

Abstract
The diagnosis of neurodegenerative diseases is made complex by the heterogenous phenotype of the patients and the regular occurrence of concomitant pathology. Studying clinicopathological correlations in autopsy series is a central approach to improve pathological prediction in clinical practice. However, such method requires a wealth of information, and the use of standard spreadsheet software is hardly suitable. To overcome this constraint, we designed a customizable and freely available neuropathology form with 456 data entry fields driven by an open‐source DataBase Management Systems (DBMS) using Structured Query Language (SQL). This approach allowed us to optimize the compilation of clinical and pathological data from our brain collection (264 autopsied patients, 22,885 data points). Information was then easily retrieved using general and specific queries, facilitating the analysis of demographics, clinicopathological correlations, and incidental and concomitant proteinopathies. Tau, amyloid‐β and α‐synuclein incidental pathology was observed in respectively 78.1%, 42.8%, and 10.7% of all the patients. These proportions increased with age, reaching 100% for Tau pathology after 80. Concomitant proteinopathy was observed in 46.4% of the patients diagnosed with neurodegenerative diseases and prion disease. We observed a particularly high rate of co‐pathology in patients with Dementia with Lewy bodies (81.3% of associated Tau and amyloid‐β pathology) and Creutzfeldt–Jakob disease (68.4% of associated Tau pathology). Finally, we used specific queries to identify old cases that could meet newly defined neuropathological criteria and revised the diagnosis of a 90‐year‐old patient to LATE Stage 2. Increasing our understanding of clinicopathological correlations in neurodegenerative diseases is crucial given the implications in clinical diagnosis, biomarker identification and targeted therapies assessment. The precise characterization of clinical and pathological data of autopsy series remains a central approach but the large amount of generated data should encourage a more systematic use of DBMS.
Keywords: clinicopathological correlations, concomitant pathology., DataBase, neurodegenerative disorders, neuropathology
1. BACKGROUND
One of the main challenges for clinicians is to predict as accurately as possible the underlying diagnosis of patients with neurodegenerative diseases. This forecasting exercise is made complex by heterogenous clinical presentation of subjects having the same disease, and further complicated by the regular occurrence of concomitant pathology in the central nervous system (CNS) of the patients [1, 2]. The stakes are high as both the management of individual cases and the success of current clinical trials for targeted therapies strongly depend on the accuracy of patients' diagnosis. To date, the analysis of clinicopathological correlations in autopsy series remains one of the most effective methods to improve pathological prediction in clinical practice [3, 4, 5]. However, this approach requires a wealth of information to be relevant, and the use of standard spreadsheet software to collect and manage such volume of data is hardly suitable, time consuming, and increases storage demands.
To overcome this constraint, the use of DataBase Management Systems (DBMS) seems to be an interesting alternative to traditional data storage techniques. DBMS are software developed to store and process large volumes of data while hiding the complexity of the operations performed and optimizing the final size of the file. Most of the DBMS use Structured Query Language (SQL) to allow logical organization of data independently of their physical organization. Data collection can be simplified by using data entry forms, allowing to fill of a large amount of information quickly and intuitively. In the same way, data extraction from raw information is facilitated by using queries that generate synthetic responses to targeted questions. Depending on its size and purpose, a DataBase file can be hosted and managed on a single computer, computer clusters or cloud storage. Such characteristics and properties should encourage the use of DBMS in neuropathology to store and retrieve clinical and pathological information for both diagnosis and research purpose. The National Alzheimer's Coordinating Center (NACC) Uniform Data Set is an example of such application: this DataBase contains clinical and pathological information from Alzheimer's Disease Research Centers (ADRCs) across the United States, collected with a standardized data entry form [6, 7]. However, the use of relational DataBases in neuropathology remains limited since DBMS often require advanced computer knowledge and coding skills, making them difficult to handle for clinicians.
In this study, we designed a simple‐to‐use and customizable neuropathology form driven by an open‐source relational DBMS to study clinicopathological correlations and concomitant pathology in patients autopsied in our neuropathology department.
2. METHODS
2.1. Cases
In the University Hospital of Angers, clinical diagnoses of neurodegenerative disorders are made according to consensus criteria by physicians from three reference centers: the resources and research center for memory disorders, the national reference center for neurogenetic diseases, and the resources and competences center for amyotrophic lateral sclerosis and motor neuron diseases. Patients affected by other neurological and neurodegenerative diseases are diagnosed and followed by clinicians from the neurology department of the University Hospital. Brain autopsies are performed in the Neurobiology and Neuropathology department of Angers by expert neuropathologists with standardized protocols, except for cases with clinically suspected Creutzfeldt–Jakob disease (patients are autopsied in Angers and the brains are sent to the national Creutzfeldt–Jakob disease surveillance unit at la Pitié‐Salpétrière, Paris). Once collected, post‐mortem brains are divided sagittally. One hemibrain is dissected fresh and frozen for genetic and biochemical analysis, the other is fixed in 10% buffered formalin for a period up to 4 months, sectioned coronally and analyzed. Tissue samples are collected from 15 standardized regions (orbitofrontal cortex, middle frontal gyrus, caudate nucleus, white matter, cingulate gyrus, nucleus basalis of Meynert, precentral gyrus, putamen, pallidum, first temporal gyrus, amygdala, thalamus, hippocampus, angular gyrus, calcarine sulcus, midbrain, pons, cerebellar cortex, dentate nucleus, medulla, and cervical spinal cord). Samples are processed into paraffin, cut at a thickness of 4 μm, and stained with hematoxylin eosin, luxol fast blue, periodic acid Schiff and Gallyas silver. Depending on the first pathological observations and the clinical context, sections are then immunostained with anti‐ubiquitine (Dako), anti‐p62 (Santa Cruz), anti‐ Phospho‐Tau (AT8, Innogenetics), anti‐A4 (Dako), anti‐phospho‐α‐synuclein (Zymed/Wako), anti‐Phospho‐TDP‐43 (Proteintech), anti‐FUS (Sigma), Anti‐SOD (Millipore), anti‐polyglutamin (Chemicon), anti‐GFAP (Agilent/Dako), anti‐neurofilament (Monozan/Dako) antibodies. Pathological diagnoses are made based on consensus criteria [8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19]. The final diagnosis is retained after discussion with the clinicians who followed the patients.
2.2. Design and management of the relational DataBase
A customizable neuropathology data entry form was created with LibreOffice software version 7.1.0 for Windows, an open‐source DBMS derived from the OpenOffice.org project. The form was designed based on the standardized examination performed on every autopsied brain in the neuropathology department of the University Hospital of Angers, described above. The form was anonymized and defined with 456 possible data entry fields per patient, divided into 9 color‐coded sections: identity number, clinical data, paraclinical data, clinical diagnosis, death and autopsy, macroscopic examination (fresh and fixed), microscopic examination (standard staining and immunohistochemistry), histological diagnosis, and final postmortem diagnosis (Figure S1). A relational DataBase was created using the form to collect clinical and pathological data from the patients autopsied in the Neuropathology department of Angers from 2005 to 2020, using the medical records of the cases. The DataBase was filled by 2 trained neurologists with basic computer skills. The query module of the DBMS was then used to query the DataBase and generate reports.
2.3. Complementary immunohistochemistry
Formalin‐fixed tissue samples embedded in paraffin were retrieved from the Brain Bank and sectioned at room temperature into 4 μm thick sections using a Leica RM125 microtome (Leica Biosystems, Wetzlar, Germany). Sections were collected on slides and immunostained with a fully automated IHC stainer Leica‐BOND III (Leica Biosystems, Wetzlar, Germany). Washes, blocking, antigen retrieval, incubation with primary antibody against TDP‐43 (Proteintech, Cat# 18280‐1‐AP, 1:6000) and phospho‐TDP‐43 Ser409/410 (Proteintech, Cat# 22309‐1‐AP, 1:6000), enzymatic detection and hematoxylin counterstain were automatically performed following manufacturer's instructions. Finally, samples were mounted and analyzed under a Zeiss Axioscop40 microscope equipped with an Axio‐cam MRC5 camera, using AxioVisio 4.6 software (Carl Zeiss, Oberkochen, Germany).
2.4. Statistical analysis
Statistical analyses were conducted using PRISM software version 5.0 for Windows (GraphPad, La Jolla, CA). Comparisons of means were performed using Student's t‐test for two groups analysis, and one‐way analysis of variance (ANOVA) for multiple groups analysis, after verifying the Normal distribution and the homoscedasticity of data using Shapiro–Wilk's test. Differences were considered significant at p < 0.05.
3. RESULTS
3.1. Relational DataBase development
A standardized and anonymized data entry form was designed using an open‐source DBMS, with 456 possible data entry fields per patient. A relational DataBase was then created and filled using the form by 2 trained neurologists with basic computer skills. There was no difference in reliability between the raters (20 cases tested). The form could be completed in approximately 10 min per patient, with an average of 100 data points collected for each patient. The same approach tested for 20 cases with a standard spreadsheet software took approximately 50–70 min per patient. The medical records of the 264 patients autopsied in the Neuropathology department of Angers from 2005 to 2020 were analyzed. In total, 22,885 data points has been collected in the DataBase in approximatively 90 h. The final size of the file was 6.1 MB. The DBMS query module was then used to query the DataBase and generate reports.
3.2. Overall demographics
We first used general queries to estimate the overall demographics of our series. Of the 264 cases, 52% were male. Mean age at onset was 60 ± 16 years, mean disease duration was 5 ± 7 years and mean age at death was 65 ± 15 years. Clinical and pathological information are detailed in Table S1. The distribution of the 264 patients according to the final diagnosis retained is reported on Figure 1. To put into perspective the number of autopsied patients, based on the activity reports of our reference centers from 2005 to 2020, 4624 patients have been diagnosed with cortical degeneration, 558 with Huntington's disease and 1542 with Amyotrophic lateral sclerosis.
FIGURE 1.
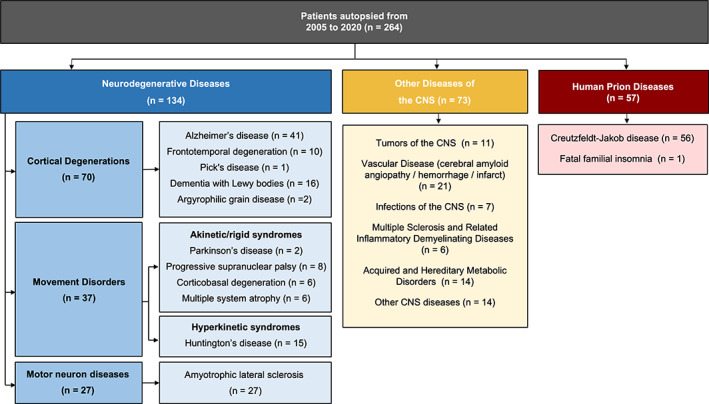
Distribution of the 264 patients autopsied in the neuropathology department of Angers from 2005 to 2020 according to the final diagnosis retained. CNS, central nervous system
3.3. Clinicopathological correlations and diagnosis accuracy
We then aimed to assess clinicopathological correlations in patients affected by neurodegenerative disorders and Creutzfeldt–Jakob disease. We used queries adapted to the specificities of the groups “neurodegenerative diseases” and “prion disease.” We also used the following query: [SELECT “CLINICAL DIAGNOSIS”, “FINAL DIAGNOSIS”, “CLINICOPATHOLOGICAL CONCORDANCE” FROM “Data_Base”] to estimate for each disease the clinical sensitivity (physician's ability to diagnose the disease) and positive predictive value (PPV, accuracy of the retained clinical diagnosis).
3.3.1. Alzheimer's disease
Among the 41 patients with a postmortem diagnosis of AD, 22 (53.7%) were male (Table 1). Mean age at symptom onset was 62 ± 12 years and mean age at death was 72 ± 12 years. The predominant symptom was memory disorders (81.8%), followed by phasic (69.7%) and praxis disorders (66.7%). Combined analysis of cerebrospinal fluid (CSF) biomarkers showed a 60% A+T+ concordance, and 20% had either an A−T+ or A+T− profile. All the patients had abnormal neuropsychological testing and more than 90% had pathological brain imaging. One patient has been diagnosed with mutation on the PSEN1 gene. The average brain weight was 1.23 ± 0.19 kg, with predominant frontal, temporal, and parietal atrophy (Figure 2). Thal phases, Braak stages and CERAD scores are detailed in Table S2. Clinical sensitivity and PPV for the diagnosis of AD were 73.2% and 85.7% respectively (Table 2).
TABLE 1.
Demographic data—Cortical degeneration
| AD | FTLD | DLB | |
|---|---|---|---|
| Description of the population | |||
| Sample size, n | 41 | 10 | 16 |
| Male, n/n tot (%) | 22/41 (53.7%) | 7/10 (70.0%) | 12/16 (75.0%) |
| Age at symptom onset (years), mean ± SD |
62.0 ± 11.5 n tot = 33 |
62.5 ± 11.4 n tot = 10 |
66.6 ± 9.3 n tot = 14 |
| Duration of disease (years), mean ± SD |
8.4 ± 5.1 n tot = 33 |
6.5 ± 4.4 n tot = 10 |
6.5 ± 4.0 n tot = 14 |
| Age at death (years), mean ± SD |
71.9 ± 11.6 n tot = 41 |
69.0 ± 12.6 n tot = 10 |
74.5 ± 8.5 n tot = 16 |
| Clinical data | |||
| Memory disorders, n/n tot (%) | 27/33 (81.8%) | 5/10 (50.0%) | 9/15 (60.0%) |
| Phasic disorders, n/n tot (%) | 23/33 (69.7%) | 8/10 (80.0%) | 7/15 (46.7%) |
| Praxis disorders, n/n tot (%) | 22/33 (66.7%) | 7/10 (70.0%) | 9/15 (60.0%) |
| Executive disorders, n/n tot (%) | 20/33 (60.6%) | 8/10 (80.0%) | 8/15 (53.3%) |
| Anxio‐depressive disorders, n/n tot (%) | 14/33 (42.4%) | 3/10 (30.0%) | 2/15 (13.3%) |
| Behavioral disorders, n/n tot (%) | 15/33 (45.4%) | 7/10 (70.0%) | 10/15 (66.7%) |
| Hallucinations/delirium, n/n tot (%) | 3/33 (9.1%) | 0/10 (0%) | 11/15 (73.3%) |
| Extrapyramidal syndrome, n/n tot (%) | 4/33 (12.1%) | 1/10 (10.0%) | 13/15 (86.7%) |
| Paraclinical data | |||
| Biomarker CSF, n/n tot (%) | |||
| A+T+ | 6/10 (60.0%) | 0 | – |
| A−T+ | 2/10 (20.0%) | 1/3 (33.3%) | – |
| A+T− | 2/10 (20.0%) | 0 | – |
| A−T− | 0 | 2/3 (66.7%) | – |
| CT‐scan/ MRI abnormalities, n/n tot (%) | 29/31 (93.5%) | 9/10 (90.0%) | 10/10 (100.0%) |
| Neuropsychological tests abnormalities, n/n tot (%) | 25/25 (100.0%) | 7/7 (100.0%) | 8/8 (100.0%) |
| PET‐scan abnormalities, n/n tot (%) | 13/14 (92.9%) | 4/4 (100.0%) | 5/5 (100.0%) |
| DAT‐scan abnormalities, n/n tot (%) | – | – | 5/5 (100.0%) |
| Genetic mutations | One PSEN1 gene mutation | One C9orf72 gene mutation | – |
| Neuropathological data | |||
| Immediate macroscopic analysis | |||
| Brain weight (kg), mean ± SD |
1.23 ± 0.19 n tot = 39 |
1.12 ± 0.13 n tot = 10 |
1.29 ± 0.13 n tot = 15 |
Note: For each subgroup, the total number of headcount (n tot) is specified in case of missing data. Cerebrospinal fluid biomarkers profiles: biomarkers of Aβ plaques (labeled “A”) are low CSF Aβ42 or Aβ42/Aβ40 ratio; biomarkers of fibrillar Tau (labeled “T”) are elevated CSF phosphorylated tau (p‐Tau). Binarizing each of the two biomarker groups into normal/abnormal (−/+) results in four possible biomarker profiles: A+T+, A−T+, A+T−, A−T [20].
Abbreviations: AD, Alzheimer disease; FTLD, frontotemporal lobar degeneration; DLB, dementia with Lewy dodies; CSF, cerebro spinal fluid; CT, computed tomography; MRI, magnetic resonance imaging; PET, positron emission tomography; IHC, immunohistochemistry.
FIGURE 2.
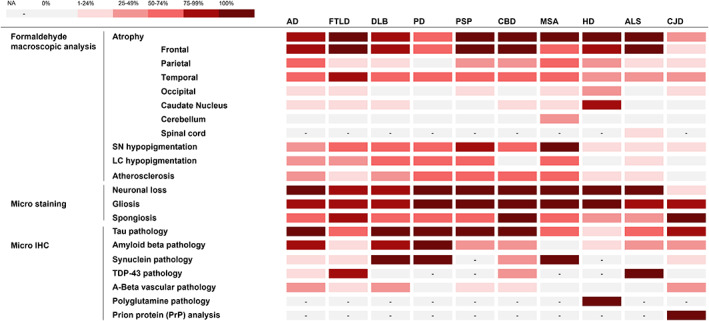
Heat map of the neuropathological observations. IHC, Immunohistochemistry; LC, Locus coeruleus; SN, Substantia nigra; NA, not applicable
TABLE 2.
Clinical and pathological diagnoses
| Clinical diagnosis | Pathological diagnosis | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AD | FTLD | DLB | PD | PSP | CBD | MSA | HD | ALS | CJD | Other | Total | |
| AD | 30 | 3 | – | – | – | 2 | – | – | – | – | – | 35 |
| FTLD | 1 | 5 | – | – | – | – | – | – | – | – | 1 | 7 |
| DLB | 1 | – | 12 | – | – | 1 | – | – | – | – | 1 | 15 |
| PD | – | – | – | 1 | – | – | – | – | – | – | – | 1 |
| PSP | 1 | – | – | – | 8 | – | 2 | – | – | 1 | – | 12 |
| CBD | 1 | 1 | – | – | – | 2 | – | – | – | – | – | 4 |
| MSA | – | – | – | – | – | – | 3 | – | – | – | – | 3 |
| HD | – | – | – | – | – | – | – | 15 | – | – | – | 15 |
| ASL | – | – | – | – | – | – | – | – | 27 | – | – | 27 |
| CJD | – | – | 1 | – | – | – | – | – | – | 54 | 4 | 59 |
| Other | 7 | 1 | 3 | 1 | – | 1 | 1 | – | – | 1 | 71 | 86 |
| Total | 41 | 10 | 16 | 2 | 8 | 6 | 6 | 15 | 27 | 56 | 76 | 264 |
Note: Pick's disease, Argyrophilic grain diseases, fatal familial insomnia, tumors of the central nervous system (CNS), vascular diseases, infections of the CNS, multiple sclerosis and related inflammatory demyelinating diseases, acquired and hereditary metabolic disorders, other CNS pathology.
Abbreviations: AD, Alzheimer disease; ALS, amyotrophic lateral sclerosis; CBD, cortico‐basal degeneration; CJD, Creutzfeldt–Jakob disease; DLB, dementia with Lewy bodies; FTLD, frontotemporal lobar degeneration; HD, Huntington's disease; MSA, multiple system atrophy; PD, Parkinson's disease; PSP, progressive supranuclear palsy.
3.3.2. Frontotemporal lobar dementia
Among the 10 patients with a postmortem diagnosis of frontotemporal lobar dementia (FTLD), 7 (70.0%) were male (Table 1). Mean age at symptom onset was 62 ± 11 years and mean age at death was 69 ± 13 years. Predominant symptoms were executive and phasic disorders (80.0%), followed by praxis and behavioral disorders (70.0%), then memory disorders (50.0%). All the patients had abnormal neuropsychological testing and 90% had pathological brain imaging. One patient has been diagnosed with mutation on the C9orf72 gene. The average brain weight was 1.12 ± 0.13 kg, with predominant frontal and temporal atrophy (Figure 2). In our series of 10 cases, 6 (60.0%) were classified as FTLD‐TDP‐43, 2 were classified as FTLD‐Tau (20.0%) and 2 cases (20.0%) had no distinct neuropathologic sign (FTLD‐U). Clinical sensitivity and PPV for the diagnosis of FTLD were 50.0% and 71.4% respectively (Table 2).
3.3.3. Dementia with Lewy bodies
Among the 16 patients with a postmortem diagnosis of dementia with Lewy bodies (DLB), 12 (75.0%) were male (Table 1). Mean age at symptom onset was 67 ± 10 years and mean age at death was 75 ± 9 years. The predominant symptom was extrapyramidal syndrome (86.7%), followed by hallucinations (73.3%) and behavioral disorders (66.7%). All the patients had pathological brain imaging and abnormal neuropsychological testing. The average brain weight was 1.29 ± 0.13 kg, with predominant frontal and temporal atrophy (Figure 2). Clinical sensitivity and PPV for the diagnosis of DLB were 75.0% and 80.0% respectively (Table 2).
3.3.4. Parkinson's disease
Two patients had a postmortem diagnosis of Parkinson's disease (PD) (Table 3). Their age at symptom onset were 62 and 82 years, and age at death 67 and 82 years. They both had extrapyramidal syndrome. No paraclinical data were available. Brain weight were 1.2 and 1.5 kg, with predominant frontal and temporal atrophy (Figure 2). Both patients were clinically diagnosed with PD before death.
TABLE 3.
Demographic data—movement disorders
| PD | PSP | CBD | MSA | |
|---|---|---|---|---|
| Description of the population | ||||
| Sample size, n | 2 | 8 | 6 | 6 |
| Male, n/n tot (%) | 0/2 (0%) | 3/8 (37.5%) | 4/6 (66.7%) | 4/6 (66.7%) |
| Age at symptom onset (years), mean ± SD |
72.0 ± 14.1 n tot = 2 |
65.5 ± 9.4 n tot = 8 |
67.7 ± 7.8 n tot = 6 |
58.6 ± 11.8 n tot = 5 |
| Duration of disease (years), mean ± SD |
2.5 ± 3.5 n tot = 2 |
8.2 ± 4.7 n tot = 8 |
9.2 ± 6.0 n tot = 6 |
4.6 ± 1.1 n tot = 5 |
| Age at death (years), mean ± SD |
74.5 ± 10.6 n tot = 2 |
73.7 ± 8.7 n tot = 8 |
76.8 ± 4.4 n tot = 6 |
63.3 ± 11.1 n tot = 6 |
| Clinical data | ||||
| Memory disorders, n/n tot (%) | 0/2 (0%) | 4/8 (50.0%) | 6/6 (100.0%) | 1/5 (20.0%) |
| Phasic disorders, n/n tot (%) | 0/2 (0%) | 6/8 (75.0%) | 4/6 (66.7%) | 0/5 (0%) |
| Praxis disorders, n/n tot (%) | 0/2 (0%) | 6/8 (75.0%) | 5/6 (83.3%) | 2/5 (40.0%) |
| Executive disorders, n/n tot (%) | 0/2 (0%) | 7/8 (87.5%) | 5/6 (83.3%) | 1/5 (20.0%) |
| Anxio‐depressive disorders, n/n tot (%) | 1/2 (50.0%) | 1/8 (12.5%) | 3/6 (50.0%) | 3/5 (60.0%) |
| Behavioral disorders, n/n tot (%) | 1/2 (50.0%) | 2/8 (25.0%) | 4/6 (66.7%) | 0/5 (0%) |
| Hallucinations/delirium, n/n tot (%) | 1/2 (50.0%) | 0/8 (0%) | 0/6 (0%) | 0/5 (0%) |
| Extrapyramidal syndrome, n/n tot (%) | 1/2 (50.0%) | 6/8 (75.0%) | 3/6 (50.0%) | 4/5 (80.0%) |
| Central motor symptoms, n/n tot (%) | 1/2 (50.0%) | 3/8 (37.5%) | 0/6 (0%) | 1/5 (20.0%) |
| Visual disturbances, n/n tot (%) | 0/2 (0%) | 7/8 (87.5%) | 0/6 (0%) | 3/5 (60.0%) |
| Paraclinical data | ||||
| Biomarker CSF, n | – | 1 (A+T−) |
1 (A−T+) 1 (A−T−) |
1 (A−T−) |
| CT‐scan/MRI abnormalities, n/n tot (%) | – | 7/7 (100.0%) | 6/6 (100.0%) | 2/3 (66.7%) |
| Neuropsychological tests abnormalities, n/n tot (%) | – | 7/8 (87.5%) | 6/6 (100.0%) | 1/1 (100.0%) |
| PET‐scan abnormalities, n/n tot (%) | – | 7/8 (87.5%) | 5/5 (100.0%) | 1/1 (100.0%) |
| DAT‐scan abnormalities, n/n tot (%) | – | – | 1/1 (100.0%) | 2/2 (100.0%) |
| Neuropathological data | ||||
| Immediate macroscopic analysis | ||||
| Brain weight (kg), mean ± SD |
1.38 ± 0.17 n tot = 2 |
1.21 ± 0.18 n tot = 8 |
1.16 ± 0.18 n tot = 6 |
1.30 ± 0.22 n tot = 5 |
Note: For each subgroup, the total number of headcount (n tot) is specified in case of missing data. Cerebrospinal fluid biomarkers profiles: biomarkers of Aβ plaques (labeled “A”) are low CSF Aβ42 or Aβ42/Aβ40 ratio; biomarkers of fibrillar Tau (labeled “T”) are elevated CSF phosphorylated tau (p‐Tau). Binarizing each of the two biomarker groups into normal/abnormal (−/+) results in four possible biomarker profiles: A+T+, A−T+, A+T−, A−T− [20].
Abbreviations: CBD, cortico basal degeneration; CSF, cerebro spinal fluid; CT, computed tomography; IHC, immunohistochemistry. MRI, magnetic resonance imaging; MSA, multiple system atrophy; PD, Parkinson's disease; PET, positron emission tomography; PSP, progressive supranuclear palsy.
3.3.5. Progressive supranuclear palsy
Among the eight patients with a postmortem diagnosis of progressive supranuclear palsy (PSP), three (37.5%) were male (Table 3). Mean age at symptom onset was 65 ± 9 years and mean age at death was 74 ± 9 years. Predominant symptoms were visual disturbances and executive disorders (87.5%), followed by extrapyramidal syndrome, phasic and praxis disorders (75.0%). All the patients had pathological brain imaging and more than 87.5% had pathological neuropsychological testing. The average brain weight was 1.21 ± 0.18 kg, with predominant frontal atrophy (Figure 2). Hypopigmentation of the Substantia nigra was noted in 87.5%. Clinical sensitivity and PPV for the diagnosis of PSP were 100% and 66.7% respectively (Table 2).
3.3.6. Corticobasal degeneration
Among the six patients with a postmortem diagnosis of corticobasal degeneration (CBD), four (66.7%) were male (Table 3). Mean age at symptom onset was 68 ± 8 years and mean age at death was 77 ± 4 years. The predominant symptom was memory disorders (100%), followed by praxis and executive disorders (83.3%), and phasic and behavioral disorders (66.7%). Half of the cases had extrapyramidal syndrome and anxio‐depressive symptoms. All the patients had pathological brain imaging and abnormal neuropsychological testing. The average brain weight was 1.16 ± 0.18 kg, with predominant frontal atrophy (Figure 2). Clinical sensitivity and PPV for the diagnosis of CBD were 33.3% and 50.0% respectively (Table 2).
3.3.7. Multiple system atrophy
Among the six patients with a postmortem diagnosis of multiple system atrophy (MSA), four (66.7%) were male (Table 3). Mean age at symptom onset was 59 ± 12 years and mean age at death was 63 ± 11 years. Extrapyramidal syndrome was predominant (80.0%), followed by anxio‐depressive symptoms, visual disturbances (60.0%), and praxis disorders (40.0%). None of the patients had phasic, behavioral disorders, or hallucination. The average brain weight was 1.30 ± 0.22 kg, with frontal, temporal, and parietal atrophy (Figure 2). Hypopigmentation of the substantia nigra was noted in 100.0%. Clinical sensitivity and PPV for the diagnosis of MSA were 50.0% and 100% respectively (Table 2).
3.3.8. Huntington's disease
Among the 15 patients with a postmortem diagnosis of Huntington's disease (HD), 6 (40.0%) were male (Table 4). Mean age at symptom onset was 36 ± 9 years, mean disease duration was 16 ± 4 years, and mean age at death was 52 ± 10 years. All the patients had movement disorders. The mean CAG triplet repeats was 47 ± 4. The average brain weight (1.09 ± 0.15 kg) was significantly lower when compared with the other groups (p < 0.001), even though the patients died at a younger age. Atrophy was dominant in the frontal cortex and the caudate nucleus (Figure 2). Von Sattel stage was grade 3 in 40.0% and grade 4 in 60.0%. Polyglutamine pathology was observed in all patients. Clinical sensitivity and PPV for the diagnosis of HD were both 100.0% (Table 2).
TABLE 4.
Demographic data—Huntington's disease, amyotrophic lateral sclerosis, Creutzfeldt–Jakob disease
| HD | ALS | CJD | |
|---|---|---|---|
| Description of the population | |||
| Sample size, n | 15 | 27 | 56 |
| Male, n/n tot (%) | 6/15 (40.0%) | 12/27 (44.4%) | 25/56 (44.6%) |
| Age at symptom onset (years), mean ± SD |
35.9 ± 9.2 n tot = 12 |
60.0 ± 10.2 n tot = 24 |
69.1 ± 9.7 n tot = 53 |
| Duration of disease (years), mean ± SD |
16.1 ± 3.9 n tot = 12 |
5.4 ± 3.8 n tot = 24 |
0.6 ± 1.5 n tot = 53 |
| Age at death (years), mean ± SD |
51.7 ± 9.8 n tot = 15 |
65.7 ± 9.9 n tot = 27 |
69.6 ± 9.8 n tot = 56 |
| Clinical data | |||
| Memory disorders, n/n tot (%) | 2/13 (15.4%) | 2/23 (8.7%) | 31/54 (57.4%) |
| Phasic disorders, n/n tot (%) | 5/13 (38.5%) | 1/23 (4.3%) | 35/54 (64.8%) |
| Praxis disorders, n/n tot (%) | 6/13 (46.1%) | 1/23 (4.3%) | 34/54 (63.0%) |
| Executive disorders, n/n tot (%) | 6/13 (46.1%) | 1/23 (4.3%) | 28/54 (51.8%) |
| Anxio‐depressive disorders, n/n tot (%) | 3/13 (23.1%) | 5/23 (21.7%) | 5/54 (9.3%) |
| Behavioral disorders, n/n tot (%) | 6/13 (46.1%) | 1/23 (4.3%) | 18/54 (33.3%) |
| Hallucinations/delirium, n/n tot (%) | 1/13 (7.7%) | 1/23 (4.3%) | 11/54 (20.4%) |
| Extrapyramidal syndrome, n/n tot (%) | 4/13 (30.8%) | – | 11/54 (20.4%) |
| Movement disorders, n/n tot (%) | 13/13 (100.0%) | – | 38/54 (70.4%) |
| Peripheral motor symptoms, n/n tot (%) | – | 21/23 (91.3%) | 0/54 (0%) |
| Central motor symptoms, n/n tot (%) | – | 22/23 (95.6%) | 9/54 (16.7%) |
| Bulbar symptoms, n/n tot (%) | – | 23/23 (100.0%) | 7/54 (13.0%) |
| Visual disturbances, n/n tot (%) | – | – | 18/54 (33.3%) |
| Paraclinical data | |||
| Triplet repeat, mean ± SD |
47.4 ± 4.0 n tot = 10 |
– | – |
| Standard biological abnormalities, n/n tot (%) | – | 2/12 (16.7%) | 15/46 (32.6%) |
| Standard CSF abnormalities, n/n tot (%) | – | – | 12/36 (33.3%) |
| 14‐3‐3 protein, n/n tot (%) | – | – | 34/45 (75.5%) |
| Biomarker CSF, n/n tot (%) | |||
| A+T+ | – | – | 2/9 (22.2%) |
| A−T+ | – | – | 7/9 (77.8%) |
| A+T− | – | – | – |
| A−T− | – | – | – |
| ENMG abnormalities, n/n tot (%) | – | 16/17 (94.1%) | – |
| EEG abnormalities, n/n tot (%) | – | – | 41/50 (82.0%) |
| CT‐scan/MRI abnormalities, n/n tot n tot (%) | – | 3/10 (30.0%) | 53/55 (96.4%) |
| Neuropsychological tests abnormalities, n/n tot (%) | – | – | 25/25 (100.0%) |
| Neuropathological data | |||
| Immediate macroscopic analysis | |||
| Brain weight (kg), mean ± SD |
1.09 ± 0.15 n tot = 13 |
1.29 ± 0.15 n tot = 26 |
1.24 ± 0.18 n tot = 56 |
Note: For each subgroup, the total number of headcount (n tot) is specified in case of missing data. Cerebrospinal fluid biomarkers profiles: biomarkers of Aβ plaques (labeled “A”) are low CSF Aβ42 or Aβ42/Aβ40 ratio; biomarkers of fibrillar Tau (labeled “T”) are elevated CSF phosphorylated Tau (p‐Tau). Binarizing each of the two biomarker groups into normal/abnormal (−/+) results in four possible biomarker profiles: A+T+, A−T+, A+T−, A−T− [20].
Abbreviations: ALS, amyotrophic lateral sclerosis; CJD, Creutzfeldt–Jakob disease; CSF, cerebro spinal fluid; CT, computed tomography; EEG, electroencephalogram; ENMG, electroneuromyogram; HD, Huntington's disease; IHC, immunohistochemistry; MRI, magnetic resonance imaging.
3.3.9. Amyotrophic lateral sclerosis
Among the 27 patients with a postmortem diagnosis of amyotrophic lateral sclerosis (ALS), 12 (44.4%) were male (Table 4). Mean age at symptom onset was 60 ± 10 years and mean age at death was 66 ± 10 years. All the patients had bulbar symptoms and more than 90% had peripheral and/or central motor symptoms. ENMG was abnormal in more than 90% of cases. The average brain weight was 1.29 ± 0.15 kg, with predominant frontal and spinal atrophy (Figure 2). TDP‐43 pathology was observed in all patients. Associated FTLD‐TDP‐43 lesions were noted in two cases. Clinical sensitivity and PPV for the diagnosis of ALS were both 100.0% (Table 2).
3.3.10. Creutzfeldt–Jakob disease
Among the 56 patients with a postmortem diagnosis of Creutzfeldt–Jakob disease (CJD), 25 (44.6%) were male (Table 4). Mean age at symptom onset was 69 ± 10 years and mean age at death was 70 ± 10 years. The mean disease duration was 0.6 ± 1.5 years. Clinical symptoms were heterogeneous: predominant symptom was movement disorders (70.4%), followed by phasic (64.8%), praxis (63.0%) and memory disorders (57.4%). Half of the cases had executive disorders, 33.3% had behavioral disorders and visual disturbances. The 14.3.3 protein was elevated in the CSF in 75.5%, and Tau in 100%. EEG abnormalities were observed in 82.0% (e.g. periodic sharp wave complexes). CT‐scan or MRI were abnormal in almost 100% and, when performed, neuropsychological tests were always pathological. The average brain weight was 1.24 ± 0.18 kg, atrophy was rarely noted (Figure 2). While neuronal loss was minimal (<5%), gliosis and spongiosis accounted for a significant proportion (>90%). Prion protein (PrP) pathology was observed in all patients. The western blot analysis of protease‐resistant prion protein (PrPres) showed several isoforms: type 1 in 32.6%, type 1/2A in 28.2%, type 2A in 36.9% and type 2B in 2.2%. One case had acquired variant of CJD and 3 cases had familial CJD (2 patients with E200K mutation, 1 with PRNPp.Val180Ile mutation and 1 with Gerstmann‐Sträussler‐Scheinker syndrome). Clinical sensitivity and PPV for the diagnosis of CJD were 96.4% and 91.5% respectively (Table 2).
3.4. Incidental proteinopathy and mixed pathologies
We then aimed to assess the proportion of incidental proteinopathy in our series, defined as protein inclusions observed in the brain of the patients but unrelated to the final diagnosis. We used the queries [SELECT “FINAL DIAGNOSIS,” “TAU PATHOLOGY” FROM “Data_Base” WHERE “TAU PATHOLOGY” = ‘Yes’], [SELECT “FINAL DIAGNOSIS”, “AMYLOID BETA PATHOLOGY” FROM “Data_Base” WHERE “AMYLOID BETA PATHOLOGY” = ‘Yes’] and [SELECT “FINAL DIAGNOSIS,” “SYNUCLEIN PATHOLOGY” FROM “Data_Base” WHERE “SYNUCLEIN PATHOLOGY” = ‘Yes’], considering all the patients from the 3 groups “neurodegenerative diseases,” “prion disease” and “other diseases of the CNS.” The proportion of TDP‐43 proteinopathy could not be estimated as TDP‐43 immunostaining was only performed for ALS or FTLD suspected cases from 2007 to 2016 (systematically performed since 2017). We then excluded of each group the patients for whom the final diagnosis was related to the considered protein. Tau incidental proteinopathy was observed in 78.1% of the patients who were not diagnosed with AD, FTLD‐Tau, PSP, CBD, argyrophilic grain disease or Pick's disease. Amyloid‐β incidental proteinopathy was observed in 42.8% of the patients who were not diagnosed with AD or amyloid angiopathy. α‐Synuclein incidental proteinopathy was observed in 10.7% of the patients who were not diagnosed with DLB, PD or MSA (Figure 3A). We then added the parameter “age at death” to the queries and could observe an increasing rate of incidental Tau and amyloid‐β incidental proteinopathy related to the age of the autopsied patients (Figure 3B).
FIGURE 3.
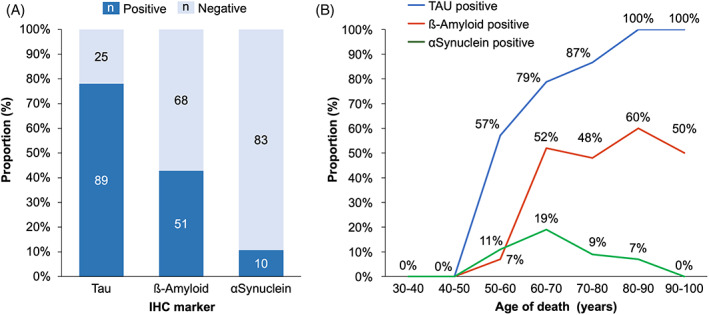
Incidental proteinopathy in autopsied patients. Tau, amyloid‐β and α‐synuclein incidental proteinopathy observed in patients for whom the final diagnosis was not related to the considered protein. (A) Overall proportions. (B) Distribution according to the age of the cases
We next aimed to assess the proportion of mixed proteinopathies in patients diagnosed with neurodegenerative diseases and prion disease, defined as the presence of protein inclusions observed in the brain in addition to the proteinopathy related to the final diagnosis. We used the query: [SELECT “FINAL DIAGNOSIS,” “TAU PATHOLOGY,” “AMYLOID BETA PATHOLOGY,” “SYNUCLEIN PATHOLOGY” FROM “Data_Base” WHERE “TAU PATHOLOGY” = ‘Yes’ OR “AMYLOID BETA PATHOLOGY” = ‘Yes’ OR “SYNUCLEIN PATHOLOGY” = ‘Yes’] considering the patients from the 2 groups “neurodegenerative diseases” and “prion disease.” The results are reported in Table 5. Nearly half of the patients (46.4%) had mixed proteinopathies. We observed a particularly high rate of concomitant pathology in patients with DLB (81.3% of associated Tau and Amyloid‐β pathology) and CJD (68.4% of associated Tau pathology), while the mean age of the two groups were not significantly higher when compared to the other groups (p = 0.343 DLB, p = 0.148 CJD).
TABLE 5.
Primary diagnoses and concomitant pathologies
| Concomitant pathologies | |||||
|---|---|---|---|---|---|
| Primary diagnoses | ADNC | Amyloid plaques | Amyloid angiopathy | α‐Synuclein pathology | |
| Tauopathy | Alzheimer's disease (41) | N.A. | N.A. | 15/41 (36.6%) | 5/22 (22.7%) |
| Frontotemporal lobar degeneration—Tau (2) | N.A. | 0 | 0 | 0 | |
| Progressive supranuclear palsy (8) | 3/8 (37.5%) | 2/6 (33.3%) | 1/6 (16.6%) | ‐ | |
| Corticobasal degeneration (6) | 3/6 (50.0%) | 2/5 (40.0%) | 1/5 (20.0%) | 1/4 (25.0%) | |
| Synucleinopathy | Dementia with lewy bodies (16) | 13/16 (81.3%) | 13/16 (81.3%) | 6/16 (37.5%) | N.A. |
| Parkinson's disease (2) | 1/2 (50.0%) | 1/2 (50.0%) | 0 | N.A. | |
| Multiple system atrophy (6) | 3/6 (50.0%) | ‐ | ‐ | N.A. | |
| TDP‐43 Proteinopathy | Frontotemporal lobar degeneration—TDP (6) | 4/6 (66.7%) | 2/6 (33.3%) | 1/6 (16.7%) | 1/4 (25.0%) |
| Amyotrophic lateral sclerosis (27) | 8/14 (57.1%) | 4/11 (36.4%) | 0 | ‐ | |
| Huntington's disease (15) | 2/6 (33.3%) | 1/6 (16.7%) | 0 | ‐ | |
| Creutzfeldt–Jakob disease (56) | 26/38 (68.4%) | 18/38 (47.4%) | 11/38 (28.9%) | 1/37 (2.7%) | |
Note: For each subgroup, the number of headcount (n) is specified.
Abbreviations: AD, Alzheimer's disease; ADNC, Alzheimer's disease neuropathologic change; FTLD, frontotemporal lobar degeneration; NA, not applicable.
3.5. Diagnosis revision following classification updates and new pathological entities
Classifications and diagnosis criteria in neuropathology are in constant evolution, following regular definitions of new entities, endophenotypes and pathological markers. Recently, a newly proposed pathology called limbic‐predominant age‐related TDP‐43 encephalopathy (LATE) has been defined as a late onset amnestic syndrome mimicking Alzheimer's disease (AD) with stereotypical TDP‐43 pathology in limbic brain structures, frequently associated with amyloid‐β and Tau pathology [21].
With the aim of identifying potential undiagnosed LATE patients in our series, we extracted from our DataBase all the cases older than age 75 who were diagnosed with AD due to progressive amnestic syndrome but presented mild to moderate post‐mortem Aβ and Tau pathology. To this end, we used the following query: [SELECT “AGE AT DEATH,” “MEMORY IMPAIRMENT,” “FINAL DIAGNOSIS,” “AMYLOID BETA PATHOLOGY THAL,” “TAU PATHOLOGY BRAAK,” “TAU PATHOLOGY CERAD” FROM “Data_Base” WHERE “MEMORY IMPAIRMENT” = ‘Yes’ AND “FINAL DIAGNOSIS” = ‘Alzheimer's disease’ HAVING (MIN(“AGE AT DEATH”) = 75 AND MAX(“AMYLOID BETA PATHOLOGY THAL”) = ‘Phase 3 (A2)’ AND MAX(“TAU PATHOLOGY BRAAK”) = ‘Stage III (B2)’ AND MAX(“TAU PATHOLOGY CERAD”) = ‘Moderate (C2)’)].
Three patients were identified, complementary immunohistochemical analysis with phosho‐TDP‐43 antibody were performed on amygdala, hippocampus, and middle frontal gyrus samples. Phospho‐TDP‐43 immunoreactive cytoplasmic inclusions could be seen in one patient, a 90‐year‐old woman who died in 2007 after a 10‐year history of progressive amnestic syndrome. The brain weight was 1.070 kg. Diffuse cortical neuronal loss was noted, more pronounced in the hippocampus (Figure 4A,B). Pathological cytosolic inclusions of phospho‐TDP‐43 were observed in neurons, predominantly confined to the limbic system (Figure 4C,D). Mild Tau pathology and no amyloid plaques were observed in the entorhinal cortex and the hippocampal CA1 subfield (Figure 4E,F). The final diagnosis was revised to LATE Stage 2.
FIGURE 4.
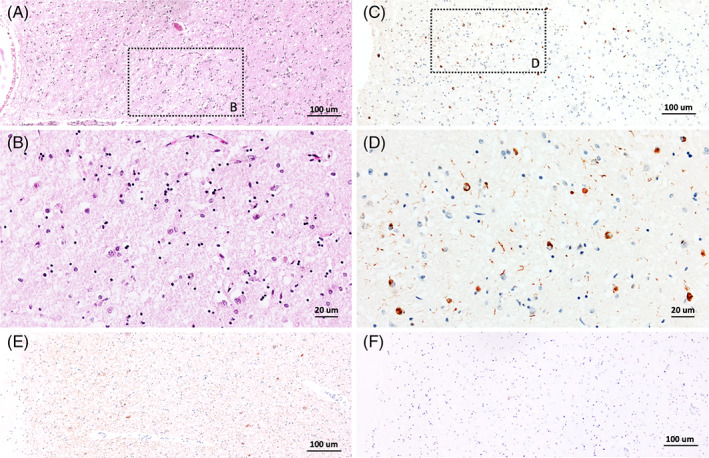
Diagnosis revision in a 90‐year‐old patient with progressive amnestic syndrome. Cortical neuronal loss and gliosis were observed in the hippocampal CA1 subfield (A,B). Phospho‐TDP‐43 cytoplasmic inclusions could be seen in neurons along with phospho‐TDP‐43 immunoreactive dystrophic neurites (C,D). Mild Tau pathology (E) and no amyloid plaques (F) were observed in the same region
4. DISCUSSION
In this study, we designed a data entry form driven by an open‐source DBMS to generate a relational DataBase in neuropathology. The form is freely available upon request to the authors, does not require advanced computer skills, and can be used and customized with open‐source software to fit protocols performed in other neuropathology departments (clinical information, sampled regions, staining, diagnoses). The use of a DBMS allowed us to optimize the compilation of clinical and pathological data from our brain collection with an average filling time of about 10 min per patient and a final size of 6.1 MB for 22,885 data points. Next, we could easily retrieve information using multiple and conditional queries, manually generated or built by the query module of the software. Using these queries, we finally analyzed data from the base to study clinicopathological correlations and to quickly identify cases for both diagnosis and research purpose.
To discuss these results, it first must be noted that the DataBase has been filled retrospectively from medical records, with variable clinical information available. Since 2021 in Angers, we are collecting data with the help of clinicians at the time of death, to ensure a more accurate filling. Another point of discussion is that the patients in our series may not be reflective of the general population affected by neurodegenerative disorders, as they were referred to reference centers for both diagnosis and management. Moreover, complex cases are usually more likely to be autopsied than typical cases, increasing this referral bias. As an illustration, the mean age at symptom onset of AD patients (62 years) in our series is lower compared to epidemiological series [22], and the mean duration of disease is shorter (8 years). In the same way, only two cases affected by Parkinson's disease have been autopsied in our department during the studied period, versus 20 “atypical” parkinsonian syndromes PSP, CBD, and MSA. This higher proportion of uncommon and complex cases undoubtedly affects diagnosis accuracy and concomitant pathology rates. The reported sensitivities and PPV in our study are in line with previous series [3, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37]. Clinical sensitivity and PPV were the highest for ALS, HD and CJD. This could be explained by the typical clinical picture at end stage for ALS, the access to genetic testing for HD, and the relatively specific paraclinical markers for CJD. It has also to be considered that the autopsies were widely performed on patients with a strongly suspected diagnosis for epidemiological (CJD) or research (ALS and HD) purposes. Clinical sensitivity and PPV were lower for cortical degenerations and movements disorders, possibly due to the autopsy indication (unknown diagnosis), the heterogeneity of the endophenotypes in these diseases and the higher rates of co‐pathologies increasing the clinical complexity.
Indeed, while current histopathological classifications of neurodegenerative diseases are mainly based on the predominant pathology found in the brain, concomitant proteinopathies and vascular lesions are frequently observed, raising the concept of mixed pathology that surely influence the phenotype of the diseases (onset, presentation, course, and prognosis), making diagnosis even more complex. In the last decade, there has been increasing interest in the epidemiology and pathophysiology of mixed proteinopathies [2, 38, 39]. The proportion of cases who presented with co‐pathologies in our study is similar to those of previous series [1, 38, 40, 41, 42, 43, 44]. Ageing is considered the main factor fostering co‐pathologies, since the risk of developing a proteinopathy increases with age. In our series Tau, Amyloid‐β and α‐Synuclein incidental pathology was observed in 78.1%, 42.8%, and 10.7% respectively, and these proportions increased with age reaching 100% for Tau after 80. Similar results have been obtained in large post‐mortem studies assessing age‐related prevalence of neurofibrillary tangles, Aβ plaques and Lewy body pathology [39]. Synergistic interaction of pathological proteins (cross‐seeding), common pathogenic pathways, and genetic and epigenetic background are also suspected to participate in the development of combined proteinopathies [45]. In line with this, we observed in our series a significantly higher rate of Tau and Amyloid‐β pathology in DLB cases and Tau pathology in CJD cases, while the patients were not older than the other cases affected by neurodegenerative disorders. These associations have also been observed in previous series [23, 42, 46, 47], supporting the hypothesis of synergistic pathomechanisms in the brain of patients affected by DLB and CJD. The elevated rate of mixed proteinopathies observed in our series and others should be considered with caution, as patients referred to reference centers and autopsied often present with complex clinical pictures that may reflect the addition of distinct or synergic proteinopathies, surely increasing the proportion of mixed pathologies in series. However, even if the rate of comorbid proteinopathies is probably lower in the general population, the concept should be further studied given the progressive increase in the mean age of the population and the implications in clinical diagnosis, biomarker identification and therapeutic trials.
Finally, in this study we used specific queries to identify old cases of the DataBase that could meet LATE criteria. Additional immunohistochemical labelling of phospho‐TDP‐43 was performed on amygdala, hippocampus, and middle frontal gyrus samples of 3 selected cases, and the AD diagnosis of a 90‐year‐old patient could be revised to LATE Stage 2 given the presence of marked TDP‐43 pathology in the limbic system. This simple approach can be used to quickly identify old cases that could meet newly defined neuropathological criteria and regularly update the diagnoses of a brain collection.
5. CONCLUSIONS
In conclusion, the systematic detection and characterization of the proteinopathies observed in autopsied patients should be promoted to increase our understanding of co‐pathologies and help to identify new neuropathological entities. This approach is crucial given the implications in clinical diagnosis, biomarker identification and trials for targeted therapies. The large amount of generated data inherent to such process should encourage a more systematic use of DBMS, with the prospect of applying supervised or unsupervised machine learning algorithms on large and reliable DataBases.
AUTHOR CONTRIBUTIONS
FEB, VC, VGP, CSG, AP, SG, AL, JC, CV, FL, and PC contributed to the organization of the autopsies. PC conceived and designed the study. PC and IJ conducted the experiments and data collection. PC, IJ and FL analyzed and interpreted the data. PC and IJ wrote the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING STATEMENT
This work was funded by the University Hospital of Angers (Grant N° 2019‐264 900_036) and the French National Institute of Health and Medical Research (INSERM Research Fellow 2017–2019).
ETHICS STATEMENT
All brain donors or their relatives had given their informed consent for the use of brain tissue for research. The use of human biological samples in this study has been given ethical approval by the regional ethics committee West II (biological resource center national identifier BB‐0033‐00038, declaration number DC‐2011‐146). The study protocol has been declared to the French commission for information technology and civil liberties (declaration number ar21‐0007v0).
Supporting information
Figure S1. Data entry form.
Table S1. General demographic data. For each subgroup, total number of headcount (n tot) is specified in case of missing data.
Table S2. AD neuropathologic changes.
ACKNOWLEDGMENTS
The authors are grateful to the donors and their families. We also thank C. Dumez, I. Viau and L. Denechaud for technical assistance.
Journe‐Mallet I, Gouju J, Etcharry‐Bouyx F, Chauvire V, Guillet‐Pichon V, Scherer‐Gagou C, et al. Design and application of a customizable relational DataBase to assess clinicopathological correlations and concomitant pathology in neurodegenerative diseases. Brain Pathology. 2023;33(3):e13138. 10.1111/bpa.13138
DATA AVAILABILITY STATEMENT
The data that support the findings of this study and the neuropathology form (working with both LibreOffice and OpenOffice software) are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Rahimi J, Kovacs GG. Prevalence of mixed pathologies in the aging brain. Alz Res Therapy. 2014;6(9):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kovacs GG. Are comorbidities compatible with a molecular pathological classification of neurodegenerative diseases? Curr Opin Neurol. 2019;32(2):279–91. [DOI] [PubMed] [Google Scholar]
- 3. Brunnstrom H. Clinicopathological concordance in dementia diagnostics. Am J Geriatr Psychiatry. 2009;17(8):664–70. [DOI] [PubMed] [Google Scholar]
- 4. Andersson EM, Hoff EJ, Waldö ML, Englund E. Clinicopathological concordance in cognitive disease diagnostics. Clin Neuropathol. 2020;39(5):99–104. [DOI] [PubMed] [Google Scholar]
- 5. Taipa R, Pinho J, Melo‐Pires M. Clinico‐pathological correlations of the most common neurodegenerative dementias. Front Neurol. 2012;3:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Besser LM, Kukull WA, Teylan MA, Bigio EH, Cairns NJ, Kofler JK, et al. The revised National Alzheimer's coordinating center's neuropathology form—available data and new analyses. J Neuropathol Exp Neurol. 2018. Aug 1;77(8):717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mock C, Teylan M, Beecham G, Besser L, Cairns NJ, Crary JF, et al. The utility of the national Alzheimer's coordinating center's database for the rapid assessment of evolving neuropathologic conditions. Alzheimer Dis Assoc Disord. 2020;34(2):105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239–59. [DOI] [PubMed] [Google Scholar]
- 9. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–86. [DOI] [PubMed] [Google Scholar]
- 10. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. [DOI] [PubMed] [Google Scholar]
- 12. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mackenzie IRA, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117(1):15–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mackenzie IRA, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;9(s3):417–23. [DOI] [PubMed] [Google Scholar]
- 16. Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24(2):197–211. [DOI] [PubMed] [Google Scholar]
- 17. Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935–46. [DOI] [PubMed] [Google Scholar]
- 18. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classifcation of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–77. [DOI] [PubMed] [Google Scholar]
- 19. Budka H, Aguzzi A, Brown P, Brucher J‐M, Bugiani O, Gullotta F, et al. Neuropathological diagnostic criteria for Creutzfeldt–Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol. 1995;5(4):459–66. [DOI] [PubMed] [Google Scholar]
- 20. Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nichols E, Szoeke CEI, Vollset SE, Abbasi N, Abd‐Allah F, Abdela J, et al. Global, regional, and national burden of Alzheimer's disease and other dementias, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019;18(1):88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grau‐Rivera O, Gelpi E, Nos C, Gaig C, Ferrer I, Saiz A, et al. Clinicopathological correlations and concomitant pathologies in rapidly progressive dementia: a brain bank series. Neurodegener Dis. 2015;15(6):350–60. [DOI] [PubMed] [Google Scholar]
- 24. Snowden JS, Thompson JC, Stopford CL, Richardson AMT, Gerhard A, Neary D, et al. The clinical diagnosis of early‐onset dementias: diagnostic accuracy and clinicopathological relationships. Brain. 2011;134(9):2478–92. [DOI] [PubMed] [Google Scholar]
- 25. Soria JA, Huisa BN, Edland SD, Litvan I, Peavy GM, Salmon DP, et al. Clinical‐neuropathological correlations of Alzheimer's disease and related dementias in Latino volunteers. J Alzheimer's Disease. 2018;66(4):1539–48. [DOI] [PubMed] [Google Scholar]
- 26. Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer disease centers, 2005–2010. J Neuropathol Exp Neurol. 2012;71(4):8–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gauthreaux K, Bonnett TA, Besser LM, Brenowitz WD, Teylan M, Mock C, et al. Concordance of clinical Alzheimer diagnosis and neuropathological features at autopsy. J Neuropathol Exp Neurol. 2020;79(5):465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Balasa M, Gelpi E, Martín I, Antonell A, Rey MJ, Grau‐Rivera O, et al. Diagnostic accuracy of behavioral variant frontotemporal dementia consortium criteria (FTDC) in a clinicopathological cohort: bvFTD criteria validation. Neuropathol Appl Neurobiol. 2015;41(7):882–92. [DOI] [PubMed] [Google Scholar]
- 29. Perry DC, Brown JA, Possin KL, Datta S, Trujillo A, Radke A, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140(12):3329–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, Logroscino G. Accuracy of clinical diagnosis of Parkinson disease: a systematic review and meta‐analysis. Neurology. 2016;86(6):566–76. [DOI] [PubMed] [Google Scholar]
- 31. Rizzo G, Arcuti S, Copetti M, Alessandria M, Savica R, Fontana A, et al. Accuracy of clinical diagnosis of dementia with Lewy bodies: a systematic review and meta‐analysis. J Neurol Neurosurg Psychiatry. 2018;89(4):358–66. [DOI] [PubMed] [Google Scholar]
- 32. Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. 2006;66(1):41–8. [DOI] [PubMed] [Google Scholar]
- 33. Osaki Y, Ben‐Shlomo Y, Lees AJ, Daniel SE, Colosimo C, Wenning G, et al. Accuracy of clinical diagnosis of progressive supranuclear palsy. Mov Disord. 2004;19(2):181–9. [DOI] [PubMed] [Google Scholar]
- 34. Miki Y, Foti SC, Asi YT, Tsushima E, Quinn N, Ling H, et al. Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study. Brain. 2019. Sep 1;142(9):2813–27. [DOI] [PubMed] [Google Scholar]
- 35. Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol. 2011;70(2):327–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tagliapietra M, Zanusso G, Fiorini M, Bonetto N, Zarantonello G, Zambon A, et al. Accuracy of diagnostic criteria for sporadic Creutzfeldt–Jakob disease among rapidly progressive dementia. J Alzheimers Dis. 2013;34(1):231–8. [DOI] [PubMed] [Google Scholar]
- 37. Iwasaki Y, Mimuro M, Yoshida M, Sobue G, Hashizume Y. Clinical diagnosis of Creutzfeldt–Jakob disease: accuracy based on analysis of autopsy‐confirmed cases. J Neurol Sci. 2009;277(1–2):119–23. [DOI] [PubMed] [Google Scholar]
- 38. Elobeid A, Libard S, Leino M, Popova SN, Alafuzoff I. Altered proteins in the aging brain. J Neuropathol Exp Neurol. 2016;75(4):316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spires‐Jones TL, Attems J, Thal DR. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017;134(2):187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Colom‐Cadena M, Grau‐Rivera O, Planellas L, Cerquera C, Morenas E, Helgueta S, et al. Regional overlap of pathologies in Lewy body disorders. J Neuropathol Exp Neurol. 2017;76:216–24. [DOI] [PubMed] [Google Scholar]
- 41. Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134(2):171–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age‐related and APOE4‐associated. Brain. 2018;141(7):2181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kovacs GG, Milenkovic I, Wöhrer A, Höftberger R, Gelpi E, Haberler C, et al. Non‐Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community‐based autopsy series. Acta Neuropathol. 2013;126(3):365–84. [DOI] [PubMed] [Google Scholar]
- 44. Robinson JL, Richardson H, Xie SX, Suh E, Van Deerlin VM, Alfaro B, et al. The development and convergence of co‐pathologies in Alzheimer's disease. Brain. 2021;144(3):953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clinton LK, Blurton‐Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic interactions between A, Tau, and ‐Synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010. May 26;30(21):7281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 2017;16(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kovacs GG, Rahimi J, Ströbel T, Lutz MI, Regelsberger G, Streichenberger N, et al. Tau pathology in Creutzfeldt–Jakob disease revisited: Tau pathology in CJD. Brain Pathol. 2017;27(3):332–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Data entry form.
Table S1. General demographic data. For each subgroup, total number of headcount (n tot) is specified in case of missing data.
Table S2. AD neuropathologic changes.
Data Availability Statement
The data that support the findings of this study and the neuropathology form (working with both LibreOffice and OpenOffice software) are available from the corresponding author upon reasonable request.