

Abstract
The p.Thr61Ile (p.T61I) mutation in coiled‐coil‐helix–coiled‐coil‐helix domain containing 2 (CHCHD2) was deemed a causative factor in Parkinson's disease (PD). However, the pathomechanism of the CHCHD2 p.T61I mutation in PD remains unclear. Few existing mouse models of CHCHD2‐related PD completely reproduce the features of PD, and no transgenic or knock‐in (KI) mouse models of CHCHD2 mutations have been reported. In the present study, we generated a novel CHCHD2 p.T61I KI mouse model, which exhibited accelerated mortality, progressive motor deficits, and dopaminergic (DA) neurons loss with age, accompanied by the accumulation and aggregation of α‐synuclein and p‐α‐synuclein in the brains of the mutant mice. The mitochondria of mouse brains and induced pluripotent stem cells (iPSCs)‐derived DA neurons carrying the CHCHD2 p.T61I mutation exhibited aberrant morphology and impaired function. Mechanistically, proteomic and RNA sequencing analysis revealed that p.T61I mutation induced mitochondrial dysfunction in aged mice likely through repressed insulin‐degrading enzyme (IDE) expression, resulting in the degeneration of the nervous system. Overall, this CHCHD2 p.T61I KI mouse model recapitulated the crucial clinical and neuropathological aspects of patients with PD and provided a novel tool for understanding the pathogenic mechanism and therapeutic interventions of CHCHD2‐related PD.
Keywords: CHCHD2, insulin‐degrading enzyme, mitochondria, mouse model, Parkinson's disease
We set out to study the biological role of the CHCHD2 p.T61I mutation in Parkinson's disease in vivo by generating a knock‐in mouse model harbouring the mutation. The mutant mice had accelerated mortality, progressive motor deficits, and the loss of dopaminergic neurons. α‐synuclein and p‐α‐synuclein aggregations were observed in the brains of the mutant mice; these mice also exhibited an aberrant mitochondrial morphology and impaired mitochondrial function. Induced pluripotent stem cell‐derived dopaminergic neurons carrying the CHCHD2 p.T61I mutation reproduced the impaired mitochondria. Proteomic and RNA sequencing analysis revealed that p.T61I mutation induced mitochondrial dysfunction in aged mice likely through repressed IDE expression, resulting in the degeneration of the nervous system.
1. INTRODUCTION
Parkinson's disease (PD) is the second most frequently occurring neurological degenerative disease after Alzheimer's disease (AD) and is characterised by motor deficits, including resting tremor, bradykinesia, muscle rigidity, and postural instability. PD also has non‐motor manifestations, such as hyposmia, depression, and cognitive deficits. The main pathological features of PD are the loss of dopaminergic (DA) neurons in the substantia nigra (SN) and the formation of Lewy bodies (LBs), which are mainly composed of α‐synuclein, in the cytoplasm of the remaining neurons. Familial forms of PD account for approximately 5% of all cases [1], with single‐gene mutations described in genes, such as SNCA [2], PINK1 [3], and LRRK2 [4]. Molecular studies on these genes have shed light on the pathomechanism of DA neuronal loss, including autophagy impairment, mitochondrial dysfunction, immunity deregulation, and endoplasmic reticulum stress [5]. Funayama et al. suggested that the p.Thr61Ile (p.T61I) mutation in coiled‐coil‐helix–coiled‐coil‐helix domain containing 2 (CHCHD2) is a causative factor for PD [6], and we also discovered this mutation in a Chinese family with autosomal dominant PD [7]. The onset age of patients in that family was between 39 and 53 years. Most had symptoms of resting tremor, bradykinesia, rigidity, and gait disturbance. However, the pathogenesis by which CHCHD2 gene mutations lead to PD remains largely unknown.
CHCHD2 is a multifunctional protein that usually localises to the intermembrane spaces of mitochondria and regulates mitochondrial metabolism, mitochondria‐mediated apoptosis, and the expression of respiratory‐chain components [8]. In the nucleus, CHCHD2 regulates the transcription of genes coding for oxygen response elements [9]. In‐vitro studies have revealed that the CHCHD2 p.T61I mutation causes α‐synuclein aggregation [10]. In other studies, the loss of CHCHD2 in Drosophila caused a higher production of reactive oxygen species (ROS), degeneration of mitochondrial cristae, and impairment of oxygen respiration, leading to DA neuronal loss and motor dysfunction with age [11]. However, according to Sato et al. [12], the disease phenotypes were absent on the CHCHD2‐knockout (KO) mouse model, which was corroborated by Nguyen et al. [13] who reported that CHCHD2‐KO mice failed to exhibit morbidity by 19 months of age. Those data did not support the loss‐of‐function mechanism of CHCHD2 mutation in PD. It is of great importance to investigate whether gain‐of‐function or dominant‐negative effect of CHCHD2 mutations are linked to a particular phenotype and the pathomechanism of PD via a CHCHD2 mutant knock‐in (KI) mice model.
We modelled a KI mouse model harbouring CHCHD2 p.T61I mutation and performed in‐depth behavioural assessments. We discovered that CHCHD2 p.T61I mutant mice died prematurely and developed motor defects. α‐synuclein and p‐α‐synuclein aggregated in the brain of CHCHD2 p.T61I mutant mice, with abnormal function and structure of mitochondria. Human induced pluripotent stem cells (iPSCs)‐derived DA neurons carrying the heterozygous (HET) CHCHD2 p.T61I mutation also exhibited impaired mitochondrial respiration and structure. Proteomics and RNA‐sequencing (RNA‐seq) suggested that these phenotypes might be correlated with insulin‐degrading enzyme (IDE)‐insulin metabolism and mitochondria. The present study provides a novel CHCHD2 p.T61I KI mouse model that results in certain PD‐like phenotypes, supporting the hypothesis that the CHCHD2 p.T61I mutation may act by means of dysregulation of IDE‐insulin metabolism and mitochondria.
2. MATERIALS AND METHODS
2.1. Animal studies
All animal studies adhered to the guidelines of and were approved by the Institutional Animal Care and Use Committee of Zhengzhou University and the experiments complied with current Chinese laws. The mouse Chchd2 gene is equivalent to the human CHCHD2 gene. Owing to the high off‐target rates of CRISPR/Cas9 genome editing of CHCHD2, we employed conventional gene targeting technology in embryonic stem cells to generate a HET KI mutation resulting in the p.T61I amino acid substitution in C57BL/6 mice (Figure (S1)). Embryonic stem cells derived from clones carrying the desired homologous recombination were injected into C57BL/6 embryos. Chimeric progeny was hybridised with C57BL/6 mice, and HET KI mice were crossbred to produce homozygous (HOM) KI mice and wild type (WT) mice. The resulting mice were genotyped via PCR with allele‐targeting primers. Littermates were used. Mice were housed with food and water provided under a 12 –12 h light–dark cycle. No significant differences were observed between male and female mice, and male mice were used for experiments. Mice were monitored weekly for their general condition and survival. The age of death was recorded to construct a Kaplan–Meier survival curve. Body weights were recorded weekly from the age of 8 weeks. Motor coordination ability was assessed by using the pole test and the gait test. In the pole test trial, mice were placed on top of a pole (60 cm tall, 10 mm diameter) with their heads facing downward. After 3 days of training, the test was performed and the time it took each mouse to descend the pole was recorded. The CatWalk XT gait analysis system (Noldus Information Technology, Wageningen, Netherlands) was used to analyse gait. The mouse was placed on the walkway, and the kinematic parameters of gait initiation were recorded. Stride length was measured as the distance between placements of the right hind paw, and the base of support was measured as the average distance between the hind paws. Each animal was trained three times to cross the runway consistently before the experiment. Mouse endurance was measured by using the rotarod test. Mice were placed on the rotarod, which was accelerated from 4 to 40 rpm within 5 min. We trained the mice for the first 3 days and recorded the latency to fall on day 4. Anxiety‐like behaviours of the mice were assessed by using the OF and the EPM. In the OF test, mice were placed in a clean, square area (50 cm × 50 cm) and allowed to explore for 5 min. The time in the central area and the total distance moved were vedio‐recorded. In the EPM, mice were placed in the middle of two perpendicular, intersecting runways (open and closed arms). We observed the mice for 5 min and recorded the time spent on the open arms by a video.
2.2. Protein extraction and Western blotting
Total protein extracts were prepared using RIPA buffer (Cat. #: R0010, Solarbio) supplemented with 1× PMSF (Cat. #: P0100, Solarbio) and 1× phosphatase inhibitor cocktail (Cat. #: HY‐K0022, MedChemExpress LLC, Monmouth Junction, NJ, USA). After 30 min of lysis on ice with frequent tapping, cells and dissected mouse tissues were centrifuged at 14,000 × g at 4°C for 15 min. The supernatant was mixed with SDS‐PAGE loading buffer (Cat. #: WB‐0091, DING GUO) at a ratio of 4:1 and incubated at 100°C for 10 min. The sample was loaded onto a 10 or 12.5% Tris‐glycine gel (Cat. #: PG213, Epizyme) and blotted onto a 0.2‐μm polyvinylidene difluoride membrane (Cat. #: ISEQ00010, Immobilon). Membranes were blocked with 5% skimmed milk at room temperature for 2 h and incubated for 12–16 h with indicated primary antibodies (Table S1). After incubation with the secondary antibody at room temperature for 2 h on the next day, membranes were washed in 1× TBST medium and imaged by using an Amersham Imager 600 (GE Healthcare Bio‐Sciences AB).
2.3. Ex vivo tissue slice preparation, histochemical staining, and confocal immunofluorescence
Mice were anaesthetized and then perfused with ice‐cold PBS and 4% paraformaldehyde, respectively. After perfused, the brains and spinal cords were rapidly removed and soaked in 4% paraformaldehyde at 4°C for 24 h. After dehydration and embedding, the slices (thickness, 15 μm) were sectioned using a vibratome (VT1200S Leica Microsystems). For immunohistochemistry, the slices were put into citrate buffer and for antigen retrieval. After blocking for 15 min in normal goat serum, sections were incubated with primary antibodies overnight at 4°C, and in species‐specific secondary antibodies at room temperature. After 3 washes of PBS, the sections were developed with DAB and dehydrated in 75–85–95–100% ethanol and xylene. Every other five brain sections were stained with tyrosine hydroxylase (TH), α‐synuclein, and p‐α‐synuclein antibodies, separately. DA neurons on every other one stained section in the ipsilateral brain were quantified using NIH ImageJ and then stacked together as the amounts of DA neurons in substantia nigra pars compacta (SNpc) and ventral tegmental area (VTA). The mean optical density (OD) of TH‐positive fibers, α‐synuclein, and p‐α‐synuclein were assessed using ImageJ software. For immunofluorescence, the slices were put into citrate buffer and for antigen retrieval. After incubated in 0.3% Triton X‐100 for 30 min and 10% BSA for 1 h, sections were incubated with primary antibodies overnight at 4°C and secondary antibodies for 2 h at room temperature. Confocal images were collected by Zeiss 980 system. For the Nissl staining, lumbar spinal cords sections of three mice per genotype at 11 months of age were quantified. All the motor neurons (diameter > 25 μm) in the ventral horn were counted bilaterally. The HE‐staining, Masson's Trichrome, and succinate dehydrogenase (SDH) activity staining were operated according to standard operating procedures. The antibodies used are listed in Table S1.
2.4. Analysis of mitochondrial respiratory chain activities
After excised, spinal cords were immediately snap‐frozen in liquid nitrogen and stored at 80°C. The activities of the mitochondria complexes were determined by the Micro Mitochondrial Respiratory Chain Complexes Assay Kit (Cat. #: BC0515, BC3235, BC3245, BC0945, BC1445, Solarbio, China) according to the manufacturer's instructions. In brief, mitochondria were isolated, and the homogenates were added into the reaction buffer. The activity of complex I‐V were determined at 340, 605, 550, 550 and 660 nm using a spectrophotometer, respectively. The enzyme activity displayed as nmol/min/mg protein.
2.5. Electron microscopy
Small (approximately 1 mm3) and fresh brain tissues were fixed by performing cardiac perfusion with 2.5% glutaraldehyde and 4% paraformaldehyde. DA neurons were fixed with 2.5% glutaraldehyde. After being fixed with osmium acid and dehydrated with acetone, the samples were embedded in acetone and resin. Ultrathin sections (70‐μm thick) were prepared and observed under an electron microscope (JEM‐1400, JEOL, Ltd, Tokyo, Japan). Abnormal mitochondria were identified based on the abnormal structure of the cristae.
2.6. Measurement of mitochondrial membrane potential
The JC‐1 mitochondrial membrane potential assay kit (Beyotime Biotechnology, China) was used to measure the Δψm of the mouse brain. Briefly, after isolation of fresh mitochondria from tissues with the Tissue Mitochondria Isolation Kit (Beyotime Biotechnology, China) according to the manufacturer's instruction, mitochondria were mixed with JC‐1 staining solution. The excitation and emission wavelengths of JC‐1 monomers were 490 and 530 nm, respectively, and those of JC‐1 aggregates were 525 and 590 nm, respectively. All fluorescence intensities were measured by using a microplate reader (Molecular Devices SpectraMax M5e, Molecular Devices, USA). The Δψm was calculated as the red: green fluorescence ratio.
2.7. Detection of ATP levels in brain tissues
ATP levels in brain tissues of mice were measured by using a firefly luciferase‐based ATP assay kit (Beyotime Biotechnology, China). Briefly, brain tissues were homogenised and centrifuged at 12,000 × g at 4°C for 5 min. Next, 20 μl of each sample was mixed with 100 μl of ATP‐detection working dilution. Luminance (in RLU) was measured by using a microplate reader (Molecular Devices SpectraMax M5e, Molecular Devices, USA). The protein concentrations of each group were determined by using the BCA Protein Assay (Solarbio, China). Total ATP content is expressed as μmol/mg protein.
2.8. ROS detection
ROS production was evaluated via dihydroethidium (DHE) staining, as previously reported [14]. Frozen sections (15‐μm thick) of fresh brain tissue were incubated with 5 μM DHE in PBS for 30 min at 37°C. After addition of a DAPI dye solution and anti‐fluorescence quencher, the sections were imaged with an LSM 880 confocal microscope (Carl Zeiss AG, Germany). DAPI‐positive cell nuclei appear blue upon ultraviolet excitation, and DHE‐positive cells were red.
2.9. Assessment of mitochondrial DNA (mtDNA) copy number and mtDNA deletion
The genomic DNA of mouse brains were extracted by the Animal Tissues/Cells Genomic DNA Extraction Kit (Cat. #: D1700, Solarbio). The total mtDNA copy number was determined by performing real‐time quantitative PCR. The primers used are listed in Table S2. Reaction volumes of 20 μl were inserted in QuantStudio 5 PCR system (Thermo Fisher Scientific, USA). The ratio of mtDNA amplicon to GAPDH amplicon was used as a measure of mtDNA content in each group. A melting curve was used to evaluate the specificity of each reaction. mtDNA deletions were detected by performing long‐extension PCR with total DNA in an Applied Biosystems PCR system (Thermo Fisher Scientific, USA) to generate full‐length mtDNA (16.3 kb) and mtDNA with deletions. The primers used are listed in Table S2. PCR products were then loaded on a 0.8% agarose gel and electrophoresed for 4 h at 130 V.
2.10. Proteomic analysis
After termination, the SN and striatum (STR) of WT and HET mice (n = 3 per group) were dissected and snap‐frozen in liquid nitrogen. After the sample was lysed by using ultrasound, it was digested with trypsin. The digested peptides were desalted with Strata‐X C18 (Phenomenex, Inc., Torrance, CA, USA), freeze‐dried in vacuo, and labelled with tandem mass tags. The peptides were fractionated by using high‐pH reversed‐phase HPLC and a ZORBAX 300Extend C18 column (5‐μm particle size, 4.6‐mm inner diameter, 250‐mm length; Agilent Technologies Inc., Santa Clara, CA, USA). After dissolving the peptides with mobile phase A during liquid chromatography, an EASY‐nLC 1000 liquid chromatograph (Thermo Fisher Scientific, USA) was used for separation. The peptides were separated by using ultra‐high‐performance liquid chromatography, injected into a nanospray ionisation source (Thermo Fisher Scientific, USA), and analysed by using a Q Exactive Plus Hybrid Quadrupole‐Orbitrap mass spectrometer (Thermo Fisher Scientific, USA). Data were acquired with a data‐dependent acquisition scan program. Secondary mass spectrometry data were retrieved by using the MaxQuant software package version 1.5.2.8.S.
2.11. Elisa
Blood was collected from the tail vein of each mouse and centrifuged to separate the serum. ELISA was performed according to the manufacturer's instructions. The concentrations of insulin and IDE in the serum were measured by using kits from Crystal Chem (Cat. #: 90080; Elk Grove Village, IL, USA) and Cloud‐Clone Corp. (Cat. #: SEB897Ra; Wuhan, China), respectively.
2.12. Real‐time quantitative PCR
Total RNA was extracted by using TRIzol solution (Cat. #: 15596–026, Ambion Inc., Austin, TX, USA). cDNA was synthesized by adding 1 μg of RNA to a HiScript III RT SuperMix (Cat. #: R323‐01, Vazyme Biotech Co., Ltd, Nanjing, China). Three biological replicates were analysed in each experiment. Real‐time quantitative PCR reactions were performed by using Taq Pro Universal SYBR qPCR Master Mix (Cat. #: Q712, Vazyme Biotech Co., Ltd) in a QuantStudio 5 PCR system (Thermo Fisher Scientific, USA). The data were analysed by using QuantStudio Design and Analysis Software (Thermo Fisher Scientific, USA). The primers used are listed in Table S2.
2.13. Generation of DA neuronal lines
This part of the study was approved by the medical research ethics committee of the First Affiliated Hospital of Zhengzhou University. We have obtained all individuals' free prior informed consents. Small pieces of skin were taken from the subjects' ankles and induced to fibroblasts in a high‐sugar medium containing 20% serum. Thereafter, the mutant iPSC line and the familial normal control iPSC line in the CHCHD2 p.T61I mutant pedigree were induced from fibroblasts via lentivirus infection. The Dopaminergic Progenitor Differentiation Kit (Cat. #: RP01017, Nuwacell Biotechnology Co., Ltd, Anhui, China) was used to induce midbrain DA progenitors from the two iPSC lines (passages 20–30). From the midbrain DA progenitors, DA neuronal lines were matured by using the Dopaminergic Neuron Maturation and Differentiation Kit (Cat. #: RP01017‐I, Nuwacell Biotechnology Co., Ltd).
2.14. Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 30 min and blocked with 0.3% BSA with 3% Triton X‐100 for 60 min. The cells were incubated overnight at 4°C with primary antibodies diluted in 0.3% BSA. Appropriate secondary antibodies were used with DAPI nuclear counterstain (Thermo Fisher Scientific, USA). A comprehensive list of antibodies and their sources is provided in Table S1. Fluorescence signals were captured by using an LSM 880 confocal microscope (Carl Zeiss AG). The percentage of TH‐positive cells in each group was determined in samples derived from three independent experiments. Images were selected for quantification via uniform random sampling. ImageJ software was used for image quantification.
2.15. Seahorse‐based analysis
Midbrain DA progenitors of the two cell lines were planted overnight at a density of 2 × 104 cells per well in 96‐well XF Cell Culture Microplates pre‐coated with Matrigel Matrix (Cat. #: 354277, Corning Inc., Corning, NY, USA). Mitochondrial oxygen consumption was measured with the XF Cell Mito Stress Test Kit (Cat. #: 103015–100, Agilent Technologies Inc.). The sensor cartridge was calibrated for further processing. Cells were washed twice with and incubated in Assay Medium (10 mM pyruvate, 10 mM glucose, and 10 mM Glutamine Agilent Seahorse XF DMEM Medium; Cat. # 103575–100; Agilent Technologies Inc.) in a 37°C non‐CO2 incubator for 1 h. Oligomycin (1.5 μM), carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (0.5 μM), and rotenone/antimycin A (0.5 μM) were added to ports A, B, and C, respectively. The OCR were measured in each well by using an Agilent Seahorse XFe Controller (Agilent Technologies Inc.). We monitored the basal OCR and the OCR following injection of oligomycin, carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone, and antimycin A. We calculated basal, ATP‐linked, proton leak (non‐ATP linked), maximal, reserve capacity, and non‐mitochondrial OCR. Data were expressed as the OCR in pmol/min, normalized to cell protein in individual wells determined by the BCA method.
2.16. siRNA transfection and RNA‐seq analyses
For the IDE knockdown experiment, an siRNA targeting IDE or a control siRNA were transfected into SHSY5Y using RNAiMAX (Thermo Fisher Scientific, USA). Transfected cells were harvested 36 h after transfection and total RNA was isolated using TRIzol Reagent (Cat. #: 15596026, Invitrogen). After DNA digestion by DNaseI, RNA quality and integrity were determined by NanodropTM OneCspectrophotometer (Thermo Fisher Scientific, USA) and 1.5% agarose gel electrophoresis. Qualified RNAs were finally quantified by Qubit3.0 with QubitTM RNA Broad Range Assay kit (Life Technologies, Q10210). Stranded RNA‐seq library were generated using KC‐DigitalTM Stranded mRNA Library Prep Kit for Illumina® (Catalog NO. DR08502, Wuhan Seqhealth Co., Ltd. China) and sequenced on Novaseq 6000 sequencer (Illumina) with PE150 model. Duplicate consensus sequences were used for standard RNA‐seq analysis, which were mapped to the mouse reference genome using STAR software (version 2.5.3a) with default settings. Reads mapped to the exon region of each gene were counted by featureCounts (Subread‐1.5.1; Bioconductor) and the RPKM was calculated. Differentially expressed genes between two groups were identified using the edgeR package (version 3.12.1). A p‐value threshold of 0.05 and a Fold‐change threshold of 2 were used to determine the statistical significance of gene expression differences. Kyoto Encyclopaedia of Genes and Genomes (KEGG) enrichment analysis are constructed with the KOBAS software (version: 2.1.1).
2.17. Statistical analysis
Data are presented as the mean ± standard error of the mean and were analysed by using IBM SPSS Statistics 21.0 (IBM, Armonk, NY, USA). The sample size for each experiment is indicated in the figure legends. Kaplan–Meier survival curves were compared using the log‐rank test. Three‐sample comparisons were performed by using the one‐way ANOVA, followed by least significant difference post‐hoc testing or Tamhane T2 testing. Two‐sample comparisons were performed by Student's t‐test with Benjamini–Hochberg correction. Statistical significance was set at p < 0.05. Graphs were created with GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA). In all figures, *, **, and *** correspond to p < 0.05, 0.01, and 0.001, respectively, for HET compared to WT; #, ##, and ### correspond to p < 0.05, 0.01, and 0.001, respectively, for HOM compared to WT; ns denotes a non‐significant result (p ≥ 0.05).
3. RESULTS
3.1. CHCHD2 p.T61I mutant mice manifested a decreased life span and body weight
To generate a novel CHCHD2 p.T61I KI mice model, we constructed a targeting vector which was assembled into the target allele (Figure (S1)). A cytosine‐to‐thymine mutation was introduced in exon 2 of the mouse gene, which changed the codon for threonine to that for isoleucine (Figure (S1)). CHCHD2 p.T61I KI mice were born at Mendelian ratio, but KI mice had a shorter lifespan than the WT group (Figure 1(A)). A decreased body weight in CHCHD2 p.T61I KI mice than WT mice was noticed starting at approximately 44 weeks of age (Figure 1(B)). CHCHD2 p.T61I mutant mice exhibited whitening and loss of hair, whereas WT mice had black hair (data not shown).
FIGURE 1.
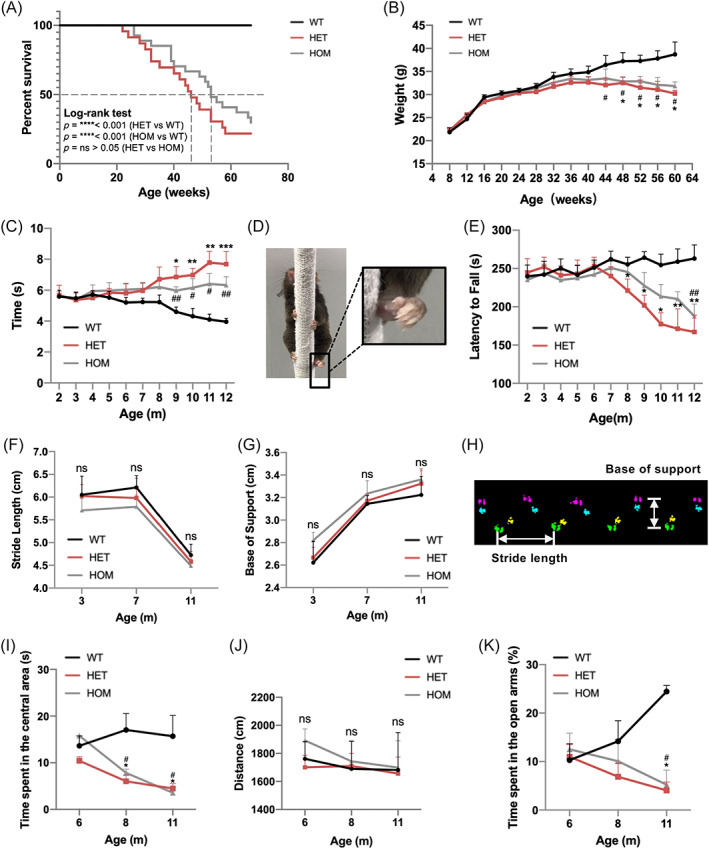
CHCHD2 p.T61I mutation results in age‐dependent motor deficits and high anxiety level. (A) Kaplan–Meier analysis of survival of mice with the indicated genotypes. Mutant mice had a shorter lifespan than WT mice. Statistical analysis was performed by using the log‐rank (Mantel–Cox) test; WT: n = 20; HET: n = 23; HOM: n = 25. (B) Body‐weight curves of mice with the indicated genotypes at different ages. n = 4–34 per genotype (mice populations dwindled with age, and the numbers of mice in the WT, HET and HOM groups were comparable in each age group). (C) The time spent on the pole climbing test. The time was recorded from 2‐ to 12‐month‐old mice. WT: n = 6–13; HET: n = 6–13; HOM: n = 6–14, and the numbers of mice in the WT, HET, and HOM groups in each age group were comparable. (D) CHCHD2 p.T61I mutant mice exhibited poor hind‐paw pole grasping (magnified images). (E) Exercise endurance measured using latency to fall during the accelerating rotarod test; WT: n = 6–15; HET: n = 6–16; HOM: n = 6–18, and the numbers of mice in the WT, HET, and HOM groups in each age group were comparable. (F–H) Gait analysis of mice measured using stride length of the right hind paws (F) and base of support of the hind paws (G). n = 6–8 per genotype. (H) Graphical representation of gait parameters with paw locations indicated. (I–J) Time spent in the centre field (I) and the total distance travelled (J) in the OF test; n = 6–8 per genotype. (K) Time spent on the open arms in the EPM test; n = 6–9 per genotype. Data are indicated as the mean ± SEM. One‐way ANOVA was performed, followed by LSD post‐hoc testing and Tamhane T2 testing. *, **, and *** indicate p < 0.05, 0.01, and 0.001, respectively, for HET compared to WT; # and ## correspond to p < 0.05 and 0.01, respectively, for HOM compared to WT.
3.2. Motor and anxiety phenotypes in CHCHD2 p.T61I mutant mice
We examined the general locomotion level and exercise endurance ability of the mice beginning at 2 months of age. The mutant mice took a longer time to descend the pole in the pole test than WT mice from 9 months of age (Figure 1(C)). Aberrant paw control was observed in CHCHD2 p.T61I mice, characterised by paw clenching (Figure 1(D)). CHCHD2 p.T61I mice also exhibited a shorter latency on the rotarod wheel than WT mice, suggesting an exercise intolerance of CHCHD2 p.T61I mice (Figure 1E). There were no statistical differences in stride length or the base of support (Figure 1(F–H)) between WT and mutant mice. As patients with PD also have non‐motor symptoms, we tested whether there were age‐dependent changes in anxiety‐like behaviour of mutant mice. In the open field test (OF), HET and HOM mice started to spend less time in the centre of the field than WT mice at 8 months of age (Figure 1(I)), indicating an anxiety‐like behaviour. There was no difference among genotypes in the distances travelled by mice (Figure 1(J)), suggesting similar spontaneous exploring behaviour. In the elevated plus maze (EPM), p.T61I mice spent less time in the open arms at 11 months of age (Figure 1(K)), consistent with the aberrant pattern of exploration in the OF test. These results suggested that, in addition to lower exercise endurance and motor coordination defect, CHCHD2 p.T61I mice also suffered from an anxiety phenotype.
3.3. CHCHD2 p.T61I mutation contributes to neuropathological alterations in mice
Pathological studies of 11‐month‐old mutant mice revealed the loss of TH‐positive DA neurons in the SNpc compared with age‐matched WT controls, which is a hallmark of PD (Figure 2(A)). CHCHD2 p.T61I mice do not exhibit obvious DA neurodegeneration in the VTA (Figure 2(A)). Consistently, a reduction in TH‐positive DA nerve terminals in STR was also found in 11‐month‐old p.T61I mutant mice (Figure 2(A)). α‐synuclein and p‐α‐synuclein accumulated and aggregated in the cerebral cortex of 11‐month‐old p.T61I mutant mice compared with WT mice (Figure 2(B)). p‐α‐synuclein depositions were also found in the SN DA neurons of 11‐month‐old mutant mice (Figure 2(C)). We did not detect obvious α‐synuclein and p‐α‐synuclein depositions in the hippocampus and STR of the mutant mice (Figure (S2)). Levels of DA and its metabolites DOPAC and HVA in the STR of 11‐month‐old mice were measured by HPLC (Figure 2(D–G)). Consistent with impaired motor function, levels of DA were decreased in p.T61I mutant mice compared with WT group (Figure 2(D)). No significantly differences in DA turnover rate [(DOPAC+HVA)/DA] or the levels of DOPAC and HVA were noted between the three genotypes (Figure 2(E–G)). Given that the mutant mice showed motor deficits, we sought to determine if the mutant mice at 11 months of age develop alterations in motor neurons of the spinal cords. The numbers of large Nissl‐stained cells representative of motor neurons in the lumbar spinal cord of the mutant mice were comparable in different groups (Figure (S2)). The analysis of respiratory chain activities confirmed the impairment of complex II and III in the spinal cords of mutant mice (Figure 2(H)). The activities of the complex I, IV and V in the spinal cords of the mutant mice were not affected (Figure (S2)).
FIGURE 2.
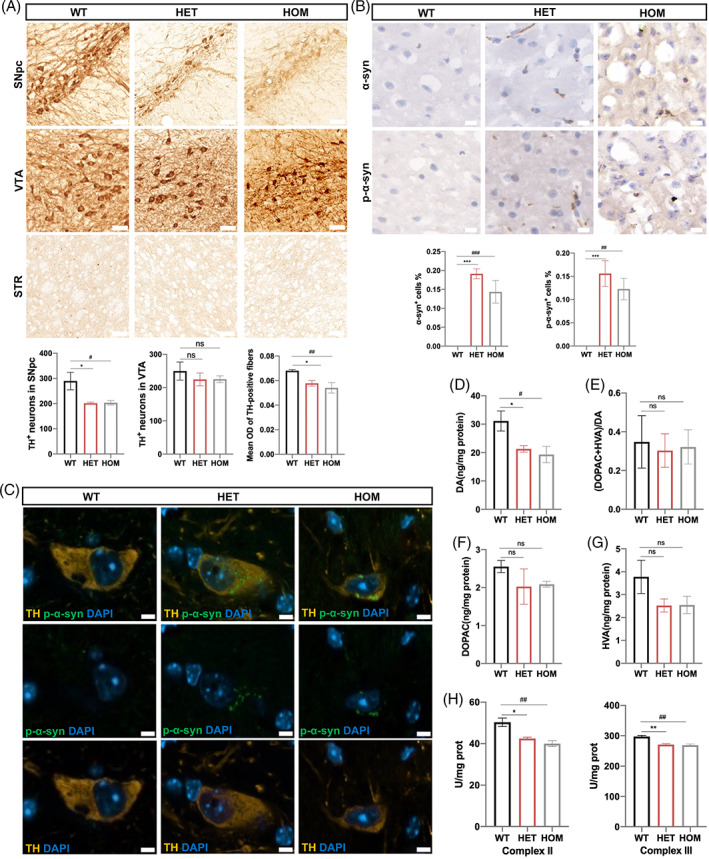
Histopathological abnormalities in CHCHD2 p.T61I mutant mice. (A) Histological analysis of TH+ DA neurons in the SNpc and VTA, and DA nerve terminals in STR of 11‐month‐old mice (n = 5–7 per genotype). Scale bars: 50 μm. (B) Cryosections of the cerebral cortex of mice at 11 months of age were immunostained for α‐synuclein and p‐α‐synuclein (n = 3 per genotype). Scale bars: 10 μm. (C) Confocal microscopy demonstrated p‐α‐synuclein (green) aggregated in the cytoplasm of TH+ DA neuron (orange) in the SN of p.T61I mutant mice (n = 3 per genotype). Scale bars: 5 μm. (D–G) concentrations of DA (D), DA turnover rate (DOPAC+HVA/DA) (E), DOPAC (F), and HVA (G) in p.T61I mice compared with WT mice at age of 11 months (n = 3 per genotype). (H) Analysis of the activity of respiratory chain complex II and III in lumbar spinal cord of mice. (n = 3 per genotype). Data are indicated as the mean ± SEM. One‐way ANOVA was performed, followed by LSD post‐hoc testing and Tamhane T2 testing. *, **, and *** indicate p < 0.05, 0.01, and 0.001, respectively, for HET compared to WT; # and ## correspond to p < 0.05 and 0.01, respectively, for HOM compared to WT.
3.4. CHCHD2 and CHCHD10 aggregated in the cytoplasm of p.T61I mutant brain
There were trends towards decreased levels of CHCHD2 in the SN or STR of 11‐month‐old mutant mice, but it did not reach significance (Figure 3(A)), consistent with what we observed in the fibroblasts of a CHCHD2 p.T61I mutant patient [15]. CHCHD10 is a homologue of CHCHD2 and is reportedly involved in the development of PD caused by the CHCHD2 p.T61I mutation via interaction with CHCHD2 [15]. The expression patterns of CHCHD10 in the SN and STR of 11‐month‐old mice were also not affected by the mutation (Figure 3(A)). However, we observed the aggregations of CHCHD2 and CHCHD10 in the SN of the mutant mice (Figure 3(B)). Previous studies have shown that CHCHD10 p.S55L mutant mice exhibited increased CHCHD10 and CHCHD2 immunoreactivity in midbrain and spinal cord and developed severe cardiomyopathy and myopathy [16, 17], which led us to wonder if CHCHD2 mutant mice also had muscle damage. We found the myofibers of the skeletal muscles of CHCHD2 p.T61I mice showed slight increased nuclear density (Figure 3(C)). The myocardium was not notably compromised in the CHCHD2 p.T61I mutant mice (Figure (S2)), which suggested that the pathogenic mechanisms of CHCHD2 and CHCHD10 may be different, although they are homologues.
FIGURE 3.
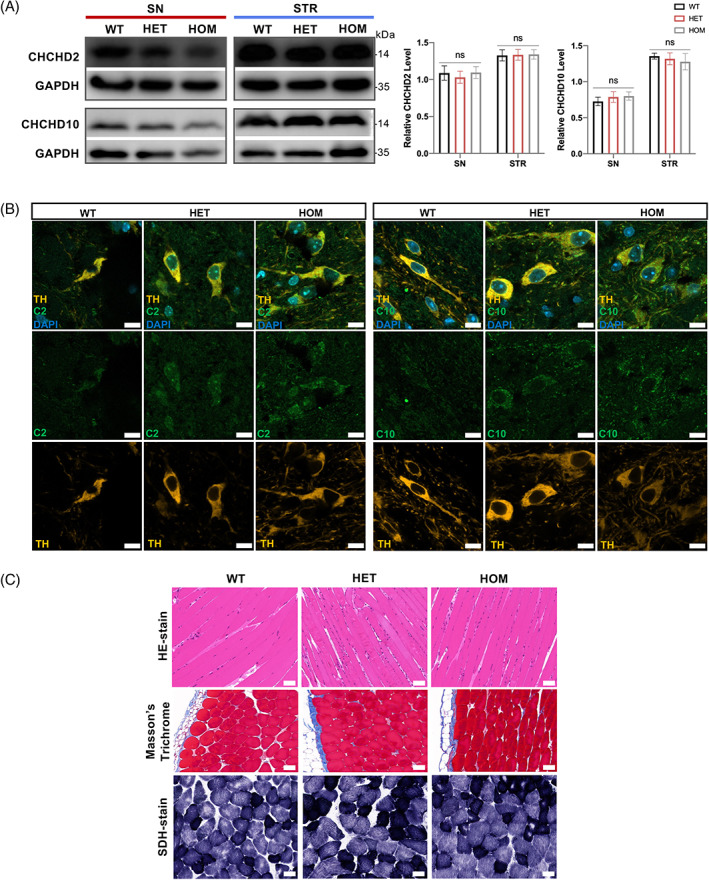
CHCHD2 and CHCHD10 aggregated in the SN of p.T61I mice. (A) Western blot analysis of brain protein homogenates with CHCHD2, CHCHD10, and GAPDH (loading control) antibodies and quantification of CHCHD2 and CHCHD10 protein levels (n = 5 per genotype). (B) Confocal microscopy demonstrated CHCHD2 (green) and CHCHD10 (green) aggregated in the cytoplasm of TH+ DA neuron (orange) in the SN of p.T61I mutant mice (n = 3 per genotype). Scale bars: 10 μm. (C) HE‐staining, Masson's trichrome, and SDH staining of the skeletal muscles (n = 5 per genotype). Scale bars: 50 μm. Data are indicated as the mean ± SEM. One‐way ANOVA was performed, followed by LSD post‐hoc testing. ns, not significant. [Correction added on 28 April 2023, after first online publication: Figure 3 has been replaced.]
3.5. Impaired mitochondrial structure and function in CHCHD2 p.T61I mutant mice
For a better understanding of the CHCHD2‐mediated PD phenotypes, we investigated mitochondrial biology in detail in 11‐month‐old mice. Initially, we performed ultrastructural analyses using electron microscopy. A larger number of mitochondria with irregular inner structures were observed in the SN and STR of CHCHD2 p.T61I mutant mice compared with those in WT mice, characterised by matrix swelling, the appearance of vacuoles, disorganisation, and the loss of cristae (Figure 4(A)). The quantification analysis revealed no differences on mitochondrial length, aspect ratio (long / short axis), and area between 11‐month‐old mutant and WT mice (Figure 4(A)). Functional changes associated with phenotypic abnormalities in mitochondria were measured with the mitochondrial membrane potential (Δψm). We observed a lower red: green fluorescence ratio in the SN and STR of mutant mice than in those of WT mice, suggesting the depolarisation of mitochondria and a lower Δψm (Figure 4(B)). A lower ATP content was detected in the STR of mutant mice than in that of WT mice (Figure 4(C)). To evaluate ROS production, we stained brain slices with DHE, which is the most specific and least problematic dye for detection of superoxide radicals. We found a higher number of DHE‐positive cells in the STR of mutant mice than WT mice (Figure 4(D)).
FIGURE 4.
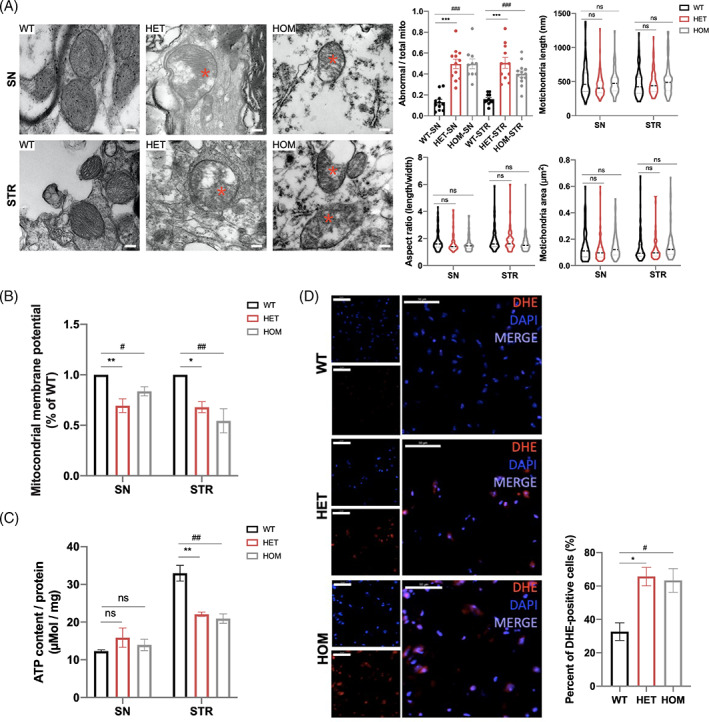
Impaired morphology and function of mitochondria in the brains of CHCHD2 p.T61I mutant mice. (A) Electron micrographs taken in the SN and STR of 11‐month‐old mice and quantification of abnormal mitochondria (% of total mitochondria), mitochondrial length, aspect ratio, and mitochondria area (80–100 mitochondria from three mice per group). Abnormal cristae and vacuolization were visible in CHCHD2 p.T61I mutant mice. The asterisks highlight the abnormal structures in the mitochondria. Scale bars: 200 nm. (B) Mitochondrial membrane potentials of the SN and STR of 11‐month‐old mice were determined by using the JC‐1 assay (n = 3 per genotype). (C) ATP concentrations in the SN and STR of 11‐month‐old mice (n = 3 per genotype). (D) DHE staining or ROS positive cells in the STR of 11‐month‐old mice (n = 3 per genotype). Scale bars = 50 μm. Data are indicated as the mean ± SEM. One‐way ANOVA was performed, followed by LSD post‐hoc testing. *, **, and *** indicate p < 0.05, 0.01 and 0.001, respectively, for HET compared to WT; #, ##, and ### correspond to p < 0.05, 0.01 and 0.001, respectively, for HOM compared to WT. ns, not significant.
The expressions of OPA1 [18], Mfn2, and Drp1, which modulate the fusion and fission of the mitochondrial membranes [19] did not differ among the three groups (Figure (S3)). Some electron transport chain protein complexes in the brain of mutant mice remained unchanged compared to those in WT mice (Figure (S3)). CHCHD2 p.T61I mutation did not affect mtDNA copy number (Figure (S3)) and mtDNA deletion (Figure (S3)) in the mouse brain. VDAC, a protein regulating mitochondrial biogenesis, also showed no difference on expressions between WT and mutant mice (Figure (S3)).
3.6. Proteome identified certain novel substrates of CHCHD2‐dependent regulation
The CHCHD2 p.T61I mutation may regulate proteins critical to brain function. We attempted to gain a higher‐level understanding of cellular processes affected by the CHCHD2 mutation by identifying differentially expressed proteins in the SN and STR lysates of WT and HET mice at 11 months of age. We identified 71 upregulated and 41 downregulated proteins in the STR (Figure 5(A); Figure (S4); Table S3); 30 upregulated and 19 downregulated proteins in the SN (Figure 5(B); Figure (S4); Table S3) in HET mice relative to WT mice. For functional annotation analysis, Eukaryotic Orthologous Groups (KOG) functional classification revealed that the “posttranslational modification, protein turnover, chaperones” was among the largest clusters in both the SN and STR (Figure (S4)). Interestingly, a mitochondrial protein in this group, IDE, and was expressed differentially in both SN and STR (Figure 5(A,B)). And a subunit of the respiratory chain complex I (NDUFB3), PD‐associated proteins (PRKACA, TRAP1), AD‐associated protein (ARF6), and autophagy‐related proteins (WDFY1, OPTN) were also differently expressed (Figure 5(A,B)).
FIGURE 5.
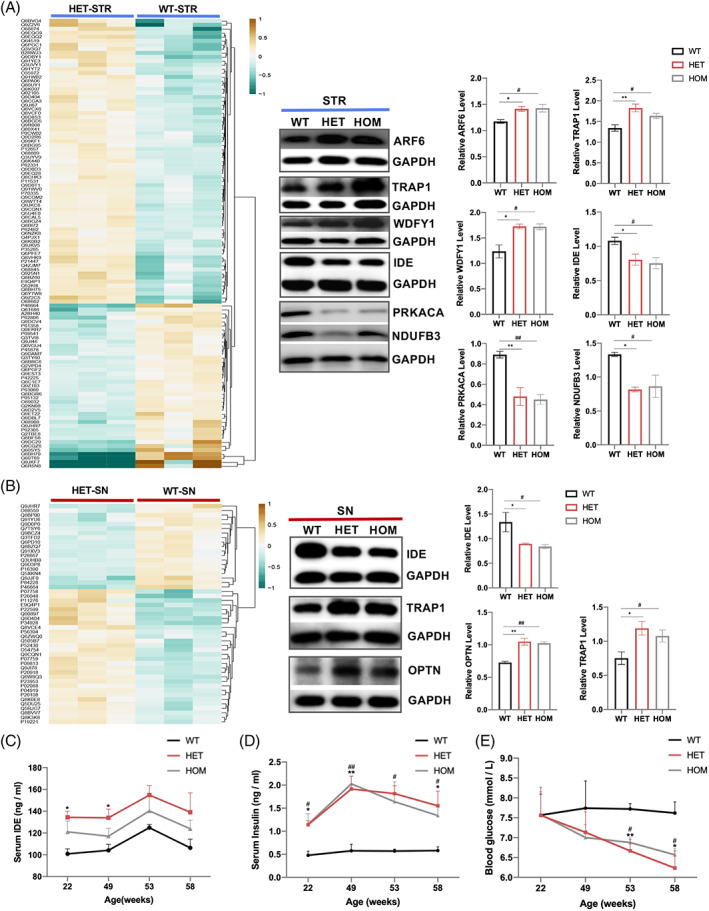
Proteome of the SN and STR of WT and HET 11‐month‐old mice. (A‐B) Heat maps (left) of STR (upper graph) and SN (bottom graph) proteins. Quantification of intensities (right) of selected proteins in the STR (upper graph) and SN (bottom graph) in mice with the indicated genotypes (n = 3 animals per tissue per genotype). (C) Average serum IDE levels of mice (n = 3–7 per genotype). (D) Average serum insulin levels in mice (n = 3–7 per genotype). (E) Average glucose levels of mice (n = 5–7 per genotype). Data are indicated as the mean ± SEM. One‐way ANOVA was performed, followed by LSD post‐hoc testing. * and ** correspond to p < 0.05 and 0.01, respectively, for HET compared to WT; # and ## correspond to p < 0.05 and 0.01, respectively, for HOM compared to WT.
IDE, especially, a chaperones‐like protein, is reportedly closely associated with AD and PD [20]. Interestingly, in contrast with its downregulation in the brain of mutant mice, plasma IDE was upregulated in mutant mice (Figure 5(C)), similar with that in patients with sporadic PD in our study (data not shown). However, the levels of serum insulin (Figure 5(D)) were higher and blood glucose levels (Figure 5(E)) were lower in mutant mice than those in WT mice. These results imply a difference in metabolic profiles of IDE between the central and peripheral nervous systems, which deserves deeper research. Clearance of circulating insulin occurs primarily in the liver and is initiated and restricted by insulin receptor (IR)‐mediated internalization. Given this functional role, reduced hepatic IR levels may lead to abnormal insulin levels. However, the expression levels of IR in the liver did not differ between WT and mutant mice (Figure (S5)). IDE has certain non‐proteolytic functions, such as chaperoning and modulating the ubiquitin‐proteasome system (UPS) as a heat shock protein, which may affect the deposition of α‐synuclein and p‐α‐synuclein. Accumulation of misfolded proteins can trigger the mitochondrial unfolded protein response (mtUPR), an adaptive transcriptional program that promotes mitochondrial recovery [21]. Prominent targets upregulated by the mtUPR include mitochondrial matrix chaperones, such as HSP60. Mitochondrial damage can also induce mitophagy which removes dysfunctional mitochondria to prevent apoptosis [22]. Nevertheless, the p.T61I mutation did not induce a stronger autophagy, apoptosis or mtUPR (Figure (S5)), as what were observed in the CHCHD2 p.T61I cell model [23], indicating that IDE may be involved in PD via other pathways.
3.7. Patient‐specific iPSCs‐derived DA neurons with CHCHD2 p.T61I mutation reproduced mitochondrial impairment and IDE downregulation
Differences in neurodegenerative phenotypes among species complicate the extrapolation of results from animal models to humans. The discovery of iPSCs enabled the replication of DA neurons from patients with PD. We generated iPSC cell lines from a CHCHD2 p.T61I mutant patient [24] and a healthy control in that patient's pedigree. DA neuronal lines were induced from those cells to investigate the pathogenic mechanism of the CHCHD2 p.T61I mutation. On day 35 of differentiation, cells began to express tyrosine hydroxylase (TH). DA neurons from all lines demonstrated co‐expression of βIII‐tubulin, a neuronal marker. The mutant cell line exhibited lower TH positive expression than control cell line, with similar βIII‐tubulin expression, which suggested a lower DA neuronal differentiation efficiency of CHCHD2 p.T61I mutant DA neuronal line (Figure 6(A)). We also observed swollen mitochondrial cristae (Figure 6(B)) in the CHCHD2 p.T61I mutant DA neuronal line. We attempted to further determine the impact of the CHCHD2 p.T61I mutation on mitochondrial function by investigating mitochondrial respiration via Seahorse analysis (Figure 6(C)). Although spare respiration capacity (Figure 6(G)) did not statistically differ between the cell lines, the basal oxygen consumption rate (OCR) (Figure 6(D)), maximal OCR (Figure 6(E)), and ATP production (Figure 6(F)) were statistically significantly lower in mutant cell line than in control cell line. In addition, we observed no statistically significant difference in CHCHD2 protein expression between the cell lines (Figure 6(H)). The IDE protein expression was lower in mutant cells than in control cells (Figure 6(H)), consistent with CHCHD2 p.T61I mice.
FIGURE 6.
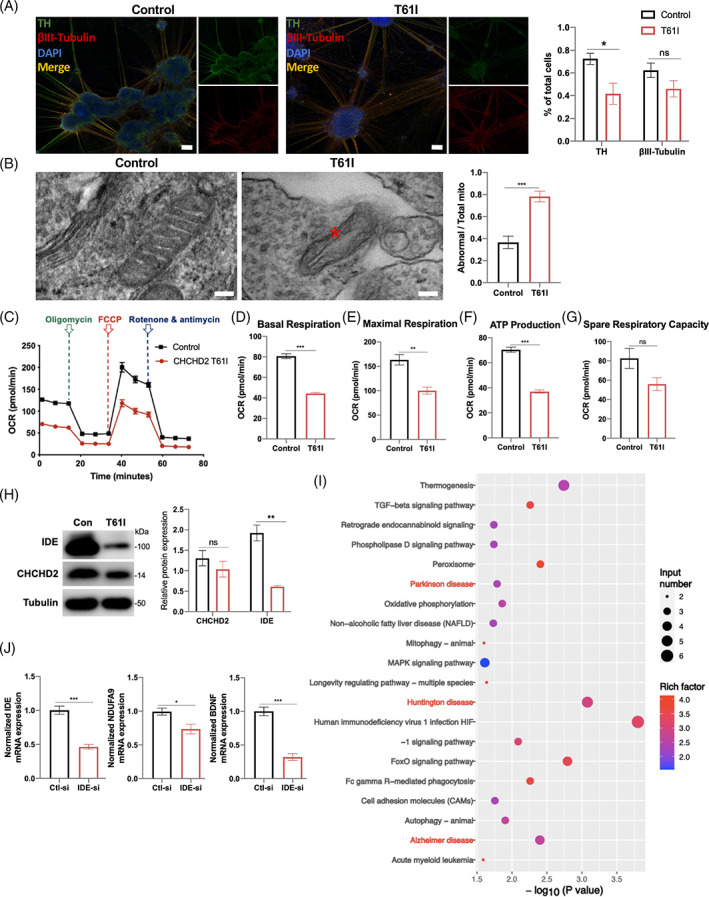
The effect of the CHCHD2 p.T61I mutation on iPSCs‐derived DA neurons. (A) Confocal images of iPSCs‐derived DA neurons with antibodies against TH (green) and βIII‐tubulin (red), and quantification of the numbers of TH+ and βIII‐tubulin + neurons. Scale bars:100 μm. (B) Electron micrographs taken in the DA neurons and quantification of abnormal mitochondria. The asterisk highlights the abnormal structure in the mitochondria. Scale bars: 100 nm. (C) Mitochondrial OCR in control and CHCHD2 p.T61I mutant DA progenitors (mDAPs) were measured by using Seahorse‐based analysis (n = 4–5 per genotype). (D–G) Basal respiration (D), maximal respiration (E), ATP production (F), and spare respiration capacity of cell lines (G) (n = 4–5 per genotype). (H) Representative western blot image of proteins in the DA neurons and quantification of intensities of proteins, relative to β‐tubulin expression. (I) KEGG pathway analysis of down‐regulated genes in IDE‐siRNA group. (J) Differentially expressed genes in IDE‐siRNA group involved in HD, AD, and PD KEGG pathways. Data are indicated as the mean ± SEM of three independent experiments. Student's t‐test with Benjamini–Hochberg correction were performed. *, **, and *** correspond to p < 0.05, 0.01, and 0.001, respectively. ns, not significant.
3.8. IDE‐knockdown impaired mitochondrial complex I
To gain a higher‐level understanding of the cellular processes affected by IDE in human cells line, we performed RNA‐seq of SHSY5Y cells transfected with IDE‐siRNA and control‐siRNA. The transcriptomic profiles of IDE‐siRNA group showed 419 significantly altered genes relative to control‐siRNA group (Table S4). Many of the significantly down‐regulated genes participated in the Huntington disease (HD), AD and PD KEGG pathways (Figure 6(I)), including IDE, BDNF and NDUFA9 (Figure 6(J)). Interestingly, NDUFA9, a subunit of the respiratory chain complex I, was repressed in all three pathways, similar with what we found in the brain of the mutant mice. These results demonstrated that IDE may cooperate with CHCHD2, causing the damage of mitochondrial complex I.
4. DISCUSSION
The CHCHD2 p.T61I mutation has been reported as a causative factor for PD in Japanese and Chinese Han individuals [6, 7], although the pathomechanism remains unclear. Genetic models are compelling approaches to examine the association of specific mutations in genetic diseases. Various CHCHD2 mutation‐related PD models have been generated to investigate the pathogenesis of PD, such as the CHCHD2‐KO mouse model, Drosophila models, and Caenorhabditis elegans models. Few existing mouse models of CHCHD2‐related PD completely reproduce the features of PD [25], and no transgenic or KI mouse models of CHCHD2 mutations have been reported. Here, we report a novel CHCHD2 p.T61I KI mouse model that recapitulates the cardinal features of human CHCHD2 p.T61I mutation‐related PD, providing a potentially useful way to unravel the pathophysiology of PD.
How does the CHCHD2 p.T61I mutation drive the pathogenesis of PD? A possible explanation is that the mutation destabilises CHCHD2 protein, effectively nullifying its function. However, we did not observe any statistically significant differences of CHCHD2 protein in the SN and STR of CHCHD2 p.T61I mutant mice and DA neuronal lines, as previously observed in fibroblasts isolated from patients with the CHCHD2 p.T61I mutation [15]. However, the aggregations of CHCHD2 and CHCHD10 were observed in the SN of CHCHD2 p.T61I mutant mice, similar with CHCHD2/CHCHD10‐KO cell lines and CHCHD2 p.T61I human cell line [23, 26]. CHCHD2 p.T61I KI mice exhibited a shorter life span and lower body weight, with a deficit in motor capacity and mitochondria, and a loss of DA neurons in the SN at the age of 11 months, compared with WT mice. Similarly, overexpression of human CHCHD2 p.T61I mutation in Drosophila resulted in mitochondrial dysfunction, dopamine neuron loss, and reduced life span [27]. In contrast, neither Sato et al. [12] nor Nguyen et al. [13] observed morbidity or DA neuronal loss by 14 and 30 months of age in CHCHD2‐KO mice, respectively. Although CHCHD2‐KO mice in Sato's study developed a mild motor deficit and a subtle decrease in TH+ neurons at 30 months of age, 50% of both control and KO mice had already died at this extremely advanced age [12]. The unique features of CHCHD2 p.T61I KI mice further support our hypothesis that disease‐causing p.T61I mutation and the total loss of CHCHD2 are not functionally equivalent. Interestingly, CHCHD10‐KO mice also exhibited no morbidity at 1 year of age [28], whereas mice carrying the CHCHD10‐KI mice exhibited disease phenotypes at early age and died at the age of 14 months [16, 17]. We suspected that the CHCHD2 p.T61I mutation may confer a toxic function, similar with that of the CHCHD10 mutation, but there is still a lot of controversy about the pathogenic mechanism of CHCHD2. In C. elegans and Drosophila, the KO of the orthologous of both CHCHD2 and CHCHD10 induced abnormal curling phenotype. WT, but not mutant protein, can rescue these phenotypes, suggesting a loss‐of‐function mutation [29, 30, 31]. However, CHCHD2‐KO mice have no obvious phenotypes [12, 13], suggesting that CHCHD2 mutation do not act as loss‐of‐function in mice, and species differences may be one of the reasons. Interestingly, the HET and HOM mutant mice in our research exhibited phenotypes of similar severity. Combined with previous studies demonstrating misfolding of the CHCHD2 mutant protein and resulting deposition of the wild‐type CHCHD2 [23], we speculate a dominant‐negative effect of mutant CHCHD2 on wild‐type CHCHD2, explaining the dominant inheritance of this form of PD. Further research needs to be supplemented.
CHCHD2 p.T61I KI mice in our research exhibited high anxiety level, with impaired mitochondria. The current diagnostic criteria for PD are based on motor symptoms, but the number of DA neurons in the SN has already been reduced by about half when patients are diagnosed with PD [32]. Non‐motor symptoms often precede motor symptoms and were deemed a clue for identifying prodromal PD, which is an excellent point for therapies. These pre‐motor symptoms included mood disturbances, loss of smell and taste, excessive sweating, fatigue and pain [33, 34] Anxiety symptom also occurs in the early stages of the disease [35, 36, 37]. It was reported that the 6‐hydroxydopamine (6‐OHDA) animal models [38, 39, 40, 41, 42] and the paraquat (PQ) induced rat model [43] of PD reproduced the anxiety and depression symptoms typically observed in PD patients, probably be due to the dopamine lesion of DA neurons [44]. The anxiety is increasingly associated with mitochondrial dysfunction [45, 46, 47]. A Ndufs4‐defecient mice model with suboptimal mitochondrial function showed more anxiety‐related behaviour following being subjected to a chronic unpredictable stress paradigm [48]. Specifically, by promoting cholesterol transport to the inner mitochondrial membrane, the mitochondria critically control the rate‐limiting step in neurosteroidogenesis [49]. Neurosteroids are allosteric modulators of GABA(A) receptor function, a neurotransmitter system well‐known for its crucial involvement in anxiety. Overexpression of the antioxidant catalase targeted to the mitochondria led to reduced anxiety‐related behaviour [50]. The deficiency of uncoupling protein 2, a member of the mitochondrial transporter superfamily, instead, leads to anxiety‐like behaviours [51, 52]. Overall, these studies suggested that perturbation of mitochondrial function in the brain may lead to increased anxiety level. The anxiety phenotype in our mouse model might be of use to study in combination emotional deficits associated to PD.
We observed the accumulation and aggregation of α‐synuclein and p‐α‐synuclein in mutant mouse brains, which was agrees with a case of extensive accumulation of aggregated α‐synuclein in a patient with the CHCHD2 p.T61I mutation and CHCHD2‐KO Drosophila model [10]. CHCHD2 and α‐synuclein co‐localised in LBs in both the brain stem and limbic regions, suggesting an interaction between them [10]. A recent study revealed down‐regulated expression of CHCHD2 in the SN of patients with sporadic PD, and that an increase of α‐synuclein level decreases the CHCHD2 level in mice with the p.A53T mutation [53]. Nevertheless, we did not observe a statistically significantly lower expression of CHCHD2 in the SN and STR of CHCHD2 p.T61I KI mice. Another finding in our study is that the mitochondria in the brain of CHCHD2 p.T61I KI mice and iPSCs‐derived DA neurons exhibited impaired structure and function, similar to those in the CHCHD2‐KO mouse model, potentially further impairing the motor coordination of mutant mice. Mitochondrial dysregulation has long been recognised as a key cause of PD [54] and affects the function of neurons in the central nervous system. The CHCHD2 p.T61I mutation is in the conserved α‐helix domain, which may change the protein's localisation and function in mitochondria [55]. Animal models of PD‐linked mutations have revealed phenotypes of mitochondrial dysfunction, including dysfunction of mitochondrial complex I, disruption of membrane potential, and increased levels of ROS [56]. Proteomic analysis in our study also revealed the downregulation of one member of mitochondrial complex I, NDUFB3, in the STR of p.T61I mutant mice, consistent with what Patricia et al. reported that the disruption of mitochondrial complex I in mouse dopaminergic neurons triggered progressive, human‐like Parkinsonism [57].
Proteomics analysis in present study revealed the possible involvement of IDE, which is closely associated with AD and PD, especially piqued our interest. IDE was down‐regulated in the SN and STR of CHCHD2 p.T61I mutant mice and the patient‐specific, iPSCs‐derived DA neurons. IDE is a regulator of the insulin level, which is reportedly associated with neurodegenerative disease [20], eating behaviour, peripheral metabolism [58], and cognitive function [59]. Epidemiological studies suggest that patients with type 2 diabetes mellitus (T2DM) have a high risk (40%) of developing PD [60]. In contrast, the blood glucose levels of CHCHD2 p.T61I KI mice in our study were lower than those of WT mice, with higher serum insulin and IDE levels, which suggested that IDE may not affect the pathogenesis of CHCHD2 mutation‐related PD through blood glucose. A previous study showed that the accumulation of α‐synuclein was observed in the pancreas of an IDE‐loss mouse model [61]. Interestingly, we also observed α‐synuclein and p‐α‐synuclein deposition in the brain of mutant mice, which raises the question of whether the loss of IDE causes α‐synuclein and p‐α‐synuclein deposition. IDE has proteolytic capacities, degrading proteins such as amyloid‐β peptide in AD [62], islet amyloid polypeptide in T2DM [63], and other substrates [64]. However, IDE reportedly cannot degrade α‐synuclein because of the size limitation of its proteolytic chamber [65]. IDE also has several non‐proteolytic roles, such as chaperoning [66], activating ubiquitin [67], and modulating the UPS as a heat shock protein [68], all of which may affect the depositions of α‐synuclein and p‐α‐synuclein, similar to the function of IDE on blocking aggregation of amyloidogenic peptides [69]. In addition, previous studies demonstrated that IDE–α‐synuclein interactions protected against α‐synuclein formation and stimulated IDE protease activity, and an avenue of research on IDE activity should be pursued in the future. Interestingly, we observed another subunit of the respiratory chain complex I, NDUFA9, was repressed after the knockdown of IDE in SHSY5Y cells, similar with what we observed in the CHCHD2 p.T61I KI mice. Taken together, these results demonstrated the damage of the mitochondria and alteration of insulin metabolism triggered by IDE reduction in CHCHD2 p.T61I mice, which may further cause increased ROS, ATP depletion and cell death.
There are also some limitations to our study. First, since previous studies did not show that the incidence of CHCHD2 p.T61I mutation is related to gender, we did not compare gender differences in the CHCHD2 p.T61I KI mouse model. In addition, some of our data showed that the HET mice developed disease phenotypes earlier than HOM group, which requires more in‐depth research to explain. Furthermore, we did not conduct a more in‐depth study on IDE‐related pathogenic mechanisms of CHCHD2 p.T61I mutation. These shortcomings will be further explored in subsequent studies.
Collectively, we developed a CHCHD2 p.T61I KI mouse model that recapitulated distinct clinical phenotypes of PD, α‐synuclein and p‐α‐synuclein deposition, and impaired mitochondria. Additionally, the downregulated expression of IDE may be involved in the pathomechanism of the damaged mitochondria and DA neuronal pathology in the CHCHD2 p.T61I mice, which may provide a further avenue of research on the pathogenic mechanism and treatment in PD.
AUTHOR CONTRIBUTIONS
Liyuan Fan, Chengyuan Mao and Changhe Shi conceived and designed the study; Liyuan Fan, Xinwei Li, Huimin Zheng and Yun Su perform the experiments; Liyuan Fan, Shuo Zhang drafted the manuscript; Liyuan Fan and Shuo Zhang analysed and interpreted the data; Liyuan Fan and Haiyang Luo acquired the genetic data; Jing Yang, Shuo Zhang, Yu Fan, Huifang Sun, Xinwei Li, Zhongxian Zhang and Jinxin Miao provide experimental guidance and procured critical reagents; Bo Song and Zongping Xia provided experimental technical guidance; Yuming Xu provided funding support; Chengyuan Mao and Yuming Xu supervised the study. All authors have read and approved the final manuscript.
FUNDING STATEMENT
This work was supported by the National Natural Science Foundation of China [Grant U1904207, 81530037, 82271277, 81974211, 81901300]; National Key R&D Program of China [Grant 2017YFA0105003]; Non‐profit Central Research Institute Fund of Chinese Academy of Medical Sciences [2020‐PT310‐01]; Scientific and Technological Project of Henan Province [Grant SBGJ202003020]; Innovative and Scientific and Technological Talents Training Project of Henan Province [Gran YXKC2021062].
CONFLICT OF INTEREST
No authors had conflicts of interests relevant to this study.
Supporting information
Figure S1 Generation of CHCHD2 p.T61I mutant mice. (A–C) A p.T61I (ACC to ATC codon) mutation was introduced into exon 2 in the 3′‐homology arm. The arrow indicates the C to T mutation in the CHCHD2 gene. (D) Pattern diagram of mouse model construction. The ES cells derived from the clones carrying the desired homologous recombination were injected into C57BL/6 mouse embryos. The chimeric offspring were crossed with C57BL/6 mice to obtain a germline. HET KI mice were interbred to obtain HOM KI and WT mice, which were subsequently genotyped with PCR using primers specific for the experiments.
Figure S2 Pathological staining of the brain, spinal cords, and myocardium. (A–B) Cryosections of different brain regions of mice at 11 months of age were immunostained for p‐α‐synuclein and α‐synuclein. (C) HE‐staining of the brain. No LBs are observed in the STR and hippocampus of p.T61I 11‐month‐old mice. Scale bars = 20 μm. (D) Nissl staining of lumbar spinal cords of mice showing large motor neurons (n = 3 per genotype). Scale bars = 20 μm. ns, not significant. (E) Analysis of the activity of respiratory chain complex I, IV and V in lumbar spinal cord of mice. (n = 3 per genotype). Data are indicated as the mean ± SEM. One‐way ANOVA was performed, followed by LSD post‐hoc testing. ns, not significant. (F) HE‐staining and Masson's Trichrome of the myocardium of mice. Scale bars = 50 μm.
Figure S3 Electron transport chain and mtDNA unaffected by the CHCHD2 p.T61I mutation. (A) Representative western blot images of Drp1, Mfn2, and OPA1 proteins in the SN and STR of 11‐month‐old mice, with quantification of protein intensities relative to GAPDH. n = 3 per genotype. (B) Representative western blot image and quantification of OXPHOS proteins in the SN and STR of 11‐month‐old mice. NDUFB8 (complex I), SDHB (complex II), UQCRC2 (complex III), MTCO2 (complex IV), ATP5A (complex V). n = 3 per genotype. (C) Quantification of mtDNA copy numbers in the tissues of 11‐month‐old mice. Data are expressed as the mtDNA: β‐actin concentration ratio. n = 3 per genotype. (D) Long‐extension PCR of mtDNA (16.3‐kb amplicon) from tissues of 11‐month‐old mice. Gastrocnemius (Gast), myocardium (Myoc), cortex (Cort), hippocampus (Hipp), cerebellum (Cere). (E) Representative western blot images and quantification of the VDAC1 protein in the SN and STR of 11‐month‐old mice. Data are summarized as the mean ± SEM. One‐way ANOVA was performed, followed by LSD post hoc testing. ns, not significant.
Figure S4 Proteomics of HET and WT mice. (A) Volcano plot indicating the altered protein expressions in HET STR compared to that in control STR of mice. (B) Volcano plot indicating the altered protein expressions in HET SN compared to that in control SN of mice. Red and blue dots indicate upregulated and downregulated proteins, respectively (fold‐change >1.2; p < 0.05). (C) KOG function classifications of SN and STR.
Figure S5 Studies on insulin‐metabolism, autophagy, apoptosis, and mtUPR pathways. Representative western blot images of proteins in the SN and STR (left) of 11‐month‐old mice. Quantification of proteins (right), relative to GAPDH, β‐actin or β‐tubulin expression. Data are indicated as the mean ± SEM (n = 3 per genotype). One‐way ANOVA was performed, followed by LSD post hoc testing. ns, not significant.
Table S1 Antibodies used in the present study.
Table S2 Primer sequences used to analyse mouse mtDNA deletions, mtDNA copy number, and RT‐qPCR.
Supplementary Table S3. Differentially expression proteins in the proteomes of the STR and SN of 11‐month‐old mice.
Supplementary Table S4 Differentially expression genes in the RNA‐seq of SHSY5Y cells.
Fan L, Zhang S, Li X, Hu Z, Yang J, Zhang S, et al. CHCHD2 p.Thr61Ile knock‐in mice exhibit motor defects and neuropathological features of Parkinson's disease. Brain Pathology. 2023;33(3):e13124. 10.1111/bpa.13124
Liyuan Fan, Shuo Zhang and Xinwei Li contributed equally to this article.
Contributor Information
Changhe Shi, Email: shichanghe@gmail.com.
Chengyuan Mao, Email: maochengyuan2015@126.com.
Yuming Xu, Email: xuyuming@zzu.edu.cn.
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1. Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. 10.1016/s0896-6273(03)00568-3 [DOI] [PubMed] [Google Scholar]
- 2. Ozansoy M, Başak AN. The central theme of Parkinson's disease: α‐synuclein. Mol Neurobiol. 2013;47:460–5. 10.1007/s12035-012-8369-3 [DOI] [PubMed] [Google Scholar]
- 3. Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol. 2010;23:228–42. 10.1177/0891988710383572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li JQ, Tan L, Yu JT. The role of the LRRK2 gene in parkinsonism. Mol. Neurodegener. 2014;9:47. 10.1186/1750-1326-9-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zeng XS, Geng WS, Jia JJ, Chen L, Zhang PP. Cellular and molecular basis of neurodegeneration in Parkinson disease. Front Aging Neurosci. 2018;10:109. 10.3389/fnagi.2018.00109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Funayama M, Ohe K, Amo T, Furuya N, Yamaguchi J, Saiki S, et al. CHCHD2 mutations in autosomal dominant late‐onset Parkinson's disease: a genome‐wide linkage and sequencing study. Lancet Neurol. 2015;14:274–82. 10.1016/s1474-4422(14)70266-2 [DOI] [PubMed] [Google Scholar]
- 7. Shi, C. H. , Mao C.Y., Zhang S.Y., Yang J., Song B., Wu P., Zuo C.T., Liu Y.T., Ji Y., Yang Z.H., Wu J., Zhuang Z.P., Xu Y.M. CHCHD2 gene mutations in familial and sporadic Parkinson's disease. Neurobiol Aging 38, 217.e219‐217.e213 (2016). 10.1016/j.neurobiolaging.2015.10.040 [DOI] [PubMed] [Google Scholar]
- 8. Liu Y, Clegg HV, Leslie PL, di J, Tollini LA, He Y, et al. CHCHD2 inhibits apoptosis by interacting with Bcl‐x L to regulate Bax activation. Cell Death Differ. 2015;22:1035–46. 10.1038/cdd.2014.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aras S, Bai M, Lee I, Springett R, Hüttemann M, Grossman LI. MNRR1 (formerly CHCHD2) is a bi‐organellar regulator of mitochondrial metabolism. Mitochondrion. 2015;20:43–51. 10.1016/j.mito.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 10. Ikeda A, Nishioka K, Meng H, Takanashi M, Hasegawa I, Inoshita T, et al. Mutations in CHCHD2 cause α‐synuclein aggregation. Hum Mol Genet. 2019;28:3895–911. 10.1093/hmg/ddz241 [DOI] [PubMed] [Google Scholar]
- 11. Liu W, Duan X, Xu L, Shang W, Zhao J, Wang L, et al. Chchd2 regulates mitochondrial morphology by modulating the levels of Opa1. Cell Death Differ. 2020;27:2014–29. 10.1038/s41418-019-0482-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sato S, Noda S, Torii S, Amo T, Ikeda A, Funayama M, et al. Homeostatic p62 levels and inclusion body formation in CHCHD2 knockout mice. Hum Mol Genet. 2021;30:443–53. 10.1093/hmg/ddab057 [DOI] [PubMed] [Google Scholar]
- 13. Nguyen MK, McAvoy K, Liao SC, Doric Z, Lo I, Li H, et al. Mouse midbrain dopaminergic neurons survive loss of the PD‐associated mitochondrial protein CHCHD2. Hum Mol Genet. 2021;31:1500–18. 10.1093/hmg/ddab329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mo J, Enkhjargal B, Travis ZD, Zhou K, Wu P, Zhang G, et al. AVE 0991 attenuates oxidative stress and neuronal apoptosis via Mas/PKA/CREB/UCP‐2 pathway after subarachnoid hemorrhage in rats. Redox Biol. 2019;20:75–86. 10.1016/j.redox.2018.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mao C, Wang H, Luo H, Zhang S, Xu H, Zhang S, et al. CHCHD10 is involved in the development of Parkinson's disease caused by CHCHD2 loss‐of‐function mutation p.T61I. Neurobiol Aging. 2019;75:38–41. 10.1016/j.neurobiolaging.2018.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anderson CJ, Bredvik K, Burstein SR, Davis C, Meadows SM, Dash J, et al. ALS/FTD mutant CHCHD10 mice reveal a tissue‐specific toxic gain‐of‐function and mitochondrial stress response. Acta Neuropathol. 2019;138:103–21. 10.1007/s00401-019-01989-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Genin EC, Madji Hounoum B, Bannwarth S, Fragaki K, Lacas‐Gervais S, Mauri‐Crouzet A, et al. Mitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10(S59L/+) mouse. Acta Neuropathol. 2019;138:123–45. 10.1007/s00401-019-01988-z [DOI] [PubMed] [Google Scholar]
- 18. Ruan Y, Hu J, Che Y, Liu Y, Luo Z, Cheng J, et al. CHCHD2 and CHCHD10 regulate mitochondrial dynamics and integrated stress response. Cell Death Dis. 2022;13:156. 10.1038/s41419-022-04602-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–67. 10.1091/mbc.E12-10-0721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Matos AM, de Macedo MP, Rauter AP. Bridging type 2 diabetes and Alzheimer's disease: assembling the puzzle pieces in the quest for the molecules with therapeutic and preventive potential. Med Res Rev. 2018;38:261–324. 10.1002/med.21440 [DOI] [PubMed] [Google Scholar]
- 21. Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta. 2013;1833:410–6. 10.1016/j.bbamcr.2012.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85:257–73. 10.1016/j.neuron.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cornelissen T, Spinazzi M, Martin S, Imberechts D, Vangheluwe P, Bird M, et al. CHCHD2 harboring Parkinson's disease‐linked T61I mutation precipitates inside mitochondria and induces precipitation of wild‐type CHCHD2. Hum Mol Genet. 2020;29:1096–106. 10.1093/hmg/ddaa028 [DOI] [PubMed] [Google Scholar]
- 24. Wang Y, Wang Z, Sun H, Mao C, Yang J, Liu Y, et al. Generation of induced pluripotent stem cell line (ZZUi007‐a) from a 52‐year‐old patient with a novel CHCHD2 gene mutation in Parkinson's disease. Stem Cell Res. 2018;32:87–90. 10.1016/j.scr.2018.08.011 [DOI] [PubMed] [Google Scholar]
- 25. Chia SJ, Tan EK, Chao YX. Historical perspective: models of Parkinson's disease. Int J Mol Sci. 2020;21. 10.3390/ijms21072464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang X, Wu BP, Nguyen D, Liu YT, Marani M, Hench J, et al. CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Hum Mol Genet. 2018;27:3881–900. 10.1093/hmg/ddy270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tio M, Wen R, Lim YL, Zukifli ZHB, Xie S, Ho P, et al. Varied pathological and therapeutic response effects associated with CHCHD2 mutant and risk variants. Hum Mutat. 2017;38:978–87. 10.1002/humu.23234 [DOI] [PubMed] [Google Scholar]
- 28. Burstein SR, Valsecchi F, Kawamata H, Bourens M, Zeng R, Zuberi A, et al. In vitro and in vivo studies of the ALS‐FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum Mol Genet. 2018;27:160–77. 10.1093/hmg/ddx397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meng H, Yamashita C, Shiba‐Fukushima K, Inoshita T, Funayama M, Sato S, et al. Loss of Parkinson's disease‐associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat Commun. 2017;8:s. 10.1038/ncomms15500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woo JA et al. Loss of function CHCHD10 mutations in cytoplasmic TDP‐43 accumulation and synaptic integrity. Nat Commun. 2017;8:15558. 10.1038/ncomms15558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Imai Y, Inoshita T, Meng H, Shiba‐Fukushima K, Hara KY, Sawamura N, et al. Light‐driven activation of mitochondrial proton‐motive force improves motor behaviors in a drosophila model of Parkinson's disease. Commun. Biol. 2019;2:424. 10.1038/s42003-019-0674-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fearnley, J. M. & Lees, A. J. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain 114 (Pt 5), 2283–2301 (1991). 10.1093/brain/114.5.2283 [DOI] [PubMed] [Google Scholar]
- 33. Chaudhuri KR, Schapira AH. Non‐motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8:464–74. 10.1016/s1474-4422(09)70068-7 [DOI] [PubMed] [Google Scholar]
- 34. Pont‐Sunyer C, Hotter A, Gaig C, Seppi K, Compta Y, Katzenschlager R, et al. The onset of nonmotor symptoms in Parkinson's disease (the ONSET PD study). Mov Disord. 2015;30:229–37. 10.1002/mds.26077 [DOI] [PubMed] [Google Scholar]
- 35. Barone P. Neurotransmission in Parkinson's disease: beyond dopamine. Eur J Neurol. 2010;17:364–76. 10.1111/j.1468-1331.2009.02900.x [DOI] [PubMed] [Google Scholar]
- 36. Chen H, Burton EA, Ross GW, Huang X, Savica R, Abbott RD, et al. Research on the premotor symptoms of Parkinson's disease: clinical and etiological implications. Environ Health Perspect. 2013;121:1245–52. 10.1289/ehp.1306967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barone P. Treatment of depressive symptoms in Parkinson's disease. Eur J Neurol. 2011;18(Suppl 1):11–5. 10.1111/j.1468-1331.2010.03325.x [DOI] [PubMed] [Google Scholar]
- 38. Masini D, Bonito‐Oliva A, Bertho M, Fisone G. Inhibition of mTORC1 signaling reverts cognitive and affective deficits in a mouse model of Parkinson's disease. Front Neurol. 2018;9:208. 10.3389/fneur.2018.00208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xie C, Prasad AA. Probiotics treatment improves hippocampal dependent cognition in a rodent model of Parkinson's disease. Microorganisms. 2020;8. 10.3390/microorganisms8111661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tadaiesky MT, Dombrowski PA, Figueiredo CP, Cargnin‐Ferreira E, da Cunha C, Takahashi RN. Emotional, cognitive and neurochemical alterations in a premotor stage model of Parkinson's disease. Neuroscience. 2008;156:830–40. 10.1016/j.neuroscience.2008.08.035 [DOI] [PubMed] [Google Scholar]
- 41. Carnicella S, Drui G, Boulet S, Carcenac C, Favier M, Duran T, et al. Implication of dopamine D3 receptor activation in the reversion of Parkinson's disease‐related motivational deficits. Transl Psychiatry. 2014;4:e401. 10.1038/tp.2014.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen L, Deltheil T, Turle‐Lorenzo N, Liberge M, Rosier C, Watabe I, et al. SK channel blockade reverses cognitive and motor deficits induced by nigrostriatal dopamine lesions in rats. Int J Neuropsychopharmacol. 2014;17:1295–306. 10.1017/s1461145714000236 [DOI] [PubMed] [Google Scholar]
- 43. Campos FL, Carvalho MM, Cristovão AC, Je G, Baltazar G, Salgado AJ, et al. Rodent models of Parkinson's disease: beyond the motor symptomatology. Front. Behav. Neurosci. 2013;7:175. 10.3389/fnbeh.2013.00175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Drui G, Carnicella S, Carcenac C, Favier M, Bertrand A, Boulet S, et al. Loss of dopaminergic nigrostriatal neurons accounts for the motivational and affective deficits in Parkinson's disease. Mol Psychiatry. 2014;19:358–67. 10.1038/mp.2013.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hollis F, Van der Kooij MA, Zanoletti O, Lozano L, Cantó C, Sandi C. Mitochondrial function in the brain links anxiety with social subordination. Proc Natl Acad Sci U S A. 2015;112:15486–91. 10.1073/pnas.1512653112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Einat H, Yuan P, Manji HK. Increased anxiety‐like behaviors and mitochondrial dysfunction in mice with targeted mutation of the Bcl‐2 gene: further support for the involvement of mitochondrial function in anxiety disorders. Behav Brain Res. 2005;165:172–80. 10.1016/j.bbr.2005.06.012 [DOI] [PubMed] [Google Scholar]
- 47. Filiou MD, Sandi C. Anxiety and brain mitochondria: a bidirectional crosstalk. Trends Neurosci. 2019;42:573–88. 10.1016/j.tins.2019.07.002 [DOI] [PubMed] [Google Scholar]
- 48. Emmerzaal TL, Preston G, Geenen B, Verweij V, Wiesmann M, Vasileiou E, et al. Impaired mitochondrial complex I function as a candidate driver in the biological stress response and a concomitant stress‐induced brain metabolic reprogramming in male mice. Transl Psychiatry. 2020;10:176. 10.1038/s41398-020-0858-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nothdurfter C, Baghai TC, Schüle C, Rupprecht R. Translocator protein (18 kDa) (TSPO) as a therapeutic target for anxiety and neurologic disorders. Eur Arch Psychiatry Clin Neurosci. 2012;262(Suppl 2):S107–12. 10.1007/s00406-012-0352-5 [DOI] [PubMed] [Google Scholar]
- 50. Olsen RH, Johnson LA, Zuloaga DG, Limoli CL, Raber J. Enhanced hippocampus‐dependent memory and reduced anxiety in mice over‐expressing human catalase in mitochondria. J Neurochem. 2013;125:303–13. 10.1111/jnc.12187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gimsa U, Kanitz E, Otten W, Aheng C, Tuchscherer M, Ricquier D, et al. Alterations in anxiety‐like behavior following knockout of the uncoupling protein 2 (ucp2) gene in mice. Life Sci. 2011;89:677–84. 10.1016/j.lfs.2011.08.009 [DOI] [PubMed] [Google Scholar]
- 52. Wang D, Zhai X, Chen P, Yang M, Zhao J, Dong J, et al. Hippocampal UCP2 is essential for cognition and resistance to anxiety but not required for the benefits of exercise. Neuroscience. 2014;277:36–44. 10.1016/j.neuroscience.2014.06.060 [DOI] [PubMed] [Google Scholar]
- 53. Liu X, Wang Q, Yang Y, Stewart T, Shi M, Soltys D, et al. Reduced erythrocytic CHCHD2 mRNA is associated with brain pathology of Parkinson's disease. Acta Neuropathol Commun. 2021;9:37. 10.1186/s40478-021-01133-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schapira AH et al. Mitochondria in the etiology and pathogenesis of Parkinson's disease. Ann Neurol. 1998;44:S89–98. 10.1002/ana.410440714 [DOI] [PubMed] [Google Scholar]
- 55. Kee TR, Espinoza Gonzalez P, Wehinger JL, Bukhari MZ, Ermekbaeva A, Sista A, et al. Mitochondrial CHCHD2: disease‐associated mutations, physiological functions, and current animal models. Front Aging Neurosci. 2021;13:660843. 10.3389/fnagi.2021.660843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ. Soluble, prefibrillar α‐synuclein oligomers promote complex I‐dependent, Ca2+−induced mitochondrial dysfunction. J Biol Chem. 2014;289:21490–507. 10.1074/jbc.M113.545749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. González‐Rodríguez P, Zampese E, Stout KA, Guzman JN, Ilijic E, Yang B, et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature. 2021;599:650–6. 10.1038/s41586-021-04059-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Heni M, Kullmann S, Preissl H, Fritsche A, Häring HU. Impaired insulin action in the human brain: causes and metabolic consequences. Nat Rev Endocrinol. 2015;11:701–11. 10.1038/nrendo.2015.173 [DOI] [PubMed] [Google Scholar]
- 59. Ott V, Benedict C, Schultes B, Born J, Hallschmid M. Intranasal administration of insulin to the brain impacts cognitive function and peripheral metabolism. Diabetes Obes Metab. 2012;14:214–21. 10.1111/j.1463-1326.2011.01490.x [DOI] [PubMed] [Google Scholar]
- 60. Yang YW, Hsieh TF, Li CI, Liu CS, Lin WY, Chiang JH, et al. Increased risk of Parkinson disease with diabetes mellitus in a population‐based study. Medicine. 2017;96:e5921. 10.1097/md.0000000000005921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Steneberg P, Bernardo L, Edfalk S, Lundberg L, Backlund F, Östenson CG, et al. The type 2 diabetes‐associated gene ide is required for insulin secretion and suppression of α‐synuclein levels in β‐cells. Diabetes. 2013;62:2004–14. 10.2337/db12-1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kurochkin IV, Goto S. Alzheimer's beta‐amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994;345:33–7. 10.1016/0014-5793(94)00387-4 [DOI] [PubMed] [Google Scholar]
- 63. Bennett RG, Duckworth WC, Hamel FG. Degradation of amylin by insulin‐degrading enzyme. J Biol Chem. 2000;275:36621–5. 10.1074/jbc.M006170200 [DOI] [PubMed] [Google Scholar]
- 64. Fernández‐Gamba A, Leal MC, Morelli L, Castaño EM. Insulin‐degrading enzyme: structure‐function relationship and its possible roles in health and disease. Curr Pharm Des. 2009;15:3644–55. 10.2174/138161209789271799 [DOI] [PubMed] [Google Scholar]
- 65. Sousa L, Guarda M, Meneses MJ, Macedo MP, Vicente Miranda H. Insulin‐degrading enzyme: an ally against metabolic and neurodegenerative diseases. J Pathol. 2021;255:346–61. 10.1002/path.5777 [DOI] [PubMed] [Google Scholar]
- 66. de Tullio MB, Castelletto V, Hamley IW, Martino Adami PV, Morelli L, Castaño EM. Proteolytically inactive insulin‐degrading enzyme inhibits amyloid formation yielding non‐neurotoxic aβ peptide aggregates. PloS One. 2013;8:e59113. 10.1371/journal.pone.0059113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Grasso G, Lanza V, Malgieri G, Fattorusso R, Pietropaolo A, Rizzarelli E, et al. The insulin degrading enzyme activates ubiquitin and promotes the formation of K48 and K63 diubiquitin. Chem Commun (Camb). 2015;51:15724–7. 10.1039/c5cc06786c [DOI] [PubMed] [Google Scholar]
- 68. Tundo GR, Sbardella D, Ciaccio C, Grasso G, Gioia M, Coletta A, et al. Multiple functions of insulin‐degrading enzyme: a metabolic crosslight? Crit Rev Biochem Mol Biol. 2017;52:554–82. 10.1080/10409238.2017.1337707 [DOI] [PubMed] [Google Scholar]
- 69. Sharma SK, Chorell E, Steneberg P, Vernersson‐Lindahl E, Edlund H, Wittung‐Stafshede P. Insulin‐degrading enzyme prevents α‐synuclein fibril formation in a nonproteolytical manner. Sci Rep. 2015;5:12531. 10.1038/srep12531 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Generation of CHCHD2 p.T61I mutant mice. (A–C) A p.T61I (ACC to ATC codon) mutation was introduced into exon 2 in the 3′‐homology arm. The arrow indicates the C to T mutation in the CHCHD2 gene. (D) Pattern diagram of mouse model construction. The ES cells derived from the clones carrying the desired homologous recombination were injected into C57BL/6 mouse embryos. The chimeric offspring were crossed with C57BL/6 mice to obtain a germline. HET KI mice were interbred to obtain HOM KI and WT mice, which were subsequently genotyped with PCR using primers specific for the experiments.
Figure S2 Pathological staining of the brain, spinal cords, and myocardium. (A–B) Cryosections of different brain regions of mice at 11 months of age were immunostained for p‐α‐synuclein and α‐synuclein. (C) HE‐staining of the brain. No LBs are observed in the STR and hippocampus of p.T61I 11‐month‐old mice. Scale bars = 20 μm. (D) Nissl staining of lumbar spinal cords of mice showing large motor neurons (n = 3 per genotype). Scale bars = 20 μm. ns, not significant. (E) Analysis of the activity of respiratory chain complex I, IV and V in lumbar spinal cord of mice. (n = 3 per genotype). Data are indicated as the mean ± SEM. One‐way ANOVA was performed, followed by LSD post‐hoc testing. ns, not significant. (F) HE‐staining and Masson's Trichrome of the myocardium of mice. Scale bars = 50 μm.
Figure S3 Electron transport chain and mtDNA unaffected by the CHCHD2 p.T61I mutation. (A) Representative western blot images of Drp1, Mfn2, and OPA1 proteins in the SN and STR of 11‐month‐old mice, with quantification of protein intensities relative to GAPDH. n = 3 per genotype. (B) Representative western blot image and quantification of OXPHOS proteins in the SN and STR of 11‐month‐old mice. NDUFB8 (complex I), SDHB (complex II), UQCRC2 (complex III), MTCO2 (complex IV), ATP5A (complex V). n = 3 per genotype. (C) Quantification of mtDNA copy numbers in the tissues of 11‐month‐old mice. Data are expressed as the mtDNA: β‐actin concentration ratio. n = 3 per genotype. (D) Long‐extension PCR of mtDNA (16.3‐kb amplicon) from tissues of 11‐month‐old mice. Gastrocnemius (Gast), myocardium (Myoc), cortex (Cort), hippocampus (Hipp), cerebellum (Cere). (E) Representative western blot images and quantification of the VDAC1 protein in the SN and STR of 11‐month‐old mice. Data are summarized as the mean ± SEM. One‐way ANOVA was performed, followed by LSD post hoc testing. ns, not significant.
Figure S4 Proteomics of HET and WT mice. (A) Volcano plot indicating the altered protein expressions in HET STR compared to that in control STR of mice. (B) Volcano plot indicating the altered protein expressions in HET SN compared to that in control SN of mice. Red and blue dots indicate upregulated and downregulated proteins, respectively (fold‐change >1.2; p < 0.05). (C) KOG function classifications of SN and STR.
Figure S5 Studies on insulin‐metabolism, autophagy, apoptosis, and mtUPR pathways. Representative western blot images of proteins in the SN and STR (left) of 11‐month‐old mice. Quantification of proteins (right), relative to GAPDH, β‐actin or β‐tubulin expression. Data are indicated as the mean ± SEM (n = 3 per genotype). One‐way ANOVA was performed, followed by LSD post hoc testing. ns, not significant.
Table S1 Antibodies used in the present study.
Table S2 Primer sequences used to analyse mouse mtDNA deletions, mtDNA copy number, and RT‐qPCR.
Supplementary Table S3. Differentially expression proteins in the proteomes of the STR and SN of 11‐month‐old mice.
Supplementary Table S4 Differentially expression genes in the RNA‐seq of SHSY5Y cells.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.