

Abstract
Based on accumulating evidence, cholesterol metabolism dysfunction has been suggested to contribute to the pathophysiological process of traumatic brain injury (TBI) and lead to neurological deficits. As a key transporter of cholesterol that efflux from cells, the ATP‐binding cassette (ABC) transporter family exerts many beneficial effects on central nervous system (CNS) diseases. However, there is no study regarding the effects and mechanisms of ABCG1 on TBI. As expected, TBI resulted in the different time‐course changes of cholesterol metabolism‐related molecules in the injured cortex. Considering ABCG1 is expressed in neuron and glia post‐TBI, we generated nestin‐specific Abcg1 knockout (Abcg1‐KO) mice using the Cre/loxP recombination system. These Abcg1‐KO mice showed reduced plasma high‐density lipoprotein cholesterol levels and increased plasma lower‐density lipoprotein cholesterol levels under the base condition. After TBI, these Abcg1‐KO mice were susceptible to cholesterol metabolism turbulence. Moreover, Abcg1‐KO exacerbated TBI‐induced pyroptosis, apoptosis, neuronal cell insult, brain edema, neurological deficits, and brain lesion volume. Importantly, we found that treating with retinoid X receptor (RXR, the upstream molecule of ABCG1) agonist, bexarotene, in Abcg1‐KO mice partly rescued TBI‐induced neuronal damages mentioned above and improved functional deficits versus vehicle‐treated group. These data show that, in addition to regulating brain cholesterol metabolism, Abcg1 improves neurological deficits through inhibiting pyroptosis, apoptosis, neuronal cell insult, and brain edema. Moreover, our findings demonstrate that the cerebroprotection of Abcg1 on TBI partly relies on the activation of the RXRalpha/PPARgamma pathway, which provides a potential therapeutic target for treating TBI.
Keywords: ATP‐binding cassette G1, bexarotene, cholesterol metabolism, neurological deficits, pyroptosis, traumatic brain injury
Brain‐specific knockout of Abcg1 disturbs cholesterol metabolism and aggravates TBI.
1. INTRODUCTION
Traumatic brain injury (TBI) can be defined as the disruption in brain structure and function, caused by an external physical force [1]. TBI, the “silent epidemic,” contributes to worldwide disability and death more than any other traumatic insults [2, 3]. However, despite the high burden of TBI, no effective pharmaceutical intervention or treatment is available for TBI patients as determined by randomized controlled trials. The crucial mechanisms that contribute to cell death and functional deficits following TBI are also still elusive [4]. During the pathophysiological process of TBI, post‐injury cascades including but not limited to apoptosis [5], pyroptosis [5], inflammation [5], and oxidative stress [6] are activated that may have systemic effects on TBI. Besides, lipid metabolism dysregulation in the central nervous system (CNS) is also found after CNS diseases and insults. There are considerable evidences that altered levels of cholesterol and its metabolites are involved in several adult CNS diseases and insults, including TBI, stroke, Huntington's disease, Niemann–Pick disease type C, and Parkinson's disease [7]. A few trials about the effects of TBI on brain cholesterol synthesis and metabolism have demonstrated that not only cholesterol metabolic pathways may provide a therapeutic target for TBI, but also cholesterol metabolism intermediates may potentially act as biomarkers to aid in the identification or severity stratification of TBI [8].
Given cholesterol's amphipathic property, it works as one of the main regulators for the lipid organization in the cytomembrane by maintaining membrane integrity and fluidity [9]. Notably, almost all brain cholesterol is a product of local synthesis, where the blood–brain barrier (BBB) efficiently protects it from exchange with lipoprotein cholesterol in the circulation [10]. Cholesterol synthesis and regulatory mechanism in the brain is quite distinct from the ones in peripheral tissues. The brain cholesterol homeostasis dysregulation markedly affects neuronal and glial functions, leading to CNS diseases [11, 12]. Both cellular loss from initial impact as well as secondary neurodegeneration post‐TBI can lead to an increase in cholesterol and lipid debris, and the excess cholesterol can be toxic and result in cell death [7]. Moreover, patients after a TBI showed an increase of cholesterol in the cerebrospinal fluid post‐TBI [13]. Thus, further research works on cholesterol metabolism after TBI contribute to the therapy of TBI.
Cholesterol metabolism in the brain is regulated by a number of molecules. For example, the ATP‐binding cassette (ABC) transporters, such as ABCA1 and ABCG1, play a critical role in controlling tissue cholesterol level by mediating the transport of cellular cholesterol to nascent high‐density lipoprotein (HDL) and mature HDL, which is called as the reverse cholesterol transport (RCT) [14]. In addition, ABCG4 facilitates the efflux of cholesterol from cells to HDL [15]. Prior studies found that both the Abcg1 and Abca1 genes are highly activated by the nuclear receptor retinoid X receptors (RXRs), peroxisome proliferator‐activated receptors (PPARs), and liver X receptors (LXRs) [16, 17]. In vitro study demonstrated that the coordinated action of ABCA1 and ABCG1 is required for optimal removal of cellular cholesterol [14]. In addition to the regulation of cholesterol metabolism in the brain, ABCA1 and ABCG1 exert neuroprotective function through inhibiting apoptosis, inflammation, and oxidative stress [9]. Previous studies found that mice lacking brain Abca1 exhibited increased neuroinflammation through mediating cholesterol efflux onto lipid‐poor apolipoprotein (Apo), including ApoE [18, 19, 20]. Meaningfully, the lack of one copy of Abca1 significantly increased the expression of microglia sensome genes only in ApoE4 TBI mice [21]. Unlike ABCA1, ABCG1 is involved in maintaining cellular or tissue cholesterol homeostasis, but over‐expression of ABCG1 alone is not sufficient to cause changes in cognition, learning, and memory or hippocampal synaptic plasticity [22]. However, it remains possible that the lack of Abcg1 may influence neuronal physiology and contribute to the cognitive deficits post‐TBI. During the RCT, the enzyme lecithin‐cholesterol acyltransferase (LCAT), localized on HDL surface, transfers a fatty acid from phospholipid to unesterified cholesterol to form cholesteryl esters. Cholesteryl esters then are internalized into the core of HDL via the activation of the scavenger receptor class B type 1 (SCARB1) and alternately shifted to low‐density lipoprotein (LDL) from HDL by the cholesteryl ester transfer protein (CETP) [23]. So cholesterol metabolism after TBI is regulated by complex molecular mechanisms.
Bexarotene (Bex), a selective BBB‐permeant RXRalpha agonist, is the most frequently applied anti‐cancer retinoid in clinical practice [24]. Bex has been shown to restore some cognitive functions through activating RXRalpha/PPARgamma heterodimers in animal models of neurodegenerative disease [24]. Bex has also been found to improve neurological dysfunctions post‐TBI, at least partially in an RXRalpha/PPARgamma/ApoE‐dependent manner [25, 26, 27]. In addition, Bex has anti‐apoptosis and anti‐inflammatory effects on TBI. Thus, Bex has logically been considered as a potential agent for the treatment of TBI. However, the mechanism of Bex on TBI is not fully understood. In particular, the effect of Bex on the reduction of excess cholesterol in brain tissues has not been confirmed in TBI model and further research to aid the clinical transformation of TBI therapy is needed.
As discussed earlier, cholesterol metabolism disturbance can cause neuroinflammation and even cell death [28]. Pyroptosis, which is also termed cell inflammatory necrosis, is a manner of programmed cell death dependent on inflammatory caspases (caspase‐1, caspase‐4, or caspase‐5 in human; caspase‐1 or caspase‐11 in mice) [29]. TBI can cause tissue destruction and hemorrhage that lead to the release of damage‐associated molecular patterns (DAMPs), which are recognized by the NOD‐like receptor protein 3 (NLRP3) and promote inflammasome formation [30]. Then, inflammasomes may activate caspase‐1 and promote the formation of functional mature bodies by cleaving the IL‐1beta precursor and IL‐18 precursor and induce the opening of cell membrane and pyroptosis by cleaving gasdermin D (GSDMD) post‐TBI [31]. Therefore, pyroptosis can lead to nuclear pyknosis, cell swelling, pore formation, membrane rupture, massive leakage of cytoplasmic components, and even cell death [30]. Nevertheless, the role and mechanism of Abcg1 in pyroptosis mobilized by TBI have not been investigated.
In this study, we investigated the effects of Abcg1 on cholesterol metabolism in TBI by establishing a brain‐specific Abcg1 knockout (Abcg1‐KO) mouse model. We found that Abcg1‐KO aggravated cholesterol homeostasis dysregulation, pyroptosis, and neurological dysfunctions post‐TBI, which may be partly regulated by RXRalpha/PPARgamma pathway.
2. MATERIALS AND METHODS
2.1. Animals and models
Adult male C57BL/6 wild‐type mice (6–8 weeks, weighting 20–25 g) were purchased from the Laboratory Animal Center of Soochow University. Mice lacking Abcg1 selectively in the brain were generated by crossing Abcg1‐floxed (Abcg1 flox/flox ) mice with Nestin‐Cre mice (Figure S1). Abcg1 flox/flox mice were purchased from the Jackson Laboratory. Nestin‐Cre transgenic mice were purchased from Shanghai Biomodel Organism. Abcg1 flox/flox mice were crossed with Nestin‐Cre transgenic mice to obtain Abcg1 Nestin−/flox mice. Then, male Abcg1 Nestin−/flox mice cohabited with the female Abcg1 Nestin−/flox mice (Figure S1A) to obtain brain‐specific Abcg1 knockout mice (Abcg1 Nestin‐/Nestin‐ or also described as Abcg1‐KO) and Abcg1 flox/flox (control) mice. Mice carrying the Abcg1 flox/flox and Nestin‐Cre alleles were genotyped by PCR (Figure S1B). The primers are presented in Table S1. Animals were housed for at least 7 days prior to surgery in a temperature (22–25°C) and humidity‐controlled (50%–55% relative humidity) animal facility with a 12‐h light/dark cycle. All mice were allowed free access to food and water except that food was withheld overnight before surgery. All animal experiments were reviewed and approved by the Ethics Committee of Soochow University.
In this study, surgical preparation and moderate TBI model was performed as previously described [32]. In brief, mice were anesthetized with sodium pentobarbital (70 mg/kg) and mounted in a stereotaxic frame (David Kopf Instruments). A sagittal incision was then made in the scalp and a circular craniotomy (4‐mm diameter) was located approximately midway between bregma and lambda lateral to the sagittal suture on the left side. The bone flap was gently removed without disruption of the dura. TBI was generated in a direction perpendicular to the brain surface, using a pneumatic cortical impact device (Amscien Instruments) with the following parameters: high pressure 200 Kpa, low pressure 100 Kpa, and the depth of 1.0 mm. The craniotomy then was closed with standard suture material immediately after TBI. Animals were anesthetized and sacrificed for biological studies at the indicated time points. In the Sham group, the mice received an identical procedure except for pneumatic cortical impact injury. To minimize the variance in operation, TBI model and drug administration were performed by the same skilled operators.
Human cortex tissue was provided by the Medicolegal Expertise Center of Soochow University. The injured cortex tissues (2–4 d post‐TBI) were harvested from traffic accident victims who died from craniocerebral injury. The normal cortex tissues were harvested from a donor who died from sudden cardiac death. The corpse research was also approved by the Ethical Committee of Soochow University. Informed consent was obtained from the relative of each victim before recruitment.
2.2. Groups and drug administrations
2.2.1. Experiment 1
To investigate the time patterns in the expression of cholesterol metabolism‐related proteins after TBI, mice were sacrificed 6 h, 12 h, 1 d, 2 d, 3 d, 7 d, and 14 d after surgery (n = 6/group), and western blotting was performed using the injured and contralateral cortex, respectively. The expressions of ABCA1 and ABCG1 in the Sham group and the injured cortex 3 d after TBI were also detected by immunofluorescence staining (n = 5/group). Distributions of ABCA1 and ABCG1 in the injured cortex 3 d after TBI were shown by double immunofluorescence staining (n = 5/group). The expressions of ABCA1 and ABCG1 in normal and injured cortex from the corpse were also shown by immunohistochemical staining (n = 5/group) (Figure 1A).
FIGURE 1.
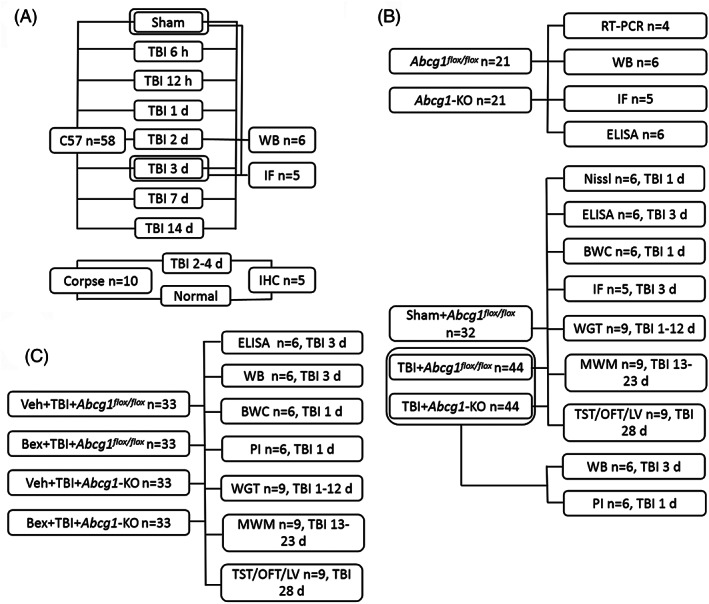
Experimental design for the current study. Bex, bexarotene; BWC, brain water content; IF, immunofluorescence; IHC, immunohistochemistry; LV, lesion volume; MWM, Morris water maze; OFT, open field test; PI, propidium iodide; TBI, traumatic brain injury; TST, tail suspension test; Veh, vehicle; WB, western blotting; WGT, wire grip test
2.2.2. Experiment 2
Various studies were performed to investigate the effect and mechanism of Abcg1 on TBI. First, the Abcg1‐KO mouse was identified by RT‐PCR (n = 4/group), western blotting (n = 6/group) and immunofluorescence staining (n = 5/group), versus the Abcg1 flox/flox mouse. The effect of Abcg1 on the cholesterol metabolism in the normal cortex was investigated by the enzyme‐linked immunosorbent assay (ELISA) kit and animals were divided into two groups: Abcg1 flox/flox group (n = 6/group) and Abcg1‐KO group (n = 6/group). Second, ELISA (n = 6/group), western blotting (n = 6/group), immunofluorescence staining (n = 5/group), Nissl staining (n = 6/group), brain water content (n = 6/group), propidium iodide (PI) labeling (n = 6/group), behavioral tests (n = 9/group), and lesion volume (n = 9/group) were performed at the indicated time points postinjury to examine the role and mechanism of Abcg1 on TBI and mice were divided into two groups: TBI + Abcg1 flox/flox group and TBI + Abcg1‐KO group, or three groups: Sham+Abcg1 flox/flox group, TBI + Abcg1 flox/flox group, and TBI + Abcg1‐KO group (Figure 1B).
2.2.3. Experiment 3
To further exploit the mechanism of Abcg1 on TBI, RXR agonist Bex was given post‐TBI. ELISA (n = 6/group), western blotting (n = 6/group), PI labeling (n = 6/group), brain water content (n = 6/group), behavioral deficits (n = 9/group), and lesion volume (n = 9/group) were assessed at the indicated time points post‐injury, and mice were divided into four groups: vehicle (Veh) + TBI + Abcgl flox/flox group, Bex + TBI + Abcgl flox/flox group, Veh + TBI + Abcgl‐KO group, and Bex + TBI + Abcgl‐KO group. Bex was prepared as follows: (1) 10 mg Bex (Abcam, ab141025) was initially dissolved in dimethyl sulfoxide (DMSO); (2) the mixed solution was prepared with polyethylene glycol (15)‐hydroxystearate (Solutol), ethanol, and water at a 15:10:75 ratio; (3) the solutions in steps 1 and 2 were mixed together to make the final concentration of Bex (0.70 mg/ml). Bex was injected intraperitoneally at a dose of 5 mg/kg at 2 h post‐TBI for the first time, and then continuously dosed once a day post‐TBI until mice execution. The solution given to the vehicle group was dosed at the same volume, same route, and same time course as Bex (Figure 1C).
2.3. Western blotting
Mice were euthanized on the corresponding time after TBI. The injured cortical tissues (2 × 2 × 2‐mm tissue block including the impact site and surrounding areas) from each mouse were isolated and fully homogenized in the RIPA lysis buffer (Beyotime Biotechnology). The homogenates were then centrifuged at 13,362g for 30 min at 4°C, and the protein concentrations were determined using NanoDrop 2000C (Thermo Scientific). Sixty micrograms of protein from each sample were separated by electrophoresis using an 8%–12% SDS‐PAGE gel and then transferred to PVDF membranes (Millipore) on a wet electrotransferred unit (Bio‐Rad). The membranes were blocked in 5% BSA in TBST for 2 h at room temperature and incubated with the primary antibodies overnight at 4°C. After washing with TBST, blots were incubated with an HRP‐conjugated secondary antibody for 2 h. The immunoreactivity of blots was detected with an ECL chemiluminescence system (Chemi Scope). The signal intensity of blots was quantified using Image J software and normalized to beta‐actin.
Anti‐ABCA1 (A16337), anti‐ABCG4 (A14596), anti‐ApoE (A16344), anti‐LCAT (A3942), anti‐LXR‐alpha (A3974), and anti‐CETP (A1355) antibodies were purchased from Abclonal Technology Inc. Anti‐IL‐1beta (sc‐52,012), anti‐apoptosis‐associated speck‐like protein containing CARD (ASC, sc‐271054), anti‐GSDMD (sc‐393581), and anti‐Caspase‐1 (sc‐56036) antibodies were purchased from Santa Cruz Company. Anti‐RXR‐alpha (ab125001), anti‐Bax (ab32503), and anti‐PPAR‐gamma (ab59256) antibodies were purchased from Abcam Company. Anti‐ABCG1 (NB400‐132) antibody was purchased from Novus Biologicals Inc. Anti‐SCARB1 (DF6479) antibody was purchased from Affinity Biosciences Inc. Anti‐Cleaved caspase‐3 (BS1518) and anti‐Bcl‐2 (BS1511) antibodies were purchased from Bioworld Technology Inc., and the anti‐beta‐actin antibody (ATF0001) was purchased from CMS1518 Inc.
2.4. Immunofluorescence and immunohistochemical staining
Animals were anesthetized on days 3 after TBI, and sequentially transcardially perfused with 30 ml of 0.1 M PBS and 30 ml of 4% paraformaldehyde (PFA). Then, brains were post‐fixed with 4% PFA for 24 h and subsequently dehydrated in serial sucrose solutions (10%, 20%, and 30%) in 0.1 M PBS for 36 h at 4°C. Brains were frozen in Tissue‐Tek OCT compound (Sakura Finetek) for 20 min and cut into coronal slices (12 μm) using a freezing microtome (Leica SM 2000R). Then, sections were rinsed with 0.1 M PBS for 10 min and incubated with a blocking solution (0.1% Triton X‐100 and 5% donkey plasma in 0.1 M PBS) for 2 h at room temperature. Sections were then incubated with ABCA1 (A16337), ABCG1 (NB400‐132), ASC (sc‐271054), Caspase‐1 (sc‐56036), GSDMD (sc‐393581), NeuN (ab104225, Abcam), GFAP (ab53554, Abcam), Iba1 (ab5076, Abcam) in blocking solution overnight at 4°C. After rinsing with 0.1 M PBS for 30 min, sections were incubated with the corresponding secondary antibodies in the blocking solution for 1 h at room temperature. To visualize the overall density of the total cells, sections were incubated with DAPI (1:10,000, Beyotime Biotechnology) in a blocking solution for 2 min at room temperature. The stained sections were examined using a Nikon fluorescence microscope (Japan). In a blinded manner, the positive cells (ASC, Caspase‐1, and GSDMD) were counted from five evenly distributed areas (regions of interest) in each brain section containing the injured cortex.
Brains of corpses were fixed with 4% PFA for 3 d and subsequently dehydrated in serial sucrose solutions (10%, 20%, and 30%) in 0.1 M PBS for 3 d at 4°C. Then brains were frozen and cut into coronal slices (12 μm). Cortical sections from the corpse were rinsed in PBS twice and incubated in 3% hydrogen peroxide for 30 min. Sections were washed three times in 0.1 M PBS and blocked 1 h in 10% FBS (Gibco) in PBS with 0.01% Triton X‐100. Slides were incubated overnight with anti‐ABCA1 (1:200) and ABCG1 (1:200) antibodies, respectively. After rinsing with 0.1 M PBS for 30 min, sections were incubated with the corresponding HRP‐labeled secondary antibodies in the blocking solution for 1 h at room temperature. DAB staining was performed using DAB kit (Beyotime Biotechnology). The cell nucleus was strained by hematoxylin. The stained sections were examined using a Nikon microscope.
2.5. Quantitative real‐time PCR
The mRNA level of Abcg1 gene was determined using real‐time quantitative PCR. Briefly, the total RNA was isolated with trizol reagent (Invitrogen) according to the manufacturer's protocol. After determining the total RNA concentration using a NanoDrop 2000C (Thermo Scientific), reverse transcription to cDNA was carried out using PrimeScript RT Master Mix (Takara Bio). The real‐time PCR thermal cycler (LightCycler, Roche Applied Science) was used to conduct quantitative real‐time PCR. Real‐time PCR of all samples was done in triplicate using the SYBR Green master mix (Roche Applied Science). The mRNA level was normalized to Gapdh for internal control. The relative expression levels of genes were quantified using the comparative △Ct method. The primers are presented in Table S2.
2.6. Measurement of cholesterol‐related indicators by ELISA
The levels of total cholesterol (T‐CHO), HDL cholesterol (HDL‐c), LDL cholesterol (LDL‐c), and triglyceride (TG) in blood plasma and brain tissue were measured using ELISA kits (Jiancheng Bioengineering).
2.7. Nissl staining
Nissl staining was performed to detect the degree of cortical peri‐injury neuron degeneration 1 d after TBI [32]. Frozen sections (12 μm) were prepared as described above and dehydrated with gradient alcohol: 75% alcohol for 15 min and 90% alcohol for 25 min. Then, the sections were washed with distilled water for 2 min. After that, the sections were incubated with Nissl staining solution (Beyotime Biotechnology, Shanghai, China) at room temperature for 5 min. Then the sections were rinsed with distilled water and observed using a Nikon microscope (Japan).
2.8. Measurement of brain water content
Brain water content was measured as described previously [32]. Briefly, mice (n = 6/group) were anesthetized and sacrificed 24 h after TBI. The removed brain was taken out and divided into three parts: the ipsilateral hemisphere, the contralateral hemisphere, and the cerebellum. Samples were immediately weighed on an electronic analytical balance to obtain the wet weight, and then dried in a 160°C oven for 24 h to obtain the dry weight. The water content of each part of the brain tissue was determined as (wet weight − dry weight)/wet weight × 100%.
2.9. PI labeling
PI labeling was performed 1 d after TBI (n = 6/group). PI (total volume: 200 μl, concentration: 0.4 mg/ml, Sigma–Aldrich Corporation) was intraperitoneally injected 1 h before sacrificing the mouse. Mouse was euthanized and the brain was quickly and carefully removed to ensure that the contusion region was intact, rapidly frozen in liquid nitrogen vapor, and then coronally sectioned at 12 μm thickness and 200‐μm intervals with a cryostat (Leica SM 2000R). Sections were rinsed with 100% ethanol and then examined with a Nikon fluorescence microscope (Japan). For each brain, five ×200 cortical fields were randomly chosen for analysis using a random number generator [33].
2.10. Analysis of neurological functions
A wire grip test was performed in this study to assess the effect of Abcg1 on motor function after TBI. The wire‐grip test was conducted as previously described [33] and performed on Days 1–12 post‐TBI. Briefly, a 45‐cm‐long metal wire was held at the height of 45 cm, and a five‐point scale was used to assess the motor function. One test included three trials and the average value was regarded as the final score each day.
The Morris Water Maze (MWM) test, including the hidden platform test and the probe test, was performed to evaluate the effect of Abcg1 on spatial learning and memory performance of mice after TBI [33]. Briefly, mice were trained in the MWM on Days 3–5 (three trials per day, to familiarize them with the environment) before TBI. The hidden platform test was carried out on Days 13–22 post‐TBI. During each of the two trials, each animal was permitted 90 s to reach the submerged platform and remained on it for 10 s. If a mouse failed to reach the platform within 90 s, it was guided to the platform and allowed to stay for 10 s. In the probe test, the mouse was permitted to swim freely for 90 s in the tank with the goal platform removed on Day 23 post‐TBI. The number of crossing platform was recorded as a measure of spatial memory. Swimming escape latency and the crossing number were recorded and analyzed with a tracking device and software (Chromotrack 3.0, San Diego Instruments).
The tail suspension test (TST) and open field test (OFT) were performed on Day 28 post‐TBI to evaluate the depression‐like phenotype after injury. Because odor cues might interfere with the behavioral performance, 75% ethanol was used to wipe the apparatus during the TST and OFT manipulations. In the TST, a mouse was acclimated to the behavior room for 1 h and hung upside down in the tail suspension chamber (DigBehv‐LG, Jiliang Software Technology). The chamber transformed the stretching force into electrical signals such that the computer could record even slight movement of the mouse, whose tail was stuck to a detecting hook. Behaviors were recorded with a video camera for 6 min. All movements with values under the threshold set at 0.75 would be determined immobile. In the OFT, animals were placed in an OFT arena (30 × 30 × 30 cm). Behaviors and parameters were recorded and analyzed using an automated behavioral tracking system (JLBehv‐OFG‐4, Shanghai Jiliang Software Technology). The parameters included the total distance, distance moved in the center, and time spent in the center of the OFT arena.
2.11. Assessment of lesion volume
To determine whether Abcg1 alleviates tissue damage caused by TBI, we evaluated lesion volume in the ipsilateral cortical hemisphere on Day 28 following TBI. The brains were harvested. Frozen brain sections (12 μm) were collected at 500‐μm intervals, and stained with hematoxylin staining solution. The areas of the injured and non‐injured hemispheres were quantitatively analyzed using ImageJ software in a blinded fashion. Data were presented as the volume percentage of the lesion versus the contralateral hemisphere.
2.12. Statistical analysis
All experiments were randomized and performed in a blinded manner. All data are presented as the means ± SEM. Wire grip test and MWM (the hidden platform test) data were analyzed using a two‐factor repeated measures analysis of variance (ANOVA, for group and time) followed by Bonferroni's post hoc test for multiple comparisons. MWM (the probe test), TST, OFT, immunofluorescence staining, ELISA, and western blotting data were analyzed using one‐way ANOVA with Dunnett's t test. The lesion volume, and PI labeling data were analyzed using Student's t test. A p value of <0.05 was considered statistically significant. The power analysis was performed using the G*Power 3.1.9.4 software. Statistical analyses were conducted with GraphPad Prism 8.0 software. All data were based on at least three independent experiments.
3. RESULTS
3.1. The expression and distribution of cholesterol metabolism‐related proteins post‐TBI
To investigate the influence of TBI on cholesterol metabolism, the expression levels of cholesterol metabolism‐related proteins, including ABCA1, ABCG1, ABCG4, SCARB1, ApoE, LCAT, RXRalpha, PPARgamma, and LXRalpha, were determined in the cortex in the Sham group and after TBI from 6 h to 14 d. The levels of ABCA1, ABCG1, and ABCG4 were increased from 3 d to 14 d, 12 h to 14 d, and 6 h to 3 d post‐TBI, respectively, compared with the Sham group (Figure 2A,B, p < 0.05). Notably, compared with the Sham group, the levels of LCAT and PPARgamma were markedly increased from 6 h to 14 d, and 1 d to 14 d, respectively, postinjury (Figure 2A,B, p < 0.05). The expression of ApoE was significantly elevated on Days 1, 3, and 14 postinjury, relative to the Sham group (Figure 2A,B, p < 0.05). The general tendency of RXRalpha was markedly decreased and reached a valley from Days 2 to 3 postinjury, then gradually increased afterward (Figure 2A,B, p < 0.05). Interestingly, although there were no statistical changes in the levels of SCARB1 in the acute stage post‐TBI, a significant upregulation of SCARB1 was found on Days 14 postinjury, relative to the Sham group (Figure 2A,B, p < 0.05). In addition, the level of LXRalpha has a marked increase on 1 d and decrease on 3 d post‐TBI. Furthermore, we examined the cholesterol metabolism‐related proteins in the contralateral cortex, and our results showed that the levels of these proteins did not significantly fluctuate (Figure S2, p > 0.05). Notably, 3 d post‐TBI was the time point when there were statistical differences in almost all these proteins. These data suggest that TBI induces diverse dynamic changes of cholesterol metabolism‐related proteins in the cortex.
FIGURE 2.
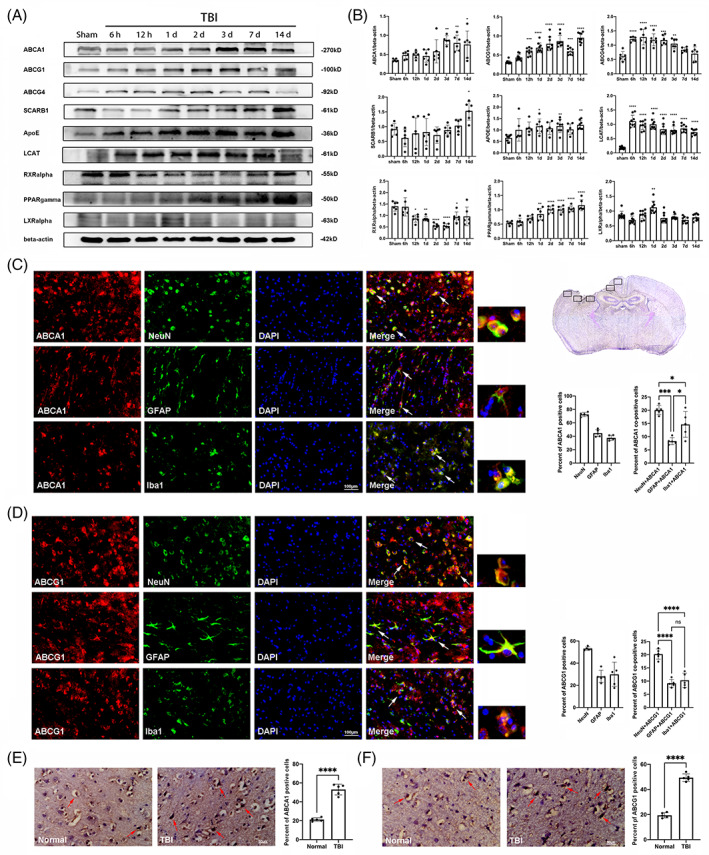
The expression of cholesterol metabolism‐related proteins in the cortex post‐TBI. (A) Representative western blotting bands of ABCA1, ABCG1, ABCG4, SCARB1, ApoE, LCAT, RXRalpha, PPARgamma, LXRalpha, and beta‐actin in the cortex at the indicated time point post‐TBI. (B) The relative optical densitometric quantification of the proteins was normalized against beta‐actin. (C, D) A panoramic view of brain slice pointed out the location of injured cortical brain regions used for staining and cell counts. Distribution and semi‐quantitative analysis of ABCA1 and ABCG1 in the injured cortex on Day 3 after TBI. Examples of co‐localization of ABCA1/ABCG1 with the neuron marker NeuN, the astrocyte marker GFAP, and the microglia marker Iba‐1 using white arrows. (E, F) The immunohistochemical staining and semi‐quantitative analysis of ABCA1 and ABCG1 in the injured and normal cortex in the human brain. Examples of ABCA1/ABCG1‐positive cell using white arrows. Data are mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, or ****p < 0.0001 compared with the sham group (for A and B), ****p < 0.0001 compared with the normal group (for E and F). ns, not significant. n = 6/group for A and B; n = 5/group for C–F. Magnification: ×400, scale bar = 100 μm for C and D, 50 μm for E and F
ABCA1 and ABCG1, the key signaling molecules, are involved in RCT and maintain cholesterol homeostasis in CNS injuries and disorders [23]. We therefore examined the expressions and distributions of ABCA1 and ABCG1 in the injured cortex on Day 3 after TBI. As the panoramic view of brain slice showed (Figure 2C), five framed sections of injured cortical brain are used for staining and cell counts. First, the expressions of ABCA1 and ABCG1 in the mice brain 3 d after TBI were detected by immunofluorescence staining. The results showed that the quantity of ABCA1 and ABCG1‐positive cells was significantly increased in the injured cortex in the mice brain 3 d after TBI, relative to the Sham group (Figure S3). Then, double immunofluorescence staining of ABCA1 and ABCG1 was performed with neuron marker NeuN, astrocyte marker GFAP and microglia marker Iba1. Semi‐quantitative analysis showed that 72.50% of neurons, 44.66% of astrocytes, and 37.65% of microglia co‐expressed ABCA1 (Figure 2C), as well as 53.16% of neurons, 28.09% of astrocytes, and 29.93% of microglia co‐expressed ABCG1 (Figure 2D) on Day 3 after TBI. Further data analysis showed that among all cells (DAPI staining) in the injured cortex, NeuN/ABCA1 co‐postive cells accounted for 20.08%, GFAP/ABCA1 co‐positive cells accounted for 8.33% and Iba1/ABCA1 co‐positive cells accounted for 14.64% (Figure 2C). Similarly, among all cells in the injured cortex, NeuN/ABCG1 co‐positive cells accounted for 20.22%, GFAP/ABCG1 co‐positive cells accounted for 8.49% and Iba1/ABCG1 co‐positive cells accounted for 10.30% (Figure 2D). These results indicate that both ABCA1 and ABCG1 predominantly present in neurons. To study the expressions of ABCA1 and ABCG1 in the human brain after TBI, immunohistochemical staining of the cortex from the corpse was performed. The results showed that the number of ABCA1 and ABCG1‐positive cells was significantly increased in the injured cortex at the acute stage (Days 2–4) post‐TBI, relative to the normal cortex region (Figure 1E,F). These data demonstrate that multiple neuronal cells undergo RCT and maintain cholesterol homeostasis after TBI.
3.2. Brain‐specific knockout of Abcg1 mice exhibits cholesterol metabolism turbulence
Given that ABCG1 is expressed in the neuron, astrocyte, and microglia, we generated Nestin‐specific Abcg1 knockout mice (Abcg1‐KO). Abcg1‐KO mice were identified using western blotting, real‐time PCR, and immunofluorescence. Decreased expressions of Abcg1 mRNA and protein from the cortex were observed in the Abcg1‐KO mice, compared with the Abcg1 flox/flox mice (Figure 3A,B, p < 0.05). As shown in Figure 3C,D, the Abcg1‐KO mice had a reduction of ABCG1 immunogenicity, relative to the Abcg1 flox/flox mice. Next, the role of Abcg1‐KO on cholesterol metabolism in blood plasma was examined. Strikingly, a decrease of HDL‐c and an increase of LDL‐c were observed in the Abcg1‐KO mice, compared with the Abcg1 flox/flox mice (Figure 3E, p < 0.05). However, there were no statistical differences in the TG and T‐CHO levels between the Abcg1 flox/flox mice and the Abcg1‐KO mice. These results suggest that Abcg1 plays an important role in maintaining cholesterol metabolism homeostasis in basal conditions.
FIGURE 3.
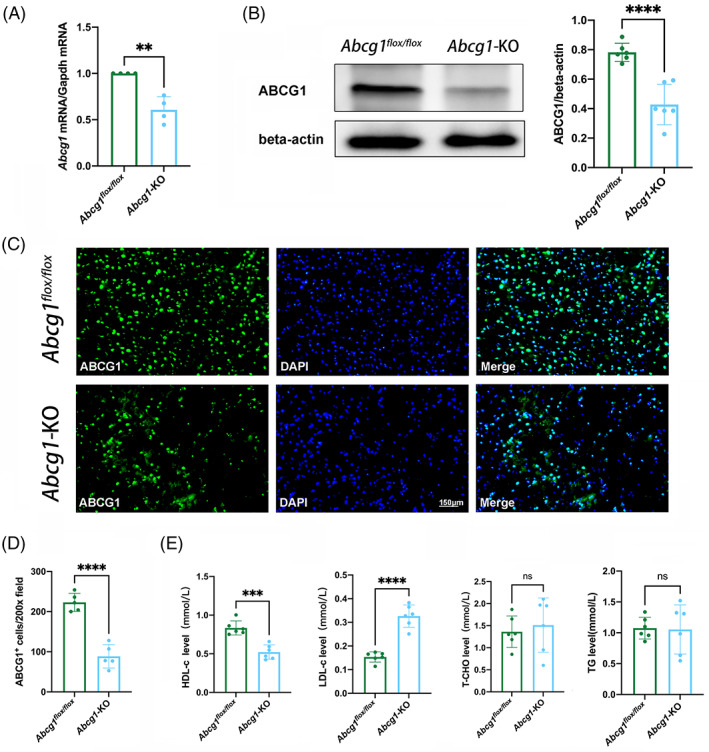
Abcg1‐KO mice developed cholesterol metabolism dyshomeostasis under basal conditions. (A) Real‐time PCR of Abcg1 mRNA from Abcg1 flox/fiox and Abcg1‐KO mice. (B) Western blotting analysis of ABCG1 protein levels from Abcg1 flox/fiox and Abcg1‐KO mice. (C, D) Representative immunofluorescence analysis of ABCG1 in the cortex from Abcg1 flox/fiox and Abcg1‐KO mice. Scale bar = 150 μm. (E–H) HDL‐c, LDL‐c, T‐CHO, and TG levels in the blood plasma from Abcg1 flox/fiox and Abcg1‐KO mice were measured by ELISA. *p < 0.05, **p < 0.01, ***p < 0.001 or ****p < 0.0001 compared with Abcg1 flox/fiox group. ns, not significant. n = 4/group for A, n = 5/group for C and D, n = 6/group for B and E
3.3. Abcg1‐KO mice are susceptible to TBI‐induced cholesterol metabolism turbulence
To examine whether Abcg1‐KO affects TBI‐induced cholesterol metabolism turbulence, the levels of HDL‐c, LDL‐c, T‐CHO, and TG were first measured. The ELISA assay was conducted using blood plasma and injured cortex. In blood plasma, HDL‐c decreased, T‐CHO, and TG increased, but LDL‐c had no change at 3 days post‐TBI, compared to the Sham group (Figure 4A, p < 0.05). Meanwhile, a significant increase of LDL‐c level, a remarkable decrease of HDL‐c level and no significant changes of TG and T‐CHO in the Abcg1‐KO group were observed at 3 days after TBI, compared with the TBI + Abcg1 flox/flox group (Figure 4A, p < 0.05). When it came to the brain tissue, the area around the injured cortex was selected as a sample to be tested. A decrease in HDL‐c level and an increased level of LDL‐c, T‐CHO, and TG were found post‐TBI, relative to the Sham group (Figure 4B, p < 0.05). In addition, Abcg1‐KO exacerbated the changes of all these indicators post‐TBI, compared with the TBI + Abcg1 flox/flox group (Figure 4B, p < 0.05). To study the role of Abcg1‐KO on TBI, cholesterol metabolism‐related proteins were measured in the injured cortex. Decreased expressions of ABCA1, ABCG4, and SCARB1 on Day 3 after TBI in the Abcg1‐KO mice were observed, relative to the Abcg1 flox/flox mice (Figure 4C,D, p < 0.05). Interestingly, a significant increase of PPARgamma, RXRalpha, LXRalpha, ApoE, LCAT, and CETP was observed on Day 3 following TBI in the Abcg1‐KO group, compared with the Abcg1 flox/flox group (Figure 4C,D, p < 0.05). These results suggest that Abcg1‐KO deteriorates TBI‐induced cholesterol metabolism turbulence.
FIGURE 4.
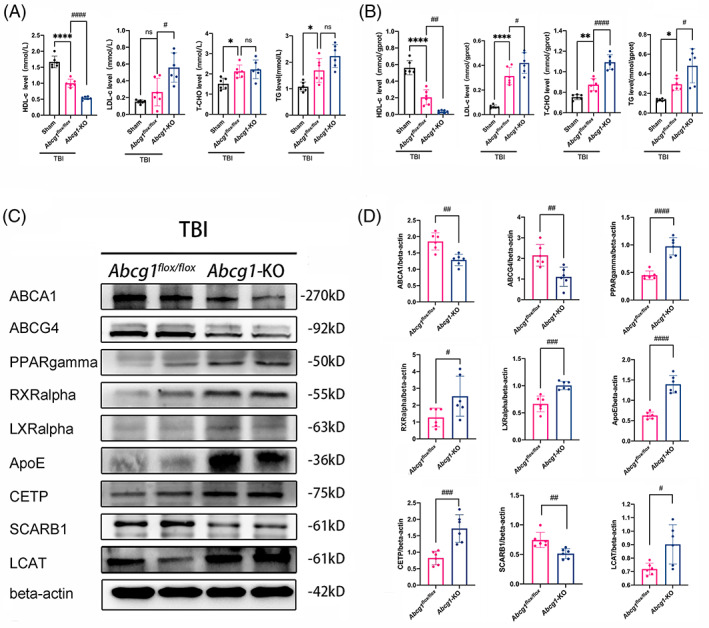
Abcg1‐KO mice developed more severe cholesterol metabolism dyshomeostasis. (A, B) HDL‐c, LDL‐c, T‐CHO, and TG levels in blood plasma and the injured cortex were measured by ELISA at 3 d post the Sham and TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. (C, D) Representative western blots and optical densitometric quantification of ABCA1, ABCG4, PPARgamma, RXRalpha, LXRalpha, ApoE, CETP, SCARB1, and LCAT in the injured cortex at 3 d post‐TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. Data are mean ± SEM; *p < 0.05, **p < 0.01, or ****p < 0.0001 compared with the Sham group. #p < 0.05, ##p < 0.01, ###p < 0.001, or ####p < 0.0001 compared with TBI + Abcg1 flox/fiox group. ns, not significant. n = 6/group
3.4. Abcg1‐KO mice are vulnerable to TBI‐induced pyroptosis and apoptosis
Cholesterol metabolism turbulence may lead to neuroinflammation and neuronal cell death post‐TBI, so pyroptosis and apoptosis‐related proteins, including IL‐1beta, ASC, GSDMD, Caspase‐1, cleaved caspase‐3, Bcl‐2, and Bax, were measured in the injured cortex using western blotting post‐TBI. We found that the levels of IL‐1beta and cleaved caspase‐3 were increased from 12 h to 14 d post‐TBI compared with the Sham group (Figure S4, p < 0.05). And the levels of ASC, GSDMD, Caspase‐1, Bcl‐2, and Bax were increased from 6 h to 14 d, relative to the Sham group (Figure S4, p < 0.05). Moreover, compared with the Abcg1 flox/flox group, Abcg1‐KO increased the levels of ASC, GSDMD, IL‐1beta, and Caspase‐1 (Figure 5A,B, p < 0.05) post‐TBI. Whereafter, TBI‐induced apoptosis in the Abcg1‐KO mice was reflected by a significant increase of cleaved caspase‐3 and Bax expression and a marked decrease of Bcl‐2 expression relative to the Abcg1 flox/flox mice (Figure 5A,B, p < 0.05). In addition, the expression of IL‐1beta, ASC, GSDMD, Caspase‐1, cleaved caspase‐3, Bcl‐2, and Bax was temporally changed in the injured cortex after TBI (Figure S3). Double immunofluorescence staining for ASC, Caspase‐1, and GSDMD with NeuN was also performed to investigate neuronal pyroptosis on Day 3 post‐TBI. There was almost no expression of ASC, Caspase‐1, and GSDMD in the Sham group (Figure 5C–E). Compared to the Sham group, TBI increased the number of ASC/NeuN, GSDMD/NeuN, and Caspase‐1/NeuN co‐positive cells in the injured cortex (Figure 5C–E, p < 0.05). Moreover, the Abcg1‐KO mice exhibited a significant increase of the number of ASC/NeuN, GSDMD/NeuN, and Caspase‐1/NeuN co‐positive cells, relative to the Abcg1 flox/flox mice (Figure 5C–E, p < 0.05). Therefore, the data indicate that Abcg1‐KO deteriorates TBI‐induced pyroptosis and apoptosis.
FIGURE 5.
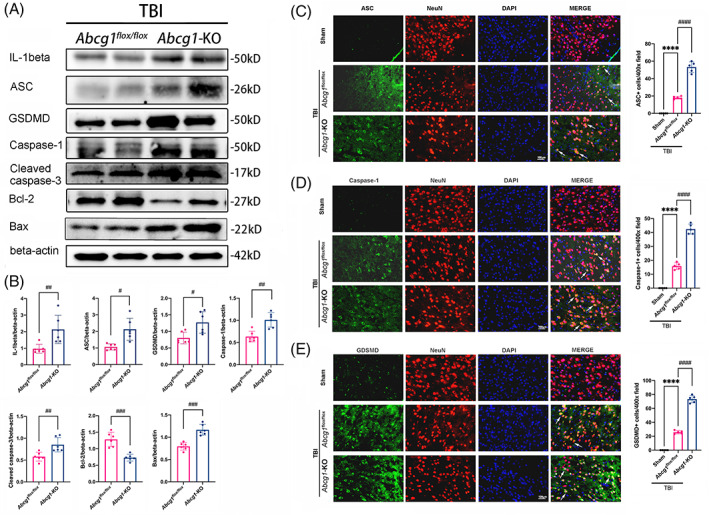
Abcg1‐KO aggravated TBI‐induced pyroptosis and apoptosis. (A, B) Representative western blots and optical densitometric quantification of IL‐1beta, ASC, GSDMD, Caspase‐1, cleaved caspase‐3, Bcl‐2, and Bax in the injured cortex at 3 d post‐TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. (C, D) Representative immunofluorescence staining of ASC/NeuN, Caspase‐1/NeuN, and GSDMD/NeuN double labeling with white arrows and statistical analysis of the positive cell number in the injured cortex at 3 d post the Sham and TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. Data are mean ± SEM; *p < 0.05, **p < 0.01, or ****p < 0.0001 compared with the Sham group. #p < 0.05, ##p < 0.01, ###p < 0.001, or #### p < 0.0001 compared with TBI + Abcg1 flox/fiox group. n = 6/group for A and B, n = 5/group for C–E. Magnification: ×400 and scale bar 100 μm for C–E
3.5. Abcg1‐KO exacerbated neuronal cell insult and brain edema after TBI
Nissl staining was measured on Day 1 postinjury to evaluate the effect of Abcg1‐KO on neurodegeneration. Microscopic examination showed that the neurons were round shape and lilac blue in the Sham group, while the damaged neurons caused by TBI were pyknosis and dark blue (Figure 6A, p < 0.05). Moreover, Abcg1‐KO exacerbated the shrinkage and hyperchromatic morphology of the Nissl bodies post‐TBI, relative to the Abcg1 flox/flox group (Figure 6A, p < 0.05). PI labeling was performed on Day 1 postinjury to further elucidate the effect of Abcg1‐KO on TBI‐induced cell plasma membrane insult. More PI‐positive cells were observed in the injured cortex of the TBI + Abcg1‐KO group than in the TBI + Abcg1 flox/flox group (Figure 6B,C, p < 0.05). As brain edema contributes to the high mortality of TBI, the brain water content was also measured on Day 1 postinjury. Compared to the Sham group, the TBI + Abcg1 flox/flox group showed significantly severe cerebral edema in the ipsilateral hemisphere after TBI (Figure 6D, p < 0.05). In addition, the brain edema was significantly more severe in the ipsilateral hemisphere of the Abcg1‐KO group than the TBI + Abcg1 flox/flox group (Figure 6D, p < 0.05). However, there were no significant differences of brain edema in control among the three groups in the contralateral hemisphere and cerebellum (Figure 6D, p > 0.05). These data show that Abcg1‐KO deteriorates TBI‐induced neuronal cell insults and brain edema.
FIGURE 6.
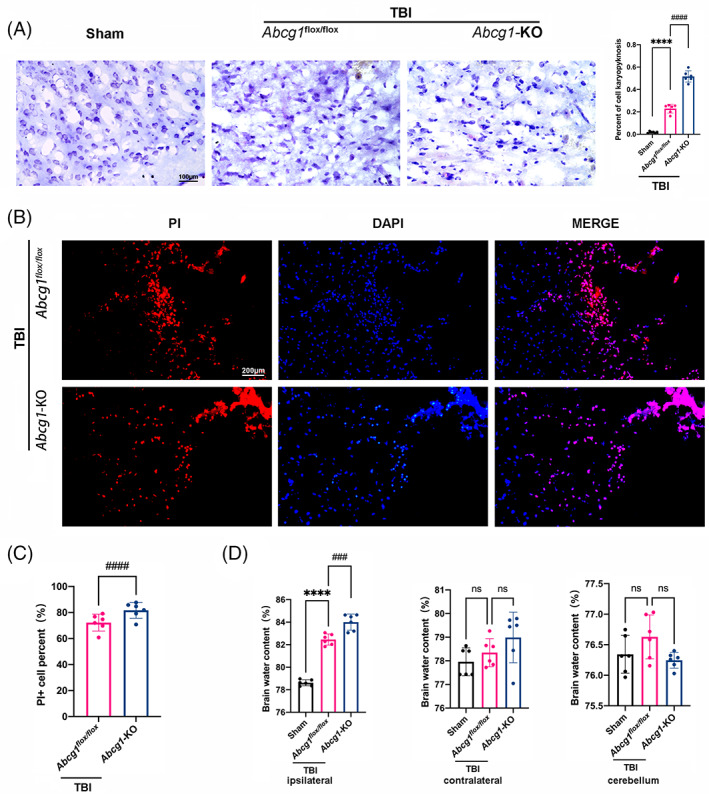
Abcg1‐KO deteriorated TBI‐induced neurodegeneration, cell plasma membrane insults, and brain edema. (A) Representative photographs showing the results of Nissl staining in the injured cortex on 1 d post the Sham and TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. (B, C) Representative photographs and quantitative analysis of PI‐positive cells in the injured cortex on 1 d post‐TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. (D) Representative histogram showing the brain water content, including the ipsilateral hemisphere, contralateral hemisphere, and cerebellum on 1 d post the Sham and TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. Data are mean ± SEM. ****p < 0.0001 compared with the Sham group. ###p < 0.001 or ####p < 0.0001 compared with TBI + Abcg1 flox/fiox group. ns, not significant. n = 6/group. Magnification: ×400 and scale bar 100 μm for A. Magnification: ×200 and scale bar 200 μm for B
3.6. Abcg1‐KO aggravates TBI‐induced motor dysfunctions, memory deficits, depression‐like behaviors, and brain lesion volume
A series of behavioral experiments were carried out to determine brain‐specific knockout of Abcg1 on the long‐term outcomes of TBI. In the wire grip test, mice of the Abcg1 flox/flox group showed motor dysfunctions from Days 1 to 9 post‐TBI, compared to the Sham group (Figure 7A, p < 0.05). Interestingly, the Abcg1‐KO group demonstrated more severe motor dysfunctions on Days 2–11 post‐TBI, relative to the Abcg1 flox/flox group (Figure 7A, p < 0.05). In addition, the MWM test was performed to estimate the effect of Abcg1 on spatial learning and memory deficits. In the hidden platform test, an increased latency was observed on Days 13–18 post‐TBI in the Abcg1 flox/flox group when compared to the Sham group (Figure 7B, p < 0.05). However, compared to the Abcg1 flox/flox group, Abcg1‐KO group prolonged the latency on Days 18–20 post‐TBI (Figure 7B, p < 0.05). The probe trial was performed on the 23rd day post‐TBI, where a decreased crossing number was observed in the Abcg1 flox/flox group compared with the Sham group, (Figure 7B, p < 0.05). However, Abcg1‐KO group demonstrated a more decreased crossing number post‐TBI, relative to the Abcg1 flox/flox group (Figure 7B, p < 0.05). The TST and the OFT were conducted on the 28th day postinjury to evaluate the depression‐like phenotype after TBI. In the TST, the TBI + Abcg1 flox/flox group presented a longer immobility time than the Sham group (Figure 7C, p < 0.05). Nevertheless, mice in the TBI + Abcg1‐KO group exhibited significantly longer immobility time than mice in the TBI + Abcg1 flox/flox group (Figure 7C, p < 0.05). In the OFT, the total distance, the distance moved in center, and the time spent in center of the TBI + Abcg1 flox/flox group were decreased significantly compared with the Sham group, while the TBI + Abcg1‐KO group exacerbated this phenomenon, showing less total distance, distance moved in center, and time spent in center (Figure 7D, p < 0.05). The brain lesion volume was examined on days 28 after TBI to estimate the effect of Abcg1 on TBI‐induced cell loss. We found that TBI induced brain lesion volume, relative to the Sham group (Figure 7E, p < 0.05). However, compared to the TBI + Abcg1 flox/flox group, the Abcg1‐KO significantly augmented brain lesion volume (Figure 7E, p < 0.05). These results support the findings that Abcg1‐KO deteriorates the recovery of neurological deficits, including motor dysfunctions, cognitive deficits, and depression‐like behaviors, and augments brain lesion volume after TBI.
FIGURE 7.
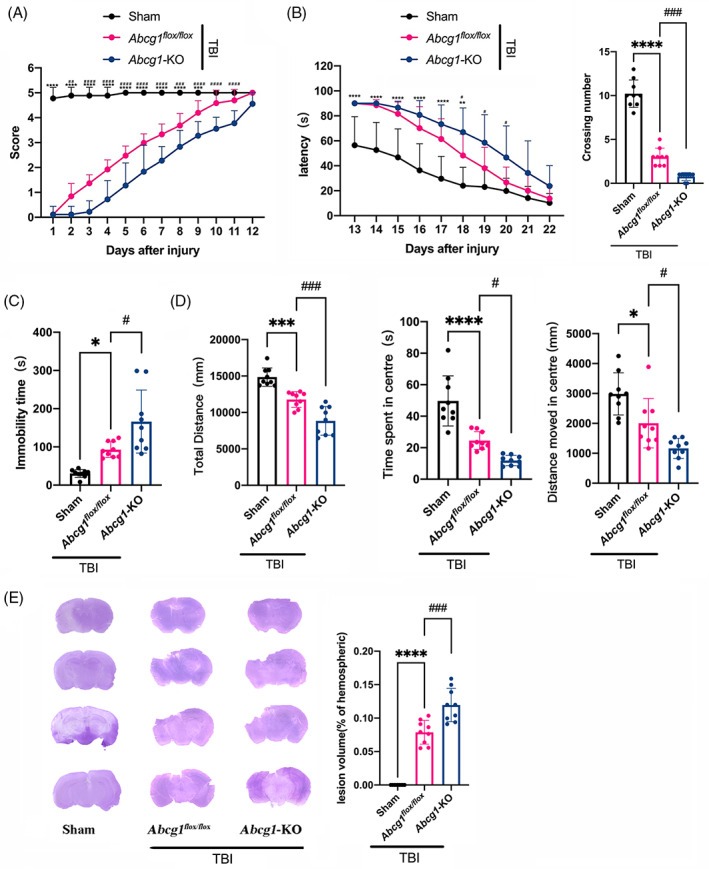
Abcg1‐KO deteriorated neurological deficits and brain lesion volume after TBI. (A) Representative scatter diagram showing the score of the wire grip test on Days 1–12 post the Sham and TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. (B) Representative scatter diagram showing the latency of the hidden platform test on Days 13–22 and the crossing number of the probe trial on Day 23 post the Sham and TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. (C) Representative scatter diagram showing the immobility time of the TST on Day 28 post the Sham and TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. (D) Representative scatter diagram showing the total distance, the distance moved in center, and the time spent in center of the OFT on Day 28 post the Sham and TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. (E) Representative hematoxylin staining photographs of brain sections and quantitative analysis of brain lesion volume on Day 28 post the Sham and TBI + Abcg1 flox/fiox and TBI + Abcg1‐KO. Data are mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, or ****p < 0.0001 compared with the Sham group at each time point. #p < 0.05, ###p < 0.001, or ####p < 0.0001 compared with TBI + Abcg1 flox/fiox group at each time point. ns, not significant. n = 9/group. OFT, open field test; TBI, traumatic brain injury; TST, tail suspension test
3.7. Bex relieved TBI‐induced cholesterol metabolism dysregulation and pyroptosis but did not inhibit apoptosis in Abcg1‐KO mice
To further study the protective mechanism of the Abcg1 on TBI, both the Abcg1 flox/flox and Abcg1‐KO mice were administrated RXRalpha agonist, Bex, post‐TBI. After Bex administration, the levels of HDL‐c, LDL‐c, T‐CHO, and TG of the injured cortex on Day 3 post‐TBI were measured. Compared to the Veh‐treated group, a markedly increase of HDL‐c, and a prominent decrease of LDL‐c, T‐CHO, and TG were observed in the Bex‐treated group post‐TBI, regardless of the Abcg1 flox/flox mice and the Abcg1‐KO mice (Figure 8A,B, p < 0.05). Cholesterol metabolism, pyroptosis, and apoptosis‐related proteins were then detected on Day 3 post‐TBI. In the Abcg1 flox/flox mice, a significant increase of RXRgamma, ABCG4, ApoE, LCAT, PPARalpha, and Bcl‐2, and a significant decrease of ABCA1, CETP, LXRalpha, ASC, cleaved caspase‐3, and Bax were found post‐TBI in the Bex‐treated group, relative to the Veh‐treated group (Figure 8C,D, p < 0.05). In the Abcg1‐KO mice, RXRgamma, and ABCA1 had a markedly increase, and CETP, PPARalpha, and ASC had a significant decrease post‐TBI in the Bex‐treated group, relative to the Veh‐treated group (Figure 8E,F, p < 0.05). Interestingly, there were no statistical differences of ABCG4, ApoE, LCAT, LXRalpha, cleaved caspase‐3, Bcl‐2, and Bax between the Bex‐treated group and Veh‐treated group (Figure 8E,F, p > 0.05). Thus, TBI‐induced cholesterol metabolism dysregulation and pyroptosis are relieved after Bex administration, regardless of in the Abcg1 flox/flox mice and the Abcg1‐KO mice. However, Bex can only inhibit apoptosis post‐TBI in the Abcg1 flox/flox mice, not in the Abcg1‐KO mice.
FIGURE 8.
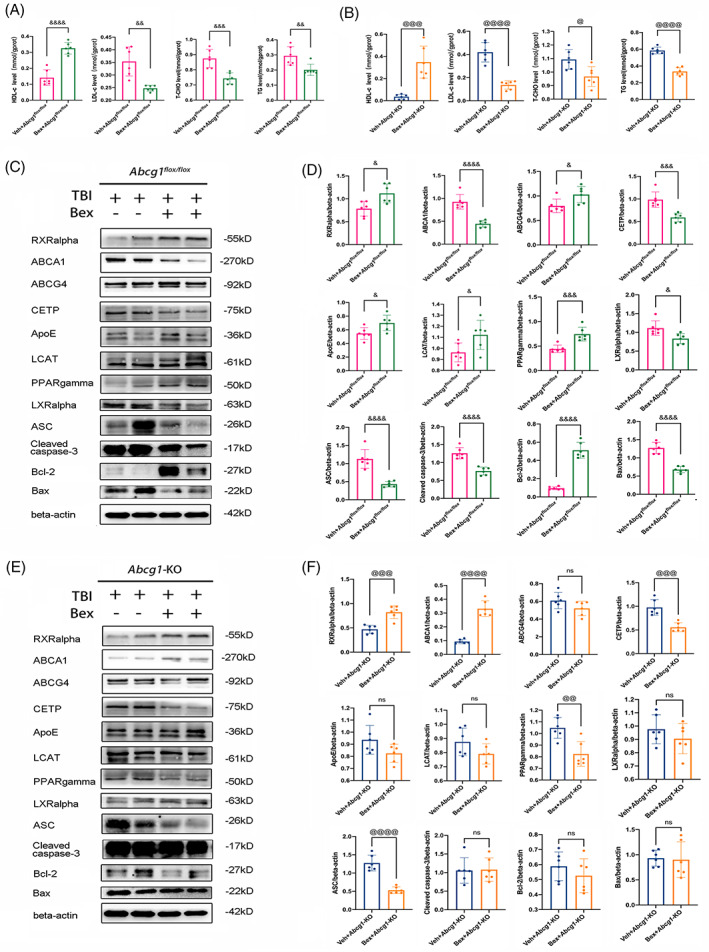
Bex partly relieved TBI‐induced cholesterol metabolism dysregulation, pyroptosis, and apoptosis under brain‐specific knockout of Abcg1. (A) HDL‐c, LDL‐c, T‐CHO, and TG levels in the injured cortex were measured by ELISA at 3 d post‐Veh + TBI + Abcg1 flox/fiox and Bex + TBI + Abcg1 flox/fiox . (B) HDL‐c, LDL‐c, T‐CHO, and TG levels in the injured cortex were measured by ELISA at 3 d post‐Veh + TBI + Abcg1‐KO and Bex + TBI + Abcg1‐KO. (C, D) Western blotting for RXRalpha, ABCA1, ABCG4, CETP, ApoE, LCAT, PPARgamma, LXRalpha, ASC, cleaved caspase‐3, Bcl‐2, and Bax was evaluated in the injured cortex at 3d post‐Veh + TBI + Abcg1 flox/fiox and Bex + TBI + Abcg1 flox/fiox . (E, F) Western blotting for RXRalpha, ABCA1, ABCG4, CETP, ApoE, LCAT, PPARgamma, LXRalpha, ASC, cleaved caspase‐3, Bcl‐2, and Bax was evaluated in the injured cortex at 3 d post‐Veh + TBI + Abcg1‐KO and Bex + TBI + Abcg1‐KO. Data are mean ± SEM; &p < 0.05, &&p < 0.01, &&&p < 0.001, or &&&&p < 0.0001 compared with the Veh + TBI + Abcg1 flox/fiox group. @p < 0.05, @@p < 0.01, @@@p < 0.001, or @@@@p < 0.0001 compared with Veh + TBI + Abcg1‐KO group. ns, not significant. n = 6/group
3.8. Bex decreases cell plasma membrane insults but do not alleviate brain edema post‐TBI in Abcg1‐KO mice
PI labeling was measured on Day 1 post‐injury to evaluate the effect of Bex on TBI‐induced cell plasma membrane insults. Less PI‐positive cells were observed in the injured cortex of the Bex + TBI + Abcg1 flox/flox group than in the Veh + TBI + Abcg1 flox/flox group (Figure 9A,B, p < 0.05). Interestingly, Bex also decreased the number of PI‐positive cells in the injured cortex on Day 1 post‐TBI in the Abcg1‐KO mice (Figure 9D,E, p < 0.05). Brain edema was also detected on Day 1 post‐TBI. Compared to the Veh + TBI + Abcg1 flox/flox group, the Bex + TBI + Abcg1 flox/flox group showed significantly reduced brain edema in the ipsilateral hemisphere after TBI (Figure 9C, p < 0.05). However, Bex cannot alleviate the brain water content in the ipsilateral hemisphere on Day 1 post‐TBI in the Abcg1‐KO mice (Figure 9F, p > 0.05). As a comparison, there were no significant differences of brain water content among the Veh‐treated and the Bex‐treated groups in contralateral hemisphere and cerebellum, regardless of in the Abcg1 flox/flox mice and the Abcg1‐KO mice (Figure 9C,F, p > 0.05). In brief, these data show that Bex decreases cell plasma membrane insults regardless of in the Abcg1 flox/flox mice and the Abcg1‐KO mice, while Bex can only alleviate brain edema post‐TBI in the Abcg1 flox/flox mice, not in the Abcg1‐KO mice.
FIGURE 9.
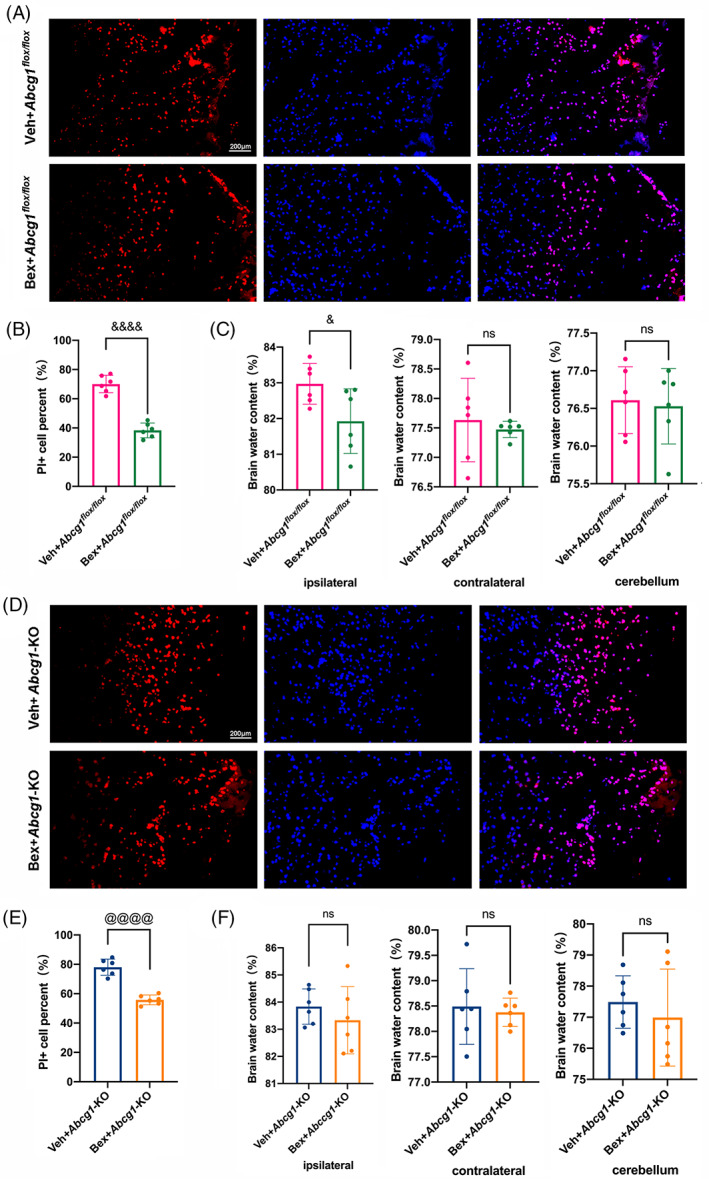
Bex decreased TBI‐induced cell plasma membrane insults but not brain edema under brain‐specific knockout of Abcg1. (A, B) Representative photographs and quantitative analysis of PI‐positive cells in the injured cortex on 1 d post‐Veh + TBI + Abcg1 flox/fiox and Bex + TBI + Abcg1 flox/fiox . (C) Representative histogram showing the brain water content, including the ipsilateral hemisphere, contralateral hemisphere, and cerebellum on 1 d post‐Veh + TBI + Abcg1 flox/fiox and Bex + TBI + Abcg1 flox/fiox . (D, E) Representative photographs and quantitative analysis of PI‐positive cells in the injured cortex on 1 d post‐Veh + TBI + Abcg1‐KO and Bex + TBI + Abcg1‐KO. (F) Representative histogram showing the brain water content, including the ipsilateral hemisphere, contralateral hemisphere, and cerebellum on 1 d post‐Veh + TBI + Abcg1‐KO and Bex + TBI + Abcg1‐KO. Data are mean ± SEM. &p < 0.05, &&p < 0.01, &&&p < 0.001, or &&&&p < 0.0001 compared with the Veh + TBI + Abcg1 flox/fiox group. @p < 0.05, @@p < 0.01, @@@p < 0.001, or @@@@p < 0.0001 compared with Veh + TBI + Abcg1‐KO group. ns, not significant. n = 6/group. Magnification: ×200 and scale bar 200 μm for A and D
3.9. Bex ameliorates TBI‐induced neurological deficits but do not decrease lesion volume in Abcg1‐KO mice
To determine the Bex‐treatment on the long‐term outcomes, behavioral experiments were carried out after TBI in the Abcg1 flox/flox and Abcg1‐KO mice. In the Abcg1 flox/flox mice, the Bex‐treatment significantly alleviated motor deficits on days 3–8 post‐TBI, compared to the Veh‐treatment (Figure 10A, p < 0.05). In the Abcg1‐KO mice, the Bex‐treatment slightly alleviated the motor disorders on Days 6, 7, 10, and 11 post‐TBI (Figure 10F, p < 0.05). When it came to the MWM test, a decreased latency was observed on days 16 and 17 in the Bex‐treatment when compared to the Veh‐treatment in Abcg1 flox/flox mice (Figure 10B, p < 0.05). In the Abcg1‐KO mice, the latency was shortened by Bex administration on Days 17–19 post‐TBI, relative to the Veh‐administration (Figure 10G, p < 0.05). On the 23rd day, the platform was removed to investigate the crossing number in the MWM test. An increase in the crossing number was observed post‐TBI in the Bex + Abcg1 flox/flox group, compared with the Veh + Abcg1 flox/flox group (Figure 10B, p < 0.05). Meaningfully, the crossing number was also mildly increased in the Bex + Abcg1‐KO mice post‐TBI, relative to the Veh‐treated Abcg1‐KO mice (Figure 10G, p < 0.05). On the 28th day post‐TBI, the results of the TST made clear that Bex can prompt mice to make more positive escape efforts than the vehicle group both in Abcg1 flox/flox mice and Abcg1‐KO mice (Figure 10C,H, p < 0.05). The OFT was also performed on the 28th day post‐TBI. We found that the total distance and time spent in center of the Bex group were increased significantly compared with the vehicle group both in Abcg1 flox/flox mice and Abcg1‐KO mice (Figure 10D,I p < 0.05), while the Bex treatment reduced distance moved in center in the Abcg1 flox/flox mice (Figure 10C, p < 0.05), but not in the Abcg1‐KO mice (Figure 10I, p > 0.05). In addition, HE staining on the tissue sections was performed. As shown in Figure 10E,J, the lesion volume in the Bex treatment was significantly less than the vehicle treatment in the Abcg1 flox/flox mice, but not in the Abcg1‐KO mice. These results illustrate that Bex promotes the recovery of neurological function post‐TBI regardless of in the Abcg1 flox/flox mice and the Abcg1‐KO mice, however, Bex can only reduce the lesion volume post‐TBI in the Abcg1 flox/flox mice, not in the Abcg1‐KO mice.
FIGURE 10.
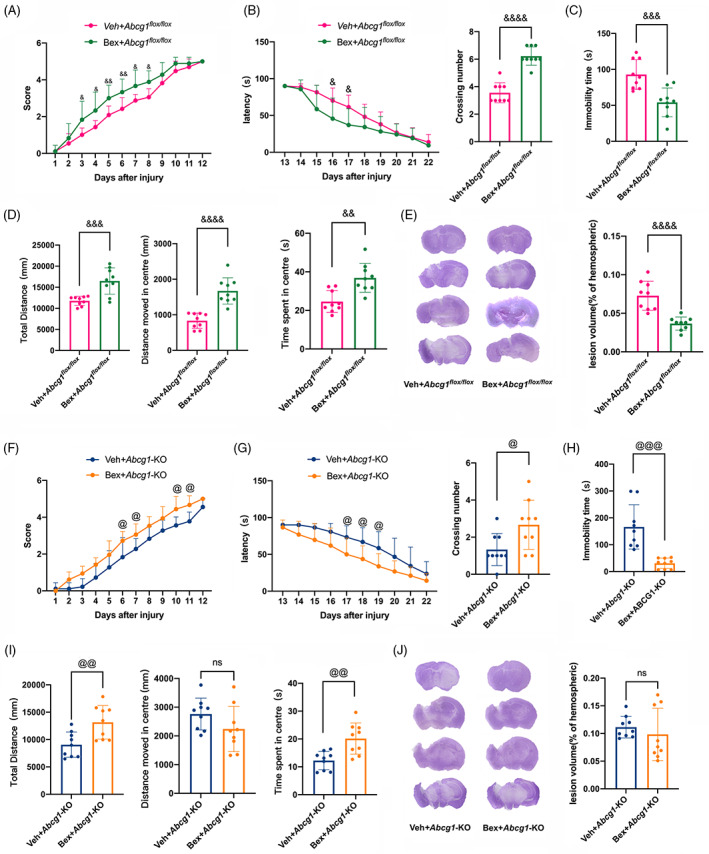
Bex improved TBI‐induced neurological deficits but not brain lesion volume under brain‐specific knockout of Abcg1. (A) Representative scatter diagram showing the score of the wire grip test on Days 1–12 post‐Veh + TBI + Abcg1 flox/fiox and Bex + TBI + Abcg1 flox/fiox . (B) Representative scatter diagram showing the latency of the hidden platform test on Days 13–22 and the crossing number of the probe trial on Day 23 post‐Veh + TBI + Abcg1 flox/fiox and Bex + TBI + Abcg1 flox/fiox . (C) Representative scatter diagram showing the immobility time of the TST on Day 28 post‐Veh + TBI + Abcg1 flox/fiox and Bex + TBI + Abcg1 flox/fiox . (D) Representative scatter diagram showing the total distance, the distance moved in center, and the time spent in center of the OFT on Day 28 post‐Veh+TBI + Abcg1 flox/fiox and Bex + TBI + Abcg1 flox/fiox . (E) Representative hematoxylin staining photographs of brain sections and quantitative analysis of brain lesion volume on Day 28 post the Veh + TBI + Abcg1 flox/fiox and Bex + TBI + Abcg1 flox/fiox . (F) Representative scatter diagram showing the score of the wire grip test on Days 1–12 post‐Veh + TBI + Abcg1‐KO and Bex + TBI + Abcg1‐KO. (G) Representative scatter diagram showing the latency of the hidden platform test on Days 13–22 and the crossing number of the probe trial on Day 23 post‐Veh + TBI + Abcg1‐KO and Bex + TBI + Abcg1‐KO. (H) Representative scatter diagram showing the immobility time of the TST on Day 28 post‐Veh + TBI + Abcg1‐KO and Bex + TBI + Abcg1‐KO. (I) Representative scatter diagram showing the total distance, the distance moved in center, and the time spent in center of the OFT on Day 28 post‐Veh + TBI + Abcg1‐KO and Bex + TBI + Abcg1‐KO. (J) Representative hematoxylin staining photographs of brain sections and quantitative analysis of brain lesion volume on Day 28 post‐Veh + TBI + Abcg1‐KO and Bex + TBI + Abcg1‐KO. Data are mean ± SEM. &p < 0.05, &&p < 0.01, &&&p < 0.001, or &&&&p < 0.0001 compared with Veh + TBI + Abcg1 flox/fiox at each time point. @p < 0.05, @@p < 0.01, @@@p < 0.001, or @@@@p < 0.0001 compared with Veh + TBI + Abcg1‐KO group at each time point. ns, not significant. n = 12/group. OFT, open field test; TBI, traumatic brain injury; TST, tail suspension test
4. DISCUSSION
In this study, we provided experimental evidence that brain‐specific knockout of Abcg1 aggravated cholesterol metabolism imbalance post‐TBI in Figure 11. Moreover, Abcg1‐KO exacerbated TBI‐induced pyroptosis, apoptosis, neuronal cell insult, brain edema, neurological deficits, and brain lesion volume. Importantly, we found that Abcg1‐KO partly abolished the protection of Bex following TBI.
FIGURE 11.
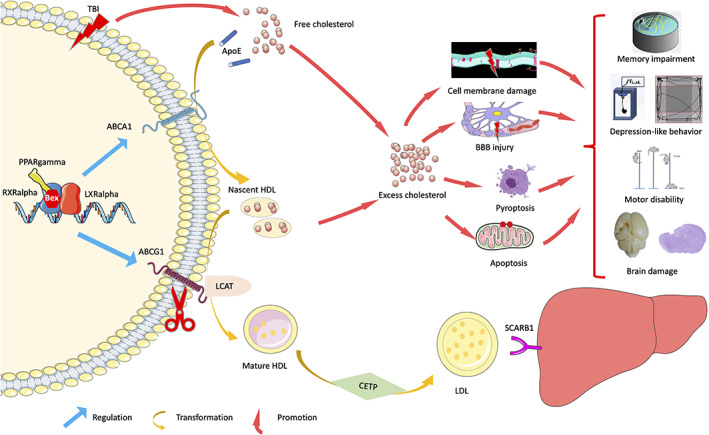
Schematic diagram representing the main findings of this study. Brain‐specific knockout of Abcg1 aggravated cholesterol metabolism imbalance and exacerbated pyroptosis, apoptosis, neuronal cell insult, brain edema, neurological deficits, and brain lesion volume post‐TBI. Importantly, Abcg1‐KO partly abolished the protection of Bex, which relies on the activation of RXRalpha/PPARgamma pathway, following TBI.
In mammals, the uptake, transport, and storage of cholesterol are tightly coordinated by various proteins and pathways to maintain cholesterol homeostasis at both the cellular and systemic levels [34]. The CNS cells, including neuron, astrocyte, and microglia, have evolved highly regulated mechanisms for controlling cellular cholesterol levels [35]. The molecules regulating cholesterol homeostasis include not limited the cholesterol‐transporter protein (ABCG1, ABCG4, ApoE, and ABCA1), RXRs, PPARs, LXRs, CETP, SCARB1, and LCAT [35]. In the present study, we found that TBI induced the diverse dynamic changes of the above cholesterol metabolism‐related proteins in the injured cortex. Further research found that the key proteins of RCT, including ABCA1 and ABCG1, were expressed in neuron, astrocyte, and microglia, which was also confirmed by the prior study [36]. Moreover, the corpse findings showed the expressions of ABCA1 and ABCG1 were increased in the injured cortex suffering from TBI in the traffic accident. ABCA1 and ABCG1 have been regarded as the vital molecules to export and remove excess cholesterol through HDL to reduce cellular damage and stress in the CNS [37, 38]. Dysregulation of cholesterol metabolism and transport in the brain following TBI can result in the accumulation of free cholesterol in various brain cells [10, 39]. Therefore, we speculated that multiple cells underwent RCT and maintained cholesterol homeostasis after TBI.
ABCG1 is a half‐transporter, which forms homodimers, mediating the transporter of cholesterol from cells to the major HDL fractions HDL‐c2 and HDL‐c3 [15, 40]. Therefore, ABCG1 plays a role in redistributing cholesterol to cell surface domains, where it becomes accessible for cholesterol removal by HDL‐c. As a key cholesterol‐transporting protein, ABCG1 might be involved in the pathophysiological process and could be a therapeutic target for patients with TBI. However, the neurological links between ABCG1 and cholesterol homeostasis regulation have not been elucidated in the TBI model.
To characterize the role of Abcg1 in the brain, we generated brain‐specific Abcg1 knockout mice in the present study. The total cholesterol pool (T‐CHO) and lipid‐storage unit (TG) were not statistically altered in the plasma of the Abcg1‐KO mice, but a lower level of HDL‐c and a higher level of LDL‐c were found under physiological condition. We speculated that transport of cholesterol from intracellular to extracellular was disrupted because of the brain‐specific Abcg1 knockout. However, little information is available on the effect of Abcg1 on the cholesterol metabolism post‐TBI. The present study found a lower level of HDL‐c and a higher level of LDL‐c in the plasma and injured cortex in the Abcg1‐KO mice post‐TBI, relative to the Abcg1 flox/flox mice. Moreover, compared with the Abcg1 flox/flox mice, a higher level of T‐CHO and TG was found in the injured cortex of the Abcg1‐KO mice. Our findings is in agreement with previous studies [41, 42], where patients with diffuse axonal injury (DAI) had significantly lower levels of HDL‐c in serum during the first week following TBI, relative to the non‐DAI group. In addition, TBI increased the total serum cholesterol level in rats with pre‐existing hypercholesterol that were fed with a high‐fat diet [43]. Moreover, plasma and cerebrospinal fluid HDL‐c are reported to be correlated and plasma HDL‐c levels that can influence brain HDL‐c level [9]. However, it is difficult to ascertain whether lower levels of plasma HDL‐c are a cause or a consequence of TBI. The accumulated cholesterol activates RXR, PPAR, and LXR, which leads to the activation of the downstream gene Abca1 [44]. This is mainly because both Abcg1 and Abca1 are post‐transcriptionally regulated by the RXR/LXR/PPAR pathway [44]. In our study, the mechanism of Abcg1 on TBI‐induced cholesterol metabolism turbulence were reflected by a significant decrease in ABCA1, ABCG4 and SCARB1 expression and a marked increase in PPARgamma, RXRalpha, LXRalpha, ApoE, LCAT, and CETP expression in Abcg1‐KO mice, relative to Abcg1 flox/flox mice. We conjectured that TBI‐induced cholesterol metabolism turbulence activated the endogenous cholesterol homeostasis mechanism, such as RXRalpha/LXRalpha/PPARgamma pathway. However, the lower levels of the key molecules of RCT, including ABCA1 and ABCG4, could not transport cholesterol to HDL‐c. Previous investigation also showed that the ABCG1 and ABCG4 transporters promote recycling of cholesterol as well as other biosynthetic precursors derived from astrocytes to neurons, via their efflux to HDL [22]. Therefore, the high level of free cholesterol may underlie its toxicity, as excessive cholesterol results in a generation of toxic ROS, an enhancement of production of neuroinflammation, and beta‐amyloid and mitochondria dysfunction, leading to pyroptosis and apoptosis [45].
Neuroinflammation plays a vital role in the pathophysiological process of TBI. Previous research showed that NLRP3 inflammasome, caspase‐1, and IL‐1beta expression was observed in animal models of TBI at different time points [46, 47, 48, 49]. Moreover, increased levels of NLRP3, IL‐1beta, IL‐18, and caspase‐1 were observed in patients of TBI [49, 50, 51]. The NLRP3 inflammasome and related molecules induced pyrotopic cell death through activating the executive protein GSDMD post‐TBI. Our TBI model showed that ASC, caspase‐1, IL‐1beta, and GSDMD have a marked increase postinjury. As a novel form of cell death, pyroptosis plays an important role in various pathological conditions, including TBI [49]. A number of recent studies have found that pharmacological inhibition of pyroptosis can improve neurological deficits following moderate‐to‐severe TBI [31]. Our research demonstrated that Abcg1‐KO mice with TBI were more susceptible to pyroptosis, apoptosis and neuronal cell insult, brain edema, neurological deficits, and brain lesion volume, relative to the Abcg1 flox/flox mice. There was emerging evidence suggesting that HDL may play a beneficial role in preventing BBB injury and preserving cognitive function under normal and pathological conditions [42, 52]. Another direct damage of cholesterol accumulation is impairing the integrity of the BBB. In turn, the injury of BBB may result in cholesterol accumulating and contribute to secondary pathological injury following TBI [42]. Therefore, abnormal cholesterol deposition in the brain is directly involved in the poor cognitive outcome. In the current study, our findings suggest that Abcg1‐KO‐induced excessive cellular cholesterol worsened secondary brain injuries and neurological deficits through aggravating pyroptosis. However, further study is needed to investigate the neurological links between the Abcg1 gene and pyroptosis.
Although ABCG1 plays a vital role in cholesterol metabolism by consuming excess cellular cholesterol to promote HDL‐c maturation, its precise functions and mechanisms in the brain remain unexplored. Prior study showed that Bex, an RXRalpha (the upstream molecule of ABCG1) agonist, exerts multiple protective effects, including anti‐tumor, anti‐oxidative, and anti‐inflammatory properties [25]. However, there is no information regarding the effect of Bex on pyroptosis after TBI. Most of the studies evaluated the effects of Bex on cellular, histopathological and behavioral outcomes following TBI to exploit whether brain‐specific loss of Abcg1 abolished Bex's protection against TBI. We found that Bex decreased the levels of LDL‐c, T‐CHO, and TG and increased the level of HDL‐c in the injured cortex, regardless of in the Abcg1 flox/flox mice and the Abcg1‐KO mice. These findings underscore Bex maintaining cholesterol metabolism homeostasis post‐TBI even the gene Abcg1 was loss in brain. In addition, Bex significantly attenuated TBI‐induced apoptosis and pyroptosis, neuronal damage, cerebral edema, lesion volume, and behavioral deficits in the Abcg1 flox/flox mice. However, although Bex can relieve pyroptosis and neurological dysfunctions, it cannot attenuate apoptosis, cerebral edema, and lesion volume post‐TBI in the Abcg1‐KO mice. Prior study showed that Bex promotes axon sprouting, preserves myelin integrity, increases BDNF expression, and promotes microglia/ macrophages toward an anti‐inflammatory state after TBI partially in a PPARγ‐dependent manner, which may facilitate functional improvements after TBI [27]. Thus, Bex as a multifunctional agent can exert its neuroprotective effects after TBI through multiple mechanisms. In addition, the Abcg1 gene is partly involved in the neuroprotective function of Bex in our TBI model.
However, further research is needed to elucidate the role and mechanism of the Abcg1 on TBI. Although apoptosis and pyroptosis are important mechanisms of cell death, other cell death pathways (e.g., necrosis, ferroptosis, necroptosis, autophagy) are simultaneously activated and modulated by TBI. Future research will need to exploit the effects of brain‐specific Abcg1 knockout on other cell death modes. In addition, the fate of excess cholesterol in the absence of Abcg1 in neurons, astrocyte, and microglia remains elusive. Moreover, it is necessary to elucidate the effects and mechanisms of Abcg1 on other brain areas, such as hippocampus and white matter zone (myelin and axon regeneration), during the pathophysiological process of TBI.
In summary, our study shows that, in addition to regulating brain cholesterol metabolism, Abcg1 improves neurological deficits and reduces brain lesion volume through inhibiting pyroptosis, apoptosis, neuronal cell insult, and brain edema. Moreover, the cerebroprotection of Abcg1 on TBI partly relies on the activation of RXRalpha/PPARgamma pathway, which provides a potential therapeutic target for treating TBI. Thus, the present study sheds new light on the understanding of the diverse biological functions of Bex and provides a path for investigating the anti‐pyroptosis actions of Bex following TBI.
AUTHOR CONTRIBUTIONS
Heng Xu, Le‐xin Zheng, and Xue‐shi Chen have contributed equally to this work. Tao Wang, Heng Xu, Le‐xin Zheng, Xue‐shi Chen, Qiu‐yu Pang, Ya‐nan Yan, Rong Liu, Yan Yang, Yuan Gao, and Lu‐yang Tao performed the experiments and analyzed data. Tao Wang, Heng Xu, Han‐mu Guo, Zhi‐yang Ren, Zhi‐ya Gu, Cheng Gao, Cheng‐liang Luo, Ying Zhao, Ying Wang, and Lu‐yang Tao were responsible for the conception and design of the study and manuscript writing and polishing.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Data S1: Supporting Information
ACKNOWLEDGMENTS
This study was supported by grants 82072110, 81971800, and 81970371 from the National Natural Science Foundation of China, SKJY2021046 from Suzhou Municipal Science and Technology Bureau, KF202201 from Shanghai Key Lab of Forensic Medicine & Key Lab of Forensic Science, Ministry of Justice, China (Academy of Forensic Science), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Xu H, Zheng L, Chen X‐S, Pang Q, Yan Y, Liu R, et al. Brain‐specific loss of Abcg1 disturbs cholesterol metabolism and aggravates pyroptosis and neurological deficits after traumatic brain injury. Brain Pathology. 2023;33(3):e13126. 10.1111/bpa.13126
Heng Xu, Le‐xin Zheng, and Xue‐shi Chen have contributed equally to this study.
Contributor Information
Tao Wang, Email: taowang@suda.edu.cn.
Lu‐yang Tao, Email: taoluyang@suda.edu.cn.
DATA AVAILABILITY STATEMENT
The data used or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Carroll L, Cassidy JD, Holm L, Kraus J, Coronado V. Methodological issues and research recommendations for mild traumatic brain injury: the WHO collaborating centre task force on mild traumatic brain injury. J Rehabil Med. 2004;36:113–25. [DOI] [PubMed] [Google Scholar]
- 2. Jiang J‐Y, Gao G‐Y, Feng J‐F, Mao Q, Chen L‐G, Yang X‐F, et al. Traumatic brain injury in China. Lancet Neurol. 2019;18(3):286–95. [DOI] [PubMed] [Google Scholar]
- 3. Khellaf A, Khan DZ, Helmy A. Recent advances in traumatic brain injury. J Neurol. 2019;266(11):2878–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rubiano AM, Carney N, Chesnut R, Puyana JC. Global neurotrauma research challenges and opportunities. Nature. 2015;527(7578):S193–7. [DOI] [PubMed] [Google Scholar]
- 5. Crane PK, Gibbons LE, Dams‐O'Connor K, Trittschuh E, Leverenz JB, Keene CD, et al. Association of traumatic brain injury with late‐life neurodegenerative conditions and neuropathologic findings. JAMA Neurol. 2016;73(9):1062–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lewén A, Fujimura M, Sugawara T, Matz P, Copin J‐C, Chan PH. Oxidative stress–dependent release of mitochondrial cytochrome c after traumatic brain injury. J Cereb Blood Flow Metabol. 2001;21(8):914–20. [DOI] [PubMed] [Google Scholar]
- 7. Dave AM, Peeples ES. Cholesterol metabolism and brain injury in neonatal encephalopathy. Pediatr Res. 2021;90(1):37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pfrieger FW, Ungerer N. Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res. 2011;50(4):357–71. [DOI] [PubMed] [Google Scholar]
- 9. Gliozzi M, Musolino V, Bosco F, Scicchitano M, Scarano F, Nucera S, et al. Cholesterol homeostasis: researching a dialogue between the brain and peripheral tissues. Pharmacol Res. 2021;163:105215. [DOI] [PubMed] [Google Scholar]
- 10. Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6(4):254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438(7068):612–21. [DOI] [PubMed] [Google Scholar]
- 12. Ikonen E. Mechanisms for cellular cholesterol transport: defects and human disease. Physiol Rev. 2006;86(4):1237–61. [DOI] [PubMed] [Google Scholar]
- 13. Kay AD, Day SP, Kerr M, Nicoll JAR, Packard CJ, Caslake MJ. Remodeling of cerebrospinal fluid lipoprotein particles after human traumatic brain injury. J Neurotrauma. 2003;20(8):717–23. [DOI] [PubMed] [Google Scholar]
- 14. Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71(1):537–92. [DOI] [PubMed] [Google Scholar]
- 15. Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP‐binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high‐density lipoproteins. Proc Natl Acad Sci. 2004;101(26):9774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Venkateswaran A, Laffitte Bryan A, Joseph Sean B, Mak Puiying A, Wilpitz Damien C, Edwards Peter A, et al. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXRα. Proc Natl Acad Sci. 2000;97(22):12097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cartagena CM, Ahmed F, Burns MP, Pajoohesh‐Ganji A, Pak DT, Faden AI, et al. Cortical injury increases cholesterol 24 S hydroxylase (Cyp46) levels in the rat brain. J Neurotrauma. 2008;25(9):1087–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karasinska JM, de Haan W, Franciosi S, Ruddle P, Fan J, Kruit JK, et al. ABCA1 influences neuroinflammation and neuronal death. Neurobiol Dis. 2013;54:445–55. [DOI] [PubMed] [Google Scholar]
- 19. Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice*. J Biol Chem. 2005;280(52):43224–35. [DOI] [PubMed] [Google Scholar]
- 20. Koldamova R, Fitz NF, Lefterov I. ATP‐binding cassette transporter A1: from metabolism to neurodegeneration. Neurobiol Dis. 2014;72:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Castranio EL, Wolfe CM, Nam KN, Letronne F, Fitz NF, Lefterov I, et al. ABCA1 haplodeficiency affects the brain transcriptome following traumatic brain injury in mice expressing human APOE isoforms. Acta Neuropathol Commun. 2018;6(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parkinson PF, Kannangara TS, Eadie BD, Burgess BL, Wellington CL, Christie BR. Cognition, learning behaviour and hippocampal synaptic plasticity are not disrupted in mice over‐expressing the cholesterol transporter ABCG1. Lipids Health Dis. 2009;8(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahley RW. Central nervous system lipoproteins. Arterioscler Thromb Vasc Biol. 2016;36(7):1305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cramer Paige E, Cirrito John R, Wesson Daniel W, Lee CYD, Karlo JC, Zinn Adriana E, et al. ApoE‐directed therapeutics rapidly clear β‐amyloid and reverse deficits in AD mouse models. Science. 2012;335(6075):1503–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhong J, Cheng C, Liu H, Huang Z, Wu Y, Teng Z, et al. Bexarotene protects against traumatic brain injury in mice partially through apolipoprotein E. Neuroscience. 2017;343:434–48. [DOI] [PubMed] [Google Scholar]
- 26. He J, Liu H, Zhong J, Guo Z, Wu J, Zhang H, et al. Bexarotene protects against neurotoxicity partially through a PPARγ‐dependent mechanism in mice following traumatic brain injury. Neurobiol Dis. 2018;117:114–24. [DOI] [PubMed] [Google Scholar]
- 27. He J, Huang Y, Liu H, Sun X, Wu J, Zhang Z, et al. Bexarotene promotes microglia/macrophages ‐ specific brain ‐ derived neurotrophic factor expression and axon sprouting after traumatic brain injury. Exp Neurol. 2020;334:113462. [DOI] [PubMed] [Google Scholar]
- 28. Chiu C‐C, Liao Y‐E, Yang L‐Y, Wang J‐Y, Tweedie D, Karnati HK, et al. Neuroinflammation in animal models of traumatic brain injury. J Neurosci Methods. 2016;272:38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin‐1 to drive NLRP3 inflammasome assembly. EMBO J. 2019;38(10):e101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu X, Chen H, Xu H, Wu Y, Wu C, Jia C, et al. Role of Pyroptosis in traumatic brain and spinal cord injuries. Int J Biol Sci. 2020;16(12):2042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Brien WT, Pham L, Symons GF, Monif M, Shultz SR, McDonald SJ. The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J Neuroinflammation. 2020;17(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen X, Gao C, Yn Y, Cheng Z, Chen G, Rui T, et al. Ruxolitinib exerts neuroprotection via repressing ferroptosis in a mouse model of traumatic brain injury. Exp Neurol. 2021;342:113762. [DOI] [PubMed] [Google Scholar]
- 33. Gao C, Chen X, Xu H, Guo H, Zheng L, Yn Y, et al. Restraint stress delays the recovery of neurological impairments and exacerbates brain damages through activating endoplasmic reticulum stress‐mediated neurodegeneration/autophagy/apopotosis post moderate traumatic brain injury. Mol Neurobiol. 2022;59(3):1560–76. [DOI] [PubMed] [Google Scholar]
- 34. Yamauchi Y, Rogers MA. Sterol metabolism and transport in atherosclerosis and cancer. Frontiers in Endocrinology. 2018;9:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Damisah Eyiyemisi C, Hill Robert A, Rai A, Chen F, Rothlin Carla V, Ghosh S, et al. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Science Advances. 2020;6(26):eaba3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kober AC, Manavalan APC, Tam‐Amersdorfer C, Holmér A, Saeed A, Fanaee‐Danesh E, et al. Implications of cerebrovascular ATP‐binding cassette transporter G1 (ABCG1) and apolipoprotein M in cholesterol transport at the blood‐brain barrier. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(6):573–88. [DOI] [PubMed] [Google Scholar]
- 37. Vaughan AM, Oram JF. ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol‐rich HDL. J Lipid Res. 2006;47(11):2433–43. [DOI] [PubMed] [Google Scholar]
- 38. Wang N, Yvan‐Charvet L, Lütjohann D, Mulder M, Vanmierlo T, Kim T‐W, et al. ATP‐binding cassette transporters G1 and G4 mediate cholesterol and desmosterol efflux to HDL and regulate sterol accumulation in the brain. FASEB J. 2008;22(4):1073–82. [DOI] [PubMed] [Google Scholar]
- 39. Martins IJ, Berger T, Sharman MJ, Verdile G, Fuller SJ, Martins RN. Cholesterol metabolism and transport in the pathogenesis of Alzheimer's disease. J Neurochem. 2009;111(6):1275–308. [DOI] [PubMed] [Google Scholar]
- 40. Vaughan AM, Oram JF. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid‐depleted Apolipoproteins*. J Biol Chem. 2005;280(34):30150–7. [DOI] [PubMed] [Google Scholar]
- 41. Zhong YH, Zheng BE, He RH, Zhou Z, Zhang SQ, Wei Y, et al. Serum levels of HDL cholesterol are associated with diffuse axonal injury in patients with traumatic brain injury. Neurocrit Care. 2021;34(2):465–72. [DOI] [PubMed] [Google Scholar]
- 42. Hottman DA, Chernick D, Cheng S, Wang Z, Li L. HDL and cognition in neurodegenerative disorders. Neurobiol Dis. 2014;72:22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chong AJ, Lim S‐W, Lee Y‐l, Chio C‐C, Chang C‐H, Kuo J‐R, et al. The neuroprotective effects of simvastatin on high cholesterol following traumatic brain injury in rats. World Neurosurg. 2019;132:e99–e108. [DOI] [PubMed] [Google Scholar]
- 44. Yan J‐q, Tan C‐z, Wu J‐h, Zhang D‐c, Chen J‐l, Zeng B‐y, et al. Neopterin negatively regulates expression of ABCA1 and ABCG1 by the LXRα signaling pathway in THP‐1 macrophage‐derived foam cells. Mol Cell Biochem. 2013;379(1):123–31. [DOI] [PubMed] [Google Scholar]
- 45. Song Y, Liu J, Zhao K, Gao L, Zhao J. Cholesterol‐induced toxicity: an integrated view of the role of cholesterol in multiple diseases. Cell Metab. 2021;33(10):1911–25. [DOI] [PubMed] [Google Scholar]
- 46. Liu H‐D, Li W, Chen Z‐R, Hu Y‐C, Zhang D‐D, Shen W, et al. Expression of the NLRP3 Inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res. 2013;38(10):2072–83. [DOI] [PubMed] [Google Scholar]
- 47. Ma J, Xiao W, Wang J, Wu J, Ren J, Hou J, et al. Propofol inhibits NLRP3 Inflammasome and attenuates blast‐induced traumatic brain injury in rats. Inflammation. 2016;39(6):2094–103. [DOI] [PubMed] [Google Scholar]
- 48. Chen Y, Meng J, Xu Q, Long T, Bi F, Chang C, et al. Rapamycin improves the neuroprotection effect of inhibition of NLRP3 inflammasome activation after TBI. Brain Res. 2019;1710:163–72. [DOI] [PubMed] [Google Scholar]
- 49. Irrera N, Russo M, Pallio G, Bitto A, Mannino F, Minutoli L, et al. The role of NLRP3 Inflammasome in the pathogenesis of traumatic. Brain Inj. 2020;21(17):6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wallisch JS, Simon DW, Bayır H, Bell MJ, Kochanek PM, Clark RSB. Cerebrospinal fluid NLRP3 is increased after severe traumatic brain injury in infants and children. Neurocrit Care. 2017;27(1):44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin C, Chao H, Li Z, Xu X, Liu Y, Bao Z, et al. Omega‐3 fatty acids regulate NLRP3 inflammasome activation and prevent behavior deficits after traumatic brain injury. Exp Neurol. 2017;290:115–22. [DOI] [PubMed] [Google Scholar]
- 52. Fellows K, Uher T, Browne RW, Weinstock‐Guttman B, Horakova D, Posova H, et al. Protective associations of HDL with blood‐brain barrier injury in multiple sclerosis patients. J Lipid Res. 2015;56(10):2010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information
Data Availability Statement
The data used or analyzed during the current study are available from the corresponding author on reasonable request.