

Abstract
Epithelial cell damage-initiated chronic obstructive pulmonary disease (COPD) is implicated in regulated cell death (RCD) including ferroptosis triggered by complex gene-environment interactions. Our data showed that iron overload and ferroptosis are associated with COPD progression in COPD patients and in experimental COPD. Furthermore, we found that, in lung tissues of COPD patients, circSAV1 was associated with COPD progression by circRNA-seq screening. Knockdown of circSAV1 reversed cigarette smoke extract (CSE)-induced ferroptosis. Mechanistically, m6A-modified circSAV1 formed an RNA-protein ternary complex of circSAV1/YTHDF1/IREB2 to facilitate the translation of IREB2 mRNA. Elevated protein levels of IREB2 disrupted iron homeostasis, resulting in accumulation of a labile iron pool (LIP) and lipid peroxidation, which contribute to ferroptosis. Here we demonstrate, by use of an experimental COPD model induced by cigarette smoke (CS), that silencing of circSAV1 and the treatment with deferoxamine (DFO) blocked CS-induced ferroptosis of lung epithelial cells, which attenuated COPD progression in mice. Our results reveal that N6-methyladenosine-modified circSAV1 triggers ferroptosis in COPD through recruiting YTHDF1 to facilitate the translation of IREB2, indicating that circSAV1 is a mediator of ferroptosis and that circSAV1-dependent ferroptosis is a therapeutic target for COPD.
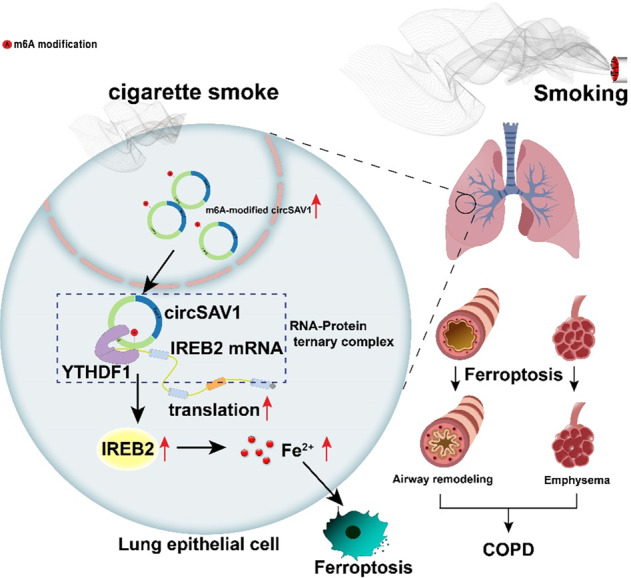
In lung epithelial cell, m6A-modified circSAV1, via recruiting YTHDF1, induces the formation of a circSAV1/YTHDF1/IREB2 mRNA protein ternary complex, which promotes translation of IREB2 mRNA. Further, elevated IREB2 contributes to the accumulation of a labile iron pool (LIP) and lipid peroxidation, then triggers ferroptosis of lung epithelial cells. The ferroptosis of airway epithelial cells and alveolar epithelial cells induces airway remodeling and emphysema, respectively, which causes COPD.
Subject terms: Epigenetics, Respiratory tract diseases, Experimental models of disease, Predictive markers
Introduction
In 2019, Chronic Obstructive Pulmonary Disease (COPD) caused more than 3.23 million deaths, and was ranked as the third leading cause of death [1]. As COPD progresses, patients present with limitation of airflow and narrow airways caused by remodeling of the small airways [2, 3]. Emphysema, caused by destruction of the alveoli, is a pathologic phenotype of COPD [2]. The pathogenesis of COPD is poorly understood but is suggested to be related to dysregulated cellular responses of epithelial cells within the airway and alveolar niches [2].
The development of COPD involves complex interactions between genes and the environment and is associated with an alpha-1 antitrypsin deficiency. Cigarette smoke (CS) remains a major environmental risk factor for COPD [4, 5]. Smoking, biofuel smoke-induced regulated cell death (RCD), and release of damage-associated molecular patterns (DAMPs), which cause inflammatory responses and organ dysfunction, contribute to the pathological process of COPD [6]. Ferroptosis, an iron-dependent, non-apoptotic RCD, is caused by hydroxyl radicals formed by reaction of iron (II)(Fe2+) with peroxides through the Fenton reaction [7, 8]. For mice, a high-iron diet causes systemic inflammation induced by DNA damage and accelerates progression of pancreatitis, a process that is reversed by liproxstatin-1, an inhibitor of ferroptosis [9]. Although ferroptosis is implicated in liver fibrosis [10], traumatic brain injury [11], and acute kidney injury [12], the pathological process of COPD is unclear.
The iron-regulatory proteins (IRPs), IREB1 and IREB2, mediators of intracellular iron homeostasis, bind to iron-response elements (IREs) located on transcripts of ferritin and other iron metabolism-related genes [13]. Moreover, the upregulation of IREB2 in breast cancer cells inhibits ferritin H translation and stabilizes TFR1 mRNA, elevating the intracellular labile iron pool (LIP) [14]. Intracellular free-radical iron initiates lipid peroxidation with the Fenton reaction and causes oxidative damage to lipids, proteins, and nucleic acids [15]. Depletion of IREB1 and IREB2 in fibroblasts blocks the iron-starvation response and reduces sensitivity to ferroptosis [16]. In addition, overexpression of IREB2 in hepatic stellate cells elevates levels of iron and leads to ferroptosis [17]. Lavage samples from smoking patients with COPD contain particulate matter, including iron, which accumulates in lung and triggers oxidative stress and an inflammatory response via a disturbance of iron homeostasis [18]. Thus, we hypothesized that IREB2 regulates iron homeostasis, resulting in elevation of radical iron and leading to ferroptosis in lung epithelial cells.
Circular RNAs (circRNAs), covalently closed circles of RNA, are distinct from miRNAs and lncRNAs in regulating gene expression [19]. In addition to their role as miRNA and protein sponges, circRNAs regulate gene expression by binding specific mRNAs [20, 21]. Some circRNAs are linked to pathological changes and some are emerging as potential therapeutic targets, owing to their tissue-specific expression and stability [19, 22]. In rhabdomyosarcoma cells, circZNF609 binds with CKAP5 mRNA and elevates CKAP5 translation by recruitment of the ELAVL1 protein, promoting cell-cycle progression [21]. In addition, m6A-modified circMET accelerates CDKN2 degradation via recruitment of the YTHDF2 protein and promotes tumor progression [23]. Thus, investigation of the post-transcriptional regulation of circRNAs in COPD is warranted.
Herein, we sought to elucidate the function of ferroptosis in the pathological process of COPD. For human COPD and an experimental COPD mouse model, we demonstrated an elevation of ferroptosis with increased levels of m6A-modified circSAV1. Further, m6A-modified circSAV1 interacted with IREB2 mRNA via complementary base pairing to promote mRNA translation by recruitment of the m6A reader YTHDF1. In lung epithelial cells, accumulation of IREB2 protein elevated the LIP, thus triggering lipid peroxidation and ferroptosis. Thus, our data show that circSAV1 is involved in ferroptosis and highlight a potential target for prevention of COPD development and for treatment.
Results
Iron overload and ferroptosis are associated with COPD progression in COPD patients and in experimental COPD
To determine if ferroptosis is involved in COPD progression, lung tissue samples were collected from non-smokers (n = 8), smokers (n = 8), and COPD smokers (n = 13) (Fig. 1A and Table S1). The results of colorimetric and Perls’ stains demonstrated higher levels of total and ferrous iron in lungs of smokers and COPD smokers (Fig. 1C, D). To evaluate lipid peroxidation for COPD patients, immunoblot and immunohistochemical analyses showed higher levels of 4-hydroxynonenal (4-HNE), a product of lipid peroxidation, in lung epithelial cells of smokers and COPD smokers (Fig. 1B, E). In lung homogenates of smokers and COPD smokers, there were, compared to non-smokers, higher levels of IREB2 and lower levels of the IREB2 target protein FTH1 (Fig. 1B).
Fig. 1. Ferroptosis, an iron homeostasis disorder, relates to COPD progression.

A The clinical information for lung tissue donors. B Representative immunoblot analysis (top) and quantification (bottom) of IREB2, FTH1, and 4-HNE levels in lung tissues of non-smokers, smokers, and COPD smokers (n = 8 per group). C Quantification of total iron (top) and ferrous iron (bottom) in homogenates of lungs of non-smokers, smokers, and COPD smokers (n = 8 per group). D Representative micrographs (left) and quantification (right) of Perls’-stained lung sections of non-smokers, smokers, and COPD smokers (n = 4 per group); scale bars, 100 μm (top), 20 μm (bottom). E Representative micrographs (left) and quantification (right) of 4-HNE immunostaining of lung sections of non-smokers, smokers, and COPD smokers (n = 4 per group); scale bars, 100 μm (top), 20 μm (bottom). F Representative immunoblot analysis (top) and quantification (bottom) of IREB2, FTH1, and 4-HNE levels in lungs of non-smokers (n = 3) and COPD smokers in GOLD stages (n = 2, 4, 4, 3). G Quantification of total iron (top) and ferrous iron (bottom) in lung homogenates of non-smokers (n = 3) and COPD smokers in GOLD stages (n = 2, 4, 4, 3). H Representative micrographs (left) and quantification (right) of Perls’-stained lung sections of non-smokers (n = 3) and COPD smokers in the GOLD stages (n = 2, 4, 4, 3); scale bars, 100 μm (top), 20 μm (bottom). I Representative micrographs (left) and quantification (right) of 4-HNE immunostaining in lung tissues of non-smokers (n = 3) and COPD smokers in GOLD stages (n = 2, 4, 4, 3); scale bars, 100 μm (top), 20 μm (bottom). Throughout, data are means ± SEM. *P < 0.05, **P < 0.01, by Student’s unpaired t-test. #P < 0.05, ##P < 0.01 by one-way analysis of variance with Bonferroni correction. n.s., not significant.
In our investigation of the involvement of ferroptosis in COPD progression, we found that IREB2 accumulated with COPD progression, along with lower expression of FTH1 (Fig. 1F). Notably, the abundance of IREB2 was higher for mice with COPD pathological changes (Fig. S1H). Consistent with the increase of IREB2 with COPD progression, there were, in human lung samples, higher levels of total and ferrous iron (Fig. 1G). Perls’ staining showed more iron deposits in lung sections of COPD patients than in lungs of non-smoker subjects, and iron deposits increased with the development of COPD (Fig. 1H). For an experimental COPD mouse model, we observed higher levels of total and ferrous iron in lungs of mice with more severe COPD pathological changes (Fig. S1E and S1F). Furthermore, with progression of COPD, the 4-HNE levels in lung epithelial cells were higher (Fig. 1F, I). Congruently, 4-HNE was elevated in lung epithelial cells of experimental COPD (Fig. S1G). Thus, these data show that disruption of iron homeostasis and ferroptosis in lung epithelial cells are relevant to COPD, and these factors increased as COPD progressed in COPD patients and in experimental COPD.
The levels of circSAV1 are elevated in lung tissues of smokers and COPD smokers, which relates to COPD progression
Pathological circRNAs are emerging as clinical targets and biomarkers [24]. To determine whether circRNAs are associated with COPD progression, we performed circRNA-Seq analysis of lung samples of healthy human donors with smoking and COPD patients with smoking, compared to healthy human donors without smoking (Fig. 2A). Volcano plots identified 46 differentially expressed circRNAs in lung samples of healthy human donors with smoking, compared with nonsmokers (Fig. 2B). Moreover, there were 68 significantly changed circRNAs in lung tissues of smokers with COPD (Fig. 2B). Six circRNAs were simultaneously up-regulated in lung tissues of smokers and smokers with COPD (Fig. 2C).
Fig. 2. circSAV1 was elevated in lung tissue of smokers and COPD smokers, and related to COPD progression.

A Heatmap showing clusters of circRNAs (top 50) differentially expressed in lung tissues of non-smokers (n = 6), smokers (n = 6), and COPD smokers (n = 10). B Top, volcano plot of dysregulated circRNAs from lung tissues of smokers as compared to lung tissues of nonsmokers. Bottom, volcano plot of dysregulated circRNAs from lung tissue of COPD smokers as compared to lung tissue of nonsmokers. Genes in upper left and right quadrants are significantly differentially expressed. C Venn diagrams of the intersection of significantly differentially expressed circRNAs of smokers as compared to lung tissues of nonsmokers and COPD smokers as compared to lung tissues of nonsmokers. D The levels of circRNAs in lung tissues of non-smokers, smokers, and COPD smokers (n = 8 per group). The sera of nonsmokers (n = 39), smokers (n = 31), and COPD smokers (n = 47) were collected, and the levels of circABCC4 (E), circPHF20 (F), and circSAV1 (G) were determined by qRT-PCR. The sera of non-smokers (n = 39) and COPD smokers at different GOLD stages (n = 15, 12, 13, 7) were collected, and the levels of circABCC4 (H), circPHF20 (I), and circSAV1 (J) were determined by qRT-PCR. Throughout, data are means ± SEM. *P < 0.05, **P < 0.01 by Student’s unpaired t-test. #P < 0.05, ##P < 0.01 by one-way analysis of variance with Bonferroni correction. n.s., not significant.
To understand the relationship between the 6 changed circRNAs and the progression of COPD, we assessed the expression levels of the 6 circRNAs in lung tissues of human subjects. We found that circABCC4 (has_circ_0030586), circPHF20 (has_circ_0007130), and circSAV1 (has_circ_0007101) were higher in lung tissues of COPD patients (Fig. 2D). circABCC4, circPHF20, and circSAV1 were higher in serum of smokers and COPD patients (Fig. 2E, G). Further, Pearson’s correlation analysis showed that serum circPHF20 and circSAV1 levels negatively correlated with FEV1/FVC (%) and with FEV1 predicted, but there was no significant correlation of circABCC4 with FEV1/FVC (%) or FEV1 predicted (Fig. S2A–F). The levels of circSAV1 in serum positively correlated with the GOLD stages of COPD; in contrast, there was no significant correlation of serum circABCC4 and circPHF20 levels with GOLD stages (Figs. 2H–J and S3A–C). These data demonstrate that circSAV1 is differently expressed in lung tissues of smokers and smokers with COPD, and that serum levels of circSAV1 are elevated with COPD progression. Furthermore, these results indicate that circSAV1 is a candidate biomarker for COPD progression.
Targeting of circSAV1 protects lung epithelial cells from ferroptosis by eliminating CSE-induced accumulation of cellular free iron
Single-cell RNA-seq of lung tissue from COPD patients reveals that redox regulation and cellular injury in lung epithelial cells are associated with COPD pathogenesis [25]. To determine a cellular role in CSE-induced cell death, after 48 h of CSE treatment of human bronchial epithelial cells (HBECs), CCK8 assays showed greater CSE-induced cell death of HBECs (Fig. S4A). Pretreatment with the ferroptosis inhibitor, DFO, reduced CSE-induced death of HBECs, but there was a negligible response to the necroptosis inhibitor, necrostatin-1, or the apoptosis inhibitor, Z-VAD-FMK (Fig. S4F). DFO eliminated the CSE-induced elevation of intracellular free iron (Fig. S4G). For HBECs, BODIPY 581/591 assays confirmed that treatment with DFO attenuated CSE-induced lipid peroxidation (Fig. S4H and S4I). Consistent with in vivo results, DFO reversed CSE-induced 4-HNE accumulation (Fig. S4J). DFO exhibited similar effects in CSE-treated BEASE-2B cells (Fig. S5F–J) and alveolar epithelial cells (HPAEpiCs, Fig. S6A–F). These data indicate that, for lung epithelial cells, ferroptosis is involved in CSE-induced cell death.
To elucidate the function of circSAV1 in CSE-induced cell death, we knocked down circSAV1 in CSE-treated BEAS-2B cells (Fig. 3A). Heatmaps identified the expression of groups of genes that were different with different circSAV1 levels (Fig. 3B, C). Analysis of the Gene Ontology database showed that many of the differentially expressed genes were related to regulation of programmed cell death and iron ion binding (Fig. 3D). Analysis of the Kyoto Encyclopedia of Genes and Genomes found that the chemical carcinogenesis-reactive oxygen species pathway was enriched (Fig. 3E). Next, in vitro experiments showed that down-regulation of circSAV1, but not down-regulation of circABCC4 or circPHF20, protected BEAS-2B cells from CSE-induced cell death (Figs. 3F and S7I). Calcein-AM assays showed that low levels of circSAV1 reduced CSE-induced accumulation of LIP (Fig. 3G). Immunofluorescence staining and flow cytometry demonstrated that CSE-induced lipid peroxidation was lower in BEAS-2B cells with circSVA1 knockdown (Fig. 3H, I). In addition, circSAV1 inhibition ameliorated 4-HNE overexpression in BEAS-2B cells treated with CSE (Fig. 3J). Thus, these results indicate that, for lung epithelial cells, circSAV1 is a primary cause of CSE-induced ferroptosis.
Fig. 3. circSAV1 enhances free iron and triggers ferroptosis in CSE-treated BEAS-2B cells.

After BEAS-2B cells were transfected with 20 nM of circSAV1 siRNA-1, circSAV1 siRNA-2, or con siRNA, cells were treated with 0 or 8% CSE for 48 h. A The levels of circSAV1 in BEAS-2B cells were determined by qRT-PCR. B Heatmap displaying the correlation of changed genes in BEAS-2B cells with different circSAV1 levels. C Circos plot of differentially expressed mRNAs (top 100) in BEAS-2B cells with various circSAV1 levels. Bulb map of GO annotations (D) and KEGG (E) enrichment analysis of the significantly dysregulated genes. F Quantification of cell viability by CCK8 assays. G The levels of LIP were measured by the calcein-AM method. H Representative immunofluorescence images obtained with staining with the lipophilic redox-sensitive dye BODIPY581/591 are shown; scale bars, 10 μm. I Left, representative images of lipid peroxidation by flow cytometry using BODIPY581/591. Right, quantification of lipid peroxidation by flow cytometry using BODIPY581/591. J Representative immunoblot analysis (left) and quantification (right) of IREB1, IREB2, FTH1, and 4-HNE expression. Throughout, data are means ± SD, n = 3 independent experiments. *P < 0.05, **P < 0.01 by Student’s unpaired t-test.
A circSAV1/YTHDF1/IREB2 ternary complex facilitates the translation of IREB2 mRNA
To further elucidate the mechanism by which circSAV1 promotes accumulation of free iron and CSE-caused ferroptosis, we focused on the regulatory protein of intracellular iron homeostasis. circSAV1 is derived from exon 2 and exon 3 of the SVA1 gene (Fig. 4A). The back splice junction site in circSAV1 was confirmed by Sanger sequencing (Fig. 4B). PCR assays demonstrated that divergent primers amplified circSAV1 in cDNA reverse-transcribed from random hexamers but not from genomic DNA (Fig. 4C). In line with nuclear and cytoplasmic fractionation analyses (Fig. 4D), fluorescence in situ hybridization (FISH) assays indicated that circSAV1 was predominantly localized in the cytoplasm and that there was low expression in the nucleus (Fig. 4E).
Fig. 4. circSAV1 interacts with YTHDF1 via its m6A motif, which contributes to IREB2 mRNA translation.

A circSAV1 generated from the second and third exon of the SAV1 gene locus. B PCR validation of circSAV1 using an outward-facing primer and Sanger sequencing of the PCR product. C Complementary DNA (cDNA) and genomic DNA (gDNA) of CSE-treated HBECs were used as templates to amplify circSAV1 and SAV1 mRNA, RT-PCR products with divergent primers and convergent primers. D Top, qRT-PCR analysis of cytoplasmic and nuclear RNA of HBECs with the circSAV1 primer. Middle & Bottom, representative immunoblot analysis of cytoplasmic and nuclear proteins of HBECs. E RNA fluorescence in situ hybridization for circSAV1. Cytoplasm was marked with an 18 s probe. Nuclei were stained with DAPI; scale bars, 10 μm. F RNA pulldown assays showing the association of circSAV1 with m6A modifications. Top, circSAV1 pulldown efficiency of the circSAV1 probe shown by RT-PCR. Middle, dot plot representing m6A levels associated with circSAV1. Bottom, methylene blue (MB) stain as loading control. G RIP assays showing the association of circSAV1 with m6A modifications. Top, m6A RIP efficiency of m6A-antibody shown by dot blots. Middle, MB stain as loading control. Bottom, RT-PCR representing circSAV1 levels associated with m6A modifications. H Top, schematic illustration showing the m6A motif located at circSAV1 and the RNA probe for RNA-EMSA assays. Bottom, RNA-EMSA assay showing the binding capacity of purified YTHDF1 with biotin-labeled oligonucleotides containing the m6A motif from circSAV1. I Sequence BLAST analysis showing that circSAV1 directly targets the 5′UTR of IREB2. J Relative luciferase mRNA of the luciferase reporter gene with IREB2-WT or IREB2-Mut in control or circSAV1-knockdown of CSE-treated BEAS-2B cells. K Relative luciferase activity of the luciferase reporter gene with IREB2-WT or IREB2-Mut in control or circSAV1-knockdown CSE-treated BEAS-2B cells. Throughout, data are means ± SD, n = 3 independent experiments. *P < 0.05, **P < 0.01 by Student’s unpaired t-test. n.s., not significant.
In eukaryotic cells, the iron-regulatory proteins, IREB1 and IREB2, are mediators of iron metabolism [13]. For CSE-treated BEAS-2B cells, down-regulation of circSAV1 reduced the elevation of IREB2 protein but not IREB1 (Fig. 3J). For CSE-treated BEAS-2B cells, there was down-regulation of circSAV1with no corresponding change in IREB2 mRNA levels (Fig. S8B). Meanwhile, overexpression of circSAV1 did not reduce IREB2 protein degradation (Fig. S12). SRAMP [26], which predicts sites of m6A modifications, was used to show that these sites are present in circSAV1 (Fig. S10). Consistent with the predicted sites, a circSAV1 pull-down assay and an m6A RNA immunoprecipitation (RIP) assay indicated that circSAV1 contains m6A methylation sites (Fig. 4F, G). Thus, we concentrated on the function of m6A-modified circSAV1 in elevation of the IREB2 protein. YTH N6-methyladenosine RNA binding protein 1 (YTHDF1) preferentially binds to the m6A motif to regulate mRNA translation [27]. RNA-EMSA confirmed that the m6A motif inside circSAV1 is required for interaction with YTHDF1, which recognized m6A modifications (Fig. 4H). Moreover, YTHDF1 RIP assay indicated that YTHDF1 interacts with circSAV1 (Fig. S11).
In cells, circRNAs compete with binding of linear mRNAs, thus altering mRNA translation [19]. RNAInter [28] analysis predicted the binding sites of circSAV1 within the IREB2 mRNA 5’UTR (Fig. 4I). Meanwhile, a circSAV1 pull-down assay indicated that circSAV1 binds with IREB2 mRNA (Fig. S13A). Next, knockdown of circSAV1 inhibited CSE-induced elevation of the luciferase activity of IREB2-WT (Fig. 4K), with no corresponding change in luciferase mRNA levels (Fig. 4J). Furthermore, overexpression of circSAV1 elevated the luciferase activity of IREB2-WT, with no corresponding change in luciferase mRNA levels (Figs. S13B and S13C). These data reveal the epigenetic function of circSAV1 in lung epithelial cells, that is, enhancing the mRNA translation of IREB2 via formation a circSAV1/YTHDF1/IREB2 ternary complex.
circSAV1 drives ferroptosis through IREB2
Given the effects of circSAV1 and IREB2 in vitro, we used pAV-CMV-circSAV1 that overexpressed circSAV1 and IREB2 siRNA, which diminished IREB2 expression in BEAS-2B cells (Fig. 5A, B). Overexpression of circSAV1 increased the levels of IREB2 protein, along with lower expression of FTH1 (Fig. 5C). Next, CCK8 assays showed that knockdown of IREB2 protected BEAS-2B cells from circSAV1-driven ferroptosis (Fig. 5D). Calcein-AM assays showed that low levels of IREB2 reduced circSAV1-induced accumulation of LIP (Fig. 5E). Immunofluorescence staining and flow cytometry demonstrated that circSAV1-induced lipid peroxidation was lower in BEASE-2B cells with IREB2 knockdown (Fig. 5F, G). In addition, IREB2 inhibition ameliorated 4-HNE overexpression in BEAS-2B cells with circSAV1 overexpression (Fig. 5C). These results indicate that, for lung epithelial cells, circSAV1 triggers ferroptosis, at least in part, by increasing IREB2.
Fig. 5. In BEAS-2B cells, circSAV1 triggers ferroptosis via IREB2.

BEAS-2B cells were transfected with OE-circSAV1 or with 20 nM of IREB2 siRNA for 48 h. The levels of circSAV1 (A) and IREB2 (B) in BEAS-2B cells were determined by qRT-PCR. C Representative immunoblot analysis (Top) and quantification (Bottom) of IREB2, FTH1, and 4-HNE expression. D Quantification of cell viability by CCK8 assays. E The levels of LIP were measured by the calcein-AM method. F Representative immunofluorescence images of staining with the lipophilic redox-sensitive dye BODIPY581/591 are shown; scale bars, 10 μm. G Representative images (left) and quantification (right) of lipid peroxidation by flow cytometry with BODIPY581/591. Throughout, data are means ± SD, n = 3 independent experiments. *P < 0.05, **P < 0.01 by Student’s unpaired t-test.
Ferroptosis is triggered in experimental COPD, and DFO treatment shows amelioration of emphysema and airway remodeling
Ferroptosis can be prevented by DFO, an iron chelator, which forms chelates with iron and excretes it, an effect that reduces pathological iron deposits [29]. We determined if regulation of iron in lung tissues of mice altered response to CS exposure in experimental COPD (Fig. 6A). Administration of DFO reduced CS-induced iron overloading (Fig. 6B, C), and 4-HNE levels were lower in lung epithelial cells of experimental COPD mice treated with DFO (Fig. 6D, E). An in vivo study confirmed that administration of DFO protected mice from CS-induced enlarged alveolar spaces (Fig. 6F), thickening of ECM around small airways (Fig. 6G), and elevation of Penh values (Fig. 6H). In summary, these data show, for the experimental COPD model, a relationship between iron overload and lipid peroxidation and demonstrate that ferroptosis is pathogenic in experimental COPD.
Fig. 6. DFO ameliorates emphysema and airway remodeling through inhibiting ferroptosis in experimental COPD.

A Schematic chart for male BALB/c mice exposed to air or to CS (300 mg/m3 TPM) with or without DFO (100 mg/kg) treatment. B Quantification of total iron (top) and ferrous iron (bottom) in lung tissues of mice. C Representative Perls’-stained lung sections (left) and quantification (right); scale bars, 100 μm (top), 20 μm (bottom). D Representative micrographs of lung sections with 4-HNE immunostaining (left) and quantification (right); scale bars, 100 μm (top), 20 μm (bottom). E Representative immunoblot analysis (top) and quantification (bottom) of IREB2, FTH1, and 4-HNE expression in mouse lungs. F Representative H&E-stained lung sections (left) and quantification of Lm (right); scale bars, 100 μm (top), 20 μm (bottom). G Representative Masson-stained lung sections (left) and quantification of the thickness of ECM protein around the small airways (right) of mice; scale bars, 100 μm (top), 20 μm (bottom). H AHR is represented as Penh of mouse response to 0, 12.5, 25, or 50 μg/mL methacholine. Throughout, data are means ± SEM, n = 6 biologically independent mice. *P < 0.05, **P < 0.01 by Student’s unpaired t-test, n.s., not significant.
Reduction of circSAV1 protects against emphysema and airway remodeling via inhibiting ferroptosis in experimental COPD
We next investigated the functional role of circSAV1 in the etiopathogenesis of experimental COPD, using an AAV-circSAV1 shRNA that diminished circSAV1 expression in mice (Fig. 7A–C). In line with in vitro experiments, IREB2 was lower in mice treated with circSAV1 shRNA compared to mice treated with NC shRNA (Fig. 7D). Meanwhile, levels of the IREB2 target protein FTH1 were enhanced (Fig. 7D). In lung tissues of mice, circSAV1 reduction was associated with attenuated iron overloading (Fig. 7E, F). For lung epithelial cells of mice with COPD, silencing of circSAV1 compromised 4-HNE accumulation (Fig. 7D, G). Moreover, circSAV1 reduction protected from CS-induced changes in diameter of alveolar spaces (Fig. 7H), thickening of the ECM around small airways (Fig. 7I), and increases of Penh values (Fig. 7J). These data support the concept that, for experimental COPD, circSAV1 contributes to ferroptosis of lung epithelial cells, to emphysema, and to airway remodeling.
Fig. 7. Downregulated circSAV1 reduces emphysema and airway remodeling through inhibiting ferroptosis in experimental COPD.

A Schematic chart of male BALB/c mice exposed to air or to CS (300 mg/m3 TPM) with or without AAV-circSAV1 shRNA treatment. B The AAV infection efficiency of mouse lung tissues shown in immunofluorescence images; scale bars, 50 μm. C The levels of circSAV1 in lung tissues of mice were determined by qRT-PCR. D Representative immunoblot analysis (left) and quantification (right) of IREB2, FTH1, and 4-HNE levels of mouse lungs. E Quantification of total (top) and ferrous (bottom) iron in lungs of mice. F Representative Perls’-stained lung sections (left) and quantification (right); scale bars, 100 μm (top), 20 μm (bottom). G Representative micrographs of lung sections with 4-HNE immunostaining (left) and quantification (right); scale bars, 100 μm (top), 20 μm (bottom). H Representative H&E-stained lung sections of mice (left) and quantification of Lm (right); scale bars, 100 μm (top), 20 μm (bottom). I Representative Masson-stained lung sections (left) and quantification of the thickness of ECM protein around the small airways (right) of mice; scale bars, 100 μm (top), 20 μm (bottom). J AHR is represented as Penh of mice response to 0, 12.5, 25, or 50 μg/mL methacholine. K Schematic of the major findings of how m6A-modified circSAV1 regulates ferroptosis in COPD by iron overload. Throughout, data are means ± SEM, n = 6 biologically independent mice. *P < 0.05, **P < 0.01 by Student’s unpaired t-test.
Discussion
COPD is an umbrella term for a variety of lung pathologic changes caused by gene-environment interactions. There are no clinically effective therapeutics for preventing the pathologic changes of COPD in airway and alveolar cells, but the impact of environmental risk factors can be probed by examination of epigenetic modifications [2, 30]. An understanding of the epigenetic mechanisms underlying COPD would be valuable for identifying new pharmacological treatments. Herein, we demonstrate that enhanced circSAV1 is related to COPD pathogenesis. By using an experimental COPD model, we established that circSAV1 promotes ferroptosis in lung epithelial cells, which contributes to COPD. Thus, we found, in lung epithelial cells, a functional interaction between circSAV1 and IREB2 mRNA. Overexpression of circSAV1 promotes the translation of IREB2 mRNA via m6A modifications to form a circSAV1/YTHDF1/IREB2 ternary complex that leads to iron overloading and ferroptosis. Moreover, serum circSAV1 is promising as a clinical biomarker for COPD.
circRNAs, as covalently closed loop RNAs, are distinct from linear RNAs in structure, stability, and immunogenicity, and they have clinical potential as noninvasive biomarkers and as drugs for treatment of pulmonary diseases [19, 31]. Although circRNAs are being explored, their roles in COPD progression remain poorly understood. In the present study, circRNA-seq revealed differentially expressed circRNAs in lung tissues of COPD patients. Serum levels of circSAV1 were higher in more severe GOLD stages, and negatively correlated with FEV1/FVC % and FEV1 predicted. Moreover, examination of the mouse model of experimental COPD revealed that diminished circSAV1 expression prevented COPD progression. RNA-based therapeutics have potential to target presently undruggable genes and to present a new paradigm for treatment of various diseases [32]. Small-interfering (siRNAs) and microRNAs (miRNAs) can be used to target protein-coding mRNAs considered to be involved in the pathophysiology of disease progression [31]. Herein, we used AAV-circSAV1 shRNA to reduce elevation of circSAV1 in experimental COPD and to mitigate the physiopathology of experimental COPD. The present results support the concepts that circSAV1 is a promising therapeutic target and could serve as a biomarker for diagnosis of COPD.
m6A is a transcriptional modification of mRNAs and ncRNAs [33]. m6A modifications affect the biological functions of circRNAs [34]. Our previous study showed that enhanced m6A methylation is involved in development of emphysema [35]. In lung tissues of mice exposed to CS, m6A levels are elevated. Enhancement of m6A modifications, by increasing iron uptake by head and neck squamous cell carcinomas, promote cell proliferation [36]. Herein, we observed that reducing m6A levels in experimental COPD alleviated iron accumulation and lipid peroxidation. Additionally, we found, for CSE-treated lung epithelial cells, that elevated m6A modifications were associated with production of IREB2 protein; post-transcriptional regulation via m6A modification may account for this. The m6A motif can regulate RNA splicing, translation, stabilization, and translocation; these effects on RNA are attributed to diverse m6A readers [33].
YTH family members regulate mRNAs in an m6A-dependent manner. YTHDF1 promotes mRNA translation by binding to the 5′ UTR m6A motif, and YTHDF3 interacts with YTHDF2 to promote the degradation of targeted mRNAs [33, 37]. In mouse brain, YTHDF1 accelerates the translational efficiency of m6A-modified neuronal mRNAs, thereby contributing to learning and memory [38]. Establishment of the biological role of YTHDF1 in lung tissue contributes to a better understanding of the effects of m6A modification during COPD progression. In bronchial epithelial cells, the elevation of IREB2 protein is due to the high YTHDF levels. SRAMP [26] analysis, which predicts m6A sites, suggested that the m6A-motif was not present in the 5’ UTR of IREB2 mRNA (Fig. S10B). We also demonstrated that, in bronchial epithelial cells exposed to CSE, a circSAV1 deficiency down-regulates expression of the IREB2 protein. We established a mechanism by which m6A-modified circSAV1 promoted the translation of IREB2 mRNA. These results show that, for COPD, elevated m6A modifications contribute to recruitment of YTHDF1 to circSAV1. Further, circSAV1 binds to the 5’ UTR of IREB2 mRNA by complementary base pairing, resulting in the formation of a circSAV1/YTHDF1/IREB2 ternary complex. By forming the complex, circSAV1 facilitates the translation of IREB2 mRNA to promote COPD progression.
IREB2 is necessary for maintenance of intracellular iron metabolism [39]. For mice overexpressing Irp2, excessive iron accumulation is associated with embryonic death [40]. UK Biobank GWAS evaluation of genetic associations shows that IREB2 may be involved in tobacco-related respiratory diseases, cardiovascular diseases, and lung cancer [41]. In addition, Cloonan and colleagues show that Ireb2-/- mice resist CS-induced experimental COPD [42]. In line with a previous study showing that CS exposure induces iron accumulation in lungs of COPD patients [18], our data showed that, for smoking COPD patients, iron deposited in lung epithelial cells was associated with elevation of IREB2 protein. Such iron overloading is in line with a previous observation that systemic inflammation in COPD is responsible for enhanced serum hepcidin, an iron-regulating peptide hormone made in the liver [43]. We found that elevated IREB2 in lung epithelial cells enhanced LIP.
Ferroptosis, a specialized form of regulated cell death, is accomplished through lethal lipid peroxidation [44]. Iron overloading acts as a trigger in the induction of ferroptosis [45]. Herein, our data demonstrated accumulation of lipid peroxides in COPD patients with iron deposition. Experiments with cultured cells showed that inhibition of ferroptosis by DFO attenuated CSE-induced death of lung epithelial cells as assessed by analysis of their viability. A study with mice showed the effectiveness of iron chelators in experimental COPD. DFO, an iron chelator that targets intracellular LIP, is used in the therapy for thalassemia major and sideroblastic anemia and is being evaluated in clinical trials [46, 47]. Thus, we provide a therapeutic strategy for the prevention of COPD via targeting aberrant iron deposition.
In summary, the present study revealed that circSAV1 is a pathogenic driver of COPD. For COPD patients and for an experimental COPD model, m6A-modified circSAV1 via recruiting YTHDF1 promotes IREB2 mRNA translation through forming an mRNA-protein ternary complex of circSAV1/YTHDF1/IREB2. Furthermore, elevated levels of IREB2 protein contribute to accumulation of LIP and trigger ferroptosis of lung epithelial cells. The ferroptosis of airway epithelial cells and alveolar epithelial cells induce airway remodeling and emphysema, respectively, which caused COPD (Fig. 7K). Thus, our results reveal a mechanism by which N6-methyladenosine-modified circSAV1 triggers ferroptosis in COPD through recruiting YTHDF1 to facilitate the translation of IREB2, which indicates that circSAV1 is a mediator of ferroptosis and that circSAV1-dependent ferroptosis is a therapeutic target for COPD.
Materials and methods
Further details of materials and methods are described in supplementary information
Human samples
Lung tissues of non-smokers, smokers, and COPD smokers were obtained from lobectomies for benign lung nodules or from lung transplants at Wuxi People’s Hospital. Human sera were obtained from medical examinations at Wuxi People’s Hospital. Informed consent was obtained from all human donors, and the protocol for human research was approved by the ethical committee of Nanjing Medical University (number: 2021-130). Physician diagnoses of COPD followed guidelines of the Global Initiative for Chronic Obstructive Lung Disease. All human samples were anonymized before use. The clinical information for serum donors and for human lung tissue donors is shown in Table 1.
Table 1.
Clinical information for serum donors.
| Non-smokers | Smokers | COPD smokers | |
|---|---|---|---|
| Number | 39 | 31 | 47 |
| Sex, male | 37 (94.9%) | 30 (96.8%) | 43 (91.5%) |
| Age, years | 61.1 ± 10.2 | 62.3 ± 8.0 | 66.0 ± 7.6* |
| BMI | 25.2 ± 2.9 | 24.8 ± 2.9 | 23.4 ± 4.2* |
| GOLD status, I/II/III/IV | - | - | 15/12/13/7 |
| Smoking history, pack-years | - | 30.4 ± 15.5 | 41.5 ± 32.9# |
| FEV1 (%) predicted | 92.6 ± 14.4 | 81.4 ± 19.8* | 59.5 ± 25.8**,## |
| FEV1/FVC (%) | 85.2 ± 7.9 | 82.7 ± 8.4 | 53.0 ± 12.0**,## |
Data are presented as means ± SD, unless otherwise stated.
Descriptive data for the intervention and comparison groups were compared using the chi-square test for categorical variables and ANOVA for quantitative data.
*P < 0.05 versus non-smokers, **P < 0.01 versus non-smokers.
#P < 0.05 versus smokers, ##P < 0.01 versus smokers.
Animals
BALB/c mice (male, 6-8-week-old) were ordered from the Animal Core Facility of Nanjing Medical University and housed under SPF at the Safety Assessment and Research Center for Drug, Pesticide and Veterinary Drug of Jiangsu Province. All experiments with animals were reviewed and approved by the animal ethics committee of Nanjing Medical University (IACUC-1907025).
Cell cultures
Primary human bronchial epithelial cells (HBECs) were obtained from healthy donors by fiberoptic bronchoalveolar lavage. Isolated HBECs were maintained in bronchial epithelial growth medium (BEGM, Lonza, CC-3170) and seeded on culture vessels coated with rat-tail collagen I (10 μg/ml) under 5% CO2 at 37 °C. The medium was changed every three days. For experiments, HBECs were used within passage 5.
Human Pulmonary Alveolar Epithelial Cells (HPAEpiCs), ordered from ScienCell Research Laboratories, were maintained in Alveolar Epithelial Cell Medium (AEpiCM, ScienCell, #3201) and seeded on culture vessels coated with poly-L-lysine stock solution (2 μg/ml) under 5% CO2 at 37 °C. The medium was changed every three days. All HPAEpiCs were used within passage 5.
BEAS-2B normal human bronchial epithelium cells, obtained from the American Type Culture Collection (ATCC), were maintained in RPMI-1640 (Life Technologies/Gibco, C11875500BT) containing 10% fetal bovine serum (FBS, Life Technologies/Gibco, 10099141C) and Penicillin-Streptomycin (Beyotime, C0222) under 5% CO2 at 37 °C. Cells were passaged at a ratio 1:3 every two days.
Statistical analyses
Data were expressed as the means ± SEM or SD taken from at least three independent experiments. Statistical analyses were performed using GraphPad Prism 9 software (Graphpad Software Inc., San Diego, CA), with P < 0.05 considered as significant. Statistical differences were determined by an unpaired Student’s t test or by one-way analysis of variance with Bonferroni correction. Pearson’s correlations were calculated between two parameters.
Supplementary information
Acknowledgements
The authors thank Donald L. Hill (University of Alabama at Birmingham, USA), an experienced, English-speaking scientific editor, for editing. This work was supported by the Natural Science Foundations of China (81973085, 82173472, 82173563, 81973005); and the Priority Academic Program Development of Jiangsu Higher Education Institutions (2022); and the Top Talent Support Program for young and middle-aged people of Wuxi Health Committee (BJ2020006).
Author contributions
QL and TB developed the hypothesis. TB and YW provided the clinical samples. HX, YW and JL performed the RNA-seq. HX, JZ, JL, CC, YY, LL and QX performed the in vivo experiments. HX, JZ and CC performed the in vitro experiments. QL and HX wrote the manuscript.
Data availability
The data that support the findings of this study are available from the corresponding authors on reasonable request.
Competing interests
The authors declare no competing interests.
Ethics statement
All human research was approved by the ethical committee of Nanjing Medical University (number: 2021-130). All experiments with animals were reviewed and approved by the animal ethics committee of Nanjing Medical University (IACUC-1907025).
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Haibo Xia, Yan Wu, Jing Zhao.
Contributor Information
Tao Bian, Email: biantaophd@126.com.
Qizhan Liu, Email: drqzliu@hotmail.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-023-01138-9.
References
- 1.Diseases GBD, Injuries C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1204–22. doi: 10.1016/S0140-6736(20)30925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang C, Zhou J, Wang J, Li S, Fukunaga A, Yodoi J, et al. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther. 2020;5:248. doi: 10.1038/s41392-020-00345-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuhn C, 3rd, Homer RJ, Zhu Z, Ward N, Flavell RA, Geba GP, et al. Airway hyperresponsiveness and airway obstruction in transgenic mice. Morphologic correlates in mice overexpressing interleukin (IL)-11 and IL-6 in the lung. Am J Respir Cell Mol Biol. 2000;22:289–95. doi: 10.1165/ajrcmb.22.3.3690. [DOI] [PubMed] [Google Scholar]
- 4.Collaborators GBDT. Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and attributable disease burden in 204 countries and territories, 1990-2019: a systematic analysis from the Global Burden of Disease Study 2019. Lancet. 2021;397:2337–60. doi: 10.1016/S0140-6736(21)01169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halpin DMG, Criner GJ, Papi A, Singh D, Anzueto A, Martinez FJ, et al. Global Initiative for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease. The 2020 GOLD Science Committee Report on COVID-19 and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2021;203:24–36. doi: 10.1164/rccm.202009-3533SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10:3145. doi: 10.1038/s41467-019-10991-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–81. doi: 10.1080/15548627.2020.1810918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu M, Tao J, Yang Y, Tan S, Liu H, Jiang J, et al. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis. 2020;11:86. doi: 10.1038/s41419-020-2299-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai E, Han L, Liu J, Xie Y, Zeh HJ, Kang R, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11:6339. doi: 10.1038/s41467-020-20154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu A, Feng B, Yu J, Yan L, Che L, Zhuo Y, et al. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021;46:102131. doi: 10.1016/j.redox.2021.102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rui T, Wang H, Li Q, Cheng Y, Gao Y, Fang X, et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J Pineal Res. 2021;70:e12704. doi: 10.1111/jpi.12704. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Quan F, Cao Q, Lin Y, Yue C, Bi R, et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J Adv Res. 2021;28:231–43. doi: 10.1016/j.jare.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004;23:386–95. doi: 10.1038/sj.emboj.7600041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Deng Z, Hatcher H, Miller LD, Di X, Tesfay L, et al. IRP2 regulates breast tumor growth. Cancer Res. 2014;74:497–507. doi: 10.1158/0008-5472.CAN-13-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He YJ, Liu XY, Xing L, Wan X, Chang X, Jiang HL. Fenton reaction-independent ferroptosis therapy via glutathione and iron redox couple sequentially triggered lipid peroxide generator. Biomaterials. 2020;241:119911. doi: 10.1016/j.biomaterials.2020.119911. [DOI] [PubMed] [Google Scholar]
- 16.Terzi EM, Sviderskiy VO, Alvarez SW, Whiten GC, Possemato R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv. 2021; 7: eabg4302 [DOI] [PMC free article] [PubMed]
- 17.Li Y, Jin C, Shen M, Wang Z, Tan S, Chen A, et al. Iron regulatory protein 2 is required for artemether -mediated anti-hepatic fibrosis through ferroptosis pathway. Free Radic Biol Med. 2020;160:845–59. doi: 10.1016/j.freeradbiomed.2020.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Ghio AJ, Hilborn ED, Stonehuerner JG, Dailey LA, Carter JD, Richards JH, et al. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am J Respir Crit Care Med. 2008;178:1130–8. doi: 10.1164/rccm.200802-334OC. [DOI] [PubMed] [Google Scholar]
- 19.Liu CX, Chen LL. Circular RNAs: Characterization, cellular roles, and applications. Cell. 2022;185:2016–34. doi: 10.1016/j.cell.2022.04.021. [DOI] [PubMed] [Google Scholar]
- 20.Gil N, Ulitsky I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat Rev Genet. 2020;21:102–17. doi: 10.1038/s41576-019-0184-5. [DOI] [PubMed] [Google Scholar]
- 21.Rossi F, Beltran M, Damizia M, Grelloni C, Colantoni A, Setti A, et al. Circular RNA ZNF609/CKAP5 mRNA interaction regulates microtubule dynamics and tumorigenicity. Mol Cell. 2022;82:75–89:e79. doi: 10.1016/j.molcel.2021.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guarnerio J, Bezzi M, Jeong JC, Paffenholz SV, Berry K, Naldini MM, et al. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell. 2016;165:289–302. doi: 10.1016/j.cell.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Chen Y, Liu N, Lu Y, Ma W, Yang Z, et al. CircMET promotes tumor proliferation by enhancing CDKN2A mRNA decay and upregulating SMAD3. Mol Cancer. 2022;21:23. doi: 10.1186/s12943-022-01497-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Misir S, Wu N, Yang BB. Specific expression and functions of circular RNAs. Cell Death Differ. 2022;29:481–91. doi: 10.1038/s41418-022-00948-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sauler M, McDonough JE, Adams TS, Kothapalli N, Barnthaler T, Werder RB, et al. Characterization of the COPD alveolar niche using single-cell RNA sequencing. Nat Commun. 2022;13:494. doi: 10.1038/s41467-022-28062-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Y, Zeng P, Li YH, Zhang Z, Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91. doi: 10.1093/nar/gkw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin D, Guo J, Wu Y, Du J, Yang L, Wang X, et al. m(6)A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2019;12:135. doi: 10.1186/s13045-019-0830-6. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Kang J, Tang Q, He J, Li L, Yang N, Yu S, et al. RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucl Acids Res. 2022;50:D326–32. doi: 10.1093/nar/gkab997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49. doi: 10.1038/s41392-020-00428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benincasa G, DeMeo DL, Glass K, Silverman EK, Napoli C. Epigenetics and pulmonary diseases in the horizon of precision medicine: a review. Eur Respir J. 2021;57:2003406 [DOI] [PubMed]
- 31.Mei D, Tan WSD, Tay Y, Mukhopadhyay A, Wong WSF. Therapeutic RNA Strategies for Chronic Obstructive Pulmonary Disease. Trends Pharmacol Sci. 2020;41:475–86. doi: 10.1016/j.tips.2020.04.007. [DOI] [PubMed] [Google Scholar]
- 32.Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35:222–9. doi: 10.1038/nbt.3802. [DOI] [PubMed] [Google Scholar]
- 33.Kumari R, Ranjan P, Suleiman ZG, Goswami SK, Li J, Prasad R, et al. mRNA modifications in cardiovascular biology and disease: with a focus on m6A modification. Cardiovasc Res. 2021;118:1680–92. [DOI] [PMC free article] [PubMed]
- 34.Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. 2019;10:4695. doi: 10.1038/s41467-019-12651-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia H, Wu Y, Zhao J, Li W, Lu L, Ma H, et al. The aberrant cross-talk of epithelium-macrophages via METTL3-regulated extracellular vesicle miR-93 in smoking-induced emphysema. Cell Biol Toxicol. 2022;38:167–83. doi: 10.1007/s10565-021-09585-1. [DOI] [PubMed] [Google Scholar]
- 36.Ye J, Wang Z, Chen X, Jiang X, Dong Z, Hu S, et al. YTHDF1-enhanced iron metabolism depends on TFRC m(6)A methylation. Theranostics. 2020;10:12072–89. doi: 10.7150/thno.51231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5’ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi H, Zhang X, Weng YL, Lu Z, Liu Y, Lu Z, et al. m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature. 2018;563:249–53. doi: 10.1038/s41586-018-0666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–14. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 40.Moroishi T, Nishiyama M, Takeda Y, Iwai K, Nakayama KI. The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab. 2011;14:339–51. doi: 10.1016/j.cmet.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 41.Dong G, Feng J, Sun F, Chen J, Zhao XM. A global overview of genetically interpretable multimorbidities among common diseases in the UK Biobank. Genome Med. 2021;13:110. doi: 10.1186/s13073-021-00927-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabon MA, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22:163–74. doi: 10.1038/nm.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tandara L, Grubisic TZ, Ivan G, Jurisic Z, Tandara M, Gugo K, et al. Systemic inflammation up-regulates serum hepcidin in exacerbations and stabile chronic obstructive pulmonary disease. Clin Biochem. 2015;48:1252–7. doi: 10.1016/j.clinbiochem.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 44.Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Li X, Ge C, Min J, Wang F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022;29:467–80. doi: 10.1038/s41418-022-00941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glickstein H, El RB, Shvartsman M, Cabantchik ZI. Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood. 2005;106:3242–50. doi: 10.1182/blood-2005-02-0460. [DOI] [PubMed] [Google Scholar]
- 47.Breuer W, Ermers MJ, Pootrakul P, Abramov A, Hershko C, Cabantchik ZI. Desferrioxamine-chelatable iron, a component of serum non-transferrin-bound iron, used for assessing chelation therapy. Blood. 2001;97:792–8. doi: 10.1182/blood.V97.3.792. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors on reasonable request.