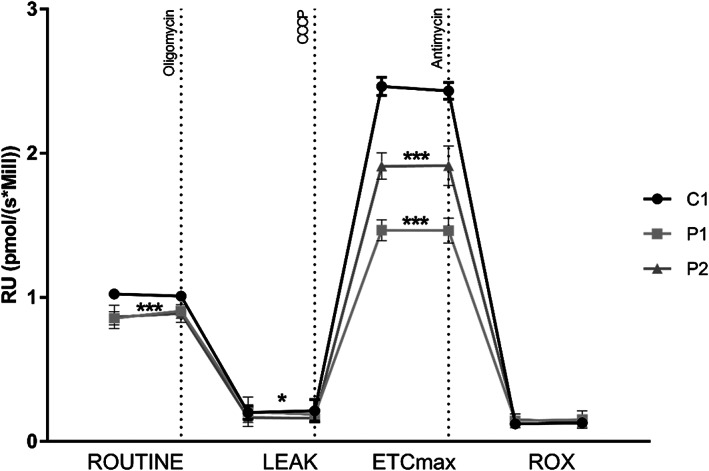
Mitochondrial respiratory capacity is reduced in PTCD3 patients. Mitochondrial respiration was analyzed in patients' fibroblasts by measuring the oxygen consumption rate (OCR). A slight decrease in basal OCR was observed in patients compared to control cells. Upon treatment with CCCP, an important reduction of the maximal respiratory capacity (ETCmax) was observed in both patients. C, control; P, patient. OCR at basal state (ROUTINE); residual oxygen consumption after oligomycin treatment (LEAK); maximum oxygen consumption induced by carbonyl cyanide 3‐chlorophenylhydrazone (CCCP) titration (ETCmax); residual oxygen consumption after antimycin A treatment (ROX). OCR was measured as pmol/(second * millions of cells). The data are expressed as relative units (RU) of control cells. Two biological replicates were performed for each sample. *p < 0.05, ***p < 0.001.
3.5. Functional complementation of PTCD3 ‐mutated fibroblasts
The pathogenic effect of the PTCD3 mutations was demonstrated by functional complementation studies in immortalized fibroblasts. Similarly to naïve fibroblasts, the levels of PTCD3 as well as NDUFB8 (complex I) and MT‐CO2 (complex IV) were reduced in immortalized P1 and P2.1 cells. Additionally, UQCRC2 (complex III) was also diminished in P1. The expression of wild‐type PTCD3 cDNA using a lentiviral vector resulted in a complete recovery of the steady‐state levels of the altered OXPHOS subunits in both patients (Figure 7A,B). Furthermore, the mitochondrial respiratory capacity, which was significantly reduced in naïve patient's cells, was recovered upon lentiviral transduction with wild‐type PTCD3 but not when cells were transduced with an empty plasmid (Figure 7C).
FIGURE 7.
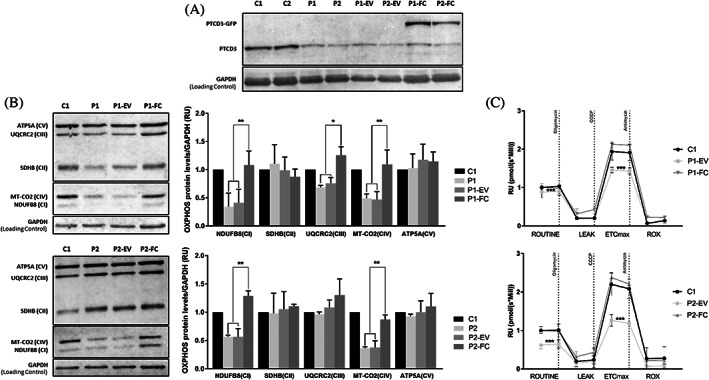
PTCD3 transduction recovers the expression of altered OXPHOS subunits and the mitochondrial respiratory capacity. (A) Lentiviral transduction with GFP‐tagged wild‐type PTCD3 restored the expression of PTCD3 in P1 and P2.1 patients. (B) The steady‐state levels of NDUFB8, MT‐CO2, and UQCRC2 in P1 and NDUFB8 and MT‐CO2 in P2.1 were recovered upon lentiviral transduction with wild‐type PTCD3. GAPDH was used as a loading control. Three biological replicates were performed for each sample. (C) Transduction with wild‐type PTCD3, but not with the empty vector, restored the mitochondrial respiratory capacity in immortalized patients' fibroblasts. Oxygen consumption rate (OCR) at basal state (ROUTINE); residual oxygen consumption after oligomycin treatment (LEAK); maximum oxygen consumption induced by carbonyl cyanide 3‐chlorophenylhydrazone (CCCP) titration (ETCmax); residual oxygen consumption after antimycin a treatment (ROX). OCR was measured as pmol/(second * millions of cells). The data are expressed as relative units (RU) of control cells. Two biological replicates were performed for each sample. C, control; EV, cells transduced with an empty vector; FC, functionally complemented cells; P, patient. *p < 0.05, **p < 0.01. ***p < 0.001.
4. DISCUSSION
Mitochondrial translation machinery defects are a continuously growing group of mitochondrial energy metabolism disorders [7, 8, 36]. To date, mutations in PTCD3, encoding a protein associated to the small subunit of the mitochondrial ribosome, have only been reported in one individual [9]. In this study, we describe three additional patients harbouring PTCD3 mutations from two unrelated families, broadening the genetic and phenotypic spectrum of this disorder and reinforcing PTCD3 as a gene whose loss of function is associated with Leigh syndrome.
The main clinical and biochemical characteristics of the newly reported individuals, as well as the case described by Borna et al. [9] are summarized in Table 1. The previously reported PTCD3 patient had intrauterine growth retardation and developed symptoms soon after birth. The disease progressed rapidly, and the patient died at the age of 1 year and 4 months. She had respiratory insufficiency, microcephaly, depressed nasal bridge, rigidity, myoclonic epilepsy, nystagmus, optic atrophy, hearing loss and psychomotor retardation [9, 37]. In contrast, none of the individuals reported here had intrauterine growth retardation, and the onset age ranged from the first weeks to 3 months of life. They survived longer than the previously reported patient, being exitus at the ages of 11 and 17 years old, or still alive at 19 years of age. Although the disease progressed more slowly in our patients, the clinical phenotype was consistent with the main features of the individual reported by Borna et al. [9], including psychomotor delay, respiratory insufficiency, feeding difficulties and tonic–clonic seizures. Nystagmus was also observed in two of our patients, but only P1 had optic atrophy and hearing loss. Remarkably, dystonia was observed in all three individuals reported here. On the other hand, P2.1 and P2.2 suffered from scoliosis, which had not been reported in the previous PTCD3 individual and was most likely caused by their older age and to the effect of the severe dystonia.
Brain MRI of the patient described by Borna et al., showed mild cerebral atrophy, punctate lesions in the brainstem, and T2W hyperintense lesions in the thalamus, caudate nucleus, and putamen [9]. The diagnosis was compatible with Leigh Syndrome, but it was questioned by some authors, that classified the patient as Leigh‐like rather than Leigh syndrome [38]. In our study, brain imaging performed on P1 and P2.1 revealed optic nerve atrophy and thalamic changes. P1 also had increased signal intensity in the globus pallidus. On the other hand, optic atrophy is a clinical sign that has been observed in patients with Leigh syndrome of other etiologies, and thus it is not useful in guiding the clinical diagnosis [39].
The clinical and outcome differences observed among patients could be caused by the different effects of the variants identified in each case. In this study, we found four novel PTCD3 compound heterozygous variants in three patients from two families. P1 harboured the c.1453‐1G>C and c.1918C>G variants, whereas P2.1 and P2.2 carried c.710del and c.902C>T substitutions. As shown in Table 2, c.1453‐1G>C and c.710del variants were classified as ‘pathogenic’. In contrast, c.902C>T and c.1918C>G variants were classified as ‘variants of uncertain significance’. The potential effect of the identified variants on the encoded protein was investigated using RT‐PCR in patient fibroblasts. We first corroborated the effect of the two variants classified as pathogenic. As expected, the c.710del resulted in a frameshift leading to a truncated PTCD3 protein, and the c.1453‐1G>C substitution disabled the consensus acceptor splice site of PTCD3 intron 18 and produced an aberrantly spliced transcript that was subjected to NMD. This effect was further confirmed by the transient expression of a minigene vector containing this substitution in HAP1 cells, demonstrating that the splicing process might be almost completely abolished as a consequence of this variant. Interestingly, cDNA analysis of the variants initially classified as VUS suggested that c.902C>T (within exon 12) and c.1918C>G (within exon 23) could also be altering mRNA processing, as evidenced by the skipping of the exons where these variants were located. The fact that both variants were found within the PTCD3 coding region rather than in conserved splicing regulatory regions prompted us to investigate whether the mRNA defects observed in patient cells were caused by these substitutions. Indeed, expression of the c.902C>T and c.1918C>G variants in HAP1 cells using a minigene system revealed an important splicing alteration for both substitutions, resulting in a leaky splicing defect leading to the generation of both exon‐skipped and properly spliced transcripts. The aberrantly spliced transcripts produced by the c.902C>T variant potentially led to a truncated PTCD3 protein, whereas the remaining pool of correctly spliced mRNA may encode a full‐length protein carrying the missense p.Thr301Ile substitution. On the other hand, the exon‐skipped transcripts caused by c.1918C>G are predicted to generate an in‐frame deletion affecting 28 amino acids close to the C‐terminal region of the protein. The fraction of properly processed transcripts led to a protein harbouring the p.Pro640Ala substitution. Although the real effect of the p.Pro640Ala and p.Thr301Ile substitutions on protein function is still uncertain, our functional data strongly suggests that the c.902C>T and c.1918C>G variants are pathogenic by altering mainly mRNA processing. The splicing leakage derived from these alleles may provide a potential explanation for the differences observed between our patients and the previously reported individual. In fact, the genotype of the patient reported by Borna et al. appears to be more severe than ours [9]. This patient was compound heterozygous for the c.415‐2A>G variant, which targeted a consensus splice site, and the c.1747_1748insCT, which caused a frameshift within the codifying region. Both variants resulted in a premature termination codon and truncated PTCD3 protein products. As a result, the absence of PTCD3 protein is nearly complete, whereas in our patients, although reduced, was not absent. Additionally, according to our functional data, we speculated that the remaining PTCD3 protein levels detected in our patients may correspond, at least in part, to PTCD3 proteins harbouring the p.Pro640Ala and p.Thr301Ile substitutions.
TABLE 2.
Effect of the identified PTCD3 variants.
| Genetic variant | Predicted effect on protein | Variant classification (ACMG) | Prediction upon cDNA and minigene functional analysis | ||
|---|---|---|---|---|---|
| Effect on protein | Variant classification (ACMG) | ||||
| P1 | c.1453‐1G>C | p.Ala485IlefsTer12 | Pathogenic | p.Ala485IlefsTer12 | Pathogenic |
| c.1918C>G | p.Pro640Ala | VUS | p.Ser608_Lys660del | Pathogenic | |
| P2.1 and P2.2 | c.710delC | p.Thr237AsnfsTer14 | Pathogenic | p.Thr237AsnfsTer14 | Pathogenic |
| c.902C>T | p.Thr301Ile | VUS | p.Ala289GlufsTer18 | Pathogenic | |
PTCD3 is associated with the small subunit of the mitochondrial ribosome and plays an important role in the regulation of mitochondrial translation [25, 26, 28]. Alterations of this highly regulated process have been shown to cause multiple OXPHOS defects and disrupted energy metabolism [6]. In fact, studies in fibroblasts from the only patient with PTCD3 deficiency reported to date revealed a combined OXPHOS system defect caused by decreased stability of the small mitoribosomal subunit and the subsequent impairment of mitochondrial translation [9]. These authors also provided a detailed characterization of the mitochondrial function in patient's cells, showing an important reduction of the steady‐state levels of complexes I and IV subunits, altered enzyme activities of these complexes and decreased mitochondrial respiration. The PTCD3 patients described here have mitochondrial deficiencies similar to the previously reported individual. Western blot analysis demonstrated a significant decrease of NDUFB8 (complex I) and MT‐CO2 (complex IV) protein levels in P1 and P2.1 fibroblasts. Additionally, a decline of UQCRC2, a complex III subunit, was detected in P1 cells. This observation was consistent with the proteomic study performed in the previously reported PTCD3 patient, which besides to a strong reduction of complexes I and IV subunits, also showed a mild but significant decrease of complex III components [9].
Mitochondrial respiratory chain analysis showed reduced activity of complex IV in one of our patients, and a combined defect of complexes I and IV in the other one. It is worth noting that, similarly to the previously reported PTCD3 patient, the activity of CII (after normalization to citrate synthetase activity) was slightly above the control range in both individuals. The nature of this change is not clear but it could rely on the fact that complex II is the only OXPHOS complex composed entirely of nuclear‐encoded subunits. Indeed, complex II is known to be largely unaltered in mitochondrial protein translation deficiencies. The functional consequences of the observed mitochondrial abnormalities were further analyzed by high‐resolution respirometry. Although the basal OCRs were only moderately affected, both PTCD3 patients had a significant reduction of the maximal respiratory capacity. In contrast, the previously reported patient had a decrease in both basal and maximal respiration rate, which could be another factor influencing the severity of the phenotype [9]. Functional complementation studies demonstrated the pathogenic effect of the PTCD3 variants as the expression of wild‐type PTCD3 in immortalized patients' fibroblasts restored the steady‐state levels of the altered OXPHOS subunits and the mitochondrial respiratory capacity.
In summary, we report two additional families carrying PTCD3 mutations, providing evidence that this deficiency is definitively associated with Leigh syndrome. Therefore, it should be considered in the diagnostic workflow of this disease. Finally, we emphasize the value of functional validation studies to demonstrate the pathogenicity when the identified variants are of unknown significance.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics committee of HCB‐IDIBAPS. Informed consent has been obtained from the families involved in this study.
Supporting information
APPENDIX S1: Supporting Information
ACKNOWLEDGMENTS
This research was funded by the Instituto de Salud Carlos III (PI19/01310, PI20/00340, and FI18/00253) and co‐funded by European Union and the Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), an initiative of the Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación, Spain). This study was also supported by the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR, 2017:SGR 1428) and the CERCA Programme/Generalitat de Catalunya and by the Departament de Salut, Generalitat de Catalunya (URDCAT project, SLT002/16/00174). The study was supported by the German Bundesministerium fur Bildung und Forschung (BMBF) through the PerMed project PerMiM (01KU2016B to VAY and JG). The authors would like to thank CNAG‐CRG for excellent technical assistance.
Muñoz‐Pujol G, Ortigoza‐Escobar JD, Paredes‐Fuentes AJ, Jou C, Ugarteburu O, Gort L, et al. Leigh syndrome is the main clinical characteristic of PTCD3 deficiency. Brain Pathology. 2023;33(3):e13134. 10.1111/bpa.13134
Gerard Muñoz‐Pujol and Juan D. Ortigoza‐Escobar contributed equally to this study.
Contributor Information
Roser Urreizti, Email: rurreizti@fsjd.org.
Frederic Tort, Email: ftort@recerca.clinic.cat.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
REFERENCES
- 1. Signes A, Fernandez‐Vizarra E. Assembly of mammalian oxidative phosphorylation complexes I‐V and supercomplexes. Essays Biochem. 2018;62(3):255–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wasilewski M, Chojnacka K, Chacinska A. Protein trafficking at the crossroads to mitochondria. Biochim Biophys Acta. 2017;864:125–37. [DOI] [PubMed] [Google Scholar]
- 3. Gustafsson CM, Falkenberg M, Larsson NG. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem. 2016;85:133–60. [DOI] [PubMed] [Google Scholar]
- 4. Ott M, Amunts A, Brown A. Organization and regulation of mitochondrial protein synthesis. Annu Rev Biochem. 2016;85:77–101. [DOI] [PubMed] [Google Scholar]
- 5. Shokolenko IN, Alexeyev MF. Mitochondrial DNA: a disposable genome? Biochim Biophys Acta. 2015;1852(9):1805–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boczonadi V, Horvath R. Mitochondria: impaired mitochondrial translation in human disease. Int J Biochem Cell Biol. 2014;48:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang F, Zhang D, Zhang D, Li P, Gao Y. Mitochondrial protein translation: emerging roles and clinical significance in disease. Front Cell Dev Biol. 2021;9:675465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Webb BD, Diaz GA, Prasun P. Mitochondrial translation defects and human disease. J Transl Genet Genom. 2020;4:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borna NN, Kishita Y, Kohda M, Lim SC, Shimura M, Wu Y, et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics. 2019;20(1):9–25. [DOI] [PubMed] [Google Scholar]
- 10. Bursle C, Narendra A, Chuk R, Cardinal J, Justo R, Lewis B, et al. COXPD9 an evolving multisystem disease; congenital lactic acidosis, sensorineural hearing loss, hypertrophic cardiomyopathy, cirrhosis and interstitial nephritis. JIMD Rep. 2017;34:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carroll CJ, Isohanni P, Pöyhönen R, Euro L, Richter U, Brilhante V, et al. Whole‐exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J Med Genet. 2013;50(3):151–921. [DOI] [PubMed] [Google Scholar]
- 12. Chen A, Tiosano D, Guran T, Baris HN, Bayram Y, Mory A, et al. Mutations in the mitochondrial ribosomal protein MRPS22 lead to primary ovarian insufficiency. Hum Mol Genet. 2018;27(11):1913–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Di Nottia M, Marchese M, Verrigni D, Mutti CD, Torraco A, Oliva R, et al. A homozygous MRPL24 mutation causes a complex movement disorder and affects the mitoribosome assembly. Neurobiol Dis. 2020;141:104880. [DOI] [PubMed] [Google Scholar]
- 14. Gardeitchik T, Mohamed M, Ruzzenente B, Karall D, Guerrero‐Castillo S, Dalloyaux D, et al. Bi‐allelic mutations in the mitochondrial ribosomal protein MRPS2 cause sensorineural hearing loss, hypoglycemia, and multiple OXPHOS complex deficiencies. Am J Hum Genet. 2018;102(4):685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jackson CB, Huemer M, Bolognini R, Martin F, Szinnai G, Donner BC, et al. A variant in MRPS14 (uS14m) causes perinatal hypertrophic cardiomyopathy with neonatal lactic acidosis, growth retardation, dysmorphic features and neurological involvement. Hum Mol Genet. 2019;28(4):639–49. [DOI] [PubMed] [Google Scholar]
- 16. Kohda M, Tokuzawa Y, Kishita Y, Nyuzuki H, Moriyama Y, Mizuno Y, et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 2016;12(1):e1005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lake NJ, Webb BD, Stroud DA, Richman TR, Ruzzenente B, Compton AG, et al. Biallelic mutations in MRPS34 Lead to instability of the small Mitoribosomal subunit and Leigh syndrome. Am J Hum Genet. 2017;101(2):239–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menezes MJ, Guo Y, Zhang J, Riley LG, Cooper ST, Thorburn DR, et al. Mutation in mitochondrial ribosomal protein S7 (MRPS7) causes congenital sensorineural deafness, progressive hepatic and renal failure and lactic acidemia. Hum Mol Genet. 2015;24(8):2297–307. [DOI] [PubMed] [Google Scholar]
- 19. Miller C, Saada A, Shaul N, Shabtai N, Ben‐Shalom E, Shaag A, et al. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol. 2004;56(5):734–8. [DOI] [PubMed] [Google Scholar]
- 20. Pulman J, Ruzzenente B, Bianchi L, Rio M, Boddaert N, Munnich A, et al. Mutations in the MRPS28 gene encoding the small mitoribosomal subunit protein bS1m in a patient with intrauterine growth retardation, craniofacial dysmorphism and multisystemic involvement. Hum Mol Genet. 2019;28(9):1445–62. [DOI] [PubMed] [Google Scholar]
- 21. Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs‐Telem D, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet. 2007;44(12):784–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Serre V, Rozanska A, Beinat M, Chretien D, Boddaert N, Munnich A, et al. Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim Biophys Acta. 2013;1832(8):1304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bakare AB, Lesnefsky EJ, Iyer S. Leigh syndrome: a tale of two genomes. Front Physiol. 2021;12:693734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 2016;79(2):190–203. [DOI] [PubMed] [Google Scholar]
- 25. Amunts A, Brown A, Toots J, Scheres SHW, Ramakrishnan V. The structure of the human mitochondrial ribosome. Science. 2015;348(6230):95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaushal PS, Sharma MR, Booth TM, Haque EM, Tung CS, Sanbonmatsu KY, et al. Cryo‐EM structure of the small subunit of the mammalian mitochondrial ribosome. Proc Natl Acad Sci U S A. 2014;20:7284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davies SM, Rackham O, Shearwood AM, Hamilton KL, Narsai R, Whelan J, et al. Pentatricopeptide repeat domain protein 3 associates with the mitochondrial small ribosomal subunit and regulates translation. FEBS Lett. 2009;583(12):1853–8. [DOI] [PubMed] [Google Scholar]
- 28. Kummer E, Leibundgut M, Rackham O, Lee RG, Boehringer D, Filipovska A, et al. Unique features of mammalian mitochondrial translation initiation revealed by cryo‐EM. Nature. 2018;560(7717):263–7. [DOI] [PubMed] [Google Scholar]
- 29. Yépez VA, Gusic M, Kopajtich R, Mertes C, Smith NH, Alston CL, et al. Clinical implementation of RNA sequencing for mendelian disease diagnostics. Genome Med. 2022;14(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Desvignes JP, Bartoli M, Delague V, Krahn M, Miltgen M, Béroud C, et al. VarAFT: a variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 2018;46:W545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brechtmann F, Mertes C, Matusevičiūtė A, Yépez VA, Avsec Ž, Herzog M, et al. OUTRIDER: a statistical method for detecting aberrantly expressed genes in RNA sequencing data. Am J Hum Genet. 2018;103(6):907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mertes C, Scheller IF, Yépez VA, Çelik MH, Liang Y, Kremer LS, et al. Detection of aberrant splicing events in RNA‐seq data using FRASER. Nat Commun. 2021;12(1):529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yépez VA, Mertes C, Müller MF, Klaproth‐Andrade D, Wachutka L, Frésard L, et al. Detection of aberrant gene expression events in RNA sequencing data. Nat Protoc. 2021;16(2):1276–96. [DOI] [PubMed] [Google Scholar]
- 34. Macías‐Vidal J, Gort L, Lluch M, Pineda M, Coll MJ. Nonsense‐mediated mRNA decay process in nine alleles of Niemann‐pick type C patients from Spain. Mol Genet Metab. 2009;97(1):60–4. [DOI] [PubMed] [Google Scholar]
- 35. Pesta D, Gnaiger E. High‐resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol. 2012;810:25–58. [DOI] [PubMed] [Google Scholar]
- 36. Tort F, Barredo E, Parthasarathy R, Ugarteburu O, Ferrer‐Cortès X, García‐Villoria J, et al. Biallelic mutations in NDUFA8 cause complex I deficiency in two siblings with favorable clinical evolution. Mol Genet Metab. 2020;131(3):349–57. [DOI] [PubMed] [Google Scholar]
- 37. Hisatomi Y, Murayama K, Ohtake A, Okazaki Y. Reply to the "Letter to the Editor" from Dr. J Finsterer and colleagues. Neurogenetics. 2019;20(1):55–6. [DOI] [PubMed] [Google Scholar]
- 38. Finsterer J, Scorza CA, Scorza FA. PTCD3 mutations cause Leigh‐like rather than Leigh syndrome. Neurogenetics. 2019;20(1):53–4. [DOI] [PubMed] [Google Scholar]
- 39. Zhu CC, Traboulsi EI, Parikh S. Ophthalmological findings in 74 patients with mitochondrial disease. Ophthalmic Genet. 2017;38(1):67–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1: Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.